

Abstract
Conformational heterogeneity is a defining hallmark of intrinsically disordered proteins and protein regions (IDRs). The functions of IDRs and the emergent cellular phenotypes they control are associated with sequence-specific conformational ensembles. Simulations of conformational ensembles that are based on atomistic and coarse-grained models are routinely used to uncover the sequence-specific interactions that may contribute to IDR functions. These simulations are performed either independently or in conjunction with data from experiments. Functionally relevant features of IDRs can span a range of length scales. Extracting these features requires analysis routines that quantify a range of properties. Here, we describe a new analysis suite SOURSOP, an object-oriented and open-source toolkit designed for the analysis of simulated conformational ensembles of IDRs. SOURSOP implements several analysis routines motivated by principles in polymer physics, offering a unique collection of simple-to-use functions to characterize IDR ensembles. As an extendable framework, SOURSOP supports the development and implementation of new analysis routines that can be easily packaged and shared.
Graphical Abstract
1. INTRODUCTION
Natively unfolded proteins or intrinsically disordered proteins and regions (collectively referred to as IDRs,) are a ubiquitous class of proteins and domains that regulate various molecular functions and cellular phenotypes1–4. Unlike folded domains, which are well-described by a few structurally similar microstates, IDRs are defined by their conformational heterogeneity4,5. As a result, the accurate description of IDRs in the solution state necessitates a statistical description of the underlying conformational ensembles6. These ensembles, which are affected by changes to solution conditions and the types of components present in the solvent, are distributions of energetically accessible protein conformations that capture the sequence-encoded conformational biases associated with a given IDR 4,7,8. Several studies have established direct connections between sequence-ensemble relationships of IDRs and the molecular functions of these conformationally heterogeneous regions8–10. Accordingly, there is a need for facile, ready-to-use methods to uncover the molecular grammars that underlie sequence-ensemble-function relationships of IDRs9.
Measurements of IDR ensembles in solution allow for quantitative mapping of sequence-ensemble relationships. Techniques that obtain statistical information on molecular conformation without pre-supposing the existence of a single dominant state are well-equipped to characterize IDR ensembles. These techniques include static and dynamic multiangle light scattering (SLS and DLS, respectively), small-angle X-ray scattering (SAXS), circular dichroism (CD), infrared spectroscopies, electron paramagentic resonance (EPR) spectrosopy, nuclear magnetic resonance (NMR) spectroscopy, multiparameter fluorescence spectroscopies, and other single-molecule techniques7,11–16. While these experimental techniques offer a window into conformational behaviors, they typically probe a single type of conformational characteristic (e.g., global ensemble average dimensions, distances between specific positions along the chain, etc.). Accirdingly, alongside these experimental approaches, all-atom and coarse-grained molecular simulations are routinely deployed to make predictions or interpret data obtained from experimental measurements. The joint application of experimental and computational methods enables the integration of multiple conformational inputs, thus affording a holistic assessment of sequence-ensemble relationships17–23.
Simulations of all stripes, but specifically all-atom simulations based on explicit or implicit representations of solvent are especially useful for describing sequence-specific conformational ensembles of IDRs24,25. If a simulation can fully explore the conformational landscape and the forcefield being used is accurate enough, then all-atom molecular simulations enable the direct prediction of ensembles from sequence. These computationally derived ensembles can be compared directly or indirectly with experiments or used in isolation to understand functional and evolutionary constraints on IDRs 19,20,25. Consequently, there has been substantial interest in developing and applying Molecular Dynamics (MD) and Monte Carlo (MC) simulations to study IDRs 26–33. It is worth noting that the length of an IDR is not an intrinsic limitation on the generation of converged results from all-atom simulations especially MC simulations based on implicit solvent models such as ABSINTH. The limitation is invariably the ruggedness of the free energy landscape, which is generally a challenge for IDRs characterized by a diverse range of energy scales.
