

Abstract
Introduction of oxygen into aromatic C–H bonds is intriguing from both fundamental and practical perspectives. Although the 3d metal-catalyzed hydroxylation of arenes by H2O2 has been developed by several prominent researchers, a definitive mechanism for these crucial transformations remains elusive. Herein, density functional theory calculations were used to shed light on the mechanism of the established hydroxylation reaction of benzene with H2O2, catalyzed by [NiII(tepa)]2+ (tepa = tris[2-(pyridin-2-yl)ethyl]amine). Dinickel(III) bis(μ-oxo) species have been proposed as the key intermediate responsible for the benzene hydroxylation reaction. Our findings indicate that while the dinickel dioxygen species can be generated as a stable structure, it cannot serve as an active catalyst in this transformation. The calculations allowed us to unveil an unprecedented mechanism composed of six main steps as follows: (i) deprotonation of coordinated H2O2, (ii) oxidative addition, (iii) water elimination, (iv) benzene addition, (v) ketone generation, and (vi) tautomerization and regeneration of the active catalyst. Addition of benzene to oxygen, which occurs via a radical mechanism, turns out to be the rate-determining step in the overall reaction. This study demonstrates the critical role of Ni-oxyl species in such transformations, highlighting how the unpaired spin density value on oxygen and positive charges on the Ni–O• complex affect the activation barrier for benzene addition.
Short abstract
During the Ni(II)-catalyzed hydroxylation process of benzene to phenol by H2O2, although dinickel dioxygen species can exist as stable structures, they cannot function as an active catalyst. DFT calculations reveal that the rate-determining step in this transformation is the addition of benzene to oxygen, which necessitates Ni–O species with high oxyl character and a significant positive charge.
Introduction
Phenol and its derivatives are a valuable class of chemical precursors used in the synthesis of various industrial products, including dyes, phenol-formaldehyde resins, bisphenol A, caprolactam, pharmaceuticals, medicines, polymers, and raw materials for numerous chemicals.1 The current industrial process for phenol production relies on three-step cumene methods, which involve the propylation of benzene or toluene, autoxidation to obtain the cumene hydroperoxide derivative, and Hock rearrangement. These reactions require a high temperature, high pressure, and strongly acidic conditions, resulting in the production of acetone as a byproduct in equal quantities during the final step. Furthermore, the overall yield of phenol from benzene is disappointingly low, reaching only 5%.2,3 Therefore, the industry desires a simpler and more efficient process that can be conducted under milder conditions. In particular, there has been considerable research focused on developing a direct aromatic oxygenation reaction using cost-effective and environmentally benign oxidants such as O2 and H2O2.4−19 Even so, introducing hydroxyl functionality into arenes using these oxidants presents challenges due to the modest reactivity of aromatic C–H bonds and the tendency for phenols to overoxidize, ultimately resulting in a diminished reaction efficiency.20−22
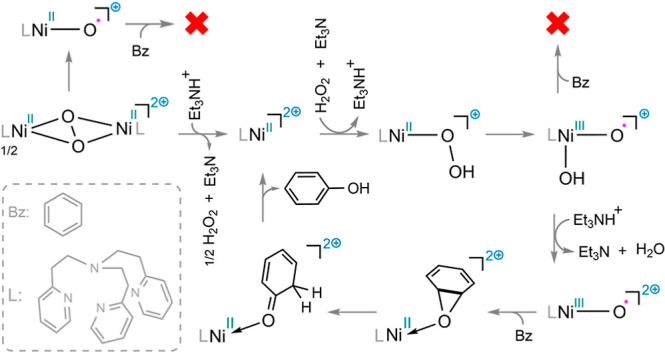
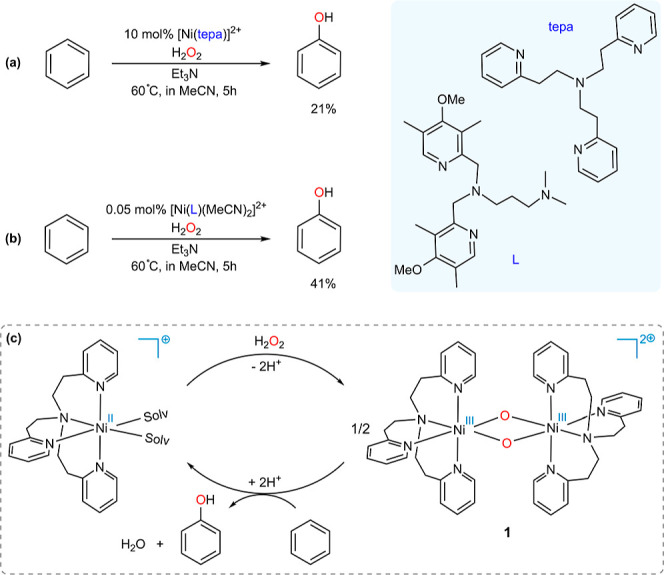
In 2015, Itoh and co-workers14 reported a remarkable study in which they described the catalytic ability of nickel(II) complexes supported by pyridylalkylamine ligands as homogeneous catalysts for the direct hydroxylation of benzene to phenol using H2O2 as the oxidant (Scheme 1a). The same approach was executed by Mayilmurugan and co-workers employing similar ligands (Scheme 1b).10 Both research groups suggested that a dinickel(III) bis(μ-oxo) species serves as an active catalyst in the hydroxylation process of benzene and its derivatives (Scheme 1c).
Scheme 1. Nickel-Catalyzed Hydroxylation of Benzene Using H2O2 Developed by (a) Itoh et al. and (b) Mayilmurugan et al. (c) Proposed Catalytic Mechanism by Itoh and Co-workers.
Although Itoh and other researchers have demonstrated that dinickel(III) bis(μ-oxo) complexes can form under such conditions,23−26 there is no experimental or computational support for their involvement in the hydroxylation of benzene. It is noteworthy that while a few instances of intramolecular oxidation involving aromatic groups embedded within the supporting ligands of dinickel(III) bis(μ-oxo) species have been reported,23,27,28 there are no known cases of intermolecular oxidation of benzene derivatives occurring through such a species. Moreover, in 2018, Itoh and co-workers reported the synthesis of the [NiIII-(dpema)2(O)2]2+ complex (dpema:N,N-(di-[2-pyridine-2-yl]ethyl)methylamine), which exhibited oxygenation reactivity toward external substrates but not aromatic oxidation.29
This outstanding oxidation system prompted us to conduct a comprehensive density functional theory (DFT) study to investigate the mechanistic aspects of benzene hydroxylation utilizing H2O2 as the oxidant and this particular type of Ni(II) complex as a catalyst. Elucidating the mechanism underlying this hydroxylation reaction can enhance our knowledge of the processes involved in other transition metal-catalyzed hydroxylation reactions of aromatic substrates using H2O2.4,5,7,9,11,12,30−37 To do so, we selected the [NiII(tepa)]2+ complex based on the study by Itoh et al., which has demonstrated its superior efficiency over other analogous nickel complexes supported by pyridylalkylamine ligands.14
Results and Discussion
Preliminary Evaluation of the Mechanism Proposed by Itoh et al.
