

Abstract
Whenever an asexual viral population evolves by adapting to new environmental conditions, beneficial mutations, the ultimate cause of adaptation, are randomly produced and then fixed in the population. The larger the population size and the higher the mutation rate, the more beneficial mutations can be produced per unit time. With the usually high mutation rate of RNA viruses and in a large enough population, several beneficial mutations could arise at the same time but in different genetic backgrounds, and if the virus is asexual, they will never be brought together through recombination. Thus, the best of these genotypes must outcompete each other on their way to fixation. This competition among beneficial mutations has the effect of slowing the overall rate of adaptation. This phenomenon is known as clonal interference. Clonal interference predicts a speed limit for adaptation as the population size increases. In the present report, by varying the size of evolving vesicular stomatitis virus populations, we found evidence clearly demonstrating this speed limit and thus indicating that clonal interference might be an important factor modulating the rate of adaptation to an in vitro cell system. Several evolutionary and epidemiological implications of the clonal interference model applied to RNA viruses are discussed.
In recent years, increasing attention has been paid to the study of RNA virus adaptive evolution from an experimental standpoint. The evolution of biological fitness (understanding it as a macroscopic characteristic that includes many components such as speed of replication, efficiency of encapsidation, ability for cell-to-cell transmission, or resistance to antiviral factors or defective interfering particles) has been studied in vitro for virus such as vesicular stomatitis virus (VSV) (4, 9, 10, 21, 24–26), foot-and-mouth disease virus (11, 12), φ6 (2), and φX174 (29). Several common features and conclusions are evident from these studies. For example, the rate of adaptation decreases with evolutionary time as the evolving population reaches an adaptive peak which usually represents the best solution to the problem imposed by the selective environmental pressures. Another important conclusion is that the rate of adaptation, as well as the final peak reached, depends on the initial fitness of the viral clone employed (10). Despite these advances in the understanding of viral evolutionary adaptation, few experiments have analyzed the effect of population size on the rate of viral adaptation. Burch and Chao (2) analyzed the effect of population size on the fixation of mutations of different magnitudes during the evolutionary process. A positive correlation was found between population size and the fitness effect that was fixed. In addition to this pioneering work, in a recent study (22) we investigated the effect of population size on the magnitude of the fitness effect fixed and in the rate of viral adaptation and showed a positive correlation between viral population size and the fitness effect fixed as well as a limit imposed by population size to the rate of adaptation.
Recently, Gerrish and Lenski (13) developed a theoretical framework to explain adaptive evolution in microbial asexual populations. Experimental support for the model was recently provided by studies with the bacterium Escherichia coli (6) and the RNA virus VSV (22), where several of the predictions made by the model were confirmed. The model is based on the phenomenon of clonal interference. Briefly, the idea of clonal interference is as follows. Imagine a beneficial mutation that spontaneously arose in a genotype. The time required for fixing this mutation in the population should be long if the population size is large. However, the frequency of the mutant in the population is low for a substantial portion of the time (18). During this period, at which the beneficial mutation is present at low frequency, there is a certain likelihood, which indeed depends on the mutation rate, that a second beneficial mutation will arise in a different genotype. If the population is sexual, both beneficial mutations can be brought together in forming a new genotype that benefits from their joint effect. If the population is asexual, the two mutations cannot recombine and so must compete with one another on their way to fixation. The result is that only the best one will become fixed, eliminating the other(s). Even if the first mutation is the best possible candidate, the time for its fixation is longer due to presence of the second beneficial mutation. Once this beneficial genotype fixes, secondary beneficial mutations can arise against the new dominant genetic background. Thus, in asexual populations, beneficial mutations must fix in a sequential manner.
The main conclusions of the clonal interference model can be summarized as follows (see reference 13 for a more detailed description of each conclusion). (i) The probability of fixation of a given beneficial mutation decreases with both population size and mutation rate. (ii) As population size or mutation rate increases, adaptive substitutions result in larger fitness increases. (iii) The rate of adaptation is an increasing but decelerating function of both population size and mutation rate. (iv) Beneficial mutations that become transiently common but do not achieve fixation due to interfering beneficial mutations are relatively abundant. (v) Transient polymorphisms may give rise to a “leapfrog” effect, where the most common genotype at a given moment might be less closely related to the immediately preceding one than with an earlier genotype.
