

Abstract
The natural product veranamine was isolated from the marine sponge Verongula rigida. It contains a unique heterocyclic scaffold and demonstrates in vivo antidepressant activity and selective affinity for 5HT2B and sigma-1 receptors. The first total synthesis of veranamine is reported. Our scalable synthesis offers veranamine in six steps and 25% yield via an unprecedented vinylogous Pictet–Gams pyridine formation strategy. Veranamine is a promising new lead compound for antidepressant drug development.
Graphical Abstract
Major depressive disorder is a debilitating illness that contributes substantially to global disease burden.1,2 Many patients benefit from a range of approved antidepressant drugs that increase the intersynaptic concentrations of serotonin, norepinephrine, or dopamine by inhibiting membrane transporter proteins or by blocking the degradation of these neurotransmitters by monoamine oxidase enzymes.3 Unfortunately, all of today’s pharmaceutical interventions are beleaguered by problems of late onset of action, troubling side effects, and low efficacy.4–6 Many researchers now seek an improved understanding of the mechanistic paradigms of depression to facilitate the selection of optimal pharmaceutical targets.7–13 In parallel, new antidepressant lead compounds with unique molecular scaffolds and mechanisms of action are needed. Cognizant of the promise of marine natural products as unique leads for drug development,14,15 we have turned to the marine chemical space in the search for new compounds that demonstrate in vivo antidepressant activity.16–18
Herein we report the isolation, characterization, in vitro and in vivo evaluations, and total synthesis of one such compound, veranamine, from the Florida sponge Verongula rigida (1, Figure 1). The structure of veranamine resembles that of the β-carboline alkaloid family, which is exemplified by the psychoactive natural product harmine (2).19 Unlike the β-carbolines, however, the tricyclic scaffold of veranamine is not completely unsaturated, and thus not flat, as it contains an sp3-hybridized nonprotonated carbon atom bearing two methyl groups in its central ring. An increase in the ratio of sp3- to sp2-hybridized carbon atoms has been associated with advantageous properties with respect to drug development.20 The veranamine scaffold constitutes a previously unknown variation of the benzo[c][2,7]naphthyridine (3) tricyclic ring system found in larger pyridoacridine alkaloids,21 such as amphimedine (4),22 which have a wide range of biological activities.23,24 The geminal dimethyl moiety of veranamine resembles an analogous feature of the well-known psychoactive natural compound cannabinol (5).25
Figure 1.
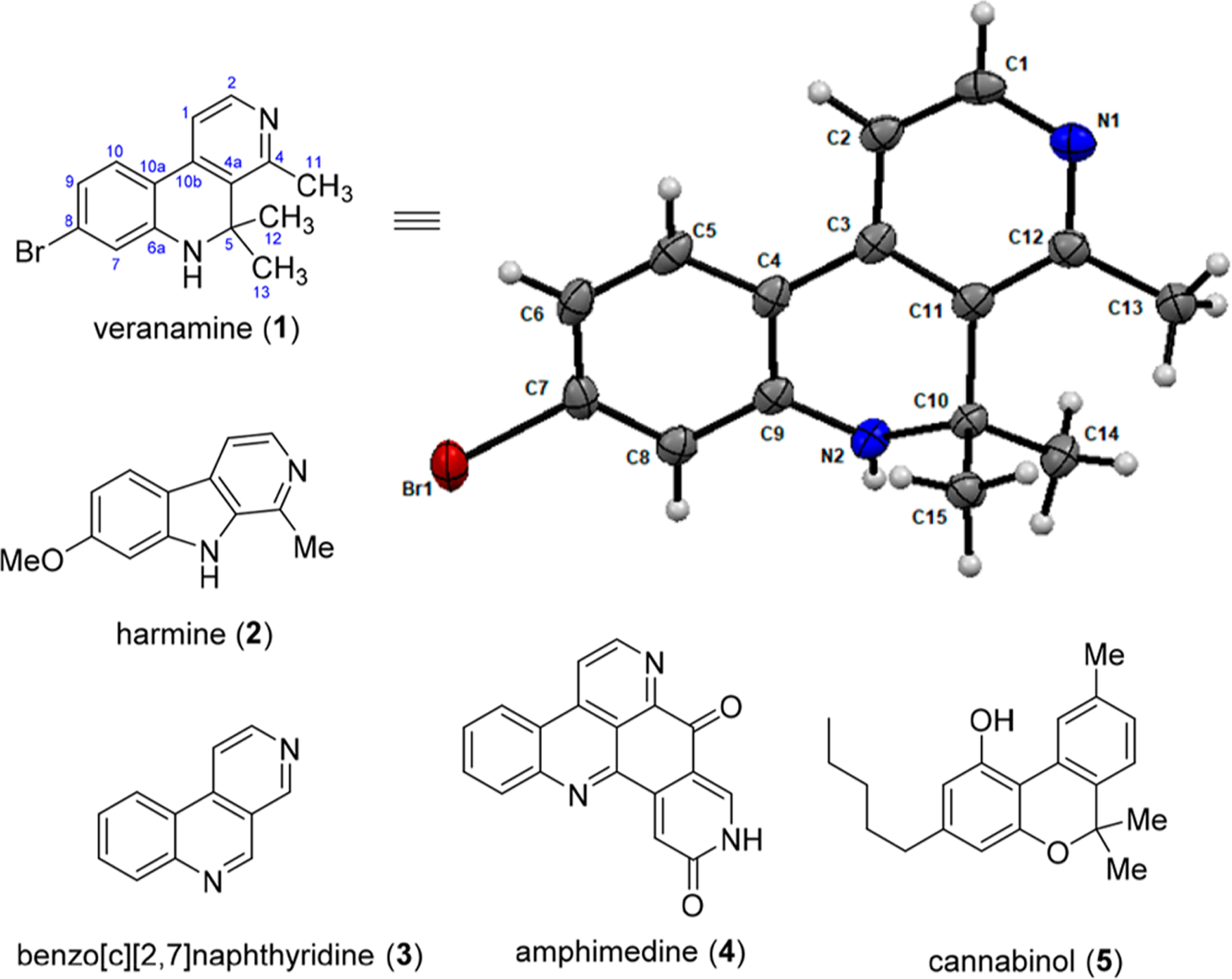
Veranamine (1), related compounds 2–5, and the crystal structure of 1 with the atom-labeling scheme. Displacement ellipsoids are drawn at the 50% probability level.
