

Abstract
Our innate immune system uses pattern recognition receptors (PRRs) as a first line of defense to detect microbial ligands and initiate an immune response. Viral nucleic acids are key ligands for the activation of many PRRs and the induction of downstream inflammatory and antiviral effects. Initially it was thought that endogenous (self) nucleic acids rarely activated these PRRs, however emerging evidence indicates that endogenous nucleic acids are able to activate host PRRs in homeostasis and disease. In fact, many regulatory mechanisms are in place to finely control and regulate sensing of self-nucleic acids by PRRs. Sensing of self-nucleic acids is particularly important in the brain, as perturbations to nucleic acid sensing commonly leads to neuropathology. This review will highlight the role of nucleic acid sensors in the brain, both in disease and homeostasis. We also indicate the source of endogenous stimulatory nucleic acids where known and summarize future directions for the study of this growing field.
1. Introduction
Pattern recognition is an essential function of the innate immune system that is responsible for the immediate sensing of microbes and the initiation of an anti-microbial inflammatory response. Pattern recognition is mediated by various germline-encoded pattern recognition receptors (PRRs) that are expressed in nearly all cell types throughout the body, not just professional immune cells. PRRs recognize distinct pathogen-associated molecular patterns (PAMPs), classically thought to be foreign in origin. This allows cells to differentiate between self and non-self ligands. Many PRRs evolved to detect the presence of foreign nucleic acids, as they are generally indicative of the viral genome invading the cell. Sensing of foreign single-stranded RNA (ssRNA), double-stranded RNA (dsRNA), or cytoplasmic DNA, all originating from viruses, induce an inflammatory response and antiviral immunity.
However, since ssRNA, dsRNA, and DNA are all also prevalent in the host cell, an intriguing question emerges: how do PRRs differentiate self vs. non-self nucleic acids? Now emerging evidence indicates that many nucleic acid-sensing PRRs can also be activated by endogenous (self) nucleic acids in both health and disease, and regulating PRR-self nucleic acid sensing is vital to prevent aberrant PRR activation and cell injury (Ablasser & Hur, 2020; Chen & Hur, 2021; Cottrell, Andrews, & Bass, 2023; Kawasaki & Kawai, 2019; Lind, Rael, Pestal, Liu, & Barton, 2021). These regulatory mechanisms include (but are not limited to) the sequestration of self nucleic acids in separate compartments from their cognate PRRs, and modifying self nucleic acids to reduce immunogenicity. When regulatory mechanisms fail, aberrant activation of PRRs can lead to various autoinflammatory and autoimmune disorders. Such ‘sterile’ activation of both DNA and RNA sensing PRRs have been observed in a wide variety of human diseases, and can either be the initial cause of the disorder (primary), or a result of disease pathology (secondary).
More recent findings suggest that PRRs can also sense self-nucleic acids during homeostasis, and such mechanisms could be critical for development, cell proliferation, and antiviral defense (Escoubas et al., 2024; Dorrity et al., 2023; Gao et al., 2021; Kim et al., 2014; Lammert et al., 2020). Therefore, regulatory mechanisms to prevent self-nucleic acid sensing may have evolved to be partly leaky, permitting host PRRs to sense low levels of self-nucleic acids in a regulated manner. Altogether, self-nucleic acid sensing by PRRs must be balanced: lack of self-nucleic acid sensing could disrupt homeostasis, while excessive self-nucleic acid sensing could cause toxic inflammation.
The central nervous system (CNS) seems particularly sensitive to changes which affect nucleic acid sensing. Gene mutations which lead to the overactivation of nucleic acid-sensing PRRs often cause diseases that primarily affect the nervous system (Bamborschke et al., 2021; Crow & Stetson, 2021; Crow et al., 2006, 2015; Rice et al., 2007, 2009, 2014). Additionally, many neurological disorders or the natural course of aging are accompanied by activation of nucleic acid sensing PRRs (Franceschi, Garagnani, Parini, Giuliani, & Santoro, 2018; Gulen et al., 2023; Roy et al., 2022). Additionally, our recent study found that neurons carry exceptionally high levels of immunostimulatory dsRNA structures that signal through PRRs to protect neurons from viral infection, but could also predispose neurons to toxic inflammation when RNA homeostasis is perturbed (Dorrity et al., 2023). Thus, nucleic acid sensing in the brain walks a tightrope, balancing between the risk of underactivation (infection) and overactivation (inflammation and autoimmunity).
Here, we will review the role of endogenous nucleic acid sensing in the CNS in both health and disease. We will overview the major nucleic acid sensing PRRs present in cells as well as their endogenous ligands, where known. We will then describe the function of these sensors in development, as well as the roles and regulation of these sensors at homeostasis. Finally, we will describe the various neuropathologies which may arise due to disruption of the finely tuned sensing of endogenous nucleic acids. We will conclude with a discussion of opportunities for continued research and therapeutic applications of these findings. Sensing of self-nucleic acids is a growing field and presents myriad opportunities for further study, as well as new avenues for treatment of a broad spectrum of neurologic disease.
2. RIG-I and MDA5
Many viruses produce long dsRNA structures as an intermediate in their replication cycle or as part of their genome. Many PRRs exist to detect viral dsRNAs and induce an immune response, such as the cytosolic RIG-I-like receptors (RLRs). While it was initially thought that long dsRNA structures were unique to viral replication, emerging evidence shows that the human transcriptome also contains relatively long dsRNAs, that can activate PRRs (Chen & Hur, 2021; Cottrell et al., 2023). Of the RLRs, both RIG-I and MDA5 bind dsRNA and signal through the adaptor protein MAVS to produce the antiviral and proinflammatory cytokine type I interferon (IFN) (Fig. 1). However, the two RLRs have distinct activating ligands. MDA5 is activated by very long and perfectly complementary dsRNA, while RIG-I is activated by comparatively shorter 5′ppp-dsRNA (Brisse & Ly, 2019). However, both proteins have been found to contribute to autoimmune disease.
Fig. 1. Canonical downstream signal pathways of nucleic acid sensing PRRs.
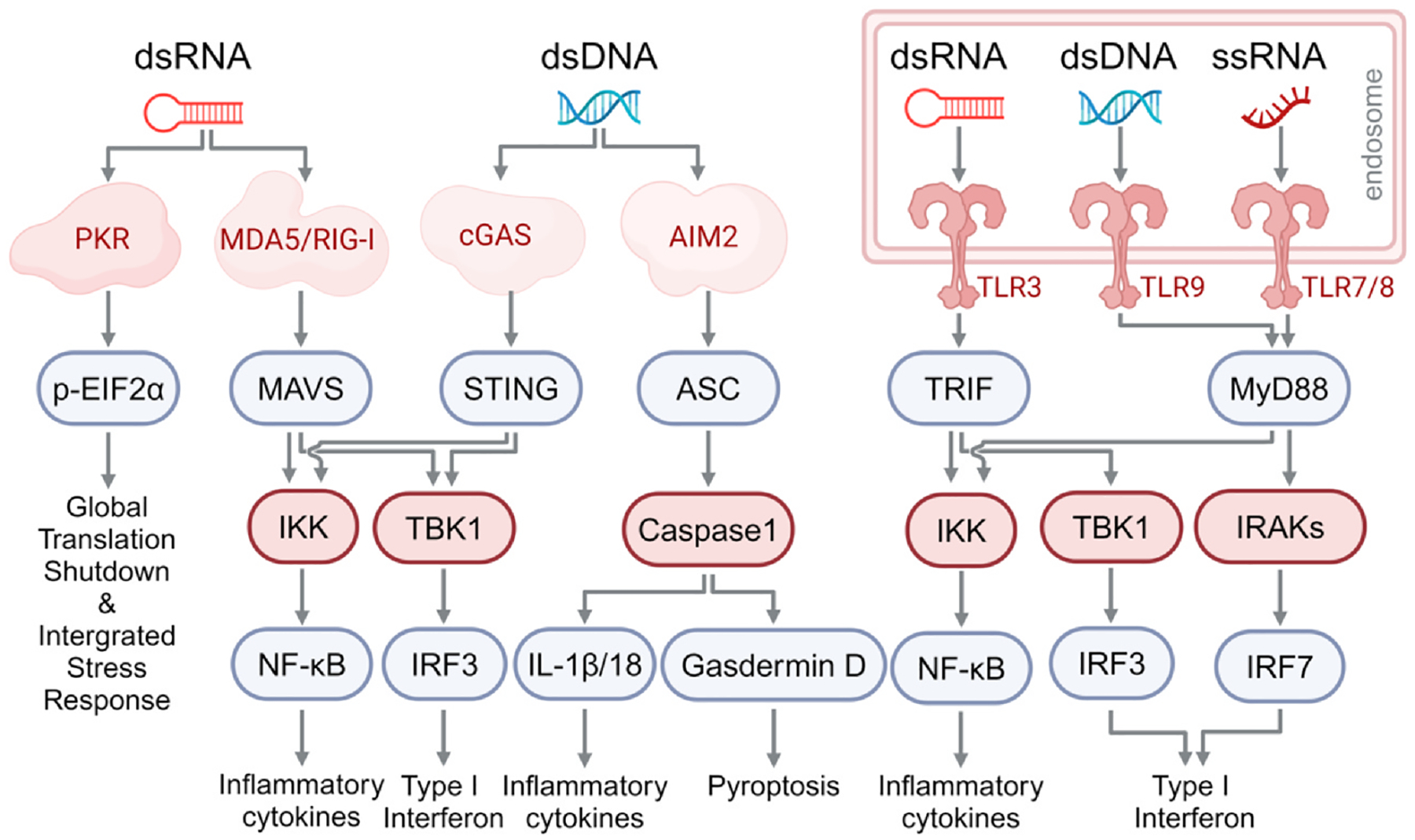
PKR is activated by cytoplasmic dsRNA to phosphorylate eIF2α, leading to global translation shutdown. MDA5/RIG-I are also activated by cytoplasmic dsRNA, and through MAVS, promote the production of inflammatory cytokines via NF-kB and type I IFN via IRF-3. Cytoplasmic dsDNA activates cGAS and AIM2. Activated cGAS induces STING activation through the production of cGAMP, which in turn induces the production of inflammatory cytokines via NF-kB and type I IFN via IRF3. Activated AIM2 induces inflammasome formation and caspase 1 activation, which further cleaves and activates inflammatory cytokines (IL-Ib, IL-18) and gasdermin D, ultimately causing pyroptosis. TLR3 detects dsRNA in the endosome and induces the production of cytokines and type I IFN via TRIF. TLR 7/8 and TLR9 detect ssRNA and dsDNA in the endosome, respectively, and promote the production of inflammatory cytokines and type I IFN via MyD88.
Pertinent to neuroinflammation, MDA5 (encoded by IFIH1) is known to be involved in Aicardi-Goutières syndrome (AGS). AGS is a type I interferonopathy in which patients exhibit spontaneous type I IFN signaling without viral infection, often resulting in childhood death (Crow & Stetson, 2021). Interestingly, AGS patients have much higher levels of type I IFN in the brain compared to the blood, suggesting that the brain is the primary site for type I IFN induction (Crow & Stetson, 2021; Lebon et al., 1988; Lodi et al., 2021). Many of the genes mutated in AGS, including MDA5 (Rice et al., 2014), are involved in either nucleic acid sensing or nucleic acid processing, such as TREX1, PNPT1, SAMHD1, ADAR1, and RNase H2 (Bamborschke et al., 2021; Crow et al., 2006, 2015; Rice et al., 2007, 2009). Some of these mutations, such as ADAR1 and PNPT1 mutants, sensitize MDA5 to sensing of endogenous dsRNAs, potentiating a type I IFN response (Ahmad et al., 2018; de Reuver & Maelfait, 2023; Dhir et al., 2018). Mutations in TREX1, SAMHD1, and RNase H2 cause AGS through DNA sensors, which will be discussed below. Gain-of-function MDA5 mutations alone are sufficient to cause AGS (Ahmad et al., 2018; Onizawa et al., 2021; Rice et al., 2014). Depending on the mutations, mutant MDA5 can either become constitutively active independently of RNA, or become sensitized to activation by host dsRNAs despite RNA modifications that prevent MDA5 activation (Ahmad et al., 2018; Oda et al., 2014). In addition, loss-of-function mutations in the RNA-editing enzyme ADAR1 can lead to aberrant MDA5 activity and cause AGS (Fig. 2B). ADAR1 introduces A-to-I edits in dsRNA substrates, and ADAR1 editing of RNA duplexes has been proposed to disrupt dsRNA structures, allowing edited RNAs to evade recognition by MDA5. The role and mechanism of ADAR1 in various human diseases have been well reviewed previously (de Reuver & Maelfait, 2023; Lamers, Van Den Hoogen, & Haagmans, 2019; Livingston & Crow, 2016; Samuel, 2019). Additionally, mutations in MDA5 are associated with systemic lupus erythematosus (SLE), which is frequently accompanied by neuropsychiatric symptoms (Brey et al., 2002; Robinson et al., 2011). This demonstrates that MDA5 dysfunction can be associated with broad neurological disorders, and more work is necessary to identify further cases of MDA5-mediated neuroinflammation.
