

Abstract
Objective
Fabry disease is caused by enzymatic defects in alpha‐galactosidase A that leads to the accumulation of glycosphingolipids throughout the body, resulting in a multisystemic disorder. The most common neurological manifestations are neuropathic pain, autonomic nervous system dysfunction and strokes, but some rarer neurological manifestations exist. Among these, aseptic meningitis is a possible complication. Our objectives were to measure the prevalence of this complication in a cohort of patients with Fabry disease, and to describe its clinical features.
Methods
We conducted a retrospective review of Fabry disease patients followed at our tertiary referral center between 1995 and September 2023 with at least one episode of meningitis, and performed a systematic review to identify similar published cases.
Results
Four patients out of 107 (3.7%) had at least one episode of aseptic meningitis. Our systematic review identified 25 other observations. The median age of these 29 patients was 29.0 years, the median cerebrospinal fluid leukocyte count was 24 cells/mm3 with a predominance of lymphocytes in 64.7% of cases. In 82.8% of the patients, the diagnosis of Fabry disease was unknown before the meningitis. Large artery stenosis was present in 17.2% of patients and 57.1% of patients had a recent stroke concomitant with the meningitis. Several differential diagnoses were evoked, such as multiple sclerosis or central nervous system vasculitis.
Interpretation
Our study suggests that Fabry disease should be considered as a cause of aseptic meningitis. The pathophysiological mechanisms underlying meningeal inflammation remain largely unknown but may reflect the dysregulation of pro‐inflammatory signaling pathways.
Introduction
Fabry disease (FD, OMIM #301 500) is a lysosomal storage disorder caused by an X‐linked inborn error of glycosphingolipid catabolism, resulting in a deficient enzyme activity of the alpha‐galactosidase A (aGalA) encoded by the GLA gene. More than 1000 mutations have been identified. 1 The lack of enzyme activity leads to the accumulation of glycosphingolipids, mainly of globotriaosylceramide (Gb3) and globotriaosylsphingosine (lyso‐Gb3), in different tissues.
Two phenotypes have been described depending on the residual enzymatic activity. The historical classic FD is characterized by the lowest aGalA activity leading to multivisceral deposits of Gb3, resulting in severe disease with various organ involvement: progressive renal failure with proteinuria, hypertrophic cardiomyopathy (HCM), angiokeratoma, cornea verticillata, cochleo‐vestibular involvement, and neurological complications such as autonomic and peripheral neuropathy, leading to acroparesthesia and hypohidrosis, and the early occurrence of strokes. The non‐classic FD is associated with higher aGalA activity and characterized by an almost exclusive HCM.
Manifestations occur not only in males but also in heterozygous females. Due to the X‐inactivation and the subsequent genetic mosaicism, organ involvement in female is difficult to predict with frequent incomplete phenotype. The same elevated frequency of ischemic cerebrovascular events has been observed in both sexes. 2 Some rarer neurological symptoms may occur in FD, including neuroinflammatory conditions. Knowledge of these atypical presentations is important for early diagnosis and for the understanding of pathophysiological mechanisms. We report here four cases of aseptic meningitis in FD and propose a systematic review of this rare condition.
Patients and Methods
Patients
We performed a retrospective review of the medical records of FD patients referred to our tertiary center in Paris, France, from 1995 to September 2023 with aseptic meningitis characterized by >5 leukocytes/mm3 in cerebrospinal fluid (CSF) with no evidence of infectious origin.
FD was defined in case of alpha‐galactosidase (aGalA) deficiency in men or known pathogenic variant of the GLA gene in women as widely accepted. 3
All patients or relatives gave their informed consent, the study was in accordance with the Helsinki declaration and French legislation.
Systematic review
We followed good practice guidelines from Cochrane Handbook. 4 This systematic review is reported in accordance with the Preferred Reporting Items for Systematic reviews and Meta‐Analyses (PRISMA) statement. 5
Our main objective was to identify other cases of aseptic meningitis associated with FD in the literature, and to determine their clinical, biological, and radiological characteristics.
The following electronic databases were searched on October 2023: MEDLINE, Embase, and Cochrane library. A dedicated search algorithm using keywords and free‐text words was developed for each database (Appendices S1–S3). We also searched the abstracts of the main international and French congresses in the field of metabolic and lysosomal diseases, neurology, and internal medicine, whose detailed list is available in Appendix S4.
We included all case reports, cases series and observational prospective or retrospective studies (cohort and case control studies) from 1970 to September 2023. We excluded articles mentioning meningitis without CSF cell count information. We excluded articles mentioning the diagnosis of meningitis without specifying the number of leukocytes in CSF.
We managed records with Zotero v5.0.
CM identified eligible studies by screening titles and abstracts and then read the full texts. In case of doubts, WM (expert in FD) read the full texts as second reviewer.
Standardized data extraction form was used. In case of missing data, we attempted to contact the authors and exclude data from analysis.
Data synthesis: Main analyses
The planned analyses were calculation of median age of patients, interquartile ranges (IQR), sex ratio, median CSF leukocytes count, prevalence of strokes, white‐matter lesions (WMLs), dolichoectasia, and macrovascular involvement.
We classified patients regarding the duration of meningitis (“acute meningitis” in case of single episode of meningitis, either because a control lumbar puncture is normal, or because there has been no control of lumbar puncture; “recurrent meningitis” in case of normalization of CSF parameters between two episodes of authenticated meningitis with >5 cells/mm3 in the CSF; “chronic meningitis” in case of persistence of cells >5/mm3 in the CSF for more than 1 month) and the presence, absence, or association of clinical meningeal syndrome (headache and stiff neck) and focal neurologic deficits. We thus obtain six theoretical groups of patients with homogeneous symptomatology.