RESULTS AND DISCUSSION
Isolation.
Veranamine was obtained from V. rigida, collected in the Florida Keys. A 3 kg amount of V. rigida was extracted exhaustively with EtOH. The extract was reduced in volume and fractionated on silica gel by vacuum liquid chromatography using a gradient of solvents from hexane to MeOH. Polar fractions were further purified by reversed-phase chromatography on a solid C18 phase extraction cartridge, followed by HPLC on a C8 column to yield 8 mg of the new alkaloid veranamine (1). The HRMS of veranamine showed two peaks of nearly equivalent intensity at m/z 303.0494 and 305.0475 [M + H], indicating the presence of a bromine atom and suggesting the formula C15H15BrN2. Veranamine’s UV spectrum had absorption maxima at λ 204, 245, and 380 nm, and its IR spectrum showed peaks at 3422 cm−1(broad) corresponding to an NH stretching band and 2331 cm−1 and 1634 cm−1, indicative of an aromatic system. Structural assignment was accomplished using 1H and 13C NMR with HMQC, HMBC, and COSY correlations (Table 1) and confirmed by X-ray analysis (Figure 1).
Table 1.
NMR Data for Veranamine in Methanol-d4 (400 MHz for 1H and 100 MHz for 13C)
| carbon | δC, type | δH (J in Hz) | COSY | HMBC |
|---|---|---|---|---|
| 1 | 115.38, CH | 7.61, d (5.2) | 8.25 | 2, 4a, 10a |
| 2 | 144.8, CH | 8.25, d (5.2) | 7.61 | 1, 4, 6a |
| 4 | 153.11, C | |||
| 4a | 134.0, C | |||
| 5 | 53.47, C | |||
| 6a | 146.0, C | |||
| 7 | 117.3, CH | 6.93, d (1.8) | 8, 9, 10a | |
| 8 | 125.2, C | |||
| 9 | 120.6, CH | 6.88, dd (8.4, 1.8) | 7.55 | 7, 8, 10 |
| 10 | 125.9, CH | 7.55, d (8.4) | 6.88 | 6a, 8, 10b |
| 10a | 116.5, C | |||
| 10b | 140.6, C | |||
| 11 | 24.14, CH3 | 2.7, s | 4, 4a | |
| 12 | 27.9, CH3 | 1.62, s | 4a, 5 | |
| 13 | 27.9, CH3 | 1.62, s | 4a, 5 |
In Vivo Evaluation.
The structural similarities between veranamine and compounds with neurological activities motivated us to evaluate the antidepressant activity of this natural product by using a forced-swim test (Figure 2A).26 Veranamine demonstrated potent antidepressant activity at a dose of 20 mg/kg, i.p., decreasing the immobility time significantly, relative to the clinically utilized drug desipramine. A locomotor activity test indicated that veranamine did not demonstrate nonspecific stimulant action, a potential cause of false-positive results in the forced-swim assay (Figure 2B).27
Figure 2.
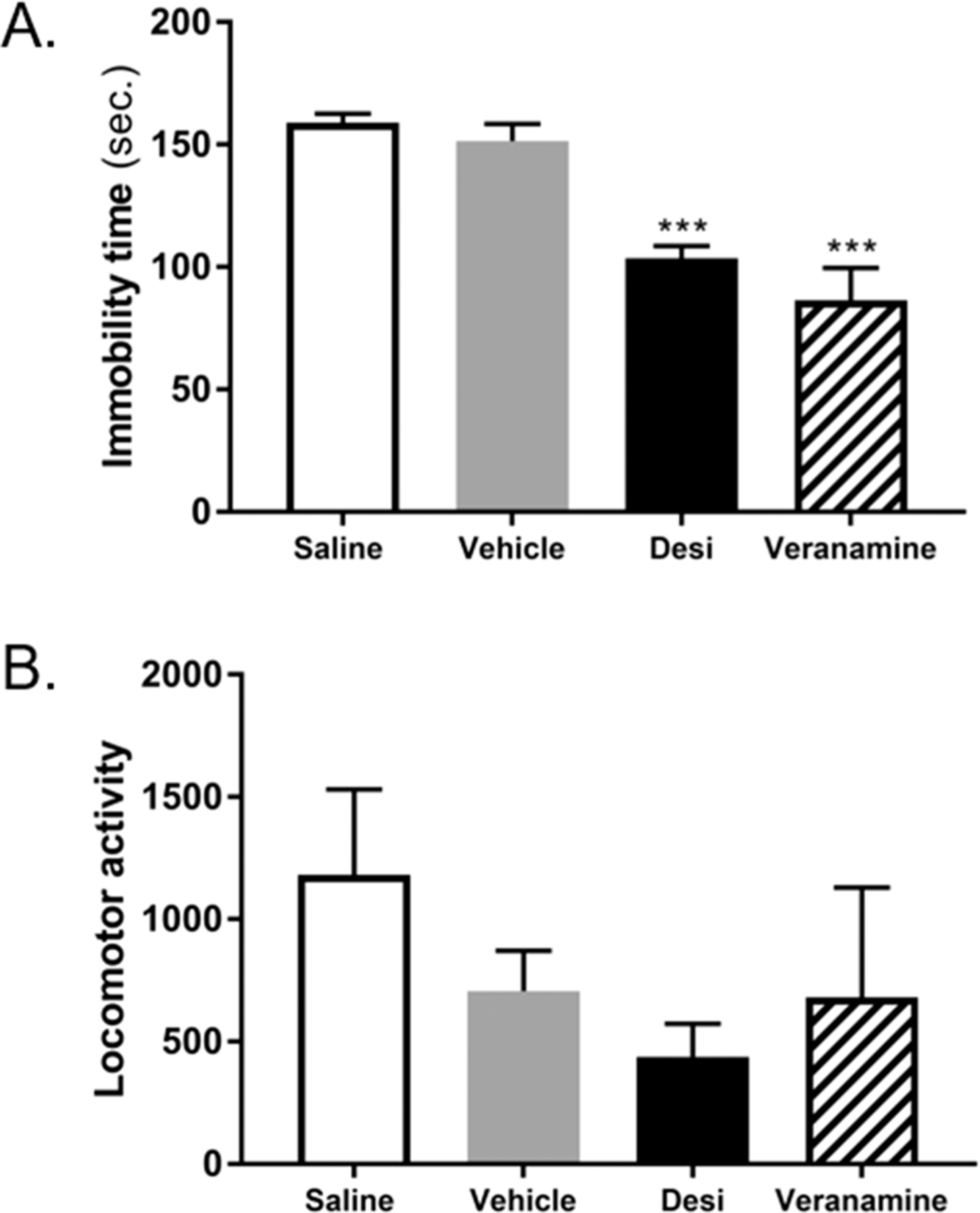
Effect of natural veranamine (20 mg/kg, i.p.) on immobility time in mouse forced-swim test (A) and locomotor activity in Swiss Webster mice (B) compared to desipramine (Desi., 20 mg/kg). ***p < 0.001 versus corresponding saline vehicle (Tukey’s posthoc test).
