

Abstract:
We report here the orchestration of molecular ion networking and a set of computationally assisted structural elucidation approaches in the discovery of a new class of pyrroloiminoquinone alkaloids that possess selective bioactivity against pancreatic cancer cell lines. Aleutianamine represents the first in a new class of pyrroloiminoquinone alkaloids possessing a highly strained multibridged ring system, discovered from Latrunculia (Latrunculia) austini Samaai, Kelly & Gibbons, 2006 (class Demospongiae, order Poecilosclerida, family Latrunculiidae) recovered during a NOAA deep-water exploration of the Aleutian Islands. The molecule was identified with the guidance of mass spectrometry, nuclear magnetic resonance, and molecular ion networking (MoIN) analysis. The structure of aleutianamine was determined using extensive spectroscopic analysis in conjunction with computationally assisted quantifiable structure elucidation tools. Aleutianamine exhibited potent and selective cytotoxicity toward solid tumor cell lines including pancreatic cancer (PANC-1) with an IC50 of 25 nM and colon cancer (HCT-116) with an IC50 of 1 μM, and represents a potent and selective candidate for advanced preclinical studies.
Graphical Abstract
■. INTRODUCTION
Pancreatic cancer is one of the most deadly cancers with a 5-year survival rate of less than 8%.1 The only curative therapy is surgery for which only 15–20% of patients are eligible and of those about 20% are long-term survivors.2 For metastatic disease there are essentially only two chemotherapy options: the multidrug regime FOLFIRINOX and gemcitabine.2 Although standard chemotherapeutic treatments for pancreatic cancer add months to a patient’s overall survival time, there is little improvement in survival rates.3 This disparity, compared to other cancer types, has led to the projection that pancreatic cancer will become the second most deadly cancer in the United States by 2030, surpassed only by lung cancer.4 Any improvements in treatment strategies will come as a much-needed reprieve to patients diagnosed with this uniquely challenging disease.
Natural products have provided a diversity of valuable drugs in addition to the identification of key targets for the control of cancer.5–8 It has been estimated that up to 50% of all present day therapeutics are derived from natural products, and in the case of cancer, the number is near 75%.9,10 The plant-derived chemotherapy paclitaxel is one of the most highly documented treatments from a natural source, and marine invertebrates have provided lead molecules such as discodermolide,11 halichondrin B, ecteinascidin 743, didemnin B, and the dolastatins, all in various stages of development.12–15 Among the unique marine-derived ring systems, the pyrroloiminoquinone alkaloids have been studied for decades.16–18 The promising biological activities for members of this class and their unique structures have made these molecules prime targets for syntheses19–21 and biosynthesis studies.22–25
Our discovery of new discorhabdins26 and related metabolites27 from the North Pacific sponge Latrunculia (Latrunculia) austini Samaai, Kelly & Gibbons, 2006 (class Demospongiae, order Poecilosclerida, family Latrunculiidae), recently reviewed and redescribed in a review of the family Latrunculiidae,28 prompted a recollection during the NOAA deep ocean survey and detailed assessment of the National Cancer Institute (NCI) repository of this genus group to facilitate the discovery of new brominated alkaloids. Identification of new molecules early in the workflow using mass spectrometry (MS), nuclear magnetic resonance (NMR), and computational tools minimizes time, effort, and the cost of large screening campaigns.29 A variety of dereplication strategies have been developed in recent years including high-performance liquid chromatography (HPLC)-MS, -NMR, and -NMR-MS or bioactivity fingerprints, such as cytological profiling or BioMAP;30,31 however, few are as efficient and useful as the recently developed network analysis tools for the visualization of observed molecules as familial groupings generated using MS fragmentation data assessed via vector correlations and displayed as an MS/MS network.32 The visualization of networks using Cytoscape enables the direct observation of similarities as well as differences between two or more samples in which similar entities within the network are clustered together while disparate or unique entities are grouped separately.33 With use of this approach, the discovery of aleutianamine and assessment of the NCI repository was strongly facilitated as aleutianamine is a minor constituent co-occurring with other highly biologically active molecules.
