

Abstract
G‐protein‐coupled receptors (GPCRs) transmit downstream signals predominantly via G‐protein pathways. However, the conformational basis of selective coupling of primary G‐protein remains elusive. Histamine receptors H2R and H3R couple with Gs‐ or Gi‐proteins respectively. Here, three cryo‐EM structures of H2R‐Gs and H3R‐Gi complexes are presented at a global resolution of 2.6‐2.7 Å. These structures reveal the unique binding pose for endogenous histamine in H3R, wherein the amino group interacts with E2065.46 of H3R instead of the conserved D1143.32 of other aminergic receptors. Furthermore, comparative analysis of the H2R‐Gs and H3R‐Gi complexes reveals that the structural geometry of TM5/TM6 determines the primary G‐protein selectivity in histamine receptors. Machine learning (ML)‐based structuromic profiling and functional analysis of class A GPCR–G‐protein complexes illustrate that TM5 length, TM5 tilt, and TM6 outward movement are key determinants of the Gs and Gi/o selectivity among the whole Class A family. Collectively, the findings uncover the common structural geometry within class A GPCRs that determines the primary Gs‐ and Gi/o‐coupling selectivity.
Keywords: cryo‐EM structure, G protein selectivity, GPCR, H2R, H3R, machine learning, signaling complex
G‐protein‐coupled receptors (GPCRs) primarily signal through G‐protein pathways. Here, three cryo‐EM structures of H2R‐Gs and H3R‐Gi are reported at 2.6–2.7 Å resolution. Comparative analysis shows TM5/TM6 geometry determines primary G‐protein selectivity in histamine receptors. Machine learning profiling reveals TM5 length, TM5 tilt, and TM6 outward movement govern Gs and Gi/o selectivity in Class A GPCRs.
1. Introduction
Comprised of over 800 members, G‐protein‐coupled receptors (GPCRs) constitute the largest family of membrane proteins in the human genome.[ 1 ] GPCR‐mediated signaling pathways are involved in virtually every physiological function and many pathologies, thus representing the targets of approximately one‐third of all medications, including antihistamines, antipsychotics, and opioid painkillers. In response to myriad extracellular stimuli, GPCRs exert adequate intracellular signaling predominantly through the engagement and activation of four major G‐protein families: Gs, Gi/o, Gq/11, and G12/13.[ 1 , 2 ] Various G‐protein subtypes elicit distinct signaling pathways and trigger versatile physiological effects by regulating different key effectors, such as adenylate cyclase, phospholipase C, and mitogen‐activated protein kinases.[ 2b ] While GPCR activation by agonists typically triggers multiple G‐protein signaling pathways, many GPCRs exhibit pronounced bias and can preferentially activate certain downstream responses.[ 3 ] In many circumstances, the beneficial effects of a drug come from one signaling pathway and the adverse effects from other pathways.[ 4 ] Understanding the molecular mechanism underlying the selective coupling of an agonist‐stimulated GPCR to an appropriate G‐protein is therefore crucial for transmembrane signal transduction.
An increasing number of studies suggested that many GPCRs exhibit a general promiscuity in their interactions with various G‐protein subfamilies.[ 2 , 5 ] The inherent dynamic nature of GPCRs enables them to sample multiple transition states between inactive and active states, contributing to the promiscuity of G‐protein selectivity.[ 5 , 6 ] Primary signaling stands out due to its remarkable coupling efficiency and rapid kinetics, emerging as the predominant signaling route for GPCRs among the spectrum of G‐protein signaling interactions. According to the GPCR database (GPCRdb), among 265 non‐olfactory GPCRs annotated on the G‐protein family level, 228 (86%) have the potential to modulate receptor‐specific cell activities by regulating adenylyl cyclase[ 2 , 5 , 7 ] (Figure 1A). Notably, among 183 GPCRs that predominantly couple with Gs‐ or Gi/o‐proteins, 169 GPCRs (92.3% of the total) exhibit a high degree of selectivity for either Gs‐ (48 GPCRs) or Gi/o‐ (121 GPCRs) proteins.[ 2a ] Therefore, Gs‐ and Gi/o‐proteins are the most prevalent G‐proteins among all the subtypes, leading to inverse cellular signaling responses through the stimulation or inhibition of adenylyl cyclase, respectively.
Figure 1.
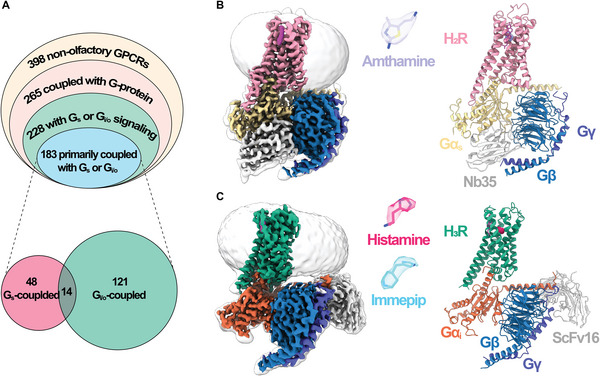
Cryo‐EM structures of H2R‐Gs and H3R‐Gi complexes. A) (Up) Proportion of Gs‐ or Gi/o‐coupled receptors in the non‐olfactory GPCRs. (Down) Number of receptors primarily coupling with Gs‐ or Gi/o‐proteins. B) The cryo‐EM density map (left) and atomic model (right) of amthamine‐bound H2R‐Gs complex. The amthamine is depicted as stick within a transparent EM density map. C) The cryo‐EM density map (left) and atomic model (right) of H3R‐Gi complex. The ligands histamine and immepip are depicted as sticks within a transparent EM density map.
GPCR structures are indispensable tools, providing snapshots at local energy minimum that aid in uncovering the determinants for the primary signaling of Gs‐ and Gi/o‐protein selectivity.[ 8 ] The resolution revolution in cryo‐electron microscopy (cryo‐EM) has led to an explosion of GPCR–G‐protein complex structures, particularly the Gs‐ and Gi/o‐coupled receptors. These structures have unveiled a common mode of GPCR–G‐protein recognition, in which the C‐terminal helix (α5) of Gα inserts into the cytosolic transmembrane (TM) cavity of GPCR.[ 1 , 9 ] The coupling determinants for GPCR in Gα proteins are widely accepted, with the selectivity barcode being the α5 helix, the αN‐β1 hinge, and the β2‐β3 loop of Gα.[ 1 , 9 ] However, the molecular basis of primary G‐protein‐coupling selectivity of GPCRs at the receptor side remains intricate. Most investigations suggest that GPCRs coupled with different G‐proteins generally adopt nearly identical conformations,[ 10 ] with the exception of the 5HT4,[ 11 ] resulting in the widespread doubts of the existence of complementary determinants at the GPCR level.[ 12 ] Additionally, the absence of evolved consensus sequences for G‐protein recognition at GPCRs makes it difficult to draw a unified conclusion regarding primary G‐protein selectivity.[ 1 , 12 , 13 ] Previous structural studies have underscored the importance of ICL2, ICL3, TM5, and TM6 of GPCRs in Gs/Gi/o‐protein selectivity.[ 9 , 14 ] However, the limitation is that these studies often focused on specific receptors or receptor subfamilies, and the observed characteristics are not universal across class A GPCR families, rendering the primary G‐protein selectivity mechanism still promiscuous and challenging.
Histamine, a biogenic amine, plays a pivotal role in various physiological and pathophysiological processes, such as allergic and inflammatory reactions, gastric acid secretion, wake‐sleep disorders, and neurotransmission in the central nervous system.[ 15 ] The histamine receptor system comprises four members (H1R, H2R, H3R, and H4R), all of which belong to the Class A GPCR family.[ 16 ] Drugs targeting H1R, H2R, and H3R have been approved to treat allergic disorders,[ 17 ] gastroduodenal ulcers,[ 18 ] and narcolepsy,[ 19 ] respectively. The four subtypes of histamine receptors deliver downstream signaling by coupling with different heterotrimeric G‐proteins: H1R mainly couples with Gq‐protein; H2R primarily couples with Gs‐protein and could alternatively couple with Gq‐protein; H3R and H4R are both capable of mediating downstream signaling via Gi/o‐protein activation primarily. Given their diverse coupling profiles, histamine receptors serve as an excellent model system for investigating the structural basis of primary G‐protein‐coupling and the preferential profile of GPCRs.
To date, structures of the inactive states of H1R, H2R, and H3R, as well as the active states of H1R and H4R, are available.[ 20 ] However, the absence of active‐state structures of H2R and H3R has impeded the understanding of the molecular basis of H2R and H3R signaling and the structural‐based drug design of histamine receptors. In this study, we report three cryo‐EM structures of H2R‐Gs and H3R‐Gi complexes at a global resolution of 2.6–2.7 Å. These structures reveal the structural basis for agonist recognition and G‐protein selectivity of H2R and H3R. A comparative analysis of the H2R‐Gs and H3R‐Gi complexes reveals that the conformation of TM5 and TM6 determines G‐protein‐selectivity in histamine receptors. Furthermore, machine learning (ML)‐based structural profiling and functional analysis of publicly available and home‐generated class A GPCR–G‐protein complexes indicate that TM5 length, TM5 tilt, and TM6 outward movement determine Gs and Gi/o selectivity. To further enhance our understanding, we develop a ML‐derived architecture classifier for Gs‐ and Gi/o‐coupled receptors using homology models from GPCRdb as a training dataset. Remarkably, the classifier achieves an accuracy up to 91%, as validated by experimentally determined GPCR‐G complex structures. Collectively, our results reveal the common structural geometries of class A GPCRs determine primary Gs‐ and Gi/o‐coupling selectivity.
2. Results
2.1. Cryo‐EM Structures of the Agonist‐Bound H2R‐Gs and H3R‐Gi Complexes
To investigate the structure of the H2R‐Gs complex, we co‐expressed full‐length H2R with the human heterotrimeric Gs protein in Sf9 insect cells. A flag tag was fused to the N‐terminus of H2R to aid purification. The H2R‐Gs complex was assembled in the presence of a highly selective agonist, amthamine, and further stabilized by a camelid antibody, Nb35.[ 21 ] Utilizing single‐particle cryo‐EM analysis, the purified H2R‐Gs complex was solved at a nominal resolution of 2.7 Å (Figure 1B; Figures S1, S2 and Table S1, Supporting Information). The H3R‐Gi complex was obtained through a co‐expression strategy, similar to that of the H2R‐Gs complex. In addition to the N‐terminal flag tag, a C‐terminal tandem maltose‐binding protein (MBP) tag was also introduced to H3R. A NanoBiT tethering strategy was used to improve the stability of the H3R‐Gi complex.[ 22 ] The endogenous agonist histamine and synthetic agonist immepip were used separately to generate the agonist‐bound H3R‐Gi complexes, and the complexes were further stabilized by a single‐chain antibody scFv16.[ 23 ] The histamine‐bound H3R‐Gi complex and immepip‐bound H3R‐Gi complex were solved at a global resolution of 2.7 Å and 3.0 Å, respectively (Figure 1C; Figures S1,S2 and Table S1, Supporting Information).
All three GPCR–G‐protein complex structures are resolved at near‐atomic resolution, and the EM density maps were clear enough to build the model of the receptors, heterotrimeric G‐protein, antibody, and the bound ligand in the receptor orthosteric pockets (Figure S2 and Table S1, Supporting Information). The N‐ and C‐termini of both receptors, as well as the ICL3 of H3R and the α‐helical domains of Gα, exhibit poor resolution in the cryo‐EM density maps, consistent with previous cryo‐EM solved GPCR–G‐protein complexes due to the high flexibility of these regions. Notably, the density of cholesterol between TM1 and TM7 is observed in both H2R‐Gs and H3R‐Gi complexes, highlighting the critical role of cholesterol molecules in maintaining conformational stability and signaling activity of GPCR.
