

SUMMARY
Traditional methods for site-specific drug delivery in the brain are slow, invasive, and difficult to interface with recordings of neural activity. Here, we demonstrate the feasibility and experimental advantages of in vivo photopharmacology using “caged” opioid drugs that are activated in the brain with light after systemic administration in an inactive form. To enable bidirectional manipulations of endogenous opioid receptors in vivo, we developed photoactivatable oxymorphone (PhOX) and photoactivatable naloxone (PhNX), photoactivatable variants of the mu opioid receptor agonist oxymorphone and the antagonist naloxone. Photoactivation of PhOX in multiple brain areas produced local changes in receptor occupancy, brain metabolic activity, neuronal calcium activity, neurochemical signaling, and multiple pain- and reward-related behaviors. Combining PhOX photoactivation with optical recording of extracellular dopamine revealed adaptations in the opioid sensitivity of mesolimbic dopamine circuitry in response to chronic morphine administration. This work establishes a general experimental framework for using in vivo photopharmacology to study the neural basis of drug action.
In brief
Light-activated drugs offer exquisite control over the timing and location of drug action in the brain. McClain et al. describe systemically available photoactivatable opioid drugs and their use to study opioid-sensitive circuits in behaving mice. This work establishes an experimental framework for the validation and application of photopharmacological probes in vivo.
INTRODUCTION
Pharmacological probes are widely used to study the nervous system. However, small molecule drugs act slowly and with spatial imprecision due to diffusion, even when applied locally in the brain through implanted cannulas. This impedes in vivo neuropharmacological studies involving electrophysiological, imaging, and behavioral tracking measurements at high temporal resolution, which benefit from the ability to correlate measurements with well-defined, time-locked stimuli that can be readily varied in intensity and duration. Photopharmacological probes, which are converted from an inactive pro-drug form into a biologically active form with light, offer a general solution to this problem.1–3 The use of light to control biological signaling provides experimental advantages due to the spatiotemporal precision of illumination and the ability to target deep brain structures in rodents using minimally invasive optical fibers. The spatiotemporal precision of photopharmacology arises from pre-equilibration of brain tissue with the inactive pro-drug, such that brief (millisecond-second) light flashes release the active drug directly at its site of action, with sub-second kinetics, and in a highly stereotyped manner. Importantly, the dose can be readily controlled by varying the amount of pro-drug administered or the amount of light delivered. These features can streamline studies involving local drug administration during ongoing animal behavior. Indeed, the first example of in vivo optogenetics involved photoactivation of a caged ligand for a foreign receptor.4 However, despite a sizable pharmacopeia of photopharmacological probes, many of which have been shown to work in vivo,5–15 their potential as tools to study biological mechanisms in vivo has yet to be fully realized. This shortcoming stems from limitations in pro-drug bioavailability, residual activity, and the requirement for long illumination periods due to poor bioavailability and/or inefficient photoactivation.
An attractive target for the development of photopharmacological probes is the mu opioid receptor (MOR), which is the primary target of morphine and other addictive analgesic drugs. Endogenous opioid neuropeptides are implicated in diverse functions, including feeding, memory, behavioral reinforcement, social interactions, and pain modulation. Our prior work with photopharmacological probes for MORs and other receptors in ex vivo preparations (e.g., acute brain slices) has provided insights into mechanisms of neuropeptide polypharmacology, ligand-receptor interactions, signaling kinetics, and cellular adaptations to chronic opioid treatment.16–21 We recently demonstrated the feasibility of using in vivo uncaging to rapidly modulate ongoing behavior using a photoactivatable derivative of the MOR agonist [d-Ala2, NMe-Phe4, Gly-ol5]enkephalin (DAMGO).22 Because peptides do not readily cross the blood-brain barrier (BBB), caged DAMGO was administered through an optofluidic cannula prior to photoactivation. However, cannulas are invasive, require disruptive animal handling for fluid delivery, and only allow for limited quantities of caged drug to be applied prior to photoactivation. Furthermore, the compatibility of in vivo photopharmacology with optical recording techniques remains unexplored.
Here, we describe a pair of brain-penetrant photopharmacological reagents that afford spatiotemporally precise, bidirectional optical control of endogenous MORs in vivo after systemic administration. These molecules can be photoactivated in the brain with sub-second flashes of light through implanted optical fibers to drive rapid changes in behavior and neural circuit function. Furthermore, we demonstrate that these reagents are compatible with commonly used in vivo optical methods, which is an important step toward achieving all-optical manipulation and recording of neural function.
RESULTS
Design, synthesis, and in vitro inactivity of PhOX and PhNX
To streamline our design and synthesis, we pursued photoactivatable analogs of the closely related agonist and antagonist oxymorphone (OXM, 1) and naloxone (NLX, 2), respectively (Figure 1A). We previously established that NLX can be readily caged via modification with a carboxynitroveratryl (CNV) group, yielding (CNV-NLX, 3), which contains a negatively charged carboxylic acid.18 To improve BBB penetrance, we used a dimethoxynitrophenethyl (DMNPE) caging group,23 which contains a neutral methyl group, affording photoactivatable oxymorphone (PhOX, 4) and photoactivatable naloxone (PhNX, 5). Although the DMNPE caging group requires ultraviolet (UV) light for photoactivation, because it does not absorb visible light, it is compatible with green and red fluorescent probes used for in vivo imaging (Figure S1A). Alkylation of OXM with 1-(1-bromoethyl)-4,5-dimethoxy-2-nitrobenzene afforded PhOX in 63% yield (Figure 1B). NLX was similarly converted to PhNX in 67% yield. Using high-pressure liquid chromatography and mass spectrometry, we confirmed that both PhOX and PhNX are stable in the dark for 24 h when dissolved in phosphate-buffered saline (PBS) and that they cleanly photoconvert back into OXM and NLX, respectively, upon exposure to UV light (Figures 1C and S1B–S1D). Similar to a recent study with a photoactivatable morphine analog,15 we also prepared a derivative of OXM using the diethylaminocoumarin (DEAC) caging group (DEAC-OXM, 6, Figure 1A). However, rather than cleanly converting to OXM, DEAC-OXM underwent a 1,4 photo-Claisen rearrangement to predominantly yield a functionally dead compound (RE-DEAC-OXM, Figure S2; Data S1), similar to other coumarin-caged phenols.24,25 We therefore focused our efforts on PhOX and PhNX.
Figure 1. Design, synthesis, and in vitro validation of PhOX and PhNX.
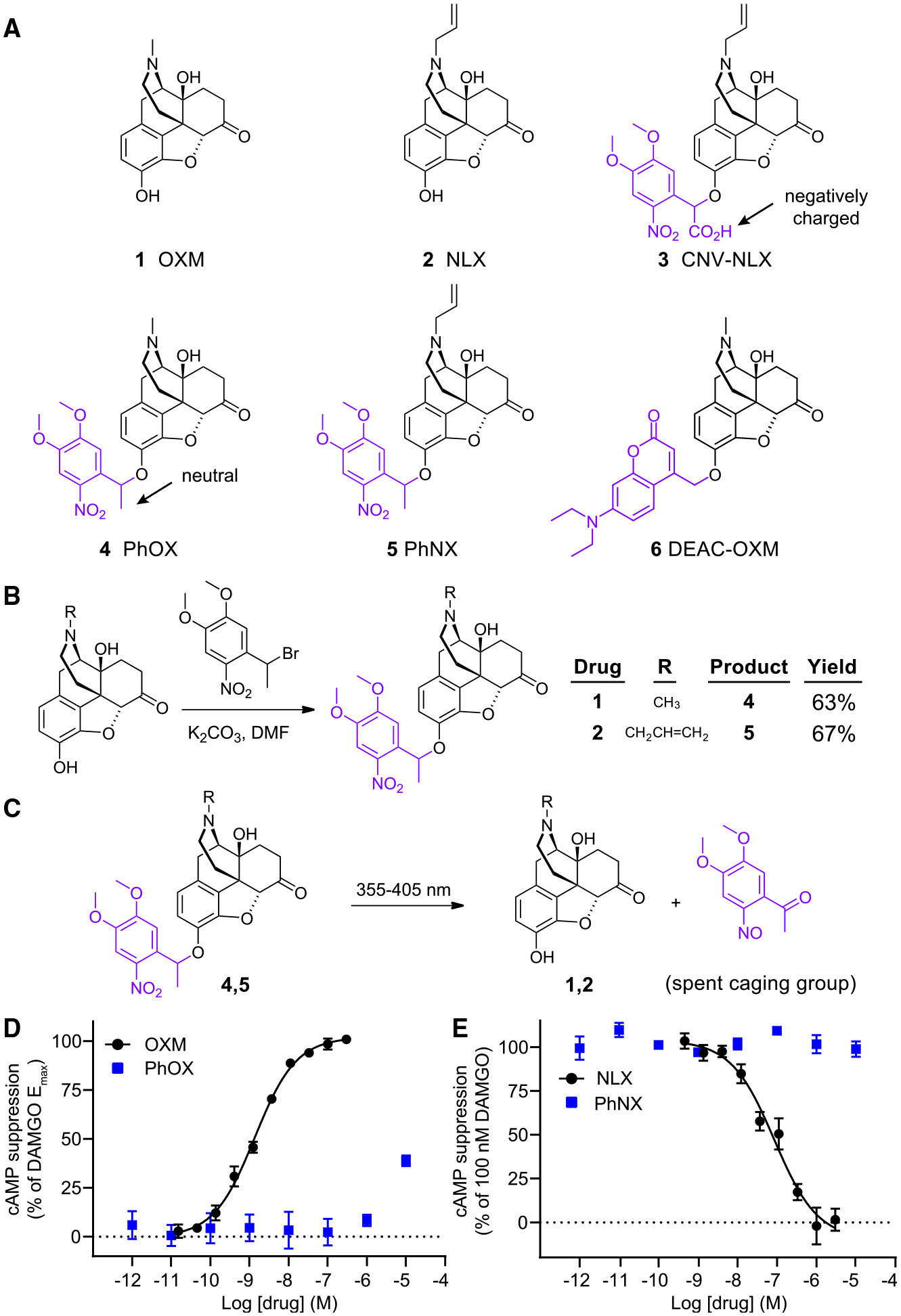
(A) Chemical structures of OXM (1), NLX (2), and the photoactivatable small molecule opioid drugs CNV-NLX (3), PhOX (4), PhNX (5), and DEAC-OXM (6). The light-removable DMNPE and DEAC caging groups are drawn in violet.
(B) Reaction scheme depicting the one-step alkylation procedure used to synthesize PhOX and PhNX.
(C) Reaction scheme depicting ultraviolet-light-driven photorelease of OXM and NLX from PhOX, and PhNX, respectively.
(D) Agonist dose-response curves at the MOR using a cAMP assay. The solid line depicts the best-fit sigmoidal function used to derive the indicated EC50 value. Data were normalized to the response produced by DAMGO (1 μM) and are expressed as mean ± SEM (n = 5 wells per concentration).
(E) Antagonist dose-response curves at the MOR in the presence of DAMGO (100 nM). Data are presented as in (D). See also Figures S1–S3.
To determine whether PhOX and PhNX exhibit reduced activity at MOR compared with OXM and NLX, we obtained dose-response curves at the murine MOR26 expressed in human embryonic kidney cells using a cyclic AMP (cAMP) assay of G protein signaling. Whereas OXM exhibited an EC50 of 1.4 ± 0.2 nM and full receptor activation compared with the MOR agonist DAMGO (1 μM), PhOX (up to 10 μM) produced only partial receptor activation (Figure 1D). Similar to CNV-NLX.18 whereas NLX antagonized MOR activation by DAMGO (100 nM) with an IC50 of 86 ± 28 nM, PhNX (up to 10 μM) was inactive (Figure 1E). Dose-response curves of DAMGO in the absence and presence of PhOX (300 nM) revealed that PhOX does not antagonize MOR at non-agonistic concentrations (Figure S3A). Further evaluation demonstrated that PhOX is not a biased agonist of the β-arrestin pathway (Figure S3B). PhOX and PhNX exhibited neither agonism nor antagonism in the cAMP assay at delta and kappa opioid receptors (Figures S3C–S3F). Of note, 300 nM OXM partially activated the delta opioid receptor, consistent with its morphine-like pharmacology. Together, these results indicate that both PhOX and PhNX are highly inactive at opioid receptors and undergo clean uncaging with UV light.
Photoactivation of PhOX and PhNX engages endogenous opioid receptors ex vivo
We next assayed PhOX and PhNX in acute brain slices taken from the CA1 region of the mouse hippocampus, where MOR activation suppresses perisomatic inhibition onto pyramidal neurons.17,27 During whole-cell voltage clamp recordings, inhibitory postsynaptic currents (IPSCs) were evoked with a small bipolar stimulating electrode (Figure 2A). Whereas bath application of OXM (3 μM) strongly suppressed IPSC amplitude, PhOX (3 μM) was inactive (Figure 2B). However, application of a brief light flash from a 365-nm light-emitting diode (LED) (50 ms, 5 mW) rapidly suppressed the IPSC in an NLX (3 μM)-sensitive manner (Figure 2C). These results are summarized in Figure 2D.
Figure 2. Evaluation of PhOX-mediated photo-agonism and PhNX-mediated photo-antagonism in acute brain slices.
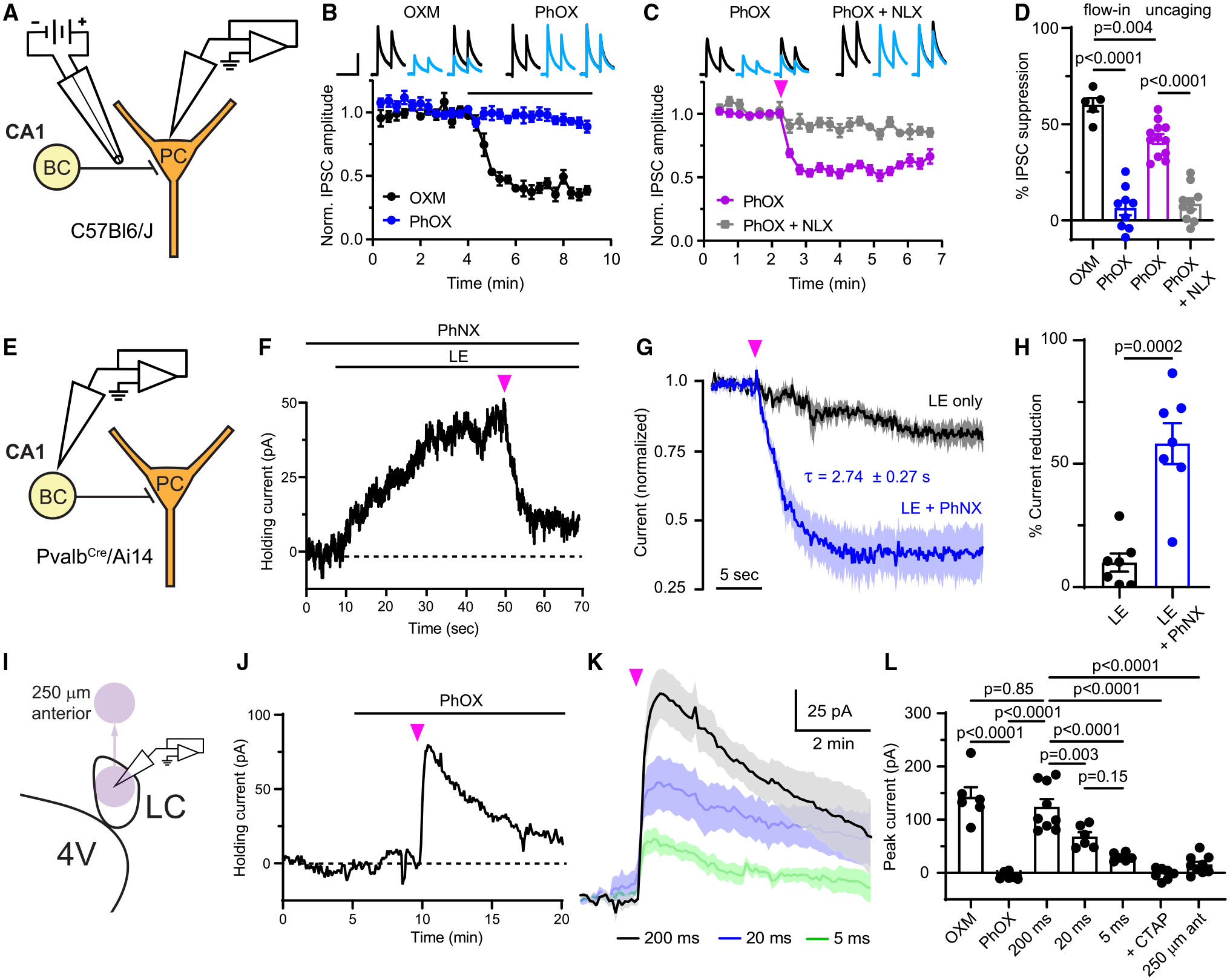
(A) Schematic depicting whole-cell voltage clamp recording of opioid-sensitive synaptic inhibition in the hippocampus. BC, basket cell; PC, pyramidal cell.
(B) Baseline-normalizedIPSCs in response to bath application of drug, as indicated by the black line (OXM: n = 5 cells from 2 mice; PhOX: n = 9 cells from 6 mice). Top insets: example average IPSCs (n = 3 sweeps from one cell) before (black) and after (blue) drug application. Scale bars, 200 pA, 80 ms.
(C) IPSC suppression in response to uncaging with a full-field flash of UV light (pink arrow) (PhOX: n = 12 cells from 7 mice; PhOX + NLX: n = 10 cells from 5 mice).
(D) Summary data for (B) and (C) (one-way ANOVA, F(3,32) = 58.2, p < 0.0001, Bonferroni’s multiple comparisons test).
(E) Schematic depicting the whole-cell voltage clamp recording of outward currents from BCs.