Computational approaches, in conjunction with spectroscopic methods, provide a powerful and emerging method for the assignment of atom connectivity, relative configuration, and absolute configuration of complex molecules.34,35 Highly noteworthy is the protocol developed recently by Martin and Williamson et al.36 incorporating anisotropic NMR parameters with computational approaches to determine the structures of complex natural products. The successful characterization of karlotoxin 2 (KmTx2) followed by KmTx8 and KmTx9 was supported by NMR chemical shift calculation tools including gauge-including atomic orbitals (GIAO) and DP4+ probability studies in conjunction with heteronuclear single quantum coherence (HSQC) spectroscopy studies.37–40 Excited-state time-dependent density functional theory (TDDFT) calculations were applied here in conjunction with experimental electronic circular dichroism (ECD) spectroscopy to determine the absolute configurations of the natural products.41–45 We also used comparison of experimental and calculated 1H and 13C NMR chemical shifts of different regioisomers matching the chemical formula of aleutianamine to help verify that the structure was correctly assigned. The protocol presented here involves orchestration of a number of computational methods in combination with spectroscopic analyses, leading to the discovery and establishment of the well-defined structure for aleutianamine.
■. RESULTS AND DISCUSSION
Computationally Assisted Discovery and Structural Elucidation for Aleutianamine.
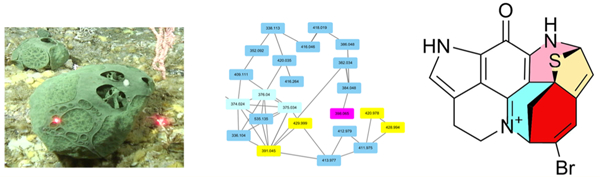
The frozen sponges were extracted with ethanol, and the extract was analyzed by LCMS. The MS/MS molecular ion networking (MoIN) data were collected from the injection of both various global extracts of Latrunculia species (Table S2, Supporting Information) and the isolated standards discorhabdin A, E, F, Y, 3-dihydrodiscorhabdin A, and 3-dihydrodiscorhabdin D standards. The organized landscape of the MoIN (Figure 1) was generated using Cytoscape (for details see Supporting Information) and showed several discorhabdin-related compounds. An unreported brominated alkaloid signal (m/z 398.065) was identified using the networking map.
Figure 1.

Molecular ion network (MoIN) cluster for alkaloids from various global Latrunculia species collected by NOAA Alaska and from the NCI Natural Products Repository.
To further explore the structure and distribution of this unknown molecule, extraction and purification of specimens of L. austini, collected during NOAA Alaska’s trawling surveys and remotely operated collections, led to isolation of the potential target mass m/z 398 that resulted from MoIN analysis (vide supra) as a green-yellow solid with the molecular formula of C18H13BrN3OS generated by high-resolution mass spectrometry analysis. The presence of 18 carbons was validated using 13C NMR data. A pyrroloiminoquinone moiety was initially proposed by comparing the aromatic region of the 13C NMR spectrum with the known natural product discorhabdin A.46,47 The presence of this moiety was confirmed by the COSY correlation of H-16 with H-17, ROESY correlations of H-14 with H-13 and H-16, and HMBC correlations indicated in Figure 2A. Interestingly, H-17b displayed an additional HMBC correlation with a secondary carbon (δC 62.8) of which the attached proton (δ 5.20, t, J = 2.8) also displayed an HMBC signal to C-17 (Figure 2B). This evidence suggested that the latter, C-3 (δ 62.8), was connected to the pyrroloiminoquinone moiety likely via the imine nitrogen (N-18). The presence of this connection was secured by the 1H-15N HMBC correlation of H-4b (vide infra) to N-18 (δ 143.2). N-18 was thereby positively charged.
Figure 2.

Elucidation of the key functionalities and 2D structure of aleutianamine.