2.2. Ligand Recognition of Histamine Receptors
In the H2R‐Gs complex, the agonist amthamine occupies the conventional orthosteric pocket of H2R. As depicted in Figure S3A (Supporting Information), this pocket is defined by TM3, 5, 6, and 7, and it is capped by the extracellular loop 2 (ECL2) of H2R. The binding of amthamine is stabilized through a combination of polar and hydrophobic interactions (Figure 2A). Specifically, the aminoethyl group of amthamine is positioned between D983.32 and Y2506.51 (we use the Ballesteros–Weinstein numbering system[ 24 ]), engendering a charge‐charge interaction with D983.32— a conserved “salt‐bridge” also observed in other available monoamine receptor structures (Figure 2A).[ 20 , 25 ] Additionally, the two amines present in the thiazolamine moiety of amthamine establish polar interactions with T1033.37, D1865.42, and T1905.46 of H2R (Figure 2A). Consequently, the polar network effectively restrains both the aminoethyl group (head) and thiazolamine group (tail) of amthamine. Consistent with these observations, alanine mutagenesis of the majority of these residues markedly attenuated the cAMP accumulation response of H2R to amthamine (Figure 2B; Figure S4A and Table S2, Supporting Information). It's important to note that the T1905.46A mutant exhibits an anomalous behavior, possibly attributed to compensatory effects involving T1033.37. The thiazole ring of amthamine is sandwiched between V993.33 on one side and F2516.52/F2546.55 on the other side, facilitating crucial hydrophobic interactions (Figure 2A). The methyl group position on the thiazole ring intensifies the hydrophobic interaction with F2546.55. Moreover, amthamine establishes van der Waals interactions with C1023.36 and L2747.39 (Figure 2A). Our mutational studies targeting these residues corroborate the substantial import of these hydrophobic interactions (Figure 2B; Figure S4A and Table S2, Supporting Information).
Figure 2.
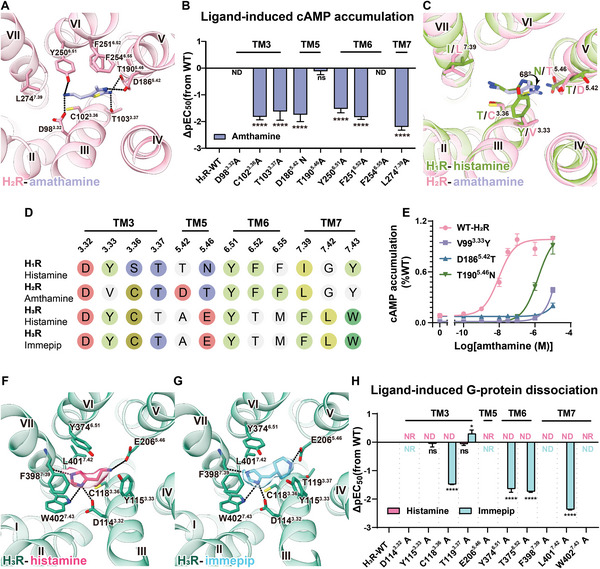
Ligand recognition of histamine receptors. A) Detailed interaction of amthamine with H2R. Dashed lines represent hydrogen bonds. B) Amthamine induced cAMP accumulation in HEK293 cells expressing H2R mutants of the residues in orthosteric pocket (n = 3, ordinary one‐way ANOVA, ****P < 0.0001, ND, not determinable, which refers to cannot be established over the tested concentration range, ns refers to no significance between the WT and mutant). C) Structural comparisons of ligand recognition between histamine‐H1R and amthamine‐H2R structures. D) Sequence alignment of the orthosteric pocket of histamine receptors. Residues that interact with ligands are highlighted with colored circles. E) Dose‐dependent curves for amthamine induced cAMP accumulation in HEK293 cells expressing the H2R mutants (V993.33Y, D1865.42T, T1905.46N) that are not conserved in H1R (n = 3). F‐G) Detailed interactions of histamine F) and immepip G) with H3R. H) Agonists induced Gi dissociation in HEK293 cells expressing H3R mutants of the residues in orthosteric pocket by NanoBiT assays (n = 3, ordinary one‐way ANOVA, *P = 0.0456, ****P < 0.0001, NR refers to no response to the ligand, ND, not determinable, which refers to cannot be established over the tested concentration range, ns refers to no significance between the WT and mutant).
H2R displays the greatest similarity to H1R among the four histamine receptors, exhibiting an approximate 73% sequence homology in the 7TM region. As anticipated, the residues within the binding pocket of both H1R and H2R manifest a high degree of conservation. Among the 12 residues within the H2R binding pocket, seven are identical and three show homogeneous to their counterparts in H1R (Figure 2D; Figure S3F, Supporting Information). Notably, despite this conservation, discernible differences emerge in the ligand pocket shapes of H1R and H2R (Figure S3B,C, Supporting Information). Y1083.33 and N1985.46, possessing substantial steric bulk, serve to constrict the ligand pocket in H1R (Figure 2C). In contrast, the structurally equivalent residues V993.33 and T1905.46 in H2R do not introduce such steric hindrance. As a consequence, the docking of amthamine into H1R would inevitably give rise to significant steric clashes, particularly involving the thiazole ring and the primary amine moiety of the ring with Y1083.33 and N1985.46 (Figure 2C). Furthermore, the substitution of D1865.42 in H2R with the corresponding T1945.42 residue in H1R diminished the polar interactions with amthamine (Figure 2C). Collectively, our structures provide a clear explanation for the high selectivity of amthamine as an H2R agonist. Furthermore, mutations of these three residues of H2R to the corresponding residues in H1R significantly decreased the potency and efficacy of amthamine, further supporting our conclusion (Figure 2E; Table S2, Supporting Information).
The H3R‐Gi complexes, bound with histamine and immepip, exhibit highly analogous overall structures, featuring a remarkably low root‐mean‐square deviation (RMSD) of 0.3 Å for the H3R Cα atoms. Both ligands reside within the orthosteric pocket, a characteristic shared with H2R and other aminergic receptors, as depicted in Figure 2. In the histamine‐bound H3R‐Gi complex, the amino group of histamine engages a pivotal hydrogen bond with E2065.46 (Figure 2F). Simultaneously, the Nπ atom within the imidazole ring establishes van der Waals forces with C1183.36, while also forming hydrogen bonds with D1143.32 and W4027.43 (Figure 2F). Furthermore, the Nτ atom establishes a hydrogen bond with the backbone of F3987.39 (Figure 2F). Through diligent alanine mutagenesis investigations, we affirmatively ascertained the indispensability of these interactions for histamine binding and the ensuing receptor activation. Notably, the alanine mutations targeting E2065.46, D1143.32, and W4027.43 unequivocally abolish the capacity for Gi protein activation (Figure 2H; Figure S4B and Table S2, Supporting Information). Additionally, the EC50 value of the C1183.36A mutant experiences an approximately 150‐fold reduction in comparison to wild‐type H3R. Furthermore, the imidazole ring of histamine securely embeds itself within the hydrophobic core constituted by Y3746.51, L4017.42, W4027.43, and F3987.39 (Figure 2F). This interaction is particularly evident in the formation of a face‐to‐face π‐π stacking association with F3987.39. The veracity of these observations is endorsed by alanine substitutions targeting the residues implicated in this critical hydrophobic core, further underscoring their pivotal role in facilitating histamine recognition (Figure 2H; Figure S4B and Table S2, Supporting Information).
Within the H3R orthosteric pocket, the imidazole rings of histamine and immepip overlap and share many interactions with H3R in a nearly identical position (Figure 2G). Immepip, a synthetic agonist derived from histamine, replaces the aminoethyl moiety with a piperidine ring. A notable departure emerges in the positioning of the amine within the piperidine ring, situated a mere 1.3 Å closer to E2065.46 than the primary amine in histamine, thus intensifying its polar interactions with E2065.46. The bulky piperidine ring also establishes hydrophobic and van der Waals interactions with Y1153.33 and T1193.37, conferring enhanced compatibility of immepip with the dumbbell‐shaped pocket characteristic of H3R (Figure S3D,E, Supporting Information). Consequently, the binding interface of immepip with H3R spans ≈290 Å2, an appreciable 20 Å2 more expansive than that of histamine. Notably, despite modifications introduced to Y1153.33 and T1193.37, the resulting effect on the pEC50 values to immepip is negligible, potentially attributable to the compensatory amplification of polar interactions between the piperidine moiety and E2065.46 (Figure 2H; Figure S4C and Table S2, Supporting Information).
2.3. Unique Histamine Binding Mode of H3R
Recent reports have detailed the structures of aminergic receptors bound to their endogenous ligands, including H1R with histamine,[ 20a ] D1R with dopamine,[ 25c ] 5‐HT1A with 5‐HT,[ 25a ] and β1AR with noradrenaline.[ 26 ] Comparison of these structures reveals that the biogenic monoamine ligands adopt a nearly identical binding pose, situated at the bottom of the receptor's orthosteric binding pockets. Notably, the amino group of these monoamine ligands forms a salt‐bridge with the residue D3.32, which is highly conserved among aminergic receptors. Intriguingly, our high‐resolution cryo‐EM Map unveils a distinctive binding pose of histamine in H3R, in which the amino group of histamine forms a salt‐bridge with E2065.46 rather than interacting with D1143.32 in other aminergic receptors (Figure 3A–F; Figure S5, Supporting Information). To further validate this surprising observation, we constructed an alternative model with histamine adopting the classical pose observed in other aminergic receptors. This alternative model, along with the non‐classical model revealed by cryo‐EM map, were subjected to molecular dynamics (MD) simulation. As anticipated, histamine in our non‐classical pose for H3R remained stable throughout a 200 ns simulation (Figure S5B, Supporting Information). In contrast, histamine in the classic pose of aminergic receptors, as seen in H1R, exhibited high instability after a 200 ns simulation, with two considerably distinct conformations in the trajectory (Figure S5C, Supporting Information). These findings conclusively establish that histamine adopts a unique pose in H3R compared to other aminergic receptors.
Figure 3.
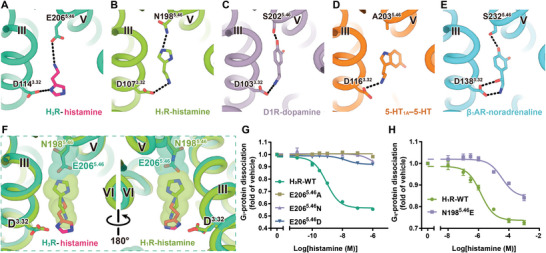
Endogenous ligand binding poses of aminergic receptors. A–E) Binding poses of H3R (A), H1R (B), D1R (C), 5‐HT1A (D), and β1AR (E) with its endogenous ligands. F) Superimposition of H3R‐hisatime and H1R‐histamine structures. G,H) Effects of histamine‐induced G‐protein dissociation in HEK293 cells expressing histamine receptor mutants at 5.46 position. Dose‐response curves for H3R E2065.46 mutants (G) and H1R N1965.46E mutant (H) were measured using the NanoBiT assay (n = 3).
Sequence alignment of all aminergic receptors reveals that the 5.46 position is mainly occupied by small side‐chain residues that are either uncharged polar or hydrophobic, such as threonine, serine, and alanine (Table S3, Supporting Information). This allows sufficient space to accommodate the bulky conjugated group of monoamine ligands. Consequently, the relatively compact imidazole group of histamine enables the inclusion of residues with bulkier side chains in the orthosteric binding pocket, as exemplified by the presence of asparagine in H1R. However, compared to A5.46 in 5‐HT1A and S5.46 in D1R, the elongated glutamine residue in H3R causes severe steric hindrance with the imidazole group in the classic pose. Consequently, histamine rotates 180‐degree along the bilayer plane, resulting in the amino group of histamine interacting with E2065.46 instead of D1143.32 (Figure 3A–F; Figure S5, Supporting Information). A comparison of the orthosteric ligand binding pockets of H1R and H3R reveals that five of the eight residues constituting the H3R ligand pocket are not conserved with H1R (Figure 2D; Figure S3F, Supporting Information). This indicates that the shape and size of histamine binding pockets are significantly distinct between the two histamine receptors. The unique histamine binding of H3R is further confirmed by the differential effects of mutations of H1R and H3R at the 5.46 position (Figure 3G,H; Table S4, Supporting Information). This position contributes to the different binding capacities to the endogenous ligand histamine, enabling the bulk discovery of selective agonists and antagonists targeting H3R.
2.4. Structural Differences Reveal the Basis of G‐Protein Selectivity in Histamine Receptors
Histamine receptors H1R, H2R, and H3R primarily couple with Gq‐, Gs‐, or Gi‐proteins, respectively. Our structures of H2R‐Gs and H3R‐Gi complexes, along with the recently determined H1R‐Gq structures,[ 20a ] enable structural comparisons among histamine receptor–G‐protein signaling complexes. Such comparative analysis holds the potential to unveil the intricate molecular determinants underlying G‐protein‐coupling preferences within this receptor subfamily, offering insights that could extend to other GPCRs. Upon superimposing the three structures focused on the receptor, we observe a congruence in the alignment of TM1, TM2, TM3, TM7, and ICL1 on the cytoplasmic side. Conversely, disparities emerge in the configuration of TM4‐6 and ICL2‐3 (Figure S6A,B, Supporting Information). This observation highlights that while the α5 of Gα inserts into the intracellular cavity of the receptors, contributing to the classical α5‐insertion interface within the complex structures, the specific manner in which the G‐protein binds diverges significantly across the three structures.