In the present contribution, which must be seen as a complement to our previously published experiments (22), we want to address the third of these conclusions, i.e., that the rate of adaptation is an increasing but decelerating function of population size. To do so, we will simulate in vitro the evolution of VSV populations of different population sizes and then measure the rate of adaptation at each population size. This approach to measuring the effect of population size in the rate of adaptation is different from and more straightforward that the one we used in our previous work (22). Regression analysis of the rate of evolution on population size will show us if the relationship between the two parameters is linear or if a maximum rate exists, as predicted by the model.
MATERIALS AND METHODS
VSV clones.
The first clone employed was a I1 monoclonal antibody (MAb)-sensitive, surrogate wild-type clone. This wild-type clone was derived from the Mudd-Summers strain of the VSV Indiana serotype and was replicated for more than two decades in J. J. Holland's laboratory on BHK-21 cells with either low-multiplicity passages or plaque-to-plaque clonal propagation to minimize interference by defective interfering particles. We obtained a clone from him a decade ago, and to avoid any further genetic change, a large volume with a high titer (∼1010 PFU/ml) was produced and kept at −80°C in 1-ml aliquots. This wild-type clone was used as a common competitor during the competition experiments. The second viral clone, MARM C (also obtained from Holland's laboratory), was generated from the wild-type clone and has an Asp259 → Ala substitution in the G surface protein. This substitution allows the mutant to replicate under MAb I1 concentrations that completely neutralize the wild-type clone (28). All the evolution experiments were done with MARM C.
Before the evolution experiments, the MARM C and wild-type clones were plated in the presence of inhibitory concentrations of MAb I1 to test for the presence or absence of the MAR phenotype. To start the experiments with a genetically homogeneous virus, a clone of MARM C was isolated and used to initiate the evolution experiments.
The fitness of MARM C relative to the wild-type has been determined many times under identical or nearly identical conditions to those employed here (4, 7, 8, 15, 20, 21), as well as 18 times during the course of the present experiment (in six independent blocks with three replicates per block). In this experiment, we obtained a fitness value not significantly different from unity (mean and standard error = 1.04 ± 0.03; Student's t test = 1.4227; P = 0.1729). Thus, the mutation conferring the MARM phenotype can be considered selectively neutral.
Cell lines and culture conditions.
Baby hamster kidney cells (BHK-K) were grown as monolayers in Dulbecco's modified Eagle's medium (DMEM) containing 5% newborn calf serum and 0.06% proteose peptone 3. Cells were grown in 25-cm2 plastic flasks (containing 5 ml of medium) for infections or in 100-cm2 plates (containing 15 ml of medium) for routine maintenance. The cell density was determined three times during the course of the experiment. To do so, cells from a 25-cm2 flask were detached (with a solution containing 0.05 mg of trypsin per ml and 0.2 mg of EDTA per ml), gently suspended in DMEM, and serially diluted. A convenient dilution was visually counted using a Neubauer hemocytometer (0.00625-mm3 grade volume). The cells grew at a density of (7.7 ± 0.5) × 106 cells/cm2.
The I1 monoclonal antibody employed in the competition experiments was produced and characterized by Lefrancois and Lyles (17) and VandePol et al. (28). We propagated hybridoma cells in DMEM containing 20% bovine calf serum, 2 μg of thymidine per ml, 0.1 μg of glycine per ml, and 14 μg of hypoxanthine per ml in large flasks to produce many liters of high-titer neutralizing MAb, which was stored at −80°C until used.
Cell were maintained in incubators at 37°C under a 5% CO2 atmosphere.
Effective viral population size.
The effective population size, Ne for a haploid asexual population under a batch transfer regimen is given by the expression Ne = N0τ (18), where N0 is the initial population size and τ is the number of generations of growth per day necessary to reach the final density. Regardless the different N0 values used in the present experiment, the viral burst from a 25-cm2 flask after complete destruction of the cell monolayer (1 day of infection in our case) was estimated to be about 3.8 × 1010 PFU. This value depends only on the available cells for infection, which was a constant throughout the experiment. Therefore, to obtain different N0 values, it is sufficient to make proper dilutions. We used the three different N0 values shown in the first column of Table 1.