Results
Description of four patients from our reference center for FD
Among 107 patients with FD followed in our center between January 1995 and September 2023, four had at least one episode of aseptic meningitis (prevalence 3.7%).
Patient 1
This female patient was first seen in 2004 at the age of 25 years for recurrent headaches with normal cerebral MRI and lymphocytic meningitis (30–70 cells/mm3; protein level up to 0.81 g/L) and elevated C‐reactive protein (CRP; 5–20 mg/L). After a 6 months empirical antituberculous treatment, MRI showed ischemic lesions in both thalami with persistent lymphocytic meningitis. Early ischemic stroke associated with acroparesthesia in childhood and renal failure in the father evoked FD that was confirmed with a pathogenic variant of the GLA gene (c.123delC).
Aspirin and enzyme replacement therapy (ERT) with agalsidase alfa (0.2 mg/kg/14 d) were introduced in 2005. After 9 months of ERT, headaches, and CSF pleocytosis remained. The patient presented intracranial hypertension with optic nerve sheaths ectasia and sellar arachnoidocele on MRI (Fig. 1A,B), which was considered secondary to the chronic meningitis. High‐dose of corticosteroids and acetazolamide were introduced, pleocytosis decreased suggesting the coexistence of an inflammatory condition leading to the introduction of azathioprine in 2006 (Fig. 2).
Figure 1.
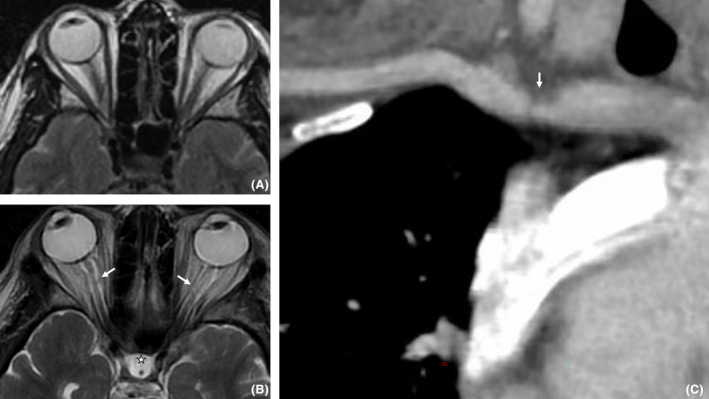
Orbital MRI axial T2W in 2008 (A) and 2020 (B) showed appearance of an enlargement of the optic nerve sheath (white arrows) and a sellar arachnoidocele (star). Curvilinear reformat of CT angiography on supra‐aortic arteries (C) showed a short stenosis of the right subclavian artery (arrow).
Figure 2.
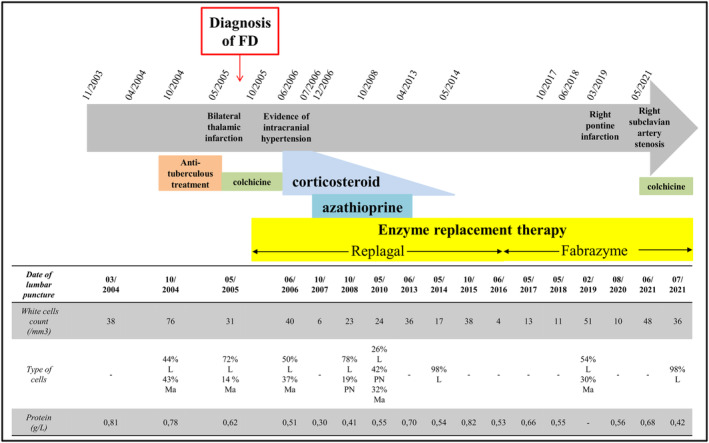
Evolution of symptoms and CSF features for Patient 1, and description of successive treatment lines. L is for lymphocytes; Ma is for macrophages; PN is for neutrophil.
Headaches, CSF pleocytosis, and inflammatory syndrome relapsed with cortidependance. Azathioprine was stopped in 2013 in front of unobvious clinical benefits and cytopenia, and corticosteroids in 2014 because of steroid‐induced diabete. FD‐specific treatment was switched to agalsidase beta 1 mg/kg/14 d in 2016 without any clear benefit. In 2019, an episode of confusion revealed a new right pontine ischemic stroke.
In 2021, a symptomatic stenosis of the right subclavian artery was discovered (Fig. 1C), 18‐fluorodeoxyglucose positron emission tomography (PET) scan and pan‐aortic angioscanner were normal. MFEV gene, HLA‐B51 alleles analyses, then an extensive screening by next‐generation sequencing (NGS, using NextSeq 500 Illumina) for known mutations responsible for auto‐inflammatory syndromes (including MVK, NOD2, NRAS, PSTPIP1, and TRAP1) were negative. Colchicine was tried unsuccessfully. Headaches finally improved with venlafaxine and removal of all painkillers in the hypothesis of medication overuse headache. Asymptomatic meningitis persists on the last CSF analysis performed in July 2021.
