

Abstract
Radical-polar crossover of organoborates is a poweful tool that enables the creation of two C-C bonds simultaneously. Small ring systems have become essential motifs in drug discovery and medicinal chemistry. However, step-economic methods for their selective functionalization remains scarce. Here we present a one-pot strategy that merges a simple preparation of strained organoboron species with the recently popularized polar radical crossover of borate derivatives to stereoselectively access tri-substituted azetidines, cyclobutanes and five-membered carbo- and heterocycles.
Subject terms: Synthetic chemistry methodology, Stereochemistry, Synthetic chemistry methodology
Strained carbo- and heterocycles are essential motifs in medicinal chemistry and drug discovery, however, their selective functionalization remains challenging. Here, the authors develop a one-pot strategy to access stereo-defined trisubstituted azetidines, cyclobutanes, and five-membered carbo- and heterocycles based on the polar radical crossover of borate derivatives.
Introduction
Strained carbo- and heterocycles have been brought to the forefront of medicinal chemistry and drug discovery programs in recent years as modulable sp3-rich 3D-isosters of diverse aromatic systems1–4. Besides exalting greater metabolic stability, it has been shown that small molecular scaffolds can help improve lipophilicity as well as pharmacokinetics5–9. The puckered conformation adopted by four-membered rings renders them ideal cores for drug discovery as they can balance both rigidity (observed in constraints systems such as propellanes10–13 or cubanes14,15) and flexibility (conformers in larger cyclic scaffolds). Azetidines and cyclobutanes can, therefore, be used towards the three-dimensionalization of pyridyl and phenyl moieties, their substitution pattern following defined exit vectors (Fig. 1).
Fig. 1. Previous work and present contribution on radical-polar crossover reactions.

Achievements reached in the diastereoselective radical-polar crossover of cyclic systems such as 4- and 5-membered carbo- and heterocycles, in contrast with previous work by Studer, Aggarwal, Morken and Renaud on acyclic systems.
Recent step-economic strategies towards substituted azetidines include the work of Baran and Gianatassio on strain-release amination16 and alkylation17 of 1-azabicyclo[1.1.0]butanes (ABB) that provide an elegant route to 3-substituted structures, as well as our contribution on 1,3-bisarylations18. The group of Aggarwal reported the strain-release of ABB through boron-homologations for the synthesis of 3,3-bis functionalized azetidines19–22. Cyclobutanes were similarly obtained from metallated bicyclo[1.1.0]butanes23–27. Aside from strain-releasing strategies, substituted azetidines and cyclobutanes are traditionally approached through [2 + 2]-cycloadditions as recently illustrated by Schindler28–31, Bach32–35, Glorius36–38 and Brown39–43 as well as cyclizations and ring contraction and expansion reactions44–50, which imply a pre-organization of the substituents around the structure of starting materials.
Aiming at the development of a synthetic toolbox that would allow diversely and selectively access functionalized four-membered building blocks, we set out to combine our expertise on the metalation of small heterocycles51–58 and 1,2-boronate rearrangements59–64 to design a simple one-pot sequence towards tri-substituted architectures. We envisioned that the inspiring work on polar radical crossover (PRC) pioneered by the groups of Studer65,66, Aggarwal67,68, Morken69 and Renaud70 could reveal fantastic opportunities to introduce three substituents at once on azetidines, cyclobutanes and other heterocycles, starting from corresponding cyclic alkenyl-metal intermediates. To the best of our knowledge, control over the stereochemical outcome of this transformation remained moderate, as the radical process was only exemplified on acyclic alkenylboronates. However, for this strategy to take a consequent step further and enable a broad scope of applications, one would have to gain control over the spatial arrangement of vicinal substituents. We predicted that the diastereoselectivity of the 1,2-metalate rearrangement would be controlled thanks to the cyclic nature of our substrates, due to a locked configuration of reactive intermediates.
Results and discussion
Optimizations of reaction conditions
The first test was performed on azetinyllithium 1 (generated in situ), providing the bisorganoborinate 2 after the addition of n-BuBpin in THF. Generation of a radical species from nonafluorobutyl iodide under UV irradiation at −40 °C—as assumed from literature precedent71— provided the expected trisubstituted structure 3 in 44% with a moderate dr of 4:1 (Fig. 2, entry 1, table 1). We started optimizing the reaction parameters by assessing the importance of the stoichiometry of perfluorinated butyl-iodide on the yield. Under similar conditions, azetidine 3 was obtained with increased yields up to 78% with 1.5 equivalents of C4F9I (entry 2), and comparable yield could be observed under blue light irradiation at −20 °C, keeping the same levels of diastereoselectivity. Solvent effects were examined next. While 1,3-dimethyl-2-imidazolidinone (DMI), dichloromethane and dichloroethane did not improve selectivity (entries 5–7), a diastereomeric ratio of 5:1 was measured in 2-methyl-THF (mTHF, entry 8).
Fig. 2. Optimizations of the polar radical crossover on in situ generated azetinyllithium species.