TABLE 1.
Parameters describing the experiment
| Initial population size, N0 (PFU) | No. of generations per day (τ) | Effective population size, Ne (PFU) | Time of evolution (no. of daily transfers) | Time of evolution (no. of generations) |
|---|---|---|---|---|
| 1.5 × 102 | 6 | 9 × 102 | 17 | 102 |
| 1.5 × 105 | 4 | 6 × 105 | 25 | 100 |
| 1.5 × 107 | 2 | 3 × 107 | 50 | 100 |
The second variable involved in the estimation of Ne, the number of generations of growth per day, τ, has been estimated by means of equation A1 (see Appendix). For the final viral density and the number of host cells given above, the number of generations per day for each initial density is given in the second column of Table 1. From these values, it is possible to compute the Ne values in our experiment (third column in Table 1). Obviously, these values are determined mostly by N0.
Experimental design.
For each Ne, two replicates were initiated by infecting flasks with MARM C at their corresponding N0. After 24 h of infection, the resulting virus was properly diluted and used to infect a fresh monolayer. This batch transfer procedure was performed for a number of consecutive days (given in the fourth column of Table 1). Virus under each Ne regimen experienced a different number of generations per day (second column in Table 1); nonetheless, evolution experiments were stopped when all lines reached 100 generations. Samples were taken periodically from the evolving populations and used to estimate the fitness at different time points. This basic experiment was performed twice to get statistical power.
Relative-fitness assays.
At the end of each evolution experiment, the MARM C evolving populations sequentially isolated were assayed for relative fitness with threefold replication (15). Relative fitness measures the degree of adaptation of viral populations to the experimental environment. Each evolving MARM C population was mixed, in three independent test tubes, with a known amount of wild-type clone, and the initial ratio for each replicate mixture, R0, was determined by performing plaque assays with and without I1 MAb in the agarose overlay medium. Incorporation of the antibody into the plaque overlay medium (after virus penetration), instead of standard virus neutralization, avoids the problem of phenotypic mixing and hiding (i.e., the encapsidation of MARM RNAs within phenotypically wild-type envelops) (14). Each competition mixture was transferred serially during a sufficient number of passages to obtain good estimates of relative fitness as follows. At each transfer, the resulting virus mixture was diluted by a factor of 104 and used to initiate the next competition passage by infection of a fresh cell monolayer. The ratio of MARM C to wild type was determined by plating with and without I1 MAb in the overlay agarose medium at different transfers. These determinations gave the proportion MARM C to wild type at transfer t, Rt. Fitness was defined as W = (Rt/R0)1/t (3) and obtained by fitting lnW to the time series data by the least-squares method (27).
Statistical analysis.
All statistical analyses described were done with the SPSS 8.0.1S for Windows package (23). To detect the diminishing-returns effect of population size in the rate of adaptation predicted by the clonal-interference model, we fitted our estimates of the rate of adaptation obtained for the three different population sizes to two models. The first is a linear model, dW/dt = aNe, in which the rate of adaptation is directly proportional to the effective population size, i.e., the supply of beneficial mutations. The second model is a hyperbolic one, dW/dt = aNe/(b + Ne), such that the rate of adaptation shows an upper limit due to clonal interference (6). The hyperbolic models was chosen on the basis of its mathematical properties and shape: faster increases for smaller Ne values, a decline in the rate of increase with increasing Ne, and the existence of a maximum dW/dt value. From a statistical point of view, the hyperbola is also convenient: it uses only two parameters, one more than the linear model, which allows us to make nested comparisons among models. In both models, the intercept to the dW/dt value is set to zero, because it is expected that an asexual population of null size, that is, with no genetic variability, cannot improve its fitness by adaptation (6).
RESULTS
Data description and homogeneity among replicates.