Fig. 2. Extracellular and intracellular self-nucleic acids.
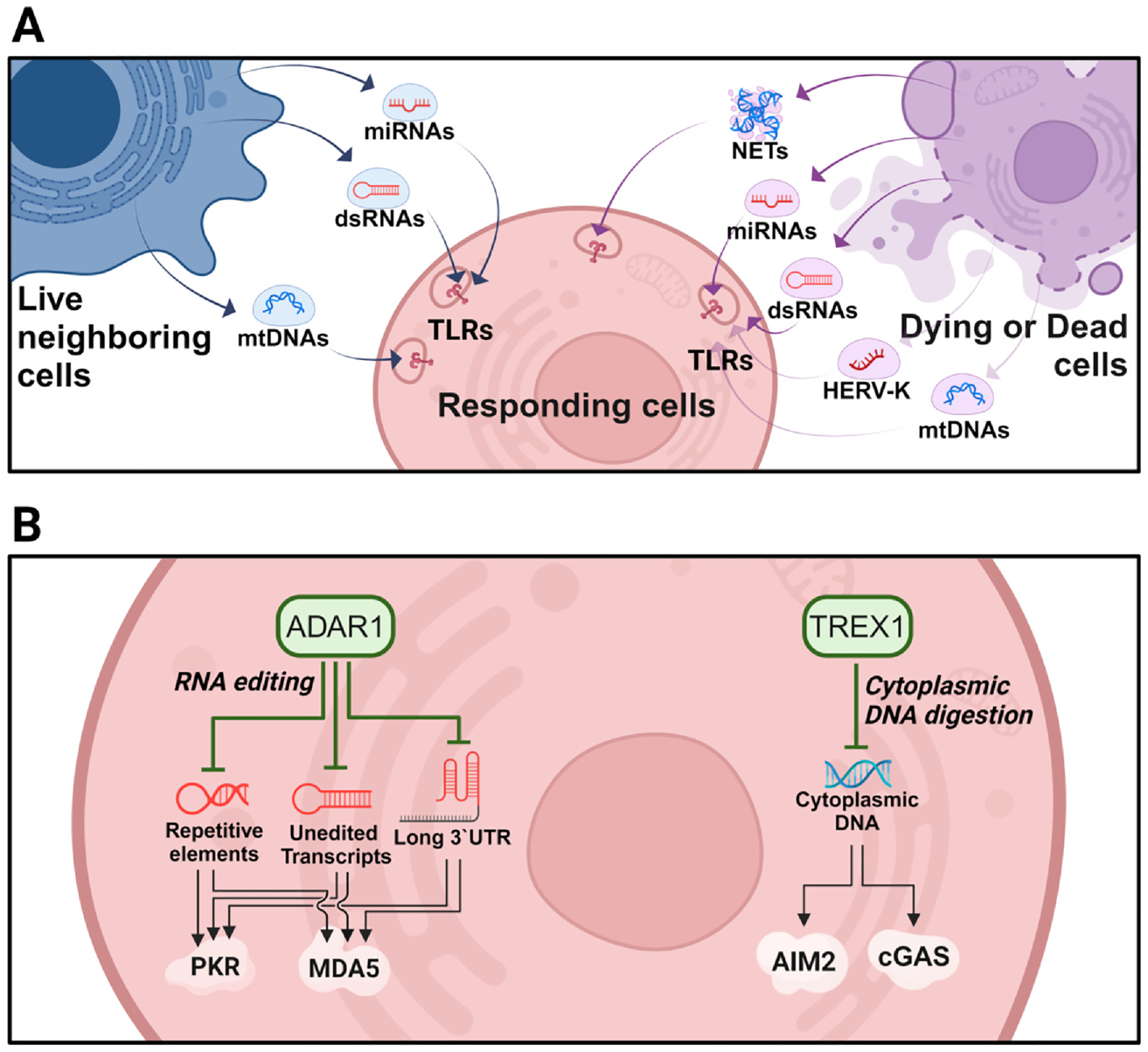
(A) Extracellular nucleic acids originating from neighboring live, dead, or dying cells. Released miRNAs, dsRNAs, HERV-K, mtDNA, and NETs can activate TLRs (TLR3, 7/8, and −9) in nearby responding cells. (B) Intracellular nucleic acids originating from the loss of regulators within cells. ADAR1 and TREX1 are major regulators of immunostimulatory self-RNA and self-DNA nucleic acids, respectively. Loss of these regulatory mechanisms can lead to hyperactivation of various PRRs and promotes neurodegeneration.
MDA5 is also able to produce tonic IFN under homeostatic conditions (Dorrity et al., 2023). MDA5-dependent tonic type I IFN is constitutively induced by the high dsRNA burden in neurons under homeostatic conditions (Dorrity et al., 2023). The role of tonic inflammation in the brain is an understudied topic, but previous studies have demonstrated that low levels of tonic type I IFN is necessary for neural homeostasis, cortical development, maintenance of plasticity, and protection from viruses (Escoubas et al., 2024; Dorrity et al., 2023; Ejlerskov et al., 2015; Gao et al., 2021; Hosseini et al., 2020). This role for MDA5 in the production of tonic type I IFN, as well as its role in AGS, demonstrates that MDA5 activity must be closely regulated to balance the needs of homeostatic IFN while avoiding toxic inflammation.
RIG-I can also be activated in non-infectious disorders. For example, mutations in the RIG-I encoding gene DDX58 are associated with the type I interferonopathy Singleton-Merten syndrome (SMS) (Jang et al., 2015), although SMS is not a neurological disorder. Within the brain, sterile RIG-I activation has been associated with both neurodegenerative diseases and injury. RIG-I expression is elevated in the temporal cortex of patients in the early stages of Alzheimer’s disease (De Rivero Vaccari et al., 2014). In primary human astrocytes, a positive feedback loop exists between amyloid β and RIG-I, in which RIG-I activation increases amyloid β expression, and increased amyloid β expression increases RIG-I expression (De Rivero Vaccari et al., 2014). In a rat model of stroke, both RIG-I and type I IFN are elevated in the hippocampus following injury, especially in astrocytes (Brand, De Rivero Vaccari, Mejias, Alonso, & De Rivero Vaccari, 2015). However, it is important to note that in both of these cases, future studies need to determine the role of RIG-I activation in neural disease progression.
For both MDA5 and RIG-I, the exact identity of the self-ligands that induce neuroinflammation is not yet known (Fig. 3). Recent data, although not in the neural context, demonstrated that double-stranded mitochondrial RNA (mtRNA) can activate MDA5 in human cells (Dhir et al., 2018; Tigano, Vargas, Tremblay-Belzile, Fu, & Sfeir, 2021). Additionally, dsRNA formed from primate-specific SINE Alu elements has been shown to be a common activating ligand for MDA5 (Ahmad et al., 2018). Many Alu elements are located in 3′ untranslated regions (3′UTRs), and this coincides with our recent data showing that elongated 3′UTRs activate MDA5 in human neurons (Dorrity et al., 2023). Additionally, it has been proposed that only a small subset of endogenous dsRNAs can activate RLRs, but the identity of immunostimulatory self-RNAs remain elusive (Levanon et al. 2024). More research should be directed to determine the RNA species that activate RLRs, including RNA ligands that may be specific to the neural compartment.
Fig. 3. Intra-and extracellular self-ligands of PRRs leading to downstream effects in the brain.
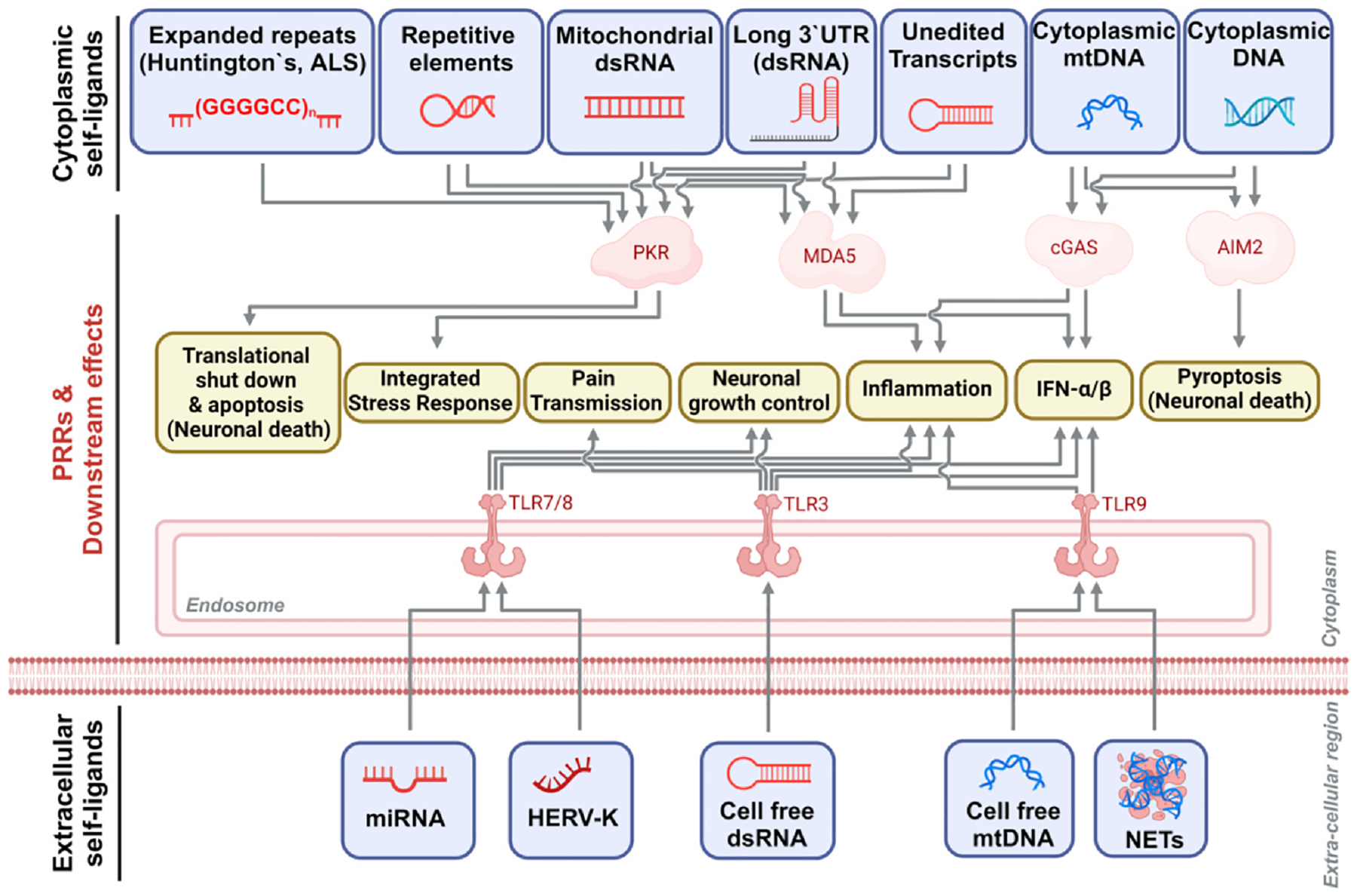
In the cytoplasm, expanded repeats, long 3′UTRs in mRNA, double-stranded mitochondrial RNAs (mtRNAs), and unedited transcripts can activate PKR and MDA5, stimulating inflammation, type I IFN production, and neuronal apoptosis triggered by translational shutdown. Cytoplasmic DNA and mitochondrial DNA (mtDNA) can stimulate cGAS and AIM2 activity to induce inflammation and pyroptosis. Extracellular miRNA, HERV-K, cell free dsRNA, mtDNA, and NETs can activate TLRs in neighboring cells leading to pain transmission, neuronal growth control, inflammation, and type I IFN production.