Further analysis indicated that the nitrogenated secondary C-3 was also connected to a methylene group C-4 (δC 31.4, δH 2.50, δHa dd, J = 2.7, 12.7, δHb 2.60, dd, J = 2.7, 12.7) as suggested by the COSY correlations among H-3, H-4a, and H-4b, and connected to a disubstituted olefinic moiety C-2 (δC 117.2) and C-1 (δC 128.4, δH 7.20, s), as suggested by the HMBC correlations of H-3 to C-2 and C-1 (Figure 2B). C-3 is vicinal to C-2 instead of C-1 since neither COSY nor ROESY correlation was observed between H-3 and H-1, but H-1 displayed HMBC correlation with C-3. This assignment was also corroborated by DP4+ calculations (vide infra). To this end, a key iminium N-18-C-3 bond that connects the pyrroloiminoquinone scaffold and a major not yet fully uncovered functionality (the eastern hemisphere) was present.
The structural elucidation continued with C-4 which was connected to C-5 (δC 48.7), a key heteroatom-containing tertiary carbon serving as a linchpin (Figure 2C) to unite the Western and Eastern hemispheres. This connection was suggested from three-bond HMBC correlations of H-3 with C-5 and H-4b with C-21, and two-bond HMBC correlations of H-4a,b with C-5. Moving further to assign the connections of the other two bonds of C-5 led us to revisit the spectroscopic information on the C-1-C-2 bond (Figure 2D). H-1 displayed HMBC correlations not only to C-5 but also to two additional carbons (δC 141.4 and 111.9), the last two unassigned carbon resonances in the downfield region of the 13C NMR spectrum. In addition, H-4a,b also showed HMBC correlations with the carbon at δC 141.4. This correlation pattern demonstrated that these two carbons comprised the Δ6(7) double bond conjugated with the C-1-C-2 bond, and C-6 connecting both C-1 and C-5. This connectivity pattern was confirmed by a strong ROESY signal of H-7 with H-1. At this stage, the new azabicyclo[3.3.1]nonane ring system fused at the pyrroloiminoquinone moiety was assigned (Figure 2D).
Advancing to the last unassigned carbon resonance (δC 64.6), which was determined by HSQC to be a methine (δH 6.02, brd), this carbon (C-8) was not only connected to C-7, as demonstrated by the COSY correlation of H-7 and H-8 (δ 6.02, brd) (Figure 2E), but also to N-9, as suggested by a three-bond HMBC correlation of H-8 with C-10. This connection was secured by ROESY correlations of H-8 with both H-9 and H-7, a two-bond HMBC correlation of H-8 with C-7, and a three-bond HMBC correlation of H-8 with C-6.
Assignment of the thio and bromo substituents was straightforward. H-8 displayed an HMBC correlation with C-5 (Figure 2F), as indicated by a three-bond correlation via a sulfur bridge between C-8 and C-5. The bromo group is therefore attached to C-2 to complete the last assignment of bond connection.
With the 2D structure of aleutianamine determined, the relative configuration was elucidated next. Because of the tetrahedral geometry of C-5, C-3, and C-8, and the resulting ring constraint, only one relative configuration is possibly stable; that is, the sulfur bridge between C-5 and C-8 and the C-4 methylene bridge are syn-periplanar relative to each other (Figure 3). This configuration (Figure 4) was modeled and subjected to computational analysis. The structure was optimized using hybrid density functional theory (DFT) calculations at the B3LYP/6–311++G(3d,3p) level with PCM (polarizable continuum solvation model) using a dielectric constant representing dimethylsulfoxide. A notable feature of this structure is the near orthogonal arrangement of the bromotetrahydrobenzo[b] thiophene and dihydropyrrolo-[4,3,2-de] quinoline-8(1H)-one moieties, which suggested a highly strained ring system. Notably, all the calculated distances between nonexchangeable protons were under the classified ROESY distance constraint ranges which were deduced from the signal integration values (Table S3, Supporting Information). Highly noteworthy, the experimental values of the signal intensities were correlated with the computed distances (Chart S1, Supporting Information) in an exponential function d = −0.355 ln(i) + 1.65 and with high agreement with the computational data. This new protocol that correlates ROESY signal intensities with calculated proton distances thereby provided an efficient quantifiable verification of the structural assignment.
Figure 3.

Relative configuration of aleutianamine.
Figure 4.