(F) Example recording demonstrating photoinhibition of the current evoked by bath application of LE upon PhNX uncaging. Scale bars, 10 pA, 10 s.
(G) Normalized response of LE-evoked currents to a UV light flash in the absence and presence of PhNX (LE only: n = 7 cells from 3 mice; LE + PhNX: n = 7 cells from 3 mice).
(H) Summary of the LE-evoked current remaining 10–15 s after application of a light flash (unpaired two-tailed t test).
(I) Schematic depicting whole-cell voltage clamp recording from noradrenergic neurons in rat LC and movement of the uncaging spot (purple circle) away from the recorded neurons.
(J) Example recording from an LC neuron in which bath application of PhOX was followed by uncaging.
(K) Average currents evoked by uncaging PhOX with light flashes of varied duration (200 ms: n = 9 cells from 3 rats; 20 ms: n = 6 cells from 3 rats; 5 ms: n = 6 cells from 3 rats).
(L) Summary of the currents evoked in LC neurons (one-way ANOVA, F(6,42) = 33.6, p < 0.0001, Sidak’s multiple comparisons test). All data are plotted as mean ± SEM. See also Figure S3.
To validate PhNX, we obtained whole-cell voltage-clamp recordings from fluorescently identified parvalbumin-expressing basket cells in slices from PvalbCre/Rosa26-lsl-tdTomato (Ai14) mice. In the presence of PhNX (3 μM), bath application of the mu/delta opioid receptor agonist [Leu5]-enkephalin (LE, 300 nM) evoked outward currents that were rapidly reversed with light (200 ms, 5 mW, 365 nm) (Figures 2E and 2F). Whereas the control LE current slowly desensitized, PhNX photoactivation rapidly suppressed it (Figures 2G and 2H). As with PhOX, unilluminated PhNX did not affect the IPSC (Figure S3G).
To gain insight into the spatiotemporal profile of photomodulation, we acquired whole-cell voltage-clamp recordings from noradrenergic neurons in horizontal slices of rat locus coeruleus (LC) (Figure 2I), where MOR agonists evoke large outward K+ currents.16,28 Bath application of OXM (1 μM) produced currents that trended toward being smaller than those produced by DAMGO (1 μM) (Figures S3H and S3I). Notably, OXM did not drive strong desensitization (Figures S3J and S3K), indicating that OXM minimally engages the β-arrestin signaling pathway, similar to morphine.29 Whereas bath-applied PhOX (1 μM) was inactive, strong photoactivation (200 ms, 40 mW, 365 nm) produced large, rapidly activating outward currents (τon = 8.36 s) that decayed over several minutes (τoff = 5.44 min) (Figures 2J and 2K). Reducing the flash duration (20 and 5 ms, 40 mW) produced a graded reduction in amplitude (Figure 2K), yet even the smallest currents decayed slowly (τoff = 2.15 min, 5 ms flash). These deactivation kinetics are much slower than those observed with caged peptides16,22 and likely reflect slow clearance of the relatively hydrophobic small molecule drug.30 The MOR-specific antagonist CTAP completely blocked the currents, and moving the uncaging site 250 μm anterior to the recorded neuron (Figure 2I) evoked only small currents. These data are summarized in Figure 2L. Collectively, these experiments demonstrate that PhOX and PhNX are inert in the dark, that their photoactivation engages endogenous opioid receptors to modulate neuronal physiology within seconds of illumination, and that receptor activation persists for minutes after uncaging.
In vivo photoactivation after systemic administration of PhOX and PhNX
We next determined whether PhOX and PhNX cross the BBB, which is presumably required for effective in vivo uncaging. Following intravenous (i.v.) administration of equimolar doses of either OXM (5 mg/kg), PhOX (8.5 mg/kg), NLX (10 mg/kg), or PhNX (14 mg/kg) in mice, drug concentrations were determined in the brain and blood plasma. As shown in Figure 3A, both PhOX and PhNX exhibited similar brain/plasma ratios to their parent drugs, which suggests that they should reach useful concentrations in the brain upon systemic administration. We also determined that PhOX exhibits a half-life of 90 min in the blood after intraperitoneal (i.p.) administration (Figure 3B), which offers a broad time window for experimentation. Notably, the HCl salts of both PhOX and PhNX are water soluble at millimolar concentrations and could be administered in saline without the addition of surfactants or co-solvents.
Figure 3. In vivo photoactivation of PhOX after systemic administration.
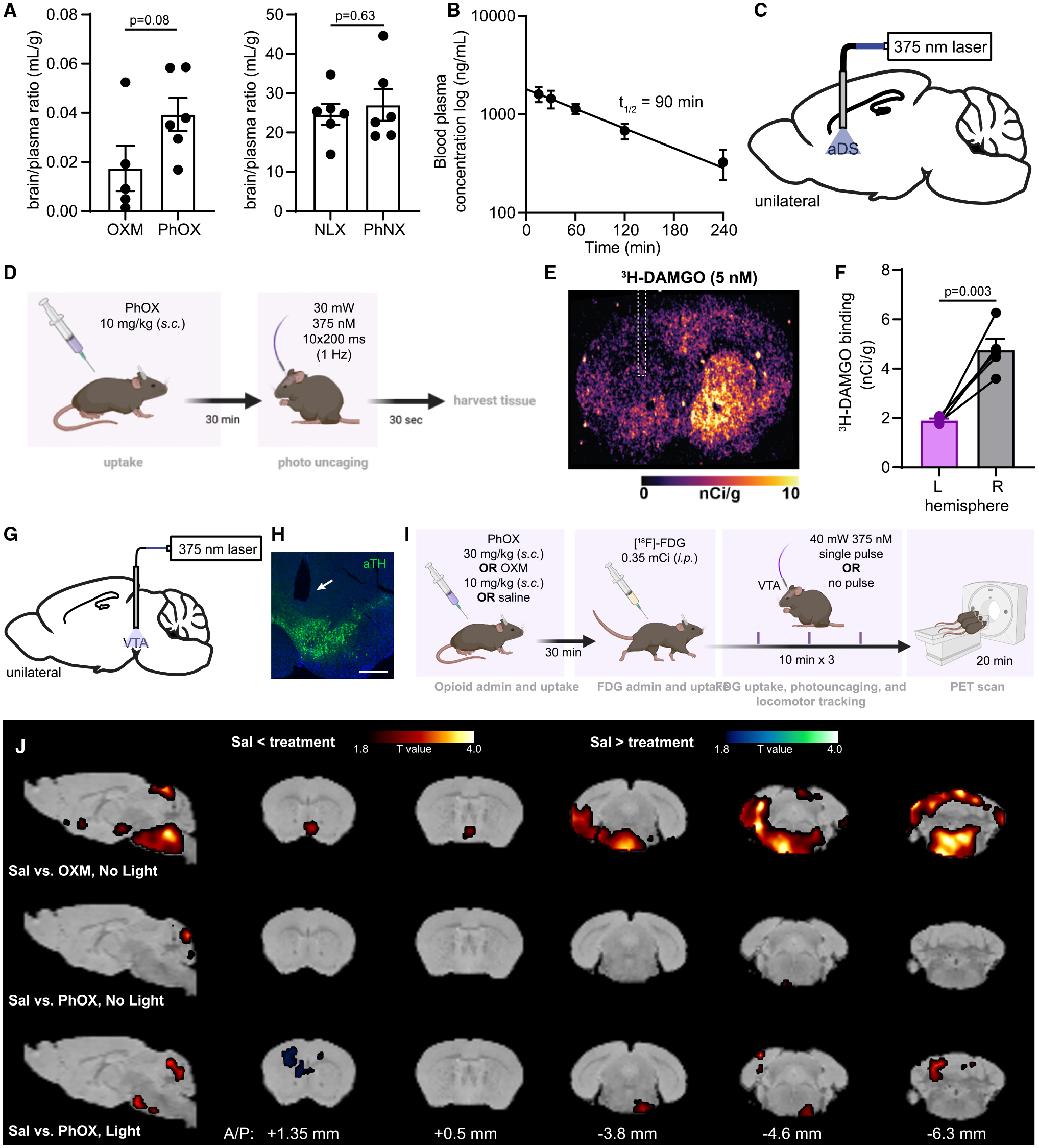
(A) Brain/plasma ratios determined 15 min after administration (n = 5–6 mice per condition, unpaired two-tailed t test). Data are plotted as mean ± SEM.
(B) Time course of PhOX clearance from the bloodstream (15 mg/kg, n = 4 mice). Data are plotted as mean ± SEM.
(C) Schematic indicating the implantation of an optical fiber in the aDS.
(D) Experimental timeline for in vivo uncaging followed by autoradiography.
(E) Autoradiographic image of the fiber implant site in mice.
(F) Quantification of [3H]-DAMGO binding in the illuminated (L) and unilluminated (R) hemispheres (n = 5 sections from 5 mice, paired two-tailed t test). Data are plotted as mean ± SEM.
(G) Schematic indicating the implantation of an optical fiber above the VTA.
(H) Representative fluorescence image of the VTA implant site (scale bars, 0.5 mm).
(I) Schematic depicting the experimental protocol for PET imaging after PhOX uncaging.
(J) Brain-wide voxel-based analysis of [18F]-FDG uptake. Color shaded areas overlaid on the corresponding sections of a brain atlas31 represent clusters of voxels (R100) with significant (p < 0.05) increases or decreases in FDG accumulation compared with saline (n = 5–6 mice per condition).
To determine whether in vivo uncaging can be achieved after systemic administration, we turned to ex vivo autoradiography, which reveals receptor occupancy after drug binding to endogenous receptors in vivo. Mice were implanted unilaterally with optical fibers in the anterior dorsal striatum (aDS) where MORs are prominent (Figure 3C). After recovery, the optical fibers were connected to a 375-nm laser and mice were administered either saline or PhOX (10 mg/kg, subcutaneously [s.c.]), followed by a train of light flashes (10 × 200 ms, 1 Hz, 30 mW, Figure 3D). Immediately after illumination, the brains were removed and flash-frozen. Sections of aDS were briefly incubated with [3H]-DAMGO (5 nM) prior to development on a phosphor screen. Quantification of [3H]-DAMGO binding revealed that PhOX photoactivation resulted in significant MOR occupancy, as evidenced by decreased [3H]-DAMGO binding at the site of illumination compared with the contralateral hemisphere (Figures 3E and 3F). These results demonstrate the feasibility of local in vivo drug uncaging using systemic administration and brief flashes of light.
PET imaging reveals spatially restricted alterations of brain metabolic activity
To assess the spatial control afforded by in vivo uncaging, we imaged the induction of metabolic activity in the brain in response to unilateral PhOX photoactivation in the ventral tegmental area (VTA, Figures 3G and 3H), where MOR agonists disinhibit dopamine neurons.32–34 Using small animal positron emission tomography (PET), we compared the brain-wide neural activity evoked by PhOX photorelease in the VTA to systemic OXM administration. As schematized in Figure 3I, mice were administered either OXM (10 mg/kg, s.c.), PhOX (30 mg/kg, s.c.), or saline (s.c.), followed 30 min later by [18F]-fluorodeoxyglucose (FDG, 0.35 mCi, i.p.) to label metabolically active cells. Mice were subsequently connected to a fiber optic cable and placed in an open field chamber. Over 30 min, half of the saline-treated and PhOX-treated mice were exposed to 375 nm light (3 × 200 ms, 40 mW, one bout every 10 min), whereas the other half received no light. Mice were then anesthetized and placed in the PET scanner to obtain whole-brain maps of uncaging-induced metabolic activity.
Using a significance threshold of p < 0.05, the maps of FDG uptake were compared across saline and drug conditions (Figure 3J). Whereas OXM increased metabolic activity in the ventral midbrain and brainstem, PhOX administration without light minimally altered FDG uptake. This suggests that PhOX is not only inactive at opioid receptors in vivo but also that any off-target actions minimally impact brain activity. In contrast, photoactivation of PhOX in the left VTA enhanced FDG uptake in a small region encompassing the right VTA. This unexpected contralateral signal may reflect compensatory circuit activity or poorly understood contralateral connectivity. Nonetheless, changes in FDG uptake were restricted to the midbrain in one hemisphere only, indicating that the uncaging-induced changes were tightly restricted in space. In all test conditions, modest levels of regional cerebellar activation were observed, but the subregion was inconsistent. These results demonstrate that PhOX is highly inactive prior to light exposure while uncaging leads to spatially confined activation of opioid-sensitive neural circuits.
In vivo PhOX photoactivation drives rapid changes in behavior
To explore the feasibility of using in vivo uncaging to study behavior, we asked whether PhOX photoactivation in the brain modulates behavioral responses to pain in mice. We first confirmed that PhOX is inactive without illumination, using a hot plate assay of thermal nociception (Figure S4A). Compared with saline, PhOX (15 mg/kg, s.c.) administration did not alter nocifensive paw withdrawal latencies nor did it alter morphine efficacy (5 mg/kg, i.p.).
We previously found that photoactivation of the MOR agonist DAMGO in the nucleus accumbens medial shell (NAc-mSh) suppresses the behavioral response to formalin-induced inflammatory pain.22 We therefore asked whether PhOX photoactivation in the NAc-mSh produces analgesia in a related assay in which capsaicin is applied to the hindpaw to trigger thermal sensitivity in the Hargreaves test (Figure 4A). After fiber implantation over the NAc-mSh, PhOX (12 mg/kg, s.c.) was administered and withdrawal latencies were measured at the hindpaw contralateral to the fiber. After application of a thin layer of capsaicin cream (0.1%) produced an increase in thermal pain sensitivity, a train of light flashes (10 × 200 ms, 1 Hz, 45 mW) transiently returned the paw withdrawal latency to baseline 5 min after uncaging (Figure 4B). As a negative control, the same mice were assayed without illumination. Notably, PhOX photoactivation in the NAc-mSh did not alter locomotor behavior (Figures S4B and S4C). These results establish that in vivo PhOX uncaging can alter behavior and reveal that opioid signaling in the NAc-mSh can reduce the thermal hypersensitivity produced by capsaicin.
Figure 4. In vivo photoactivation of PhOX suppresses pain-related behavior and drives behavioral reinforcement.
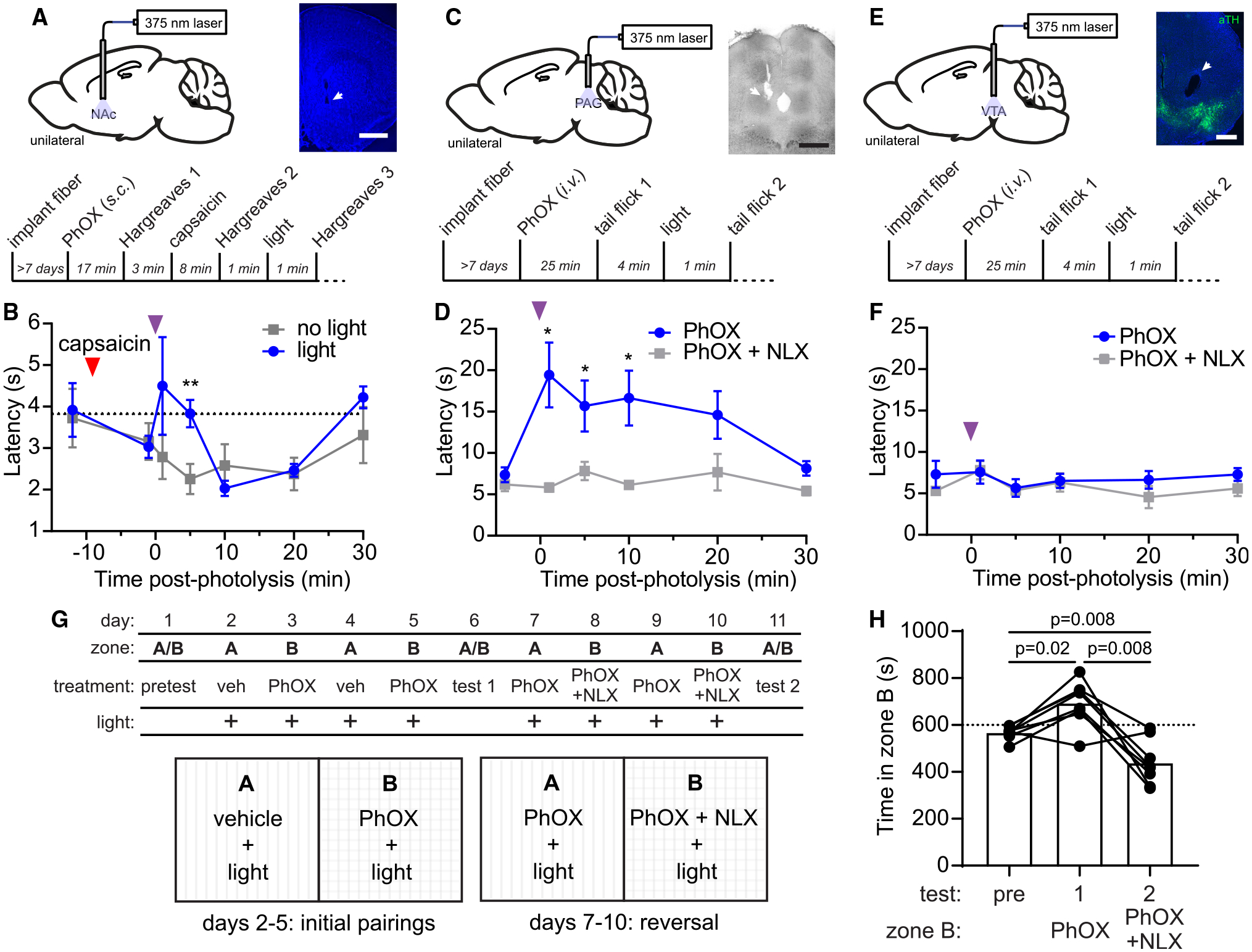
(A) Schematic indicating the implantation of an optical fiber in the NAc-mSh (top left), representative fluorescence image of the optical fiber implant site (scale bars, 1 mm) (top right), and experimental timeline (bottom).