3. PKR
Many PRRs induce inflammation in an indirect manner by inducing proinflammatory cytokines such as type I IFN. In contrast, Protein Kinase R (PKR) is a cytosolic and ubiquitously expressed PRR in all cell types, and is capable of direct anti-viral action. Upon binding dsRNA, PKR homodimerizes, autophosphorylates, and becomes active, ultimately phosphorylating eIF2α and leading to global translational shutdown and an integrated stress response (ISR) (Fig. 1) (Gal-Ben-Ari, Barrera, Ehrlich, & Rosenblum, 2019). While this is critical for inhibiting viral replication, it also has a multitude of effects on cells both in health and disease. The role of PKR in neuroinflammation was initially described in Huntington’s disease, where it was shown that PKR was activated in patient brain tissue, specifically in hippocampal neurons, and that repeat expansions in huntingtin transcripts were able to activate PKR (Bando et al., 2005; Peel et al., 2001). More recently, mtRNAs have been proposed to activate PKR in a Huntington’s disease model (Lee et al., 2020).
In patients with Alzheimer’s disease, it was found that neurons had high levels of activated PKR (phosphorylated PKR) and phosphorylated eIF2α (Chang, Wong, Ng, & Hugon, 2002). It was shown that amyloid β plaques was able to activate PKR, and experimental vaccines against amyloid β were able to reduce PKR activation in neurons (Chang et al., 2002; Paquet et al., 2015). In mouse models of Alzheimer’s disease, depletion of PKR drastically improved memory and cell health, linking PKR activation with disease pathology (Mouton-Liger et al., 2015; Tible et al., 2019).
In addition, PKR activation has been observed in other neurodegenerative diseases, such as amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). One of the driving causes of familial ALS-FTD is the expansion of the G4C2 repeat sequences in the C9orf72 gene (Balendra & Isaacs, 2018). Recently, enhanced PKR activation was confirmed in the frontal cortex of ALS patient tissue, and RNA repeat expansions in C9orf72 have been proposed to form dsRNAs and activate PKR in primary neurons (Parameswaran et al., 2023; Rodriguez et al., 2021; Zu et al., 2020). Additionally, in a rat model of ALS, downregulation of TAR DNA-binding protein 43 (TDP-43), an RNA binding protein implicated in ALS, resulted in accumulation of repetitive elements and dsRNAs that activated PKR in astrocytes and contributed to disease progression (LaRocca, Mariani, Watkins, & Link, 2019). Furthermore, inhibition of PKR in a mouse model of ALS/FTD, drastically improved cognitive outcomes (Zu et al., 2020). PKR has even been shown to modulate memory. Inhibition of PKR in the mouse brain improved memory scores, and increased PKR activity in the hippocampus of healthy aged mice correlated with lower cognitive scores (Gal-Ben-Ari et al., 2019; Lu et al., 2022). Overall, PKR contributes to the progression of several neurodegenerative diseases, and warrants further study as a key mediator of neurodegeneration in general. The role of PKR in memory function is a more recent discovery, and more research should be directed at this topic to explore the potential of PKR as a target for pharmacological intervention.
In addition to its role in neurodegeneration, mutations in PKR and regulators of PKR have been directly implicated in several other neurological disorders. Initially, mutations in protein activator of PKR (PACT), a protein modulator of PKR, were identified in patients with dystonia, a disorder of the nervous system characterized by uncontrolled movements (Camargos et al., 2008). These PACT mutations increased PKR activation and induced apoptosis in patient derived cells (Burnett, Vaughn, Sharma, Kulkarni, & Patel, 2020; Vaughn et al., 2015). More recently, apparent gain-of-function mutations in the PKR gene EIF2AK2 were identified in patients exhibiting dystonia, further supporting PKR’s role in this phenomenon (Kuipers et al., 2021).
PKR is also involved in AGS, as is demonstrated in various ADAR1 depletion models. In neural cells, loss of ADAR1 led to cell death induced by PKR activation (Fig. 2B) (Chung et al., 2018; Dorrity et al., 2023). In an ADAR1 defective mouse model for AGS, inhibition of the integrated stress response induced by PKR activity was sufficient to prevent pathology (Maurano et al., 2021). Additionally, depletion of both MDA5 and PKR in ADAR1p150−/− mice (ADAR1p150 is an isoform of ADAR1) was sufficient to rescue lethality of ADAR1p150−/− mice and allow the mice to live to adulthood (Hu et al., 2023). These data indicate that PKR could be a major driver of cell death in AGS (Fig. 3).
Beyond disease, emerging data suggest that PKR may also play a role in healthy brain tissue. Potential loss-of-function mutations in EIF2AK2 were identified in patients with a wide array of neural abnormalities, most prominently leukoencephalopathy (Mao et al., 2020). This demonstrates a potential role of PKR in neural development, although this idea warrants further exploration. Additionally, low grade PKR activation in neurons may contribute to the tonic antiviral state induced by neuronal dsR NAs (Dorrity et al., 2023). Indeed, mice lacking PKR are extremely susceptible to viral infection (Balachandran et al., 2000). PKR activation therefore must be tightly regulated, balancing antiviral effects with the risk of sustained translational shutdown.
4. TLR3
TLR3 is a toll-like receptor family PRR that detects dsRNA in endosomes. In contrast to the cytosolic PRRs, TLR3 is mainly found in the endocytic compartment. Activated TLR3 transmits signals through the TRIF pathway, activating and translocating IRF3 into the nucleus, leading to the production of type I IFN (Fig. 1). TLR3 is broadly expressed in the CNS across various neural cell types (Bsibsi, Ravid, Gveric, & Van Noort, 2002; Jack et al., 2005). TLR3 is especially highly expressed in glial cells such as astrocytes. TLR3 on astrocytes is significantly induced in response to viral infection or external cytokines (Bsibsi et al., 2006). Additionally, in the blood-brain barrier, TLR3 on astrocytes detects danger signals and mediates neuroprotection (Bsibsi et al., 2006).
Two recent studies demonstrated that TLR3 expressed on neurons is capable of inducing low levels of constitutive (‘tonic’) type I IFN in non-infected, homeostatic conditions (Dorrity et al., 2023; Gao et al., 2021). Further corroborating these findings, tonic type I IFN is also detected in non-diseased human brain tissue (Dorrity et al., 2023), and activation of TLR3 (and MDA5) was shown to be driven by neuronal dsRNAs derived from long 3′UTRs (Fig. 3) (Dorrity et al., 2023). This tonic type I IFN produced from neurons has been proposed to pre-emptively protect the CNS from viral infection, circumventing the need for a more extreme inflammatory response (Paludan & Mogensen, 2021). Supporting this idea, a series of human genetic studies found that loss-of-function mutations in the TLR3 pathway cause increased susceptibility to herpes simplex encephalitis (Casrouge et al., 2006; Gao et al., 2021; Zhang et al., 2007). Moreover, our recent study suggested that constitutive type I IFN made by neurons leads to resistance to infection by HSV-1, Zika virus, and Sindbis virus (Dorrity et al., 2023). Therefore, constitutive TLR3 activation in neurons is likely to be critical to preemptively protect neurons from infection with diverse viruses.
In addition to neurons, sterile activation of TLR3 was also identified in neural progenitor cells (NPCs), the common precursor cell type in the CNS. TLR3 knockout in NPCs increased proliferation, while activation of TLR3 decreased proliferation (Lathia et al., 2008). In adult mice, TLR3 knock-out increases neuronal abundance and leads to increased hippocampal volume and structural changes (Okun et al., 2010). As such, the persistent TLR3 activation in neurons not only promotes immune activity but may also contribute to neuronal development.
The dysregulation of TLR3 has been widely observed in various brain pathologies. A histologic study confirmed that TLR3 expression was increased in the brains of patients with Alzheimer’s disease compared to healthy controls (Bsibsi et al., 2002; Walker, Tang, & Lue, 2018), although interestingly, increased TLR3 expression is not found in the neurons of these samples. Moreover, other studies showed that TLR3 plays a key role in producing high levels of pro-inflammatory cytokines, leading to neuronal death in Alzheimer’s, Parkinson’s, prion, and chronic inflammatory disease models in diverse organisms (Deleidi, Hallett, Koprich, Chung, & Isacson, 2010; Fiebich, Batista, Saliba, Yousif, & de Oliveira, 2018; Field, Campion, Warren, Murray, & Cunningham, 2010; Walker et al., 2018; Zhang et al., 2023).
Currently, the self-RNA ligands that activate TLR3 are not yet well identified. Recently, it was reported that peripheral chronic injury in mice causes TLR3-mediated cognitive decline in the CNS (Zhang et al., 2023). The authors analyzed mediators linking peripheral nervous system pain signals to inflammatory responses in the CNS. Interestingly, chronic stress promoted the release of self-dsRNAs from Schwann cells, a type of glial cell. Accumulated external dsRNAs activated TLR3 in neurons, confirming inflammatory signal transduction by intercellular RNA exchange. Similarly, several studies have shown that dsRNAs released from damaged tissue activate TLR3 on neighboring cells (Chen et al., 2014; Nelson et al., 2015), although the exact identity of dsRNAs that activate TLR3 are not well defined (Figs. 2A and 3). Our recent study proposed that long 3′UTRs in neurons are a major source for dsRNAs that activate TLR3 on neurons, but it is unclear how intracellular long 3′UTRs reach TLR3 in the endosome (Dorrity et al., 2023). It is imperative that more research be conducted into identifying the self ligands that activate TLR3, and further determine how these ligands enter the endosome to activate TLR3.
5. TLR 7/8
Both TLR7 and TLR8 recognize GU-rich single-stranded RNAs (ssRNA) in endosomes. Once either of these receptors are activated, the signal is transmitted via MyD88 to ultimately induce immune gene activation and the production of IFN and NF-κB induced pro-inflammatory cytokines (Fig. 1) (Diebold, Kaisho, Hemmi, Akira, Reis, & Sousa, 2004; Heil et al., 2013). Infection with RNA viruses triggers TLR7/8 activation and initiates an anti-viral response (Lund et al., 2004). Within the CNS, TLR7/8 is expressed in microglia and neurons beginning in the postnatal stage (Bsibsi et al., 2002; Kaul et al., 2012; Ma, Haynes, Sidman, & Vartanian, 2007; Olson & Miller, 2004).
Aside from infection models, TLR7 activation in B cells is also well-characterized as key to the development of SLE (Brown et al., 2022; Fillatreau, Manfroi, & Dörner, 2020). Neuropsychiatric SLE is a common manifestation of SLE, comprised of various of cognitive impairment, seizures, and psychiatric disorders (Brey et al., 2002). The pathogenesis of Neuropsychiatric SLE is complex, comprising of blood-brain barrier disruption (Kamintsky et al., 2020), antibody deposition in the brain (Cocco et al., 2023), and vascular lesions and complement activation (Cohen et al., 2017) in the brain. In addition, TLR7 activation has been found to worsen neural autoimmunity by downregulating T regulatory cell levels in a mouse model of Experimental Autoimmune Encephalitis, a model of multiple sclerosis (Lalive et al., 2014). Aberrant TLR7 signaling can, therefore ultimately lead to severe autoimmune disease in the brain, even if the proximal signaling is apparently in the periphery.
At homeostasis, TLR7/8 has been found to affect neuronal growth in mouse in vivo and ex vivo models. TLR7-deficient mice and mouse neurons demonstrated significantly increased axonal growth (Liu et al., 2013). Conversely, treatment of cultured primary mouse neurons with R848, a TLR7/8 agonist, reduced neurite outgrowth and promoted apoptosis (Ma et al., 2006). Independently, several studies confirmed a key role of TLR7/8 in neural remodeling (Hung et al., 2018; Liu et al., 2013; Liu, Huang, Hung, & Hsueh, 2015; Ma et al., 2006; Mukherjee et al., 2015).
Micro RNAs (miRNAs) are small nucleotide noncoding RNA that are 19–24nt in length. The canonical function of miRNAs is controlling the stability of mRNAs and ultimately the expression of the encoded genes (Kim, 2005). It has been shown that some GU-rich miRNAs can directly act as ligands that activate TLR7/8 in the absence of infection (Fig. 3) (Chen, Liang, Zhang, Zen, & Zhang, 2013; Fabbri et al., 2012; Fabbri, Paone, Calore, Galli, & Croce, 2013; Feng et al., 2017; He et al., 2014; Liu et al., 2015; Zhang et al., 2018). miRNAs can be found in exosomes and secreted to the extracellular space, where they can then fuse with other cells (Fig. 2A) (Rabinowits, Gerçel-Taylor, Day, Taylor, & Kloecker, 2009; Zhang et al., 2015). During this process, GU-rich miRNAs can enter the endosome and trigger TLR7/8 activation (Fabbri et al., 2013; Feng et al., 2017; Liu et al., 2015; Zhang et al., 2018), potentially acting as an inter-cell message in noninfectious states.