ROESY correlations and the DFT-calculated distances (Å) between the protons (ROESY correlations are indicated as red arrows; carbon atoms are colored in gray; nitrogen atoms in blue; oxygen atoms in red; bromine atoms in dark red; and hydrogen atoms in cyan).
With the relative configuration established, the absolute configuration was the next focus. ECD calculations were performed for the determination of the absolute configuration via excited-state calculations using the TDDFT method. The calculated excitation energies and rotatory strengths were fitted with Gaussian functions to generate the simulated ECD spectra. By comparison of the calculated spectra to the experimental spectra (Figure 5), the absolute configuration of aleutianamine was assigned as (3R, 5R, 8S).
Figure 5.

Overlaid experimental ECD spectra with the TDDFT computed ECD curves at the B3LYP/6–31G(d,p) level.
Computational Validation of the Structure of Aleutianamine.
Assigning the position of the Br-substituted sp2 and the non-Br-substituted sp2 carbons, based solely on NMR spectroscopic analysis, is challenging.48 However, in optimal cases, a Br-substituted carbon can be identified by 1H and 13C NMR chemical shift calculations using the GIAO49–51 method at the PCM/mPW1PW91/6–311+G(d,p) level of theory51 for DP4+ calculations.52 Two possible regioisomers of aleutianamine having the bromo substituent at C-1 [aleutianamine-1′ (3R, 5R, 8S)] or C-2 [aleutianamine-1 (3R, 5R, 8S)], respectively, and a regioisomer with swapped positions of NH and S [aleutianamine-1″ (3R, 5R, 8R)] were employed for DFT and DP4+ calculations to validate the correct structure of aleutianamine and to further validate the utility of the technique in the assignment of regiochemistry (cf. Supporting Information, section 3.2, for details on the methods, Tables S9–S11 for the relative energies of the different conformations, and Figures S24–S26 showing the 3D structure of the different conformations of the regioisomers). The calculated chemical shifts of aleutianamine-1, aleutianamine-1′, and aleutianamine-1″ were compared to the experimental 1H and 13C NMR chemical shifts via the corrected mean absolute error (CMAE), the corrected total absolute deviation (CTAD), and DP4+ probability chemical shift analysis (Tables S12–S17, Figure S27, Supporting Information). The CMAE and CTAD results showed that aleutianamine-1 was the most probable isomer (Figure 6). These results were corroborated by the DP4+ statistical analysis, which predicted aleutianamine-1 to be the correct regioisomer with a probability of 100%, on the basis of utilization of both 1H and 13C NMR data (Figure 6 and Figure S27, Supporting Information). Collectively, the excellent agreement between NMR spectroscopic and computational data strongly supported the (3R, 5R, 8S) absolute configuration of aleutianamine and its 2-bromo substituent.
Figure 6.

Corrected total absolute deviation (CTAD), corrected mean absolute error (CMAE), and DP4+ probability analysis (sarotti-NMR.weebly.com) for 1 (aleutianamine-1) and two of its regioisomers (Gibbs free energies at the PCM/mPW1PW91/6–311+G(d,p) level were used for the analysis).
Biosynthetic Pathway of Aleutianamine.
It is surprising that the complex molecular architecture of aleutianamine appears to have arisen from only two standard amino acids l-tryptophan and l-tyrosine, generating a strained ring system without the rearrangement of carbon. From a biosynthetic perspective, the phenolic moiety of makaluvamine F may serve as a suitable precursor and could undergo oxidation to form quinone methide A (Figure 7).
Figure 7.

Proposed biosynthesis of aleutianamine.
A Stork-type enamine Michael addition/cyclization followed by an imine cyclization would lead to B with the novel azabicyclo[3.3.1]nonane ring system constructed. Reduction of the latter would furnish the structure of aleutianamine. Another possible precursor is discorhabdin A, found in abundance in this species, and the proposed biosynthetic conversion from discorhabdin A to aleutianamine is shown in Figure S28 of the Supporting Information.
Bioassay.
