

Before 1990 the role of inherited factors in the aetiology of adult cancer was relatively poorly understood and aroused little interest among doctors and the public alike—although familial adenomatous polyposis (the autosomal dominant colon cancer syndrome) was an exception in this respect. In the past decade, however, interest has increased markedly. In the West Midlands, for example, familial cancer referrals constituted <1% (<20 cases) of all clinical genetic referrals in 1991, whereas now they represent over 30% of cases (>1000).
Despite the estimate that 5-10% of colorectal cancer has an inherited basis, only a small percentage of referred families have mutations in one of the currently identified genes. Furthermore, mutation studies are usually possible only if DNA is available from an affected patient, so molecular investigation will facilitate the management of only a small minority of cases. The remaining referrals must be managed with clinically derived strategies. This article discusses the clinical features and management of dominant colon cancer syndromes and provides referral guidelines and screening protocols for more common familial clusters.
Genetic counselling for families with a history of cancer requires a full and accurate family history. When possible, histological confirmation of the reported tumours should be obtained. It should then be possible to recognise the specific cancer syndromes. It is important to emphasise to families that however extensive the family history of cancer (unless present on both sides), a person will always have a greater than 50% chance of not developing that particular tumour. This may surprise but greatly reassure many families.
Familial adenomatous polyposis
Familial adenomatous polyposis, previously called polyposis coli (or Gardner's syndrome if extra colonic manifestations were present), is the best recognised of the colorectal cancer syndromes but accounts for less than 1% of all colorectal cancers and has an incidence of 1 in 10 000. It is characterised by the presence of 100 or more tubovillous adenomas in the colon, with intervening microadenoma on histological examination. The mean age of diagnosis of polyps is during teenage years, and almost all of gene carriers have polyps by the age of 40. If these polyps are left untreated, malignant transformation is inevitable, with a mean age of colorectal cancer occurring during the patients' mid-30s, often with synchronous tumours.
This condition is an autosomal dominant disorder, with the offspring of affected individuals at 50% risk of being gene carriers. The diagnosis of familial adenomatous polyposis should always result in a careful and full evaluation of the family history. Wherever possible, parents should have at least one colonoscopy, irrespective of age. In most cases without a family history, parental examination will be negative and the proband will probably be one of 30% of cases that represent new mutations. The siblings of all probands, however, should be offered annual flexible sigmoidoscopy up to the age of 40 or until proved to be non-gene carriers.
The cloning of the causative gene (APC) on chromosome 5 in 1991 dramatically changed the management of familial adenomatous polyposis. If DNA is available from an affected individual, mutation detection is possible in about 70% of families. In these families first degree relatives should be offered predictive testing with appropriate genetic counselling. In families with no identified mutation, linkage studies to identify the “high risk” chromosome 5 are possible in many cases. Non-gene carriers should be reassured and surveillance stopped. Gene carriers should be offered annual flexible sigmoidoscopy from the age of 12. Once several polyps have been identified, the timing and type of surgery available should be discussed (a sensitive issue in teenagers and young adults). The two most common options are ileal-rectal anastomosis with annual surveillance of the remaining rectal tissue; and ileal-anal anastomosis with reconstruction of a rectal pouch using terminal small bowel.
Molecular testing is usually offered to “at risk” children at age 10-14 before starting annual sigmoidoscopy. However, parental pressure for earlier testing (before the child can give consent) is not uncommon, and ongoing studies may help to clarify when to proceed with testing.
Early and late extracolonic tumours in familial adenomatous polyposis
Hepatoblastoma (early)
Adrenal adenoma (early or late)
Desmoid disease (early or late)
Papillary thyroid cancer—females only (late)
Periampullary carcinoma (late)
Cloning APC explained several clinical features and aided studies of genotypes and phenotypes. For example, congenital hypertrophy of the retinal pigment epithelium, an attenuated phenotype (that is, fewer than 100 polyps or late onset), and non-malignant but debilitating and potentially lethal desmoid disease each show an association with mutations in specific exon regions. The cloning also confirmed clinical findings that familial adenomatous polyposis and Gardner's syndrome were different manifestations of the same disease spectrum that could coexist within the same family.
With greater clinical awareness, regular surveillance, and the advent of molecular investigation, almost all colorectal cancer deaths in inherited cases of familial adenomatous polyposis can be avoided. Increased survival has revealed later complications, in particular periampullary or duodenal adenocarcinoma (present in about 12% of postcolectomy cases). Also important are aggressive desmoid disease and other rare malignant disease.