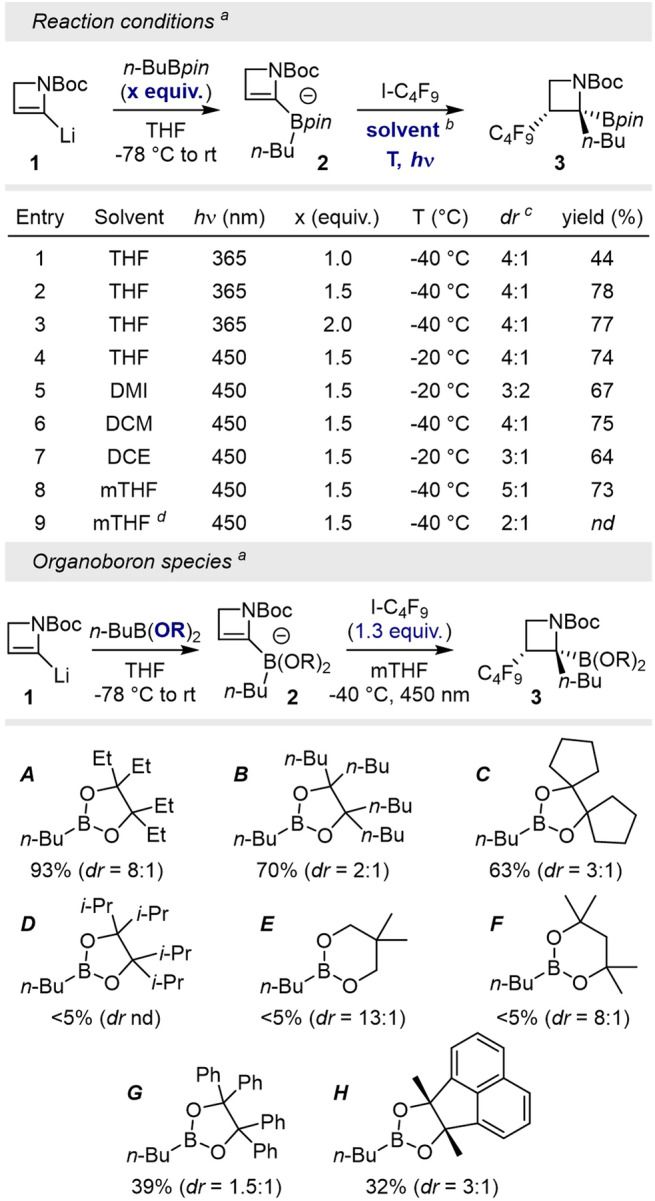
a Indicated yields have been assessed by GC-analysis of the crude mixture; C12H26 (1 vol%, 30 mL) was used as standard. b for reactions performed in other solvents than THF, a solvent switch was performed after the removal of THF for the crude mixture. c indicated dr were measured on the crude mixture by 19F-NMR. d The reaction was performed in the absence of C12 standard.
It is interesting to note that the reaction performed in the absence of dodecane (C12H26) as standard only resulted in product formation with poor diastereoselectivity (entry 9, dr = 2:1).
With these encouraging results in hands, the influence of steric effects was evaluated by changing the ligand structure on the boron atom. The process was reiterated employing organoboron species A–H under the conditions displayed in entry 8. While reagents A, B and C gave similarly high yields (63–93%), the groups present on the pinacol scaffold allowed for a broad modulation of dr values, a maximum being reached for reagent A (n-BuBEpin, dr = 8:1). Only traces of products were observed with 1,3-propyldiols (E and F) and the isopropyl-pinacol structure D. Surprisingly low dr were obtained using phenyl-pinacol derivative G or “Bmac” H72, products being additionally isolated only in moderate yields.
With a fair adequation between conversion and diastereoselectivity, ethyl-pinacol (Epin) ligands A were further employed to explore the scope of the reaction. In addition to positively influencing the dr, products obtained with ligand A showed high stability on silica (avoids protodenoronation) when compared to classical pinacols.
From azetines to trisubstituted azetidines
The scope of the transformation was first assessed on azetinyllithium species 1, in situ generated from 3-methoxyazetidines.[15f] Coordination to an organoboron derivative R1-BEpin primarily gives borinate 2, which was then engaged in PRC after the solvent switch to mTHF and addition of the radical precursor R2-I under blue light irradiation (Fig. 3). Products 3a–e were synthesized in moderate to good yields from alkylboron reagents and perfluorinated radical precursors with good dr values (8:1–20:1), except for methylboronic ester (3e, dr = 1:1), which might come from a lack of steric effects (vide infra).
Fig. 3. Evaluation of the PRC sequence scope for the synthesis of trisubstituted azetidines from azetinyllithium species.
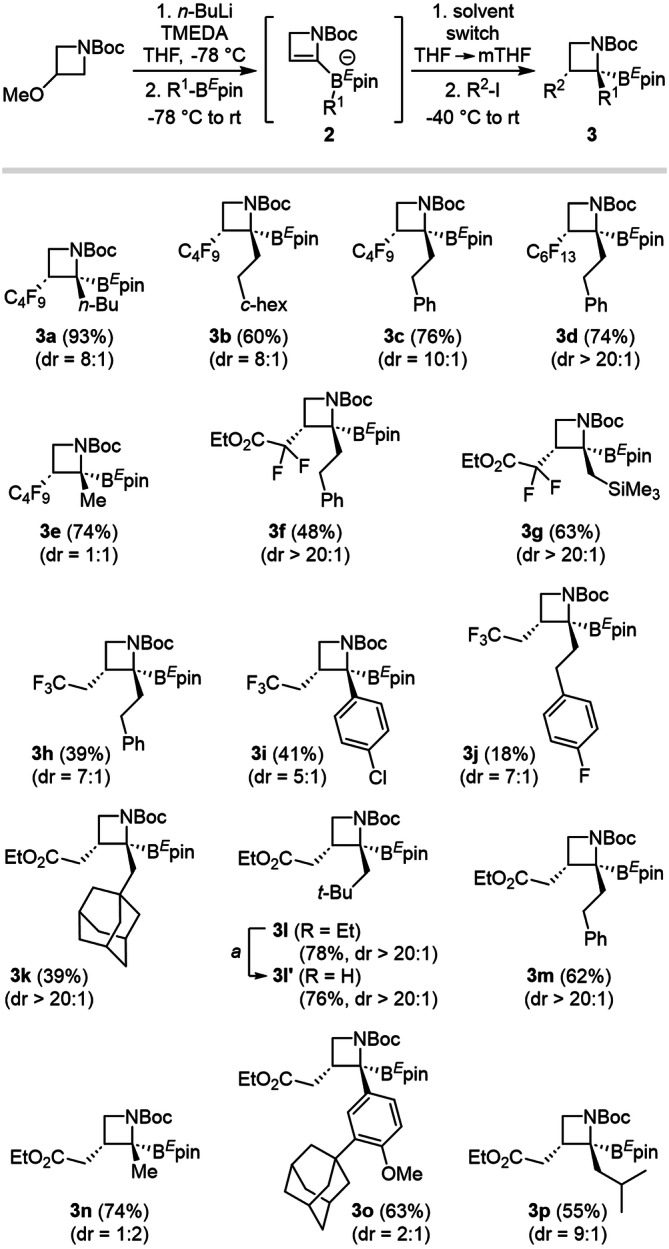
Azetinyllithium 1 is generated in situ by α-lithiation/elimination/α-lithiation, starting from commercially available 3-methoxyazetidines. The first steps are conducted in THF, but the best distereoselectivities are achieved by performing the last rearrangement step in Me-THF.