(B) Paw withdrawal latency in the Hargreaves assay (n = 6 mice, ** indicates p < 0.005, paired two-tailed t test).
(C) Same as (A) for the PAG (scale bars, 0.5 mm).
(D) Withdrawal latency in the tail flick assay (n = 6 mice, * indicates p < 0.05, Wilcoxon signed-rank test).
(E) Same as (A) for the VTA (scale bars, 0.5 mm).
(F) Withdrawal latency in the tail flick assay (n = 4 mice, no significant differences detected, Mann-Whitney test).
(G) Schematic depicting the CPP protocol.
(H) Time spent in zone B on test days (n = 8 mice, repeated measures one-way ANOVA, F(1.13,7.88) = 19.4, p = 0.002, Bonferroni’s multiple comparisons test). All data are plotted as mean ± SEM. See also Figure S4.
We next attempted PhOX uncaging in the periaqueductal gray (PAG), where MOR activation produces strong antinociception35 (Figure 4C). Mice were administered either PhOX (15 mg/kg, i.v.) or PhOX + NLX (PhOX, 15 mg/kg; NLX, 10 mg/kg, i.v.) prior to monitoring nocifensive behavior using a tail-flick assay. 1 min after illumination (10 × 200 ms, 1 Hz, 45 mW), tail-flick latency was significantly elevated compared with NLX-treated mice (Figure 4D) and remained so for at least 10 min after uncaging. Notably, the initial increased latency was similar to that observed 30 min after systemic morphine administration (10 mg/kg, i.p.) (Figure S4D). In contrast, PhOX uncaging in the VTA, 1.6 mm anteroventral to the PAG, was ineffective (Figures 4E and 4F). Together, these results validate the use of PhOX to study pain modulation and indicate that in vivo uncaging provides good spatial and temporal control over drug action.
MOR signaling in the VTA drives behavioral reinforcement.36 We therefore asked whether VTA PhOX uncaging produces a conditioned place preference (CPP). We first established that PhOX is neither reinforcing nor aversive without illumination (Figures S4E and S4F). Next, the effect of illumination was examined in a two-phase experiment (Figure 4G). During phase 1, after administration of either vehicle or PhOX (15 mg/kg, i.v.), light (10 × 200 ms, one bout every 5 min, 20 mW) was applied during conditioning. This conditioning paradigm resulted in a preference for the PhOX-paired chamber on test day 1 (Figure 4H). In phase 2, mice were administered both PhOX and NLX (PhOX, 15 mg/kg; NLX, 10 mg/kg, i.v.) in the chamber previously paired with PhOX, and PhOX+vehicle (PhOX, 15 mg/kg, saline, i.v.) in the previously vehicle-paired chamber. This pairing reversal led to a strong preference for the newly PhOX-paired chamber on test day 2.
Local administration of MOR agonist in the VTA also increases locomotor behavior in mice,37 which provides a temporally resolved assay for evaluating the behavioral response to in vivo uncaging.22 After either PhOX (15 mg/kg i.v.) or PhOX+NLX (PhOX, 15 mg/kg; NLX, 10 mg/kg, i.v.) administration, mice were connected to a 375-nm laser via a commutator and placed in an open field chamber (Figures 5A–5C). After 15 min, PhOX uncaging (10 × 200 ms, 1 Hz, 30 mW) triggered a rapid increase in locomotion that was abolished by NLX. Strikingly, locomotion increased within 5 s of the first light flash and peaked within 5 s of the last flash (Figure S5A). Similar to the PhOX uncaging-induced antinociception, the resulting locomotor activation persisted for at least 15 min (Figure 5B). The total distance traveled in the 15 min before and after uncaging is summarized in Figure 5C. These results reveal a rapid behavioral response to drug action that supports the use of photopharmacology to drive mechanistic studies during ongoing behavior.
Figure 5. In vivo photoactivation of PhOX in the VTA drives rapid locomotor activation.
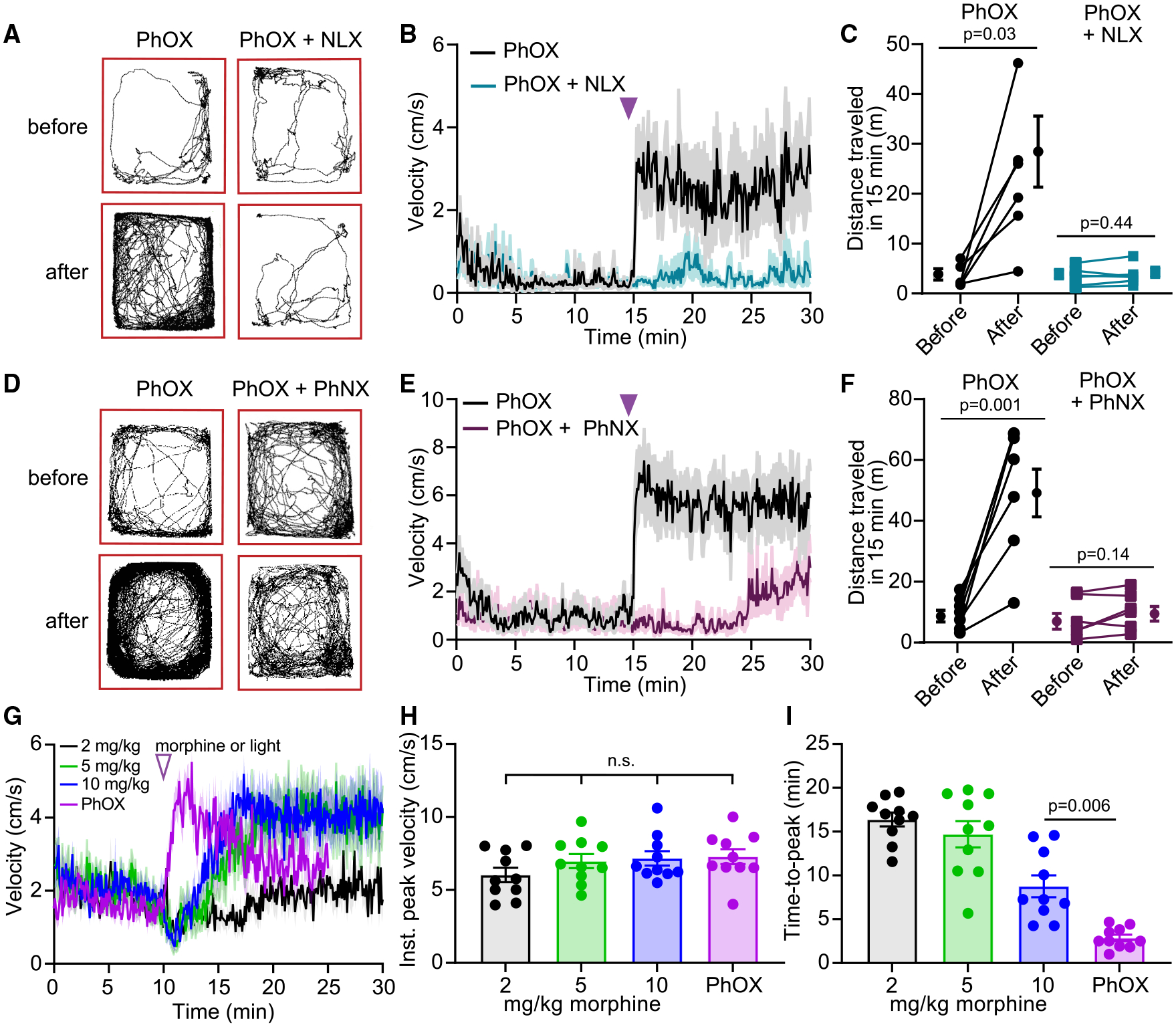
(A) Example maps of open field locomotor activity.
(B) Plot of average velocity over time (n = 6 mice).
(C) Summary plot of the total distance traveled before or after photoactivation (n = 6 mice, Wilcoxon signed-rank test).
(D) Same as (A).
(E) Same as (B) (n = 7 mice).
(F) Same as (C) (n = 7 mice, paired two-tailed t test).
(G) Same as (B) but comparing PhOX photoactivation to systemic morphine (n = 10 mice).
(H) Summary plot of the instantaneous peak velocity reached after morphine injection or PhOX photoactivation (n = 10 mice, repeated measures one-way ANOVA, F(2.2, 19.3) = 2.33, p = 0.12).
(I) Summary plot of the time to reach the peak locomotor response (n = 10 mice, repeated measures one-way ANOVA, F(1.9, 16.9) = 36.12, p < 0.0001, Tukey’s multiple comparisons test). All data are plotted as mean ± SEM. See also Figure S5.
To evaluate the potential for in vivo PhNX uncaging, we repeated this experiment while replacing NLX with PhNX (30 mg/kg), such that illumination would simultaneously release both OXM and NLX. As shown in Figures 5D–5F, inclusion of PhNX completely prevented the initial locomotor response, although it increased after 10 min, which suggests that NLX may be cleared more rapidly than OXM after photorelease.
In order to benchmark PhOX photoactivation to systemic morphine, we compared the locomotor response evoked by multiple doses of morphine (2, 5, 10 mg/kg, i.p.) to that produced by unilateral uncaging in the VTA (10 mg/kg, s.c., 10 × 200 ms, 1 Hz, 20 mW) (Figure 5G). Whereas the response to light was immediate and rapid (τon = 0.8 min), the response to morphine injection was delayed by several minutes and slower to rise, presumably due to morphine’s pharmacokinetics (5 mg/kg τon = 3.7 min, 10 mg/kg τon = 2.9 min). Although the instantaneous peak velocity was similar across conditions (Figure 5H), the time to reach peak velocity was shorter for PhOX uncaging than for morphine (10 mg/kg) (Figure 5I). These results demonstrate that unilateral PhOX uncaging recapitulates the locomotor effect of 5–10 mg/kg morphine, but with exceptional temporal precision and kinetics.
Using this assay, we also found that low-cost LEDs (365 and 385 nm), despite being power limited (~2 mW fiber output), are able to evoke modest behavioral responses, though inferior to a 375-nm laser (15 mW) (Figures S5B–S5D). Exploring the relationship between the timing and dose of PhOX administration revealed that photoactivation experiments can begin 15 min after PhOX (s.c.) administration (Figures S5E–S5G), but that the response to light is smaller 90 min post-injection compared with 30 min, and greatly diminished after 3 h, consistent with PhOX’s 90 min half-life (Figures S5H–S5J). We also observed that even the smallest dose tested (2.5 mg/kg, s.c.) affords a behavioral response (Figures S5K–S5M) and that the response to 10 flashes (200 ms, 1 Hz, 30 mW) trends toward being larger than the response to 3 flashes (Figures S5N–S5P). We also assessed the extent to which PhOX uncaging causes tissue damage (Figure S6). Immunohistochemical analysis of dorsal striatum tissue sections just below the fiber tip did not reveal changes in the expression of cellular markers for neuroinflammation at 4 h (Iba1) and 24 h (Iba1 and GFAP) after application of a repetitive uncaging stimulus (four bouts of 10 × 200 ms, 1 Hz, 20 mW) in the presence of PhOX (15 mg/kg, i.v.) and NLX (10 mg/kg, i.v., to prevent MOR signaling). Together, these results establish a range of working conditions for achieving reliable behavioral responses to PhOX photoactivation in vivo.
Photopharmacology interfaces with fiber photometry
Photopharmacology offers the opportunity to integrate pharmacological manipulations into experimental frameworks for all-optical interrogation of neural circuits.38 To explore this possibility, we combined PhOX uncaging with fiber photometry. Because VTA opioid-evoked locomotor activation and behavioral reinforcement are associated with dopamine release in the NAc,39,40 we monitored extracellular dopamine using the genetically encoded dopamine sensor dLight1.3b while photo-activating PhOX in the VTA (Figures 6A and 6B). After viral transduction in the NAc-mSh, mice were implanted with two fibers: one over the NAc-mSh to measure dopamine and another over the ipsilateral VTA for PhOX uncaging. After recovery, mice were administered either PhOX (15 mg/kg, i.v.) or PhOX + NLX (PhOX, 15 mg/kg; NLX, 10 mg/kg, i.v.), fitted with optical fibers, and then placed into an open field. After a baseline period, photoactivation with a single light flash (200 ms, 50 mW) produced a large, rapid increase in extracellular dopamine that was abolished by NLX (Figures 6C, 6D, S7A, and S7B). Dopamine release began within 3 s of the flash, reached 90% of the maximum value within 10 s (τon = 4.25 s) (Figure S7C), and decayed over the course of several minutes (τoff = 5.8 min). This optical stimulus was ineffective in the absence of PhOX (Figure S7D), indicating that light does not activate dopamine neurons via tissue heating.41 Furthermore, in the presence of PhOX, VTA irradiation at 473 nm (200 ms, 50 mW) was also ineffective, verifying that imaging light does not produce significant uncaging (Figure S7E). Photoactivation of PhOX (15 mg/kg, i.v.) using single flashes of varied intensity (5–20 mW) yielded sub-saturating dopamine responses (Figures 6E and 6F). Notably, 1 mW was sufficient to produce detectable dopamine release, indicating relatively high light sensitivity under these conditions. Furthermore, despite the small amplitude, the 1 mW uncaging response lasted for several minutes, which aligns well with the kinetics observed in LC slices. Application of two flashes several minutes apart drove dopamine release repeatedly, albeit with reduced efficacy on the 2nd flash (Figure S7F). These results demonstrate the compatibility of in vivo photopharmacology with fluorescence-based measurements of neural function and establish a robust, spatiotemporally precise assay for probing the opioid sensitivity of mesolimbic dopamine circuitry.
Figure 6. Interfacing in vivo photopharmacology with fiber photometry.
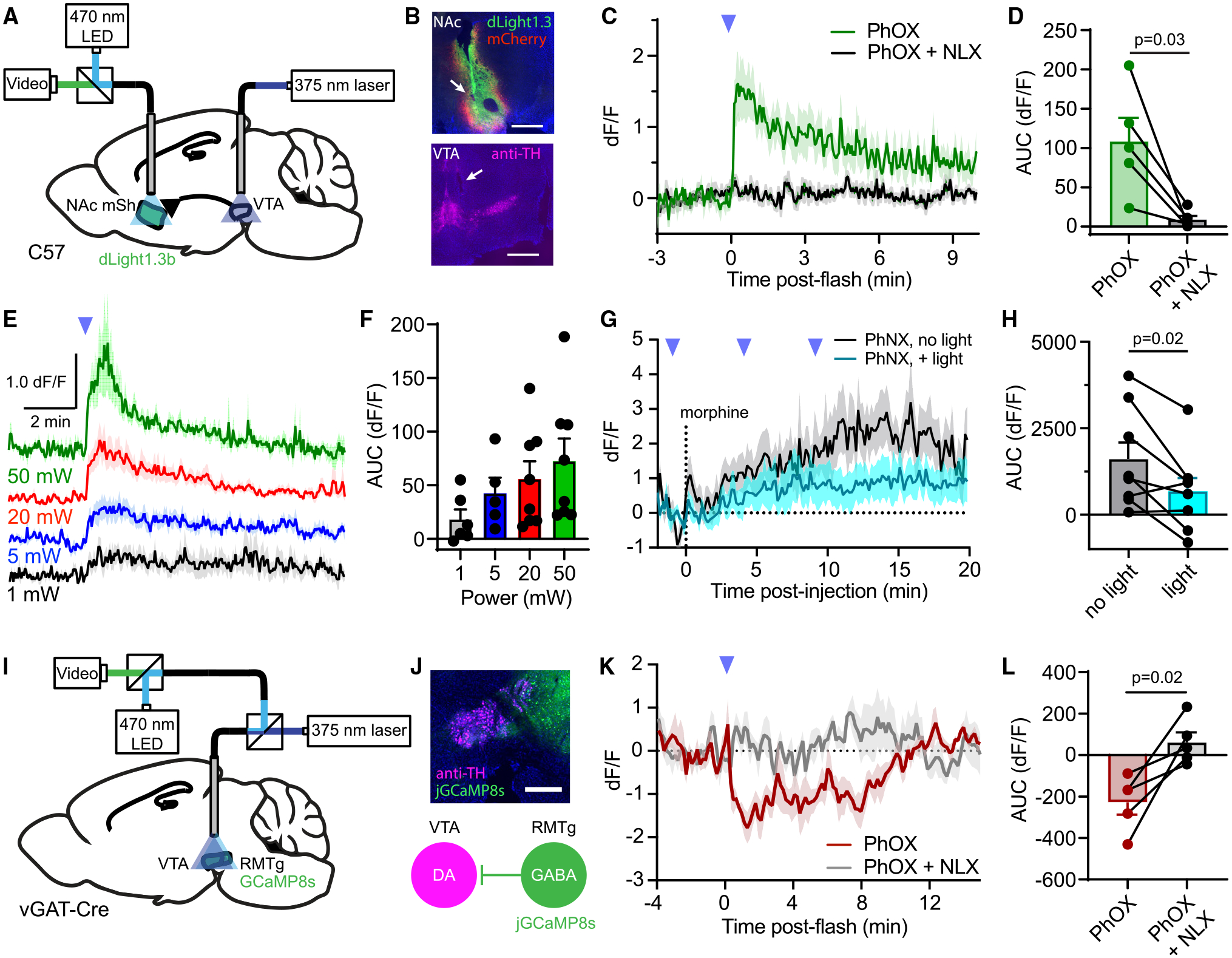
(A) Schematic indicating fiber implantation in the VTA for uncaging and fiber photometry recording of extracellular dopamine in the ipsilateral NAc-mSh.
(B) Example images of viral expression and fiber implantation site for fiber photometry in the NAc-mSh, and uncaging site in the VTA by immunostaining for tyrosine hydroxylase (TH, magenta). Scale bars, 0.5 mm (NAc) and 0.75 mm (VTA).