Aleutianamine, with its unique ring system, showed solid tumor selectivity in a differential cytotoxicity zone assay used for discovery of solid tumor selective anticancer leads.25 Selectivity of cell killing was noted for murine colon cancer 38, human breast cancer MCF-7, human prostate cancer LNCaP, and human pancreatic cancer PANC-1 compared to those of both murine and human leukemia cells which are typically more sensitive. This breadth of selectivity is unusual for active compounds which usually demonstrate selectivity to only one of the cell types assayed. The IC50 value for aleutianamine tested against human HCT-116 colon cancer cells was 1 μM and against PANC-1 pancreatic cancer cells was 25 nM, indicating significant potency and selectivity. Clonogenic studies were carried out using HCT-116 cells to define the concentration-survival relationship as a function of exposure time.26 The end point used, tS10 (10% survival of clonogenic cells for exposure time t) provides information essential for future preclinical therapeutic studies. These clonogenic studies yielded a 2S10 value of >5 μM, a 24S10 value of 0.75 μM, and a 168S10 value of 0.1 μM. The clonogenic data indicated that, depending upon the pharmacokinetics and maximum tolerated dose of aleutianamine, a chronic treatment might be expected to be efficacious. The principal structural feature of discorhabdins is the core of a planar iminoquinone moiety which has been shown to intercalate and cleave DNA as well as inhibit the action of topoisomerase II.16 The makaluvamines have been shown to be topoisomerase II inhibitors acting via cleavable complex formation, or via the direct induction of DNA double-strand breaks. Makaluvamines A and C are reported to decrease tumor size in a solid human tumor model.53 However, discorhabdins A and C exhibited no inhibition of topoisomerase II despite their significant cytotoxicity with the same core iminoquinone skeleton, supporting a dual mechanism against cancer.16 On the basis of the data generated thus far, aleutianamine showed significantly higher cytotoxicity than discorhabdin A toward cancer cell lines and demonstrated selective inhibition to pancreatic cancer cell lines, making it a promising candidate for synthesis, biosynthesis, and further development.
■. CONCLUSION
Extensive spectroscopic data analysis in conjunction with advanced computational approaches established a quantifiable structure elucidation protocol that led to assignment of the sulfur-bridged seven-membered ring incorporated with a bromine-substituted eight-membered moiety, yielding a new class of pyrroloiminoquinone alkaloid with potent bioactivity toward solid tumor cell lines. This special type of ring arrangement formed a rigid and highly fused system which is fully constructed by multibridge rings, unlike the typical spiro ring system previously established for the discorhabdins and the other reported pyrroloiminoquinone alkaloids. A similar constrained bridge ring system with an olefinic carbon on the bridge head can also be found in paclitaxel. Such strain may feasibly contribute to the overall electrophilicity and potent bioactivity of this unique molecule, thus necessitating future SAR studies. A biogenesis map was proposed (Figure S28, Supporting Information) to infer a possible biomimetic total synthesis and biosynthetic study of aleutianamine and other pyrroloiminoquinone alkaloids.
Supplementary Material
■. ACKNOWLEDGMENTS
We thank the Mississippi Center for Supercomputing Research and NSF MRI 1338056 for providing access to supercomputer resources, Frank Wiggers from the National Center for Natural Products Research for the NMR spectra data acquisition, NOAA/RACE/AFSC for the assistance in the collection of sponge material during the Aleutian Island deep ocean surveys, the NCCIH (R01AT007318), NSF CHE-1460568, China Scholarship Council, the Cooper Family, and The Abney Foundation for financial support.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b11403.
1H and 13C NMR, DEPT135, COSY, HMQC, HMBC, ROESY, 1H-15N HMBC, MoIN, computational chemistry methods and data, extraction and isolation procedures, and proposed biosynthetic pathway of aleutianamine (PDF)
Notes
The authors declare no competing financial interest.