In Vitro Evaluation.
In order to elucidate its biochemical target and mechanism of action, natural veranamine was screened using receptor binding assays of a panel of 41 neuroreceptors and transporters at the NIMH Psychoactive Drug Screening Program (Table 2).28 Veranamine demonstrated binding affinity to two targets, 5HT2B and sigma-1, with Ki values of 390 and 560 nM, respectively. The compound showed no affinity (Ki > 10 μM) for any of the adrenergic, dopamine, histidine, muscarinic acetylcholine, or opioid receptors in the panel and did not bind to the sigma-2 receptor or to any of the other 11 serotonin receptors evaluated. Furthermore, unlike the majority of clinically used antidepressants today, veranamine showed no affinity for serotonin, norepinephrine, or dopamine transporters. Given the antidepressant phenotype observed in our animal assays, we were excited by this selective and unique binding profile, with affinity to just two of the 40 protein targets that were evaluated.
Table 2.
In Vitro Binding Affinity of Veranamine against a Panel of Receptors and Transporters
| receptor type | receptor | binding affinity (μM) |
|---|---|---|
| adrenergic receptors | α1-A | >10 |
| α1-B | >10 | |
| Alpha2A | >10 | |
| Alpha2B | >10 | |
| Alpha2C | >10 | |
| Beta1 | >10 | |
| dopamine receptors | D1 | >10 |
| D2 | >10 | |
| D3 | >10 | |
| D4 | >10 | |
| D5 | >10 | |
| histamine receptors | H1 | >10 |
| H2 | >10 | |
| H3 | >10 | |
| H4 | >10 | |
| GABA | GABA-A | |
| muscarinic acetylcholine receptors | M1 | >10 |
| M2 | >10 | |
| M3 | >10 | |
| M4 | >10 | |
| M5 | >10 | |
| neurotransmitter transporters | SERT | >10 |
| NET | >10 | |
| DAT | >10 | |
| opioid receptors | MOR | >10 |
| DOR | >10 | |
| KOR | >10 | |
| serotonin receptors | 5HT1A | >10 |
| 5HT1B | >10 | |
| 5HT1D | >10 | |
| 5HT1E | >10 | |
| 5HT2A | >10 | |
| 5HT2B | 0.39 | |
| 5HT2C | >10 | |
| 5HT3 | >10 | |
| 5HT5A | >10 | |
| 5HT6 | >10 | |
| 5HT7 | >10 | |
| 5HT7a | >10 | |
| sigma receptors | σ 1 | 0.56 |
| σ 2 | >10 |
The sigma-1 receptor is a well-studied prospective pharmaceutical target that has been associated with depression,29 as well as addiction,30–32 neuropathic pain,33–35 and neurodegenerative disorders.36–38 The 5HT2B receptor is a potentially problematic target in terms of drug development. Antagonists of 5HT2B are pharmaceutically relevant in the treatment of migraine pain,39 but activators of this receptor are linked to cardiopathy.40–42 Martoteaux and co-workers recently showed that 5HT2B knockout mice demonstrated increased stress-induced depression symptoms43 and were resistant to treatment by selective serotonin reuptake inhibitors.44
Total Synthesis.
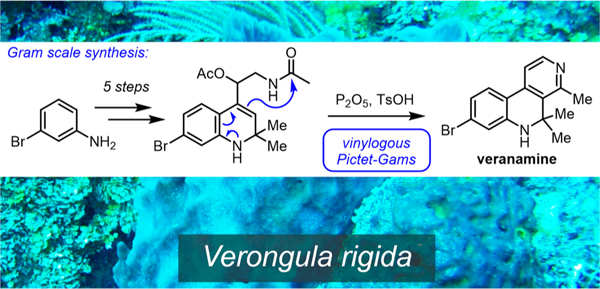
Given its in vivo antidepressant activity, unique receptor binding profile, and distinctive molecular architecture, we were interested in further preclinical antidepressant investigations of veranamine and a medicinal chemistry program based on this new scaffold. To enable this research, a scalable synthesis of this natural product was required. Soon after our disclosure of veranamine in a review article16 and patent,45 a synthesis of its debromo analogue (8) was reported by Liang and co-workers in 2015.46 Their efficient three-step sequence featured an elegant inverse electron-demand hetero-Diels–Alder reaction between the enamine of ketone 6 and 3-methyl-1,2,4-triazine (7) to form the pyridine ring of the target compound (8) in 53% yield, along with 14% of the undesired regioisomeric cycloadduct 9 (Scheme 1). Veranamine itself could not be readily accessed using this strategy because compound 8 is not electronically predisposed for electrophilic bromination at the desired site (Scheme 1). The alternative strategy of preinstallation of the requisite bromine atom would incur problematic regioselectivity in the initial Sonogashira cross-coupling reaction.
Scheme 1.