In both H1R‐Gq and H3R‐Gi structures, TM6 exhibits comparable outward displacements at their cytoplasmic ends. However, in the H2R‐Gs complex, a significant TM6 movement of 4.7 Å further outward than in H3R, as measured at Cα atoms of residue 6.30 (Figure S6C, Supporting Information). Notably, this represents the most significant TM6 outward displacement among the histamine receptor subfamilies. Consequently, the C‐terminus of α5 in Gαs shifts by 4.0 Å toward TM6 in comparison to Gαi within the H3R‐Gi complex, as measured at the conserved Cα atoms of residue H5.25 (common Gα numbering system)[ 27 ] in Gα (Figure S6C, Supporting Information). Additionally, beyond the dynamic TM6, we observed significant variations in the conformation of TM4, as well as their corresponding ICL2 regions, on the cytoplasmic side. Notably, the intracellular tip of TM4 in H3R exhibits a noteworthy 4.9 Å shift toward TM2 in comparison to H2R, as determined by measuring the Cα atoms of residue 4.40 (Figure S6B, Supporting Information). This specific movement induces the ICL2 of H3R to rotate approximately 64‐degree compared to H2R (Figure S6B, Supporting Information). Besides, our H2R‐Gs and H3R‐Gi structures also reveal that the ICL2s form polar and hydrophobic interactions with the αN helix, the β2‐β3 loop, and the α5 helix of Gα, which are critical for G‐protein coupling.
The most notable distinctions among the three GPCR–G‐protein complexes are prominently evident within the TM5/TM6 regions (Figure 4A,B). In the H2R‐Gs complex, the length TM5 of H2R extends ≈2.5 helical turns (from position 5.67 to 5.75, encompassing nine residues) compared with H1R (Figure 4B; Figure S2, Supporting Information). Furthermore, the extended TM5 in H2R tilts toward TM6 from the Y2025.58 residue for an 18‐degree angle (Figure 4B). Subsequently, the cytosolic tip of TM5 inserts into the cavity of Gαs, which is formed by the αG‐α4 loop, the α4 helix, and the α4‐β6 turn, resulting in additional interactions with the α4 of Gαs (Figure 4C). In the H3R‐Gi structure, both TM5 and TM6 of H3R exhibit unexpected elongations comprising nine and eight residues (2.5 and 2.2 helical turns) compared to H1R, respectively (Figure 4B). This elongation of TM5/TM6 regions in H3R establishes close contacts with the α4‐β6 turn of Gαi, thereby strengthening the H3R‐Gi coupling effectiveness (Figure 4D). Consequently, the interfaces of H2R‐Gs (1610 Å2) and H3R‐Gi (1381 Å2) are both significantly larger than that of H1R‐Gq (1144 Å2). In conclusion, the elongations observed in TM5/TM6 regions within H2R and H3R constitute additional yet distinctive interfaces between the receptor and G‐proteins, hereafter referred to as non‐canonical receptor‐Gα interface. We anticipated that the differential non‐canonical receptor‐Gα interactions resulting from the extended TM5/TM6 could be the key determinants for the primary Gs or Gi/o selectivity. To validate our hypothesis, we analyzed the structural geometries of TM5/TM6 among available GPCR–G‐protein structures. This exploration aimed to unravel potential the correlations between the Gs‐ or Gi/o‐coupling selectivity within class A receptors.
Figure 4.
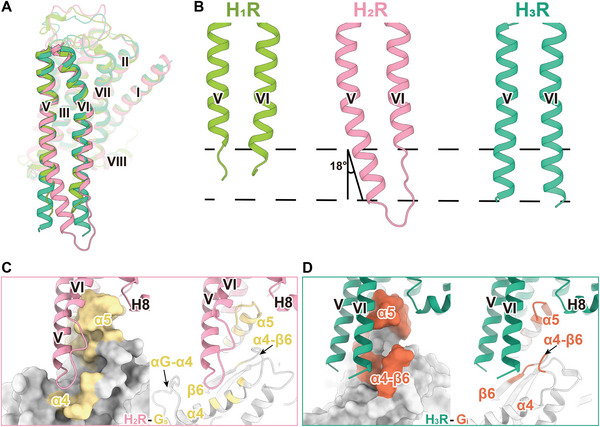
Structural comparison of histamine receptor–G‐protein complexes. A) Structural superposition of the active models of H1R, H2R, and H3R. B) Comparison of TM5 and TM6 in H1R‐Gq, H2R‐Gs, and H3R‐Gi complexes. C,D) Surface (left) and cartoon (right) representation of binding area of H2R (C) and H3R (D) with their respective coupled Gα subunit. Residues within 4Å of H2R or H3R in Gαs or Gαi are highlighted in yellow or orange, respectively.
2.5. TM5 and TM6 are Responsible for Gs and Gi/o Selectivity
Plentiful GPCR–G‐protein structures have been determined with increasing speed since we resolved the first two cryo‐EM structures of GPCR–G‐protein complexes in 2017.[ 9a,b ] Currently, ≈170 structures of class A GPCRs coupled to their corresponding G‐protein are available (as of July 2022). Among them, 13 Gs‐ and 30 Gi/o‐coupled signaling complex structures (Table S5, Supporting Information) of unique class A receptors, whose structures have been resolved with their corresponding primary G‐protein, are chosen for further analysis. We found that the extended TM5/TM6 structural geometries observed in H2R‐Gs and H3R‐Gi complexes are widespread in above structures. More than half of receptors in Gs‐coupled state, the length of TM5 is remarkably longer than TM6 (the median length of TM5 and TM6 is 25 and 21, respectively (Table S5, Supporting Information). Helix lengths of the intracellular region are measured from the residue x.50 to the cytosolic tip of TM5 or TM6), including three β‐adrenergic receptors, D1R, MC1R, MC4R, GPBAR, GPR52, and H2R (Figure 5A).[ 14 , 28 ] In addition, similar to H2R, the cytosolic halve of TM5 in Gs‐coupled receptors exhibits a curved conformation toward TM6, with a range of tilting angles spanning from 8‐ to 26‐degree (Figure 5A; Table S5, Supporting Information). Nevertheless, the situation for Gi/o‐coupled structures is more complex. For instance, certain receptors, including H3R, µOR, S1PR3, 5‐HT1A, 5‐HT1B, and 5‐HT1D, possess a TM6 length exceeding 26 residues.[ 14 , 25 , 29 ] Notably, for 5‐HT1A, 5‐HT1B, and 5‐HT1D, the length of TM6 is 35, 31, and 31 residues, respectively. A significant proportion of GPCR‐Gi/o structures, including MT1, M2R, 5‐HT1F, chemokine receptors, etc., exhibit short helices of both TM5 and TM6, with the length of TM6 similar or only slightly longer than TM5 (Figure 5B).[ 30 ] It is noteworthy that in the majority of Gi/o‐coupled receptor structures, the TM5 is straight, as in H3R, indicating that the cytosolic ends of TM5 are not curved toward TM6, as in H2R in Gs‐coupled structures (Figure 5A,B). Therefore, the length of TM5 and TM6, together with the tilt of the TM5 cytosolic end, exhibits significant differences between Gs‐ and Gi/o‐coupled receptors, likely responsible for Gs and Gi/o selectivity.
Figure 5.
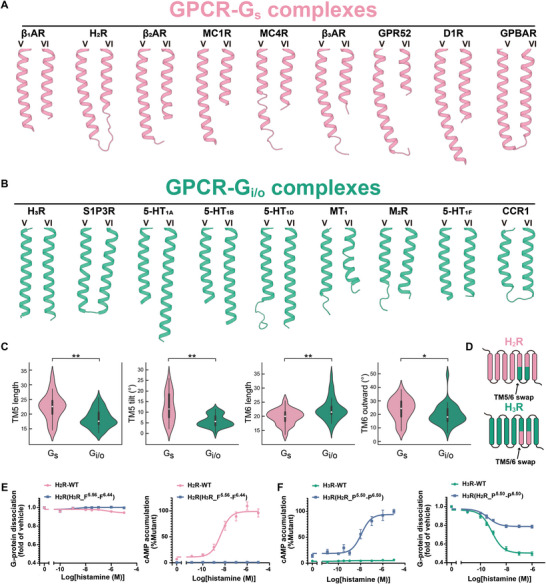
TM5 and TM6 are responsible for Gs and Gi selectivity. A) Representative of the TM5/TM6 structural geometry of Gs‐coupled receptors. B) Representation of the TM5/TM6 structural geometry of Gi‐coupled receptors. C) Violin plots depicting four distinct features of TM5/TM6 in Gs‐ and Gi/o‐coupled receptors. The white dot denotes the median. The interquartile range is shown by the broad black bar in the middle. Except for points considered to be “outliers” using an interquartile range‐based technique, the thin line reflects the remainder of the distribution. n = 54, ns P > 0.05, *P < 0.05, **P < 0.01 by Mann‐Whitney U test. D) Schematic representation of the “TM5/TM6 swap” experiments. E–F) Dose‐dependent curves for histamine induced G‐protein dissociation and cAMP accumulation in HEK293 cells expressing chimera receptors (n = 3). E) Replacement of I5.56‐F6.44 of H2R with F5.56‐F6.44 of H3R did not confer the ability to dissociate Gi‐protein (left), but resulted in loss of the ability to activate the Gs signal pathway (right). F) Replacement of P5.50‐P6.50 of H3R with P5.50‐P6.50 of H2R resulted in gain of the ability to activate the Gs signal pathway (left), but loss of the ability to dissociate Gi‐protein (right).
Previous research has established that the outward movement of TM6 is the hallmark of receptor activation, thus plays a crucial role in receptor Gs/Gi/o‐coupling selectivity. Specifically, Gs‐coupled structures exhibit larger outward movements of TM6 compared to Gi/o‐coupled receptors.[ 9c ] To evaluate the significance of four structural geometries of TM5 and TM6 in Gs and Gi/o selectivity (the lengths of TM5 and TM6, the tilt of the TM5 cytosolic end, and the outward movement of TM6), we analyzed currently available Gs‐ and Gi/o‐coupled structures of class A receptors, along with our unpublished structures (including a total of 20 Gs‐ and 34 Gi/o‐coupled receptors) (Table S5, Supporting Information). The lengths of TM5 and TM6 were measured from the cytosolic tip to 5.50 and 6.50, respectively, and the lengths and angles of TM5/TM6 of receptors were extracted as illustrated in Figure 6A (see Experimental Section). Consistent with our speculation, significant differences were observed in the geometries of TM5 and TM6 between Gs‐ and Gi/o‐coupled receptors. The average length of TM5 in Gs‐coupled receptors is four residues longer than that of Gi/o‐coupled receptors (23.1 ± 4.6 and 18.9 ± 3.1, respectively) (Figure 5C; Table S5, Supporting Information). The tilt of TM5 in Gs‐coupled structures was distributed into two major clusters (12.3 ± 7.1°), while the majority of the Gi/o‐coupled receptors exhibit smaller tilts (5.90 ± 2.9°) (Figure 5C; Table S5, Supporting Information). The average outward movement of TM6 in Gs‐coupled receptors was slightly larger than that of Gi/o‐coupled structures (23.8 ± 8.4° and 19.7 ± 7.6°, respectively), consistent with previous findings (Figure 5C; Table S5, Supporting Information). However, the TM6 lengths of Gs‐coupled receptors and Gi/o‐coupled receptors were indistinguishable, with comparable length distributions and medians (Figure 5C; Table S5, Supporting Information). In conclusion, the structural geometries of TM5/TM6, particularly the length of TM5, the tilt of cytosolic TM5, and the outward movement of TM6, may be responsible for primary Gs and Gi/o selectivity.
Figure 6.