Patient 2
A 50‐year‐old female patient was referred to a tertiary neurology center in 2016 for fever, headache and diplopia. CRP was elevated up to 80 mg/L. MRI showed multiple lacunar strokes, leptomeningeal enhancement, and intramural vascular enhancement. Cerebral angiography revealed short stenosis of multiple arteries of the posterior fossa (Fig. 3). CSF analysis revealed lymphocytic meningitis with 250 cells/mm3 (89% of lymphocytes). Diagnosis of idiopathic cerebral vasculitis of the central nervous system (CNS) was made.
Figure 3.
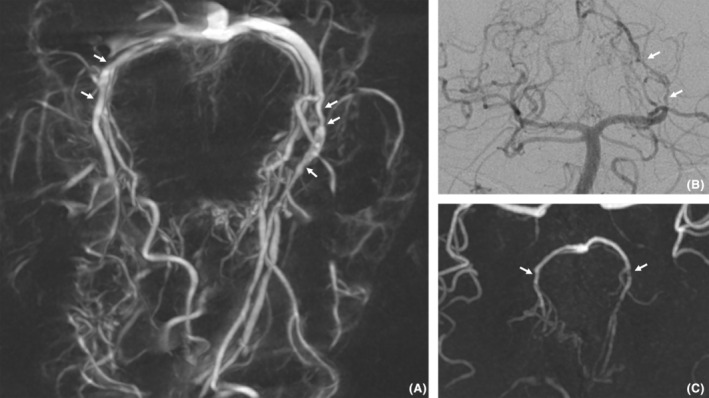
Angiography (A and B) and time‐of‐flight MRI (C) images of Patient 2. Multiple short stenoses (white arrows) of large and medium caliber arteries (posterior cerebral arteries and perforators) are seen here. The association with multiple ischemic events and the progressive nature of the stenoses suggested a diagnosis of vasculitis.
The patient received corticosteroid with cyclophosphamide. After six courses of cyclophosphamide (600 mg/m2), fever disappeared, but headache and diplopia persisted, with persistent meningitis on lumbar puncture. Brain MRI showed new ischemic lesions. Cyclophosphamide was switched to rituximab (Fig. 4).
Figure 4.
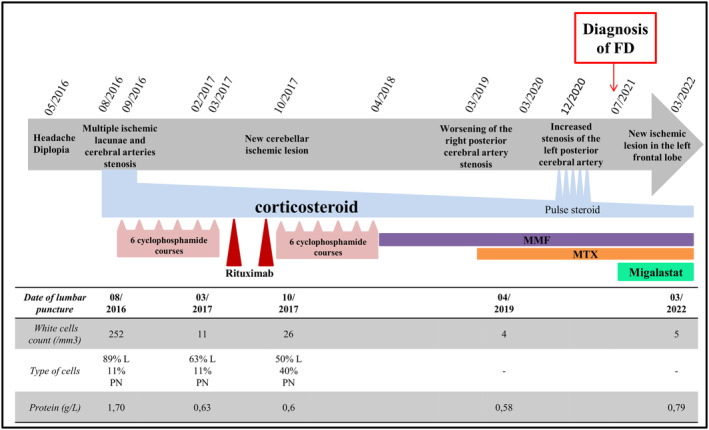
Evolution of symptoms and CSF features for Patient 2, and description of successive treatment lines. L is for lymphocytes; PN is for neutrophil; MMF is for mycophenolate mofetil; MTX is for methotrexate.
After two courses of rituximab, brain MRI revealed new cerebellar ischemic lesion and the appearance of new vascular stenoses of the left anterior cerebral artery and the right posterior cerebral artery. Lymphocytic meningitis persisted. Rituximab was considered ineffective and switched back to cyclophosphamide. MRI appeared stable. Maintenance immunosuppressive therapy with mycofenolate mofetil (MMF; 2 g/ day) was initiated, in combination with corticosteroids.
One year later, brain MRI showed a worsening of the right posterior cerebral artery stenosis. Methotrexate was added to MMF. Imagery remains stable for one year before the description of new stenoses in the posterior cerebral and the basilar arteries on MRI accompanied by paresthesia of the face. Methylprednisolone infusions were administered with unobvious benefit.
Concurrently, HCM was described, with late gadolinium enhancement in the inferolateral region of the left ventricle on cardiac MRI suggestive of FD. Genetic analysis confirmed in 2021 the presence of a c.950T>C variant of GLA gene. Other conditions were retrospectively attributed to FD such as acroparesthesia, proteinuria, and angiokeratoma. Looking closely at the family history, four ascendants on the maternal side died prematurely of sudden death, presumably from Fabry cardiomyopathy.
Chaperone therapy with migalastat was introduced. After 9 months of migalastat, the patient complained of a worsening of depressive mood. MRI showed new ischemic lesion in the left frontal lobe. These signs of progression contrasted with the absence of meningitis.
In 2023, the patient was admitted for disorders of consciousness. Brain MRI showed a new left frontal ischemic lesion. The patient had stopped taking migalastat a few weeks earlier, but continued methotrexate and corticosteroids. During hospitalization, the patient unexpectedly died from complicated diverticular perforation.
Patient 3
A 30‐year‐old male patient was referred to a tertiary neurology center because of recurrent ischemic strokes. The first stroke occurred 8 years ago, 2 years after a second stroke occurred in the vertebro‐basilar territory, and then the patient subsequently had three transient ischemic attacks with hemiparesis. He was treated with vitamin K antagonists and aspirin for 1 year.