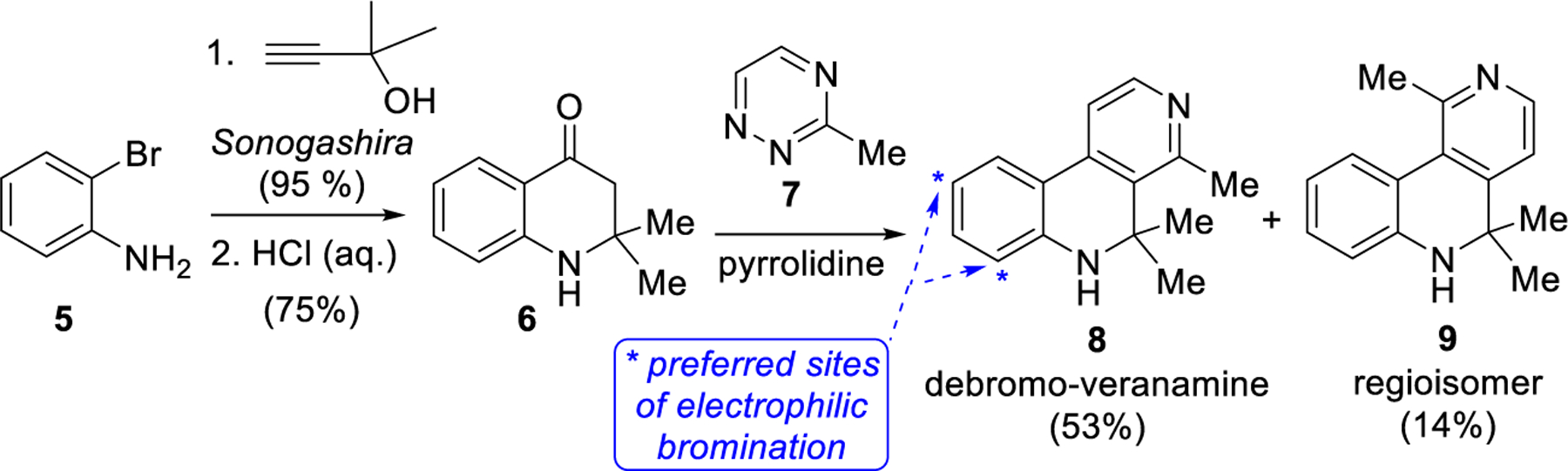
Liang’s Synthesis of Debromoveranamine
We now report an alternative approach to this scaffold and the total synthesis of veranamine in six steps and 25% overall yield, from m-bromoaniline (Scheme 2). We have employed this procedure to prepare more than 10 g of veranamine to date.
Scheme 2.

Gram-Scale Synthesis of Veranamine
We began with the preparation of dihydroquinoline 11 using a modified Skraup cyclocondensation reaction of bromoaniline 10 with acetone in 78% yield by adaptation of a previously reported procedure.47 Benzylic oxidation of 11 to aldehyde 12 was achieved in 72% yield using conditions similar to those recently reported by Batchu and Batra.48 Aldehyde 12 was treated with trimethylsilylcyanide in the presence of ZnI2, and the resulting cyanohydrin49 was reduced with lithium aluminum hydride to the corresponding vicinal amino alcohol, 14, which was diacetylated to offer acetamide 15 in 85% yield over three steps, without purification of the intermediates. At this point, considerable optimization was required to achieve our desired cyclization and dehydration of 15 directly to veranamine (1) via a vinylogous version of the Pictet–Gams reaction.50 The Pictet–Gams reaction is a derivative of the better-known Bischler–Napieralski cyclization,51 distinguished by the use of β-hydroxy-β-arylethylamine derivatives that can aromatize to isoquinolines, after cyclization, by means of dehydration rather than dehydrogenation.52 Ultimately, our preferred reaction conditions for this transformation consisted of treatment of 14 with a combination of P2O5 (7 equiv) and TsOH (2 equiv) in dimethylacetamide under microwave irradiation at 200 °C for 15 min. In this manner, veranamine was obtained in 52% yield. This constitutes the first example of a vinylogous Pictet–Gams transformation. The detailed development of this total synthesis and of the optimization of the last reaction will be reported in a forthcoming article.
CONCLUSIONS
In summary, veranamine is a marine natural product with in vivo antidepressant activity, a novel and now synthetically accessible lead-like scaffold, and a unique receptor binding profile. This compound may be a useful lead in the search for neuropsychiatric medications with novel mechanisms of action and fewer side effects. A medicinal chemistry effort is now also underway in our laboratory with the goal of enhancing potency and selectivity between 5HT2B and sigma-1 receptors for this scaffold.
EXPERIMENTAL SECTION
General Experimental Procedures.
Melting points are uncorrected. Infrared spectra were obtained as thin films on ZnSe disks. NMR experiments were performed on 500 and 300 MHz (400 and 100 MHz for natural veranamine) instruments, and samples were recorded in CDCl3 (referenced to 7.26 ppm for 1H and 77.0 ppm for 13C) and methanol-d4 (referenced to 4.85 ppm for 1H and 47.6 ppm for 13C to match referencing of natural veranamine spectra). MALDI-HRMS spectra of compounds were recorded on a Q-TOF mass spectrometer. Flash chromatography was performed using silica gel (particle size 40–63 μm). All substrates, reagents, and solvents were obtained from commercial suppliers and used as purchased without further purification. Chloroform, diethyl ether, CH2Cl2, and N,N-dimethylacetamide (DMA) were dried over activated molecular sieves (4 Å MS). All reactions were open to the atmosphere unless otherwise stated. Reaction progress was monitored by thin-layer chromatography on silica gel plates (60-F254), visualized under UV light, and plates were developed using p-anisaldehyde or potassium permanganate stains.
Collection of Sponge Material, Extraction, and Isolation of Natural Veranamine.
The sponge, Verongula rigida, was collected from shallow coral reef habitat between 3 and 21 m depth at Key Largo, Florida, in July and August 2005. Voucher specimens have been deposited in the Natural History Museum, London (BMNH 2007.4.23.3 [University of Mississippi voucher 05FL-020(2)]; BMNH 2007.4.23.4 [University of Mississippi voucher 05FL-089]). Three kilograms of the frozen sponge V. rigida was extracted four times with 2000 mL of EtOH in a sonicator. The combined extracts were filtered and concentrated in vacuo until dried. The extract (211 g) was then subjected to vacuum-liquid chromatography using a gradient solvent system from hexanes through acetone to MeOH, yielding 20 fractions. The acetone/methanol fraction (1:1) was further purified by flash column chromatography (C18 cartridge) with a water–methanol solvent system, yielding five fractions. Further purification of polar fractions on reversed-phase HPLC columns yielded 8 mg (0.0038% yield) of veranamine (1).
Veranamine (1): small, dark yellow crystals; UV λmax 204, 245, 380 nm; IR νmax 3422, 2331, 1634 cm−1; 1H and 13C NMR (MeOD, 400 and 100 MHz, respectively) Table 1; HR-ESI m/z 303.0494 [M + H]+ (calcd for C15H15BrN2, 303.0506).