Let-7 miRNA family and miR-21 are well-characterized miRNAs that can act as TLR7/8 ligands. In a mouse model, Let-7c and miR-21 were found to be abundantly expressed in the brain and secreted in exosomes. These secreted miRNAs were found to inhibit axonal growth by activating TLR7 in neighboring neurons (Liu et al., 2015). In addition to inhibiting axonal growth, these signals have a neuroprotective effect in a mouse model of glioblastoma multiforme (Buonfiglioli et al., 2019), again demonstrating a tension between the risks of over-activation and the danger of an insufficient immune response.
MicroRNA-TLR7 interactions appear to cause or exacerbate a range of neurologic disease. Upregulation of TLR7 has been found in the brains of patients with Alzheimer’s disease (Bsibsi et al., 2002; Lehmann et al., 2012; Letiembre et al., 2009). Coupled with that, levels of the microRNA let-7b were also higher in the cerebrospinal fluid of patients with Alzheimer’s disease compared to healthy controls. This led to the suggestion that in the brains of patients with Alzheimer’s disease, let-7b-TLR7 interactions were abnormally amplified leading to neuron demise (Lehmann et al., 2012). Similar let-7b mediated interactions were also found in a rat model of alcohol-induced neurotoxicity (Coleman, Zou, & Crews, 2017). In addition, in a murine model of sepsis, an increased level of extracellular miR-146a was observed, and it induced hyperactivation of neuronal TLR7 and neuronal death (Zou et al., 2022). Lastly, miR-340 and miR-132, both associated with brain injury, has been proposed to activate TLR7/8 (Wallach et al., 2020).
Human endogenous retroviruses-K (HERV-K) RNA is another type of self-RNA that has been shown to interact with TLR7/8 (Fig. 3). Like let-7b, it is more abundant in the CSF of Alzheimer’s patients and leads to neurodegeneration when introduced to the CSF of mice (Dembny et al., 2020). Taken together, some miRNAs and HERV-K activate TLR7/8 in the CNS. These self-RNA-mediated sterile activation of TLR7/8 regulate neuronal growth under normal conditions. However, it can also cause neurodegeneration during various disease conditions, such as Alzheimer’s disease, alcoholism, and sepsis.
6. cGAS
In non-mitotic cells, DNA is sequestered in the nucleus. DNA in the cytoplasm is a common sign of pathogenic infection or cellular damage, and many PRRs have evolved to detect cytosolic DNA. Cyclic GMP-AMP synthase (cGAS) is a PRR that recognizes double-stranded DNA (dsDNA). Upon binding dsDNA, cGAS produces the second messenger cGAMP from GTP and ATP. cGAMP then binds to stimulator of interferon genes (Ishikawa et al. Nature 2009), inducing the production of type I IFN and other proinflammatory cytokines (Fig. 1) (Hopfner & Hornung, 2020). The ability for cGAS to induce sterile inflammation is well studied. For instance, DNA damage or mitochondrial stress has been shown to cause nuclear DNA or mitochondrial DNA (mtDNA), to leak into the cytoplasm and activate cGAS, respectively (Fig. 3) (Li & Chen, 2018). Age-related DNA damage and cellular injury can both lead to cGAS-induced sterile neuroinflammation (Gulen et al., 2023). Additionally, certain retroelements, such as LINE-1, could be retrotranscribed at higher rates during aging, and can activate cGAS in the cytoplasm (Gorbunova et al., 2021).
“Inflamm-aging” is a term describing the observation that chronic inflammation is induced by aging, yet the pathways that cause this inflammation are poorly understood (Franceschi et al., 2018). Recently, the cGAS-STING pathway has emerged as a major participant of inflamm-aging (Gulen et al., 2023). It was also shown that cGAS is activated in the brains of healthy aged mice and older humans (Xie et al., 2023). More specifically, the cGAS-STING pathway is activated in aged murine microglia by release of mtDNA into the cytoplasm, and this cGAS signaling is sufficient to induce age-associated transcriptional changes (Gulen et al., 2023). Thus, cGAS activity in the brain can be induced via normal aging processes.
Aging is a major risk factor for many neurodegenerative disorders that are accompanied by inflammation. Abnormal cGAS activation in the brain has been observed in murine models of Alzheimer’s disease (Udeochu et al., 2023; Xie et al., 2023). Cytoplasmic mtDNA was demonstrated to activate cGAS in the murine hippocampus, and inhibition of cGAS was sufficient to ameliorate amyloid β-induced neurotoxicity (Xie et al., 2023). It was also shown that tau protein was responsible for inducing mtDNA leakage in microglia (Udeochu et al., 2023). Additionally, it has also been shown that microglia sense tau protein through the cGAS-accessory protein PQBP1, and this induces cGAS-dependent inflammation in mice (Jin et al., 2021). Sustained cGAS activity in microglia also causes memory impairment and neurotoxicity (Gulen et al., 2023). cGAS activity is also associated with other neurodegenerative disorders. In human organoid models of ataxia-telangiectasia, cGAS was shown to induce inflammation and senescence in response to micronuclei formation, and cGAS inhibition reduces ataxia-related neuropathology (Aguado et al., 2021). Lastly, accumulation of TDP-43 is a disease hallmark for ALS, and in ALS, inflammation can be driven by the cGAS-STING pathway when TDP-43 invades the mitochondria and releases mtDNA (Yu et al., 2020). Therefore, cGAS is activated in multiple neurodegenerative disorders and contributes to disease pathology.
cGAS-induced inflammation has also been shown to contribute to AGS pathology. While cGAS mutations have not been identified in AGS patients, mutations have been found in the cytosolic exonuclease TREX1 (Fig. 2B) (Crow et al., 2015; Rice et al., 2007; Wu, Fang, Huang, & Ying, 2021). TREX1 degrades cytosolic DNA from all sources, and thus inhibits cGAS activation (Thomas et al., 2017; Hemphill et al., 2021; Wang, Du, Hua, & Zhao, 2022; Yang, Lindahl, & Barnes, 2007). Loss of TREX1 in mice results in AGS symptoms which is caused by excessive cGAS activation, and co-deletion of cGAS reduces pathology (Crow & Stetson, 2021; Gao et al., 2015).
cGAS-induced neuroinflammation has also been studied in relation to traumatic brain injury (TBI). TBI induced DNA damage and cGAS activation in the cortex of mice (Barrett et al., 2021). This cGAS activation is likely induced by cytoplasmic mtDNA, and loss of cGAS is neuroprotective (Fritsch et al., 2022). Similarly, repetitive mild TBI causes cGAS activation, which induces a senescent phenotype in both neurons and astrocytes (Schwab et al., 2022). Cellular damage induced by stroke also induces cGAS-mediated neuroinflammation, likely in response to DNA damage (Li et al., 2020). Inhibition of cGAS in mouse models of stroke, specifically in microglia, results in lowered inflammation, lower pyroptosis, and less neuronal death (Jiang et al., 2021; Li et al., 2020).
Due to its involvement in many age and injury-related neurological disorders, cGAS is a growing target for pharmacological intervention. Although cGAS has been discovered relatively recently (Sun, Wu, Du, Chen, & Chen, 2013; Wu et al., 2013), cGAS specific inhibitors have been developed and new inhibitors are actively under development. These cGAS inhibitors are broadly characterized into those that block DNA binding and those that inhibit cGAMP synthesis (Decout, Katz, Venkatraman, & Ablasser, 2021; Li et al., 2021; Wang et al., 2023).
7. AIM2 inflammasome
The inflammasome is a multi-protein immunological complex that induces the production of cytokines and can initiate pyroptosis. The inflammasome can be activated by several different PRRs, one of which is the cytoplasmic dsDNA sensor absent in melanoma 2 (AIM2) (Hornung et al., 2009). AIM2 binds DNA in a sequence-independent manner, and once activated, recruits ASC via its pyrin domain (Lugrin & Martinon, 2018). ASC allows for caspase-1 to be recruited to the inflammasome. Once assembled, the AIM2 inflammasome complex further enables the proteolytic activity of caspase-1 on pro-IL-1β, pro-IL-18, and gasdermin D (GSDMD), leading to the maturation of cytokines and GSDMD mediated pro-inflammatory form of cell death called pyroptosis (Fig. 1). Host DNA can activate AIM2 upon weakening or disruption of the nuclear envelope, which causes DNA to mislocalize to the cytosol (Lugrin & Martinon, 2018). Additionally, AIM2 can sense radiation-induced DNA damage in the nucleus to mediate inflammasome activation and cell death (Hu et al., 2016).
AIM2 has been shown to be critical during homeostasis in order to promote normal brain development and function. In murine neurons, loss of AIM2 led to abnormal neural growth and neurite formation due to insufficient IL-1β signaling (Wu, Liu, Huang, & Hsueh, 2016). Additionally, AIM2 knockout mice exhibited memory defects and abnormal behavior associated with increased anxiety (Lammert et al., 2020; Wu et al., 2016). Neurodevelopment is characterized by rapid rates of neural cell proliferation and death, a process that naturally leads to large amounts of DNA damage and cellular debris. Such DNA damage has been shown to activate AIM2 in the developing brain, and this is crucial for inducing pyroptosis and clearing unnecessary cells and neurons that accumulate too much genetic damage (Fig. 3) (Lammert et al., 2020). Altogether, this demonstrates that homeostatic function of AIM2 is pivotal for proper function in neuronal tissues.
However, overactivation of AIM2 in the brain can inhibit healing and induce neurodegeneration. Cellular injury can induce DNA mislocalization and DNA damage, both of which induce AIM2 activation. AIM2 activation in the cortex and hippocampus following stroke leads to neural death via pyroptosis, restricting recovery (Kim et al., 2020). Loss of AIM2, or of the critical inflammasome component ASC, reduces signs of injury in a murine model of stroke (Denes et al., 2015). Drug inhibition of AIM2 or caspase 1 attenuated cognitive defects following stroke (Kim et al., 2020; Zhang et al., 2020). In addition to stroke, AIM2 activation is observed following TBI. Specifically, AIM2 activation was observed in brain microvascular endothelial cells of the blood-brain barrier, and inhibition of AIM2 resulted in lessened pathology (Ge et al., 2018). Thus, AIM2 activation following brain injury is deleterious.
Finally, the AIM2 inflammasome likely contributes to various forms of dementia. In a mouse model of vascular dementia, AIM2 inflammasome activation was observed in hippocampal neurons and various glial cells (Poh et al., 2021). Concurrent knockout of AIM2 allowed mice to resist demyelination and improved cognitive scores (Poh et al., 2021). AIM2 activation has also been observed in a murine model of Alzheimer’s disease. Interestingly though, while loss of AIM2 decreased amyloid β deposition, inflammation remained high and memory function did not improve (Wu, Hung, Liu, & Hsueh, 2017). Additionally, the exact ligands that activate AIM2 during neurodegeneration remains unclear. Importantly, AIM2 expression increases with age in the mouse brain (Kim et al., 2020), indicating that the threshold to activate AIM2 may decrease over the course of aging, leading to aberrant AIM2 activation. Regulation of AIM2 activation is especially critical, as inflammasome activation can lead to rapid cell death. Yet as AIM2 activation is also involved in normal brain development, a strict regulatory balance must be achieved.
8. TLR9
TLR9 is mainly located in the endosome and recognizes unmethylated CpG DNA motifs, as viral DNA is unmethylated (Hemmi et al., 2000). Once activated, TLR9 generates an innate immune response via the MyD88 pathway to eventually produce pro-inflammatory cytokines and type I IFN (Fig. 1) (Kumagai, Takeuchi, & Akira, 2008). Within the CNS, it is predominantly expressed in microglia and neurons (Bsibsi et al., 2002). Mammalian DNA CpG motifs derived from the nucleus do not activate TLR9 as it is methylated. However, mtDNA can potentially activate TLR9 (Costa et al., 2022; Garcia-Martinez et al., 2016; Rodríguez-Nuevo et al., 2018). Under normal conditions, mtDNA is tightly sequestered from PRRs by the mitochondrial double membrane. However, various stressors such as aging, tissue injury, metabolic stress, etc., lead to mitochondrial instability and the subsequent release of mtDNA into the cytosol or extracellular space, eventually making their way into endosomes. Extracellular mtDNA, otherwise known as cell-free mtDNA (cf-mtDNA), is known to be a ligand that induces sterile activation of TLR9 (Figs. 2A and 3).