At admission, the patient reported severe headaches and CSF analysis showed 24 cells/mm3 (lymphocytes 66%) with 0.81 g/L protein. Several angiokeratoma were observed on scrotum. Hypohidrosis was reported. Proteinuria was elevated up to 1 g/day. Brain MRI showed a pontine left paramedian infarction and a left lenticular infarction. Cardiac investigations including MRI were normal. FD was diagnosed thanks to decreased aGalA levels (4.3 nmol/h/mg protein [N: 20–68]) and the identification of a c.279C>G pathogenic variant of GLA gene.
ERT with agalsidase was introduced along with warfarine, aspirin 100 mg/day and ramipril 1.25 mg/day. The patient has been treated for 6 months with headaches improvement and no recurrence of stroke, but was lost to follow‐up.
Patient 4
A 31‐year‐old male patient was referred for recurrent vertigo. Brain and spinal MRI were normal. A video‐nystagmogram confirmed a left vestibular syndrome. He had a history of headaches 2 years ago that revealed meningitis (8 cells/mm3, protein level of 0.7 g/L) with no identified cause. His mother was being followed for HCM.
One year later, the patient presented ophthalmoplegia, left hemiparesis and nystagmus. Brain MRI revealed multiple small bilateral brain stem, pontine and thalamic lesions. CRP was elevated at 6.3 mg/L. CNS vasculitis was suspected. Cerebral angiography found a mild stenosis of both posterior cerebral arteries. High‐dose IV corticosteroid pulses were administered with no efficacy.
Diagnosis of FD was finally considered in front of acroparesthesia, cornea verticillata, hypohidrosis, angiokeratoma (Fig. 5A), sensorineural hearing loss and vestibular syndrome, HCM, glomerular nephropathy, and early ischemic stroke. New brain MRI showed dolichoectasia of the basilar artery (Fig. 5B). Alpha‐galactosidase A blood level was low at 3.6 nmol/h/mg protein (N: 20–68).
Figure 5.
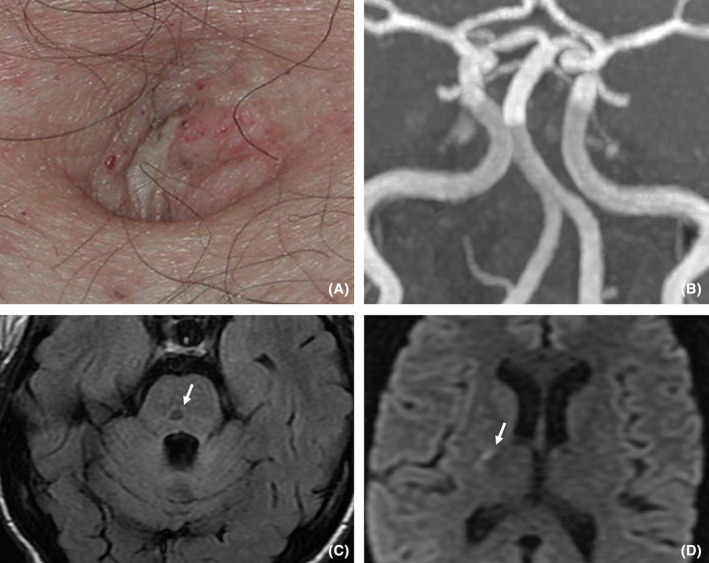
Clinical features (A) and MRI (B, C and D) of Patient 4. Presence of discrete angiokeratomas in the periumbilical region is seen on (A). Dolichoectatic aspect of the basilar artery is seen on (B) of time‐of‐flight MRI, coronal MIP (maximum intensity projection) reconstruction. On (C) (FLAIR T2 weighted imaging) and (D) (diffusion weighted imaging, DWI), we note the coexistence of lacunar infarcts (C, pontine lacuna with gliosis border, white arrow) and an acute stroke (D, DWI hyperintensity of internal capsule).
Two years after FD diagnosis, the patient suffered from a new stroke in right internal capsule. ERT was introduced with agalsidase alfa 0.2 mg/kg biweekly with ramipril and aspirin. The patient has been treated for 16 years without clinical or radiological recurrence of stroke, but intermittent headaches persisted. CSF analysis was performed 2 years after ERT introduction revealing the persistence of a mild inflammation with 9 cells/ mm3 and protein level of 0.83 g/L.
Systematic literature review
Our systematic review of the literature identified 477 articles from databases and 3 articles from selected conferences. We finally selected 23 of them. Selection process is detailed in Figure 6. We did not retain the article of Becker et al. of a 51‐year‐old woman with multiple WMLs and high cellularity in CSF, 6 because the variant identified on the GLA gene was c.937G>T [p.D313Y], whose pathogenicity is debated. 7
Figure 6.
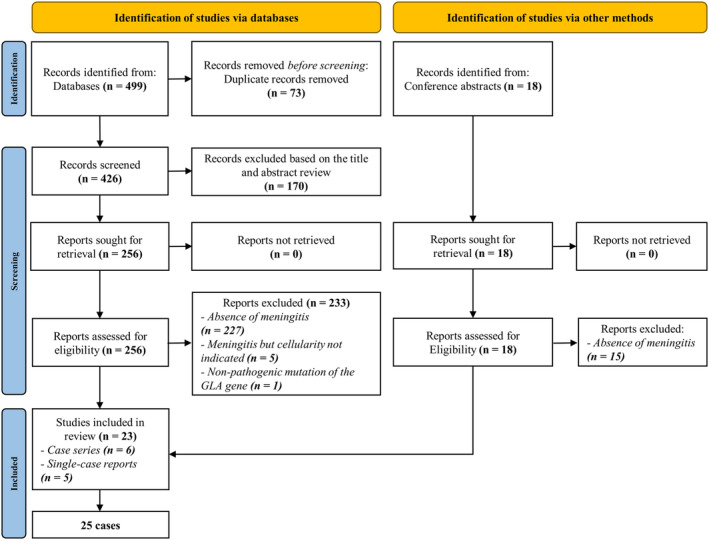
Flow chart detailing the selection process of articles for the systematic review. Due to the presence of redundant cases and case series among these 23 articles, the total number of patients finally analyzed is 25.