X-ray Crystal Structure Analysis and Crystallographic Data of Compound 1.
Veranamine was crystallized by slow evaporation from MeOH at room temperature. Veranamine crystallized in the monoclinic space group P21/n, with 4 molecules in the asymmetric unit. Cell dimensions are a = 10.7086(2) Å, b = 9.7764(2) Å, c = 13.0365(3) Å, α = γ = 90°, β = 107.060 (1)°, and V = 1304.76(5) Å3. Goodness of fit (GOF) = 1.059, data collection, indexing, and initial cell refinements were all carried out using APEX II software. Frame integration and final cell refinements were done using SAINT software. The final cell parameters were determined from least-squares refinement on 2638 reflections, with R = 0.039, wR(F2) = 0.105. Structure solution, refinement, graphics, and generation of publication materials were performed by using SHELXTL, V6.12. Hydrogen atoms were placed in their expected chemical positions using the HFIX command. CCDC 1883525 contains the supplementary crystallographic data for this paper. The data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures. More detailed crystallography data can be found in the Supporting Information.
7-Bromo-2,2,4-trimethyl-1,2-dihydroquinoline (11).
m-Bromoaniline 10 (10.0 g, 58.13 mmol), iodine (2.95 g, 11.63 mmol), and acetone (150 mL) were added into a 500 mL flask charged with a stir bar. The mixture was heated at reflux for 5 days. Acetone was periodically added to keep the solvent at a constant volume throughout the reaction time period. The solvent was removed in vacuo, and the residue was subjected to flash column chromatography (EtOAc/hexanes, gradient elution 5/95 to 30/70) to give dihydroquinoline 11 as a yellow, low-melting solid (11.45 g, 45.41 mmol, 78%): Rf (EtOAc/Hex, 30:70) = 0.24; FTIR (νmax, thin film, cm−1) 3371, 3020, 2970, 2929, 1652, 737; 1H NMR (500 MHz, CDCl3) δ 6.87 (d, J = 8.1 Hz, 1H), 6.72 (dd, J = 8.1, 1.9 Hz, 1H), 6.57 (d, J = 1.9 Hz, 1H), 5.31 (d, J = 1.5 Hz, 1H), 3.72 (s, 1H), 1.95 (d, J = 1.5 Hz, 3H), 1.27 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 144.7, 128.7, 128.0, 125.1, 121.9, 120.5, 119.9, 115.4, 52.3, 31.3, 18.6; HRESIMS 252.0382 [M + H]+ (calcd for C12H14BrN, 252.0388).
7-Bromo-2,2-dimethyl-1,2-dihydroquinoline-4-carbaldehyde (12).
To a solution of 11 (10.0 g, 39.7 mmol) in dioxane (250 mL) were added SeO2 (4.39 g, 39.6 mmol, 2.0 equiv) and H2O (3.5 mL). The mixture was heated to reflux and stirred for 4 h. The reaction was cooled to room temperature (rt), washed with H2O (200 mL), and extracted with EtOAc (2 × 200 mL). The solvent was removed in vacuo, and the resulting residue was subjected to flash column chromatography (EtOAc/hexanes, gradient elution 5/95 to 30/70) to give 12 (7.64 g, 28.7 mmol, 72% yield) as a dark orange solid: Rf (SiO2, EtOAc/hexanes, 30:70) = 0.53; FTIR (νmax, thin film, cm−1) 3308, 3060, 2962, 2918, 2848, 1680, 1630, 737; 1H NMR (500 MHz, CDCl3) δ 9.60 (s, 1H), 8.05 (d, J = 8.4 Hz, 1H), 6.80 (dd, J = 8.4, 2.0 Hz, 1H), 6.66 (d, J = 2.0 Hz, 1H), 6.28 (s, H), 3.74 (s, 3H), 1.40 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 191.9, 151.2, 144.4, 132.6, 127.5, 123.7, 120.9, 116.2, 114.3, 52.8, 29.9; HRESIMS 266.0173 [M + H]+ (calcd for C12H12BrNO, 266.0181).
2-Acetamido-1-(7-bromo-2,2-dimethyl-1,2-dihydroquinolin-4-yl)ethyl acetate (15).
In an oven-dried 500 mL round-bottom flask, aldehyde 12 (1.00 g, 3.76 mmol) was dissolved in 20 mL of CHCl3. TMSCN (1.41 mL, 11.28 mmol, 3 equiv) was added via syringe followed by addition of ZnI2 (15.0 mg, 0.047 mmol, 0.0125 equiv). The reaction was left stirring under argon (balloon) for 24 h at rt. Excess TMSCN and solvent were removed by warming the open reaction flask to 70 °C under a stream of N2 (inside a closed fume hood) for approximately 30 min. [Caution: TMSCN is highly toxic by inhalation and by skin absorption.] The crude product (13, Rf = 0.82, EtOAc/hexanes, 30:70) was then dissolved in 200 mL of anhydrous diethyl ether. The reaction was cooled to 0 °C, and LAH (0.285 g, 7.52 mmol) was added in several portions over 5 min. The reaction was left stirring for 2 h under argon. LiAlH4 was quenched by slow addition of H2O (∼10 mL). Then 2 M NaOH (∼100 mL) was added followed by extraction with EtOAc (3 × 200 mL). The combined organic layers were dried over Na2SO4. The solvent was removed in vacuo. The crude amino alcohol 14 was obtained as a brown solid (1.11 g, Rf = baseline in 100% EtOAc) and was immediately dissolved in 50 mL of CH2Cl2 followed by addition of Ac2O (1.48 mL, 15.62 mmol) and Et3N (1.81 mL, 13.02 mmol) via syringe. The reaction was stirred for 24 h at rt followed by aqueous workup (CH2Cl2, water, NaHCO3, and brine). The organic layer was dried with Na2SO4, and solvent removed in vacuo. The residue was subjected to flash column chromatography (EtOAc/hexanes, gradient elution 20/80 to 90/10) to give 15 (1.23 g, 3.23 mmol, 85% yield over 3 steps) as an off-white solid. Compound 13: Rf (SiO2, EtOAc/Hex, 3:7) = 0.82; 1H NMR (300 MHz, CDCl3) δ 7.10 (d, J = 8.3 Hz, 1H), 6.75 (dd, J = 8.3, 1.9 Hz, 1H), 6.62 (d, J = 1.9 Hz, 1H), 5.76 (d, J = 1.0 Hz, 1H), 5.26 (d, J = 1.0 Hz, 1H), 3.74 (s, 1H), 1.33 (s, 3H), 1.32 (s, 3H), 0.22 (s, 9H). Compound 14: 