As discussed in the Introduction, the dinickel(III) bis(μ-oxo) complex has been surmised to be the key intermediate responsible for the catalytic hydroxylation of benzene (Scheme 1c). This species can be produced when hydrogen peroxide is added to the two molecules of [NiII(tepa)]2+. Hence, we commenced our mechanistic inquiry by assessing the capability of 1 to oxidize benzene. It should be pointed out that side-on dioxygen 3d metal complexes commonly adopt either a bis(μ-oxo)24,26,29,38−43 or μ–η2:η2-peroxo isomeric form.44−47 In the present case, the μ–η2:η2-peroxo form (s2), which is an antiferromagnetic substance with two spin-up electrons on one nickel atom and two spin-down electrons on the other nickel atom, was found to exhibit greater stability than that of the bis(μ-oxo) form (1) (see Supporting Information for details).
As depicted in Figure 1a,48 the substitution reaction between benzene and s2, yielding (tepa)Ni–O–Bz (Bz = benzene) and (tepa)Ni–O species, exhibits a pronounced thermodynamic unfavorability, with a ΔGrxn = 36.8 kcal/mol, rendering it infeasible. Similarly, an alternate pathway for the substitution reaction leading to the production of (tepa)Ni–O–O-Bz and (tepa)Ni is extremely endothermic at ΔGrxn = 57.4 kcal/mol (Figure 1b). Also, the addition of benzene to oxygen without the full cleavage of s2 is highly unfavorable by ΔGrxn = 47.5 kcal/mol, suggesting that it is unlikely (Figure 1c). On the basis of these results, it can be deduced that complex s2 is inactive toward the oxidation of benzene due to the energetically unfavorable nature of the substitution reactions involving benzene.
Figure 1.

DFT calculated reaction potential pathways at the SMD/B3LYP-D3/def2-TZVP//SMD/B3LYP-D3/6-31G(d),SDD level of theory for the (a) and (b) substitution reactions between benzene and s2 and (c) addition of benzene to s2. The most stable ground state for each structure has been taken into account to calculate the free energies of the reactions. The superscripts “s”, “d”, “q”, and “qnt” represent the singlet, doublet, quartet, and quintet ground states, respectively. Free energies are given in kcal/mol (in blue).
Other Potential Pathways for Benzene Hydroxylation Starting from [NiII(tepa)]2+
We then extended our investigation by exploring potential mechanistic pathways underlying benzene hydroxylation, which are discussed in the following. [NiII(tepa)]2+ (t8), an octahedral complex with two vacant coordination sites, is determined to have a slightly lower free energy than those with one or two coordinated molecules of acetonitrile as the solvent (see Supporting Information for details). Here, commencing from t8, we examine potential pathways for benzene hydroxylation:
-
A
Oxidative addition of H2O2 to [NiII(tepa)]2+: The Ni(IV) species is expected to exhibit a higher oxidation potential for driving benzene hydroxylation compared to that of Ni(II). However, to proceed with the oxidative addition of H2O2 to the Ni center, an activation energy barrier of 35.0 kcal/mol must be overcome, which is inaccessible (path i in Figure 2).
-
B
Nucleophilic attack on coordinated H2O2: The nucleophilic attack of benzene on coordinated H2O2 in t9 is concerted with the breaking of the O–O bond, leading to the formation of carbocation (11) alongside Ni(II) species (t12). The generated carbocation, due to its high acidity, would readily undergo proton donation to convert into phenol. Nevertheless, the calculations demonstrate that this pathway is impeded by a 36.5 kcal/mol activation energy barrier (path ii in Figure 2).
Figure 2.

Calculated reaction pathways for oxidative addition of H2O2 to [NiII(tepa)]2+ (path i); nucleophilic attack on coordinated H2O2 by benzene (path ii) and [NiII(tepa)]2+ (path iii). The superscripts “s”, “d”, “t”, and “qnt” represent the singlet, doublet, triplet, and quintet ground states, respectively. Free energies calculated at the SMD/B3LYP-D3/def2-TZVP//SMD/B3LYP-D3/6-31G(d),SDD level of theory are given in kcal/mol (in red).
We also examined an alternative route in which t8 undergoes nucleophilic attack on coordinated H2O2 instead of benzene (path iii in Figure 2). Nonetheless, the transition structure for this transformation lies 38.7 kcal/mol above the reactants. It follows that this activation barrier is high enough to hinder the progression of the reaction.
Catalytic Process Uncovered by DFT Calculations
Generation of Oxyl
In the wake of the aforementioned results, we have once again been compelled to seek a different route for this process. Triethylamine (Et3N) is added to the reaction mixture during the hydroxylation of benzene and its derivatives.14 According to our calculations, when complex t9 is treated with Et3N, it can undergo deprotonation in a barrierless manner, resulting in the production of t14 with a relative free energy of −1.2 kcal/mol. Indeed, the coordination of H2O2 with Ni(II) greatly enhances its acidity to the extent that it can readily donate a proton to a weak base such as Et3N. The resulting complex (t14) is now more prone to the oxidative addition of the OOH– ligand, with a relative free energy barrier of 22.7 [21.5 – (−1.2)] kcal/mol.
The greater tendency of t14 toward oxidative addition (ΔG⧧ = 22.7) relative to t9 (ΔG⧧ = 27.4) can be attributed to the following factors. (1) t14 carries less positive charge than t9, resulting in an elevated electron density at the Ni center, as confirmed by natural population analysis (NPA), which indicated an increase of 0.124 units. This heightened electron density at the Ni center renders t14 more prone to oxidation. (2) During the oxidative addition, two electrons are transferred from the d orbitals of Ni to the σ★(O–O) orbital, which cleaves the O–O bond. Moving toward a more electron-rich Ni center somewhat diminishes the effective nuclear charge experienced by electrons in the d shell, which, in turn, increases their energy levels. As a result, transferring electrons to the σ★(O–O) orbital in t14 is more facilitated than that in t9. (3) There is an intrinsic instability associated with Ni(IV) species. In s10, the nickel center is confined to the +4 oxidation state, whereas in t15, nickel can adapt to the +3 oxidation state by accepting one electron from oxygen, resulting in the formation of the oxyl species. This will make the generation of t15 more favorable than that of s10.