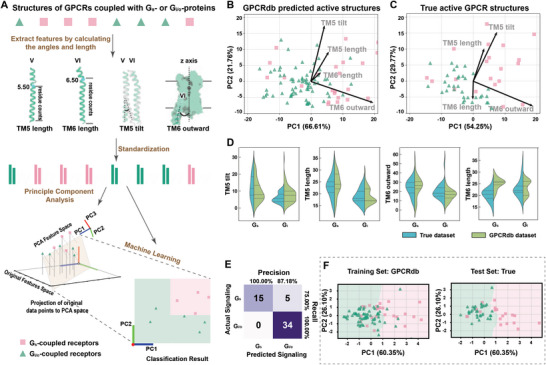
Machine learning‐based classification of GPCRs into Gs and Gi signaling pathway. A) Workflow for GPCR classification utilizing machine learning and feature pre‐processing. The length and tilt of TM5, as well as the length and outward movement of TM6 are extracted from active GPCR structures obtained from GPCRdb homology models and True structures. The resulting data of these four geometries are standardized, and then subjected to PCA. Subsequently, a Random Forest classifier is employed to classify GPCRs based on the PCA results. B,C) PCA biplots were generated for the four geometries of GPCRdb homology models (B) and True GPCR structures (C) individually. The contributions of each variable to the principal components are depicted as vectors on the plot, where the vertical component of a vector on a given PC illustrates the respective contribution of that variable to the PC. The angle between two vectors reflects the correlation between the corresponding features, while the length of a vector represents the significance of the corresponding feature. D) Comparison of violin plots depicting four geometries of TM5/TM6 in GPCRdb structures and True GPCR structures. Here the median and interquartile range are depicted as dashes. E) Confusion matrix for the True GPCR structures given by the best classifier. F) Decision boundary visualization via PCA given by the best classifier and scatter plot of GPCRdb homology models (left) and True GPCR structures (right) based on GPCRdb homology models.
To validate the hypothesis that the preference selection of G‐proteins for class A receptors is determined by TM5/TM6, we conducted “TM5/TM6 swap” experiments to reverse the Gs and Gi/o selectivity of receptors (Figure 5D). In the first set of experiments, we replaced the intracellular half of TM5, TM6, and ICL3 (P5.50 to P6.50) of Gi/o‐coupled receptors with that of Gs‐coupled receptors. As expected, these chimeric receptors induced cAMP accumulation, including H3R(H2R_P5.50‐P6.50), H3R(D1R_P5.50‐P6.50), H3R(MC4R_P5.50‐P6.50), and H3R(β2AR_P5.50‐P6.50), and D2R(H2R_P5.50‐P6.50) (Figure 5E; Figure S7A and Table S6, Supporting Information). In the second set of experiments, we attempted to switch Gs‐coupled receptors to Gi/o‐coupled receptors. Although these engineered receptors could not recruit Gi proteins, their ability to stimulate cAMP accumulation was almost lost (Figure 5F; Figure S7B and Table S6, Supporting Information). In conclusion, these “TM5/TM6 swap” experiments could alter the Gs‐ or Gi/o‐coupling selectivity of receptors, supporting the notion that the conformational architecture of TM5/TM6 determines the Gs and Gi/o selectivity of a GPCR.
2.6. Classification of Gs‐ and Gi/o‐Coupled Receptors by Machine Learning
Can the structural determinants of Gs and Gi/o selectivity identified in this study be generalized to all Class A GPCRs? To address this question, a machine learning approach was utilized to categorize primary Gs‐ and Gi/o‐coupled receptors, based on the structural geometries of TM5/TM6 of receptors (Figure 6A). To augment the generalization of the predictive model, we incorporated predicted structures in the structural datasets (see Experimental Section). The training set comprised 98 (24 Gs‐ and 74 Gi/o‐coupled receptors) GPCRdb‐predicted homology models (Table S7, Supporting Information),[ 31 ] while the test set consisted of 54 (20 Gs‐ and 34 Gi/o‐coupled receptors) true (experimental) structures (Table S5, Supporting Information). In light of potential correlations among the four structural geometries of TM5/TM6, we opted to apply Principal Component Analysis (PCA)[ 32 ] to effectively decorrelate and compress these characterizes, thereby facilitating a reduction of the TM5/TM6 geometry space from a 4D to a 2D construct (Figure 6B,C). The first two Principle Component (PCs) are reserved for further analysis (PC1 and PC2), contributing more than 86% of the variance (Figure 6B,C). The individual biplots of the training and true datasets revealed that the TM5 length, TM5 tilt, and TM6 outward movement are the primary contributors to the principal components (The contributions of each variable to the principal components are depicted as vectors on the plot, where the vertical component of a vector on a given PC illustrates the respective contribution of that variable to the PC) (Figure 6B,C). Specifically, the TM6 outward movement significantly influences PC1, whereas the length and tilt of TM5 play a critical role in PC2 (Figure 6B,C). Moreover, these three vectors exhibit similarities in the principal component plots between the training and true datasets, particularly in terms of TM5 tilt and TM6 outward movement (Figure 6B,C). The significance of TM5 length, TM5 tilt, and TM6 outward movement in determining the selectivity of Gs‐ and Gi/o‐protein coupling in class A receptors is suggested by these findings.
Remarkably, the biplots indicate that between the two datasets, the TM6 length has the largest discrepancy: its eigenvector directions are almost opposite in the two biplots, whereas the remaining vectors have similar directions (Figure 6B,C). In the training dataset, the impact of the TM6 length vector on each principal component appears to be minimal and positively correlated to both PCs (Figure 6B,C). In contrast, it exerts a negligible effect on PC1 and displays a negative correlation with PC2 in the true dataset (Figure 6B,C). Subsequent violin plots reveal a symmetry of the distributions of TM5 tilt and TM6 outward movement in the two datasets (Figure 6D). Nevertheless, there is a notable disparity in the distribution pattern of TM6 length between the two datasets (Figure 6D). Further correlation analysis of four geometries of TM5/TM6 elucidates similar inconsistency. The TM6 length from the training dataset reveals a mildly positive correlation of 0.20 for the TM5 tilt (Figure S8, Supporting Information). Conversely, the TM6 length from the true dataset unveils a negative correlation for TM5 tilt, with a magnitude of −0.29 (Figure S8, Supporting Information). In conclusion, these results imply that the TM6 length poses a challenge for model training, concurrently indicating that TM6 length marginally contributes to G‐protein selectivity.
Aimed at deriving a more precise conclusion about the underlying mechanism, we trained a classifier for primarily Gs‐ and Gi/o‐coupled receptors through a machine‐learning approach directly on the three standardized geometries of TM5/TM6 (TM6 length excluded). We employed a variant of the Random Forest Classifier[ 33 ] with constraints to the number of estimators and their maximum depth to decrease the risk of overfitting. Validation of the model using the true dataset achieved an accuracy of 90.74%, F1‐score of 0.8943, Area Under Curve (AUC) of 0.8162, and Matthews Correlation Coefficient (MCC) of 0.8086 (Figure 6E; Tables S5,S8, Supporting Information). This further illustrates the effectiveness of these three geometries for selectivity. Interestingly, certain falsely predicted receptors (PE2R2, PE2R4, V2R) in our classifier exhibited comparable Gs and Gi/o‐protein selectivity, and LSHR had limited proof to support its primary coupling according to the GPCRdb/GproteinDb.[ 31 ] In brief, the predictive model has fulfilled good performance.
Besides, with the aid of PCA visualization, we can depict a probable decision boundary given by our best classifier to derive a simpler formula (Figure 6F). The decision boundary elucidates that GPCRs with extended TM5 length, significant TM5 tilt, and prominent TM6 outward movement are indicative of Gs‐coupling receptors. In contrast, GPCRs featuring shorter TM5 length, minimal TM5 tilt, and constrained TM6 outward movement are more likely associated with Gi/o‐coupling receptors (Figure 7F).
Figure 7.
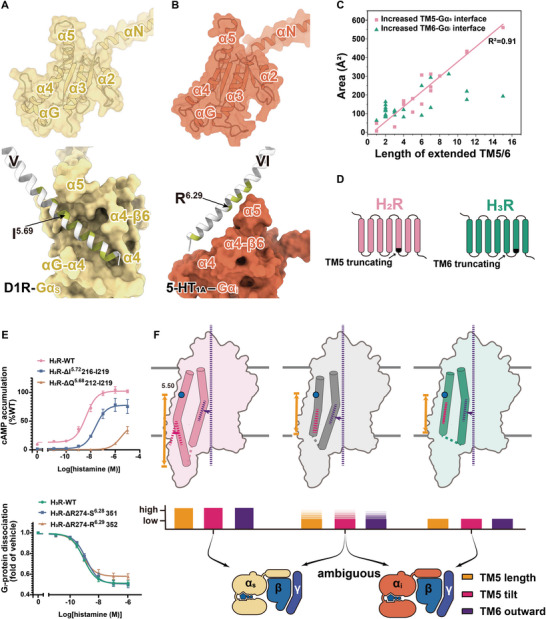
Contributions to G‐protein signaling of extended TM5/TM6. A) (Up) Surface and cartoon representations of Gαs are shown, with the surface of Gαs depicted as transparent. (Down)The binding surface of TM5 with Gαs of D1R is shown, with TM5 beyond I5.69 noted as extended helix. Residues that interact with Gα are highlighted in green. B) (Up)Surface and cartoon representations of Gαi are shown, with the surface of Gαi depicted as transparent. (Down) The binding surface of TM6 with Gαi/o of 5‐HT1A. TM6 beyond R6.29 was considered as extended helix. C) The additional contact areas of Gs‐ or Gi/o‐coupled receptors between the extended TM5/TM6 with Gαs/Gαi/o, respectively. D) Schematic representation of the TM5‐ (up) or TM6‐ (down) breaking mutants. E) Dose‐dependent curves for histamine induced cAMP accumulation or G‐protein dissociation in HEK293 cells expressing the truncated receptors (n = 3). (Up) The TM5 of H2R was respectively truncated by four amino acids and eight amino acids. The cAMP assay validated that the length of TM5 was closely related to the activation of the Gs signal pathway. (Down) The TM6 of H3R was respectively truncated by eight amino acids and nine amino acids. The NanoBit assay validated that the length of TM6 did not correlate with the dissociation of the Gi‐protein. F) Schematic representation of the geometry determinants for G‐protein selectivity of GPCRs.
2.7. TM6 Length is not Significant for Gs/Gi/o Selectivity
Structuromic analysis and ML analysis of GPCR‐G‐protein complexes suggested that the length of TM6 was not responsible for Gs and Gi/o selectivity. Therefore, further investigation is required to elucidate the mechanisms underlying the differential effects of TM5 and TM6 length in G‐protein selectivity. In the GPCR‐Gs complex, TM5 extends to insert into the groove formed by the αG‐α4 loop, the α4 helix, and the α4‐β6 loop of Gαs (Figure 7A), leading to increased interactions with Gαs. To achieve this, extended TM5 tilts toward TM6 to fit into the groove in Gαs. Failure to accomplish this could result in severe steric hindrance with the αG‐α4 loop of Gαs, as observed in D1R (Figure 7A),[ 28d ] indicating a correlation between the length of TM5 and its tilt in the Gs‐coupling receptors. Consistent with this, biplot analyses of GPCRdb‐predicted structures and true structures consistently reveal a high correlation between the length and tilt of TM5 in Gs‐coupling receptors, with coefficients of 0.54 and 0.63, respectively (Figure S8, Supporting Information). However, the groove in Gαs is absent in Gαi/o due to the absence of 13 residues in the αG‐α4 loop (Figure 7B). Consequently, in the GPCR‐Gi/o complex, the extended TM5 and TM6 can only rest on the flattened surface of Gαi/o, resulting in smaller contact areas and fewer interactions compared to the Gs‐coupled structure.
To validate this hypothesis, we investigated whether the extended TM5 in Gs‐coupled structures or extended TM6 in Gi/o‐coupled structures contributed equivalently to the increase in the contact area. Among the 54 true GPCR‐G structures, the median length of the entire TM5 is 23 in 20 Gs‐coupled structures and 18 in 34 Gi/o‐coupled structures (Table S5, Supporting Information). The median length of TM6 is 20.5 in Gs‐coupled receptors and 22 in Gi/o‐coupled structures. Consequently, we considered TM5 longer than 18 (below 5.69) in Gs‐coupled receptors and TM6 longer than 20.5 (below 6.29) in Gi/o‐coupled receptors as extended TMs. Interestingly, we found that the additional contact area between the extended TM5 and Gαs exhibited a linear correlation with the extended residue amount, ≈40±3.0 Å2 per residue (R2 = 0.91) (Figure 7C; Figure S9 and Table S9, Supporting Information). For example, the contact area between the 15‐residue‐extended TM5 of D1R with Gαs reached 561 Å2 (Figure 7A). In contrast, the increased contact area between TM6 and Gαi/o did not show a significant correlation with the length of the extended TM6. For instance, the contact area between the 15‐residue‐extended TM6 of 5‐HT1A and Gαi was only 192.9 Å2 (Figure 7B).