1H NMR (300 MHz, CDCl3) δ 6.91 (d, J = 8.3 Hz, 1H), 6.68 (dd, J = 8.3, 2.0 Hz, 1H), 6.59 (d, J = 2.0 Hz, 1H), 5.66 (s, 1H), 4.58 (ddd, J = 6.8, 3.7, 1.3 Hz, 1H), 3.76 (s, 1H), 2.97 (dd, J = 12.8, 3.7 Hz, 1H), 2.72 (dd, J = 12.8, 6.8 Hz, 1H), 2.33 (brs, 2H), 1.29 (s, 3H), 1.25 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 145.2, 132.5, 128.0, 124.5, 122.1, 120.1, 117.9, 116.2, 70.2, 52.2, 46.4, 31.3, 30.8. Compound 15: Rf (SiO2, EtOAc) = 0.49; FTIR (νmax, thin film, cm−1) 3295, 3080, 2964, 1730, 1651, 744; 1H NMR (500 MHz, CDCl3) δ 7.07 (d, J = 8.3 Hz, 1H), 6.70 (dd, J = 8.3, 2.0 Hz, 1H), 6.58 (d, J = 2.0 Hz, 1H), 5.94 (br, 1H), 5.78 (ddd, J = 8.3, 3.8, 1.1 Hz, 1H), 5.54 (d, J = 1.1 Hz, 1H), 3.88 (s, 1H), 3.68 (ddd, J = 14.4, 6.2, 3.8 Hz, 1H), 3.37 (ddd, J = 14.4, 8.3, 6.0, 1H), 2.09 (s, 3H), 1.94 (s, 3H), 1.26 (s, 3H), 1.23 (s, 3H); 13C NMR (125 MHz, CDCl3) δ δ 170.4, 170.3, 144.9, 129.2, 129.2, 124.8, 122.5, 120.2, 116.2, 77.4, 77.2, 76.9, 71.4, 51.9, 43.0, 30.8, 30.6, 23.2, 21.2; HRESIMS 381.0810 [M + H]+ (calcd for C17H21BrN2O3, 381.0814).
Veranamine (1).
To an oven-dried 25 mL microwave reaction tube equipped with a stir bar were added P2O5 (1.044 g, 7.35 mmol), p-toluenesulfonic acid monohydrate(400 mg, 2.10 mmol), compound 14 (400 mg, 1.05 mmol), and DMA (12 mL). After purging the reaction tube of oxygen with argon, the tube was sealed and the mixture was heated to 200 °C under microwave irradiation. After 15 min, the mixture was washed with H2O, using NaOH to adjust the pH of the mixture to 10; then the mixture was extracted by EtOAc (3 × 150 mL). The combined organic fractions were washed with brine and dried over Mg2SO4. The solvent was removed in vacuo. The resulting residue was subjected to column chromatography (EtOAc/hexanes, gradient elution 30/70 to 80/20) to give veranamine (1) as a yellow solid (0.1661 g, 0.55 mmol, 52%): Rf (SiO2, EtOAc) = 0.49; FTIR (νmax, thin film, cm−1) 3237, 3022, 2969, 1602, 790; 1H NMR (500 MHz, CDCl3) δ 8.37 (d, J = 5.3 Hz, 1H), 7.47 (d, J = 8.3 Hz, 1H), 7.38 (d, J = 5.3 Hz, 1H), 6.92 (dd, J = 8.3, 2.0 Hz, 1H,), 6.79 (d, J = 2.0 Hz, 1H), 3.75 (s, 1H), 2.70 (s, 3H), 1.63 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 154.3, 147.3, 145.5, 138.4, 133.9, 125.9, 124.7, 121.8, 118.1, 117.6, 115.1, 54.3, 29.8, 26.9; 1H NMR (500 MHz, methanol-d4, referenced to 4.85 ppm) δ 8.23 (d, J = 5.3 Hz, 1H), 7.53 (d, J = 6.4 Hz, 1H), 7.51 (d, J = 8.4 Hz, 1H), 6.91 (d, J = 2.0 Hz, 1H), 6.83 (dd, J = 8.4, 2.0 Hz, 1H), 2.67 (s, 3H), 1.61 (s, 6H); 13C NMR (125 MHz, methanol-d4, referenced to 47.6 ppm) δ 153.7, 146.5, 145.9, 139.4, 134.5, 125.6, 124.6, 120.5, 117.2, 116.9, 115.0, 53.4, 28.0, 24.7; HRESIMS 303.04897 [M + H]+ (calcd for C15H15BrN2, 303.0506).
In Vivo Assay Procedures.
Forced Swim Test and Locomotor Activity Test. To evaluate the isolated compound for antidepressant-like activity, male Swiss Webster mice (Harlan) (25–30 g weight) were used. Animals were housed in groups of five with a 12 h light/12 h dark cycle. Food and water were provided ad libitum. All procedures involving animals were performed as approved by the Institutional Animal Care and Use Committee of the University of Mississippi. Animals were randomly divided into groups (n = 6–10/group). Each group was injected i.p. with either the compound (1–20 mg/kg), desipramine (20 mg/kg), or vehicle (saline, 10% ethanol, or 10% ethanol/1% DMSO). Following injection, locomotor activity was monitored using an automated activity monitoring system (San Diego Instruments). Each mouse was placed in a Plexiglas enclosure, and locomotor activity was recorded as the number of photobeam interruptions for 30 min after drug injection. The activity for the last 10 min was quantified and analyzed. Immediately at the end of the locomotor session, individual mice were subjected to the forced swim test. The mice were individually placed in a clear plastic cylinder (23 cm high, 10 cm internal diameter) filled with deionized water (8 cm high) at 25 °C. The mice were recorded with a video camera (positioned at about 30 cm above the cylinder) for a total of 6 min. The total period of immobility during the last 4 min was timed by three independent observers. The mean immobility time was then calculated. A mouse was judged to be immobile when it remained afloat, making only minimal movements to keep its head above water.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by an NIH NCCAM Grant 1R01AT007318. The authors would like to acknowledge NOAA for supporting the marine collections in Key Largo, Florida. Research was conducted pursuant to the Florida Fish and Wildlife Conservation Commission Division of Marine Fisheries Management Special Activity License (Scientific Research Activities). Receptor binding assays were generously provided by the National Institute of Mental Health’s Psychoactive Drug Screening Program (NIMH PDSP), directed by B. L. Roth MD, PhD, at the University of North Carolina at Chapel Hill. H.C.A. and S.D.H. received support from an IDeA award from the NIGMS (Grant No. P20GM103408). X.Z. was supported by the Chinese Scholarship Council.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jnatprod.9b01107.