There is a strong correlation between psychological stress and cf-mtDNA levels (Cai et al., 2015; Lindqvist et al., 2018; Trumpff et al., 2021). cf-mtDNA has been shown to be induced by acute episodes of psychological stress. Examination of cf-mtDNA in the blood of 236 patients with various psychopathologies revealed a distinct increase compared to healthy controls (Trumpff et al., 2021). Strikingly, up to 46.8 times more cf-mtDNA was measured in the blood of those who attempted suicide than in controls (Trumpff et al., 2021). Significant increases in cf-mtDNA were also seen in response to acute stresses such as anger and anxiety, with the authors directly observing that treatment of primary human fibroblasts with the stress hormone glucocorticoid resulted in secretion of mtDNA (Trumpff et al., 2019). A recent study confirmed in a mouse model that neuroinflammation by mtDNA-TLR9 signaling contributes to stress-induced depression (Tripathi, Bartosh, Whitehead, & Pillai, 2023). However, the exact source of stress-induced cf-mtDNA requires further study.
Neutrophil extracellular traps (NETs) are networks of extracellular fibers, primarily composed of DNA from neutrophils that enucleate after activation (Rada, 2019). Recently, activation of TLR9 by NETs was found in primary human tissue samples (Nie et al., 2019). It has been shown that NET formation is increased in human brains following TBI, which induce neuronal apoptosis by activating TLR9 (Fig. 2A) (Mi et al., 2023). Yet more research is necessary to link NET formation with TLR9 activity in the brain.
9. Discussion
PRRs recognize a variety of different nucleic acids arising from both host- and non-host sources. In addition to protecting against pathogens, PRRs have critical roles to play in ensuring proper development and neural homeostasis (Escoubas et al., 2024; Dorrity et al., 2023; Gao et al., 2021; Lammert et al., 2020; Liu et al., 2015; Ma et al., 2006). However, as summarized in this review overactive PRR signaling can be detrimental. In the brain, aberrant activation of PRRs is associated with a variety of neurodegenerative conditions, including Alzheimer’s disease, ALS, Parkinson’s disease, as well as many neurological disorders. Hence, it is likely that PRRs need to strike a fine balance between homeostatic signaling and acute inflammation (for defense) to maintain healthy neural tissue. When this balance is perturbed, unusually low or high PRR signaling may cause and aggravate disease in the brain. We propose that activation of PRRs by self-nucleic acids must be viewed in a spectrum, rather than an all-or-nothing phenomenon.
Nucleic acid sensing PRRs are emerging as critical therapeutic targets, since PRR mediated inflammation is observed in many neurodegenerative diseases, aging, and brain injury. Additionally, various mouse models for neural disease have shown that genetically deleting PRRs or inhibiting PRRs can prevent or slow down disease, indicating that PRR signaling plays a causal role in brain pathogenesis. PRRs are attractive targets, because they are upstream triggers of immunity and ubiquitously present in all cell types, including neurons and glial cells, not just in immune cells. Given their upstream role, PRRs are likely among the first innate immune sensors to detect abnormal nucleic acid metabolism (e.g., increase in dsRNA or dsDNA levels, mtDNA and mtRNA leakage to the cytoplasm etc.), and further activate and amplify adaptive immunity, ultimately inducing downstream immune cell infiltration to the brain and gross neuropathology. Thus, in some disease contexts, targeting PRRs may be more effective at reducing neuroinflammation than targeting downstream adaptive immune pathways, as targeting PRRs can suppress the root cause of inflammation. However, there are also challenges for targeting PRRs therapeutically. First, inhibition of any given PRR will impair the immune response, risking infection. For many PRRs, specific inhibitors are not yet available, and any inhibitor developed must be capable of passing the blood brain barrier. Additionally, PRR signaling can play critical homeostatic functions in the brain. For instance, TBK1 lies downstream of many PRR signaling pathways (Fig. 1), and loss-of-function TBK1 mutations are a frequent cause for ALS and FTD although the underlying mechanism is unclear (Ahmad, Zhang, Casanova, & Sancho-Shimizu, 2016).
Another challenge going forward is determining the ligands that activate PRRs in the brain. While PRR activation and the role of PRRs in various brain diseases have been well characterized, the identities of the specific ligands that activate PRRs in the brain are unclear. A systematic identification of self-nucleic acid ligands for each PRR, can potentially transform our understanding of how nucleic acid dysregulation arises in the brain, and which nucleic acids may be targeted to reduce inflammation. While several specific transcripts have been identified that activate the ssRNA sensors TLR7/8, the identity of the self-ligands that activate the dsRNA sensors MDA5, TLR3, and PKR remains relatively obscure. It seems likely that these transcripts will be diverse in identity, as many divergent RNA families may form dsRNA structures capable of activating PRRs. A recent study utilized formaldehyde crosslinking and PKR protein pull-down to characterize the PKR-binding dsRNAs (Kim et al., 2018). Additionally, in vitro screening was utilized to identify MDA5 activating dsRNA species (Ahmad et al., 2018). Similar techniques may be leveraged to determine the RNA or DNA ligands that are predisposed towards activating other nucleic acid-sensing PRRs. Identifying and characterizing the self-DNA or self-RNA ligands of PRRs can uncover fundamental principles of innate immunity, and can also serve as a basis for developing targeted therapies against specific immunostimulatory self-nucleic acids to treat a variety of CNS diseases.
Finally, there exists the opportunity to examine the function of lesser-characterized PRRs in various neuropathologies. For example, members of the OAS protein family are PRRs that detect dsRNA and cause RNA degradation in the cell. Given that OAS proteins are upregulated by IFNs, there is a strong possibility that they are contributing to the neuropathology of various diseases where IFN signaling is known to be increased. ZBP1 is another sensor not well-characterized in neuroinflammation and pathology. ZBP1 detects Z-form nucleic acids and induces various cell death pathways, including necroptosis (Kuriakose & Kanneganti, 2018; Maelfait & Rehwinkel, 2023). Recently, the long non-coding RNA MEG3 was shown to activate necroptosis in a human neuron model of Alzheimer’s disease (Balusu et al., 2023). Necroptotic pathways are routinely induced by ZBP1 activation, but more work is necessary to draw any links between ZBP1 activation and neural disease.
Balancing PRR activity is key to maintaining a healthy brain. Insufficient PRR activation, or overactivation of PRRs can both cause disease. Self-nucleic acids have now emerged as major ligands of PRRs, and modulating self-nucleic acid-PRR signaling in the CNS holds great promise for the management of a variety of diseases, especially many age-related neurodegenerative diseases. Going forward it will be critical to further elucidate the mechanism of how self-nucleic acids interact with PRRs in the brain, and the role of self-nucleic acid sensing in the brain in health and disease. While much progress has been made, our understanding of the mechanism and function of self-nucleic acid-PRR sensing is still at its infancy. Importantly, collaborative efforts at the intersection of nucleic acid biology, immunology, and neurobiology will be critical for advancing the field.
Acknowledgments
This work is supported by NIH/National Institute of Neurological Disorder and Stroke grants R01NS127802 (to H.C.) and NIH training grants T32AR076953 (to T.J.D.) and T32GM145440 (to J.A.G.).
References
- Ablasser A, & Hur S (2020). Regulation of cGAS- and RLR-mediated immunity to nucleic acids. Nature Immunology, 21, 17–29. 10.1038/S41590-019-0556-1. [DOI] [PubMed] [Google Scholar]
- Aguado J, Chaggar HK, Gómez-Inclán C, Shaker MR, Leeson HC, Mackay-Sim A, & Wolvetang EJ (2021). Inhibition of the cGAS-STING pathway ameliorates the premature senescence hallmarks of Ataxia-Telangiectasia brain organoids. Aging Cell, 20. 10.1111/acel.13468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad L, Zhang SY, Casanova JL, & Sancho-Shimizu V (2016). Human TBK1: A gatekeeper of neuroinflammation. Trends in Molecular Medicine, 22, 511–527. 10.1016/J.MOLMED.2016.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad S, Mu X, Yang F, Greenwald E, Park JW, Jacob E, … Hur S (2018). Breaching self-tolerance to alu duplex RNA underlies MDA5-mediated inflammation. Cell, 172. 10.1016/j.cell.2017.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balachandran S, Roberts PC, Brown LE, Truong H, Pattnaik AK, Archer DR, & Barber GN (2000). Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity, 13. 10.1016/S1074-7613(00)00014-5. [DOI] [PubMed] [Google Scholar]
- Balendra R, & Isaacs AM (2018). C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nature Reviews Neurology, 14, 544–558. 10.1038/S41582-018-0047-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balusu S, Horré K, Thrupp N, Craessaerts K, Snellinx A, Serneels L, … De Strooper B (2023). MEG3 activates necroptosis in human neuron xenografts modeling Alzheimer’s disease. Science (New York, N. Y.), 381, 1176–1182. 10.1126/SCIENCE.ABP9556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamborschke D, Kreutzer M, Koy A, Koerber F, Lucas N, Huenseler C, … Cirak S (2021). PNPT1 mutations may cause Aicardi-Goutières-Syndrome. Brain & Development, 43. 10.1016/j.braindev.2020.10.005. [DOI] [PubMed] [Google Scholar]
- Bando Y, Onuki R, Katayama T, Manabe T, Kudo T, Taira K, & Tohyama M (2005). Double-strand RNA dependent protein kinase (PKR) is involved in the extrastriatal degeneration in Parkinson’s disease and Huntington’s disease. Neurochemistry International, 46. 10.1016/j.neuint.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Barrett JP, Knoblach SM, Bhattacharya S, Gordish-Dressman H, Stoica BA, & Loane DJ (2021). Traumatic brain injury induces cGAS activation and type I interferon signaling in aged mice. Frontiers in Immunology, 12. 10.3389/fimmu.2021.710608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand FJ, De Rivero Vaccari JC, Mejias NH, Alonso OF, & De Rivero Vaccari JP (2015). RIG-I contributes to the innate immune response after cerebral ischemia. Journal of Inflammation (United Kingdom), 12. 10.1186/s12950-015-0101-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brey RL, Holliday SL, Saklad AR, Navarrete MG, Hermosillo-Romo D, Stallworth CL, … McGlasson D (2002). Neuropsychiatric syndromes in lupus: Prevalence using standardized definitions. Neurology, 58. 10.1212/WNL.58.8.1214. [DOI] [PubMed] [Google Scholar]
- Brisse M, & Ly H (2019). Comparative structure and function analysis of the RIG-I-like receptors: RIG-I and MDA5. Frontiers in Immunology, 10. 10.3389/FIMMU.2019.01586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GJ, Cañete PF, Wang H, Medhavy A, Bones J, Roco JA, … Vinuesa CG (2022). TLR7 gain-of-function genetic variation causes human lupus. Nature, 605. 10.1038/s41586-022-04642-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bsibsi M, Ravid R, Gveric D, & Van Noort JM (2002). Broad expression of Toll-like receptors in the human central nervous system. Journal of Neuropathology and Experimental Neurology, 61. 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- Bsibsi M, Persoon-Deen C, Verwer RWH, Meeuwsen S, Ravid R, & Van Noort JM (2006). Toll-like receptor 3 on adult human astrocytes triggers production of neuroprotective mediators. Glia, 53. 10.1002/glia.20328. [DOI] [PubMed] [Google Scholar]
- Buonfiglioli A, Efe IE, Guneykaya D, Ivanov A, Huang Y, Orlowski E, … Lehnardt S (2019). let-7 MicroRNAs regulate microglial function and suppress glioma growth through toll-like receptor 7. Cell Reports, 29. 10.1016/j.celrep.2019.11.029. [DOI] [PubMed] [Google Scholar]
- Burnett SB, Vaughn LS, Sharma N, Kulkarni R, & Patel RC (2020). Dystonia 16 (DYT16) mutations in PACT cause dysregulated PKR activation and eIF2α signaling leading to a compromised stress response. Neurobiology of Disease, 146. 10.1016/j.nbd.2020.105135. [DOI] [PubMed] [Google Scholar]