Figure 1 shows the fitness trajectories for all three effective population sizes and replicates. Although fitness of the ancestral MARM C clone was measured in six independent blocks (with three replicates per block), no significant block effect was detected (nested analysis of variance, F2,3 = 0.1476; P = 0.8687). However, from a statistical point of view, it is more appropriate to use each particular estimate, obtained along with the other time point data, in the regression analysis rather than to use just a single average value.
FIG. 1.

Fitness trajectories followed by the MARM C VSV populations for the three different effective population sizes (indicated in each panel) and replicates. The fitness data have been log-transformed to obtain a linear regression. Error bars represent standard errors.
Clearly, under all three different Ne values, the fitness significantly increased in an exponential fashion (note the log transformation on the y axis). These exponential increases in fitness reflect an improvement in the ability of the virus to replicate in the in vitro cell system, in other words, an increase in viral adaptation. It is a basic principle in evolutionary biology that adaptation always happens by means of natural selection acting on genetic variability (i.e., availability of beneficial mutations). Therefore, the observed improvements in adaptation are due to beneficial mutations arising during the experiment. Thus, a requisite of the model (i.e., the presence of beneficial mutations) holds.
This result is in accord with previously reported findings for the same MARM C clone (25) after short periods of evolution. In experiments involving different VSV clones (9, 25, 26), as well as in experiments with the bacterium E. coli (18, 19), trajectories showing two phases have been observed: fast initial adaptation followed by a decline in the rate of adaptation. This reflects the fact that the speed of evolution slows as the degree of adaptation increases as a consequence of a lower availability of beneficial mutations for the fine-tuning. (An alternative explanation, based on the action of random genetic drift in large populations, has been proposed to explain this deceleration [26], although the argument is flawed because it is based on an incorrect statistical analysis.)
Despite the clear parallel observed between the trajectories followed under the three different effective population sizes (Ne), an analysis of variance (Table 2) showed a significant effect associated with Ne (P < 0.0001). However, no significant differences were found between the two replicates of the experiment (P = 0.1355). A significant effect of Ne on the adaptation of viral populations is predicted by the clonal-interference model. However, it is necessary to test whether the way in which Ne affects the rate of adaptation is exactly that predicted by the model, i.e., an increasing but decelerating function. We will gain a deeper insight into this question in the next two sections.
TABLE 2.
Analysis of variance for the evolution-of-fitness dataa
| Source of variation | SS | df | MS | F | P |
|---|---|---|---|---|---|
| Time (generations) | 7.1086 | 1 | 7.1086 | 171.7856 | <0.0001 |
| Ne | 1.2067 | 2 | 0.6034 | 14.5809 | <0.0001 |
| Replicate | 0.0940 | 1 | 0.0940 | 2.2722 | 0.1355 |
| Ne by replicate | 0.2552 | 2 | 0.1276 | 3.0833 | 0.0511 |
| Error | 3.4346 | 83 | 0.0414 | ||
| Total | 29.5450 | 90 |
SS, sum of squares; df, degrees of freedom; MS, mean squares; F, Snedecor's F statistic.
Rate of adaptation.
The fitness data shown in Fig. 1 were fitted to both the log-linear model described in reference 31 and the log-hyperbolic model described in reference 15. In all six cases, the log-linear model provides a significantly better fit to the data than did the log-hyperbolic one. Table 3 shows the corresponding statistics comparing the fit to both models (16). This result is not surprising, since we precisely chose MARM C for this experiment based on the previous observation that its rate of adaptation was constant during the early stages of evolution. The derivative of the linear model, dln W/dt, i.e., its slope, provides an estimate of the rate of adaptation on a logarithmic scale (last column of Table 3). Note that the logarithmic transformation has no effect on the conclusions we draw below.
TABLE 3.