As all-atom simulations have become increasingly routine, various software packages have emerged to perform and analyze molecular simulations. Major packages for performing all-atom simulations (so-called simulation engines) include, but are not limited to, Amber, CAMPARI, CHARMM, Desmond, GROMACS, LAMMPS, OpenMM, and NAMD30,34–40. Alongside the development of simulation engines, there has also been an emergence of stand-alone packages for simulation analysis. Although most simulation engines contain their analysis routines, stand-alone analysis packages provide an alternative that, in principle, can be relatively lightweight, customizable, and unburdened by coding practices or conventions of the inevitably larger simulation engines. General-purpose analysis packages include Bio3D, CPPTRAJ, ENSPARA, LOOS, MDAnalysis, MDTraj, ST-Analyzer, VMD, and others41–43 (see Supplemental Table S1 for a more extensive list). While some packages are general-purpose libraries for analyzing simulation trajectories, others are developed with a specific goal in mind 44–46. Decoupling analysis from performing simulations allows for ease of use, installation, and portability to be prioritized in analysis packages, while performance can be prioritized in simulation engines. It also enables familiarity with a single analysis framework that can be applied across different simulation engines.
All-atom simulations of IDRs are becoming increasingly common17,19,47. Despite this, there is a lack of stand-alone analysis packages specifically catering to the analysis of IDR conformational ensembles. Given their inherently heterogeneous ensembles and the lack of a relevant single reference structure, many of the structure-centric analyses commonly employed in the context of folded may be poorly suited for characterizing IDR ensembles. In contrast, concepts and principles from polymer physics have been taken and applied to interpret and understand disordered and unfolded proteins to great effect6,11,19,48–51.
Here we introduce SOURSOP (Simulation analysis Of Unfolded RegionS Of Proteins), a Python-based software package for the analysis of all-atom simulations of disordered and unfolded proteins. SOURSOP combines both analysis routines commonly found for folded proteins with a range of IDR-centric analyses that have been used extensively across many publications over the last half-decade. In the remainder of this article, we lay out the software architecture of SOURSOP, provide several examples of analysis that can be performed, and offer a discussion of practical and conceptual features associated with the software.
2. METHODS
SOURSOP is written in Python 3.7+ and built atop the general-purpose simulation analysis package MDTraj41. SOURSOP uses MDTraj as a backend for parsing simulation trajectories and can accept trajectories in a wide variety of file formats. Although trajectory files are parsed into SOURSOP-specific objects, the underlying mdtraj.topology and mdtraj.trajectory objects remain user-facing and accessible. In this way, any analysis written to work with MDTraj is directly applicable to SOURSOP objects.
SOURSOP reads a simulation trajectory into a SSTraj object. The SSTraj object automatically extracts individual protein chains into their SSProtein objects. SSProtein objects are the base object upon which single-chain analysis routines are applied as object functions. In addition, peripheral modules that include ssnmr and sspre, provide modular, protein-independent analyses that work in conjunction with an SSProtein object. In this way, SOURSOP abides by the software principle of loose coupling, facilitating maintainability and future extension. The overall architecture of SOURSOP is shown in Fig. 1A.
Figure 1: Architecture and example code for SOURSOP.
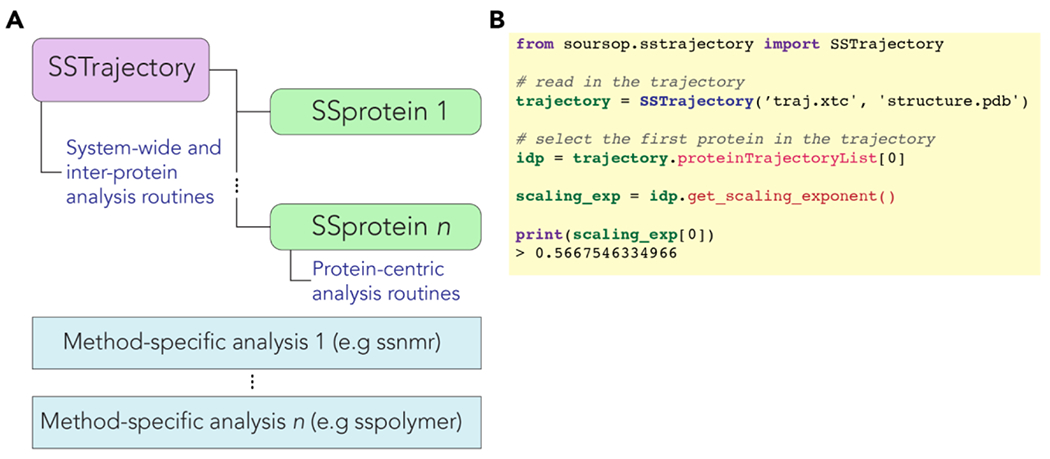
(A) Trajectory files are read into an SSTrajectory object. This object automatically parses each polypeptide chain into separate SSProtein objects. Each SSProtein object has a set of object-based analyses associated with them. Each trajectory must have between 1 and protein chains in it. In addition, various stateless method-specific analysis modules exist for certain types of analysis. Additional stateless methods can be extended to allow new analysis routines to be incorporated in a way that does not alter the SSProtein or SSTrajectory code. (B) Example code illustrating how the apparent scaling exponent can be calculated from an ensemble.