- Cai N, Chang S, Yihan Li, Li Q, Jingchu Hu, Liang J, … Flint J (2015). Molecular signatures of major depression. Current Biology, 25. 10.1016/j.cub.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camargos S, Scholz S, Simón-Sánchez J, Paisán-Ruiz C, Lewis P, Hernandez D, … Singleton AB (2008). DYT16, a novel young-onset dystonia-parkinsonism disorder: Identification of a segregating mutation in the stress-response protein PRKRA. Lancet Neurology, 7. 10.1016/S1474-4422(08)70022-X. [DOI] [PubMed] [Google Scholar]
- Casrouge A, Zhang SY, Eidenschenk C, Jouanguy E, Puel A, Yang K, … Casanova JL (2006). Herpes simplex virus encephalitis in human UNC-93B deficiency. Science (New York, N. Y.), 314, 308–312. 10.1126/SCIENCE.1128346. [DOI] [PubMed] [Google Scholar]
- Chang RCC, Wong AKY, Ng HK, & Hugon J (2002). Phosphorylation of eukaryotic initiation factor-2α (eIF2α) is associated with neuronal degeneration in Alzheimer’s disease. Neuroreport, 13. 10.1097/00001756-200212200-00011. [DOI] [PubMed] [Google Scholar]
- Chen C, Feng Y, Zou L, Wang L, Chen HH, Cai JY, … Chao W (2014). Role of extracellular RNA and TLR3-Trif signaling in myocardial ischemia-reperfusion injury. Journal of the American Heart Association, 3. 10.1161/JAHA.113.000683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Liang H, Zhang J, Zen K, & Zhang CY (2013). microRNAs are ligands of Toll-like receptors. RNA (New York, N. Y.), 19, 737. 10.1261/RNA.036319.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YG, & Hur S (2021). Cellular origins of dsRNA, their recognition and consequences. Nature Reviews Molecular Cell Biology, 23(4), 286–301. 10.1038/s41580-021-00430-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H, Calis JJA, Wu X, Sun T, Yu Y, Sarbanes SL, … Rice CM (2018). Human ADAR1 prevents endogenous RNA from triggering translational shutdown. Cell, 172. 10.1016/j.cell.2017.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocco C, Manca E, Corda G, Angioni MM, Noli B, Congia M, … Piga M (2023). Brain-reactive autoantibodies in neuropsychiatric systemic lupus erythematosus. Frontiers in Immunology, 14. 10.3389/fimmu.2023.1157149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen D, Rijnink EC, Nabuurs RJA, Steup-Beekman GM, Versluis MJ, Emmer BJ, … Bajema IM (2017). Brain histopathology in patients with systemic lupus erythematosus: Identification of lesions associated with clinical neuropsychiatric lupus syndromes and the role of complement. Rheumatology (Oxford, England), 56. 10.1093/rheumatology/kew341. [DOI] [PubMed] [Google Scholar]
- Coleman LG, Zou J, & Crews FT (2017). Microglial-derived miRNA let-7 and HMGB1 contribute to ethanol-induced neurotoxicity via TLR7. Journal of Neuroinflammation, 14. 10.1186/s12974-017-0799-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa TJ, Potje S,R, Fraga-Silva TFC, da Silva-Neto JA, Barros PR, … Tostes RC (2022). Mitochondrial DNA and TLR9 activation contribute to SARS-CoV-2-induced endothelial cell damage. Vascular Pharmacology, 142. 10.1016/j.vph.2021.106946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottrell KA, Andrews RJ, & Bass BL (2023). The competitive landscape of the dsRNA world. Molecular Cell. 10.1016/J.MOLCEL.2023.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, & Stetson DB (2021). The type I interferonopathies: 10 years on. Nature Reviews Immunology 2021, 22(8), 471–483. 10.1038/s41577-021-00633-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, … Jackson AP (2006). Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutières syndrome and mimic congenital viral brain infection. Nature Genetics, 38. 10.1038/ng1842. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GMA, Gornall HL, … Rice GI (2015). Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. American Journal of Medical Genetics. Part A, 167. 10.1002/ajmg.a.36887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Reuver R, & Maelfait J (2023). Novel insights into double-stranded RNA-mediated immunopathology. Nature Reviews. Immunology. 10.1038/s41577-023-00940-3. [DOI] [PubMed] [Google Scholar]
- De Rivero Vaccari JP, Brand FJ, Sedaghat C, Mash DC, Dietrich WD, & Keane RW (2014). RIG-1 receptor expression in the pathology of Alzheimer’s disease. Journal of Neuroinflammation, 11. 10.1186/1742-2094-11-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decout A, Katz JD, Venkatraman S, & Ablasser A (2021). The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nature Reviews Immunology, 21(9), 548–569. 10.1038/s41577-021-00524-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleidi M, Hallett PJ, Koprich JB, Chung CY, & Isacson O (2010). The toll-like receptor-3 agonist polyinosinic:polycytidylic acid triggers nigrostriatal dopaminergic degeneration. Journal of Neuroscience, 30. 10.1523/JNEUROSCI.2400-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dembny P, Newman AG, Singh M, Hinz M, Szczepek M, Krüger C, … Lehnardt S (2020). Human endogenous retrovirus HERVK(HML-2) RNA causes neurodegeneration through Toll-like receptors. JCI Insight, 5. 10.1172/JCI.INSIGHT.131093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denes A, Coutts G, Lénárt N, Cruickshank SM, Pelegrin P, Skinner J, … Brough D (2015). AIM2 and NLRC4 inflammasomes contribute with ASC to acute brain injury independently of NLRP3. Proceedings of the National Academy of Sciences of the United States of America, 112. 10.1073/pnas.1419090112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhir A, Dhir S, Borowski LS, Jimenez L, Teitell M, Rötig A, … Proudfoot NJ (2018). Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature, 560. 10.1038/s41586-018-0363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold SS, Kaisho T, Hemmi H, Akira S, Reis E, & Sousa C (2004). Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science (New York, N. Y.), 303, 1529–1531. 10.1126/SCIENCE.1093616. [DOI] [PubMed] [Google Scholar]
- Dorrity TJ, Shin H, Wiegand KA, Aruda J, Closser M, Jung E, … Chung H (2023). Long 3′UTRs predispose neurons to inflammation by promoting immunostimulatory double-stranded RNA formation. Science Immunology. 10.1126/sciimmunol.adg2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejlerskov P, Hultberg JG, Wang JY, Carlsson R, Ambjørn M, Kuss M, … Issazadeh-Navikas S (2015). Lack of neuronal IFN-β-IFNAR causes lewy body- and Parkinson’s disease-like dementia. Cell, 163. 10.1016/j.cell.2015.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escoubas CC, Dorman LC, Nguyen PT, Lagares-Linares C, Nakajo H, Anderson SR, … Molofsky AV (2024). Type-I-interferon-responsive microglia shape cortical development and behavior. Cell. 10.1016/j.cell.2024.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbri M, Paone A, Calore F, Galli R, & Croce CM (2013). A new role for microRNAs, as ligands of Toll-like receptors. RNA Biology, 10. 10.4161/rna.23144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabbri M, Paone A, Calore F, Galli R, Gaudio E, Santhanam R, … Croce CM (2012). MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proceedings of the National Academy of Sciences of the United States of America, 109. 10.1073/pnas.1209414109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Zou L, Yan D, Chen H, Xu G, Jian W, … Chao W (2017). Extracellular microRNAs induce potent innate immune responses via TLR7/MyD88-dependent mechanisms. The Journal of Immunology, 199. 10.4049/jimmunol.1700730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiebich BL, Batista CRA, Saliba SW, Yousif NM, & de Oliveira ACP (2018). Role of microglia TLRs in neurodegeneration. Frontiers in Cellular Neuroscience, 12. 10.3389/fncel.2018.00329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field R, Campion S, Warren C, Murray C, & Cunningham C (2010). Systemic challenge with the TLR3 agonist poly I: C induces amplified IFNα/β and IL-1β responses in the diseased brain and exacerbates chronic neurodegeneration. Brain, Behavior, and Immunity, 24. 10.1016/j.bbi.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillatreau S, Manfroi B, & Dörner T (2020). Toll-like receptor signalling in B cells during systemic lupus erythematosus. Nature Reviews Rheumatology, 17(2), 98–108. 10.1038/s41584-020-00544-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschi C, Garagnani P, Parini P, Giuliani C, & Santoro A (2018). Inflammaging: A new immune–metabolic viewpoint for age-related diseases. Nature Reviews Endocrinology, 14(10), 576–590. 10.1038/s41574-018-0059-4. [DOI] [PubMed] [Google Scholar]
- Fritsch LE, Ju J, Gudenschwager Basso EK, Soliman E, Paul S, Chen J, … Pickrell AM (2022). Type I interferon response is mediated by NLRX1-cGAS-STING signaling in brain injury. Frontiers in Molecular Neuroscience, 15. 10.3389/fnmol.2022.852243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal-Ben-Ari S, Barrera I, Ehrlich M, & Rosenblum K (2019). PKR: A kinase to remember. Frontiers in Molecular Neuroscience, 11, 425641. 10.3389/FNMOL.2018.00480/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Li T, Li XD, Chen X, Li QZ, Wight-Carter M, & Chen ZJ (2015). Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proceedings of the National Academy of Sciences of the United States of America, 112. 10.1073/pnas.1516465112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Ciancanelli MJ, Zhang P, Harschnitz O, Bondet V, Hasek M, … Zhang SY (2021). TLR3 controls constitutive IFN-β antiviral immunity in human fibroblasts and cortical neurons. Journal of Clinical Investigation, 131. 10.1172/JCI134529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Martinez I, Santoro N, Chen Y, Hoque R, Ouyang X, Caprio S, … Mehal WZ (2016). Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. Journal of Clinical Investigation, 126. 10.1172/JCI83885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge X, Li W, Huang S, Yin Z, Xu X, Chen F, … Lei P (2018). The pathological role of NLRs and AIM2 inflammasome-mediated pyroptosis in damaged blood-brain barrier after traumatic brain injury. Brain Research, 1697. 10.1016/j.brainres.2018.06.008. [DOI] [PubMed] [Google Scholar]
- Gorbunova V, Seluanov A, Mita P, McKerrow W, Fenyö D, Boeke JD, … Sedivy JM (2021). The role of retrotransposable elements in ageing and age-associated diseases. Nature, 596(7870), 43–53. 10.1038/s41586-021-03542-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulen MF, Samson N, Keller A, Schwabenland M, Liu C, Glück S, … Ablasser A (2023). cGAS–STING drives ageing-related inflammation and neurodegeneration. Nature, 620, 374–380. 10.1038/s41586-023-06373-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He WA, Calore F, Londhe P, Canella A, Guttridge DC, & Croce CM (2014). Microvesicles containing miRNAs promote muscle cell death in cancer cachexia via TLR7. Proceedings of the National Academy of Sciences of the United States of America, 111. 10.1073/pnas.1402714111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heil F, Hemmi H, Hochrein H, Ampenberger F, Akira S, Lipford G, … Bauer S (2013). Species-specific recognition of single-stranded RNA via Toll-like receptor 7 and 8. Science (New York, N. Y.), 303. [DOI] [PubMed] [Google Scholar]
- Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, … Akira S (2000). A Toll-like receptor recognizes bacterial DNA. Nature, 408. 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- Hemphill WO, Simpson SR, Liu M, Salsbury FR, Hollis T, Grayson JM, & Perrino FW (2021). TREX1 as a novel immunotherapeutic target. Frontiers in Immunology, 12, 660184. 10.3389/FIMMU.2021.660184/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfner KP, & Hornung V (2020). Molecular mechanisms and cellular functions of cGAS–STING signalling. Nature Reviews Molecular Cell Biology, 21(9), 501–521. 10.1038/s41580-020-0244-x. [DOI] [PubMed] [Google Scholar]
- Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR, … Fitzgerald KA (2009). AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature, 458. 10.1038/nature07725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseini S, Michaelsen-Preusse K, Grigoryan G, Chhatbar C, Kalinke U, & Korte M (2020). Type I interferon receptor signaling in astrocytes regulates hippocampal synaptic plasticity and cognitive function of the healthy CNS. Cell Reports, 31. 10.1016/j.celrep.2020.107666. [DOI] [PubMed] [Google Scholar]