The ensuing nickel complex (t15) may undergo benzene attack at its oxygen atom. Considering two metal-oxy and metal-oxyl structures,49 there are two distinct pathways through which benzene can attack Ni–O (Figure 3b): nucleophilic attack on the oxy group, which occurs via the singlet ground state sTS15, or a radical mechanism targeting the oxyl group through the triplet ground state tTS15. DFT calculations indicate a substantial energy barrier of 44.9 [43.7 – (−1.2)] kcal/mol for the nucleophilic attack mechanism [Figure 3c (top)]. Although the radical mechanism exhibits a comparatively lower activation energy of 33.3 [32.1 – (−1.2)] kcal/mol, it is still fairly high. It is, however, possible for Et3NH+ to, after forming a hydrogen-bonded adduct (t16), protonate the hydroxide ligand via the transition state tTS16–17, leading to the elimination of water and the production of intermediate t17. The ensuing complex is prone to undergo benzene attack via the radical mechanism (tTS17–18) as the key step of this transformation (ΔG⧧ = 26.6 [25.4 – (−1.2)] kcal/mol). It is noteworthy that, similar to s15, the nucleophilic attack of benzene on the oxy group at s17, which should be performed at the singlet ground state (sTS17), is hindered by a high activation energy barrier of 43.2 [42.0 – (−1.2)] kcal/mol. [Figure 3c (bottom)]. It is notable that species t8, t14, t15, and t17 and transition structures tTS15 and tTS17–18 exhibit squared total spin angular momentum operator ⟨S2⟩ values of 2.006, 2.006, 2.020, 2.012, 2.088, and 2.093, respectively, based on our calculations. These values indicate that they bear minimal spin contamination.
Figure 3.

(a) Calculated energy profile for [NiII(tepa)]2+-catalyzed hydroxylation of benzene; spin density distribution plots for t15, t17, and tTS17–18 at an isosurface value of 0.006. (b) Equilibria between oxy and oxyl forms. (c) Calculated energy barrier for the nucleophilic attack of benzene to the oxy group of s15 and s17. (d) Schematic illustration of the effect of the formal charge of the nickel complex on the ease of conversion from oxy to oxyl. The superscripts “s” and “t” represent the singlet and triplet ground states, respectively. The relative Gibbs free energy values obtained from the SMD/B3LYP-D3/def2-TZVP//SMD/B3LYP-D3/6-31G(d),SDD calculations are given in kcal/mol (in red) and spin density values in e/Å3 (in pink).
Why Does [NiII(tepa)(O)]2+ Have a Greater Tendency than That of [NiII(tepa)(O)(OH)]+ toward Addition of Benzene through the Radical Mechanism?
Investigating why the free energy barriers differ for tTS15 (ΔG⧧ = 33.1 kcal/mol) and tTS17–18 (ΔG⧧ = 26.6 kcal/mol) can provide insight into these processes. t15 and t17 possess respective spin density values of 0.942 and +1.022 e/Å3 on oxygen, respectively (noted in pink in Figure 3a). Certainly, the spin density is a decisive parameter in the radical mechanism. This difference in spin densities on oxygen can be rationalized on the basis of the orbital energies. As mentioned above, two oxy and oxyl forms of Ni–O can coexist, and only the oxyl form, which is a diradical, can react with benzene via the radical mechanism. The oxyl form is the result of transferring an electron from the occupied oxygen pz orbital to the unoccupied nickel dz2 orbital (Figure 3b). Thus, the energy level of the dz2 orbital significantly influences the contribution of the oxyl structure. t17, which carries more positive charges than t15, has lower orbital energy levels for nickel. Consequently, the diminished energy level for the vacant dz2 orbital facilitates O(pz) → Ni(dz2) electron transfer, resulting in a greater contribution of the oxyl form (Figure 3d). Therefore, t17 is more likely to participate in the radical mechanism.
Formation of the Second C–O Bond
Intrinsic reaction coordinate (IRC) calculations at the B3LYP-D3/6-31G(d),SDD level of theory (Figure 4) show that the transition structure tTS17–18 connects t17 to t18, passing through a nonlocal minimum structure t18′, where a C–O bond has been formed. Subsequently, in the zwitterionic benzene-O component in t18′, oxygen nucleophilically attacks the generated carbocation to form a new C–O bond, furnishing t18 with a relative free energy of −13.0 kcal/mol (Figure 3a).
Figure 4.

Plot of SMD/B3LYP-D3/6-31G(d),SDD energies for points along the IRC of tTS17–18. The superscript “t” represents the triplet ground state. Interatomic distances are shown in Å (in brown) and spin density values for Ni atoms, O atoms, and benzene components for selected structures along the IRC path in e/Å3 (in red).
Features of this step were also sought in terms of the spin density distribution on the nickel, oxygen, and benzene components (Figure 4). The Mulliken spin density analysis indicates that initially, the two single electrons are predominantly located on the nickel and oxygen atoms. On approaching tTS17–18, spin density on oxygen decreases and that on nickel increases. The changes in Mulliken spin densities indicate that as the unpaired electron on oxygen pairs with one π electron from the benzene ring, the remaining unpaired electron in benzene is transferred to the nickel center.
Generation of the Ketone
The conversion of benzene oxide to phenol has been documented in various catalytic systems.13,50−54 Here, we describe a pathway in which nickel complex t8 plays a catalytic role in the conversion of phenol from benzene oxide as well. Intermediate t18 is reactive toward ring opening (breaking of the O–C bond) of its epoxide moiety via transition structure tTS18–19 by overcoming an energy barrier of 14.5 kcal/mol. A hydride shift occurs immediately after ring opening to give intermediate t19 in a highly exothermic fashion (Figure 3a). Indeed, DFT results suggest that tTS18–19 is directly connected to t19, and this coordinated ketone to the nickel center is the only stationary point on the potential energy surface beyond this transition structure.
Tautomerization to Phenol and Regeneration of the Active Catalyst
Coordination of the ketone to the Ni(II) in t19 causes the β-hydrogen to become remarkably acidic,55,56 leading to its facile deprotonation by Et3N or H2O. Due to the significantly higher concentration of water compared to that of Et3N, water was used as the proton shuttle in this context. A cluster of three water molecules (H2O)357−59 then drives this keto–enol tautomerization via tTS19–8, yielding the phenol product and regenerating the active catalyst t8 (Figure 3a).
Production of the Dinickel Dioxygen Complex
As mentioned earlier, few dinickel dioxygen complexes have been characterized thus far. Therefore, we conducted an investigation into the formation of such a complex in the present case. After the coordination of H2O2 to t8 and the first deprotonation, the resulting intermediate (t14) can bind to the Ni center of another species t8 to form qnt20. Et3N can deprotonate complex qnt20 with a trivial energy barrier of 2.7 kcal/mol. The ensuing species undergoes a rearrangement from the end-on to the side-on binding mode, resulting in complex s2. The reaction free energy for the process of 2[NiII(tepa)]2+ + H2O2 + 2Et3N → [NiII(tepa)2O2]2+s2 + 2Et3NH+ is −8.8 kcal/mol. In fact, although complex s2 lies outside the catalytic cycle, it serves as the resting state of the catalyst; therefore, we designated it as the new energy reference point. From the foregoing computational data, we conclude that the rate-determining step of the whole process is tTS17–18, with a barrier height of 29.8 kcal/mol (as shown in Figure 5b), which is in line with the experiment. The catalytic cycle shown in Scheme S1 summarizes our calculation results related to the mechanism of the title reaction.
Figure 5.