Chemoprophylactic approaches to reduce polyp growth (for example, aspirin and non-digestible starch) are the subject of multicentre trials.
Hereditary non-polyposis colon cancer
Hereditary non-polyposis colon cancer (also known as Lynch syndrome) became more widely recognised about 30 years ago in families manifesting mainly colorectal cancer segregating in an autosomal dominant fashion. Many families also exhibit extracolonic tumours, particularly gynaecological, small bowel, or urinary tract carcinomas, and these became known as Lynch type 2 to distinguish them from site specific colorectal cancers, designated Lynch type 1. The subsequent name change to hereditary non-polyposis colon cancer is potentially misleading as many gene carriers will develop a small number of tubovillous adenomas, but not more than 100, as seen in familial adenomatous polyposis. The proportion of colorectal cancers due to hereditary non-polyposis colon cancer is controversial, and estimates range from 1% to 20%; most observers, however, suggest about 2%.
Modified Amsterdam criteria
Three or more cases of colorectal cancer in a minimum of two generations
One affected individual must be a first degree relative of the other two (or more) cases
One case must be diagnosed before age 50
Colorectal cancer can be replaced by endometrial or small bowel adenocarcinoma
Familial adenomatous polyposis must be excluded
The diagnosis of hereditary non-polyposis colon cancer is made on the family history as the appearance of the bowel, unlike in familial adenomatous polyposis, is not diagnostic. To improve the recognition of hereditary non-polyposis colon cancer, diagnostic criteria were devised in Amsterdam in 1991 and were subsequently modified to include non-colonic tumours.
No large series of patients fulfilling the Amsterdam criteria has a mutation detection rate >70%. The figure is much lower for families that do not meet the criteria described here. Case selection using tumour DNA instability or immunohistochemical studies can improve mutation detection rates
• Screening of other organ systems has not yet been proved beneficial• It is prudent to screen for gynaecological tumours in mutation positive families, irrespective of family history, as 40% of female gene carriers develop endometrial carcinomas• If tumours have been identified in the gynaecological or urinary tract in the family, surveillance should be offered (see the West Midlands guidelines)
In 1994 the first of the genes for hereditary non-polyposis colon cancer (hMSH2 on chromosome 2) was cloned, and since then four further genes have been identified; all are mismatch repair genes. If both copies of the genes are mutated, as postulated in Knudson's “two hit” hypothesis, that cell and all its daughter cells are missing a vital mechanism for repair of DNA in somatic tissue. Failure to repair mutations in tumour suppressor genes will in some instances result in initiation of the adenoma carcinoma sequence. Molecular studies showed that about 30% of colorectal cancers with early onset (under age 35) are due to the mismatch repair genes, and the typical age of onset and the spectrum of tumours in families with hereditary non-polyposis colon cancer were revised.
The limited available evidence suggests that screening for colorectal cancer in hereditary non-polyposis colon cancer is beneficial. In 1999 Vasen et al published figures showing clinical benefit and cost effectiveness of screening in hereditary non-polyposis colon cancer after a Finnish study reporting reduced morbidity and mortality in a non-randomised observational study of colonoscopy versus no screening.
West Midlands site specific screening strategies in hereditary non-polyposis colon cancer
Colorectal (all cases)—colonoscopy every two years at age 25-65
Endometrial*—annual pipelle biopsy (suction curettage) and ultrasound at age 30-65
Ovarian†—annual transvaginal ultrasound and serum Ca125 concentration at age 30-65
Transitional cell carcinoma in the urinary tract—annual haematuria test at age 25-40; annual urine cytology at age 40-65 (with or without cystoscopy every one to two years); annual renal ultrasound at age 40-65
*Families with history of endometrial cancer and mutation positive families. †Families with history of ovarian cancer.
The optimal surveillance frequency has not yet been defined in families with hereditary non-polyposis colon cancer. The method of choice, however, is colonoscopy rather than flexible sigmoidoscopy as 80% of cancers are proximal to the rectum (compared with only 57% in sporadic colorectal cancer). The screening interval should be at most three years and probably every 18-24 months in gene carriers. Failure to reach the caecum should be followed by barium enema examination, although surveillance using radiological techniques should probably be used sparingly owing to the theoretical mutagenic consequences in patients with DNA repair defects. Patients should understand that the strategy of colonoscopy is the removal of polyps and prevention of tumours or early diagnosis, but that complete prevention is impossible.