■ REFERENCES
- (1).Jemal A; Siegel R; Xu J; Ward E Cancer Statistics, 2010. Ca-Cancer J. Clin. 2010, 60, 277. [DOI] [PubMed] [Google Scholar]
- (2).Castellanos JA; Merchant NB Intensity of follow-up after pancreatic cancer resection. Ann. Surg. Oncol. 2014, 21, 747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Garrido-Laguna I; Hidalgo M Pancreatic cancer: from state-of-the-art treatments to promising novel therapies. Nat. Rev. Clin. Oncol. 2015, 12, 319. [DOI] [PubMed] [Google Scholar]
- (4).Rahib L; Smith BD; Aizenberg R; Rosenzweig AB; Fleshman JM; Matrisian LM Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913; Erratum in Cancer Res. 2014, 74, 4006. [DOI] [PubMed] [Google Scholar]
- (5).Cragg GM, Kingston DGI, Newman DJ, Eds. Anticancer Agents from Natural Products, 2nd ed.; CRC Press/Taylor & Francis Group: Boca Raton, FL, 2012. [Google Scholar]
- (6).Rayan A; Raiyn J; Falah M Nature is the best source of anticancer drugs: Indexing natural products for their anticancer bioactivity. PLoS One 2017, 12, e0187925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Beck A; Goetsch L; Dumontet C; Corvaïa N Strategies and challenges for the next generation of antibody–drug conjugates. Nat. Rev. Drug Discovery 2017, 16, 315. [DOI] [PubMed] [Google Scholar]
- (8).Xiao H; Verdier-Pinard P; Fernandez-Fuentes N; Burd B; Angeletti R; Fiser A; Horwitz SB; Orr GA Insights into the mechanism of microtubule stabilization by Taxol. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Newman DJ; Cragg GM Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Newman DJ; Cragg GM Natural products as sources of new drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629. [DOI] [PubMed] [Google Scholar]
- (11).Prota AE; Bargsten K; Redondo-Horcajo M; Smith AB III; Yang CPH; McDaid HM; Paterson I; Horwitz SB; Fernando Díaz J; Steinmetz MO Structural basis of microtubule stabilization by discodermolide. ChemBioChem 2017, 18, 905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12) (a).Chakraborty TK; Das S Chemistry of potent anti-cancer compounds, amphidinolides. Curr. Med. Chem.: Anti-Cancer Agents 2001, 1, 131. [DOI] [PubMed] [Google Scholar]; (b) Rinehart KL Antitumor compounds from tunicates. Med. Res. Rev. 2000, 20, 1. [DOI] [PubMed] [Google Scholar]
- (13).Mayer AM; Gustafson KR Marine pharmacology in 2003–2004: anti-tumour and cytotoxic compounds. Eur. J. Cancer 2006, 42, 2241. [DOI] [PubMed] [Google Scholar]
- (14).Alonso D; Khalil Z; Satkunanthan N; Livett B Drugs from the sea: conotoxins as drug leads for neuropathic pain and other neurological conditions. Mini-Rev. Med. Chem. 2003, 3, 785. [DOI] [PubMed] [Google Scholar]
- (15).Grimes DJ Oceans and human health: risks and remedies from the sea; National Institute of Environmental Health Sciences: 2009. [Google Scholar]
- (16).Hu J-F; Fan H; Xiong J; Wu S-B Discorhabdins and pyrroloiminoquinone-related alkaloids. Chem. Rev. 2011, 111, 5465. [DOI] [PubMed] [Google Scholar]
- (17).Antunes EM; Copp BR; Davies-Coleman MT; Samaai T Pyrroloiminoquinone and related metabolites from marine sponges. Nat. Prod. Rep. 2005, 22, 62. [DOI] [PubMed] [Google Scholar]
- (18).Lin S; McCauley EP; Lorig-Roach N; Tenney K; Naphen CN; Yang A-M; Johnson TA; Hernadez T; Rattan R; Valeriote FA; et al. Another look at pyrroloiminoquinone alkaloids–perspectives on their therapeutic potential from known structures and semisynthetic analogues. Mar. Drugs 2017, 15, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Tohma H; Harayama Y; Hashizume M; Iwata M; Kiyono Y; Egi M; Kita Y The first total synthesis of discorhabdin A. J. Am. Chem. Soc. 2003, 125, 11235. [DOI] [PubMed] [Google Scholar]
- (20).Aubart KM; Heathcock CH A biomimetic approach to the discorhabdin alkaloids: total syntheses of discorhabdins C and E and dethiadiscorhabdin D. J. Org. Chem. 1999, 64, 16. [DOI] [PubMed] [Google Scholar]