Where possible and appropriate, SOURSOP engages in memoization, a dynamic programming approach where expensive calculations are saved after being executed once52. This offers a general strategy that avoids repeated recalculation of (for example) the same sets of distances. In addition to intramolecular analysis codified in the SSProtein object, intermolecular and multi-chain analysis routines are included in the SSTraj object. In this way, a simple and standardized interface for working with protein ensemble data is provided. Ensembles to be analyzed could be generated through standard all-atom simulations, but PDB ensembles from NMR or ensemble selection procedures are also directly analyzable.
A major goal in developing SOURSOP is to make simulation analysis easy and intuitive, both for the user and developers. For example, Fig. 1B offers a simple example of computing a protein’s apparent scaling exponent () for a protein in a simulation trajectory. While a straightforward user experience is an obvious goal for any software package, providing a consistent, well-defined, and accessible software architecture is essential for long-term maintenance and extendibility. Well-structured software is also necessary to enable productive and sustainable open-source contributions.
The current working version can be found at https://github.com/holehouse-lab/soursop, with documentation at https://soursop.readthedocs.io/. SOURSOP uses PyTest (https://docs.pytest.org/en/stable/) for unit testing, Sphinx (https://www.sphinx-doc.org/en/master/), and readthedocs (https://readthedocs.org/) for documentation, and Git (https://git-scm.com/) and GitHub (https://github.com/) for version control. The original repository structure was generated using cookiecutter (https://github.com/cookiecutter/cookiecutter). Explicit dependencies include MDTraj41, SciPy53, NumPy54, Pandas55, and Cython56.
In addition to the analyses shown here, we provide a collection of Jupyter notebooks along with the full trajectories (where possible) that offer examples of more general IDR-centric analysis that can be performed on the ensembles studied here (https://github.com/holehouse-lab/supportingdata/tree/master/2023/lalmansingh_2023). The SOURSOP code is consistent and heavily commented. The documentation also provides specific guidance for the development and integration of new analysis routines into SOURSOP.
3. RESULTS
To demonstrate the analyses available in SOURSOP, we have analyzed a collection of ensembles generated by various methods. The trajectories analyzed were generated using CAMPARI (an all-atom Monte Carlo simulation engine) or Desmond (an all-atom MD simulation engine)26,57,58. The analyses performed here are offered as convenient examples of the types of analyses and insight enabled by SOURSOP. All of the analyses described in the results section are based on functions defined in the documentation at https://soursop.readthedocs.io/ and we note in-line the associated function names the first time a specific analysis is referenced. In addition, examples and tutorials for SOURSOP are available at https://soursop.readthedocs.io/en/latest/usage/examples.html.
3.1. IDR global dimensions show extensive sequence-dependent conformational biases
A challenge in studying IDRs is the absence of an obvious reference state. While folded proteins are typically associated with a native conformation which can serve as a reference point for further analysis, the structural heterogeneity of an IDR means that no single state serves this purpose. Conveniently, polymer physics offers analytical tools that can serve as reference states for disordered and unfolded protein ensembles 30,48,59–63. As a result, dimensionless polymeric parameters can be computed, which allows the conformational behavior of very different proteins to be quantitatively and directly compared. SOURSOP implements the calculation of many of these parameters, facilitating ensemble analysis.