Patient characteristics
Table S1 summarizes characteristics of the 29 patients with FD and aseptic meningitis.
Median age of these 29 patients was 29.0 years (IQR 26–41), and 58.6% were male. Median age at first abnormal lumbar puncture was 28.0 years (IQR 26–40) in men and 36.5 years (IQR 25.8–41) in women. In terms of clinical presentation and course, 41.4% of patients had acute meningitis with meningeal syndrome, 24.1% acute meningitis with focal neurologic deficits but without meningeal syndrome, 13.8% chronic meningitis with meningeal syndrome, 13.8% chronic meningitis with focal neurologic deficits but without meningeal syndrome, and 6.9% recurrent meningitis with meningeal syndrome. Table S2 summarizes clinical, radiological, cytological, and biochemical characteristics for each subgroup of patients.
Pleocytosis was usually mild with a median count of 24 cells/mm3 (IQR 17–36) and only five patients with >50 cells/mm3. Meningitis was lymphocytic in 64.7% of the cases with CSF formula available. Median proteins level in CSF was 0.76 g/L (IQR 0.66–0.97). Brain MRI showed a recent stroke in 57.1% of cases, and at least 62.1% of patients had at least one episode of stroke during follow‐up. Among patients with recent stroke, 75% had a CSF leukocyte count ≥20/mm3. Large artery stenosis was present in 17.2% of patients (n = 5), and 40.7% of patients had WMLs. Basilar artery was dolichoectatic in only two patients (7.7%).
Corticosteroids were administered in 62.1% of patients. Of the 14 patients treated with ERT, 2 had a new stroke during follow‐up. Nine patients treated with ERT had a control lumbar puncture, and meningitis persisted in 5 of them. Only one (Patient 2) was treated with migalastat, and the initiation of treatment did not prevent further strokes. CSF had already normalized prior to its introduction and remained normal under migalastat.
In 82.8% of patients, FD was unknown prior to the discovery of meningitis. Classic form of FD concerned 82.8% of patients. The association of meningitis with inflammatory or ischemic brain parenchymal lesions raises the possibility of many differential diagnoses, among which the more frequently mentioned were multiple sclerosis (MS) (in 31.0% of patients), viral or bacterial infections (22.2%), and CNS vasculitis (17.9%).
Discussion
FD neurological complications: Background elements
FD is responsible for a wide variety of manifestations, among which neurological complications play an important part, due to their potential morbidity and mortality. Neurological phenotype includes both peripheral nervous system and CNS involvement. Peripheral nervous system involvement combines autonomic dysfunction and small fiber neuropathy, characterized by neuropathic pain. 8
CNS damages in FD classically refer to strokes, transient ischemic attacks, neurosensory impairment like sensorineural hearing loss, vertigo. CNS symptoms affect hemizygous men but also heterozygous women. 9 Mean age at first ischemic stroke is 39.2 years in men and 51 years in women. 10 The frequency of stroke in young men with FD (aged 25–44 years) is almost 12 times higher than the general population and 10 times higher in young women with FD. 11 Various radiological abnormalities of the brain parenchyma and intracranial vessels can be observed in FD. The most common abnormality found are severe and progressive WMLs, occurring at an early age. In a retrospective study of 50 patients with FD, with a mean age of 33 years, Crutchfield et al. showed that 52% of the patients had WMLs, and that these abnormalities were systematically present after 55 years. 12 T1 high‐signal intensity of the pulvinar thalami, microbleeds and dolichoectasia, that is, tortuous, ectatic blood vessels, are classically seen in FD but are not specific. 13
Apart from cerebral strokes related to the cardiac rhythm disorders secondary to the HCM, pathophysiology of CNS involvement in FD remains partly misunderstood. Strokes are probably mainly related to accumulation of Gb3 in the vascular endothelium resulting in reduced vascular compliance and leading to cerebral hypoperfusion. 14
Nevertheless, cause of neurological impairment in FD is probably not limited to vascular thickening. Some evidence suggest that excess Gb3 also accumulates directly in brain tissue resulting in neuronal ballooning and gliosis. 15 , 16 Variations in the nitric oxide‐pathway, and increase in pro‐inflammatory could also play a determining role in the genesis of brain damage. 14 , 17 , 18 Numerous modifications in blood constituents have been described in FD, including leucocyte adhesion molecules 19 and homocysteine concentrations, 20 which may contribute to intracranial vasculopathy.
Two specific treatments are available for FD. ERT, which has been available for the past 20 years, consists in biweekly infusions of recombinant aGalA enzyme with agalsidase alfa, (Replagal, Shire‐Takeda) or agalsidase beta (Fabrazyme, Sanofi‐Genzyme). While the benefits in stabilizing the nephropathy and the HCM are now accepted, 21 , 22 the benefit in reducing the risk of stroke and progression of WMLs is less clear. One explanation could be that ERT does not cross the blood–brain barrier.