Visualizing cryo‐EM density map of Gi/o‐coupled complexes further revealed that the extended TM6 is not significant for G‐protein coupling. The densities for the extended TM6 in several Gi‐coupled complex including 5‐HT1A, 5‐HT1B, and 5‐HT1D are dramatically weaker and lack high‐resolution features compared with the rest of the receptor, suggesting the instable interactions between the extended TM6 and Gi‐protein (Figure S10, Supporting Information).[ 25 , 29 ] Consistently, the removal of the extended TM6 of H3R posed negligible effect on its Gi activity even when nine residues were removed from the cytosolic extremity of TM6 (Figure 7D,E; Table S10, Supporting Information). In contrast, cAMP accumulation experiments confirmed that the removal of the extended TM5 of H2R significantly reduced its Gs‐protein activity (Figure 7D,E; Table S10, Supporting Information). In conclusion, our findings indicate that a long TM5 is crucial for Gs‐coupled receptors, whereas the TM6 length is not significant for Gs and Gi/o selectivity.
3. Discussion
The four histamine receptors serve essential roles in pathophysiological and signaling events, rendering them significant drug targets. In this study, we presented the structures of one H2R‐Gs and two H3R‐Gi complexes, offering profound insights into the orthosteric pocket of histamine receptors and G‐protein selectivity. Through our investigation of the amthamine‐bound H2R structure and mutagenesis studies, we revealed the discrepancy in the ligand pocket of H1R and H2R that contributed to H2R selectivity for the agonist amthamine. Notably, our findings uncovered a unique binding pose for endogenous histamine in H3R, deviating from other monoamine receptors, as the amino group interacts with E2065.46 in H3R instead of the conserved D1143.32 found in other aminergic receptors. This observation coincided with the behavior of the antipsychotic drug quetiapine, which exhibits high or modest affinities for many aminergic receptors with the exception of H3R and H4R.[ 34 ] Importantly, the structural geometry differences observed in TM5/TM6 between H2R‐Gs and H3R‐Gi structures led us to propose the determinants of primary Gs‐ and Gi/o‐coupling selectivity.
GPCRs predominantly regulate downstream signaling through the engagement and activation of four major G‐protein subtypes. Among these four members, Gs and Gi/o are two principal signaling pathways, for about 86% GPCRs are annotated to couple with Gs‐ or Gi/o‐protein.[ 2a ] The selective coupling of GPCRs to the G‐proteins is crucial for effective signal transduction, especially those primarily coupled with Gs‐ and Gi/o‐proteins with remarkable coupling efficiency and rapid kinetics. The explosion of GPCR–G‐protein structures in recent years has significantly enhanced our comprehension on GPCR coupling mechanisms. Receptors and G‐proteins interact through a common mode, wherein the ligand‐activated GPCR opens its intracellular cavity and accommodates the α5 of Gα, which is conserved through the family/class A, B1, B2, and F GPCRs.[ 9 , 35 ] From the perspective of the G‐protein, the selectivity‐determining positions in Gα predominantly attribute to the α5 helix, along with the αN‐β1 loop, the β2‐β3 loop, and the α4 helix of Gα.[ 1b ] However, the selectivity signatures of receptors are more complicated due to the great divergence in the amino acid sequences and structural conformations of GPCRs. In contrast to the 16 members of Gα proteins which adopt similar conformations, the intracellular parts of GPCRs are more diverse, particularly in the regions of TM5, TM6, H8, intracellular loops, and the receptor C‐terminal tail. By combining reported GPCR–G‐protein structures with unpublished structures from our laboratory, we obtained a number of non‐redundant Gs‐coupled or Gi/o‐coupled structures of class A receptors, totaling 20 and 34, respectively. These structures revealed that the selectivity of Gs and Gi/o is determined by TM5/TM6 geometries, including the length and tilt of TM5, and the outward movement of TM6. The length and tilt of TM5 exhibit a high correlation in true GPCR‐Gs complexes, consistent with the extended and tilted TM5 (e.g., H2R) inserting into the groove formed by the αG‐α4 loop, the α4 helix, and the α4‐β6 loop of Gαs. However, the αG‐α4 loop is 13 residues shorter in Gαi/o than in Gs, which cannot constitute the same groove as Gαs in Gαi/o. Hence, the extended TM6 (e.g., H3R) leads to less contact with Gαi in Gi/o‐coupled structures. This phenomenon indicates that the interactions of GPCR‐Gs are stronger than that of GPCR‐Gi/o complexes, which is consistent with Matic et al.’s report that the binding energy of Gs complexes is stronger than that of Gi/o complexes.[ 13 ] Furthermore, our “TM5/6‐breaking” experiments also confirmed that TM6 length is not significant for Gs and Gi/o selectivity.
The intricate molecular mechanism underlying primary G‐protein coupling selectivity in GPCRs remains complex and not fully elucidated. The structures of GPCR–G‐protein complexes indicate that the ICL2, ICL3, TM5, and TM6 of receptors are associated with G‐protein‐coupling selectivity. However, these cytoplasmic regions of receptors rarely exhibit common patterns in terms of the sequence or amino‐acid properties related to G‐protein selectivity. In our prior study, we proposed that the TM5 and TM6 helices synergistically alternate lengths to determine the selectivity between Gs‐ and Gi/o‐proteins, defined as “a macro‐switch”.[ 11 ] Comparative sequence analyses revealed that specific amino acids function as “micro‐switches” uniquely localized in the complementary pocket. However, these two switches only partially determine the selectivity between Gs and Gi/o. Aided by the drastic increasing number and improved qualities of the structures of GPCRs in complexes with different G‐proteins in house and in the public data server, in the present study, we conducted a systematic evaluation to thoroughly explore the intricate relationship between the four geometries of TM5/TM6 and their impact on primary G‐protein selectivity by using machine learning‐based structuromic profiling. Using the GPCRdb homology models as the training dataset, we generated a Gs‐ or Gi/o‐coupling classifier that achieved an impressive 91% accuracy in the true dataset based on the TM5/TM6 geometries. Based on our classifier, we find that GPCRs with a long TM5 length and large TM5 tilt and TM6 outward movement are Gs‐coupling receptors, whilst GPCRs with a short TM5 length and small TM5 tilt and TM6 outward movement are more likely to be Gi/o‐coupling receptors. We anticipate that GPCRs that fail to satisfy the aforementioned conditions probably have comparable or promiscuous activity in stimulating Gs and Gi/o signaling (Figure 7F). Interestingly, class B1 receptors, primarily coupled to Gs‐protein, exhibit large TM5 tilt and TM6 outward movement. These structural geometries also conform to the G‐protein selectivity features observed in Class A receptors, further demonstrating the universality of our proposed G‐protein selectivity mechanism (Table S11, Supporting Information). In summary, our studies revealed that the structural geometry of TM5/TM6 serves as the determinant of the primary G‐protein signaling selectivity in GPCRs. These findings have the potential to aid efforts in engineering GPCR selectivity, especially by modifying promiscuous GPCRs to improve affinity to specific G‐proteins.
4. Experimental Section
Constructs
The wild‐type human H2R gene was cloned into the pFastBac1 vector. The N‐terminus of H2R was fused with the hemagglutinin signal peptide (HA) to enhance receptor expression, followed by a Flag tag (DYKDDDK) to facilitate complex purification. The wild‐type human H3R445 isoform was cloned into a modified pFastBac1 vector with a hemagglutinin (HA) signal sequence at the N‐terminus and a PreScission protease site followed by a Flag tag. The C‐terminus of H3R was fused with the LgBiT,[ 22 ] followed by a TEV protease site and a double MBP tag to facilitate expression and purification. The dominant‐negative bovine Gαs (DNGαs) and human Gαi1 (DNGαi1) were generated by site‐directed mutagenesis, as previously described, to stabilize the interaction with the βγ subunits,[ 36 ] and were cloned into the pFastBac1 vector. Human Gβ1 was fused with a 10× His tag at the N‐terminus and HiBiT at the C‐terminus, and together with Gγ2, was cloned into the pFastBac dual vector.
Protein Complex Expression and Purification
For the H2R‐Gs complex, the H2R, DNGαs, and Gβ1γ2 constructs were co‐expressed in Spodoptera frugiperda (Sf9) cells using the Bac‐to‐Bac Baculovirus Expression System (Invitrogen). Cells were infected at a density of 2.4 × 106 cells per mL and then co‐infected with three separate viruses at a ratio of 2:1:1 for H2R, DNGαs, and Gβ1γ2. Cells were collected 48 hours post‐infection and stored at −80 °C until use. Sf9 cell pellets expressing H2R and G‐protein trimer were resuspended in a buffer containing 20 mm HEPES pH 7.4, 100 mm NaCl, 3 mm MgCl2, 5 mm CaCl2, 2.5 mg mL−1 leupeptin, and 0.2 mg mL−1 benzamidine. The complex was assembled on the membrane by adding 100 µm amthamine, 10 mg mL−1 Nb35, 25 mU mL−1 apyrase, and further incubated at RT for 2 h. The complex was then extracted from the cell membrane using 0.5% (w/v) lauryl maltose neopentylglycol (LMNG) (Anatrace) and 0.1% (w/v) cholesteryl hemisuccinate (CHS) (Anatrace) at 4 °C for 2 h. After that, the supernatant was collected by centrifugation at 25 000 rpm for 30 min, and the solubilized complex was incubated with M1 anti‐FLAG resin for 2.5 h at 4 °C. Afterwards, the resin was collected and washed with a buffer containing 20 mm HEPES pH 7.4, 100 mm NaCl, 3 mm MgCl2, 5 mM CaCl2, 0.1% (w/v) LMNG, 0.02% (w/v) CHS, 100 µM amthamine, 2.5 mg mL−1 leupeptin, and 0.2 mg mL−1 benzamidine. Subsequently, the detergent was changed to 0.01% (w/v) LMNG, 0.005% (w/v) CHS. Then, the H2R‐Gs complex was eluted with 20 mm HEPES pH 7.4, 100 mm NaCl, 0.01% (w/v) LMNG, 0.001% (w/v) CHS, 5 mm EGTA, 0.2 mg mL−1 FLAG peptide, 100 µM amthamine, 2.5 mg mL−1 leupeptin, and 0.2 mg mL−1 benzamidine. The eluate was collected and injected onto a Superdex 6 Increase 10/300 GL column (GE Healthcare) with buffer containing 20 mm HEPES pH 7.4, 100 mm NaCl, 0.00025% (w/v) LMNG, 0.0002% (w/v) CHS, 0.00075% (w/v) GDN (Anatrace), and 100 µm amthamine. The complex fractions were concentrated with a 100 kDa MWCO Millipore concentrator to 8 mg mL−1 for making a cryo‐EM grid.
For the H3R‐Gi complex, the H3R445 isoform, DNGαi1, and Gβ1γ2 constructs were co‐expressed in Spodoptera frugiperda (Sf9) cells using the Bac‐to‐Bac Baculovirus Expression System (Invitrogen). Cells were infected at a density of 2.4 × 106 cells per mL and then co‐infected with three separate viruses at a ratio of 1:1:1 for H3R, DNGαi1, and Gβ1γ2. Cells were collected 48 h post‐infection and stored at −80 °C until use. The co‐expressed H3R‐Gi complex cell pellets were lysed in a buffer containing 20 mm HEPES pH 7.5, 100 mm NaCl, 2 mm MgCl2, adequate agonist (1 mm histamine or 10 µm immepip), and supplemented with EDTA‐free protease inhibitor cocktail (Bimake) using a Dounce homogenizer. The complex formation was initiated by adding 20 µg mL−1 scFv16 and 25mU mL−1 apyrase (NEB) and subsequently incubated for 1.5 h at RT. Cell membranes were solubilized by 1% (w/v) n‐Dodecyl‐β‐D‐Maltopyranoside (DDM, Anatrace) and 0.2% (w/v) cholesterol hemisuccinate (CHS, Anatrace) for 2.5 h at 4 °C. After centrifugation at 30 000 g for 30 min, the supernatant was isolated and incubated with amylose resin (NEB) for 1 hours at 4 °C. Then, the resin was washed with a wash buffer containing 20 mm HEPES pH 7.5, 100 mm NaCl, 2 mm MgCl2, 0.1% (w/v) LMNG, 0.02% (w/v) CHS, and agonist (1 mm histamine or 10 µm immepip). Subsequently, the detergent was changed to 0.01% (w/v) LMNG, 0.005% (w/v) CHS, and eluted with an elution buffer containing 20 mm HEPES pH 7.5, 100 mm NaCl, 2 mm MgCl2, 0.01% (w/v) LMNG, 0.005% (w/v) CHS, 10 mm Maltose, and agonist (1 mm histamine or 10 µm immepip). The double MBP tag of H3R was removed by incubating with TEV protease for 1 hour at RT. Then, the protein sample was further purified by size‐exclusion chromatography with a Superose 6 10/300 GL column (GE Healthcare) equilibrated with a running buffer containing 20 mm HEPES pH 7.5, 100 mm NaCl, 2 mm MgCl2, 0.00225% (w/v) LMNG, 0.0006% (w/v) CHS, 0.00075% (w/v) GDN (Anatrace), and agonist (1 mM histamine or 10 µM immepip).