(a) Calculated pathway for the formation of s2. (b) Calculated relative free energy for the key transition structures (tTS17–18) and the final product starting from s2. (c) Calculated free energies of reactions between q4 and benzene. (d) Calculated free energies for the transition structures of the oxygen-rebound and σ-complex mechanisms. The superscripts “s”, “t”, “q”, and “qnt” represent the singlet, triplet, quartet, and quintet ground states, respectively. Free energies calculated at the SMD/B3LYP-D3/def2-TZVP//SMD/B3LYP-D3/6-31G(d),SDD level of theory are given in kcal/mol and spin density value in e/Å3 (in pink).
Is the Oxygen Spin Density the Sole Crucial Feature for Benzene Addition to Oxygen?
We assumed that q4 is directly generated from the homolytic cleavage of s2 in Figure 5c (the heterolytic cleavage of s2 is provided in Figure S4, Supporting Information). While molecule q4 possesses a high spin density of 1.138 e/Å3 on oxygen, the calculated energy difference between it and the corresponding transition structure for the attack of benzene on its oxyl group (qTS4–3) is calculated to be 21.2 kcal/mol. In contrast, the analogous energy difference between t17 and tTS17–18 is 11.6 kcal/mol. This can be attributed to their different abilities to generate carbocations. Both routes proceed through a carbocation structure, although after tTS17–18, the structure involving the carbocation (t18′) is not a local minimum and readily transforms into t18. Ni(III) in t17 has a higher propensity for carbocation generation compared to that of Ni(II) in q4. The endothermicity of the reaction q4 + benzene → q3 confirms this statement. qTS4–3 is also a late transition state, as evident from the short C–O distance (1.90 Å), making it more sensitive to the stability of q3. Overall, it follows that not only is the spin density on the oxyl group an important feature for the addition of benzene to oxygen but also that the positive charges on the Ni–O• complex, or in other words, the oxidation state of the Ni center, is crucial.
Is qnt20 Capable of the Hydroxylation of Benzene?
The oxygen-rebound mechanism is commonly proposed in metal–oxygen-mediated C–H activation processes.60−62 This mechanism involves the initial abstraction of a hydrogen atom from the R–H substrate by M–O•, generating a radical R• and a metal hydroxide intermediate. Subsequently, the organic radical attacks the M–OH center, leading to the formation of an alcohol group.
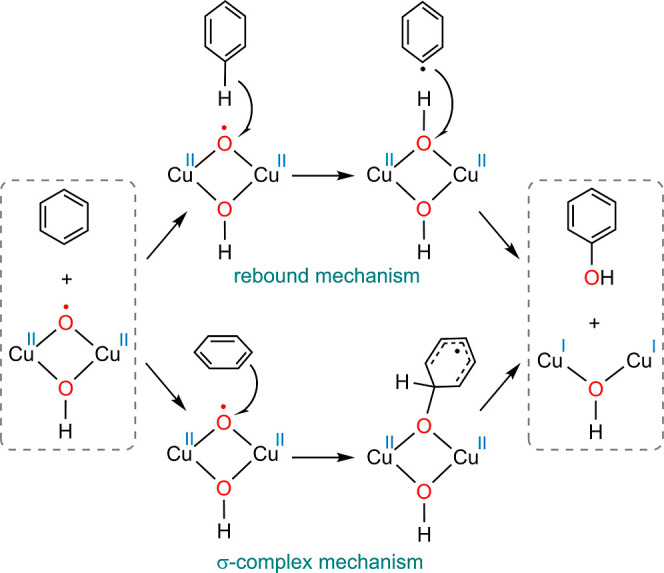
Lledós and co-workers recently computationally demonstrated that a CuII(μ–O•)(μ–OH)CuII complex is capable of oxidizing benzene into phenol in a stoichiometric reaction via the oxygen-rebound mechanism as well as a σ-complex mechanism (Scheme 2a).63 The σ-complex mechanism begins with the attack of an oxyl species on the π system of benzene, leading to the formation of a σ complex. In a subsequent step, a proton shuttle facilitates the transfer of a proton from the ipso carbon to the oxygen, producing phenol. We calculated these possibilities starting from complex qnt20 (Figure 5d) and found that the addition of benzene requires activation barriers as high as 38.2 and 43.2 kcal/mol via the transition states qntTS20i and qntTS20ii for the oxygen-rebound and σ-complex mechanisms, respectively. In both cases, the O–O distance in the hydroperoxo ligand elongates spontaneously without an additional transition state. This unreactivity of qnt20 toward the oxidation of benzene could be attributed to the lack of oxyl character of the oxygen atom. In the copper complex studied by Lledós et al., the oxygen atom possesses a spin density of 1.18 e/Å3,63 whereas this value for the analogous oxygen in qnt20 is only 0.094 e/Å3.
Scheme 2. Hydroxylation of Benzene into Phenol by a CuII(μ–O•)(μ–OH)CuII Complex via Two Different Mechanisms Reported by Lledós et al.63.
Benchmark Calculations
To compare the results obtained from the SMD/B3LYP-D3/def2-TZVP//SMD/B3LYP-D3/6-31G(d),SDD level of theory with that of other established methods for organometallic transformations, we recalculated the reaction pathway using single-point calculations by M06, M06-D3, M06-L, and M06-L-D3 DFT methods along with the def2-TZVP basis set (Table 1). Furthermore, many studies have demonstrated that a modified exact exchange fraction of 15% (15% HF) for the B3LYP method, originally set at 20% (20% HF),64 leads to the best agreement with experiments in calculating the energy of organometallic compounds.65−69 Therefore, we assigned a row of benchmark calculations to the B3LYP method with an exact exchange fraction of 15%. Different levels of theory demonstrate that transition structures tTS16–17 and tTS17–18 lie at energies significantly lower than those of transition structure tTS15. This confirms that water elimination precedes the addition of benzene to the oxyl group.
Table 1. Calculated Free Energies (kcal/mol) of the Key Intermediates and Transition Structures Relative to Those of s2 Using Different DFT Methods.