Fit to alternative log-linear and log-hyperbolic models of fitness evolution and estimates of the rate of adaptation for each line in Fig. 1
| Ne | Replicate | Model | R2 | SSRa | df | Partial Fb | P | dln W/dtc |
|---|---|---|---|---|---|---|---|---|
| 9 × 102 | 1 | Log-linear | 0.5324 | 0.4582 | 13 | 3.0526 | 0.1061 | 0.005 ± 0.001 |
| Log-hyperbolic | 0.6272 | 0.3652 | 12 | |||||
| 2 | Log-linear | 0.6329 | 0.3308 | 13 | 0.6474 | 0.4367 | 0.005 ± 0.001 | |
| Log-hyperbolic | 0.6504 | 0.3138 | 12 | |||||
| 6 × 105 | 1 | Log-linear | 0.7624 | 0.3528 | 13 | 0.2856 | 0.6028 | 0.008 ± 0.001 |
| Log-hyperbolic | 0.7679 | 0.3446 | 12 | |||||
| 2 | Log-linear | 0.7253 | 0.3174 | 13 | 0 | 1 | 0.007 ± 0.001 | |
| Log-hyperbolic | 0.7253 | 0.3174 | 12 | |||||
| 3 × 107 | 1 | Log-linear | 0.7783 | 0.5326 | 13 | 0 | 1 | 0.010 ± 0.001 |
| Log-hyperbolic | 0.7783 | 0.5326 | 12 | |||||
| 2 | Log-linear | 0.8287 | 0.6202 | 13 | 0 | 1 | 0.013 ± 0.002 | |
| Log-hyperbolic | 0.5324 | 0.5850 | 12 |
Residual sum of squares.
The degrees of freedom for the partial F test (15) are always unity for the numerator and those corresponding to the more complex model for the denominator.
Values are given with standard errors.
Diminishing-returns effect of population size.
The rates of adaptation calculated above were then regressed with Ne. As described in Materials and Methods, two different models were fitted. The first (linear) model implies that the larger the Ne value, the faster evolution takes place. The second (hyperbolic) model implies the existence of an upper limit for increasing Ne, as predicted for the clonal-interference model: the larger the number of beneficial mutations that coexist at a given time, the greater the competition, thus slowing adaptation. Figure 2 shows the fit of both models to the experimental data. The fit to both the linear (P = 0.0334) and hyperbolic (P = 0.0026) models was significant. Nevertheless, a partial-F test showed that the fit to the hyperbolic model significantly increased the goodness of fit, despite the use of an extra degree of freedom (F1,4 = 25.1180; P = 0.0074). This result clearly confirms the predictions made by the clonal-interference model.
FIG. 2.

Rate of adaptation versus effective population size. The solid line represents the fit to the hyperbolic model that implies the existence of clonal interference (R2 = 0.5039; F2,4 = 37.2301; P = 0.0026). The dashed line represents the fit to a linear model (R2 = 0.5545; F1,5 = 8.4693; P = 0.0334). Note that the Ne axis has been log-transformed to separate the data points. As a consequence, both regressions show this peculiar aspect. Error bars represent standard errors.
DISCUSSION
Our results clearly confirm the important role played by clonal interference in the evolution of asexual RNA viruses, as previously demonstrated (22). As expected from their large population sizes and high genomic mutation rates (more than one per genome and replication round), the number of possible beneficial mutations coexisting at a given time in a population must certainly be large. (The true number of beneficial mutations out of the total number of mutations produced is still an open question; however, our results [22] suggest that approximately 1/108 mutations should be beneficial.) This coexistence of beneficial mutations in different genomes implies that they must compete with each other on their way to fixation. This competition among beneficial mutations has the effect of slowing the rate of adaptation: the larger the number of beneficial mutations competing, the smaller the increase in the rate of adaptation. Acceptance of the clonal-interference model also allows us to infer some other properties of the adaptive evolution of RNA viruses. In particular, we discuss three important implications of the theory for viral evolution: the class of mutations that gets fixed, the existence of transient polymorphisms, and the reason why a high-fitness clone can be eliminated from a population of low-fitness genotypes.
The mutation fixed is necessarily the best possible candidate for the present circumstances.
Regardless of the availability of beneficial mutations (i.e., the product of beneficial-mutation rate and population size), beyond a certain population size (usually large), the adaptive substitutions appear as discrete, rare events. This implies that a clone harboring a given beneficial mutation that has a low frequency in the population or is on its way to fixation has a extremely low probability of undergoing a second beneficial mutation. This affirmation is based upon our previous estimates of the beneficial-mutations rate (22), i.e., μb = 6.39 × 10−8 beneficial mutations per genome and generation. The likelihood that such a clone undergoes two beneficial mutations is just μb2 = 4.08 × 10−15; thus, the required population size for the positive clone (not for the entire population within which it exists) must be greater than 1/4.08 × 10−15 = 2.45 × 1014 to make the second mutational event likely. This number is far larger than the population sizes reached during our experiments.