- (21).Roberts D; Joule JA; Bros MA; Alvarez M Synthesis of pyrrolo [4, 3, 2-de] quinolines from 6, 7-dimethoxy-4-methylquinoline. Formal total syntheses of damirones A and B, batzelline C, isobatzelline C, discorhabdin C, and makaluvamines A– D. J. Org. Chem. 1997, 62, 568. [DOI] [PubMed] [Google Scholar]
- (22).Lill RE; Major DA; Blunt JW; Munro MH; Battershill CN; McLean MG; Baxter RL Studies on the biosynthesis of discorhabdin B in the New Zealand sponge Latrunculia sp. B. J. Nat. Prod. 1995, 58, 306. [Google Scholar]
- (23).Miyanaga A; Janso JE; McDonald L; He M; Liu H; Barbieri L; Eustáquio AS; Fielding EN; Carter GT; Jensen PR; et al. Discovery and assembly-line biosynthesis of the lymphostin pyrroloquinoline alkaloid family of mTOR inhibitors in Salinispora bacteria. J. Am. Chem. Soc. 2011, 133, 13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Amos GC; Awakawa T; Tuttle RN; Letzel A-C; Kim MC; Kudo Y; Fenical W; Moore BS; Jensen PR Comparative transcriptomics as a guide to natural product discovery and biosynthetic gene cluster functionality. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 11121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Valeriote FA; Tenney K; Media J; Pietraszkiewicz H; Edelstein M; Johnson TA; Amagata T; Crews P Discovery and development of anticancer agents from marine sponges: perspectives based on a chemistry-experimental therapeutics collaborative program. J. Expl. Ther. Oncol. 2002, 2, 119. [PubMed] [Google Scholar]
- (26).Watts KR; Morinaka BI; Amagata T; Robinson SJ; Tenney K; Bray WM; Gassner NC; Lokey RS; Media J; Valeriote FA; Crews P Biostructural features of additional jasplakinolide (jaspamide) analogues. J. Nat. Prod. 2011, 74, 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zou Y; Hamann MT Atkamine: A new pyrroloiminoquinone scaffold from the cold water Aleutian Islands Latrunculia sponge. Org. Lett. 2013, 15, 1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kelly M; Sim-Smith C; Stone R; Samaai T; Reiswig H; Austin W New taxa and arrangements within the family Latrunculiidae (Demospongiae, Poecilosclerida). Zootaxa 2016, 4121, 1. [DOI] [PubMed] [Google Scholar]
- (29).Gerwick WH; Moore BS Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem. Biol. 2012, 19, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Perlman ZE; Slack MD; Feng Y; Mitchison TJ; Wu LF; Altschuler SJ; Altschuler SJ Multidimensional drug profiling by automated microscopy. Science 2004, 306, 1194. [DOI] [PubMed] [Google Scholar]
- (31).Wong WR; Oliver AG; Linington RG Development of antibiotic activity profile screening for the classification and discovery of natural product antibiotics. Chem. Biol. 2012, 19, 1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Wang M; Carver JJ; Phelan VV; Sanchez LM; Garg N; Peng Y; Nguyen DD; Watrous J; Kapono CA; Luzzatto-Knaan T; et al. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Watrous J; Roach P; Alexandrov T; Heath BS; Yang JY; Kersten RD; van der Voort M; Pogliano K; Gross H; Raaijmakers JM; et al. Mass spectral molecular networking of living microbial colonies. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Abe H; Yamasaki T; Fujiwara I; Sasaki S Computer-aided structure elucidation methods. Anal. Chim. Acta 1981, 133, 499. [Google Scholar]
- (35).Jaspars M Computer assisted structure elucidation of natural products using two-dimensional NMR spectroscopy. Nat. Prod. Rep. 1999, 16, 241. [Google Scholar]
- (36).Liu Y; Saurí J; Mevers E; Peczuh MW; Hiemstra H; Clardy J; Martin GE; Williamson RT Unequivocal determination of complex molecular structures using anisotropic NMR measurements. Science 2017, 356, eaam5349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Smith SG; Goodman JM Assigning stereochemistry to single diastereoisomers by GIAO NMR calculation: The DP4 probability. J. Am. Chem. Soc. 2010, 132, 12946. [DOI] [PubMed] [Google Scholar]