Crystallography procedures, NMR comparison of natural and synthetic veranamine, and NMR spectra of veranamine and all synthetic compounds (PDF)
X-ray data (CIF)
The authors declare no competing financial interest.
CCDC 1883525 contains the supplementary crystallographic data for this paper. The data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures.
Contributor Information
Anna J. Kochanowska-Karamyan, Departments of Pharmacognosy and Pharmacology, National Center for Natural Products Research, University of Mississippi, University Park, Mississippi 38677, United States; Department of Pharmaceutical Sciences, School of Pharmacy, Texas Tech University HSC, Amarillo, Texas 79106, United States
Hugo C. Araujo, Department of Chemistry, University of Idaho, Moscow, Idaho 83844, United States
Xiong Zhang, Department of Biochemistry and Biomedical Sciences, McMaster University, Hamilton, ON L8N 3Z5, Canada; Department of Chemistry, University of Idaho, Moscow, Idaho 83844, United States.
Abir El-Alfy, Departments of Pharmacognosy and Pharmacology, National Center for Natural Products Research, University of Mississippi, University Park, Mississippi 38677, United States; Department of Biopharmaceutical Sciences, Medical College of Wisconsin, Milwaukee, Wisconsin 53226, United States.
Paulo Carvalho, Departments of Pharmacognosy and Pharmacology, National Center for Natural Products Research, University of Mississippi, University Park, Mississippi 38677, United States; Feik School of Pharmacy, University of the Incarnate Word, San Antonio, Texas 78209, United States.
Mitchell A. Avery, Departments of Pharmacognosy and Pharmacology, National Center for Natural Products Research, University of Mississippi, University Park, Mississippi 38677, United States
Stephen D. Holmbo, Department of Chemistry, University of Idaho, Moscow, Idaho 83844, United States
Jakob Magolan, Department of Biochemistry and Biomedical Sciences, McMaster University, Hamilton, ON L8N 3Z5, Canada; Department of Chemistry, University of Idaho, Moscow, Idaho 83844, United States.
Mark T. Hamann, Departments of Pharmacognosy and Pharmacology, National Center for Natural Products Research, University of Mississippi, University Park, Mississippi 38677, United States; Department of Drug Discovery and Biomedical Sciences, Medical University of South Carolina, Charleston, South Carolina 29425, United States
REFERENCES
- 1.Ferrari AJ; Charlson FJ; Norman RE; Patten SB; Freedman G; Murray CJ; Vos T; Whiteford HA PLOS Medicine 2013, 10, No. e1001547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kessler RC; Berglund P; Demler O; Jin R; Merikangas KR; Walters EE Arch. Gen. Psychiatry 2005, 62, 593–602. [DOI] [PubMed] [Google Scholar]
- 3.Krishnan V; Nestler EJ Nature 2008, 455, 894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reeves RR; Ladner ME CNS Neurosci. Ther 2010, 16, 227–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rush AJ; Trivedi MH; Wisniewski SR; Nierenberg AA; Stewart JW; Warden D; Niederehe G; Thase ME; Lavori PW; Lebowitz BD Am. J. Psychiatry 2006, 163, 1905–1917. [DOI] [PubMed] [Google Scholar]
- 6.Thase ME; Haight BR; Richard N; Rockett CB; Mitton M; Modell JG; Van Meter S; Harriett AE; Wang YJ Clin. Psychiatry 2005, 66, 974. [DOI] [PubMed] [Google Scholar]
- 7.Han M-H; Nestler EJ Neurotherapeutics 2017, 14, 677–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levinstein MR; Samuels BA Front. Behav. Neurosci 2014, 8, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zarate C; Duman RS; Liu G; Sartori S; Quiroz J; Murck H Ann. N. Y. Acad. Sci 2013, 1292, 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Soczynska JK; Mansur RB; Brietzke E; Swardfager W; Kennedy SH; Woldeyohannes HO; Powell AM; Manierka MS; McIntyre RS Behav. Brain Res 2012, 235, 302–317. [DOI] [PubMed] [Google Scholar]
- 11.Duman RS; Aghajanian GK Science 2012, 338, 68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Berton O; Nestler EJ Nat. Rev. Neurosci 2006, 7, 137. [DOI] [PubMed] [Google Scholar]
- 13.Thompson SM; Kallarackal AJ; Kvarta MD; Van Dyke AM; LeGates TA; Cai X Trends Neurosci 2015, 38, 279–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Molinski TF; Dalisay DS; Lievens SL; Saludes JP Nat. Rev. Drug Discovery 2009, 8, 69. [DOI] [PubMed] [Google Scholar]
- 15.Newman DJ; Cragg GM J. Nat. Prod 2016, 79, 629–661. [DOI] [PubMed] [Google Scholar]
- 16.Kochanowska-Karamyan AJ; Hamann MT Chem. Rev 2010, 110, 4489–4497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kochanowska AJ; Rao KV; Childress S; El-Alfy A; Matsumoto RR; Kelly M; Stewart GS; Sufka KJ; Hamann MT J. Nat. Prod 2008, 71, 186–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ibrahim MA; El-Alfy AT; Ezel K; Radwan MO; Shilabin AG; Kochanowska-Karamyan AJ; Abd-Alla HI; Otsuka M; Hamann MT Mar. Drugs 2017, 15, 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cao R; Peng W; Wang Z; Xu A Curr. Med. Chem 2007, 14, 479–500. [DOI] [PubMed] [Google Scholar]