We re-analyzed a series of conformational ensembles using two such dimensionless reference parameters. Specifically, we computed instantaneous asphericity () (.get_asphericity()), which measures the shape of a given conformation64. The instantaneous asphericity is defined as
| (1) |
Where , , and are the eigenvalues of the gyration tensor (T), which in turn is defined as,
| (2) |
Where the T is calculated for every conformation, is the number of atoms in the system, is the position vector of atom in the conformation of interest, is the centroid of all atoms positions in that conformation, and reflects the tensor product (also known as the dyadic product). The gyration tensor itself also accessible via the .get_gyration_tensor() function.
In addition to the instantaneous asphericity, we can calculate , (.get_t()) a dimensionless parameter that quantifies global dimensions effectively via a normalized radius of gyration as originally defined by Vitalis and Pappu as
| (3) |
where is the number of residues in the sequence, is the contour length of the polypeptide in Angstroms (3.6×N), and , , and , are parameters used to ensure remains in the interval of 0 to 165. , , and , are defined as 2.5, 1.75 and 4.0, respectively65. By generating 2-dimensional density plots that quantify the simultaneous evaluation of and for each conformation, a quantitative and length-normalized representation of IDR global conformational preferences can be easily visualized.
Both and are transformations of the eigenvalues from the gyration tensor T. They represent global order parameters to describe the size and shape of a given conformation. An alternative normalization approach is using polymer models as reference states, or considering additional polymeric parameters that report on the global chain dimensions (e.g., hydrodynamic radius, end-to-end distance, or the apparent polymer scaling exponent)66–69. All of these can be calculated in SOURSOP (get_radius_of_gyration(), get_hydrodynamic_radius(), get_end_to_end_distance(), get_scaling_exponent()). For convenience, we focus here on , and on the normalized radius of gyration, although other metrics would likely report similar conclusions. To normalize the radius of gyration, we make use of an analytical model (the Analytical Flory Random Coil, AFRC) that provides the expected radius of gyration if the chain behaved as a polymer in a theta solvent66.
We analyzed conformational ensembles with over 3 × 104 distinct conformers obtained from previously published simulations that have been directly benchmarked against experiments to compare how ensemble size and shape vary across different IDRs (Fig. 2A, Table S1) 26,57,58,68,70–72. This analysis revealed a wide array of global conformational behaviors for IDRs. Our observations highlight properties ranging from heterogeneous compact ensembles to highly expanded self-avoiding random chains commensurate with polypeptides under strongly denaturing conditions. To contextualize these global dimensions, we also calculated normalized radii of gyration using the dimensions of a sequence-matched chain under conditions in which chain-chain and chain-solvent interaction are counterbalanced, with similar results (Fig. 2B)66.