- Hu B, Jin C, Li HB, Tong J, Ouyang X, Cetinbas NM, … Flavell RA (2016). The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science (New York, N. Y.), 354, 765–768. 10.1126/SCIENCE.AAF7532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S-B, Heraud-Farlow J, Sun T, Liang Z, Goradia A, Taylor S, … Li JB (2023). ADAR1p150 prevents MDA5 and PKR activation via distinct mechanisms to avert fatal autoinflammation. Molecular Cell. 10.1016/j.molcel.2023.09.018. [DOI] [PubMed] [Google Scholar]
- Hung YF, Chen CY, Shih YC, Liu HY, Huang CM, & Hsueh YP (2018). Endosomal TLR3, TLR7, and TLR8 control neuronal morphology through different transcriptional programs. Journal of Cell Biology, 217. 10.1083/jcb.201712113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, & Barber GN (2009). STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature, 461(7265), 788–792. 10.1038/nature08476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CS, Arbour N, Manusow J, Montgrain V, Blain M, McCrea E, … Antel JP (2005). TLR signaling tailors innate immune responses in human microglia and astrocytes. The Journal of Immunology, 175. 10.4049/jimmunol.175.7.4320. [DOI] [PubMed] [Google Scholar]
- Jang MA, Kim EK, Now H, Nguyen NTH, Kim WJ, Yoo JY, … Ki CS (2015). Mutations in DDX58, which encodes RIG-I, Cause atypical singleton-merten syndrome. American Journal of Human Genetics, 96. 10.1016/j.ajhg.2014.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang GL, Yang XL, Zhou HJ, Long J, Liu B, Zhang LM, & Lu D (2021). cGAS knockdown promotes microglial M2 polarization to alleviate neuroinflammation by inhibiting cGAS-STING signaling pathway in cerebral ischemic stroke. Brain Research Bulletin, 171. 10.1016/j.brainresbull.2021.03.010. [DOI] [PubMed] [Google Scholar]
- Jin M, Shiwaku H, Tanaka H, Obita T, Ohuchi S, Yoshioka Y, … Okazawa H (2021). Tau activates microglia via the PQBP1-cGAS-STING pathway to promote brain inflammation. Nature Communications, 12. 10.1038/s41467-021-26851-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamintsky L, Beyea SD, Fisk JD, Hashmi JA, Omisade A, Calkin C, … Hanly JG (2020). Blood-brain barrier leakage in systemic lupus erythematosus is associated with gray matter loss and cognitive impairment. Annals of the Rheumatic Diseases, 79. 10.1136/annrheumdis-2020-218004. [DOI] [PubMed] [Google Scholar]
- Kaul D, Habbel P, Derkow K, Krüger C, Franzoni E, Wulczyn FG, … Lehnardt S (2012). Expression of toll-like receptors in the developing brain. PLoS One, 7. 10.1371/journal.pone.0037767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki T, & Kawai T (2019). Discrimination between self and non-self-nucleic acids by the innate immune system. International Review of Cell and Molecular Biology. 10.1016/bs.ircmb.2018.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Seo JS, Lee SY, Ha KT, Choi BT, Shin Y, … Shin HK (2020). AIM2 inflammasome contributes to brain injury and chronic post-stroke cognitive impairment in mice. Brain, Behavior, and Immunity, 87. 10.1016/j.bbi.2020.03.011. [DOI] [PubMed] [Google Scholar]
- Kim VN (2005). MicroRNA biogenesis: Coordinated cropping and dicing. Nature Reviews Molecular Cell Biology, 6(5), 376–385. 10.1038/nrm1644. [DOI] [PubMed] [Google Scholar]
- Kim Y, Lee JH, Park JE, Cho J, Yi H, & Kim VN (2014). PKR is activated by cellular dsRNAs during mitosis and acts as a mitotic regulator. Genes & Development, 28. 10.1101/gad.242644.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Park J, Kim S, Kim MA, Kang MG, Kwak C, … Kim VN(2018). PKR senses nuclear and mitochondrial signals by interacting with endogenous double-stranded RNAs. Molecular Cell, 71. 10.1016/j.molcel.2018.07.029. [DOI] [PubMed] [Google Scholar]
- Kuipers DJS, Mandemakers W, Lu CS, Olgiati S, Breedveld GJ, Fevga C, … Bonifati V (2021). EIF2AK2 missense variants associated with early onset generalized dystonia. Annals of Neurology, 89. 10.1002/ana.25973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai Y, Takeuchi O, & Akira S (2008). TLR9 as a key receptor for the recognition of DNA. Advanced Drug Delivery Reviews, 60, 795–804. 10.1016/J.ADDR.2007.12.004. [DOI] [PubMed] [Google Scholar]
- Kuriakose T, & Kanneganti TD (2018). ZBP1: Innate sensor regulating cell death and inflammation. Trends in Immunology, 39, 123–134. 10.1016/j.it.2017.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalive PH, Benkhoucha M, Tran NL, Kreutzfeldt M, Merkler D, & Santiago-Raber ML (2014). TLR7 signaling exacerbates CNS autoimmunity through downregulation of Foxp3+ Treg cells. European Journal of Immunology, 44. 10.1002/eji.201242985. [DOI] [PubMed] [Google Scholar]
- Lamers MM, Van Den Hoogen BG, & Haagmans BL (2019). ADAR1: “Editor-in-chief” of cytoplasmic innate immunity. Frontiers in Immunology, 10, 1763. 10.3389/FIMMU.2019.01763/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammert CR, Frost EL, Bellinger CE, Bolte AC, McKee CA, Hurt ME, … Lukens JR (2020). AIM2 inflammasome surveillance of DNA damage shapes neurodevelopment. Nature, 580. 10.1038/s41586-020-2174-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRocca TJ, Mariani A, Watkins LR, & Link CD (2019). TDP-43 knockdown causes innate immune activation via protein kinase R in astrocytes. Neurobiology of Disease, 132. 10.1016/j.nbd.2019.104514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lathia JD, Okun E, Tang SC, Griffioen K, Cheng A, Mughal MR, … Mattson MP (2008). Toll-like receptor 3 is a negative regulator of embryonic neural progenitor cell proliferation. Journal of Neuroscience, 28. 10.1523/JNEUROSCI.2140-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebon P, Badoual J, Ponsot G, Goutières F, Hémeury-Cukier F, & Aicardi J (1988). Intrathecal synthesis of interferon-alpha in infants with progressive familial encephalopathy. Journal of the Neurological Sciences, 84. 10.1016/0022-510X(88)90125-6. [DOI] [PubMed] [Google Scholar]
- Lee H, Fenster RJ, Pineda SS, Gibbs WS, Mohammadi S, Davila-Velderrain J, … Heiman M (2020). Cell type-specific transcriptomics reveals that mutant huntingtin leads to mitochondrial RNA release and neuronal innate immune activation. Neuron, 107. 10.1016/j.neuron.2020.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann SM, Krüger C, Park B, Derkow K, Rosenberger K, Baumgart J, … Lehnardt S (2012). An unconventional role for miRNA: Let-7 activates Toll-like receptor 7 and causes neurodegeneration. Nature Neuroscience, 15. 10.1038/nn.3113. [DOI] [PubMed] [Google Scholar]
- Letiembre M, Liu Y, Walter S, Hao W, Pfander T, Wrede A, … Fassbender K (2009). Screening of innate immune receptors in neurodegenerative diseases: A similar pattern. Neurobiology of Aging, 30. 10.1016/j.neurobiolaging.2007.08.018. [DOI] [PubMed] [Google Scholar]
- Levanon EY, Cohen-Fultheim R, & Eisenberg E (2024). In search of critical dsRNA targets of ADAR1. Trends Genet. 40(3), 250–259. 10.1016/j.tig.2023.12.002. [DOI] [PubMed] [Google Scholar]
- Li Q, Tian S, Liang J, Fan J, Lai J, & Chen Q (2021). Therapeutic development by targeting the cGAS-STING pathway in autoimmune disease and cancer. Frontiers in Pharmacology, 12. 10.3389/FPHAR.2021.779425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Cao Y, Dang C, Han B, Han R, Ma H, … Wang L (2020). Inhibition of double-strand DNA-sensing cGAS ameliorates brain injury after ischemic stroke. EMBO Molecular Medicine, 12. 10.15252/emmm.201911002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, & Chen ZJ (2018). The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. The Journal of Experimental Medicine, 215, 1287–1299. 10.1084/JEM.20180139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind NA, Rael VE, Pestal K, Liu B, & Barton GM (2021). Regulation of the nucleic acid-sensing Toll-like receptors. Nature Reviews Immunology, 22(4), 224–235. 10.1038/s41577-021-00577-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindqvist D, Wolkowitz OM, Picard M, Ohlsson L, Bersani FS, Fernström J, … Mellon SH (2018). Circulating cell-free mitochondrial DNA, but not leukocyte mitochondrial DNA copy number, is elevated in major depressive disorder. Neuropsychopharmacology: Official Publication of the American College of Neuropsychopharmacology, 43. 10.1038/s41386-017-0001-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HY, Huang CM, Hung YF, & Hsueh YP (2015). The microRNAs Let7c and miR21 are recognized by neuronal Toll-like receptor 7 to restrict dendritic growth of neurons. Experimental Neurology, 269. 10.1016/j.expneurol.2015.04.011. [DOI] [PubMed] [Google Scholar]
- Liu HY, Hong YF, Huang CM, Chen CY, Huang TN, & Hsueh YP (2013). TLR7 negatively regulates dendrite outgrowth through the Myd88-c-Fos-IL-6 pathway. Journal of Neuroscience, 33. 10.1523/JNEUROSCI.5566-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livingston JH, & Crow YJ (2016). Neurologic phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR1, and IFIH1: Aicardi-Goutières syndrome and beyond. Neuropediatrics, 47, 355–360. 10.1055/S-0036-1592307. [DOI] [PubMed] [Google Scholar]
- Lodi L, Melki I, Bondet V, Seabra L, Rice GI, Carter E, … Frémond ML (2021). Differential expression of interferon-alpha protein provides clues to tissue specificity across type i interferonopathies. Journal of Clinical Immunology, 41. 10.1007/s10875-020-00952-x. [DOI] [PubMed] [Google Scholar]
- Lu W, Tang S, Li A, Huang Q, Dou M, Zhang Y, … Huang C (2022). The role of PKC/PKR in aging, Alzheimer’s disease, and perioperative neurocognitive disorders. Frontiers in Aging Neuroscience, 14. 10.3389/fnagi.2022.973068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugrin J, & Martinon F (2018). The AIM2 inflammasome: Sensor of pathogens and cellular perturbations. Immunological Reviews, 281, 99–114. 10.1111/IMR.12618. [DOI] [PubMed] [Google Scholar]
- Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, … Flavell RA (2004). Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proceedings of the National Academy of Sciences of the United States of America, 101. 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Haynes RL, Sidman RL, & Vartanian T (2007). An innate immune receptor in brain, neurons and axons. Cell Cycle (Georgetown, Tex.), 6, 2859. 10.4161/CC.6.23.5018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Li J, Chiu I, Wang Y, Sloane JA, Lü J, … Vartanian T (2006). Toll-like receptor 8 functions as a negative regulator of neurite outgrowth and inducer of neuronal apoptosis. Journal of Cell Biology, 175. 10.1083/jcb.200606016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maelfait J, & Rehwinkel J (2023). The Z-nucleic acid sensor ZBP1 in health and disease. The Journal of Experimental Medicine, 220. 10.1084/jem.20221156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao D, Reuter CM, Ruzhnikov MRZ, Beck AE, Farrow EG, Emrick LT, … Thiffault I (2020). De novo EIF2AK1 and EIF2AK2 variants are associated with developmental delay, leukoencephalopathy, and neurologic decompensation. American Journal of Human Genetics, 106. 10.1016/j.ajhg.2020.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano M, Snyder JM, Connelly C, Henao-Mejia J, Sidrauski C, & Stetson DB (2021). Protein kinase R and the integrated stress response drive immunopathology caused by mutations in the RNA deaminase ADAR1. Immunity, 54. 10.1016/j.immuni.2021.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi L, Min X, Shi M, liu L, Zhang Y, Zhu Y, … Chen X (2023). Neutrophil extracellular traps aggravate neuronal endoplasmic reticulum stress and apoptosis via TLR9 after traumatic brain injury. Cell Death & Disease, 14. 10.1038/s41419-023-05898-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouton-Liger F, Rebillat AS, Gourmaud S, Paquet C, Leguen A, Dumurgier J, … Hugon J (2015). PKR downregulation prevents neurodegeneration and β-amyloid production in a thiamine-deficient model. Cell Death & Disease, 6. 10.1038/cddis.2014.552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee P, Winkler CW, Taylor KG, Woods TA, Nair V, Khan BA, & Peterson KE (2015). SARM1, Not MyD88, mediates TLR7/TLR9-induced apoptosis in neurons. The Journal of Immunology, 195. 10.4049/jimmunol.1500953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson AM, Reddy SK, Ratliff TS, Hossain MZ, Katseff AS, Zhu AS, … Garza LA (2015). dsRNA released by tissue damage activates TLR3 to drive skin regeneration. Cell Stem Cell, 17. 10.1016/j.stem.2015.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie M, Yang L, Bi X, Wang Y, Sun P, Yang H, … Jiang W (2019). Neutrophil extracellular traps induced by IL8 promote diffuse large B-cell lymphoma progression via the TLR9 signaling. Clinical Cancer Research, 25. 10.1158/1078-0432.CCR-18-1226. [DOI] [PubMed] [Google Scholar]