Excellent dr (>20:1) was observed when employing ethyl 2,2-difluoro-2-iodoacetate (3f, g), and we noticed a general trend for the iodoacetates to give increased dr (3k–m, dr > 20:1) in comparison with other radical precursors (3h–j, up to 7:1 dr). The lack of sterical hindrance in the case of methyl boronic ester (3n, dr = 2:1). Arylboronic ester also tended to decrease the selectivity (3i and 3o, 2:1 to 5:1 dr). In all cases, the 1,2-metallate rearrangement proceeded in a trans-selective fashion (R1 vs. R2), as supported by thorough analytic measurements and experimental data (vide infra). Furthermore, substituted azetidines proved to be stable under basic conditions, and we were able to hydrolyze the ester moiety into the corresponding carboxylic acid 3l’ in good yield (76%), keeping the dr value above 20:1.
From cyclobutenes to trisubstituted cyclobutanes
The stereoselective synthesis of cyclobutanes through PRC was envisioned next from readily available starting cyclobutenylboronic esters 4 (Fig. 4), applying previously optimized conditions (Fig. 2). The scope of the transformation was explored by varying both radical precursors (perfluorinated alkyl iodides and iodoacetates) and nature of the organolithium species (alkyl- and aryl-lithium). A range of functionalized cyclobutanes (6a–f) was isolated in good yields and excellent dr (up to 20:1) with the exception of cyclopropylboronic ester (6c, dr = 6:1). Interestingly, an iodomethylketone proved to be an efficient radical precursor for this reaction (6e, dr = 18:1), as well as unprotected iodoacetamide (6f), although with slightly decreased diastereoselectivity (dr = 7:1). It is important to note that the diastereoselectivity of the metallate rearrangement step on cyclobutyl-intermediates is generally superior to the one on azetidinyl-species.
Fig. 4. Application of the PRC sequence to the tris-functionalization of cyclobutanes from isolated cyclobutenylboronic ester derivatives 4.

The scope of the reaction (R1) is evaluated for both aryl and alkyl substituents, showing a generally higher trend in diastereoselectivities than for azetidines.
Given the high levels of diastereoselectivity observed in cyclobutenylboron species, two derivatives possessing the thiophene sub-unit typically found in Canagliflozin (a drug used in the treatment of type 2 diabetes)73 were synthesized, employing iodoacetate (6g) and trifluoromethyliodide (6h).
Scope of the transformation on cyclopentenes
Larger carbocyclic systems were explored next, starting from stable, storable cyclopentenylboronic ester 7a (Fig. 5). Coordination of an organolithium reagent (R1-Li) to 7 promotes the formation of the bisorganoborinate 8. Radical crossover was further initiated under blue light irradiation at −40 °C after the solvent switch and addition of iodoacetates as radical precursors. Trisubstituted cyclopentanes 9a–e were obtained with high yields and stereochemical ratio (up to 20:1 dr), except for the addition of aryllithium species (9d, dr = 2:1), as previously observed. While switching the radical precursor for a tert-butyl ester gave similar results, both in efficiency and diastereoselectivity (9a’ and 9b”), trifluoromethyl iodide furnished product 9e with slightly lower levels of selectivity (7:1 dr).
Fig. 5. Generalization of the PRC sequence to larger rings.

Evaluation of the scope for the diastereoselective formation of tris-functionalized cyclopentanes from preformed cyclopentenylboronic esters 7. Steroide-derivatives are also described, although with a lower diastereomeric ratio.
These compounds also proved stable under basic conditions, product 9b being hydrolyzed into 9b’ in excellent yield (91%). Interestingly, we demonstrated the applicability of this stereoselective method to the functionalization of estron scaffolds. The stable alkenylboronic ester substrate 7b was first accessed in few steps from (+)-estrone 3-methyl ether and further engaged in PRC, leading to products 9f and 9g in high yields, although the diastereoselectivity could only reach 3:1. It is, however, important to note that addition of the in situ generated radical species occurred on the least hindered face of the cyclopentenylboronates (cis to the methyl substituent).
Scope of the transformation on heterocycles and norbornenes
Although heterocyclic five-membered rings were efficiently engaged in PRC to provide trisubstituted pyrrolidine 12a and THF 12b, c (up to 96% yield), the diastereomeric ratio could only reach up to 3:1 (Fig. 6).
Fig. 6. Stereoselective functionalization of pyrrolidines, THF (top) and norbornanes (bottom) via PRC.