Familial clusters with no recognisable single gene disorder
Families whose cancers do not meet the diagnostic criteria of familial adenomatous polyposis, hereditary non-polyposis colon cancer, or rarer colorectal cancer syndromes (such as syndromes related to the PTEN gene, Turcot's syndrome, Peutz-Jeghers syndrome, or juvenile polyposis) make up the largest and most difficult group of patients requesting management. There is rarely any indication of the aetiological basis of the cluster of colorectal cancer, and many instances will be coincidental occurrences. Other tumours clusters may be due to common environmental exposures, the effect of multiple low penetrance genes, or an interaction of both these elements. The risk of colorectal cancer may be assessed with empirical risk figures. These figures are estimates, however, and do not take into account factors such as the number of unaffected relatives. Further enquiry is usually justified if features such as multiple relatives with the same tumour or early onset of tumours are present in a family.
Lifetime risk of colorectal cancer in first degree relatives of patient with colorectal cancer (from Houlston et al, 1990)
| Population risk | 1 in 50 |
|---|---|
| One first degree relative affected (any age) | 1 in 17 |
| One first degree and one second degree relative affected | 1 in 12 |
| One first degree relative affected (age <45) | 1 in 10 |
| Two first degree relatives affected | 1 in 6 |
| Autosomal dominant pedigree | 1 in 2 |
Four pointers to recognition of familial cancer clusters
High frequency of the same tumour in the family
Early age of onset of tumours
Multiple primary tumours
Recognised associations—for example, colorectal and endometrial adenocarcinomas
Concerns about not having precise risk figures may be misguided as many patients have difficulty interpreting risk figures and are often requesting only general guidance on risk and a discussion of management options. Many different screening protocols have been suggested in the past, however, and the lack of consistency and long term audit in these families is a problem.
To manage familial cancer in the West Midlands (population 5.2 million), a protocol has been developed that builds on the Calman-Hine model for cancer services and maximise the role of primary care. The protocol provides clear inclusion and screening guidance for cancer units and centres. This has promoted a consistency of management in and between families and is now allowing data collection for audit.
West Midlands inclusion and screening criteria for a family history of colorectal cancer
| Inclusion criteria | Screening method | Age range (years) for surveillance |
| One first degree relative aged >40 | Reassure, plus general advice on symptoms | Not applicable |
| One first degree relative aged <40 | Colonoscopy every 5 years; appointment at local screening unit | 25-65, or 5 years before tumour if later |
| Two first degree relatives average age >70 | Reassure, plus general advice on symptoms | Not applicable |
| Two first degree relatives average age 60-70 | Single colonoscopy; appointment at local screening unit | About 55 |
| Two first degree relatives average age 50-60 | Colonoscopy every 5 years; appointment at local screening unit | 35-65 |
| Two first degree relatives average age <50 | Colonoscopy every 3-5 years; referral to genetics unit | 30-65 |
| Three close relatives but not meeting Amsterdam criteria | Colonoscopy every 3-5 years; referral to genetics unit | Begin age 30-40; stop at 65 |
| Three close relatives meeting Amsterdam criteria | Colonoscopy every 2 years; referral to genetics unit | 25-65 |
| Familial adenomatous polyposis | Annual sigmoidoscopy; referral to genetics unit | 12-40 |
Further reading
Foulkes W. A tale of four syndromes: familial adenomatous polyposis, Gardner syndrome, attenuated APC and Turcot syndrome. Q J Med 1995;88:853-63.
Vasen HF, van Ballegooijen, Buskens E, Kleibeuker JK, Taal BG, Griffioen G, et al. A cost-effectiveness analysis of colorectal screening of hereditary nonpolyposis colorectal carcinoma gene carriers. Cancer 1998;82:1632-7.
Burke W, Petersen G, Lynch P, Bokin J, Daly M, Garber J, et al. Recommendations for follow-up care of individuals with an inherited predisposition to cancer. 1: Hereditary nonpolyposis colon cancer. JAMA 1997;277:915-9.
Houlston RS, Murday V, Harocopos C, Williams CB, Slack J. Screening and genetic counselling for relatives of patients with colorectal cancer in a family cancer clinic. BMJ 1990;301:366-8.