| B3LYP-D3 | B3LYP-D3, 15% HF | M06 | M06-D3 | M06-L | M06-L-D3 | |
|---|---|---|---|---|---|---|
| t8 | 4.4 | 5.8 | 1.8 | 3.9 | 8.4 | 11.1 |
| t14 | 3.2 | 3.1 | 3.7 | 4.5 | 5.8 | 8.5 |
| t15 | 18.1 | 14.1 | 18.7 | 19.4 | 8.9 | 11.6 |
| tTS15 | 36.5 | 31.1 | 43.8 | 41.6 | 29.4 | 32.1 |
| tTS16–17 | 27.6 | 21.6 | 33.5 | 29.1 | 21.4 | 24.1 |
| t17 | 18.2 | 14.5 | 15.1 | 16.5 | 11.5 | 14.2 |
| tTS17–18 | 29.8 | 24.1 | 30.8 | 28.7 | 21.6 | 24.3 |
Conclusions
In this study, we conducted DFT calculations to interrogate the mechanistic features of the hydroxylation of benzene to phenol using H2O2 catalyzed by [NiII(tepa)]2+. This investigation indicates that the dinickel dioxygen species, which has been proposed as the active catalyst for this transformation, is inactive toward the oxidation of benzene. Although our findings establish dinickel(II) μ–η2:η2-peroxo (s2) as the most stable catalyst form, its inactivity toward the hydroxylation of benzene rules this resting state out of the catalytic cycle. s2 could be in equilibrium with [NiII(tepa)]2+. Then, coordinated H2O2, after deprotonation, undergoes oxidative addition at the Ni(II) center, resulting in the formation of the oxyl radical (t15). Addition of benzene to oxyl is the key step in this process, where the spin density value of oxygen determines the ease of this addition through a radical mechanism. Protonation of the hydroxide ligand in t15 renders it more susceptible to binding benzene to oxygen, so benzene is added through a radical mechanism to t17. Subsequently, as the epoxide ring opens, hydrogen from the cleaved carbon migrates to a neighboring carbon, forming a ketone structure. [NiII(tepa)]2+ can catalyze the tautomerization step to produce phenol as the final product, while water molecules can serve as a proton-shuttling agent.
The indispensable nature of this DFT study becomes evident in its ability to shed light on elusive intermediates with high energy levels, such as t15, t16, and t17, whose experimental detection would be deemed impracticable due to their low abundance. The information provided in this research contributes to our understanding of the mechanism underlying this valuable nickel-catalyzed reaction and may guide scientists in developing novel catalytic processes for arene hydroxylation.
Computational Details
Gaussian 1670 was used to fully optimize all the structures reported in this paper at the B3LYP level of theory.71 For all the calculations, solvent effects were considered using the SMD solvation model72 with acetonitrile as the solvent. The SDD basis set73,74 with effective core potential was chosen to describe nickel. The 6-31G(d) basis set was used for other atoms.75 This basis set combination will be referred to as BS1. We also employed the D3 empirical dispersion correction for all of the calculations. Frequency calculations were carried out at the same level of theory as those for structural optimization. Transition structures were located by using the Berny algorithm. IRC calculations were used to confirm the connectivity between transition structures and minima.76,77 To further refine the energies obtained from the SMD/B3LYP-D3/SDD,6-31G(d) calculations, we carried out single-point energy calculations using the B3LYP-D3 functional method with the SMD solvation model in acetonitrile along with a larger basis set (BS2) for all of the optimized structures. BS2 utilizes the def2-TZVP basis set78 on all atoms. The tight convergence criterion and ultrafine integral grid were exploited to increase the accuracy of the calculations. The free energy for each species in solution was calculated using the following formula
| 1 |
where ΔG1atm→1M = 1.89 kcal/mol is the free-energy change for compression of 1 mol of an ideal gas from 1 atm to the 1 M solution phase standard state. It is worth noting that different spin states have been computed in the energy calculations of nickel complexes, and the most stable ones have been reported.
Acknowledgments
This project has received funding from the European Union—NextGenerationEU instrument and is funded by the Academy of Finland under grant number 348327. The authors gratefully acknowledge the Digipower project, supported by Teknologiateollisuuden 100v säätiö and Jane ja Aatos Erkon säätiö. We also thank the Finland CSC-IT Center for generous grants of computer time.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.3c04461.
Additional computational results and Cartesian coordinates of all calculated species (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Weissermel K.; Arpe H.-J.. Industrial Organic Chemistry; John Wiley & Sons, 2008. [Google Scholar]
- Schmidt R. J. Industrial catalytic processes-phenol production. Appl. Catal. 2005, 280, 89–103. 10.1016/j.apcata.2004.08.030. [DOI] [Google Scholar]
- Weber M.; Weber M.; Kleine-Boymann M.. Phenol. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley, 2000. [Google Scholar]
- Bu F.; Chen C.; Yu Y.; Hao W.; Zhao S.; Hu Y.; Qin Y. Boosting Benzene Oxidation with a Spin-State-Controlled Nuclearity Effect on Iron Sub-Nanocatalysts. Angew. Chem., Int. Ed. 2023, 62, e202216062 10.1002/anie.202216062. [DOI] [PubMed] [Google Scholar]
- Sarkar A.; Sarkar A.; Karmodak N.; Dhar B. B.; Adhikari S. Bio-inspired Cu(ii) amido-quinoline complexes as catalysts for aromatic C–H bond hydroxylation. Dalton Trans. 2023, 52, 540–545. 10.1039/d2dt03242b. [DOI] [PubMed] [Google Scholar]
- Rajeev A.; Balamurugan M.; Sankaralingam M. Rational Design of First-Row Transition Metal Complexes as the Catalysts for Oxidation of Arenes: A Homogeneous Approach. ACS Catal. 2022, 12, 9953–9982. 10.1021/acscatal.2c01928. [DOI] [Google Scholar]