Evaluation of the reaction scope using readily accessible lithiated dihydropyrroles and dihydrofuranes 10, as well as norbornenylboronic esters 13.
Norbornenylboronic ester 13 was readily prepared in a few steps from commercially available norbornene and proved to be a suitable building block for PRC. It efficiently provided substituted structures 15a–c in both high yields (72–92%) and stereoselectivities (dr > 20:1). The formation of the major diastereoisomer can be explained by the addition of the radical species on the least hindered bridged side of the norbornenyl-substrate, followed by an antiperiplanar 1,2-metallate rearrangement.
Determination of the relative configuration
Stereochemical relationships between newly introduced substituents were studied first by NOE and HOE on compounds 3e, 3d and 3a (Fig. 7, see supporting information). The poor diastereomeric ratio (ca. 1:1) obtained for 3e allowed us to separate both diastereoisomers (3esyn and 3eanti) in sufficient quantities for NMR experiments. In the case of 3eanti, while a strong NOE was observed between the H-atom at position 3 and the methyl group at position 2, we could not detect any significant HOE (1H–19F), supporting the anti-configuration of R1 and R2 groups. Inversed observations were made for compound 3esyn, for which a very weak NOE was detected, but with a strong HOE (1H–19F) between the perfluorinated chain at position 3 and the methyl group at position 2. Analogy was then made for compound 3d, isolated with a higher dr (>20:1), for which we assigned the anti-configuration through both detections of a strong H–CH3 NOE and the absence of significant HOE (1H-19F). Similarly, strong NOE on compound 3a (dr = 8:1) allowed us to assign its anti-configuration.
Fig. 7. Nuclear Overhauser effect (NOE) and heteronuclear Overhauser effect (HOE) on synthesized azetidines to support the assigned relative configuration of the three substituents (top).
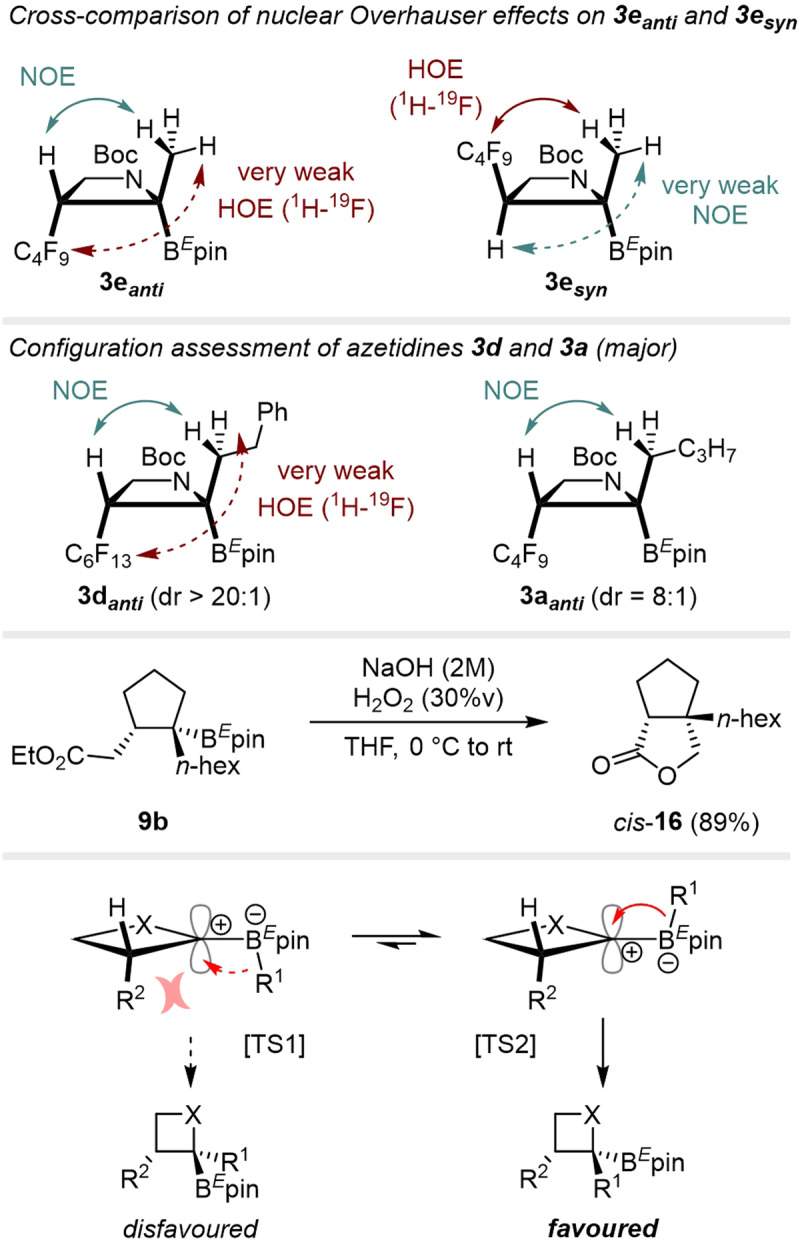
Experimental assessment of the relative configuration through oxidative C–B bond cleavage followed by lactonization (middle). Proposed model for the stereoselectivity and elements of support for the observed relative configuration (bottom).
Brown oxidation (NaOH, H2O2) on 9b led to the bicyclic compound cis-16 by subsequent lactonization, which brought additional support to the assigned stereochemistry. We propose that the R2-chain introduced from the radical precursor shields one of the two diastereotopic faces of the cyclic intermediate, disfavoring the 1,2-metallate rearrangement of R1 from the same face [TS1], to favor an antiperiplanar addition [TS2].