The second treatment option is an oral chaperone therapy with migalastat (Galafold, Amicus Therapeutics), which is only effective in patients with “amenable” missense variants of the GLA gene. 23 Pharmacological data shows that migalastat diffuses across the blood–brain barrier, which makes speculate on its effectiveness in preventing CNS complications. However, hindsight is still too limited to assess this possible benefit.
Aseptic meningitis in FD: Diagnostic approach
There are many causes of aseptic meningitis: lymphoma, autoimmune diseases, drug‐induced, CNS vasculitis, etc. FD is not commonly cited, due to the scarceness of this condition. Some authors have reported frequent headaches in patients with FD that could possibly be the manifestation of under‐diagnosed meningitis episodes. 24 Aseptic meningitis in FD tends to occur in young patients (median age of 29 year), earlier than the first stroke as reported in large retrospective series. 10 It should be stressed that aseptic meningitis is also seen in heterozygous women. The fact that a large majority of these patients were the propositus in their family, that is, the first member diagnosed, underlines the diagnostic difficulty of FD. Nevertheless, in three of the four patients from our center, a meticulous interrogation allowed the identification of a family history suggestive of FD: end‐stage renal failure before 50 year old in Patient 1s father, several cases of sudden death in the family of Patient 2, and HCM in Patient 4s mother.
Brain MRI usually shows deep WMLs, infarction and vertebrobasilar ectasia, without specific features making them distinguishable from other causes of small vessel impairment. 25 When WML burden is high, FD can mimic other WM disease, especially MS. Nonetheless, there is conflicting evidence in the literature about the risk of confusing FD with MS. 26 , 27
The case of our Patient 2 perfectly illustrates the difficulty of the diagnostic approach: cerebral arterial injury was observed with an angiographic appearance typical for CNS vasculitis but multiple lines of immunosuppressive therapy did not prevent recurrent stroke. Until her death, the coexistence of an inflammatory pathology of the CNS was still debated in this patient. With exception of dolichoectasia, macrovascular involvement is a poorly described entity in FD. In two out of our four patients however, we observed stenoses in large intracranial arteries, including basilar artery and proximal segments of the posterior arteries.
Pathophysiological hypotheses
Several nonexclusive pathophysiological hypotheses can be proposed to explain the occurrence of meningitis in FD.
A first hypothesis is that meningitis is an aspecific reaction to the rupture of blood–brain barrier during stroke. Indeed, in 57.1% of the 29 patients studied, a recent or semi‐recent stroke was present, which is much higher than in the general population of patients followed for FD (about 20%). However, several studies have shown that the increase in CSF cellularity during stroke is rare and usually minimal. Ramirez‐Lassepas et al. showed that, among 98 patients with acute ischemic stroke, only 13% had ≥5 cells/mm3, with a maximum cell count of 15/mm3. 28 More recently, Schulte‐Mecklenbeck et al. 29 analyzed the CSF of 90 patients with recent stroke, and found a median CSF cell count of 1.76/mm3 (SD 3.06), with only 6 of the 90 patients having a count >5 cells/mm3, and none exceeding 20 cells/mm3. As we observed in our cohort, the majority of patients with recent stroke had more than 20 cells /mm3 in CSF and we therefore believe that the recent onset of stroke is not sufficient to explain the elevated CSF cell count in the majority of patients.
A second hypothesis is that the accumulation of sphingolipids within the pia and arachnoid membranes may induce local lymphocytic influx. In physiological conditions, CSF contains glycosphingolipids. The enzyme deficiency in FD could lead to their accumulation, which is confirmed in some postmortem studies. 30
A third hypothesis is that meningitis reflects an inappropriate autoinflammatory reaction. Higher proinflammatory cytokine expression has been shown during FD. 31 , 32 Excess of Gb3 could be the source of pro‐inflammatory signals by disturbing lipid rafts' organization and dysregulating the activation of toll‐like receptor 4 pathway, 33 leading to a pro‐inflammatory cascade and the secretion of TNF and IL‐1beta, 34 , 35 or to appearance of reactive oxygen species in the endothelial cell by inhibiting the NO synthase enzyme. 36 Another major pro‐inflammatory mechanism in FD is the dysregulation of autophagy. 37 Numerous clinical observations in FD support these different models of disruption of inflammatory pathways such as the existence of AA amyloidosis, 38 histological description of myocarditis on autopsy series, 39 or reports of unexplained and prolonged fever. 3 , 40
Limitations
Our study has several limitations. The majority of the patients studied are published cases, and not followed up in our center, leading to missing data, in particular concerning the exhaustiveness of the etiological assessment of meningitis. Of notes, lumbar puncture was not systematically rechecked in patients with stroke concomitant with the diagnosis of meningitis, possibly because the elevated CSF leukocyte count had been attributed to the stroke. The possibility of an associated inflammatory or infectious neurological disease cannot be completely excluded in these patients. Nevertheless, based on the absence of an alternative diagnosis, we assume the high probability of a causal relationship between FD and aseptic meningitis.
Conclusion
The association of aseptic meningitis with cerebral strokes and macrovascular stenoses should raise the suspicion of FD. Our systematic review of the literature allowed us to analyze 29 cases with this rare condition. These cases highlight the fact that FD could trigger CNS inflammation in both men and women, and should be considered as a cause of chronic lymphocytic meningitis, especially when strokes, HCM, or proteinuria are present. Although rare, macrovascular stenosis should be looked as a possible complication of FD, and discussed as a differential diagnosis of a putative CNS vasculitis. The pathophysiological mechanisms underlying the pleocytosis remain partly misunderstood, but could constitute further evidence of the multisystemic pro‐inflammatory nature of FD.