Figure 2:
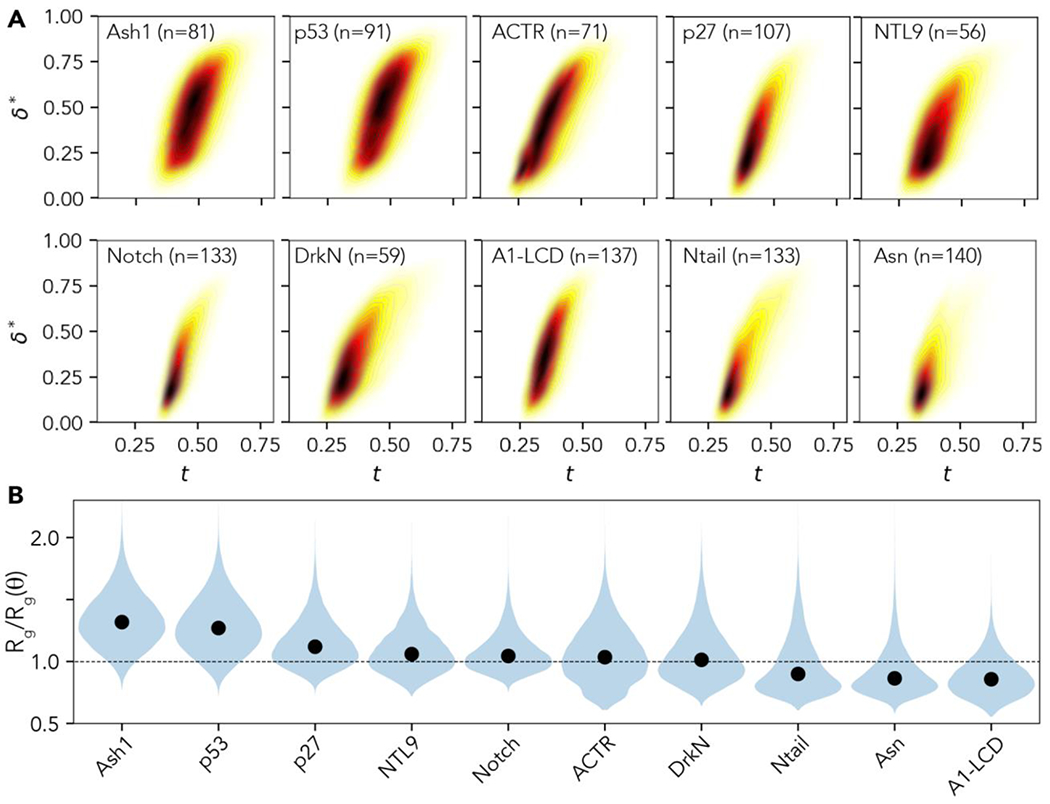
Global conformational analysis of 10 disordered protein ensembles analyzed with SOURSOP. (A) The two-dimensional density plots for instantaneous asphericity () and normalized dimensions () reveal a broad range of conformational landscapes. Ash158, p5373, p2772, NTL968, Notch70, and A1-LCD57 are ensembles generated by Monte Carlo ensembles with the ABSINTH implicit solvent model60. ACTR, drkN, NTail, and Asn (alpha synuclein) are ensembles generated by molecular dynamics simulations with Amber99-disp forcefield26. Note that NTL9 is not an IDP, but the ensemble reported here represents an unfolded-state ensemble obtained under native conditions68. (B) Normalized chain dimensions were calculated by normalizing the instantaneous radius of gyration from ensembles by the expected radius of gyration from a sequence-matched chain in the theta state, whereby chain-chain and chain-solvent interactions are counterbalanced 6,62,73.
The diversity in global IDR properties (size and shape), as illustrated in Fig. 2A, is often masked by ensemble-average properties. As a result, two IDRs may appear, on average, to be highly similar. The simulation analysis uncovers differences using the full distribution of conformations, which is evident even for relatively simple order parameters such as and , in agreement with prior work showing ensemble-average properties can mask complexities in the underlying conformational ensemble 74–76.
Aromatic residues, charged residues, and proline play an outsized role in dictating the conformational behavior of disordered proteins
Next, we applied SOURSOP to identify key sequence determinants of the attractive and repulsive intramolecular interactions that determine global and local conformational biases in IDR ensembles. To evaluate local chain interactions, we computed the radius of gyration over a sliding window of 14 residues to generate a linear profile of local density, normalizing for steric effects via an atomistic excluded volume (EV) model (Fig. 3, [get_local_collapse()] see Supplemental Information). We note the window size was chosen to reflect approximately 3x the polypeptide blob length, but the window size is a free parameter that can be passed to the function77. To assess long-range interactions, we computed scaling maps (Fig. 4, [get_distance_map()]). Scaling maps report inter-residue distances (distance maps) normalized by the expected distances from some reference polymer model, in this case, the EV model. The use of scaling maps accounts for the intrinsic contribution that chain connectivity has to inter-residue distances. While we use numerical simulations of EV polymers to generate the reference state for our scaling maps here, scaling maps can, more broadly, involve any convenient reference state, which may include theoretical models or even sequences of different compositions66,78. Moreover, SOURSOP also provides the ability to fit an ensemble to an apparent homopolymer model and then calculate deviations from that model across intra-molecular distances (get_polymer_scaled_distance_map()].