Further applications
Finally, we evaluated the robustness and the configurational stability of our cyclic organoboron systems under different conditions (Fig. 8). Switching the Boc protecting group on the nitrogen atom (3a) for a benzyl group in a two-step sequence led to N-benzyl product 17a, which was isolated in 67% yield with retained diastereomeric ratio (8:1). 17b was obtained through the same reaction sequence from its NBoc derivative parent in 56%, without the need for intermediate purification. Ligand exchange on 17b also proceeded with stereorentention towards the potassium trifluoroborate salt 19 in good yields (75%). Using a modified Brown oxidation sequence in which the BEpin group was transiently transformed into the more reactive BCl2 derivative, the bicyclic azetidine-based aminoacetal 20 was isolated in 84% with retention of the stereochemistry (dr = 9:1). A similar procedure than on the azetidines was used to stereoretentively transform the cyclopentyl-compound 9c into the corresponding trifluoroborate salt 21 with high efficiency (85%). Surprisingly, when compound 9a’ was treated with BCl3, bicyclic oxaborinanone 22 was obtained upon the addition of water and concentration in vacuo, supporting once again the relative stereochemistry observed for polar radical crossover on cyclic systems.
Fig. 8. Post-functionalization of cyclic boronic esters.

Including manipulation of N-protecting groups, ligand exchanges on the boron atom and oxidation / lactonization on azetidines, ligand exchange and oxidative formation of oxaborinanone on borylated cyclopentanes.
Conclusion
We have developed a robust, efficient and highly diastereoselective sequence based on polar radical crossover that allows to access stereodefined trisubstituted azetidines, cyclobutanes, cyclopentanes, THFs and pyrrolidines. Fine tuning of reaction conditions revealed the importance of diol ligands on the boron to maximize the stereoselectivity, opening thereof new opportunities in boron-based synthetic methodologies.
Methods
See Supplementary methods.
See Supplementary Data 1.
Supplementary information
Description of Additional Supplementary Files
Acknowledgements
D.D., F.T., C.M.T., and J.R. are grateful to the Deutsche Forschungsgemeinschaft (Heisenberg fellowship: DI 2227/4-1), to the Ludwig-Maximilians University (LMU Excellence) and the Technical University of Darmstadt for Ph.D. funding and financial support. R.G. thanks the ERASMUS program.
Author contributions
Conceptualization and supervision: D.D. Experimental investigation: F.T., R.G. Structure analysis: J.R., C.M.T. Writing: D.D., C.M.T. Supporting information: F.T., R.G., J.R. Proofreading: D.D., C.M.T., F.T., R.G., and J.R.
Peer review
Peer review information
Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Data availability
Data for this manuscript has been deposited in figshare: 10.6084/m9.figshare.25907506. “Supplementary Methods” contains detailed protocols for the preparation of substrates, reaction optimizations, scope evaluation and description of analytical data (1H and 13C NMR, HRMS). “Supplementary Data 1” contains all 1H and 13C NMR spectra.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s42004-024-01221-3.
References
- 1.Burkhard JA. Synthesis of azaspirocycles and their evaluation in drug discovery. Angew. Chem. Int. Ed. 2010;49:3524. doi: 10.1002/anie.200907108. [DOI] [PubMed] [Google Scholar]
- 2.Wuitschik G, Rogers-Evans M. Oxetanes as promising modules in drug discovery. Angew. Chem. Int. Ed. 2006;45:7736. doi: 10.1002/anie.200602343. [DOI] [PubMed] [Google Scholar]
- 3.Wuitschik G, et al. Spirocyclic oxetanes: synthesis and properties. Angew. Chem. Int. Ed. 2008;47:4512. doi: 10.1002/anie.200800450. [DOI] [PubMed] [Google Scholar]
- 4.Lopchuk JM, et al. Strain-release heteroatom functionalization: development, scope, and stereospecificity. J. Am. Chem. Soc. 2017;139:3209. doi: 10.1021/jacs.6b13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang D, Gao S. Sonogashira coupling in natural product synthesis. Org. Chem. Front. 2014;1:556. doi: 10.1039/C3QO00086A. [DOI] [Google Scholar]
- 6.Lamberth C. Alkyne chemistry in crop protection. Bioorg. Med. Chem. 2009;17:4047. doi: 10.1016/j.bmc.2008.11.037. [DOI] [PubMed] [Google Scholar]
- 7.Carreira EM, Fessard TC. Four-membered ring-containing spirocycles: synthetic strategies and opportunities. Chem. Rev. 2014;114:8257. doi: 10.1021/cr500127b. [DOI] [PubMed] [Google Scholar]
- 8.Simlandy AK, Lyu M-Y, Brown MK. Catalytic arylboration of spirocyclic cyclobutenes: rapid access to highly substituted spiro[3.n]alkanes. ACS Catal. 2021;11:12815. doi: 10.1021/acscatal.1c03491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reidl TW, Anderson LL. Divergent functionalizations of azetidines and unsaturated azetidines. Asian J. Org. Chem. 2019;8:931. doi: 10.1002/ajoc.201900229. [DOI] [Google Scholar]