(C) Average NAc-mSh dLight1.3b fluorescence in response to VTA PhOX uncaging with a single light flash (n = 5 mice).
(D) Summary plot of the data shown in (C) (AUC [area under the curve], n = 5 mice, paired two-tailed t test).
(E) Average dLight1.3b fluorescence in response to a single light flash at the indicated light powers (n = 6–8 mice).
(F) Summary plot of the data shown in (E) (n = 6–8 mice).
(G) Average dLight1.3b fluorescence in the NAc-mSh in response to systemic morphine, with or without PhNX uncaging in the ipsilateral VTA (n = 8 mice).
(H) Summary plot of the morphine-evoked fluorescence changes shown in (G) (n = 8 mice, paired two-tailed t test).
(I) Schematic indicating implantation of a fiber over the VTA coupled to both a 375-nm laser and a fiber photometry recording system to detect changes in Ca2+ activity, with jGCaMP8s expressed in RMTg GABA neurons.
(J) (Top) Example image of the fiber implant site along with jGCaMP8s expression in RMTg GABA neurons. Scale bars, 0.5 mm. (Bottom) Diagram depicting inhibition of VTA dopamine (DA) neurons by jGCaMP8s-expressing RMTg GABA neurons.
(K) Average normalized jGCaMP8s fluorescence in response to PhOX uncaging with a single light flash (n = 5 mice).
(L) Summary plot of the data shown in (K) (n = 5 mice, paired two-tailed t test). All data are plotted as mean ±SEM. See also Figure S7.
Using the same experimental configuration, we assessed the ability of unilateral PhNX uncaging to suppress activation of the mesolimbic dopamine circuit by systemic morphine (Figures 6G and 6H). As expected, morphine (5 mg/kg, i.p.) produced a sustained increase in NAc-mSh dopamine. However, photoactivation of PhNX in the ipsilateral VTA (15 mg/kg, i.v., injected 15 min prior to morphine, 10 × 200 ms, 1 Hz, 20 mW) attenuated the morphine-evoked dopamine release. This result further validates PhNX for in vivo applications.
Thus far, we photoactivated opioid drugs and optically recorded the physiological response at discrete sites, which minimizes potential artifacts due to sensor bleaching and/or UV-evoked photostates. Yet, photoactivation at the recording site enables unequivocal assignment of experimental outcomes to receptor engagement in that brain region. For example, MOR agonists are thought to activate VTA dopamine neurons through a dis-inhibitory mechanism involving the suppression of GABA release. In vivo and ex vivo electrophysiology experiments implicate opioid-sensitive GABA neurons located in the adjacent rostromedial tegmental nucleus (RMTg),42–44 but this has been difficult to directly observe in vivo due to challenges associated with site-specific drug delivery during cell-type-specific recording of neural activity. We reasoned that direct measurement of the impact of PhOX photoactivation on Ca2+ activity in RMTg GABA neuron terminals in the VTA would be a convenient and advantageous approach for addressing this question.
To enable same-site photouncaging and fiber photometry, we constructed a relay system that feeds the 375-nm laser into the photometry output pathway (Figures 6I and S7G). We transduced the RMTg in Slc32a1-cre (vGAT-IRES-Cre) mice with the genetically encoded Ca2+ indicator jGCaMP8s,45 along with mCherry for motion correction, and then implanted an optical fiber over the VTA (Figure 6J). This allowed us to record Ca2+ activity in RMTg GABA neuron terminals in the VTA while locally photoreleasing OXM with a single light flash (200 ms, 20 mW), while also recording green and red fluorescence through the fiber, before and after the administration of PhOX (30 mg/kg, s.c.). In the absence of PhOX, the UV flash caused a transient increase in green fluorescence that decayed over several minutes (τoff = 3.1 min, Figure S7H). This artifact was consistent, which allowed us to correct for it by subtracting the average of 3 pre-injection trials from post-drug administration recordings. This analysis revealed that PhOX (15 mg/kg, i.v.) photoactivation suppressed intracellular Ca2+ signaling in RMTg GABA neuron terminals located within the VTA (Figures 6K and 6L). Co-administration of NLX (10 mg/kg) blocked the observed suppression, confirming that it results from opioid receptor activation. Together, these results demonstrate that in vivo uncaging is compatible with fiber photometry and establish that RMTg GABA neuron terminals are suppressed by MOR agonists locally within the VTA in vivo.
In vivo opioid uncaging reveals mesolimbic dopamine circuit adaptations to sustained morphine exposure
Sustained consumption of opioid drugs produces tolerance. This greatly complicates their clinical use for pain treatment and contributes to the escalation of drug-seeking behavior in the context of opioid addiction. Although MORs are known to desensitize on multiple timescales upon sustained activation,46 how the opioid sensitivity of the mesolimbic dopamine circuit is altered by prolonged MOR activation is not known. Because PhOX uncaging restricts drug action to the VTA while producing a highly stereotyped dopamine transient in the NAc, in vivo photopharmacology provides a unique opportunity to probe for changes within the VTA without confounds due to opioid action in other brain areas, including the NAc itself.47–49
To ask how VTA opioid-NAc-mSh dopamine coupling might be altered by sustained MOR activation, we measured the NAc-mSh dopamine response to VTA PhOX uncaging (3 × 200 ms, 1 Hz, 20 mW) before, during, and several days after administering mice either chronic saline as a control, or escalating doses of chronic morphine (twice a day with 10, 20, 30, 40, and 50 mg/kg over 5 days) (Figures 7A and 7B). Importantly, photometry measurements were taken ~8 h after morphine administration, such that any observed changes would not be attributable to occlusion, as the half-lives of morphine and its glucuronide adducts are on the order of 30 min in mice.50 As expected, chronic saline treatment did not alter NAc-mSh dopamine release in response to VTA PhOX uncaging (Figure 7C). In contrast, compared with the morphine-naive state, chronic morphine attenuated VTA opioid-NAc dopamine coupling, reflected as a decrease in the area under the curve (Figure 7D). After several days of abstinence, the dopamine response showed a trend toward recovery. These results indicate that the opioid sensitivity of the mesolimbic dopamine circuit is reduced by prolonged MOR activation with morphine.
Figure 7. Combined in vivo uncaging and fiber photometry reveal drug-dependent changes in the opioid sensitivity of mesolimbic dopamine signaling.
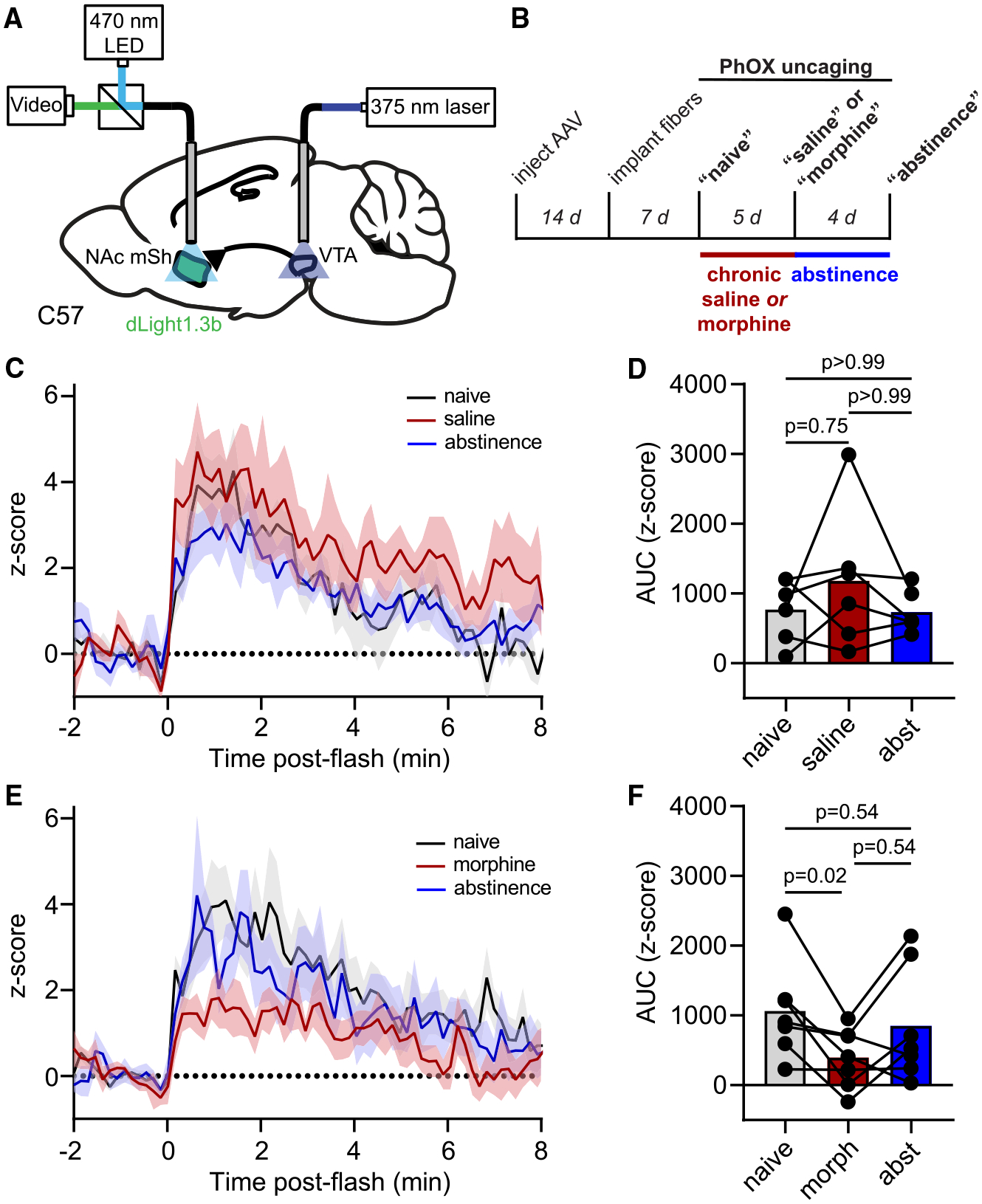
(A) Schematic indicating fiber implantation in the VTA for uncaging and fiber photometry recording of extracellular dopamine in the ipsilateral NAc-mSh.
(B) Experimental timeline.
(C) Average Z scored dLight1.3b fluorescence in the NAc-mSh in response to PhOX uncaging before, during, and after chronic saline administration (n = 6 mice).
(D) Summary plot of the data shown in (C) (n = 6 mice, Friedman test, p = 0.57, Dunn’s multiple comparisons test).
(E) Same as (C) but for chronic morphine (n = 7 mice).
(F) Same as (D) but for chronic morphine (n = 7 mice, Friedman test, p = 0.027, Dunn’s multiple comparisons test). All data are plotted as mean ± SEM.
DISCUSSION
In this study, we have demonstrated that in vivo photopharmacology interfaces with temporally resolved behavioral and neurophysiological experiments that are commonly employed to investigate the mammalian nervous system. Importantly, we showed that careful molecular design can produce water soluble, systemically active, BBB-penetrant caged drugs that can be effectively released in the brain through implanted optical fibers upon illumination with well-tolerated optical stimuli (375 nm, 200-ms flashes, 1–20 mW). Because the DMNPE caging group does not absorb blue and green light, photoactivation could be selectively achieved with UV light during one-photon optical recordings of intracellular Ca2+ and extracellular dopamine. Furthermore, we showed that optical recording and drug photoactivation can be conducted through the same optical fiber, which provides important experimental advantages. Finally, we showed that the robust stimulus-response relationship afforded by drug photoactivation can be used as the basis of longitudinal studies that track experimentally induced changes in stimulus-response coupling. In the process of establishing an experimental framework for using photopharmacology to study the brain, we also validated two reagents (PhOX and PhNX) that can be used to bidirectionally control endogenous MOR signaling in vivo.
In response to PhOX photoactivation in the VTA, we reliably observed dopamine release in the NAc using fiber photometry, consistent with numerous in vivo studies in rodents.32,51,52 However, PET imaging studies of [11C]-raclopride displacement from D2 dopamine receptors in humans are more conflicted. Although two studies found no increase in NAc dopamine after systemic administration of MOR agonists in heroin addicts,53,54 a more recent study observed morphine-evoked NAc dopamine release in non-addicted subjects.55 Our finding that VTA opioid-evoked NAc dopamine release is reduced after chronic opioid exposure offers a mechanistic explanation for this apparent conundrum. We also found that MOR activation locally suppresses output from GABAergic RMTg neurons in the VTA. In conjunction with ex vivo brain slice electrophysiology studies,43 our results are consistent with opioid tolerance in the mesolimbic dopamine pathway resulting from diminished opioid sensitivity of RMTg GABA neuron input to VTA dopamine neurons. These circuit adaptations are likely to contribute to the tolerance to the rewarding effects of opioids that occurs after sustained drug use, a hall-mark of opioid addiction.56
Most prior in vivo photopharmacology studies involve photoswitchable drugs,9 which, in principle, offer several advantages over caged drugs that are photoreleased irreversibly. Most importantly, they can be turned off after photoactivation, either with light3,57 or by using photoswitches that rapidly convert back to the inactive state in the dark.58 In contrast, caged drugs and transmitters must rely on dilution via diffusion and cellular metabolism for deactivation—this likely accounts for the slow reversal kinetics we observed after PhOX photoactivation. Although freely diffusing caged drugs and photoswitchable ligands target endogenous receptors and are applicable across species without genetic engineering, they do not offer cell-type specificity. Tethered photoswitches are genetically targetable and less prone to diffusion when anchored to a cell membrane, which provides both cell-type specificity and subcellular control over receptor activation. Encouragingly, tethered drugs have achieved cell-type-specific engagement of endogenous receptors in vivo (e.g., DARTs).59 Tethered photoswitchable6,60,61 and caged ligands62 have also been shown to target endogenous receptors in a cell-specific manner. However, a major drawback to photoswitchable drugs is their broad wavelength sensitivity, which limits their applicability in imaging experiments. The insensitivity of UV-sensitive caging groups to visible light is a primary advantage of caged ligands demonstrated in our study. Because such caging groups exhibit poor two-photon cross sections, they are likely compatible with two-photon imaging as well. Furthermore, photoswitchable ligands are often hindered by a limited photodynamic range that results from incomplete photoconversion between isomers and a limited impact of photoisomerization on drug efficacy (see Acosta-Ruiz et al.5 for a notable exception). In contrast, adding a caging group to a key site on a small molecule drug readily diminishes residual activity, often with minimal synthetic effort. Although photoswitchable ligands warrant further investment, caged drugs offer a reliable and convenient route to spatiotemporally resolved behavioral photopharmacology experiments. In either case, the experimental framework and performance bench-marks established in our study will help guide the development of optimal photopharmacological tools for in vivo applications.
Finally, the therapeutic potential of in vivo photopharmacology should not be overlooked, as light provides a means for spatially targeted drug delivery that minimizes side effects due to drug action at other sites, as well as for restricting drug action to only the time at which it is needed.2 These features are particularly desirable in the context of opioid analgesics, which, in addition to suppressing pain at multiple sites in the nervous system, can have undesirable adverse effects, such as severe respiratory depression and constipation, and are extremely addictive.15 Systemically available photoactivatable opioids such as PhOX open the door to the tantalizing possibility of restricting opioid action to sites that produce pain relief only when pain is felt or anticipated, using an external or implanted light source.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Matthew Banghart (mbanghart@ucsd.edu).
Materials availability
Samples of PhOX and PhNX are available upon request. For large quantities, Dr. Banghart will help coordinate the custom synthesis and characterization from contract resource organizations. Potential users should also express their interests to the NIDA and NIMH Drug Supply Programs so that PhOX and PhNX may be incorporated into their catalog for free distribution.
Data and code availability
Source data for all figures are available in the supporting information (Data S2). Raw data are available upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Cell lines
The commercially available HEK293T cell line (sex: female) was used in this study. Detailed growth conditions are reported in the methods details section.
Mice
Animal experimentation: All procedures were performed in accordance with protocols approved by the University of California, San Diego and the National Institute on Drug Abuse Intramural Research Program’s Institutional Animal Care and Use Committees (IACUC) following guidelines described in the US National Institutes of Health Guide for Care and Use of Laboratory Animals (UCSD IACUC protocol S16171, NIDA IACUC protocol 21-NRB-43). All surgery was performed under isoflurane anesthesia. For ex vivo brain slice electrophysiology, male and female P15–35 C57/Bl6 and PValb-Cre/Ai14 mice were used for hippocampus experiments, and male and female P8–14 Long-Evans rats were used for locus coeruleus experiments. For in vivo photoactivation experiments, 2–6 month old wild-type C57/Bl6 or vGAT-Cre mice were used either from Jackson Labs or bred in house on a C57/bl6j background. Mice were maintained on a reverse light dark cycle (12:12 dark:light) with ad libitum access to food and water and nesting material for environmental enrichment. Both male and female mice were used. All experiments were performed during the dark cycle.
METHOD DETAILS
Chemical synthesis and characterization
Commercial reagents were used as received. Oxymorphone free base was obtained from Noramco (516340913), naloxone HCl was obtained from Sigma Aldrich (N7758), 4-(bromomethyl)-7-(diethylamino) coumarin was obtained from TCI (B5008), and 1-(1-bromoethyl)-4,5-dimethoxy-2-nitrobenzene was synthesized according to a published procedure.64 All solvents were purchased as septum-sealed bottles stored under an inert atmosphere. All reactions were sealed with septa through which a nitrogen atmosphere was introduced unless otherwise noted. Reactions were conducted in round-bottomed flasks or septum-capped amber screw-cap vials containing Teflon-coated magnetic stir bars. Room lights were covered with Roscolux Canary Yellow #312 film (Rosco Laboratories, Stamford, CT) to filter out wavelengths of light that could lead to unintentional photolysis during purification and handling.