Cryo‐Grid Preparation and EM Data Collection
Three microliters of the purified GPCR–G‐protein complexes were applied onto a glow‐discharged holey carbon grid (Quantifoil R1.2/1.3) at ≈20 mg mL−1. The grids were plunge‐frozen in liquid ethane using Vitrobot Mark IV (Thermo Fisher Scientific). The frozen grids were then transferred to liquid nitrogen and stored for data collection.
Cryo‐EM imaging was performed on a Titan Krios at 300 kV using the Gatan K2 Summit detector at the Center of Cryo‐Electron Microscopy, Zhejiang University (Hangzhou, China). Movies were recorded at a dose rate of approximately 8.0 e/Å2/s with a defocus ranging from −1.0 to −2.2 µm using the SerialEM software in counting mode for the three receptor‐G‐protein complexes, respectively. The total exposure time was 8 seconds and 40 frames were recorded per micrograph. A total of 3122, 3008, and 3079 movies were collected for the amthamine‐H2R‐Gs, histamine‐H3R‐Gi, and immepip‐H3R‐Gi complexes, respectively, for structure reconstruction.
Image Processing and 3D Reconstructions
Image stacks were aligned using MotionCor 2.1.[ 37 ] Contrast transfer function (CTF) parameters were estimated using Gctf v1.18.[ 38 ] The following data processing was performed using RELION‐3.0‐beta2.
For the amthamine‐H2R‐Gs complex, automated particle picking yielded 3361930 particles. The particles extracted from the dataset were downscaled two times and subjected to 2D classification. The map of the GPBAR‐Gs complex (EMD‐30344),[ 14c ] low‐pass filtered to 40 Å, was used as an initial reference model for two rounds of 3D classification, resulting in a well‐defined subset of 857906 particles. The particles were then re‐extracted with the original pixel size and performed further 3D classification focusing on the receptor‐Gs complex, which produced a good subset accounting for 368447 particles. After 3D refinement, CTF refinement, and Bayesian polishing of the final particles, the final refinement map was generated with a global resolution of 2.7 Å at a Fourier shell correlation of 0.143.
For the histamine‐H3R‐Gi complex, template‐based particle selection produced 2487329 particles. After 2D classification and two rounds of 3D classification using the 5‐HT1D‐Gi complex (EMD‐30974)[ 25a ] low‐pass filtered map as an initial reference model, a well‐defined subset with 742110 particles was selected. Further 3D classifications focusing on the alignment of the receptor‐Gi complex produced a good subset accounting for 234203 particles, which were subsequently subjected to 3D refinement, CTF refinement, and Bayesian polishing. The final refinement generated a map with an indicated global resolution of 2.7 Å at a Fourier shell correlation of 0.143.
For the immepip‐H3R‐Gi complex, template‐based particle selection produced 2050983 particles. After 2D classification and two rounds of 3D classification using the histamine‐H3R‐Gi complex low‐pass filtered map as an initial reference model, two well‐defined subsets with 697323 particles were selected. Further 3D classifications focusing on alignment of the receptor produced a good subset accounting for 330449 particles, which were subsequently subjected to 3D refinement, CTF refinement, and Bayesian polishing. The final refinement generated a map with an indicated global resolution of 3.0 Å at a Fourier shell correlation of 0.143.
Model Building and Refinement
For the H2R‐Gs complexes, the initial model of H2R was downloaded from the activated homology models of H2R from GPCRdb.[ 31 ] The initial Gs and Nb35 complex was generated from the GPBAR‐Gs complex (PDB ID: 7CFM).[ 14c ] For the H3R‐Gi complex, the initial model of H3R was downloaded from GPCRdb as H2R. The initial Gi and scFv16 complex was generated from the 5‐HT1D‐Gi complex (PDB ID: 7E32).[ 25a ] Agonist and lipid coordinates and geometry restraints were generated using phenix.elbow. Then, the models were docked into the cryo‐EM density map using Chimera. After the initial docked models were refined using Rosetta, the models were subjected to iterative rounds of manual adjustment and auto‐refinement in Coot and Phenix, respectively. The final refinement scores were validated by the module “comprehensive validation (cryo‐EM)” in Phenix. Structure figures were prepared using PyMOL, Chimera, and ChimeraX.
Molecular Dynamics Simulations
First, the histamine bound to H3R model was subtracted from histamine‐H3R‐Gi complex. Second, the histamine was also positioned in a similar direction to the histamine‐H1R structure and fitted to the cryo‐EM density. The orientations of the receptors were calculated by the Positioning of Proteins in Membranes (PPM) Web Server.[ 39 ] Following these steps, the whole systems were prepared using the CHARMM‐GUI and embedded in a bilayer consisting of 200 1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phosphocholine (POPC) lipids using replacement methods.[ 40 ] The membrane systems were then solvated into a periodic TIP3P water box supplemented with 0.15 m NaCl. The CHARMM36m Force Field was used to model protein molecules, and the CHARMM General Force Field (CGenFF) was used for the agonist histamine.
Subsequently, two systems were subjected to minimization for 10 000 steps using the conjugate gradient algorithm. They were then heated and equilibrated at 310.13 K and 1 atm for 200 ps with 10.0 kcal mol−1 Å−2 harmonic restraints in the NAMD 2.13 software. This was followed by 5 cycles of equilibration for 2 ns each at 310.13 K and 1 atm, during which the harmonic restraints were sequentially reduced to 5.0, 2.5, 1.0, 0.5, 0.1 kcal mol−1 Å−2. Production simulations were run at 310.13 K and 1 atm in the NPT ensemble using the Langevin thermostat and Nose‐Hoover method for 250 ns. Electrostatic interactions were calculated using the particle mesh Ewald (PME) method with a cutoff of 12 Å. Throughout the final stages of equilibration and production, 5.0 kcal mol−1 Å−2 harmonic restraints were placed on the backbone of the receptors due to the flexibility of the second extracellular loop (ICL2) of H3R. Trajectories were visualized and analyzed using Visual Molecular Dynamics (VMD) version 1.9.3.
GloSensor cAMP Assay
The agonist‐induced cAMP accumulation was measured using the GloSensor™ cAMP assay kit (Promega). HEK293T cells were co‐transfected with a 3:1 ratio of WT or mutant and the pGloSensor™−22F plasmid by using LIPO (YEASON). After a minimum of 6 h, transfected cells were seeded onto cell adherent reagent (Applygen) coated 384‐well culture plates and incubated for more than 12 h at 37 °C in 5% CO2. The culture medium was removed and PBS was added to starve the cells. After 20 min later, the PBS was removed, and the cells were incubated with the equilibration medium (CO2‐Independent Medium (Gibco) with 10% FBS) containing a 4% dilution of the GloSensor™ cAMP reagent stock solution for 30 minutes at 37 °C and then 10 min at RT. To obtain the dose‐response curves, serially diluted agonists were added to each well to stimulate the cells. Luminance signal was measured using 200 ms intervals then (TECAN, 25 °C). Dose‐responses were generated from the peak response. cAMP accumulation was analyzed by a standard dose‐response curve using GraphPad Prism 8.0 (GraphPad Software). EC50 and pEC50± SEM were calculated using nonlinear regression (curve fit). Data are means ± SEM from at least three independent experiments performed in technical triplicates.
NanoLuc Binary Technology (NanoBiT) Assay
For the measurement of G‐protein dissociation, HEK293T cells were co‐transfected with WT or mutant, Gαi‐LgBit, Gβ, and Gγ‐SmBit plasmids at a 6:2:5:5 ratio. Following transfection, cells were seeded onto 96‐well culture plates and incubated for over 12 h at 37 °C in 5% CO2. Then, the cells were rinsed twice with D‐PBS and incubated with 4 nm coelenterazine‐400a (Maokangbio) in HBSS supplemented with 5 mm HEPES pH 7.4 and 0.1% BSA for 1 h. The baseline luminance signal was read immediately for 5 cycles (TECAN, 25 °C). To obtain the dose‐response curves, serially diluted agonists were added to each well to stimulate the cells. The luminance signal was then measured for another 15 cycles using 500 ms intervals (TECAN, 25 °C). Dose‐responses were generated from the 5th response, and the row data was standardized by the baseline and the vehicle group. G‐protein activation was analyzed by a standard dose‐response curve using GraphPad Prism 8.0 (GraphPad Software). EC50 and pEC50± SEM were calculated using nonlinear regression (curve fit). Data are means ± SEM from at least three independent experiments performed in technical triplicates.
Enzyme Linked Immunosorbent (ELISA) assay
To confirm the cell surface expression of H3R and its mutants, the ELISA was performed 24 h after transfection using cells plated on 96‐white plates. The cells were fixed with 4% formaldehyde for five minutes at RT and then washed once with PBS. Following fixation, the cells were blocked with blocking buffer (1% FBS in PBS) for 2 h at RT. Afterwards, the plates were incubated with a 1:10000 dilution of monoclonal ANTI‐FLAG M2‐Peroxidase (HRP) antibody (Sigma Aldrich) in blocking buffer for another 0.5 h at RT. After careful washing, 80 µL per well of SuperSignal ELISA Femto Maximum Sensitivity Substrate (ThermoFisher Scientific) solution was added. The luminance signal was measured using 500 ms intervals. The data are presented as means ± SEM from at least three independent experiments performed in technical triplicates.
Collections of Activated GPCR Data Sets
For the true GPCR datasets, structures were collected from reported distinct class A GPCR–G‐protein complex structures that are primarily coupled to Gs‐ or Gi/o‐proteins. Several Gs‐ or Gi/o‐coupled receptors that are primarily coupled to Gq protein, such as CCK1R, CCK2R, GASR, NK1R, and NTSR1, were excluded. The true datasets were enlarged to 20 Gs‐ and 34 Gi/o‐coupled structures by adding the four Gs‐coupled and one Gi‐coupled complexes from our unpublished data.
For the GPCRdb homology model datasets, the primary Gs‐ or Gi/o‐coupled activated class A GPCR homology models were collected from GPCRdb. However, the accuracy of orphan class A receptors homology models is poor due to the lack of true homologous structures. After removing the orphan receptors, 98 (24 Gs‐ and 74 Gi/o‐coupled receptors) GPCRdb homology models were used as training datasets.
Extractions of Length and Angle
To conveniently extract the length and angle geometries of receptors, the reference H3R model was positioned on the lipid bilayer using the PPM server. The 7TMs of H3R were perpendicular to the membrane plane. Then, the structures of true GPCR dataset and GPCRdb homology model dataset were superimposed onto H3R. The Z‐axis of all receptor models was perpendicular to the membrane plane. The lengths of TM5 and TM6 were measured from the cytosolic tip to 5.50 and 6.50, respectively. The angle of TM5 and TM6 was calculated between the orientation of a helix and the reference direction. The orientation of a helix was obtained by averaging the C = O bond orientation of each amino acid on the helix segment. Considering that an alpha helix turn consists of 3.6 amino acids, we selected 11 amino acids from the cytoplasmic tip of TM5 or TM6 to calculate the average orientation and eliminate the component perpendicular to the helix axis. The reference orientation for TM5 angle calculation is the TM5 orientation of H3R, in which the average C = O bond orientation is 22 amino acids instead of 11. As the TM6 angle indicates the TM6 outward movement during receptor activation, the reference orientation for the TM6 angle is the central axis of the receptors, which is the Z‐axis that is perpendicular to the membrane plane.
Data Pre‐Processing and Machine Learning Classification
We utilize the Sklearn package[ 41 ] to perform the data pre‐processing and machine learning tasks. First, the TM6 length is excluded due to the statistical reason previously stated, followed by standardizing, namely, centering and scaling the three‐geometries dataset. From above, we construct a training set of (3,n) dimensions where n = 98 (Gs:24, Gi:74) is the number of data points in the training set.
For the cross‐validation, the stratified 10‐fold validation is used on the GPCRdb set. After that, we train the Random Forest classifier on the whole training set with the parameters as follows (unspecified parameters set as default): RandomForestClassifier(max_depth = 1, n_estimators = 15, max_features = 1, class_weight = {0:74, 1:24}), in which the bagging strategy is used with sample ratio as 0.7. Note that the standardization fit with the training dataset is applied to the true dataset as well. The best model is trained with random seed as 2784 for both bagging and Random Forest.