- (38).Willoughby PH; Jansma MJ; Hoye TR A guide to small-molecule structure assignment through computation of (1H and 13C) NMR chemical shifts. Nat. Protoc. 2014, 9, 643. [DOI] [PubMed] [Google Scholar]
- (39).Peng J; Place AR; Yoshida W; Anklin C; Hamann MT Structure and absolute configuration of karlotoxin-2, an ichthyotoxin from the marine dinoflagellate Karlodinium veneficum. J. Am. Chem. Soc. 2010, 132, 3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Waters AL; Oh J; Place AR; Hamann MT Stereochemical studies of the karlotoxin class using NMR spectroscopy and DP4 chemical-shift analysis: insights into their mechanism of action. Angew. Chem. 2015, 127, 15931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Nugroho AE; Morita H Circular dichroism calculation for natural products. J. Nat. Med. 2014, 68, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Li X-C; Ferreira D; Ding Y Determination of absolute configuration of natural products: theoretical calculation of electronic circular dichroism as a tool. Curr. Org. Chem. 2010, 14, 1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Breton RC; Reynolds WF Using NMR to identify and characterize natural products. Nat. Prod. Rep. 2013, 30, 501. [DOI] [PubMed] [Google Scholar]
- (44).Wang X; Liu J; Pandey P; Fronczek FR; Doerksen RJ; Chen J; Qi X; Zhang P; Ferreira D; Valeriote FA; et al. Computationally assisted assignment of the Kadsuraols, a class of chemopreventive agents for the control of liver cancer. Org. Lett. 2018, 20, 5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Wang X; Liu J; Pandey P; Chen J; Fronczek FR; Parnham S; Qi X; Doerksen RJ; Ferreira D; Sun H; Hamann MT; Li S Assignment of the absolute configuration of hepatoprotective highly oxygenated triterpenoids using X-ray, ECD, NMR J-based configurational analysis and HSQC overlay experiments. Biochim. Biophys. Acta, Gen. Subj. 2017, 1861, 3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Kobayashi JI; Cheng J-F; Ishibashi M; Nakamura H; Ohizumi Y; Hirata Y; Sasaki T; Lu H; Clardy J Prianosin A, a novel antileukemic alkaloid from the okinawan marine sponge Prianos melanos. Tetrahedron Lett. 1987, 28, 4939. [Google Scholar]
- (47).Perry NB; Blunt JW; Munro MH Cytotoxic pigments from New Zealand sponges of the genus Latrunculia: discorhabdins A, B and C. Tetrahedron 1988, 44, 1727. [Google Scholar]
- (48).Wang X; Duggan BM; Molinski TF Mollenynes B–E from the Marine Sponge Spirastrella mollis. Band-selective heteronuclear single quantum coherence for discrimination of bromo–-chloro regioisomerism in natural products. J. Am. Chem. Soc. 2015, 137, 12343. [DOI] [PubMed] [Google Scholar]
- (49).Cheeseman JR; Trucks GW; Keith TA; Frisch MJ A comparison of models for calculating nuclear magnetic resonance shielding tensors. J. Chem. Phys. 1996, 104, 5497. [Google Scholar]
- (50).McWeeny R Perturbation theory for the Fock-Dirac density matrix. Phys. Rev. 1962, 126, 1028. [Google Scholar]
- (51).Wolinski K; Hinton JF; Pulay P Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251. [Google Scholar]
- (52).Grimblat NS; Zanardi MM; Sarotti AM Beyond DP4: An improved probability for the stereochemical assignment of isomeric compounds using quantum chemical calculations of NMR shifts. J. Org. Chem. 2015, 80, 12526. [DOI] [PubMed] [Google Scholar]
- (53).Radisky DC; Radisky ES; Barrows LR; Copp BR; Kramer RA; Ireland CM Novel cytotoxic topoisomerase II inhibiting pyrroloiminoquinones from Fijian sponges of the genus Zyzzya. J. Am. Chem. Soc. 1993, 115, 1632. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.