- 10.Frank N, et al. Synthesis of meta-substituted arene bioisosteres from [3.1.1]propellane. Nature. 2022;611:721. doi: 10.1038/s41586-022-05290-z. [DOI] [PubMed] [Google Scholar]
- 11.Pickford HD, Nugent J, Owen B. Twofold radical-based synthesis of N,C-difunctionalized bicyclo[1.1.1]pentanes. J. Am. Chem. Soc. 2021;143:9729. doi: 10.1021/jacs.1c04180. [DOI] [PubMed] [Google Scholar]
- 12.Nugent J, Arroniz C, Shire BR. A general route to bicyclo[1.1.1]pentanes through photoredox catalysis. ACS Catal. 2019;9:9568. doi: 10.1021/acscatal.9b03190. [DOI] [Google Scholar]
- 13.Makarov IS, Brocklehurst CE. Synthesis of bicyclo[1.1.1]pentane bioisosteres of internal alkynes and para-disubstituted benzenes from [1.1.1]propellane. Angew. Chem. Int. Ed. 2017;56:12774. doi: 10.1002/anie.201706799. [DOI] [PubMed] [Google Scholar]
- 14.Okude R, Mori G, Yagi A, Itami K. Programmable synthesis of multiply arylated cubanes through C–H metalation and arylation. Chem. Sci. 2020;11:7672. doi: 10.1039/D0SC01909G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reekie TA, Williams CM, Rendina LM, Kassiou M. Cubanes in medicinal chemistry. J. Med. Chem. 2019;62:1078. doi: 10.1021/acs.jmedchem.8b00888. [DOI] [PubMed] [Google Scholar]
- 16.Lopchuk JM, Wang J, Pan C-M, Malins LR. Strain-release amination, R. Gianatassio. Science. 2016;351:241. doi: 10.1126/science.aad6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gianatassio R, Kadish D. Direct alkylation of 1-azabicyclo[1.1.0]butanes. Org. Lett. 2019;21:2060. doi: 10.1021/acs.orglett.9b00321. [DOI] [PubMed] [Google Scholar]
- 18.Trauner F, Reiners F, Apaloo-Messan K-E, Nißl B. Strain-release arylations for the bis-functionalization of azetidines. Chem. Commun. 2022;58:2564. doi: 10.1039/D1CC07053C. [DOI] [PubMed] [Google Scholar]
- 19.Fawcett A, Murtaza A, Gregson CHU, Aggarwal VK. Strain-release-driven homologation of boronic esters: application to the modular synthesis of azetidines. J. Am. Chem. Soc. 2019;141:4573. doi: 10.1021/jacs.9b01513. [DOI] [PubMed] [Google Scholar]
- 20.Tyler J, Noble A, Aggarwal VK. Four-component strain-release-driven synthesis of functionalized azetidines. Angew. Chem. Int. Ed. 2022;61:e202214049. doi: 10.1002/anie.202214049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gregson CHU, Noble A, Aggarwal VK. Divergent, strain-release reactions of azabicyclo[1.1.0]butyl carbinols: semipinacol or spiroepoxy azetidine formation. Angew. Chem. Int. Ed. 2021;60:7360. doi: 10.1002/anie.202100583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fawcett A, Biberger T, Aggarwal VK. Carbopalladation of C–C σ-bonds enabled by strained boronate complexes. Nat. Chem. 2019;11:117. doi: 10.1038/s41557-018-0181-x. [DOI] [PubMed] [Google Scholar]
- 23.Tyler JL, Aggarwal VK. Synthesis and applications of bicyclo[1.1.0]butyl and azabicyclo[1.1.0]butyl organometallic. Chem. Eur. J. 2023;29:e202300008. doi: 10.1002/chem.202300008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wölfl B, Winter N, Li J, Noble A, Aggarwal VK. Strain-release driven epoxidation and aziridination of bicyclo[1.1.0]butanes via palladium catalyzed σ-bond nucleopalladation. Angew. Chem. Int. Ed. 2023;62:e202217064. doi: 10.1002/anie.202217064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lewis–Borrell L, Sneha M. Direct observation of reactive intermediates by time-resolved spectroscopy unravels the mechanism of a radical-induced 1,2-metalate rearrangement. J. Am. Chem. Soc. 2021;143:17191. doi: 10.1021/jacs.1c07964. [DOI] [PubMed] [Google Scholar]
- 26.Bennett SH, et al. Difunctionalization of C–C σ-bonds enabled by the reaction of bicyclo[1.1.0]butyl boronate complexes with electrophiles: reaction development, scope, and stereochemical origins. J. Am. Chem. Soc., 2020;142:16766. doi: 10.1021/jacs.0c07357. [DOI] [PubMed] [Google Scholar]
- 27.Gregson CHU, Ganesh V. Strain release of donor–acceptor cyclopropyl boronate complexes. Org. Lett. 2019;21:3412. doi: 10.1021/acs.orglett.9b01152. [DOI] [PubMed] [Google Scholar]
- 28.Gatazka M, McFee E. New strategies for the synthesis of 1- and 2-azetines and their applications as value-added building blocks. Org. Biomol. Chem. 2022;20:9052. doi: 10.1039/D2OB01812H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rykaczewski K, Becker M. Photochemical strategies enable the synthesis of tunable azetidine-based energetic materials. J. Am. Chem. Soc. 2022;144:19089. doi: 10.1021/jacs.2c08191. [DOI] [PubMed] [Google Scholar]
- 30.Blackmun DE, Chamness SA. Intramolecular, visible-light-mediated aza Paternò–Büchi reactions of unactivated alkenes. Org. Lett. 2022;24:3053. doi: 10.1021/acs.orglett.2c01008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wearing ER, Blackmun DE, Becker MR, Schindler CS. 1- and 2-Azetines via visible light-mediated [2 + 2]-cycloadditions of alkynes and oximes. J. Am. Chem. Soc. 2021;143:16235. doi: 10.1021/jacs.1c07523. [DOI] [PubMed] [Google Scholar]
- 32.Sarkar, D., Bera, N. & Ghosh, S. [2+2] Photochemical cycloaddition in organic synthesis. Eur. J. Org. Chem. 1310 (2020).