Subsequently, we apply PCA to fit the training dataset and get a PCA transformation for visualization. The technique of linear dimensionality reduction, known as Principal Component Analysis (PCA), employs the Singular Value Decomposition method to project the data into a subspace of reduced dimensions. We use the PCA implementation in sklearn.decomposition and it can be used with the default parameters. The first two principal components are reserved, which explain over 87% variance of the data.
The resultant principle can be distilled from the decision boundary for approximate categorization. The comprehensive transformation equation transitioning from the raw domain to Principal Component (PC) space is as follows:
X = 0.649*1/4.164*(tm5_len – 20.276) + 0.569*1/5.264*(tm5_tilt – 8.474) + 0.506*1/9.156*(tm6_outward‐ 18.749)
Y = −0.081*1/4.164*(tm5_len – 20.276) + −0.609*1/5.264*(tm5_tilt – 8.474) + 0.789*1/9.156*(tm6_outward‐ 18.749)
Upon abstracting standardization, we obtain:
X = 0.649*tm5_len_std+0.569*tm5_tilt _std+0.506*tm6_outward_std,
Y = 0.081*tm5_len_std+ 0.609*tm5_tilt _std −0.789*tm6_outward_std
Herein, [0.649 0.569 0.506] and [−0.081 −0.609 0.789] are the corresponding eigenvectors for PC1 and PC2.
The following formula facilitates the direct calculation of the corresponding values:
X = 0.156*tm5_len + 0.108*tm5_tilt + 0.055*tm6_outward – 5.110
Y = −0.020*tm5_len – 0.116*tm5_tilt + 0.086*tm6_outward – 0.239
When X>0.7, it is highly probable that GPCR will couple with Gs, indicative of an elongated TM length, a substantial TM tilt, and a significant outward shift in TM6 orientation. Conversely when X falls below or equals 0.7, there's an increased likelihood of GPCR coupling with Gi which comprises instances that are either Y.
Statistical Analysis
Data were represented as the mean ± SEM values of ≥3 independent experiments. The raw data of the NanoBiT assay was standardized by the baseline and the vehicle group, while the raw data of cAMP and ELISA assay was standardized by the corresponding WT group. The sigmoidal curves of the reporter luciferase assay were analyzed using a standard dose‐response curve in GraphPad Prism version 8.02 (GraphPad Software, San Diego, CA, USA). Data sets with two groups were analyzed by Student's t‐tests. Data with ≥3 groups were analyzed by one‐way analysis of variance (ANOVA), while the violin plots depicting four distinct features of TM5/TM6 in Gs‐ and Gi/o‐coupled receptors are calculated using Mann‐Whitney U test taking advantage of scikit learn python package. A significance level of p < 0.05 was considered, and the details are provided in the figure legends.
Conflict of Interest
The authors declare no conflict of interest.
Author Contributions
Q.S., X.T., X.W., and S.C. contributed equally to this work. Y.Z. initiated and conceived the study. Y.Z., Z.C., J.‐P.S., W.H., and M.Z. supervised the project. Q.S. designed the expression constructs, optimized the sample purification, and prepared the final samples for cryo‐EM studies for the H3R‐Gi complexes. X.W., P.X., and J.‐P.S. designed the expression constructs and prepared the final samples for cryo‐EM studies for the H2R‐Gs complex. C.M., D.‐D.S., and H.Z. evaluated the specimen by negative‐stain EM, screened the cryo‐EM conditions, prepared the cryo‐EM grids, and collected cryo‐EM images. Q.S. performed density map calculations, model building, refinement of final models. Y.Z. and Q.S. analyzed the structure, designed the mutations, and functional assays. X.T., X.W., P.X., H.X., L.J., and Y.Z. performed the functional assays. M.Z., Y.Z., S.C., and Q.S. performed machine learning analyses. Q.S. and S.‐K.Z. performed molecular dynamics simulations. Q.S., X.T., S.C., and X.W. prepared the figures. Y.Z. and Q.S. wrote the manuscript. Z.C., J.‐P.S., W.H., and M.Z. provided important discussions and essential revisions.
Supporting information
Supporting Information
Acknowledgements
The authors thank the Cryo‐Electron Microscopy Center of Liangzhu laboratory for the cryo‐EM data collection. Cryo‐EM specimens were examined with the help of Shenghai Chang at the Center of Cryo‐Electron Microscopy, Zhejiang University. Sample preparation for cryo‐EM studies was supported by Cheng Ma from the Protein Facility, Zhejiang University School of Medicine. This project was partially supported by the Ministry of Science and Technology (2019YFA050880 to Y.Z.), the National Natural Science Foundation of China (92353303 and 32141004 to Y.Z., 32000845 to Q.S., U21A20418 to Z.C., 81973302 to W.H., and 62202426 to M.Z.), National Science Fund for Distinguished Young Scholars Grant (81825022 to J.‐P.S.), STI2030‐Major Projects (2022ZD0205400 to Q.S.), the “Pioneer” and “Leading Goose” R&D Program of Zhejiang (2024C03147 to Y.Z.), the Leading Innovative and Entrepreneur Team Introduction Program of Zhejiang (2020R01006 to Y.Z.), the Key R&D Projects of Zhejiang Province (2021C03039 to Y.Z.), and the Fundamental Research Funds for the Central Universities (Y.Z.).
Shen Q., Tang X., Wen X., Cheng S., Xiao P., Zang S.‐K., Shen D.‐D., Jiang L., Zheng Y., Zhang H., Xu H., Mao C., Zhang M., Hu W., Sun J.‐P., Zhang Y., Chen Z., Molecular Determinant Underlying Selective Coupling of Primary G‐Protein by Class A GPCRs. Adv. Sci. 2024, 11, 2310120. 10.1002/advs.202310120
Contributor Information
Min Zhang, Email: min_zhang@zju.edu.cn.
Weiwei Hu, Email: huww@zju.edu.cn.
Jin‐Peng Sun, Email: sunjinpeng@sdu.edu.cn.
Yan Zhang, Email: zhang_yan@zju.edu.cn.
Zhong Chen, Email: chenzhong@zju.edu.cn.
Data Availability Statement
The Electron Microscopy Data Bank accession numbers and Protein Data Bank identifiers are EMD‐39582 and PDB ID 8YUT for the amthamine‐H2R‐Gs complex, EMD‐39583 and PDB ID 8YUU for the histamine‐H3R‐Gi complex, and EMD‐39584 and PDB ID 8YUV for the immepip‐H3R‐Gi complex, respectively. Other data that support the findings of this study are available in the supplementary material of this article.
References
- 1.a) Bjarnadottir T. K., Gloriam D. E., Hellstrand S. H., Kristiansson H., Fredriksson R., Schioth H. B., Genomics 2006, 88, 263; [DOI] [PubMed] [Google Scholar]; b) Flock T., Hauser A. S., Lund N., Gloriam D. E., Balaji S., Babu M. M., Nature 2017, 545, 317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Hauser A. S., Avet C., Normand C., Mancini A., Inoue A., Bouvier M., Gloriam D. E., Elife 2022, 11, 74107; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Neves S. R., Ram P. T., Iyengar R., Science 2002, 296, 1636. [DOI] [PubMed] [Google Scholar]
- 3. Wacker D., Stevens R. C., Roth B. L., Cell 2017, 170, 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wisler J. W., Rockman H. A., Lefkowitz R. J., Circulation 2018, 137, 2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Inoue A., Raimondi F., Kadji F. M. N., Singh G., Kishi T., Uwamizu A., Ono Y., Shinjo Y., Ishida S., Arang N., Kawakami K., Gutkind J. S., Aoki J., Russell R. B., Cell 2019, 177, 1933; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sandhu M., Cho A., Ma N., Mukhaleva E., Namkung Y., Lee S., Ghosh S., Lee J. H., Gloriam D. E., Laporte S. A., Babu M. M., Vaidehi N., Nat. Commun. 2022, 13, 7428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Du Y., Duc N. M., Rasmussen S. G. F., Hilger D., Kubiak X., Wang L., Bohon J., Kim H. R., Wegrecki M., Asuru A., Jeong K. M., Lee J., Chance M. R., Lodowski D. T., Kobilka B. K., Chung K. Y., Cell 2019, 177, 1232; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Anantakrishnan S., Naganathan A. N., Nat. Commun. 2023, 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Avet C., Mancini A., Breton B., Gouill C. Le, Hauser A. S., Normand C., Kobayashi H., Gross F., Hogue M., Lukasheva V., St‐Onge S., Carrier M., Heroux M., Morissette S., Fauman E. B., Fortin J. P., Schann S., Leroy X., Gloriam D. E., Bouvier M., Elife 2022, 11, 74101; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Harding S. D., Armstrong J. F., Faccenda E., Southan C., Alexander S. P. H., Davenport A. P., Pawson A. J., Spedding M., Davies J. A., Nc I., Nucleic Acids Res. 2022, 50, D1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Latorraca N. R., Venkatakrishnan A. J., Dror R. O., Chem. Rev. 2017, 117, 139. [DOI] [PubMed] [Google Scholar]
- 9.a) Zhang Y., Sun B., Feng D., Hu H., Chu M., Qu Q., Tarrasch J. T., Li S., Sun Kobilka T., Kobilka B. K., Skiniotis G., Nature 2017, 546, 248; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liang Y. L., Khoshouei M., Radjainia M., Zhang Y., Glukhova A., Tarrasch J., Thal D. M., Furness S. G. B., Christopoulos G., Coudrat T., Danev R., Baumeister W., Miller L. J., Christopoulos A., Kobilka B. K., Wootten D., Skiniotis G., Sexton P. M., Nature 2017, 546, 118; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Glukhova A., Draper‐Joyce C. J., Sunahara R. K., Christopoulos A., Wootten D., Sexton P. M., ACS Pharmacol. Transl. Sci. 2018, 1, 73; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Garcia‐Nafria J., Tate C. G., Mol. Cell. Endocrinol. 2019, 488, 1. [DOI] [PubMed] [Google Scholar]
- 10.a) Mao C., Xiao P., Tao X. N., Qin J., He Q. T., Zhang C., Guo S. C., Du Y. Q., Chen L. N., Shen D. D., Yang Z. S., Zhang H. Q., Huang S. M., He Y. H., Cheng J., Zhong Y. N., Shang P., Chen J., Zhang D. L., Wang Q. L., Liu M. X., Li G. Y., Guo Y., Xu H. E., Wang C., Zhang C., Feng S., Yu X., Zhang Y., Sun J. P., Science 2023, 380, eadd6220; [DOI] [PubMed] [Google Scholar]; b) Ji Y., Duan J., Yuan Q., He X., Yang G., Zhu S., Wu K., Hu W., Gao T., Cheng X., Jiang H., Xu H. E., Jiang Y., Nat. Commun. 2023, 14, 1268; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Liu Q., Yang D., Zhuang Y., Croll T. I., Cai X., Dai A., He X., Duan J., Yin W., Ye C., Zhou F., Wu B., Zhao Q., Xu H. E., Wang M. W., Jiang Y., Nat. Chem. Biol. 2021, 17, 1238; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Alegre K. O., Paknejad N., Su M., Lou J. S., Huang J., Jordan K. D., Eng E. T., Meyerson J. R., Hite R. K., Huang X. Y., Nat. Struct. Mol. Biol. 2021, 28, 936; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Zhang X., He C., Wang M., Zhou Q., Yang D., Zhu Y., Feng W., Zhang H., Dai A., Chu X., Wang J., Yang Z., Jiang Y., Sensfuss U., Tan Q., Han S., Reedtz‐Runge S., Xu H. E., Zhao S., Wang M. W., Wu B., Zhao Q., Nat. Chem. Biol. 2021, 17, 1230. [DOI] [PubMed] [Google Scholar]
- 11. Huang S., Xu P., Shen D. D., Simon I. A., Mao C., Tan Y., Zhang H., Harpsøe K., Li H., Zhang Y., You C., Yu X., Jiang Y., Zhang Y., Gloriam D. E., Xu H. E., Mol. Cell 2022, 82, 2681. [DOI] [PubMed] [Google Scholar]