- Oda H, Nakagawa K, Abe J, Awaya T, Funabiki M, Hijikata A, … Heike T (2014). Aicardi-goutières syndrome is caused by IFIH1 mutations. American Journal of Human Genetics, 95. 10.1016/j.ajhg.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okun E, Griffioen K, Barak B, Roberts NJ, Castro K, Pita MA, … Mattson MP (2010). Toll-like receptor 3 inhibits memory retention and constrains adult hippocampal neurogenesis. Proceedings of the National Academy of Sciences of the United States of America, 107. 10.1073/pnas.1005807107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson JK, & Miller SD (2004). Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. The Journal of Immunology, 173. 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- Onizawa H, Kato H, Kimura H, Kudo T, Soda N, Shimizu S, … Fujita T (2021). Aicardi-Goutières syndrome-like encephalitis in mutant mice with constitutively active MDA5. International Immunology, 33. 10.1093/intimm/dxaa073. [DOI] [PubMed] [Google Scholar]
- Paludan SR, & Mogensen TH (2021). Constitutive and latent immune mechanisms exert ‘silent’ control of virus infections in the central nervous system. Current Opinion in Immunology, 72, 158–166. 10.1016/J.COI.2021.05.004. [DOI] [PubMed] [Google Scholar]
- Paquet C, Amin J, Mouton-Liger F, Nasser M, Love S, Gray F, … Boche D (2015). Effect of active A β immunotherapy on neurons in human Alzheimer’s disease. Journal of Pathology, 235. 10.1002/path.4491. [DOI] [PubMed] [Google Scholar]
- Parameswaran J, Zhang N, Braems E, Tilahun K, Pant DC, Yin K, … Jiang J (2023). Antisense, but not sense, repeat expanded RNAs activate PKR/eIF2α-dependent ISR in C9ORF72 FTD/ALS. Elife, 12. 10.7554/eLife.85902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peel AL, Rao RV, Cottrell BA, Hayden MR, Ellerby LM, & Bredesen DE (2001). Double-stranded RNA-dependent protein kinase, PKR, binds preferentially to Huntington’s diseases (HD) transcripts and is activated in HD tissue. Human Molecular Genetics, 10. 10.1093/hmg/10.15.1531. [DOI] [PubMed] [Google Scholar]
- Poh L, Fann D, Wong P, Rajeev V, Selvaraji S, Chen C, … Arumugam TV (2021). AIM2 inflammasome mediates hallmark neuropathological alterations and cognitive impairment in a mouse model of vascular dementia. Alzheimer’s & Dementia: The Journal of the Alzheimer’s Association, 17. 10.1002/alz.051709. [DOI] [PubMed] [Google Scholar]
- Rabinowits G, Gerçel-Taylor C, Day JM, Taylor DD, & Kloecker GH (2009). Exosomal microRNA: A diagnostic marker for lung cancer. Clinical Lung Cancer, 10. 10.3816/CLC.2009.n.006. [DOI] [PubMed] [Google Scholar]
- Rada B (2019). Neutrophil extracellular traps. Methods in Molecular Biology. 10.1007/978-1-4939-9424-3_31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice G, Newman WG, Dean J, Patrick T, Parmar R, Flintoff K, … Crow YJ (2007). Heterozygous mutations in TREX1 cause familial chilblain lupus and dominant Aicardi-Goutières syndrome. American Journal of Human Genetics, 80. 10.1086/513443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, … Crow YJ (2009). Mutations involved in Aicardi-Goutières syndrome implicate SAMHD1 as regulator of the innate immune response. Nature Genetics, 41. 10.1038/ng.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Del Toro Duany Y, Jenkinson EM, Forte GMA, Anderson BH, Ariaudo G, … Crow YJ (2014). Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type i interferon signaling. Nature Genetics, 46. 10.1038/ng.2933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson T, Kariuki SN, Franek BS, Kumabe M, Kumar AA, Badaracco M, … Niewold TB (2011). Autoimmune disease risk variant of IFIH1 is associated with increased sensitivity to IFN-α and serologic autoimmunity in lupus patients. The Journal of Immunology, 187. 10.4049/jimmunol.1100857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez S, Sahin A, Schrank BR, Al-Lawati H, Costantino I, Benz E, … Albers MW (2021). Genome-encoded cytoplasmic double-stranded RNAs, found in C9ORF72 ALS-FTD brain, propagate neuronal loss. Science Translational Medicine, 13. 10.1126/scitranslmed.aaz4699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Nuevo A, Díaz-Ramos A, Noguera E, Díaz-Sáez F, Duran X, Muñoz JP, … Zorzano A (2018). Mitochondrial DNA and TLR9 drive muscle inflammation upon Opa1 deficiency. The EMBO Journal, 37. 10.15252/embj.201796553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy ER, Chiu G, Li S, Propson NE, Kanchi R, Wang B, … Cao W (2022). Concerted type I interferon signaling in microglia and neural cells promotes memory impairment associated with amyloid β plaques. Immunity, 55. 10.1016/j.immuni.2022.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel CE (2019). Adenosine deaminase acting on RNA (ADAR1), a suppressor of double-stranded RNA–triggered innate immune responses. Journal of Biological Chemistry, 294. 10.1074/jbc.TM118.004166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab N, Taskina D, Leung E, Innes BT, Bader GD, & Hazrati LN (2022). Neurons and glial cells acquire a senescent signature after repeated mild traumatic brain injury in a sex-dependent manner. Frontiers in Neuroscience, 16. 10.3389/fnins.2022.1027116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, & Chen ZJ (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science (New York, N. Y.), 339, 786–791. 10.1126/SCIENCE.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CA, Tejwani L, Trujillo CA, Negraes PD, Herai RH, Mesci P, … Muotri AR (2017). Modeling of TREX1-Dependent Autoimmune Disease using Human Stem Cells Highlights L1 Accumulation as a Source of Neuroinflammation. Cell Stem Cell. 21(3), 319–331. 10.1016/j.stem.2017.07.009 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tible M, Mouton Liger F, Schmitt J, Giralt A, Farid K, Thomasseau S, … Hugon J (2019). PKR knockout in the 5xFAD model of Alzheimer’s disease reveals beneficial effects on spatial memory and brain lesions. Aging Cell, 18. 10.1111/acel.12887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tigano M, Vargas DC, Tremblay-Belzile S, Fu Y, & Sfeir A (2021). Nuclear sensing of breaks in mitochondrial DNA enhances immune surveillance. Nature, 591. 10.1038/s41586-021-03269-w. [DOI] [PubMed] [Google Scholar]
- Tripathi A, Bartosh A, Whitehead C, & Pillai A (2023). Activation of cell-free mtDNA-TLR9 signaling mediates chronic stress-induced social behavior deficits. Molecular Psychiatry. 10.1038/s41380-023-02189-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trumpff C, Marsland AL, Basualto-Alarcón C, Martin JL, Carroll JE, Sturm G, … Picard (2019). Acute psychological stress increases serum circulating cell-free mitochondrial DNA. Psychoneuroendocrinology, 106. 10.1016/j.psyneuen.2019.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trumpff C, Michelson J, Lagranha CJ, Taleon V, Karan KR, Sturm G, … Picard M (2021). Stress and circulating cell-free mitochondrial DNA: A systematic review of human studies, physiological considerations, and technical recommendations. Mitochondrion, 59. 10.1016/j.mito.2021.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udeochu JC, Amin S, Huang Y, Fan L, Torres ERS, Carling GK, … Gan L (2023). Tau activation of microglial cGAS–IFN reduces MEF2C-mediated cognitive resilience. Nature Neuroscience, 26. 10.1038/s41593-023-01315-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughn LS, Bragg DC, Sharma N, Camargos S, Cardoso F, & Patel RC (2015). Altered activation of protein kinase PKR and enhanced apoptosis in dystonia cells carrying a mutation in PKR activator protein PACT. Journal of Biological Chemistry, 290. 10.1074/jbc.M115.669408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DG, Tang TM, & Lue LF (2018). Increased expression of toll-like receptor 3, an anti-viral signaling molecule, and related genes in Alzheimer’s disease brains. Experimental Neurology, 309. 10.1016/j.expneurol.2018.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallach T, Wetzel M, Dembny P, Staszewski O, Krüger C, Buonfiglioli A, … Lehnardt S (2020). Identification of CNS injury-related microRNAs as novel Toll-like receptor 7/8 signaling activators by small RNA sequencing. Cells, 9. 10.3390/cells9010186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Du J, Hua S, & Zhao K (2022). TREX1 plays multiple roles in human diseases. Cellular Immunology, 375, 104527. 10.1016/J.CELLIMM.2022.104527. [DOI] [PubMed] [Google Scholar]
- Wang X, Wang Y, Cao A, Luo Q, Chen D, Zhao W, … Quan J (2023). Development of cyclopeptide inhibitors of cGAS targeting protein-DNA interaction and phase separation. Nature Communications, 14. 10.1038/s41467-023-41892-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Fang L, Huang T, & Ying S (2021). Case report: Aicardi-Goutières syndrome caused by novel TREX1 variants. Frontiers in Pediatrics, 9. 10.3389/fped.2021.634281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, & Chen ZJ (2013). Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science (New York, N. Y.), 339, 826–830. 10.1126/SCIENCE.1229963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu PJ, Liu HY, Huang TN, & Hsueh YP (2016). AIM 2 inflammasomes regulate neuronal morphology and influence anxiety and memory in mice. Scientific Reports, 6. 10.1038/srep32405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu PJ, Hung YF, Liu HY, & Hsueh YP (2017). Deletion of the inflammasome sensor Aim2 mitigates Aβ deposition and microglial activation but increases inflammatory cytokine expression in an alzheimer disease mouse model. Neuroimmunomodulation, 24. 10.1159/000477092. [DOI] [PubMed] [Google Scholar]
- Xie X, Ma G, Li X, Zhao J, Zhao Z, & Zeng J (2023). Activation of innate immune cGAS-STING pathway contributes to Alzheimer’s pathogenesis in 5×FAD mice. Nature Aging, 3. 10.1038/s43587-022-00337-2. [DOI] [PubMed] [Google Scholar]
- Yang YG, Lindahl T, & Barnes DE (2007). Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell, 131, 873–886. 10.1016/j.cell.2007.10.017. [DOI] [PubMed] [Google Scholar]
- Yu CH, Davidson S, Harapas CR, Hilton JB, Mlodzianoski MJ, Laohamonthonkul P, … Masters SL (2020). TDP-43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell, 183. 10.1016/j.cell.2020.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Li S, Li L, Li M, Guo C, Yao J, & Mi S (2015). Exosome and exosomal microRNA: Trafficking, sorting, and function. Genomics, Proteomics & Bioinformatics/Beijing Genomics Institute, 13, 17. 10.1016/J.GPB.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang MJ, Zhao QC, Xia MX, Chen J, Chen YT, Cao X, … Xu Y (2020). The HDAC3 inhibitor RGFP966 ameliorated ischemic brain damage by downregulating the AIM2 inflammasome. FASEB Journal, 34. 10.1096/fj.201900394RRR. [DOI] [PubMed] [Google Scholar]
- Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, … Casanova JL (2007). TLR3 deficiency in patients with herpes simplex encephalitis. Science (New York, N. Y.), 317, 1522–1527. 10.1126/SCIENCE.1139522. [DOI] [PubMed] [Google Scholar]
- Zhang X, Gao R, Zhang C, Teng Y, Chen H, Li Q, … Chen C (2023). Extracellular RNAs-TLR3 signaling contributes to cognitive impairment after chronic neuropathic pain in mice. Signal Transduction and Targeted Therapy, 8. 10.1038/s41392-023-01543-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZJ, Guo JS, Li SS, Wu XB, Cao DL, Jiang BC, … Gao YJ (2018). TLR8 and its endogenous ligand miR-21 contribute to neuropathic pain in murine DRG. Journal of Experimental Medicine, 215. 10.1084/jem.20180800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, He J, Gu L, Shahror RA, Li Y, Cao T, … Chao W (2022). Brain innate immune response via miRNA-TLR7 sensing in polymicrobial sepsis. Brain, Behavior, and Immunity, 100. 10.1016/j.bbi.2021.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T, Guo S, Bardhi O, Ryskamp DA, Li J, Tusi SK, … Ranum LPW (2020). Metformin inhibits RAN translation through PKR pathway and mitigates disease in C9orf72 ALS/FTD mice. Proceedings of the National Academy of Sciences of the United States of America, 117. 10.1073/pnas.2005748117. [DOI] [PMC free article] [PubMed] [Google Scholar]