- Yamada Y.; Teoh C.-M.; Toyoda Y.; Tanaka K. Direct catalytic benzene hydroxylation under mild reaction conditions by using a monocationic μ-nitrido-bridged iron phthalocyanine dimer with 16 peripheral methyl groups. New J. Chem. 2022, 46, 955–958. 10.1039/D1NJ05369H. [DOI] [Google Scholar]
- Targhan H.; Evans P.; Bahrami K. A review of the role of hydrogen peroxide in organic transformations. J. Ind. Eng. Chem. 2021, 104, 295–332. 10.1016/j.jiec.2021.08.024. [DOI] [Google Scholar]
- Masferrer-Rius E.; Borrell M.; Lutz M.; Costas M.; Klein Gebbink R. J. Aromatic C- H hydroxylation reactions with hydrogen peroxide catalyzed by bulky Manganese complexes. Adv. Synth. Catal. 2021, 363, 3783–3795. 10.1002/adsc.202001590. [DOI] [Google Scholar]
- Muthuramalingam S.; Anandababu K.; Velusamy M.; Mayilmurugan R. One step phenol synthesis from benzene catalysed by nickel(II) complexes. Catal. Sci. Technol. 2019, 9, 5991–6001. 10.1039/C9CY01471C. [DOI] [Google Scholar]
- Kwong H.-K.; Lo P.-K.; Yiu S.-M.; Hirao H.; Lau K.-C.; Lau T.-C. Highly selective and efficient ring hydroxylation of alkylbenzenes with hydrogen peroxide and an osmium(VI) nitrido catalyst. Angew. Chem., Int. Ed. 2017, 56, 12260–12263. 10.1002/anie.201705986. [DOI] [PubMed] [Google Scholar]
- Tsuji T.; Zaoputra A. A.; Hitomi Y.; Mieda K.; Ogura T.; Shiota Y.; Yoshizawa K.; Sato H.; Kodera M. Specific enhancement of catalytic activity by a dicopper core: selective hydroxylation of benzene to phenol with hydrogen peroxide. Angew. Chem., Int. Ed. 2017, 56, 7779–7782. 10.1002/anie.201702291. [DOI] [PubMed] [Google Scholar]
- Ramu R.; Wanna W. H.; Janmanchi D.; Tsai Y.-F.; Liu C.-C.; Mou C.-Y.; Yu S. S.-F. Mechanistic study for the selective oxidation of benzene and toluene catalyzed by Fe(ClO4)2 in an H2O2-H2O–CH3CN system. Mol. Catal. 2017, 441, 114–121. 10.1016/j.mcat.2017.08.006. [DOI] [Google Scholar]
- Morimoto Y.; Bunno S.; Fujieda N.; Sugimoto H.; Itoh S. Direct hydroxylation of benzene to phenol using hydrogen peroxide catalyzed by nickel complexes supported by pyridylalkylamine ligands. J. Am. Chem. Soc. 2015, 137, 5867–5870. 10.1021/jacs.5b01814. [DOI] [PubMed] [Google Scholar]
- Raba A.; Cokoja M.; Herrmann W. A.; Kühn F. E. Catalytic hydroxylation of benzene and toluene by an iron complex bearing a chelating di-pyridyl-di-NHC ligand. Chem. Commun. 2014, 50, 11454–11457. 10.1039/C4CC02178A. [DOI] [PubMed] [Google Scholar]
- Shoji O.; Kunimatsu T.; Kawakami N.; Watanabe Y. Highly Selective Hydroxylation of Benzene to Phenol by Wild-type Cytochrome P450BM3 Assisted by Decoy Molecules. Angew. Chem., Int. Ed. 2013, 52, 6606–6610. 10.1002/anie.201300282. [DOI] [PubMed] [Google Scholar]
- Tani M.; Sakamoto T.; Mita S.; Sakaguchi S.; Ishii Y. Hydroxylation of benzene to phenol under air and carbon monoxide catalyzed by molybdovanadophosphoric acid. Angew. Chem. 2005, 117, 2642–2644. 10.1002/ange.200462769. [DOI] [PubMed] [Google Scholar]
- Niwa S.-i.; Eswaramoorthy M.; Nair J.; Raj A.; Itoh N.; Shoji H.; Namba T.; Mizukami F. A one-step conversion of benzene to phenol with a palladium membrane. Science 2002, 295, 105–107. 10.1126/science.1066527. [DOI] [PubMed] [Google Scholar]
- Bianchi D.; Bortolo R.; Tassinari R.; Ricci M.; Vignola R. A novel iron-based catalyst for the biphasic oxidation of benzene to phenol with hydrogen peroxide. Angew. Chem. 2000, 112, 4491–4493. 10.1002/1521-3757(20001201)112:23<4491::AID-ANGE4491>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Strukul G.Catalytic Oxidations with Hydrogen Peroxide as Oxidant; Springer Science & Business Media, 2013; Vol. 9. [Google Scholar]
- Cruse R. W.; Kaderli S.; Karlin K. D.; Zuberbuehler A. D. Kinetic and thermodynamic studies on the reaction of oxygen with two dinuclear copper(I) complexes. J. Am. Chem. Soc. 1988, 110, 6882–6883. 10.1021/ja00228a046. [DOI] [Google Scholar]
- Sheldon R. A.; Kochi J. K.. Metal-Catalyzed Oxidations of Organic Compounds in the Liquid Phase: A Mechanistic Approach. In Advances in Catalysis; Eley D., Pines H., Weisz P. B., Eds.; Academic Press, 1976; Vol. 25, pp 272–413. [Google Scholar]
- Tano T.; Doi Y.; Inosako M.; Kunishita A.; Kubo M.; Ishimaru H.; Ogura T.; Sugimoto H.; Itoh S. Nickel(II) Complexes of tpa Ligands with 6-Phenyl Substituents (Phntpa). Structure and H2O2-Reactivity. Bull. Chem. Soc. Jpn. 2010, 83, 530–538. 10.1246/bcsj.20090346. [DOI] [Google Scholar]
- Shiren K.; Ogo S.; Fujinami S.; Hayashi H.; Suzuki M.; Uehara A.; Watanabe Y.; Moro-oka Y. Synthesis, Structures, and Properties of Bis(μ-oxo)nickel(III) and Bis(μ-superoxo)nickel(II) Complexes: An Unusual Conversion of a Ni 2IIIμ-O2 Core into a Ni 2II(μ-OO)2 Core by H2O2 and Oxygenation of Ligand. J. Am. Chem. Soc. 2000, 122, 254–262. 10.1021/ja990311d. [DOI] [Google Scholar]
- Itoh S.; Bandoh H.; Nagatomo S.; Kitagawa T.; Fukuzumi S. Aliphatic hydroxylation by a bis(μ-oxo)dinickel(III) complex. J. Am. Chem. Soc. 1999, 121, 8945–8946. 10.1021/ja991326e. [DOI] [Google Scholar]
- Hikichi S.; Yoshizawa M.; Sasakura Y.; Akita M.; Moro-oka Y. First synthesis and structural characterization of dinuclear M(III) bis(μ-oxo) complexes of nickel and cobalt with hydrotris(pyrazolyl)borate ligand. J. Am. Chem. Soc. 1998, 120, 10567–10568. 10.1021/ja981837l. [DOI] [Google Scholar]
- Honda K.; Cho J.; Matsumoto T.; Roh J.; Furutachi H.; Tosha T.; Kubo M.; Fujinami S.; Ogura T.; Kitagawa T.; et al. Oxidation Reactivity of Bis(μ-oxo) Dinickel(III) Complexes: Arene Hydroxylation of the Supporting Ligand. Angew. Chem., Int. Ed. 2009, 48, 3304–3307. 10.1002/anie.200900222. [DOI] [PubMed] [Google Scholar]
- Kunishita A.; Doi Y.; Kubo M.; Ogura T.; Sugimoto H.; Itoh S. Ni(II)/H2O2 reactivity in bis[(pyridin-2-yl)methyl]amine tridentate ligand system. Aromatic hydroxylation reaction by bis(μ-oxo)dinickel(III) complex. Inorg. Chem. 2009, 48, 4997–5004. 10.1021/ic900059m. [DOI] [PubMed] [Google Scholar]