1-methyl-4,5-dimethoxy-2-nitrobenzene oxymorphone (PhOX)
To a stirred solution of oxymorphone (80 mg, 0.265 mmol) and 1-(1-bromoethyl)-4,5-dimethoxy-2-nitrobenzene (92 mg, 0.317 mmol) in anhydrous DMF (1 mL) in an amber glass vial, anhydrous K2CO3 (73 mg, 0.528 mmol) was added. The mixture was stirred at 22 °C. After 16 h, TLC showed the completion of the reaction and water (1 mL) was added. The mixture washed with EtOAc (5 mL × 2). The combined organic phases were washed with 5% LiCl (10 mL) and brine (10 mL). The organic phase was dried over Na2SO4 and concentrated under vacuum. The residual was purified by column chromatography (SiO2, EtOAc → EtOAc/MeOH (9:1)) to give PhOX as a light-yellow oil (85 mg, 63%).
1H NMR (300 MHz, CDCl3) δ 7.58 (m, 1H), 7.38 – 7.24 (m, 1H), 6.62 – 6.19 (m, 3H), 5.02 (s, 1H), 4.62 (m, J = 11.2 Hz, 1H), 4.05 – 3.87 (m, 6H), 3.12 – 2.88 (m, 2H), 2.81 (m, 1H), 2.52 – 2.30 (m, 6H), 2.26 – 2.15 (m, 1H), 2.06 (m 1H), 1.88 – 1.77 (m, 1H), 1.72 (m, 3H), 1.59 – 1.34 (m, 2H).
13C NMR (75 MHz, CDCl3) δ 208.10, 207.61, 154.25, 153.77, 147.75, 147.61, 145.73, 144.99, 140.32, 140.28, 139.84, 139.76, 135.24, 134.75, 129.78, 129.50, 126.13, 125.17, 119.62, 119.41, 118.47, 115.58, 108.86, 108.53, 107.67, 107.42, 90.41, 90.35, 73.92, 71.81, 70.25, 70.23, 64.48, 64.41, 56.69, 56.42, 56.26, 50.36, 50.10, 45.17, 45.12, 42.66, 36.05, 35.96, 31.50, 31.37, 30.57, 30.37, 23.57, 23.46, 21.93, 21.86.
LR-MS (ESI) m/z 511 [(M+H)+, 70%], 533 [(M+Na)+, 100%].
HR-MS m/z 533.1889 [M+Na]+ (calcd for C27H30N2O8Na, 533.1884).
1-methyl-4,5-dimethoxy-2-nitrobenzene naloxone (PhNX)
To a stirred solution of naloxone hydrochloride dihydrate (106 mg, 0.265 mmol) and 1-(1-bromoethyl)-4,5-dimethoxy-2-nitrobenzene (92 mg, 0.317 mmol) in anhydrous DMF (1 mL) in an amber glass vial 22 °C, anhydrous K2CO3 (110 mg, 0.796 mmol) was added. After 16 h, TLC showed the completion of the reaction and water (1 mL) was added. The mixture was washed with EtOAc (5 mL × 2). The combined organic phases were washed with 5% LiCl (10 mL) and brine (10 mL). The organic phase was dried over Na2SO4 and concentrated under vacuum. The residual was purified by column chromatography (SiO2, hexane → hexane/EtOAc (9:1)) to give PhNX as a light-yellow oil (95 mg, 67%).
1H NMR (300 MHz, Methanol-d4) δ 7.58 (m, 1H), 7.34 (m, 1H), 6.78 – 6.50 (m, 2H), 6.35 – 6.23 (m, 1H), 5.88 (m, 1H), 5.20 (m, 2H), 4.71 (m, 1H), 3.91 (m, 6H), 3.26 – 2.84 (m, 5H), 2.65 – 2.30 (m,3H), 2.23 – 1.94 (m, 2H), 1.88 – 1.64 (m, 4H), 1.59 – 1.47 (m, 1H), 1.45 – 1.15 (m, 1H).
13C NMR (75 MHz, Methanol-d4) δ 208.66, 153.81, 147.90, 145.55, 140.15, 139.97, 135.31, 134.40, 129.95, 126.86, 119.48, 119.12, 116.88, 108.82, 107.59, 90.16, 73.92, 70.18, 61.92, 57.19, 55.42, 55.35, 50.62, 43.02, 35.29, 31.29, 30.22, 22.30, 22.22.
LR-MS (ESI) m/z 537 [(M+H)+, 100%], 559 [(M+Na)+, 35%].
HR-MS m/z 537.2230 [M+H]+ (calcd for C29H33N2O8, 537.2231).
diethylaminocoumarin-oxymorphone (DEAC-OXM)
To a solution of oxymorphone (7.2 mg, 0.024 mmol) in dry DMF (2 ml) was carefully added compound 4-(bromomethyl)-7-(diethylamino) coumarin (9.2 mg, 0.026 mmol) and K2CO3 (9.9 mg, 0.072 mmol) at 22 °C. After 16 h, TLC showed the completion of the reaction and water (1 mL) was added. The aqueous layer was extracted with ethyl ether (2 × 10 mL). The combined organic phases were washed with 5% LiCl (10 mL) and brine (10 mL). The organic phase was dried over Na2SO4 and concentrated under vacuum. The organic phase was dried over Na2SO4 and concentrated under vacuum. The residual was purified by column chromatography (SiO2, hexane → hexane/EtOAc (9:1)) to give DEAC-OXM (10 mg, 81%) as a yellow oil.
1H NMR (600 MHz, Methanol-d4) δ 7.62 (d, J = 9.0 Hz, 1H), 6.83 (d, J = 8.2 Hz, 1H), 6.76 (dd, J = 9.1, 2.6 Hz, 1H), 6.70 (dd, J = 8.2, 0.9 Hz, 1H), 6.55 (d, J = 2.6 Hz, 1H), 6.24 (d, J = 1.2 Hz, 1H), 5.50 (dd, J = 15.0, 1.3 Hz, 1H), 5.38 (dd, J = 15.0, 1.3 Hz, 1H), 4.83 (s, 1H), 3.49 (q, J = 7.1 Hz, 4H), 3.06 (td, J = 14.4, 5.1 Hz, 1H), 2.96 (d, J = 5.8 Hz, 1H), 2.62 (dd, J = 18.8, 5.8 Hz, 1H), 2.58–2.47 (m, 2H), 2.45 (s, 3H), 2.25 – 2.12 (m, 2H), 1.94 – 1.85 (m, 1H), 1.65 – 1.56 (m, 1H), 1.54 – 1.46 (m, 1H), 1.23 (t, J = 7.1 Hz, 6H).
13C NMR (150 MHz, Methanol-d4) δ 209.27, 163.31, 156.13, 152.99, 150.98, 145.32, 140.76, 130.21, 127.34, 125.19, 119.72, 118.80, 109.02, 106.09, 105.02, 96.81, 90.66, 70.36, 67.94, 64.41, 50.16, 45.05, 44.21, 41.53, 35.40, 31.43, 29.90, 21.63, 11.36.
LR-MS (ESI) m/z 531 [(M+H)+, 100%].
HRMS (m/z): calculated for [C31H35N2O6]+ : 531.2490, found 531.2485.
RE-DEAC-OXM
At room temperature, 500 mL DEAC-OXM HCl (50 uM in PBS) was stirred vigorously and exposed 405 nm (13.5 mW). After 16 h, then the pH of the solution was adjusted to ~8 and extracted with EtOAc (500 mL × 2). Combined organic layers were washed with brine (500 mL), dried by Na2SO4, and concentrated under vacuum. The residue was purified by C18 flash chromatography (water/CH3CN, 95%/5%→ 100%) to give RE-DEAC-OXM (4 mg, 31%) as a yellow solid.
1H NMR (600 MHz, Methanol-d4) δ 8.49 (s,1H), 7.63 (d, J = 9.1 Hz, 1H), 6.69 (dd, J = 9.1, 2.6 Hz, 1H), 6.66 (s, 1H), 6.49 (d, J = 2.6 Hz, 1H), 5.71 (s, 1H), 4.10 (d, J = 15.9 Hz, 1H), 3.93 (d, J = 15.9 Hz, 1H), 3.57 (d, J = 5.9 Hz, 1H), 3.45 (q, J = 7.1 Hz, 4H), 3.35 (d, J = 19.3 Hz, 1H), 3.11 (dd, J = 12.7, 4.2 Hz, 1H), 3.00(dd, J = 19.3, 5.9 Hz, 1H), 2.84 (s, 3H), 2.72 (m, 2H), 2.26 (dt, J = 19.3, 3.2 Hz, 1H), 1.99 (dd, J = 14.2, 3.2 Hz, 1H), 1.67 (m, 2H), 1.18 (t, J = 7.1 Hz, 6H).
13C NMR (150 MHz, Methanol-d4) 210.22, 165.07, 158.82, 157.42, 152.34, 145.65, 139.48, 129.33, 127.60, 127.23, 122.59, 122.13, 110.38, 109.39, 108.09, 98.08, 91.02, 71.53, 68.01, 50.15, 48.35, 45.58, 41.82, 32.32, 28.93, 24.30, 12.75.
LR-MS (ESI) m/z 531 [(M+H)+, 100%].
HRMS (m/z): calculated for [C31 H35N2O6]+: 531.2490, found 531.2493.
Authentication and data analysis
Reactions were monitored by liquid chromatography-mass spectrometry (LC-MS) using C-18 column (4.6 × 50 mm, 1.8 μm, Agilent) with a linear gradient (water/MeCN 5%/95% → MeCN 100%, 0–8 min with 0.1% formic acid, 1 ml/min flow, electrospray ionization, positive ion mode, UV detection at 210 nm, 254 nm, and 350 nm). High-resolution mass spectrometry data were obtained at the UCSD Chemistry and Biochemistry Mass Spectrometry Facility on an Agilent 6230 time-of-flight mass spectrometer (TOFMS). Proton (1H) and carbon (13C) NMR spectra were recorded at room temperature in base-filtered CDCl3 on a Bruker AVA-300 spectrometer operating at 300 MHz for proton and 75 MHz for carbon nuclei. For 1H NMR spectra, signals arising from the residual protioforms of the solvent were used as the internal standards. 1H NMR data are reported as follows: chemical shift (δ) [multiplicity, coupling constant(s) J (Hz), relative integral] where multiplicity is defined as: s = singlet; d = doublet; t = triplet; q = quartet; m = multiplet or combinations of the above. All NMR spectra were processed using MestReNova 14.2.1. UV-visible spectra were recorded on a NanoDrop 2000 UV-VIS spectrophotometer (Thermo-Fisher).
In vitro uncaging and dark stability
To determine dark stability, PhOX and PhNX (1 mM) were dissolved phosphate-buffered saline (PBS, pH 7.2) and left in the dark for 24 h. Comparison of samples taken at 0 and 24 h by HPLC-MS (1260 Affinity II, Agilent Technologies, Santa Clara, CA, USA) revealed no obvious decomposition or conversion to oxymorphone or naloxone. In addition to determining the chemical composition of the uncaging product by HPLC-MS, the initial photolysis rate of PhNX and PhOX was compared using HPLC in response to illumination with 375 nm light. Solutions of PhOX and PhNX (1 mM) dissolved in PBS buffer (pH 7.2) were placed in 1 mL glass vials with stir bars and illuminated at a light intensity of 20 mW from the output of a 375 nm laser (LBX-375–400-HPE-PPA, Oxxius, France) via an optical fiber (FT200UMT, 200 μm, 0.39 NA). The solutions were illuminated in 15 sec periods.
Data analysis
Samples were removed and analyzed by LC-MS using a linear gradient (water/MeCN 5%/95% → MeCN 100%, 0–8 min with 0.1% formic acid) and a (C-18 column (4.6 × 50 mm, 1.8 μm) (Agilent). The photoproducts of DEAC-OXM were generated and analyzed in an analogous manner. The integrals of the remaining caged molecule from each sample were normalized to the integral of the unilluminated sample, averaged and plotted against time for the first two minutes of illumination. Linear regression provided a measurement of slope, and the ratio of the two slopes was used to determine the relative photolysis curve.
G-protein signaling assay
GloSensor assay of G-protein signaling. Human embryonic kidney 293T (HEK293T) cells were grown in Complete DMEM (Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) containing 5% fetal bovine serum (Corning), 50 U/mL Penicillin-Streptomycin (Invitrogen), and 1 mM sodium pyruvate (Corning)) and maintained at 37 °C in an atmosphere of 5% CO2 in 10 cm TC dishes. Media in 10 cm TC dishes with HEK 293T cells (at around 70% confluence) were replaced with Opti-MEM (Invitrogen) followed by addition of a mixture of the GPCR plasmid, the GloSensor (Promega) 22F cAMP dependent reporter plasmid (Promega), and Lipofectamine 2000 (Invitrogen) in Opti-MEM. The dishes with transfection media were incubated at 37°C in an atmosphere of 5% CO2 for 6 h before replacing media with complete DMEM. After incubating at 37°C in an atmosphere of 5% CO2 for 16 h, transfected cells were plated in ploy-D-lysine coated 96-well plates at ~40,000 cells/well and incubated at 37 °C in an atmosphere of 5% CO2 for 16 h. On the day of assay, medium in each well was replaced with 50 μL of assay buffer (20 mM HEPES, 1x HBSS, pH 7.2, 2 g/L d-glucose), followed by addition of 25 μL of 4x drug solutions for 15 min at room temperature. To measure the activation of Gi-coupled opioid receptors, 25 μL of 4 mM luciferin supplemented with isoproterenol at a final concentration of 200 nM was added, and, following gentle mixing, luminescence counting was performed using a plate reader (iD5, Molecular Devices) after 25 min. For antagonism experiments, the candidate antagonist (naloxone, PhNX, PhOX) was added to the assay buffer (50 uL/well) at 2x the final concentration and allowed to incubate for 5 minutes prior to the addition of the agonist.
Data analysis
MOR activation was expressed as % of DAMGO maximal effect and concentration-response curves were fitted using Prism 9 (Graphpad Software, La Jolla, CA, USA).
β-arrestin recruitment assay
HEK293T cells were seeded on 6-well culture plates at 3 × 106 cells/well and grown in Dulbecco modified Eagle medium (DMEM; Thermo Fisher Scientific, Pittsburg, PA, United States) supplemented with L-Glutamine 200 mM, Sodium Pyruvate 100 mM and MEM non Essential Amino Acids 100X (Biowest). 10% fetal bovine serum (FBS; Merck KgaA, Darmstadt, Germany), streptomycin (100 μg/mL), and penicillin (100 μg/mL) in a controlled environment (37°C, 98% humidity, and 5% CO2). 24 h after seeding, cells were transfected the split NanoBiT vectors NB MCS1 (Promega, Madison, WI, United States) fused to β-arrestin2 or the human MOR (0.1 μg β-arrestin2-LgBIT cDNA, 2 μg of the hMOR-SmBIT cDNA) using polyethylenimine (PEI; Polysciences EuropeGmbH, Hirschberg an der Bergstrasse, Germany) in a 1:3 DNA:PEI ratio. 48 h after transfection, cells were rinsed, harvested, and resuspended in 4 ml/well of Hanks’ Balanced Salt solution (HBSS, Sigma Aldrich, Switzerland). Cells (80 μl/well) were then plated in 96-well white plates (PO-204003, BIOGEN) and immediately treated with increasing concentrations of DAMGO or PhOX (0.1 nM to 10 μM), 5 minutes later at 2 μM elenterazine (Prolume Ltd) was added and luminescence (490 to 410 nm) was measured during 6 min using a CLARIOstar (BMG Labtech) plate reader.
Data analysis
β-arrestin recruitment was expressed as % of DAMGO maximum effect and concentration-response curves were fitted using Prism 9 (Graphpad Software, La Jolla, CA, USA).
Surgeries
For behavior, FDG PET or binding assay experiments, 200 μm, 0.37 NA optical fibers (Neurophotometrics) were implanted at the following coordinates (in mm, from bregma), for unilaterally in the left VTA at AP 3.3; ML −0.5 with a 10° angle; DV 4.15, bilaterally in the PAG at AP −4.60, ML ±0.32; DV −2.75 with a 10° angle, left NAc-mSh at AP −1.54, ML −0.48, and DV −4.28, or left aDS AP: 1.2; ML: −1.4; DV: −3.00. For experiments assessing the effect of UV light on neuroinflammatory markers, 200 μm, 0.37 NA optical fibers were implanted bilaterally at the following coordinates (in mm, from bregma) in the dorsal striatum: AP 1.1; ML ±1.4; −2.5. Implants were secured with light-cured dental cement. For site-separated fiber photometry experiments: 400 nl of AAV1-hsyn1-dLight1.3b or AAV9-CAG-dLight1.3b mixed 1:9 with AAV5-hSyn-mCherry were injected into the left NAc-mSh at AP 1.54 mm from bregma;ML 0.48 mm; DV −4.28 mm. After at least 3 weeks after virus injection mice were implanted with 200 μm, 0.37 NA optical fibers. Left NAc-msh implants were fluorescence guided at approximately 0.3 mm above viral coordinates and left VTA implants were at AP − 3.3 mm from bregma; ML −0.5 mm with a 10° angle; DV −4.15 mm or left PAG implants at AP −4.60 mm from bregma, ML 0.32 mm; DV −2.75 mm with a 10° angle. For same-site fiber photometry experiments 400 nl AAV1-syn-FLEX-jGCaMP8s-WPRE mixed 9:1 with AAV5-hSyn-mCherry was injected into left RMTg of vGAT-cre mice at AP −3.9 mm from bregma; ML −0.4 mm; DV 4.5 mm. After allowing for at least 3 weeks of expression, 200 μm, 0.37 NA optical fibers were implanted into the left VTA using fluorescence guidance, aiming for at AP −3.3 mm from bregma; ML −0.5 mm with a 10° angle; DV −4.15 mm. All mice were given at least one week to recover after fiber implant surgeries.