Author Contributions
CM and WM conceived and designed the study. CM, AG, and WM performed the analysis. CM, MZ, FV, J‐FA, WM, and OL collected and participated in the analysis of results. CM and WM wrote the paper first draft. CM, AG, MZ, FV, J‐FA, WM, and OL wrote the final version of the manuscript.
Funding Information
No funding information provided.
Conflict of Interest
WM and OL received consulting fees and participated to advisory boards organized by Sanofi‐Genzyme, Shire‐Takeda, Amicus Therapeutics, and Chiesi Farmaceutici. FV participates to the Fabry Outcome Survey observational registry (no personal incentive). CM, AG, MZ, and JFA do not declare any conflict of interest.
Supporting information
Appendix S1.
Table S1.
Table S2.
Acknowledgements
We acknowledge patients for their participation in this study.
Contributor Information
Camille Montardi, Email: camille.montardi@aphp.fr.
Wladimir Mauhin, Email: wmauhin@hopital-dcss.org.
References
- 1. Germain DP, Levade T, Hachulla E, et al. Challenging the traditional approach for interpreting genetic variants: lessons from Fabry disease. Clin Genet. 2022;101(4):390‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fellgiebel A, Müller MJ, Mazanek M, et al. White matter lesion severity in male and female patients with Fabry disease. Neurology. 2005;65(4):600‐602. [DOI] [PubMed] [Google Scholar]
- 3. Lidove O, Kaminsky P, Hachulla E, et al. Fabry disease “the new great imposter”: results of the French Observatoire in Internal Medicine Departments (FIMeD). Clin Genet. 2012;81(6):571‐577. [DOI] [PubMed] [Google Scholar]
- 4. Higgins JPT, Thomas J, Chandler J, et al., eds. Cochrane Handbook for Systematic Reviews of Interventions Version 6.3 (Updated February 2022). Cochrane; 2022. www.training.cochrane.org/handbook [Google Scholar]
- 5. Moher D, Liberati A, Tetzlaff J, Altman DG, The PRISMA Group . Preferred reporting items for systematic reviews and meta‐analyses: the PRISMA statement. PLoS Med. 2009;6(7):e1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Becker J, Rolfs A, Karabul N, Berlit P, Kraemer M. D313Y mutation in the differential diagnosis of white matter lesions: experiences from a multiple sclerosis outpatient clinic. Mult Scler J. 2016;22(11):1502‐1505. [DOI] [PubMed] [Google Scholar]
- 7. Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab. 2018;123(4):416‐427. [DOI] [PubMed] [Google Scholar]
- 8. Siedler G, Káhn A‐K, Weidemann F, Wanner C, Sommer C, Üçeyler N. Dyshidrosis is associated with reduced amplitudes in electrically evoked pain‐related potentials in women with Fabry disease. Clin Neurophysiol. 2019;130(4):528‐536. [DOI] [PubMed] [Google Scholar]
- 9. Bird TD, Lagunoff D. Neurological manifestations of Fabry disease in female carriers. Ann Neurol. 1978;4(6):537‐540. [DOI] [PubMed] [Google Scholar]
- 10. Beck M, Ramaswami U, Hernberg‐Ståhl E, et al. Twenty years of the Fabry outcome survey (FOS): insights, achievements, and lessons learned from a global patient registry. Orphanet J Rare Dis. 2022;17(1):238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mehta A, Ginsberg L, FOS Investigators . Natural history of the cerebrovascular complications of Fabry disease. Acta Paediatr Suppl. 2005;94(447):24‐27. discussion 9–10. [DOI] [PubMed] [Google Scholar]
- 12. Crutchfield KE, Patronas NJ, Dambrosia JM, et al. Quantitative analysis of cerebral vasculopathy in patients with Fabry disease. Neurology. 1998;50(6):1746‐1749. [DOI] [PubMed] [Google Scholar]
- 13. Takanashi J, Barkovich AJ, Dillon WP, Sherr EH, Hart KA, Packman S. T1 hyperintensity in the pulvinar: key imaging feature for diagnosis of Fabry disease. AJNR Am J Neuroradiol. 2003;24(5):916‐921. [PMC free article] [PubMed] [Google Scholar]
- 14. Rombach SM, Twickler TB, Aerts JMFG, Linthorst GE, Wijburg FA, Hollak CEM. Vasculopathy in patients with Fabry disease: current controversies and research directions. Mol Genet Metab. 2010;99(2):99‐108. [DOI] [PubMed] [Google Scholar]
- 15. deVeber GA, Schwarting GA, Kolodny EH, Kowall NW. Fabry disease: immunocytochemical characterization of neuronal involvement. Ann Neurol. 1992;31(4):409‐415. [DOI] [PubMed] [Google Scholar]
- 16. Okeda R, Nisihara M. An autopsy case of Fabry disease with neuropathological investigation of the pathogenesis of associated dementia. Neuropathology. 2008;28(5):532‐540. [DOI] [PubMed] [Google Scholar]
- 17. Altarescu G, Moore DF, Schiffmann R. Effect of genetic modifiers on cerebral lesions in Fabry disease. Neurology. 2005;64(12):2148‐2150. [DOI] [PubMed] [Google Scholar]