- Morimoto Y.; Takagi Y.; Saito T.; Ohta T.; Ogura T.; Tohnai N.; Nakano M.; Itoh S. A Bis(μ-oxido)dinickel(III) Complex with a Triplet Ground State. Angew. Chem. 2018, 130, 7766–7769. 10.1002/ange.201802779. [DOI] [PubMed] [Google Scholar]
- Qi H.; Xu D.; Lin J.; Sun W. Copper-catalyzed direct hydroxylation of arenes to phenols with hydrogen peroxide. Mol. Catal. 2022, 528, 112441. 10.1016/j.mcat.2022.112441. [DOI] [Google Scholar]
- Anandababu K.; Muthuramalingam S.; Velusamy M.; Mayilmurugan R. Single-step benzene hydroxylation by cobalt(II) catalysts via a cobalt(III)-hydroperoxo intermediate. Catal. Sci. Technol. 2020, 10, 2540–2548. 10.1039/C9CY02601K. [DOI] [Google Scholar]
- Muthuramalingam S.; Anandababu K.; Velusamy M.; Mayilmurugan R. Benzene hydroxylation by bioinspired copper(II) complexes: Coordination geometry versus reactivity. Inorg. Chem. 2020, 59, 5918–5928. 10.1021/acs.inorgchem.9b03676. [DOI] [PubMed] [Google Scholar]
- Kumari S.; Muthuramalingam S.; Dhara A. K.; Singh U. P.; Mayilmurugan R.; Ghosh K. Cu(I) complexes obtained via spontaneous reduction of Cu(II) complexes supported by designed bidentate ligands: Bioinspired Cu(I) based catalysts for aromatic hydroxylation. Dalton Trans. 2020, 49, 13829–13839. 10.1039/D0DT02413A. [DOI] [PubMed] [Google Scholar]
- Vilella L.; Conde A.; Balcells D.; Díaz-Requejo M. M.; Lledós A.; Pérez P. J. A competing, dual mechanism for catalytic direct benzene hydroxylation from combined experimental-DFT studies. Chem. Sci. 2017, 8, 8373–8383. 10.1039/C7SC02898A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L.; Zhong W.; Xu B.; Wei Z.; Liu X. Synthesis and characterization of copper(II) complexes with multidentate ligands as catalysts for the direct hydroxylation of benzene to phenol. Dalton Trans. 2015, 44, 8013–8020. 10.1039/C5DT00575B. [DOI] [PubMed] [Google Scholar]
- Xu B.; Zhong W.; Wei Z.; Wang H.; Liu J.; Wu L.; Feng Y.; Liu X. Iron(III) complexes of multidentate pyridinyl ligands: synthesis, characterization and catalysis of the direct hydroxylation of benzene. Dalton Trans. 2014, 43, 15337–15345. 10.1039/C4DT02032D. [DOI] [PubMed] [Google Scholar]
- Conde A.; Mar Díaz-Requejo M.; Pérez P. J. Direct, copper-catalyzed oxidation of aromatic C–H bonds with hydrogen peroxide under acid-free conditions. Chem. Commun. 2011, 47, 8154–8156. 10.1039/c1cc12804c. [DOI] [PubMed] [Google Scholar]
- Besalu-Sala P.; Magallon C.; Costas M.; Company A.; Luis J. M. Mechanistic Insights into the ortho-Defluorination-Hydroxylation of 2-Halophenolates Promoted by a Bis(μ-oxo)dicopper(III) Complex. Inorg. Chem. 2020, 59, 17018–17027. 10.1021/acs.inorgchem.0c02246. [DOI] [PubMed] [Google Scholar]
- Serrano-Plana J.; Garcia-Bosch I.; Miyake R.; Costas M.; Company A. Selective Ortho-Hydroxylation–Defluorination of 2-Fluorophenolates with a Bis(μ-oxo)dicopper(III) Species. Angew. Chem., Int. Ed. 2014, 53, 9608–9612. 10.1002/anie.201405060. [DOI] [PubMed] [Google Scholar]
- York J. T.; Llobet A.; Cramer C. J.; Tolman W. B. Heterobimetallic dioxygen activation: Synthesis and reactivity of mixed Cu- Pd and Cu- Pt bis (μ-oxo) complexes. J. Am. Chem. Soc. 2007, 129, 7990–7999. 10.1021/ja071744g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagore R.; Chen; Crabtree R. H.; Brudvig G. W. Determination of μ-oxo exchange rates in di-μ-oxo dimanganese complexes by electrospray ionization mass spectrometry. J. Am. Chem. Soc. 2006, 128, 9457–9465. 10.1021/ja061348i. [DOI] [PubMed] [Google Scholar]
- Ghosh A.; Tangen E.; Gonzalez E.; Que L. Models of High-Valent Intermediates of Non-Heme Diiron Alkane Monooxygenases: Electronic Structure of a Bis(μ-oxo)diron(IV) Complex with Locally Low-Spin Metal Centers. Angew. Chem., Int. Ed. 2004, 43, 834–838. 10.1002/anie.200351768. [DOI] [PubMed] [Google Scholar]
- Taki M.; Itoh S.; Fukuzumi S. Oxo-Transfer Reaction from a Bis(μ-oxo)dicopper(III) Complex to Sulfides. J. Am. Chem. Soc. 2002, 124, 998–1002. 10.1021/ja016023a. [DOI] [PubMed] [Google Scholar]
- Park G. Y.; Qayyum M. F.; Woertink J.; Hodgson K. O.; Hedman B.; Narducci Sarjeant A. A.; Solomon E. I.; Karlin K. D. Geometric and electronic structure of [{Cu(MeAN)}2(μ-η2:η2 (O 22-))]2+ with an unusually long O–O bond: O–O bond weakening vs activation for reductive cleavage. J. Am. Chem. Soc. 2012, 134, 8513–8524. 10.1021/ja300674m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto T.; Ohkubo K.; Honda K.; Yazawa A.; Furutachi H.; Fujinami S.; Fukuzumi S.; Suzuki M. Aliphatic C- H Bond Activation Initiated by a (μ-η2:η2-Peroxo) dicopper(II) Complex in Comparison with Cumylperoxyl Radical. J. Am. Chem. Soc. 2009, 131, 9258–9267. 10.1021/ja809822c. [DOI] [PubMed] [Google Scholar]
- Funahashi Y.; Nishikawa T.; Wasada-Tsutsui Y.; Kajita Y.; Yamaguchi S.; Arii H.; Ozawa T.; Jitsukawa K.; Tosha T.; Hirota S.; et al. Formation of a bridged butterfly-type μ-η2:η2-Peroxo Dicopper Core structure with a carboxylate group. J. Am. Chem. Soc. 2008, 130, 16444–16445. 10.1021/ja804201z. [DOI] [PubMed] [Google Scholar]
- Matsumoto T.; Furutachi H.; Kobino M.; Tomii M.; Nagatomo S.; Tosha T.; Osako T.; Fujinami S.; Itoh S.; Kitagawa T.; et al. others Intramolecular arene hydroxylation versus intermolecular olefin epoxidation by (μ-η2:η2-peroxo)dicopper(II) complex supported by dinucleating ligand. J. Am. Chem. Soc. 2006, 128, 3874–3875. 10.1021/ja058117g. [DOI] [PubMed] [Google Scholar]
- See Supporting Information, Figure S2, for full details regarding the various possible charge distributions on the generated fragments in substitution reactions between benzene and s2.