Ex vivo receptor occupancy
Mice with optical fiber implants in the anterior dorsal striatum were injected with vehicle or PhOX (10 mg/kg, s.c.). 30-min post-injection, photoactivation was performed using an Arduino-controlled 375 nm laser (Vortran, 10 × 200 ms, 1 Hz, 30 mW). Brains were harvested 30-seconds post-laser stimulation and stored at −80 °C. Frozen tissue was sectioned (20 μM) on a cryostat (Leica, Germany) and thaw mounted onto Superfrost Plus glass slides (Avantor, USA). Brain slices were incubated for 10 minutes in buffer (50 mM Tris-HCL, 10mM MgCl2) containing [3H]DAMGO (5 nM). Following incubation, slides were air dried and apposed to a BAS-TR2025 Phosphor Screen (Fujifilm) for 5–10 days and imaged using a phosphorimager (Typhoon FLA 7000).
Data analysis
Quantification of 3H-DAMGO was performed in ImageJ (NIH). Images were calibrated using a C-14 reference to convert grey pixel values to nCi/g. ROIs were then drawn around the dorsal striatum of sections proximal to the fiber placement. Average mean measurements per mouse (n = 5 mice, 5–7 sections each) were plotted to compare ipsilateral (left) to contralateral (right) hemispheres.
[18F]-Fluorodeoxyglucose PET
This procedure was based on previous studies.65,66 One week before the PET procedure, surgeries to implant a fiber optic in the VTA were performed as described. Mice were habituated to experimenter handling, patch cord tethering, and the open field arena prior to experiment day. Mice were fasted 16 h before the experiment. On the day of the experiment, mice (n = 6–8 per condition) received a subcutaneous injection of vehicle (buffered saline), PhOX (30 mg/kg), or Oxymorphone (10 mg/kg). Thirty minutes after drug injection, mice were injected intraperitoneally with 0.35 mCi of 2-deoxy-2-[18F]fluoro-D-glucose (FDG; Cardinal Health) and placed into open-field chambers for drug photouncaging during uptake. Immediately after FDG administration single pulses of 375 nm light (200 ms, 30 mW) were delivered every 10 min (“LIGHT ON”), or no light was delivered (“LIGHT OFF”). After 30 min, mice were anesthetized with 1.5% isoflurane, placed on a custom-made bed of a nanoScan small animal PET/CT scanner (Mediso Medical Imaging Systems) and scanned for 20 min on a static acquisition protocol, followed by a CT scan.
Data analysis
The PET data were reconstructed and corrected for deadtime and radioactive decay. (PMOD Technologies, Zurich, Switzerland). The PET data were coregistered to an MRI template31 using the PMOD software environment (PMOD Technologies, Zurich, Switzerland). The coregistered PET data was analyzed in a voxel-based statistical analyses using a one-way ANOVA with four levels: vehicle LIGHT ON, Oxymorphone, PhOX LIGHT ON and PhOX LIGHT OFF. The values of the statistical parameter T for each contrast between conditions were mapped on the MRI template and filtered for clusters of voxels containing more than 100 contiguous voxels with statistical significance above the threshold (p < 0.05). All statistical parametric mapping analyses were performed using Matlab R2021a (Mathworks) and SPM12 (University College London). All qualitative and quantitative assessments of PET images were performed using PMOD. After the PET experiment animals were sacrificed and fiber placement was evaluated as described below, no animals were excluded from the analysis based on fiber placement.
Brain slice preparation
For synaptic transmission experiments, postnatal day 15−35 mice of both sexes on a C57/Bl6 background were used while GIRK experiments were performed on mice of the PValbCre/Ai14 background. Mice were sacrificed by gas anesthetic followed by rapid decapitation. Brains were removed in ice-cold choline solution equilibrated with 95% O2/5% CO2 (carbogen) containing (in mM) 25 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 7 MgCl2, 25 glucose, 0.5 CaCl2, 110 choline chloride, 11.6 ascorbic acid, and 3.1 pyruvic acid, and chilled for 3 minutes. Brains were then blocked to generate sections from the horizontal plane and mounted in a VT1000S vibratome (Leica Instruments). Slices (300 μm) were cut in ice-cold choline solution perfused with carbogen. Slices were recovered for 30 minutes at 34 °C in a holding chamber containing carbogenated artificial cerebrospinal fluid (ACSF) composed of (in mM) 125 NaCl, 2.5 KCl, 25 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, and 15 glucose, osmolarity 295, after which the holding chamber was moved to room temperature until use in experiments. Horizontal slices of locus coeruleus (220 μm) were prepared from postnatal day 8−14 Long-Evans rats using the same protocol. Slices were recovered at 32 °C for 30 min and then left at room temperature until recordings were performed.
Electrophysiology
Recordings were performed on slices in a submerged slice chamber perfused with 32 °C ACSF equilibrated with 95% O2/5% CO2. Whole-cell voltage clamp recordings were made with an Axopatch 700B amplifier (Axon Instruments). Data were filtered at 3 kHz, sampled at 10 kHz, and acquired using National Instruments acquisition boards and a custom version of ScanImage written in MATLAB (Mathworks). Cells were rejected if holding currents exceeded −200 pA or if the series resistance (<20 MΩ) changed during the experiment by more than 20%.
All recordings were performed within 5 hours of slice cutting in a submerged slice chamber perfused with ACSF warmed to 32°C and equilibrated with carbogen. The hippocampal CA1 region was identified by morphology. Pyramidal cells were patched with pipets (open pipet resistance 2.8–3.5 MΩ) containing cesium low chloride internal solution composed of (in mM) 135 CsMeSO3, 3.3 QX314-Cl, 10 HEPES, 4 MgATP, 0.3 NaGTP, 8 Na2-phosphocreatine, 1 EGTA (CsOH). For synaptic transmission experiments, excitatory transmission was blocked by the addition of CPP (10 μM) and NBQX (10 μM) to the bath. Once the whole cell configuration was obtained, pyramidal cells were voltage clamped at 0 mV to facilitate detection of outward currents. IPSCs were electrically evoked (two pulses, 0.5 ms, 50–300 μA, 50 ms interval) every 20 seconds by a bipolar theta glass stimulating electrode (5 μm tip diameter) placed at the border between stratum pyramidale and stratum oriens approximately 50–150 μm from the patched~cell. After establishing a stable baseline for 3–5 minutes, drugs were added to the bath at the times and concentrations indicated in their corresponding figure.
For analysis of GIRK currents in mice, parvalbumin (PV) interneurons within the CA1 were identified by reporter expression in ParvCre/TdTomato mice. PV cells were voltage clamped at −55 mV and GIRK currents were isolated by the addition of CPP (10 μM), NBQX (10 μM), picrotoxin (10 μM), and tetrodotoxin (1 uM) to the bath. A K+-based internal was used that contained (in mM) 135 KMeSO3, 5 KCl, 5 HEPES, 4 MgATP, 0.3 NaGTP, 10 phospho-creatine, 1.1 EGTA (KOH).
In both experiments, after recordings stabilized, a 50 ms flash of UV light was applied from the 365 nm-UV channel of a pE-300white LED (CoolLED) reflected through a 60× LUMPLANFL 1.0 NA objective (Olympus) on SliceScope Pro 6000 microscope (Scientifica) with a 405 nm long-pass dichroic mirror (Di02-R405–25×36, Semrock) mounted in the fluorescence turret. Light power was set to 5 mW in the sample plane (~120 mW of an ~20 mm diameter “beam” at the back aperture). The resulting illumination field was ~0.02 mm2.
For analysis of GIRK currents in rat LC, noradrenergic neurons were visually identified by their large soma size and tight packing, leading to a translucent cluster of cells adjacent to the lateral corner of the 4th ventricle. Patch pipettes (open pipette resistance 1.6–2.2 MΩ) were filled with the K+-based internal solution. Cells were held at −55mV in voltage-clamp mode. Cells were rejected if holding currents exceeded −150 pA. Synaptic blockers were not added to the ACSF. Light power from a 365 nm LED was set to 40 mW at the back aperture of the objective and delivered using the same optics described above at variable flash durations (5, 20, 200 ms), as described in the text.
Data analysis
Data from electrophysiology experiments were analyzed in Igor Pro (version 6.02A, Wavemetrics). For synaptic transmission experiments, IPSCs were calculated by averaging a 2 ms window around the peak, and normalized to the average of the three sweeps (one minute) prior to uncaging or drug addition. To determine percent suppression, the average IPSC amplitude from minutes 2–3 post uncaging flash was compared to the minute prior to uncaging. For GIRK analysis in mouse hippocampus, peak drug effect was determined by comparing the average of three sweeps (15 seconds) to the minute prior to drug addition. For determination of uncaging effect, the 4 seconds prior to the uncaging flash were used as the baseline, and the current at seconds 5–10 post uncaging were analyzed to calculate suppression. Time constants were calculated by a mono-exponential fit to time 0–10 seconds post uncaging.
Blood-brain barrier penetrance and pharmacokinetics
Pharmacological studies were conducted at the UCSD Translational Pharmacology and Bioanalysis Laboratory. For blood-brain barrier studies, mice were anesthetized briefly with isoflurane and injected intravenously (retro-orbital) with either PhOX (8.5 mg/kg), Oxymorphone freebase (5 mg/kg), PhNX (15 mg/kg) or NLX HCl (10 mg/kg). 15 minutes post-injection, mice were anesthetized again and a retro-orbital heparinized blood sample was removed (55–65 μl), followed by cardiac perfusion with room temperature PBS (20–25 mL). Brains were then collected, immediately frozen on dry ice, and stored at −80C. Subsequently the brain tissue was homogenized through a series of extraction-filtration procedures. For half-life determination, mice were injected i.p. with PhOX and retro-orbital blood samples were taken at the following time points: 5 minutes, 15 minutes, 1 hour, and 2 hours post-dose. Plasma samples were extracted from blood samples via centrifugation at 5,000 rpm for 5 minutes.
Data analysis
Tissue and plasma concentrations were determined using LC-MS/MS. Standards were used to generate an external calibration curve in blank plasma or tissue homogenate using a linear regression algorithm to plot the peak area ratio vs concentration with 1/x weighting, over the full dynamic range of analyte concentrations.
Assessment of neuroinflammatory markers after UV photoactivation
Mice were implanted bilaterally with 200 μm optical fibers in the dorsal striatum and allowed to recover for 14 days prior to experimentation. Mice were injected with PhOX (15 mg/kg i.v.) in the presence of NLX (10 mg/kg, i.v.) in an attempt to minimize any confounding effects of direct MOR activation on neuroimmune cells.67 15 minutes after injection, mice were tethered unilaterally on the left hemisphere to a fiber optic cable attached to a 375 nm laser via a commutator. Uncaging stimuli (10 × 200 ms, 1Hz, 20 mW) were applied every 5 minutes for 20 minutes while being allowed to freely roam. Mice were then returned to their home cage and sacrificed at 4 hours and 24 hours post-photoactivation. Brains were harvested for histology and processed for immunohistochemical staining against Iba1 and GFAP.
Histology
Mice were transcardially perfused with 25 mL ice-cold PBS followed by 25 mL ice-cold 4% paraformaldehyde (PFA). Brains were harvested and post-fixed in 4% PFA overnight at 4°C, then transferred to a 30% sucrose solution and stored at 4°C until sectioning. Brains were sliced into 40 μm sections using a freezing microtome and stored in PBS at 4°C until processing for IHC. Non-immuno-stained sections were immediately mounted on coverslips in DAPI-contining Vectashield mounting medium. For immunohistochemical staining, free-floating sections were blocked in PBS-TritonX 0.01% + 5% Donkey Serum for 2 hr at RT with gentle shaking. Sections were imaged using a BZ-X100 fluorescence microscope (Keyence) and images were processed using ImageJ.
For sections of the VTA, immunohistochemical staining for tyrosine hydroxylase (TH) was performed as follows: free-floating brain slices were blocked in 5% normal donkey serum (NDS) in PBS supplemented with 0.1% TritonX (0.1% PBST) for 2 hours at RT and then transferred to a solution containing rabbit aTH primary antibody (1:1,000) and 1% NDS in 0.1% PBST, and shaken gently overnight at 4°C. Sections were then washed 3x for 10 min in PBS at RT and transferred to a solution containing Alexa 647-conjugated donkey anti-rabbit secondary antibody (1:1,000) and 1% NDS in 0.1% PBST, and then shaken gently for 1.5 hr at RT. Next they were washed 3x for 10 min in PBS at RT and mounted with DAPI-containing mounting medium.
For sections of the dorsal striatum stained for neuroinflammatory markers, mice were transcardially perfused with 25 mL ice-cold PBS followed by 25 mL ice-cold 4% PFA either 4 or 24 hours post-exposure to light. Brains were processed as described above. Following block, guinea pig anti-GFAP (1:500) and/or rabbit anti-Iba1 (1:300) were applied to wells containing blocked sections and allowed to incubate for 48 hr at 4°C with gentle shaking. Following primary incubation, sections were washed 3x with 0.01% PBST for 10 min at RT with gentle shaking and then transferred to a solution containing Alexa488-conjugated goat anti-guinea pig and/or Alexa488-conjugated goat anti-rabbit secondary antibodies in 0.01% PBST + 5% Donkey Serum for 2 hr at RT. Sections were washed 3x with 0.01% PBST for 10 min at RT with gentle shaking, and then mounted with DAPI-contining Vectashield mounting medium.
Image acquisition and data analysis
Images were taken using an epifluorescence Keyence BZX700 fluorescence microscope using a 10x objective. For Iba1 and GFAP signal quantification, mean pixel density within a small ROI just below the histologically identified implanted optical fiber lesion was calculated using ImageJ. For analysis, the mean pixel density of the illuminated hemisphere was compared to the unilluminated hemisphere.
Behavior
Tail-flick
Tail-flick analgesia experiments were conducted using a TF-2 Tail-Flick Apparatus (Columbus Instruments). Mice implanted with optical fibers in the vlPAG or VTA were pre-handled and then manually restrained during the experiment. Mice were tested twice on two consecutive days (with and without NLX); the conditions were counterbalanced across days. 15 minutes after injection of PhOX (15 mg/kg i.v.) or PhOX + NLX (15 mg/kg, 10 mg/kg, i.v.), the tail was positioned flat 15 mm from 6 W heat beam and latency to tail flick or time out (30 s) was measured. A baseline measurement was taken at 25 min after drug injection and multiple measurements were taken after application of a train of 375 nm light flashes (10 × 200 ms, 1 Hz, 30 mW). The experimenter was blind to drug treatment conditions.
Hargreaves with capsaicin hyperalgesia
Hargreaves analgesia experiments were performed mice implanted with optical fibers in their left NAc-mSh. Mice were injected with PhOX (12 mg/kg, s.c.) and set on a hargreaves heated glass platform (32°C) to acclimate for 17 minutes. The Hargreaves apparatus (Model 400, IITC life sciences) heat source was set to 50% threshold with a cutoff time of 20 seconds. Heat was applied to the right contralateral paw for all latency measurements. Paw withdrawal latencies were characterized as a sudden, responsive lifting, licking, or shaking of paw and measurements were not taken when the subject was moving or grooming. To induce, capsaicin hyperalgesia for thermal nociception68 a pharmaceutical grade 0.1% capsaicin cream was applied to the hindpaw. A pre-capsaicin baseline was measured at 17 minutes post-injection. Capsaicin was lightly applied to the hindpaw at 20 minutes post-injection, a post capsaicin baseline was taken at 28 minutes post-injection, photolysis (10 × 200 ms, 1 Hz, 45 mW) occurred at 29 minutes post-injection of PhOX. The first post-uncaging latency was 1 minute after photolysis, subsequent latencies were taken at 5, 10, 20, and 30 minutes. In the no light control experiment, mice were fiber tethered with all other parameters identical except omission of photolysis. The 375 nm laser was coupled to a commutator to allow mice freedom of movement.
Hot plate
Mice were injected with either saline or PhOX (15 mg/kg, i.v.) and tested on the hot plate 15 minutes later. The average latency of 3 individual trials (1 every 5 min) is reported for each condition. Mice were next injected with morphine (5 mg/kg, s.c.) and 30 minutes after injection mice were again tested on the hot plate 3 times.
Locomotion
For open field experiments with intravenous PhOX, mice on a on a C57Bl/6J background were implanted unilaterally on the VTA and allowed to recover. All mice were handled and acclimated to tethering to the fiber optic cable prior to photoactivation experiments. For intravenous (i.v.) injections, mice were briefly anaesthetized with 3–5% isoflurane and given retro-orbital injections of either PhOX (15 mg/kg) or PhOX + naloxone (15 mg/kg + 10 mg/kg) or PhOX + PhNX (15 mg/kg + 30mg/kg). After recovery from anesthesia, mice were tethered to an optical fiber (200 μm, 0.39 NA) coupled to a commutator connected to an Arduino-controlled 375 nm laser (Vortran or Oxxius). They were placed into a square enclosure (18 × 18 cm) where their position was video recorded at a 30 fps using a webcam (Logitech) fed into Smart 3.0 video tracking software (Panlab). After a 15-minute baseline, PhOX was photoactivated in the VTA (30mW power, 200ms, 1Hz, 10 flashes). Locomotion was recorded 15 minutes post stimulation.
For experiments assessing the locomotor response to various morphine doses, mice implanted unilaterally in the VTA were similarly tethered as in experiments with PhOX, but they were given a 10-minute baseline and recorded 20 minutes post morphine injection (s.c.) at the following doses: 2 mg/kg, 5 mg/kg, and 10 mg/kg. Each dose of morphine were assessed on a separate day and the order of dosage was individually counterbalanced to minimize effects of tolerance.