As a consequence of the interference among beneficial mutations, the one that finally gets fixed corresponds to the best possible alternative, since on its way to fixation it had to outcompete all other coexisting beneficial mutations (13). This fact has important implications, such as in the dynamics of resistance to antiviral drugs: a certain set of mutations conferring resistance to a given antiviral does not necessarily represent the entire set of possible resistance-conferring mutations but simply a subset formed by the better competitors. Therefore, under different circumstances, a different set of mutations could be present.
Common transitory polymorphisms imply viral plasticity.
The clonal-interference model also has another interesting epidemiological consequence. Beneficial mutations that become common but do not achieve fixation due to their interference with the one finally fixed are abundant. This implies that a high polymorphism is expected. As we demonstrated in our previous study (22), the larger the population size, the stronger the effect of clonal interference and hence the more beneficial mutations competing at a given time. This increased polymorphism could be important for the adaptiveness of RNA viruses if the environment fluctuates or rapidly changes, since any of these suboptimal beneficial mutations, which are destined to vanish in the existing environment, could potentially the best one in an alternative environment.
A way to self-protect resident viral population from outsiders.
de la Torre and Holland (5) previously observed that a high-fitness VSV clone seeded at low frequency within a population of lower fitness variants was unexpectedly displaced from the mixture. It was necessary to introduce the high-fitness clone above a certain threshold for it to fix (5, 15). This observation, which was difficult to explain from the classical population genetics theory (other than trivial loss by drift), is easily explained if clonal interference is taken into consideration. Imagine that the superior clone is initially present at a very low frequency. There is a high probability that beneficial mutations will arise in the most common genotype, improving its fitness, interfering with the intruder, and eventually removing the intruder from the population. However, if the initial frequency of the fitter clone is high enough, its frequency in the population will deterministically increase before any low-fitness variant has a chance to find the appropriate beneficial mutation. As a consequence, the outsider gets fixed.
ACKNOWLEDGMENTS
This work was supported by grant PM97-0060-C02-02 from the Spanish Dirección General de Enseñanza Superior. R.M. was supported by a fellowship from the Ministerio de Educación y Cultura.
We thank Paul E. Turner for insightful comments and critical reading of the manuscript and Olga Cuesta for excellent technical assistance.
Appendix
Number of generations of viral replication in a batch culture.
Let us define Ct as the number of available cells at the instant t. Similarly, we define Vt as the number of viral particles produced at the instant t.
At the beginning of the infection, the number of available cells is defined as C0 and the number of viruses transferred from the previous day is defined as V0. At the end of the process of viral infection and cell lysis, the density of viruses will be Vf and no living cells will be present (i.e., Cf = 0).
If all the initially seeded viruses efficiently attach to and infect a cell, the number of available cells at t = 1 will be C0 − V0. If each infected cell produces, on average, a progeny of R viruses, the number of virus produced at t = 1 will be RV0. If all these RV0 viruses efficiently diffuse, as expected in a liquid culture, and attach to the remaining C0 − V0 living cells (RV0 < C0 − V0), a second round of infection and lysis will occur.
At the end of the second round of infection, RV1 = R2V0 viruses will be produced, and therefore, C0 − V0 − V1 = C0 − V0 − RV0 cells would still be alive.