- 33.Gilbert A, Bach T. The awakening of a sleeping beauty: the ortho photocycloaddition in the total synthesis of protoilludane- and prezizaene-type sesquiterpenes. Synlett. 2023;34:1343. doi: 10.1055/s-0042-1751354. [DOI] [Google Scholar]
- 34.Rigotti T, Schwinger DP, Graßl R, Jandla C, Bach T. Enantioselective crossed intramolecular [2 + 2] photocycloaddition reactions mediated by a chiral chelating Lewis acid. Chem. Sci. 2022;13:2378. doi: 10.1039/D2SC00113F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rigotti T, Bach T. Bicyclo[2.1.1]hexanes by visible light-driven intramolecular crossed [2 + 2] photocycloadditions. Org. Lett. 2022;24:8821. doi: 10.1021/acs.orglett.2c03606. [DOI] [PubMed] [Google Scholar]
- 36.Kleinmans R, et al. Intermolecular [2π + 2σ]-photocycloaddition enabled by triplet energy transfer. Nature. 2022;605:477. doi: 10.1038/s41586-022-04636-x. [DOI] [PubMed] [Google Scholar]
- 37.Ma J, et al. Photochemical intermolecular dearomative cycloaddition of bicyclic azaarenes with alkenes. Science. 2021;371:1338. doi: 10.1126/science.abg0720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Strieth-Kalthoff F. Discovery of unforeseen energy-transfer-based transformations using a combined screening approach. Chem. 2019;5:2183. doi: 10.1016/j.chempr.2019.06.004. [DOI] [Google Scholar]
- 39.Wang W, Cai Y, Guo R, Brown MK. Synthesis of complex bicyclic scaffolds by intermolecular photosensitized dearomative cycloadditions of activated alkenes and naphthalenes. Chem. Sci. 2022;13:13582. doi: 10.1039/D2SC04789F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu Y, Ni D, Brown MK. Boronic ester enabled [2 + 2]-cycloadditions by temporary coordination: synthesis of artochamin J and piperarborenine B. J. Am. Chem. Soc. 2022;144:18790. doi: 10.1021/jacs.2c08777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Guo R, Adak S. Photochemical dearomative cycloadditions of quinolines and alkenes: scope and mechanism studies. J. Am. Chem. Soc. 2022;144:17680. doi: 10.1021/jacs.2c07726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo R, Chang Y-C, Herter L, Salome C, Braley SE. Strain-release [2π + 2σ] cycloadditions for the synthesis of bicyclo[2.1.1]hexanes Initiated by energy transfer. J. Am. Chem. Soc. 2022;144:7988. doi: 10.1021/jacs.2c02976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu Y, Ni D, Stevenson BG. Photosensitized [2 + 2]-cycloadditions of alkenylboronates and alkenes. Angew. Chem. Int. Ed. 2022;61:e202200725. doi: 10.1002/anie.202200725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schmid SC, Guzei IA. A stereoselective [3+1] ring expansion for the synthesis of highly substituted methylene azetidines. Angew. Chem. Int. Ed. 2017;56:12229. doi: 10.1002/anie.201705202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Han J-Q, Zhang H-H, Xu P-F, Luo Y-C. Lewis acid and (hypo)iodite relay catalysis allows a strategy for the synthesis of polysubstituted azetidines and tetrahydroquinolines. Org. Lett. 2016;18:5212. doi: 10.1021/acs.orglett.6b02430. [DOI] [PubMed] [Google Scholar]
- 46.Malik, S. & Nadir, U. K. A facile synthesis of 1-arenesulfonylazetidines through reaction of 1-arenesulfonylaziridines with dimethylsulfoxonium methylide generated under microwave irradiation. Synlett 108–110 (2008).
- 47.Kern N, Felten A-S. Robust synthesis of N-sulfonylazetidine building blocks via ring contraction of α-bromo N-sulfonylpyrrolidinones. Org. Lett. 2014;16:6104. doi: 10.1021/ol5029496. [DOI] [PubMed] [Google Scholar]
- 48.Quinodoz P, Drouillat B, Wright K, Marrot J, Couty F. N-Arylazetidines: preparation through anionic ring closure. J. Org. Chem. 2016;81:2899. doi: 10.1021/acs.joc.6b00169. [DOI] [PubMed] [Google Scholar]
- 49.de Figueiredo RM, Fröhlich R, Christmann M. N,N‘-Carbonyldiimidazole-mediated cyclization of amino alcohols to substituted azetidines and other N-heterocycles. J. Org. Chem. 2006;71:4147. doi: 10.1021/jo060130b. [DOI] [PubMed] [Google Scholar]
- 50.Fritz SP, Moya JF, Unthank MG. An efficient synthesis of azetidines with (2-bromoethyl)sulfonium triflate. Synthesis. 2012;44:1584. doi: 10.1055/s-0031-1290951. [DOI] [Google Scholar]
- 51.Eisold M, Didier D. Highly diastereoselective synthesis of methylenecyclobutanes by merging boron-homologation and boron-allylation strategies. Angew. Chem. Int. Ed. 2015;54:15884. doi: 10.1002/anie.201507444. [DOI] [PubMed] [Google Scholar]
- 52.Baumann A, Music A, Karaghiosoff K, Didier D. Highly diastereoselective approach to methylenecyclopropanes via boron-homologation/allylboration sequences. Chem. Commun., 2016;52:2529. doi: 10.1039/C5CC09904H. [DOI] [PubMed] [Google Scholar]
- 53.Eisold M, Kiefl GM, Didier D. Single-pot asymmetric approach toward enantioenriched quaternary stereocenter-containing alkylidenecyclobutanes. Org. Lett. 2016;18:3022. doi: 10.1021/acs.orglett.6b01432. [DOI] [PubMed] [Google Scholar]
- 54.Eisold M, Baumann AN. Unsaturated four-membered rings: efficient strategies for the construction of cyclobutenes and alkylidenecyclobutanes. Chem. Eur. J. 2017;23:1634. doi: 10.1002/chem.201604585. [DOI] [PubMed] [Google Scholar]