Hodgson SV, Maher ER, eds. A practical guide to human cancer genetics. Cambridge: Cambridge University Press, 1999.
It may be wise for general practitioners to use a reactive approach to patient enquiries until evidence exists to support a proactive approach. In the West Midlands, patients requesting advice are asked to complete a data collection sheet at home. This form and the inclusion criteria are available at www.bham.ac.uk/ich/clingen.htm. Completion of the form in the patient's own time, at home, facilitates discussion with relatives to clarify the relevant information and has saved time in primary care if a referral is required.
After histological confirmation in suspected familial cases, the data are evaluated centrally to identify high risk families requiring specialist investigation or referral to a cancer unit.
In a pilot study (population 200 000) the protocol reduced referrals from primary care by 50%, with a greater reduction in screening owing to a fall in low risk referrals to cancer units. This was associated with an increased referral rate for high risk referrals to clinical genetics departments. Central coordination prevents unnecessary investigations for different branches of any one family and facilitates audit.
Reports from general practitioners and patients suggest that individuals at no or minimal increased risk of cancer avoid unnecessary outpatient appointments and screening and for the most part are reassured by standardised protocols with explanations on the data collection forms. Such systems need to be studied further but seem to be preferable to continuing the current exponential rise of ad hoc responses from individual clinicians without long term audit.
Figure.

Number of referrals of patients with cancer (except familial adenomatous polyposis) to West Midlands regional clinical genetics service, 1988-98
Figure.


Features of familial adenomatous polyposis: colon with multiple polyps (top) and jaw cysts (bottom)
Figure.
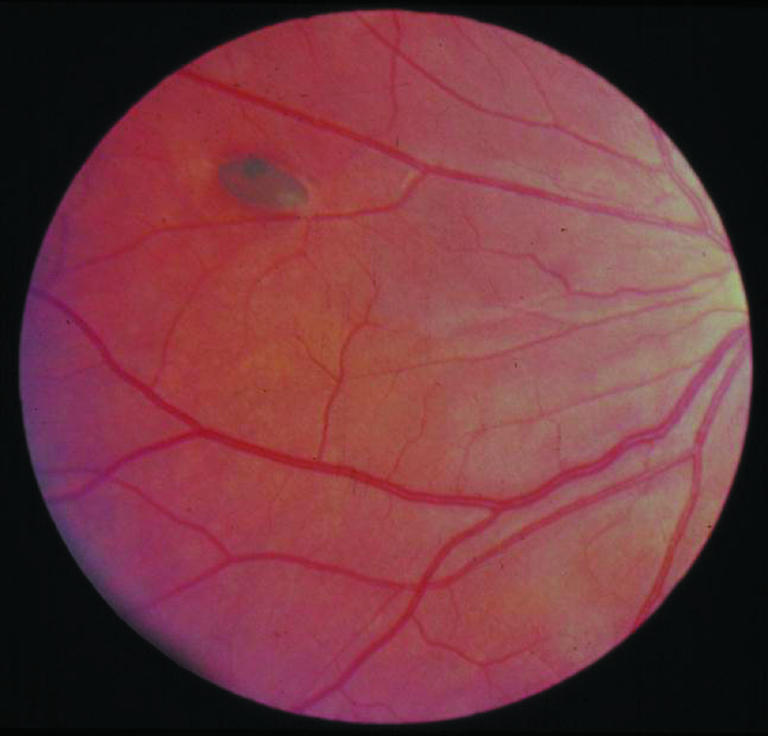
Congenital hypertrophy of the retinal pigmentary epithelium is a feature of familial adenomatous polyposis
Figure.

Site of tumours and frequency in hereditary non-polyposis colon cancer (upper figures in ranges may be overestimates owing to ascertainment bias)
Figure.

Algorithm for West Midlands family cancer strategy
Acknowledgments
Professor Eamonn Maher gave helpful comments and support.
Footnotes
T R P Cole is consultant in clinical and cancer genetics and H V Sleightholme is regional cancer coordinator in the department of clinical genetics, Birmingham Women's Hospital, Birmingham.
The ABC of colorectal cancer is edited by D J Kerr, professor at the Institute for Cancer Studies, University of Birmingham; Annie Young, research fellow at the School of Health Sciences, University of Birmingham; and F D Richard Hobbs, professor in the department of primary care and general practice, University of Birmingham. The series will be published as a book by the end of 2000.