It is possible to obtain a general expression for the above recursion process for any number, t, of these rounds of infection and lysis. The respective densities of virus and number of live cells are given by the following set of equations:
 |
 |
We can solve this set of equation with the final conditions Cf = 0 and Vf and obtain an estimate of the total number of cycles of infection and lysis that occurred within a culture bottle, g, to grow the viral population from V0 to Vf:
 |
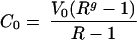 |
where g and R are the two unknown variables of the system. Solving for them, we get
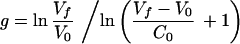 |
and
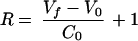 |
Remember that this g does not represents real generations but instead the number of cell infection cycles. To determine the number of generations per infection cycle, we must take into consideration the mechanism that VSV uses to replicate. The infection starts with a negative-sense RNA, which is copied to a few intermediate positive-sense RNAs, which are never encapsidated. These positive-sense RNAs are then used as templates to generate the final negative-sense offspring (1). Therefore, according to this replication model, we estimated the number of generations per cycle of cell infection as 2: the first for generating the positive-sense RNAs from the parental negative-sense RNAs and the second for copying them to the final negative-sense strains. We define generations in this way to take into consideration the fact that there are two moments during each cell infection in which mutations can appear: during the synthesis of the positive strains and later during the synthesis of the negative ones. Whether subsequent positive strains are synthesized (and from them fourth-generation negative strains) is a total mystery. So, conservatively, we can say that only two generations of RNA replication per cell occurred. Thus, the total number of RNA replication generations per infected flask will be
 |
A1 |
REFERENCES
- 1.Banerjee A K. Transcription and replication of rhabdoviruses. Microbiol Rev. 1987;51:66–87. doi: 10.1128/mr.51.1.66-87.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burch C L, Chao L. Evolution by small steps and rugged landscapes in the RNA virus φ6. Genetics. 1999;151:921–927. doi: 10.1093/genetics/151.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chao L. Fitness of RNA virus decreased by Muller's ratchet. Nature. 1990;348:454–455. doi: 10.1038/348454a0. [DOI] [PubMed] [Google Scholar]
- 4.Clarke D K, Duarte E A, Moya A, Elena S F, Domingo E, Holland J J. Genetic bottlenecks and population passages cause profound fitness differences in RNA viruses. J Virol. 1993;67:222–228. doi: 10.1128/jvi.67.1.222-228.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de la Torre J C, Holland J J. RNA virus quasispecies populations can suppress vastly superior mutant progeny. J Virol. 1990;64:6278–6281. doi: 10.1128/jvi.64.12.6278-6281.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Visser J A G M, Zeyl C W, Gerrish P J, Blanchard J L, Lenski R E. Diminishing returns from mutation supply rate in asexual populations. Science. 1999;283:404–406. doi: 10.1126/science.283.5400.404. [DOI] [PubMed] [Google Scholar]
- 7.Duarte E A, Clarke D K, Moya A, Domingo E, Holland J J. Rapid fitness losses in mammalian RNA virus clones due to Muller's ratchet. Proc Natl Acad Sci USA. 1992;89:6018–6019. doi: 10.1073/pnas.89.13.6015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duarte E A, Clarke D K, Elena S F, Moya A, Domingo E, Holland J J. Many-trillion fold amplification of single RNA virus particles fails to overcome the Muller's ratchet effect. J Virol. 1993;67:3620–3623. doi: 10.1128/jvi.67.6.3620-3623.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elena S F, Dávila M, Novella I S, Holland J J, Domingo E, Moya A. Evolutionary dynamics of fitness recovery from the debilitating effects of Muller's ratchet. Evolution. 1998;52:309–314. doi: 10.1111/j.1558-5646.1998.tb01633.x. [DOI] [PubMed] [Google Scholar]