- Shimoyama Y.; Kojima T. Metal–Oxyl Species and Their Possible Roles in Chemical Oxidations. Inorg. Chem. 2019, 58, 9517–9542. 10.1021/acs.inorgchem.8b03459. [DOI] [PubMed] [Google Scholar]
- Xie J.; Li X.; Guo J.; Luo L.; Delgado J. J.; Martsinovich N.; Tang J. Highly selective oxidation of benzene to phenol with air at room temperature promoted by water. Nat. Commun. 2023, 14, 4431. 10.1038/s41467-023-40160-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karich A.; Kluge M.; Ullrich R.; Hofrichter M. Benzene oxygenation and oxidation by the peroxygenase of Agrocybe aegerita. Amb. Express 2013, 3, 5. 10.1186/2191-0855-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudrik E. V.; Sorokin A. B. N-Bridged Diiron Phthalocyanine Catalyzes Oxidation of Benzene with H2O2 via Benzene Oxide with NIH Shift Evidenced by Using 1,3,5-[D3] Benzene as a Probe. Chem.—Eur. J. 2008, 14, 7123–7126. 10.1002/chem.200800504. [DOI] [PubMed] [Google Scholar]
- Shiota Y.; Suzuki K.; Yoshizawa K. Mechanism for the direct oxidation of benzene to phenol by FeO+. Organometallics 2005, 24, 3532–3538. 10.1021/om050136b. [DOI] [Google Scholar]
- Vogel E.; Günther H. Benzene Oxide-Oxepin Valence Tautomerism. Angew. Chem., Int. Ed. 1967, 6, 385–401. 10.1002/anie.196703851. [DOI] [Google Scholar]
- Farshadfar K.; Chipman A.; Hosseini M.; Yates B. F.; Ariafard A. A modified cationic mechanism for PdCl2-catalyzed transformation of a homoallylic alcohol to an allyl ether. Organometallics 2019, 38, 2953–2962. 10.1021/acs.organomet.9b00276. [DOI] [Google Scholar]
- Chipman A.; Gouranourimi A.; Farshadfar K.; Olding A.; Yates B. F.; Ariafard A. A computational mechanistic investigation into reduction of gold(III) complexes by amino acid glycine: A new variant for amine oxidation. Chem.—Eur. J. 2018, 24, 8361–8368. 10.1002/chem.201800403. [DOI] [PubMed] [Google Scholar]
- Song L.; Tian X.; Farshadfar K.; Shiri F.; Rominger F.; Ariafard A.; Hashmi A. S. K. An unexpected synthesis of azepinone derivatives through a metal-free photochemical cascade reaction. Nat. Commun. 2023, 14, 831. 10.1038/s41467-023-36190-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farshadfar K.; Bird M. J.; Olivier W. J.; Hyland C. J.; Smith J. A.; Ariafard A. Computational investigation into the mechanistic features of bromide-catalyzed alcohol oxidation by PhIO in water. J. Org. Chem. 2021, 86, 2998–3007. 10.1021/acs.joc.0c02903. [DOI] [PubMed] [Google Scholar]
- Hu C.; Farshadfar K.; Dietl M. C.; Cervantes-Reyes A.; Wang T.; Adak T.; Rudolph M.; Rominger F.; Li J.; Ariafard A.; et al. Gold-Catalyzed [5,5]-Rearrangement. ACS Catal. 2021, 11, 6510–6518. 10.1021/acscatal.1c01108. [DOI] [Google Scholar]
- Huang X.; Groves J. T. Beyond ferryl-mediated hydroxylation: 40 years of the rebound mechanism and C–H activation. J. Biol. Inorg. Chem. 2017, 22, 185–207. 10.1007/s00775-016-1414-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.; Ståhlberg J.; Sandgren M.; Paton R. S.; Beckham G. T. Quantum mechanical calculations suggest that lytic polysaccharide monooxygenases use a copper-oxyl, oxygen-rebound mechanism. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 149–154. 10.1073/pnas.1316609111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves J. T.; McClusky G. A. Aliphatic hydroxylation via oxygen rebound. Oxygen transfer catalyzed by iron. J. Am. Chem. Soc. 1976, 98, 859–861. 10.1021/ja00419a049. [DOI] [Google Scholar]
- Borrego E.; Tiessler-Sala L.; Lázaro J. J.; Caballero A.; Pérez P. J.; Lledós A. Direct Benzene Hydroxylation with Dioxygen Induced by Copper Complexes: Uncovering the Active Species by DFT Calculations. Organometallics 2022, 41, 1892–1904. 10.1021/acs.organomet.2c00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becke A. D. Density-functional thermochemistry. I. The effect of the exchange-only gradient correction. J. Chem. Phys. 1992, 96, 2155–2160. 10.1063/1.462066. [DOI] [Google Scholar]
- Song Y.-T.; Li X.-C.; Siegbahn P. E. Is there a different mechanism for water oxidation in higher plants?. J. Phys. Chem. B 2023, 127, 6643–6647. 10.1021/acs.jpcb.3c03029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegbahn P. E. A quantum chemical approach for the mechanisms of redox-active metalloenzymes. RSC Adv. 2021, 11, 3495–3508. 10.1039/D0RA10412D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegbahn P. E.; Blomberg M. R. A systematic DFT approach for studying mechanisms of redox active enzymes. Front. Chem. 2018, 6, 644. 10.3389/fchem.2018.00644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomberg M. R.; Borowski T.; Himo F.; Liao R.-Z.; Siegbahn P. E. Quantum chemical studies of mechanisms for metalloenzymes. Chem. Rev. 2014, 114, 3601–3658. 10.1021/cr400388t. [DOI] [PubMed] [Google Scholar]
- Reiher M.; Salomon O.; Artur Hess B. Reparameterization of hybrid functionals based on energy differences of states of different multiplicity. Theor. Chem. Acc. 2001, 107, 48–55. 10.1007/s00214-001-0300-3. [DOI] [Google Scholar]
- Frisch M. J.; et al. Gaussian 16, Version 16, Revision A.03; Gaussian, Inc.: Wallingford CT, 2016. https://gaussian.com/gaussian16/.
- Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- Dolg M.; Wedig U.; Stoll H.; Preuss H. Energy-adjusted ab initio pseudopotentials for the first row transition elements. J. Chem. Phys. 1987, 86, 866–872. 10.1063/1.452288. [DOI] [Google Scholar]
- Bergner A.; Dolg M.; Küchle W.; Stoll H.; Preuß H. Ab initio energy-adjusted pseudopotentials for elements of groups 13–17. Mol. Phys. 1993, 80, 1431–1441. 10.1080/00268979300103121. [DOI] [Google Scholar]
- Hariharan P. C.; Pople J. A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. 10.1007/BF00533485. [DOI] [Google Scholar]
- Fukui K. The path of chemical reactions-the IRC approach. Acc. Chem. Res. 1981, 14, 363–368. 10.1021/ar00072a001. [DOI] [Google Scholar]
- Fukui K. Formulation of the reaction coordinate. J. Phys. Chem. 1970, 74, 4161–4163. 10.1021/j100717a029. [DOI] [Google Scholar]
- Weigend F.; Furche F.; Ahlrichs R. Gaussian basis sets of quadruple zeta valence quality for atoms H–Kr. J. Chem. Phys. 2003, 119, 12753–12762. 10.1063/1.1627293. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.