- 55.Eisold M, Didier D. Stereoselective access to alkylidenecyclobutanes through γ-selective cross-coupling strategies. Org. Lett. 2017;19:4046. doi: 10.1021/acs.orglett.7b01803. [DOI] [PubMed] [Google Scholar]
- 56.Baumann AN, et al. Methods for the synthesis of substituted azetines. Org. Lett. 2017;19:5681. doi: 10.1021/acs.orglett.7b02847. [DOI] [PubMed] [Google Scholar]
- 57.Eisold M, Müller-Deku A, Reiners F, Didier D. Parallel approaches for the functionalization of thietes: α-metalation versus C–H activation. Org. Lett. 2018;20:4654. doi: 10.1021/acs.orglett.8b01961. [DOI] [PubMed] [Google Scholar]
- 58.Reiners F, Joseph E. Stereoselective access to azetidine-based α-amino acids and applications to small peptide synthesis. Org. Lett. 2020;22:8533. doi: 10.1021/acs.orglett.0c03131. [DOI] [PubMed] [Google Scholar]
- 59.Music A, Hoarau C. Catalyst-free enantiospecific olefination with in situ generated organocerium species. Angew. Chem. Int. Ed. 2019;58:1188. doi: 10.1002/anie.201810327. [DOI] [PubMed] [Google Scholar]
- 60.Music A, Baumann AN. Single-pot access to bisorganoborinates: applications in zweifel olefination. Org. Lett. 2019;21:2189. doi: 10.1021/acs.orglett.9b00493. [DOI] [PubMed] [Google Scholar]
- 61.Music A, et al. Electrochemical synthesis of biaryls via oxidative intramolecular coupling of tetra(hetero)arylborates. J. Am. Chem. Soc. 2020;142:4341. doi: 10.1021/jacs.9b12300. [DOI] [PubMed] [Google Scholar]
- 62.Baumann AN, Music A, Dechent J. Electro-olefination—a catalyst free stereoconvergent strategy for the functionalization of alkenes. Chem. Eur. J. 2020;26:8382. doi: 10.1002/chem.202001394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Music A, Baumann AN, Boser F. Photocatalyzed transition-metal-free oxidative cross-coupling reactions of tetraorganoborates. Chem. Eur. J. 2021;27:4322. doi: 10.1002/chem.202005282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Music A, Nuber CM, Lemke Y, Spieß P, Didier D. Electro-alkynylation: intramolecular rearrangement of trialkynylorganoborates for chemoselective C(sp2)–C(sp) bond formation. Org. Lett. 2021;23:4179. doi: 10.1021/acs.orglett.1c01126. [DOI] [PubMed] [Google Scholar]
- 65.Kischkewitz M, Okamoto K, Mück-Lichtenfeld C, Studer A. Radical-polar crossover reactions of vinylboron ate complexes. Science. 2017;355:936. doi: 10.1126/science.aal3803. [DOI] [PubMed] [Google Scholar]
- 66.Gerleve C, Kischkewitz M. Synthesis of α-chiral ketones and chiral alkanes using radical polar crossover reactions of vinyl boron ate complexes. Angew. Chem. Int. Ed. 2018;57:2441. doi: 10.1002/anie.201711390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Silvi M, Sandford C, Aggarwal VK. Merging photoredox with 1,2-metallate rearrangements: the photochemical alkylation of vinyl boronate complexes. J. Am. Chem. Soc. 2017;139:5736. doi: 10.1021/jacs.7b02569. [DOI] [PubMed] [Google Scholar]
- 68.Silvi M, Schrof R, Noble A, Aggarwal VK. Enantiospecific three-component alkylation of furan and indole. Chem. Eur. J. 2018;24:4279. doi: 10.1002/chem.201800527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lovinger GJ, Morken JP. Ni-catalyzed enantioselective conjunctive coupling with C(sp3) electrophiles: a radical-ionic mechanistic dichotomy. J. Am. Chem. Soc. 2017;139:17293. doi: 10.1021/jacs.7b10519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tappin NDC, Gnägi-Lux M, Renaud P. Radical-triggered three-component coupling reaction of alkenylboronates, α-halocarbonyl compounds, and organolithium reagents: the inverse ylid mechanism. Chem. Eur. J. 2018;24:11498. doi: 10.1002/chem.201802384. [DOI] [PubMed] [Google Scholar]
- 71.Brace NO. Syntheses with perfluoroalkyl radicals from perfluoroalkyl iodides. A rapid survey of synthetic possibilities with emphasis on practical applications. Part one: alkenes, alkynes and allylic compounds. J. Fluor. Chem. 1999;93:1. doi: 10.1016/S0022-1139(98)00255-3. [DOI] [Google Scholar]
- 72.Myhill JA, Wilhelmsen CA, Zhang L. Diastereoselective and enantioselective conjunctive cross-coupling enabled by boron ligand design. J. Am. Chem. Soc. 2018;140:15181. doi: 10.1021/jacs.8b09909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nomura S, Sakamaki S, Hongu M. Discovery of canagliflozin, a novel C-glucoside with thiophene ring, as sodium-dependent glucose cotransporter 2 inhibitor for the treatment of type 2 diabetes mellitus. J. Med. Chem. 2010;53:6355. doi: 10.1021/jm100332n. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of Additional Supplementary Files
Data Availability Statement
Data for this manuscript has been deposited in figshare: 10.6084/m9.figshare.25907506. “Supplementary Methods” contains detailed protocols for the preparation of substrates, reaction optimizations, scope evaluation and description of analytical data (1H and 13C NMR, HRMS). “Supplementary Data 1” contains all 1H and 13C NMR spectra.