- 18. Hughes DA, Mehta AB. Vascular complications of Fabry disease: enzyme replacement and other therapies. Acta Paediatr Suppl. 2005;94(447):28‐33. discussion 9–10. [DOI] [PubMed] [Google Scholar]
- 19. DeGraba T, Azhar S, Dignat‐George F, et al. Profile of endothelial and leukocyte activation in Fabry patients. Ann Neurol. 2000;47(2):229‐233. [PubMed] [Google Scholar]
- 20. Müller KB, Galdieri LC, Pereira VG, Martins AM, D'Almeida V. Evaluation of oxidative stress markers and cardiovascular risk factors in Fabry disease patients. Genet Mol Biol. 2012;35(2):418‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kampmann C, Perrin A, Beck M. Effectiveness of agalsidase alfa enzyme replacement in Fabry disease: cardiac outcomes after 10 years' treatment. Orphanet J Rare Dis. 2015;10:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Feriozzi S, Torras J, Cybulla M, et al. The effectiveness of long‐term agalsidase alfa therapy in the treatment of Fabry nephropathy. Clin J Am Soc Nephrol. 2012;7(1):60‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Weidemann F, Jovanovic A, Herrmann K, Vardarli I. Chaperone therapy in Fabry disease. Int J Mol Sci. 2022;23(3):1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sawada J, Nakagawa N, Kano K, et al. Characteristics of neurological symptoms in adult Japanese patients with Fabry disease. Intern Med. 2021;60(12):1819‐1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fazekas F, Enzinger C, Schmidt R, et al. Brain magnetic resonance imaging findings fail to suspect Fabry disease in young patients with an acute cerebrovascular event. Stroke. 2015;46(6):1548‐1553. [DOI] [PubMed] [Google Scholar]
- 26. Böttcher T, Rolfs A, Tanislav C, et al. Fabry disease ‐ underestimated in the differential diagnosis of multiple sclerosis? PLoS One. 2013;8(8):e71894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Russo C, Cocozza S, Riccio E, et al. Prevalence of GLA gene mutations and polymorphisms in patients with multiple sclerosis: a cross‐sectional study. J Neurol Sci. 2020;412:116782. [DOI] [PubMed] [Google Scholar]
- 28. Ramirez‐Lassepas M, Patrick BK. Cerebrospinal fluid cellular response in uncomplicated acute ischemic stroke. J Stroke Cerebrovasc Dis. 1992;2(3):168‐172. [DOI] [PubMed] [Google Scholar]
- 29. Schulte‐Mecklenbeck A, Kleffner I, Beuker C, et al. Immunophenotyping of cerebrospinal fluid cells in ischaemic stroke. Eur J Neurol. 2019;26(6):919‐926. [DOI] [PubMed] [Google Scholar]
- 30. Schiffmann R, Rapkiewicz A, Abu‐Asab M, et al. Pathological findings in a patient with Fabry disease who died after 2.5 years of enzyme replacement. Virchows Arch. 2006;448(3):337‐343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. De Francesco PN, Mucci JM, Ceci R, et al. Fabry disease peripheral blood immune cells release inflammatory cytokines: role of globotriaosylceramide. Mol Genet Metab. 2013;109(1):93‐99. [DOI] [PubMed] [Google Scholar]
- 32. Biancini GB, Vanzin CS, Rodrigues DB, et al. Globotriaosylceramide is correlated with oxidative stress and inflammation in Fabry patients treated with enzyme replacement therapy. Biochim Biophys Acta. 2012;1822(2):226‐232. [DOI] [PubMed] [Google Scholar]
- 33. Köberlin MS, Snijder B, Heinz LX, et al. A conserved circular network of Coregulated lipids modulates innate immune responses. Cell. 2015;162(1):170‐183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Villar VAM, Cuevas S, Zheng X, Jose PA. Localization and signaling of GPCRs in lipid rafts. Methods Cell Biol. 2016;132:3‐23. [DOI] [PubMed] [Google Scholar]
- 35. Rozenfeld P, Feriozzi S. Contribution of inflammatory pathways to Fabry disease pathogenesis. Mol Genet Metab. 2017;122(3):19‐27. [DOI] [PubMed] [Google Scholar]
- 36. Joly DA, Grünfeld J‐P. 3‐Nitrotyrosine as a biomarker for vascular involvement in Fabry disease. Kidney Int. 2014;86(1):5‐7. [DOI] [PubMed] [Google Scholar]
- 37. Shibutani ST, Saitoh T, Nowag H, Münz C, Yoshimori T. Autophagy and autophagy‐related proteins in the immune system. Nat Immunol. 2015;16(10):1014‐1024. [DOI] [PubMed] [Google Scholar]
- 38. Terré A, Knebelmann B, Buob D, et al. AA amyloidosis associated with Fabry disease. Int J Clin Pract. 2020;74(10):e13577. [DOI] [PubMed] [Google Scholar]
- 39. Sheppard MN, Cane P, Florio R, et al. A detailed pathologic examination of heart tissue from three older patients with Anderson‐Fabry disease on enzyme replacement therapy. Cardiovasc Pathol. 2010;19(5):293‐301. [DOI] [PubMed] [Google Scholar]
- 40. Verrecchia E, Zampetti A, Antuzzi D, et al. The impact of fever/hyperthermia in the diagnosis of Fabry: a retrospective analysis. Eur J Intern Med. 2016;32:26‐30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1.
Table S1.
Table S2.