For open field experiments comparing the locomotor response to PhOX photoactivation 15 or 30 min post-injection, PhOX (30 mg/kg s.c.) was injected and light was applied unilaterally to the VTA (375 nm, 10 × 200ms, 1 Hz, 70mW). For the experiments comparing the locomotor response to PhOX photoactivation 30 min, 90 min, or 3 hr post-injection, PhOX (10 mg/kg s.c.) was injected and light was applied unilaterally to the VTA (375 nm, 10 × 200ms, 1 Hz, 20 mW). For open field experiments comparing doses of subcutaneous PhOX, the following dose of PhOX were injected: 2.5 mg/kg, 5 mg/kg or 10 mg/kg s.c. 15 minutes post-injection, photoactivation in the VTA was conducted (375 nm, 10 × 200 ms, 1 Hz, 30 mW) and mice were recorded for 15 minutes post photoactivation. For open field experiments comparing the locomotor response to VTA PhOX at 3 vs. 10 light flashes, mice were injected with PhOX (10 mg/kg, s.c.) and placed into an open field. 15 minutes post-injection, photoactivation in the VTA was conducted (375 nm, 3 or 10 × 200 ms, 1 Hz, 30 mW).
For comparison of LED and UV locomotor behavior after VTA PhOX activation, mice were injected with PhOX (30 mg/kg s.c.) and after 10 minutes connected to light sources set to deliver the following light powers for photoactivation: 375 nm laser (15 mW), 385 nm LED (2.5 mW), or 365 nm LED (1.4 mW). The reported power levels in this experiment were measured from the tip of a ferrule-connected optical fiber similar to those used for the implants. Optical fibers connected to mice were coupled directly to light sources without the use of a commutator in these experiments to maximize LED light power output. Mice were placed into the open field and locomotor behavior was recorded using Smart 3.0 (Panlab) video tracking software. After an initial baseline of 20 minutes photolysis protocol was initiated (10 × 200 ms, 1 Hz, one bout every 5 min) and locomotion post-photoactivation was recorded for 20 minutes.
Data analysis
Velocity and total distance traveled was quantified using Smart 3.0 video automated tracking software (Panlab). Velocity (cm/s) was calculated by quantifying distance traveled in time bins of 5 seconds and dividing by seconds.
Conditioned place preference
For conditioned place preference experiments, a custom built 2-chamber apparatus (26 × 26 cm, UCSD Machine Shop) was used with both visual and tactile cues. Before conditioning, mice were habituated for 20 minutes by being allowed to freely roam both chambers. Pre-preference was recorded and PhOX + light was paired with the least preferred chamber. Prior to conditioning sessions, mice were injected with PhOX (15 mg/kg, i.v.) or vehicle (saline, i.v.) under anesthesia and allowed to recover for 15 minutes. For conditioning without light stimulation: Conditioning took place over 4 days, where PhOX injection was paired with one context and vehicle was paired with the other on alternating days. Conditioning sessions took place over 20 minutes. On test day, mice were allowed to roam both chambers for 20 minutes. For conditioning with light stimulation in the VTA, mice were implanted with 200 μm optical fibers as previously described. During conditioning sessions mice were tethered to a fiber optic cable attached to a 375 nm laser via a commutator and exposed to a uncaging stimulus (10 × 200 ms, 1Hz) every 5 minutes. For reversal conditioning with VTA stimulation, PhOX plus light stimulation was paired with the previously least preferred chamber from conditioning test day of Phase 1 and PhOX plus naloxone (with light) was paired with the previously most preferred chamber.
Data analysis
Place preference was quantified as time spent in each zone using Smart3.0 (Panlab) tracking software.
Two-site fiber photometry and uncaging
For fiber photometry with dLight1.3b, mice were injected with dLight1.3b +mCherry in the left NAc-mSh and after at least 3 weeks of expression, they were implanted with 200 μm optical fibers into the left NAc-mSh and left VTA. Mice were allowed to recover at least one week post fiber implantation. Fiber photometry recordings were done using a commercially available fiber photometry system FP3002 (Neurophotometrics). For dLight1.3b experiments with PhOX, mice were injected with either PhOX (15 mg/kg, i.v.) or PhOX + NLX (15 mg/kg, 10 mg/kg, i.v.). 5 minutes after injection, mice were connected to two fiber optic cables, one coupling the NAc-mSh optical fiber implant to the fiber photometry system (470 nm LED for dLight1.3b and 560 nm LED, mCherry control wavelength) and the other coupling the VTA optical fiber to a 375 nm Arduino-controlled laser (Vortran). After a 10 minute baseline, photoactivation in the VTA was applied with a single 375 nm light pulse (200 ms, 50 mW). dLight1.3b fluorescence was recorded 20 minutes post photoactivation. For fiber photometry experiments with dLight1.3b testing for the effects of off-target wavelength photostimulation in the VTA, fiber photometry with PhOX was performed as described using a 473 nm laser (Optoengine) instead of a 375 nm laser. Fiber photometry experiments without PhOX injection were performed as described except no PhOX injection was administered. Experiments examining the power-response relationship between VTA PhOX photactivation and NAc-mSh dLight1.3b were conducted on separate days as described for the following light powers: 1 mW, 5 mW, 20 mW, and 50 mW.
For repeated uncaging with VTA PhOX and NAc-mSh dLight1.3b, mice were injected with PhOX (10 mg/kg, s.c.) and after 30 minutes post-injection a 375 nm light pulse (20 mW, 200 ms) was delivered to the VTA. After 10 minutes, a 2nd light pulse was delivered. dLight1.3b signal was recorded 10 minutes post each uncaging event.
For fiber photometry experiments involving morphine injection and PhNX photoactivation, mice were injected with dLight1.3b and mCherry (control) in the NAc-mSh and implanted with optical fibers in the NAc-mSh and VTA. Mice were anaesthetized and injected with PhNX (15 mg/kg, i.v.). After recovery, mice were connected to two fiber optic cables, one coupling the NAc-mSh optical fiber implant to the fiber photometry system (470 nm LED for dLight1.3b and 560 nm LED, mCherry control wavelength) and the other coupling the VTA optical fiber to a 375 nm Arduino-controlled laser. Baseline fluorescence was recorded for 10 minutes. PhNX photoactivation in the VTA (10 × 200 ms, 1 Hz, 20 mW) occurred 1 minute prior to morphine (5 mg/kg, i.p.) injection, and again 5 and 10 minutes after the first flash sequence. Post-photoactivation, fluorescence was recorded for 20 minutes.
Same-site fiber photometry and uncaging
For same-site fiber photometry with PhOX, vGAT-Cre mice were transduced with cre-dependent GCaMP8s in the left RMTg and implanted with an optical fiber implanted over the left VTA. Mice were tethered to an optical fiber attached to a custom-built optical relay (Figure S7G) that was connected to both the fiber photometry system and a 375 nm laser (Oxxius). Prior to PhOX injection, laser stimulation was applied to the VTA to control for the effect of light delivery alone on fluorescence signal. A 10-minute baseline was recorded, then a single, 20 mW, 200 ms laser pulse was delivered every 10 minutes for a total of 3 flashes. After the preinjection test session, PhOX (30 mg/kg, s.c.) was injected while mice were still tethered to the fiber optic cable. 20 minutes after injection, Fiber Photometry recording was resumed. A 10 minute baseline was recorded, and then a 200 ms, 20 mW flash was delivered, and 20 minutes was recorded post-flash.
Data analysis
Same-site fiber photometry analysis was conducted as described for site-separated fiber photometry with the addition of laser light artifact correction. For light-artifact correction, the average ΔF/F of 3 preinjection flashes was subtracted from the post-injection ΔF/F.
Fiber photometry with chronic morphine
For chronic morphine experiments, mice were injected with dLight1.3b in the left NAc-mSh and implanted with a 400 μm optical fiber in the left NAc-mSh and a 200 μm optical fiber above the left VTA. Prior to chronic morphine treatment, mice were handled, habituated to tethering, and their baseline response to PhOX were tested. For PhOX photoactivation in the VTA, PhOX (12 mg/kg, s.c.) was injected 30 minutes prior to laser stimulation. A 5 minute baseline of dLight1.3b signal in the NAc-mSh was recorded prior to photostimulation (10 × 200 ms, 1 Hz, 20 mW). For chronic morphine treatment, mice received two daily injections of morphine or saline (11:00 am and 7:00 pm) using an intermittent escalating dose protocol,69 of 10, 20, 30, 40 and 50 mg/kg i.p. morphine. On day 5, fiber photometry recording in the NAc-mSh with PhOX uncaging was performed as described above in the morning and mice received their 2 daily morphine injections afterwards. Following 4 days of abstinence, fiber photometry with PhOX uncaging was performed again.
Data analysis
Data analysis including peri-event time histograms of ΔF/F and z-score values surrounding photoactivation events, AUC, and tau calculations were conducted using Matlab R2020a (Mathworks) and custom scripts in Igor Pro (Wavemetrics). Analysis in Matlab was conducted using pMAT an open source fiber photometry analysis package.63 The signal (470 nm) and control (560 nm) channel were individually smoothed and downsampled and bleach corrected. A scaled control channel was calculated using a least-squares regression to normalize the scale of the channels. ΔF/F values were calculated using the following equation: ΔF/F = (Signal Channel-Scaled Control Channel)/Scaled Control Channel. A robust z-score was generated using the following formula: [ΔF/F Event – median (ΔF/F baseline)]/ median absolute deviation (MAD) of baseline.
QUANTIFICATION AND STATISTICAL ANALYSIS
For all comparisons in this manuscript, we describe the number of n’s, what the n represents, the statistical test used, and the definition of error bars in the figure legend. For every comparison related to a figure, the details of the statistics are in the figure legend. For every other comparison, they are listed in the results section. Comparisons were considered to have reached statistical significance if the p-value was less than 0.05, unless otherwise stated. The p-values that correspond to asterisks are listed in the figure legends. When appropriate, exclusion criteria are listed in the corresponding methods details section.
D’Agostino & Pearson and/or Shapiro-Wilk tests were used to determine whether data were normally distributed. For comparison between two groups, either an unpaired or paired two-sided t-test was used for normally distributed datasets, and either a Mann-Whitney U-test or Wilcoxon matched-pairs signed rank test was used for non-normally distributed data. For comparison between two or more groups, differences were detected using either a One-way ANOVA or a Friedman test, and interactions were identified using Two-way ANOVA. A post-hoc comparison was used to determine differences between specific conditions (Tukey’s, Bonferroni’s, Sidak’s or Dunn’s). The source data spreadsheet available in the supporting information include the details of all statistical tests used in each figure.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-tyrosine hydroxylase | Millipore-Sigma | Cat#AB152; RRID:AB_390204 |
| Alexa-Fluor 647-conjugated donkey anti-rabbit | Invitrogen | Cat#A-32795; RRID:AB_2762835 |
| Guinea pig anti-GFAP | Synaptic Systems | Cat. # 173004; RRID:AB_10641162 |
| Rabbit anti-Iba1 | Wako Chemicals | Cat. # 019–19741; RRID:AB_839504 |
| Alexa594-conjugated goat anti-guinea pig | Life Technologies | Cat.#A-11076; RRID:AB_2534120 |
| Alexa488-conjugated goat anti-rabbit | Invitrogen | Cat. # A-11034; RRID:AB_2576217 |
| Donkey Serum | Sigma | Cat. # D9663 |
| Bacterial and virus strains | ||
| AAV1-hSyn-dLight1.3b | Lin Tian Lab | N/A |
| AAV9-CAG-dLight1.3b | Addgene | Cat#125560 |
| AAV5-hSyn-mCherry | Addgene | Cat#114472 |
| AAV1-syn-FLEX-jGCaMP8s-WPRE | Addgene | Cat#162377 |
| Chemicals, peptides, and recombinant proteins | ||
| α-methyl-4,5-dimethoxy-2-nitrobenzene oxymorphone (PhOX) | This paper | N/A |
| α-methyl-4,5-dimethoxy-2-nitrobenzene naloxone (PhNX) | This paper | N/A |
| Oxymorphone | Noramco | Cat#516340913 |
| Naloxone hydrochloride dihydrate | Sigma Aldrich | Cat#N7758 |
| Morphine sulfate | Spectrum | Cat#M1167 |
| DAMGO | Hello Bio | Cat#HB2409 |
| CTAP | Tocris | Cat#1560 |
| Critical commercial assays | ||
| GloSensor™ cAMP Assay | Promega | Cat#E1290 |
| NanoBiT® barrestin Recruitment Assays | Promega | Cat#N2014 |
| Deposited data | ||
| Source data | This paper | N/A |
| Experimental models: Cell lines | ||
| HEK 293T | ATCC | Cat#CRL-3216; RRID:CVCL_0063 |
| Experimental models: Organisms/strains | ||
| C57Bl/6 mice | Jackson Labs | Strain # 000664; RRID:IMSR_JAX:000664 |
| PvalbCre mice | Jackson Labs | Strain # 017320; RRID:IMSR_JAX:017320 |
| Rosa-Lsl-Td-tomato (Ai14) mice | Jackson Labs | Strain # 007914; RRID:IMSR_JAX:007914 |
| Slc32a1Cre mice (vGAT-cre) | Jackson Labs | Strain # 028862; RRID:IMSR_JAX:028862 |
| Long-Evans rats | Charles River | Strain Code: 006; RRID:RGD_2308852 |
| Recombinant DNA | ||
| pGloSensor −22F | Promega | Cat#E2301 |
| SSF-MOR (Mus musculus mu opioid receptor) | Banghart Lab and Williams Lab | Tanowitz and Von Zastrow26 |
| DOR (Mus musculus delta 1 opioid receptor) | Genscript | OMu22648D, Accession No. NM_013622.3 |
| KOR (Mus musculus kappa 1 opioid receptor) | cDNA Resource Center, www.cdna.org | Cat #OPRK100000 |
| β-arrestin2-LgBIT | Promega | N/A |
| hMOR-SmBIT | Promega | N/A |
| Software and algorithms | ||
| Matlab r2020a | MathWorks | RRID: SCR_001622 |
| Igor Pro 6 | Wavemetrics | RRID:SCR_000325 |
| GraphPad Prism 9 | Graphpad | RRID: SCR_002798 |
| pMAT | Barker Lab | Bruno etal.,202163 |
| Other | ||
| 375 nm laser | Vortran | Part#: 12084/B1 |
| 375 nm laser | Oxxius | Part#: LBX-375-HPE-PPA |
Highlights.