- 12. Seyedabadi M., Gharghabi M., Gurevich E. V., Gurevich V. V., Trends Biochem. Sci. 2022, 47, 570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Matic M., Miglionico P., Tatsumi M., Inoue A., Raimondi F., Nat. Commun. 2023, 14, 4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Sadler F., Ma N., Ritt M., Sharma Y., Vaidehi N., Sivaramakrishnan S., Nature 2023, 615, 734; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Teng X., Chen S., Wang Q., Chen Z., Wang X., Huang N., Zheng S., Sci. Adv. 2022, 8, eabo4158; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Yang F., Mao C., Guo L., Lin J., Ming Q., Xiao P., Wu X., Shen Q., Guo S., Shen D. D., Lu R., Zhang L., Huang S., Ping Y., Zhang C., Ma C., Zhang K., Liang X., Shen Y., Nan F., Yi F., Luca V. C., Zhou J., Jiang C., Sun J. P., Xie X., Yu X., Zhang Y., Nature 2020, 587, 499; [DOI] [PubMed] [Google Scholar]; d) Kumar K. K., Shalev‐Benami M., Robertson M. J., Hu H., Banister S. D., Hollingsworth S. A., Latorraca N. R., Kato H. E., Hilger D., Maeda S., Weis W. I., Farrens D. L., Dror R. O., Malhotra S. V., Kobilka B. K., Skiniotis G., Cell 2019, 176, 448; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Koehl A., Hu H., Maeda S., Zhang Y., Qu Q., Paggi J. M., Latorraca N. R., Hilger D., Dawson R., Matile H., Schertler G. F. X., Granier S., Weis W. I., Dror R. O., Manglik A., Skiniotis G., Kobilka B. K., Nature 2018, 558, 547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Hattori Y., Hattori K., Matsuda N., Handb Exp Pharmacol 2017, 241, 239; [DOI] [PubMed] [Google Scholar]; b) Haas H. L., Sergeeva O. A., Selbach O., Physiol. Rev. 2008, 88, 1183. [DOI] [PubMed] [Google Scholar]
- 16. Obara I., Telezhkin V., Alrashdi I., Chazot P. L., Br. J. Pharmacol. 2020, 177, 580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Simons F. E., N. Engl. J. Med. 2004, 351, 2203. [DOI] [PubMed] [Google Scholar]
- 18. Jiang L., Cheng L., Chen H., Dai H., An D., Ma Q., Zheng Y., Zhang X., Hu W., Chen Z., J. Exp. Med. 2021, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.a) Dauvilliers Y., Bassetti C., Lammers G. J., Arnulf I., Mayer G., Rodenbeck A., Lehert P., Ding C. L., Lecomte J. M., Schwartz J. C., group H. I. s., Lancet Neurol. 2013, 12, 1068; [DOI] [PubMed] [Google Scholar]; b) Lin J. S., Dauvilliers Y., Arnulf I., Bastuji H., Anaclet C., Parmentier R., Kocher L., Yanagisawa M., Lehert P., Ligneau X., Perrin D., Robert P., Roux M., Lecomte J. M., Schwartz J. C., Neurobiol Dis 2008, 30, 74. [DOI] [PubMed] [Google Scholar]
- 20.a) Xia R., Wang N., Xu Z., Lu Y., Song J., Zhang A., Guo C., He Y., Nat. Commun. 2021, 12, 2086; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Peng X., Yang L., Liu Z., Lou S., Mei S., Li M., Chen Z., Zhang H., Nat. Commun. 2022, 13, 6105; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Robertson M. J., Papasergi‐Scott M. M., He F., Seven A. B., Meyerowitz J. G., Panova O., Peroto M. C., Che T., Skiniotis G., Nat. Struct. Mol. Biol. 2022, 29, 1188; [DOI] [PubMed] [Google Scholar]; d) Shimamura T., Shiroishi M., Weyand S., Tsujimoto H., Winter G., Katritch V., Abagyan R., Cherezov V., Liu W., Han G. W., Kobayashi T., Stevens R. C., Iwata S., Nature 2011, 475, 65; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Im D., Kishikawa J. I., Shiimura Y., Hisano H., Ito A., Fujita‐Fujiharu Y., Sugita Y., Noda T., Kato T., Asada H., Iwata S., Nat. Commun. 2023, 14, 6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rasmussen S. G., DeVree B. T., Zou Y., Kruse A. C., Chung K. Y., Kobilka T. S., Thian F. S., Chae P. S., Pardon E., Calinski D., Mathiesen J. M., Shah S. T., Lyons J. A., Caffrey M., Gellman S. H., Steyaert J., Skiniotis G., Weis W. I., Sunahara R. K., Kobilka B. K., Nature 2011, 477, 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Duan J., Shen D. D., Zhou X. E., Bi P., Liu Q. F., Tan Y. X., Zhuang Y. W., Zhang H. B., Xu P. Y., Huang S. J., Ma S. S., He X. H., Melcher K., Zhang Y., Xu H. E., Jiang Y., Nat. Commun. 2020, 11, 4121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maeda S., Koehl A., Matile H., Hu H., Hilger D., Schertler G. F. X., Manglik A., Skiniotis G., Dawson R. J. P., Kobilka B. K., Nat. Commun. 2018, 9, 3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ballesteros J. A., Weinstein H., in Methods in Neurosciences, Vol.25, Elsevier, Amsterdam, Netherlands 1995, pp. 366–428.
- 25.a) Xu P., Huang S., Zhang H., Mao C., Zhou X. E., Cheng X., Simon I. A., Shen D. D., Yen H. Y., Robinson C. V., Harpsoe K., Svensson B., Guo J., Jiang H., Gloriam D. E., Melcher K., Jiang Y., Zhang Y., Xu H. E., Nature 2021, 592, 469; [DOI] [PubMed] [Google Scholar]; b) Yuan D., Liu Z., Kaindl J., Maeda S., Zhao J., Sun X., Xu J., Gmeiner P., Wang H. W., Kobilka B. K., Nat. Chem. Biol. 2020, 16, 507; [DOI] [PubMed] [Google Scholar]; c) Xiao P., Yan W., Gou L., Zhong Y. N., Kong L., Wu C., Wen X., Yuan Y., Cao S., Qu C., Yang X., Yang C. C., Xia A., Hu Z., Zhang Q., He Y. H., Zhang D. L., Zhang C., Hou G. H., Liu H., Zhu L., Fu P., Yang S., Rosenbaum D. M., Sun J. P., Du Y., Zhang L., Yu X., Shao Z., Cell 2021, 184, 943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xu X., Kaindl J., Clark M. J., Hubner H., Hirata K., Sunahara R. K., Gmeiner P., Kobilka B. K., Liu X., Cell Res. 2021, 31, 569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Flock T., Ravarani C. N. J., Sun D., Venkatakrishnan A. J., Kayikci M., Tate C. G., Veprintsev D. B., Babu M. M., Nature 2015, 524, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.a) Lin X., Li M., Wang N., Wu Y., Luo Z., Guo S., Han G. W., Li S., Yue Y., Wei X., Xie X., Chen Y., Zhao S., Wu J., Lei M., Xu F., Nature 2020, 579, 152; [DOI] [PubMed] [Google Scholar]; b) Zhang H., Chen L. N., Yang D., Mao C., Shen Q., Feng W., Shen D. D., Dai A., Xie S., Zhou Y., Qin J., Sun J. P., Scharf D. H., Hou T., Zhou T., Wang M. W., Zhang Y., Cell Res. 2021, 31, 1163; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Ma S., Chen Y., Dai A., Yin W., Guo J., Yang D., Zhou F., Jiang Y., Wang M. W., Xu H. E., Cell Res. 2021, 31, 1061; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhuang Y., Xu P., Mao C., Wang L., Krumm B., Zhou X. E., Huang S., Liu H., Cheng X., Huang X. P., Shen D. D., Xu T., Liu Y. F., Wang Y., Guo J., Jiang Y., Jiang H., Melcher K., Roth B. L., Zhang Y., Zhang C., Xu H. E., Cell 2021, 184, 931; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Su M., Zhu L., Zhang Y., Paknejad N., Dey R., Huang J., Lee M. Y., Williams D., Jordan K. D., Eng E. T., Ernst O. P., Meyerson J. R., Hite R. K., Walz T., Liu W., Huang X. Y., Mol. Cell 2020, 80, 59; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Yang F., Ling S., Zhou Y., Zhang Y., Lv P., Liu S., Fang W., Sun W., Hu L. A., Zhang L., Shi P., Tian C., National Science Review 2020, 8; [Google Scholar]; g) Nagiri C., Kobayashi K., Tomita A., Kato M., Kobayashi K., Yamashita K., Nishizawa T., Inoue A., Shihoya W., Nureki O., Mol. Cell 2021, 81, 3205. [DOI] [PubMed] [Google Scholar]
- 29.a) Zhao C., Cheng L., Wang W., Wang H., Luo Y., Feng Y., Wang X., Fu H., Cai Y., Yang S., Fu P., Yan W., Shao Z., Cell Res. 2022, 32, 218; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Garcia‐Nafria J., Nehme R., Edwards P. C., Tate C. G., Nature 2018, 558, 620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.a) Okamoto H. H., Miyauchi H., Inoue A., Raimondi F., Tsujimoto H., Kusakizako T., Shihoya W., Yamashita K., Suno R., Nomura N., Kobayashi T., Iwata S., Nishizawa T., Nureki O., Nat. Struct. Mol. Biol. 2021, 28, 694; [DOI] [PubMed] [Google Scholar]; b) Maeda S., Qu Q., Robertson M. J., Skiniotis G., Kobilka B. K., Science 2019, 364, 552; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Huang S., Xu P., Tan Y., You C., Zhang Y., Jiang Y., Xu H. E., Cell Res. 2021, 31, 1036; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Shao Z., Shen Q., Yao B., Mao C., Chen L. N., Zhang H., Shen D. D., Zhang C., Li W., Du X., Li F., Ma H., Chen Z. H., Xu H. E., Ying S., Zhang Y., Shen H., Nat. Chem. Biol. 2022, 18, 264; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Shao Z., Tan Y., Shen Q., Hou L., Yao B., Qin J., Xu P., Mao C., Chen L. N., Zhang H., Shen D. D., Zhang C., Li W., Du X., Li F., Chen Z. H., Jiang Y., Xu H. E., Ying S., Ma H., Zhang Y., Shen H., Cell Discov. 2022, 8, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pandy‐Szekeres G., Caroli J., Mamyrbekov A., Kermani A. A., Keseru G. M., Kooistra A. J., Gloriam D. E., Nucleic Acids Res. 2023, 51, D395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wold S., Esbensen K., Geladi P., Chemom. Intell. Lab. Syst. 1987, 2, 37. [Google Scholar]
- 33. Kam H. T., in Proceedings of 3rd International Conference on Document Analysis and Recognition, 1995, 1, pp. 278–282. [Google Scholar]
- 34. Jensen N. H., Rodriguiz R. M., Caron M. G., Wetsel W. C., Rothman R. B., Roth B. L., Neuropsychopharmacology 2008, 33, 2303. [DOI] [PubMed] [Google Scholar]
- 35.a) Ping Y. Q., Mao C., Xiao P., Zhao R. J., Jiang Y., Yang Z., An W. T., Shen D. D., Yang F., Zhang H., Qu C., Shen Q., Tian C., Li Z. J., Li S., Wang G. Y., Tao X., Wen X., Zhong Y. N., Yang J., Yi F., Yu X., Xu H. E., Zhang Y., Sun J. P., Nature 2021, 589, 620; [DOI] [PubMed] [Google Scholar]; b) Qi X., Friedberg L., De Bose‐Boyd R., Long T., Li X., Nat. Chem. Biol. 2020, 16, 1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liang Y. L., Zhao P., Draper‐Joyce C., Baltos J. A., Glukhova A., Truong T. T., May L. T., Christopoulos A., Wootten D., Sexton P. M., Furness S. G. B., ACS Pharmacol. Transl. Sci. 2018, 1, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zheng S. Q., Palovcak E., Armache J. P., Verba K. A., Cheng Y., Agard D. A., Nat. Methods 2017, 14, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang K., J. Struct. Biol. 2016, 193, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lomize M. A., Pogozheva I. D., Joo H., Mosberg H. I., Lomize A. L., Nucleic Acids Res. 2012, 40, D370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lee J., Cheng X., Swails J. M., Yeom M. S., Eastman P. K., Lemkul J. A., Wei S., Buckner J., Jeong J. C., Qi Y., Jo S., Pande V. S., Case D. A., Brooks C. L. 3rd, MacKerell A. D. Jr., Klauda J. B., Im W., J. Chem. Theory Comput. 2016, 12, 405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V. J. t. J. o. m. L. r., 2011, 12, 2825. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information
Data Availability Statement
The Electron Microscopy Data Bank accession numbers and Protein Data Bank identifiers are EMD‐39582 and PDB ID 8YUT for the amthamine‐H2R‐Gs complex, EMD‐39583 and PDB ID 8YUU for the histamine‐H3R‐Gi complex, and EMD‐39584 and PDB ID 8YUV for the immepip‐H3R‐Gi complex, respectively. Other data that support the findings of this study are available in the supplementary material of this article.