- 10.Elena S F, González-Candelas F, Novella I S, Duarte E A, Clarke D K, Domingo E, Holland J J, Moya A. Evolution of fitness in experimental populations of vesicular stomatitis virus. Genetics. 1996;142:673–679. doi: 10.1093/genetics/142.3.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Escarmís C, Dávila M, Charpentier N, Bracho A, Moya A, Domingo E. Genetic lesions associated with Muller's ratchet in an RNA virus. J Mol Biol. 1996;264:255–267. doi: 10.1006/jmbi.1996.0639. [DOI] [PubMed] [Google Scholar]
- 12.Escarmís C, Dávila M, Domingo E. Multiple molecular pathways for fitness recovery of an RNA virus debilitated by operation of Muller's ratchet. J Mol Biol. 1999;285:495–505. doi: 10.1006/jmbi.1998.2366. [DOI] [PubMed] [Google Scholar]
- 13.Gerrish P J, Lenski R E. The fate of competing beneficial mutations in an asexual population. Genetica. 1998;102/103:127–144. [PubMed] [Google Scholar]
- 14.Holland J J, de la Torre J C, Steinhauer D A, Clarke D K, Duarte E A, Domingo E. Virus mutation frequencies can be greatly underestimated by monoclonal antibody neutralization of virions. J Virol. 1989;63:5030–5036. doi: 10.1128/jvi.63.12.5030-5036.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holland J J, de la Torre J C, Clarke D K, Duarte E A. Quantitation of relative fitness and great adaptability of clonal populations of RNA viruses. J Virol. 1991;65:2960–2967. doi: 10.1128/jvi.65.6.2960-2967.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kleinbaum D G, Kupper L L. Applied regression analysis and other multivariate methods. North Scitute, Mass: Duxbury Press; 1978. [Google Scholar]
- 17.Lefrancois L, Lyles D. The interaction of antibody with the major surface glycoproteins of vesicular stomatitis virus. II. Monoclonal antibodies to nonneutralizing and cross-reactive epitopes. Virology. 1982;121:168–174. doi: 10.1016/0042-6822(82)90126-x. [DOI] [PubMed] [Google Scholar]
- 18.Lenski R E, Rose M R, Simpson S C, Tadler S C. Long-term experimental evolution in Escherichia coli. I. Adaptation and divergence during 2000 generations. Am Nat. 1991;138:1315–1341. [Google Scholar]
- 19.Lenski R E, Travisano M. Dynamics of adaptation and diversification: a 10000-generation experiment with bacterial populations. Proc Natl Acad Sci USA. 1994;91:6808–6814. doi: 10.1073/pnas.91.15.6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miralles R, Moya A, Elena S F. Is group selection a factor modulating the virulence of RNA viruses? Gen Res. 1997;69:165–172. doi: 10.1017/s0016672397002735. [DOI] [PubMed] [Google Scholar]
- 21.Miralles R, Moya A, Elena S F. Effect of population patchiness and migration rates on the adaptation and divergence of vesicular stomatitis virus quasispecies populations. J Gen Virol. 1999;80:2051–2059. doi: 10.1099/0022-1317-80-8-2051. [DOI] [PubMed] [Google Scholar]
- 22.Miralles R, Gerrish P J, Moya A, Elena S F. Clonal interference and the evolution of RNA viruses. Science. 1999;285:1745–1747. doi: 10.1126/science.285.5434.1745. [DOI] [PubMed] [Google Scholar]
- 23.Norusis M J. SPSS for Windows: advanced statistics, release 5. Chicago, Ill: SPSS Inc.; 1992. [Google Scholar]
- 24.Novella I S, Cilnis M, Elena S F, Kohn J, Moya A, Domingo E, Holland J J. Large-population passages of vesicular stomatitis virus in interferon-treated cells select variants of only limited resistance. J Virol. 1996;70:6414–6417. doi: 10.1128/jvi.70.9.6414-6417.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Novella I S, Duarte E A, Elena S F, Moya A, Domingo E, Holland J J. Exponential increases of RNA virus fitness during large population transmissions. Proc Natl Acad Sci USA. 1995;92:5841–5844. doi: 10.1073/pnas.92.13.5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Novella I S, Quer J, Domingo E, Holland J J. Exponential fitness gains of RNA virus populations are limited by bottleneck effects. J Virol. 1999;73:1668–1671. doi: 10.1128/jvi.73.2.1668-1671.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sokal R R, Rohlf F J. Biometry. 3rd ed. New York, N.Y: W. H. Freeman & Co.; 1995. [Google Scholar]
- 28.VandePol S B, Lefrancois L, Holland J J. Sequences of the major antibody binding epitopes of the Indiana serotype of vesicular stomatitis virus. J Virol. 1986;148:312–325. doi: 10.1016/0042-6822(86)90328-4. [DOI] [PubMed] [Google Scholar]
- 29.Wichman H A, Badgett M R, Scott L A, Boulianne C M, Bull J J. Different trajectories of parallel evolution during viral adaptation. Science. 1999;285:422–424. doi: 10.1126/science.285.5426.422. [DOI] [PubMed] [Google Scholar]