PhOX and PhNX are photoactivatable mu opioid receptor ligands
PhOX and PhNX can be photoactivated in the mouse brain after systemic drug delivery
In vivo photopharmacology is compatible with fiber photometry
PhOX reveals mesolimbic opioid-dopamine decoupling in response to chronic morphine
ACKNOWLEDGMENTS
We thank the National Institute on Drug Abuse Drug Supply Program (NDSP) for pharmacological reagents; J. Momper for bioavailability experiments; G. Or and L. Tian for dLight1.3b AAV; E. Berg for technical assistance; and T. Gremel, K. Tye, T. Hnasko, and B.K. Lim for helpful discussions. The project was supported by the Brain & Behavior Research Foundation (M.R.B.); the Esther A. & Joseph Klingenstein Fund and Simons Foundation (M.R.B.); NIH grants R00DA034648 (M.R.B.), U01NS113295 (M.R.B. and diversity supplements to D.A.J. and A.E.L.), RF1NS126073 (M.R.B.), R35GM133802 (M.R.B.), T32GM007240 (X.J.H.), and ZIA000069 (M.M.), and Plan Nacional Sobre Drogas, Ministerio de Sanidad, Spain 2021I070 (J.B.). Some figures were generated in part using BioRender.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in their field of research or within their geographical location. One or more of the authors of this paper received support from a program designed to increase minority representation in their field of research. We worked to ensure sex balance in the selection of non-human subjects.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.neuron.2023.09.017.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Ellis-Davies GCR (2007). Caged compounds: photorelease technology for control of cellular chemistry and physiology. Nat. Methods 4, 619–628. 10.1038/nmeth1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Velema WA, Szymanski W, and Feringa BL (2014). Photopharmacology: beyond proof of principle. J. Am. Chem. Soc 136, 2178–2191. 10.1021/ja413063e. [DOI] [PubMed] [Google Scholar]
- 3.Broichhagen J, Frank JA, and Trauner D (2015). A roadmap to success in photopharmacology. Acc. Chem. Res 48, 1947–1960. 10.1021/acs.accounts.5b00129. [DOI] [PubMed] [Google Scholar]
- 4.Lima SQ, and Miesenböck G (2005). Remote control of behavior through genetically targeted photostimulation of neurons. Cell 121, 141–152. 10.1016/j.cell.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 5.Acosta-Ruiz A, Gutzeit VA, Skelly MJ, Meadows S, Lee J, Parekh P, Orr AG, Liston C, Pleil KE, Broichhagen J, et al. (2020). Branched photoswitchable tethered ligands enable ultra-efficient optical control and detection of G protein-coupled receptors in vivo. Neuron 105, 446–463.e13. 10.1016/j.neuron.2019.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Donthamsetti P, Winter N, Hoagland A, Stanley C, Visel M, Lammel S, Trauner D, and Isacoff E (2021). Cell specific photoswitchable agonist for reversible control of endogenous dopamine receptors. Nat. Commun 12, 4775. 10.1038/s41467-021-25003-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gomila AMJ, Rustler K, Maleeva G, Nin-Hill A, Wutz D, Bautista-Barrufet A, Rovira X, Bosch M, Mukhametova E, Petukhova E, et al. (2020). Photocontrol of endogenous glycine receptors in vivo. Cell Chem. Biol 27, 1425–1433.e7. 10.1016/j.chembiol.2020.08.005. [DOI] [PubMed] [Google Scholar]
- 8.Font J, López-Cano M, Notartomaso S, Scarselli P, Di Pietro P, Bresolí-Obach R, Battaglia G, Malhaire F, Rovira X, Catena J, et al. (2017). Optical control of pain in vivo with a photoactive mGlu5 receptor negative allosteric modulator. eLife 6. 10.7554/eLife.23545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hüll K, Morstein J, and Trauner D (2018). In vivo photopharmacology. Chem. Rev 118, 10710–10747. 10.1021/acs.chemrev.8b00037. [DOI] [PubMed] [Google Scholar]
- 10.Levitz J, Popescu AT, Reiner A, and Isacoff EY (2016). A toolkit for orthogonal and in vivo optical manipulation of ionotropic glutamate receptors. Front. Mol. Neurosci 9, 2. 10.3389/fnmol.2016.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Durand-de Cuttoli RD, Mondoloni S, Marti F, Lemoine D, Nguyen C, Naudé J, D’izarny-Gargas T, Pons S, Maskos U, Trauner D, et al. (2018). Manipulating midbrain dopamine neurons and reward-related behaviors with light-controllable nicotinic acetylcholine receptors. eLife 7. 10.7554/eLife.37487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gutzeit VA, Acosta-Ruiz A, Munguba H, Häfner S, Landra-Willm A, Mathes B, Mony J, Yarotski D, Börjesson K, Liston C, et al. (2021). A fine-tuned azobenzene for enhanced photopharmacology in vivo. Cell Chem. Biol 28, 1648–1663.e16. 10.1016/j.chembiol.2021.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berry MH, Holt A, Levitz J, Broichhagen J, Gaub BM, Visel M, Stanley C, Aghi K, Kim YJ, Trauner D, et al. (2017). Restoration of patterned vision with an engineered photoactivatable G protein-coupled receptor. Nat. Commun 8, 1862. 10.1038/s41467-017-01990-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caporale N, Kolstad KD, Lee T, Tochitsky I, Dalkara D, Trauner D, Kramer R, Dan Y, Isacoff EY, and Flannery JG (2011). LiGluR restores visual responses in rodent models of inherited blindness. Mol. Ther 19, 1212–1219. 10.1038/mt.2011.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.López-Cano M, Font J, Aso E, Sahlholm K, Cabré G, Giraldo J, De Koninck Y, Hernando J, Llebaria A, Fernández-Dueñas V, et al. (2023). Remote local photoactivation of morphine produces analgesia without opioid-related adverse effects. Br. J. Pharmacol 180, 958–974. 10.1111/bph.15645. [DOI] [PubMed] [Google Scholar]
- 16.Banghart MR, and Sabatini BL (2012). Photoactivatable neuropeptides for spatiotemporally precise delivery of opioids in neural tissue. Neuron 73, 249–259. 10.1016/j.neuron.2011.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.He XJ, Patel J, Weiss CE, Ma X, Bloodgood BL, and Banghart MR (2021). Convergent, functionally independent signaling by mu and delta opioid receptors in hippocampal parvalbumin interneurons. eLife 10. 10.7554/eLife.69746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Banghart MR, Williams JT, Shah RC, Lavis LD, and Sabatini BL (2013). Caged naloxone reveals opioid signaling deactivation kinetics. Mol. Pharmacol 84, 687–695. 10.1124/mol.113.088096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Williams JT (2014). Desensitization of functional μ-opioid receptors increases agonist off-rate. Mol. Pharmacol 86, 52–61. 10.1124/mol.114.092098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Banghart MR, He XJ, and Sabatini BL (2018). A caged enkephalin optimized for simultaneously probing Mu and Delta opioid receptors. ACS Chem. Neurosci 9, 684–690. 10.1021/acschemneuro.7b00485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Layden AE, Ma X, Johnson CA, He XJ, Buczynski SA, and Banghart MR (2023). A biomimetic C-terminal extension strategy for photocaging amidated neuropeptides. J. Am. Chem. Soc 145, 19611–19621. 10.1021/jacs.3c03913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma X, Johnson DA, He XJ, Layden AE, McClain SP, Yung JC, Rizzo A, Bonaventura J, and Banghart MR (2023). In vivo photopharmacology with a caged mu opioid receptor agonist drives rapid changes in behavior. Nat. Methods 20, 682–685. 10.1038/s41592-023-01819-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wootton JF, and Trentham DR (1989). “Caged” compounds to probe the dynamics of cellular processes: synthesis and properties of some novel photosensitive P-2-nitrobenzyl esters of nucleotides. In Photochemical Probes in Biochemistry, Nielsen PE, ed. (Kluwer Academic; ), pp. 277–296. [Google Scholar]
- 24.Schaal J, Kotzur N, Dekowski B, Quilitz J, Klimakow M, Wessig P, and Hagen V (2009). A novel photorearrangement of (coumarin-4-yl) methyl phenyl ethers. J. Photochem. Photobiol. A 208, 171–179. 10.1016/j.jphotochem.2009.09.012. [DOI] [Google Scholar]
- 25.Wong PT, Roberts EW, Tang S, Mukherjee J, Cannon J, Nip AJ, Corbin K, Krummel MF, and Choi SK (2017). Control of an unusual photo-Claisen rearrangement in coumarin caged tamoxifen through an extended spacer. ACS Chem. Biol 12, 1001–1010. 10.1021/acschembio.6b00999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanowitz M, and Von Zastrow M (2003). A novel endocytic recycling signal that distinguishes the membrane trafficking of naturally occurring opioid receptors. J. Biol. Chem 278, 45978–45986. 10.1074/jbc.M304504200. [DOI] [PubMed] [Google Scholar]
- 27.Glickfeld LL, Atallah BV, and Scanziani M (2008). Complementary modulation of somatic inhibition by opioids and cannabinoids. J. Neurosci 28, 1824–1832. 10.1523/JNEUROSCI.4700-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Williams JT, Egan TM, and North RA (1982). Enkephalin opens potassium channels on mammalian central neurones. Nature 299, 74–77. 10.1038/299074a0. [DOI] [PubMed] [Google Scholar]
- 29.Alvarez VA, Arttamangkul S, Dang V, Salem A, Whistler JL, Von Zastrow M, Grandy DK, and Williams JT (2002). μ-opioid receptors: ligand-dependent activation of potassium conductance, desensitization, and internalization. J. Neurosci 22, 5769–5776. 10.1523/JNEUROSCI.22-13-05769.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spivak CE, Oz M, Beglan CL, and Shrager RI (2006). Diffusion delays and unstirred layer effects at monolayer cultures of Chinese hamster ovary cells: radioligand binding, confocal microscopy, and mathematical simulations. Cell Biochem. Biophys 45, 43–58. 10.1385/CBB:45:1:43. [DOI] [PubMed] [Google Scholar]
- 31.Ma Y, Hof PR, Grant SC, Blackband SJ, Bennett R, Slatest L, Mcguigan MD, and Benveniste H (2005). A three-dimensional digital atlas database of the adult C57BL/6J mouse brain by magnetic resonance microscopy. Neuroscience 135, 1203–1215. 10.1016/j.neuroscience.2005.07.014. [DOI] [PubMed] [Google Scholar]
- 32.Di Chiara G, and Imperato A (1988). Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc. Natl. Acad. Sci. USA 85, 5274–5278. 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson SW, and North RA (1992). Opioids excite dopamine neurons by hyperpolarization of local interneurons. J. Neurosci 12, 483–488. 10.1523/JNEUROSCI.12-02-00483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gysling K, and Wang RY (1983). Morphine-induced activation of A10 dopamine neurons in the rat. Brain Res 277, 119–127. 10.1016/0006-8993(83)90913-7. [DOI] [PubMed] [Google Scholar]
- 35.Yaksh TL, Yeung JC, and Rudy TA (1976). Systematic examination in the rat of brain sites sensitive to the direct application of morphine: observation of differential effects within the periaqueductal gray. Brain Res 114, 83–103. [DOI] [PubMed] [Google Scholar]
- 36.Olmstead MC, and Franklin KBJ (1997). The development of a conditioned place preference to morphine: effects of microinjections into various CNS sites. Behav. Neurosci 111, 1324–1334. 10.1037//0735-7044.111.6.1324. [DOI] [PubMed] [Google Scholar]
- 37.Joyce EM, and Iversen SD (1979). The effect of morphine applied locally to mesencephalic dopamine cell bodies on spontaneous motor activity in the rat. Neurosci. Lett 14, 207–212. 10.1016/0304-3940(79)96149-4. [DOI] [PubMed] [Google Scholar]
- 38.Packer AM, Russell LE, Dalgleish HWP, and Häusser M (2015). Simultaneous all-optical manipulation and recording of neural circuit activity with cellular resolution in vivo. Nat. Methods 12, 140–146. 10.1038/nmeth.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leone P, Pocock D, and Wise RA (1991). Morphine-dopamine interaction: ventral tegmental morphine increases nucleus accumbens dopamine release. Pharmacol. Biochem. Behav 39, 469–472. 10.1016/0091-3057(91)90210-S. [DOI] [PubMed] [Google Scholar]
- 40.Corre J, van Zessen R, Loureiro M, Patriarchi T, Tian L, Pascoli V, and Lüscher C (2018). Dopamine neurons projecting to medial shell of the nucleus accumbens drive heroin reinforcement. eLife 7. 10.7554/eLife.39945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Owen SF, Liu MH, and Kreitzer AC (2019). Thermal constraints on in vivo optogenetic manipulations. Nat. Neurosci 22, 1061–1065. 10.1038/s41593-019-0422-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matsui A, and Williams JT (2011). Opioid-sensitive GABA inputs from rostromedial tegmental nucleus synapse onto midbrain dopamine neurons. J. Neurosci 31, 17729–17735. 10.1523/JNEUROSCI.4570-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matsui A, Jarvie BC, Robinson BG, Hentges ST, and Williams JT (2014). Separate GABA afferents to dopamine neurons mediate acute action of opioids, development of tolerance, and expression of withdrawal. Neuron 82, 1346–1356. 10.1016/j.neuron.2014.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jalabert M, Bourdy R, Courtin J, Veinante P, Manzoni OJ, Barrot M, and Georges F (2011). Neuronal circuits underlying acute morphine action on dopamine neurons. Proc. Natl. Acad. Sci. USA 108, 16446–16450. 10.1073/pnas.1105418108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Y, Rózsa M, Liang Y, Bushey D, Wei Z, Zheng J, Reep D, Broussard GJ, Tsang A, Tsegaye G, et al. (2023). Fast and sensitive GCaMP calcium indicators for imaging neural populations. Nature 615, 884–891. 10.1038/s41586-023-05828-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Williams JT, Ingram SL, Henderson G, Chavkin C, von Zastrow M, Schulz S, Koch T, Evans CJ, and Christie MJ (2013). Regulation of μ-opioid receptors: desensitization, phosphorylation, internalization, and tolerance. Pharmacol. Rev 65, 223–254. 10.1124/pr.112.005942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pentney RJW, and Gratton A (1991). Effects of local delta and mu opioid receptor activation on basal and stimulated dopamine release in striatum and nucleus accumbens of rat: an in vivo electro-chemical study. Neuroscience 45, 95–102. 10.1016/0306-4522(91)90106-X. [DOI] [PubMed] [Google Scholar]
- 48.Borg PJ, and Taylor DA (1997). Involvement of μ- and δ-opioid receptors in the effects of systemic and locally perfused morphine on extracellular levels of dopamine, DOPAC and HVA in the nucleus accumbens of the halothane-anaesthetized rat. Naunyn Schmiedebergs Arch. Pharmacol 355, 582–588. 10.1007/PL00004987. [DOI] [PubMed] [Google Scholar]
- 49.Britt JP, and McGehee DS (2008). Presynaptic opioid and nicotinic receptor modulation of dopamine overflow in the nucleus accumbens. J. Neurosci 28, 1672–1681. 10.1523/JNEUROSCI.4275-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Handal M, Grung M, Skurtveit S, Ripel A, and Mørland J (2002). Pharmacokinetic differences of morphine and morphine-glucuronides are reflected in locomotor activity. Pharmacol. Biochem. Behav 73, 883–892. 10.1016/S0091-3057(02)00925-5. [DOI] [PubMed] [Google Scholar]
- 51.Di Chiara G, and Imperato A (1988). Opposite effects of mu and kappa opiate agonists on dopamine release in the nucleus accumbens and in the dorsal caudate of freely moving rats. J. Pharmacol. Exp. Ther 244, 1067–1080. [PubMed] [Google Scholar]
- 52.Spanagel R, Herz A, and Shippenberg TS (1990). The effects of opioid peptides on dopamine release in the nucleus accumbens: an in vivo microdialysis study. J. Neurochem 55, 1734–1740. 10.1111/j.1471-4159.1990.tb04963.x. [DOI] [PubMed] [Google Scholar]
- 53.Daglish MRC, Williams TM, Wilson SJ, Taylor LG, Eap CB, Augsburger M, Giroud C, Brooks DJ, Myles JS, Grasby P, et al. (2008). Brain dopamine response in human opioid addiction. Br. J. Psychiatry 193, 65–72. 10.1192/bjp.bp.107.041228. [DOI] [PubMed] [Google Scholar]
- 54.Watson BJ, Taylor LG, Reid AG, Wilson SJ, Stokes PR, Brooks DJ, Myers JF, Turkheimer FE, Nutt DJ, and Lingford-Hughes AR (2014). Investigating expectation and reward in human opioid addiction with [11C]raclopride PET. Addict. Biol 19, 1032–1040. 10.1111/adb.12073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Spagnolo PA, Kimes A, Schwandt ML, Shokri-Kojori E, Thada S, Phillips KA, Diazgranados N, Preston KL, Herscovitch P, Tomasi D, et al. (2019). Striatal dopamine release in response to morphine: a [11C]Raclopride positron emission tomography study in healthy men. Biol. Psychiatry 86, 356–364. 10.1016/j.biopsych.2019.03.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Christie MJ (2008). Cellular neuroadaptations to chronic opioids: tolerance, withdrawal and addiction. Br. J. Pharmacol 154, 384–396. 10.1038/bjp.2008.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gorostiza P, and Isacoff EY (2008). Optical switches for remote and noninvasive control of cell signaling. Science 322, 395–399. 10.1126/science.1166022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mourot A, Kienzler MA, Banghart MR, Fehrentz T, Huber FME, Stein M, Kramer RH, and Trauner D (2011). Tuning photochromic ion channel blockers. ACS Chem. Neurosci 2, 536–543. 10.1021/cn200037p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shields BC, Kahuno E, Kim C, Apostolides PF, Brown J, Lindo S, Mensh BD, Dudman JT, Lavis LD, and Tadross MR (2017). Deconstructing behavioral neuropharmacology with cellular specificity. Science 356, eaaj2161. 10.1126/science.aaj2161. [DOI] [PubMed] [Google Scholar]
- 60.Lin WC, Tsai MC, Davenport CM, Smith CM, Veit J, Wilson NM, Adesnik H, and Kramer RH (2015). A comprehensive optogenetic pharmacology toolkit for in vivo control of GABA(A) receptors and synaptic inhibition. Neuron 88, 879–891. 10.1016/j.neuron.2015.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Donthamsetti PC, Broichhagen J, Vyklicky V, Stanley C, Fu Z, Visel M, Levitz JL, Javitch JA, Trauner D, and Isacoff EY (2019). Genetically targeted optical control of an endogenous G protein-coupled receptor. J. Am. Chem. Soc 141, 11522–11530. 10.1021/jacs.9b02895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tobias JM, Rajic G, Viray AEG, Icka-Araki D, and Frank JA (2021). Genetically-targeted photorelease of endocannabinoids enables optical control of GPR55 in pancreatic β-cells. Chem. Sci 12, 13506–13512. 10.1039/d1sc02527a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bruno CA, O’Brien C, Bryant S, Mejaes JI, Estrin DJ, Pizzano C, and Barker DJ (2021). pMAT: An open-source software suite for the analysis of fiber photometry data. Pharmacol Biochem Behav 201, 173093. 10.1016/j.pbb.2020.173093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ren W, Ji A, and Ai H (2015). Light activation of protein splicing with a photocaged fast intein. J Am Chem Soc 137, 2155–2158. 10.1021/ja508597d. [DOI] [PubMed] [Google Scholar]
- 65.Cai N-S, Quiroz C, Bonaventura J, Bonifazi A, Cole TO, Purks J, Billing AS, Massey E, Wagner M, Wish ED, et al. (2019). Opioid-gal-anin receptor heteromers mediate the dopaminergic effects of opioids. J Clin Invest 129, 2730–2744. 10.1172/JCI126912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bonaventura J, Lam S, Carlton M, Boehm MA, Gomez JL, Solís O, Sánchez-Soto M, Morris PJ, Fredriksson I, Thomas CJ, et al. (2021). Pharmacological and behavioral divergence of ketamine enantiomers: implications for abuse liability. MOl Psychiatry 26, 6704–6722. 10.1038/s41380-021-01093-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cuitavi J, Andrés-Herrera P, Meseguer D, Campos-Jurado Y, Lorente JD, Caruana H, and Hipólito L (2023). Focal mu-opioid receptor activation promotes neuroinflammation and microglial activation in the mesocorticolimbic system: Alterations induced by inflammatory pain. Glia 71, 1906–1920. 10.1002/glia.24374. [DOI] [PubMed] [Google Scholar]
- 68.Rashid MH, Inoue M, Kondo S, Kawashima T, Bakoshi S, and Ueda H (2003). Novel expression of vanilloid receptor 1 on capsaicin-insensitive fibers accounts for the analgesic effect of capsaicin cream in neuropathic pain. J Pharmacol Exp Ther 304, 940–948. 10.1124/jpet.102.046250. [DOI] [PubMed] [Google Scholar]
- 69.Taylor AMW, Castonguay A, Ghogha A, Vayssiere P, Pradhan AA, Xue L, Mehrabani S, Wu J, Levitt P, Olmstead MC, et al. (2016). Neuroimmune Regulation of GABAergic Neurons Within the Ventral Tegmental Area During Withdrawal from Chronic Morphine. Neuropsychopharmacology 41, 949–959. 10.1038/npp.2015.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data for all figures are available in the supporting information (Data S2). Raw data are available upon request.