- 20.Lovering F; Bikker J; Humblet CJ Med. Chem 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]
- 21.Molinski TF Chem. Rev 1993, 93, 1825–1838. [Google Scholar]
- 22.Brahic C; Darro F; Belloir M; Bastide J; Kiss R; Delfourne E Bioorg. Med. Chem 2002, 10, 2845–2853. [DOI] [PubMed] [Google Scholar]
- 23.Marshall KM; Barrows LR Nat. Prod. Rep 2004, 21, 731–751. [DOI] [PubMed] [Google Scholar]
- 24.Kim K-H; Wissner A; Floyd MB; Fraser HL; Wang YD; Dushin RG; Hu Y; Olland A; Guo B; Arndt K Bioorg. Med. Chem. Lett 2009, 19, 5225–5228. [DOI] [PubMed] [Google Scholar]
- 25.Marsicano G; Moosmann B; Hermann H; Lutz B; Behl CJ Neurochem 2002, 80, 448–456. [DOI] [PubMed] [Google Scholar]
- 26.Castagné V; Moser P; Roux S; Porsolt RD Curr. Protoc. Pharmacol 2010, 49, 58.1–5.8.14. [DOI] [PubMed] [Google Scholar]
- 27.Curzon P; Zhang M; Radek RJ; Fox GB Frontiers in Neuroscience: The Behavioral Assessment of Sensorimotor Processes in the Mouse: Acoustic Startle, Sensory Gating, Locomotor Activity, Rotarod, and Beam Walking. In Methods of Behavior Analysis in Neuroscience; Buccafusco JJ, Ed.; CRC Press/Taylor & Francis Group, LLC.: Boca Raton, FL, 2009. [PubMed] [Google Scholar]
- 28.Besnard J; Ruda GF; Setola V; Abecassis K; Rodriguiz RM; Huang X-P; Norval S; Sassano MF; Shin AI; Webster LA; Simeons FRC; Stojanovski L; Prat A; Seidah NG; Constam DB; Bickerton GR; Read KD; Wetsel WC; Gilbert IH; Roth BL; Hopkins AL Nature (London, U. K.) 2012, 492, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stahl SM CNS Spectr 2005, 10, 319–323. [DOI] [PubMed] [Google Scholar]
- 30.Sabino V; Cottone P; Zhao Y; Iyer MR; Steardo L Jr; Steardo L; Rice KC; Conti B; Koob GF; Zorrilla EP Neuropsychopharmacology 2009, 34, 1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nguyen EC; McCracken KA; Liu Y; Pouw B; Matsumoto RR Neuropharmacology 2005, 49, 638–645. [DOI] [PubMed] [Google Scholar]
- 32.Kaushal N; Seminerio MJ; Shaikh J; Medina MA; Mesangeau C; Wilson LL; McCurdy CR; Matsumoto RR Neuropharmacology 2011, 61, 992–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vidal-Torres A; De La Puente B; Rocasalbas M; Touriño C; Bura SA; Fernańdez-Pastor B; Romero L; Codony X; Zamanillo D; Buschmann H Eur. J. Pharmacol 2013, 711, 63–72. [DOI] [PubMed] [Google Scholar]
- 34.Díaz JL; Christmann U; Fernańdez A; Torrens A; Port A; Pascual R; Álvarez I. s.; Burgueño J; Monroy X; Montero A J. Med. Chem 2015, 58, 2441–2451. [DOI] [PubMed] [Google Scholar]
- 35.Lan Y; Chen Y; Cao X; Zhang J; Wang J; Xu X; Qiu Y; Zhang T; Liu X; Liu B-FJ Med. Chem 2014, 57, 10404–10423. [DOI] [PubMed] [Google Scholar]
- 36.Niitsu T; Iyo M; Hashimoto K Curr. Pharm. Des 2012, 18, 875–883. [DOI] [PubMed] [Google Scholar]
- 37.Marrazzo A; Caraci F; Salinaro ET; Su T-P; Copani A; Ronsisvalle G NeuroReport 2005, 16, 1223–1226. [DOI] [PubMed] [Google Scholar]
- 38.Hashimoto KJ Pharmacol. Sci. (Tokyo, Jpn.) 2015, 127, 6–9. [DOI] [PubMed] [Google Scholar]
- 39.Segelcke D; Messlinger K Cephalalgia 2017, 37, 365–371. [DOI] [PubMed] [Google Scholar]
- 40.Roth BL N. Engl. J. Med 2007, 356, 6–9. [DOI] [PubMed] [Google Scholar]
- 41.Hutcheson JD; Setola V; Roth BL; Merryman WD Pharmacol. Ther 2011, 132, 146–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fitzgerald LW; Burn TC; Brown BS; Patterson JP; Corjay MH; Valentine PA; Sun J-H; Link JR; Abbaszade I; Hollis JM Mol. Pharmacol 2000, 57, 75–81. [PubMed] [Google Scholar]
- 43.. D’Andrea I; Maroteaux L; Roumier A Eur. Neuropsychopharmacol 2017, 27, S676. [Google Scholar]
- 44.Diaz SL; Narboux-Nême N; Boutourlinsky K; Doly S; Maroteaux L Eur. Neuropsychopharmacol 2016, 26, 265–279. [DOI] [PubMed] [Google Scholar]
- 45.Hamann MT; Kochanowska AJ; El-Alfy A; Matsumoto RR; Boujos A Method using marine sponge-derived compounds having antidepressant, anxiolytic and other neurological activity, and compositions of matter. US 20090093513 A1, 2009. [Google Scholar]
- 46.Liang D; Wang Y; Wang Y; Di DJ Chem. Res 2015, 39, 105–107. [Google Scholar]
- 47.Tian K; Hu D; Hu R; Wang S; Li S; Li Y; Yang G Chem. Commun 2011, 47, 10052–10054. [DOI] [PubMed] [Google Scholar]
- 48.Batchu H; Batra S Eur. J. Org. Chem 2012, 2012, 2935–2944. [Google Scholar]
- 49.Evans DA; Carroll GL; Truesdale LK J. Org. Chem 1974, 39, 914–917. [Google Scholar]
- 50.Pictet A; Gams A Ber. Dtsch. Chem. Ges 1909, 42, 2943–2952. [Google Scholar]
- 51.Bischler A; Napieralski B Ber. Dtsch. Chem. Ges 1893, 26, 1903–1908. [Google Scholar]
- 52.Li JJ Pictet-Gams isoquinoline synthesis. In Name Reactions; Springer, 2009; pp 432–433. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.