Figure 3:
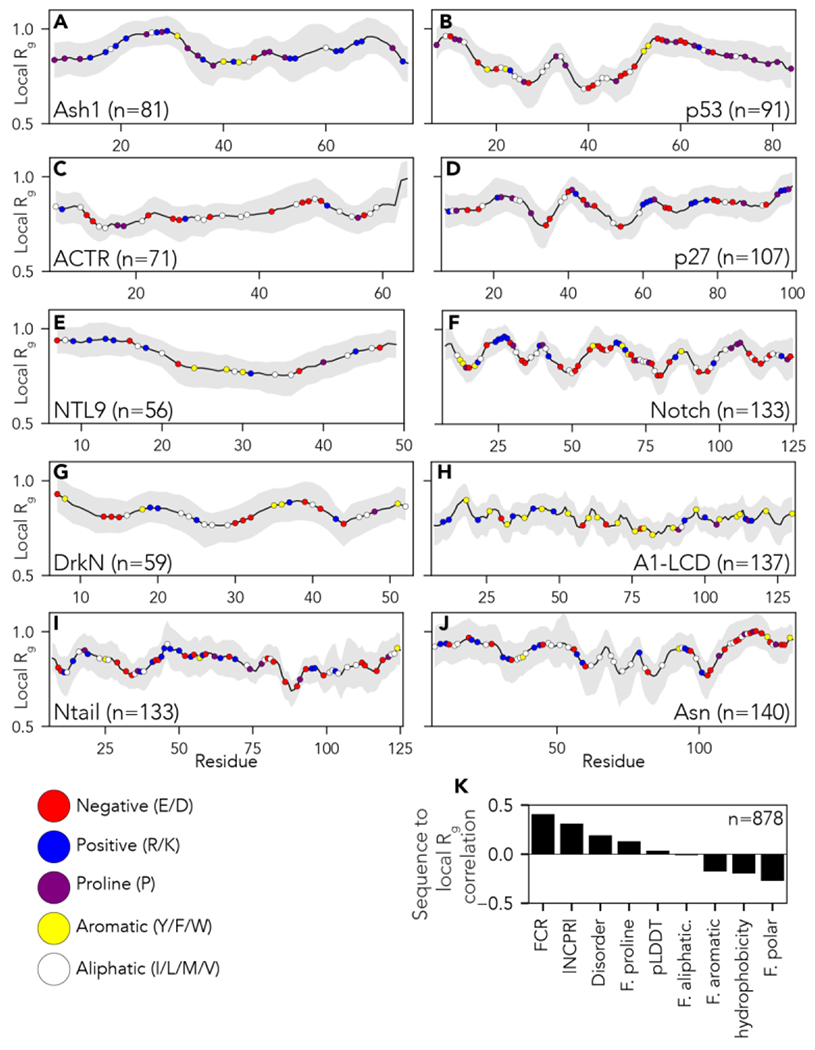
Local chain compaction with residue chemistry superimposed over the local radius of gyration (Rg). (A-J) Individual plots showing analysis for each protein ensemble as introduced in Figure 2. Local Rg is calculated using a 14-residue sliding window. Colored circles on each plot represent different amino acid chemistry groups, highlighted in the legend below panel I. (K) Pearson’s correlation coefficient between local Rg obtained for each windowed fragment reported in panels A-J and the amino acid chemistry within the window in question (see also Fig. S2). Specific sequence properties reported are the Fraction of Charged residues (FCR), absolute net charge per residue (|NCPR|), mean disorder score as predicted by metapredict (Disorder), fraction of proline residues (F. proline), mean predicted Local Distance Difference Test (pLDDT - a measure of predicted AlphaFold2 structure confidence), fraction of aliphatic residues (F. aliphatic), fraction of aromatic residues (F. aromatic), Kyte Doolitle hydrophobicity (hydrophobicity) and fraction of polar residues (F. polar). The fraction of charged residues (FCR) is the strongest positive determinant of expansion, closely followed by the absolute net charge per residue (|NCPR|). While polar residues, in principle, correlate as negative determinants of expansion, the negative correlation is driven by subregions deficient in charged residues and enriched in only polar residues.
Figure 4:
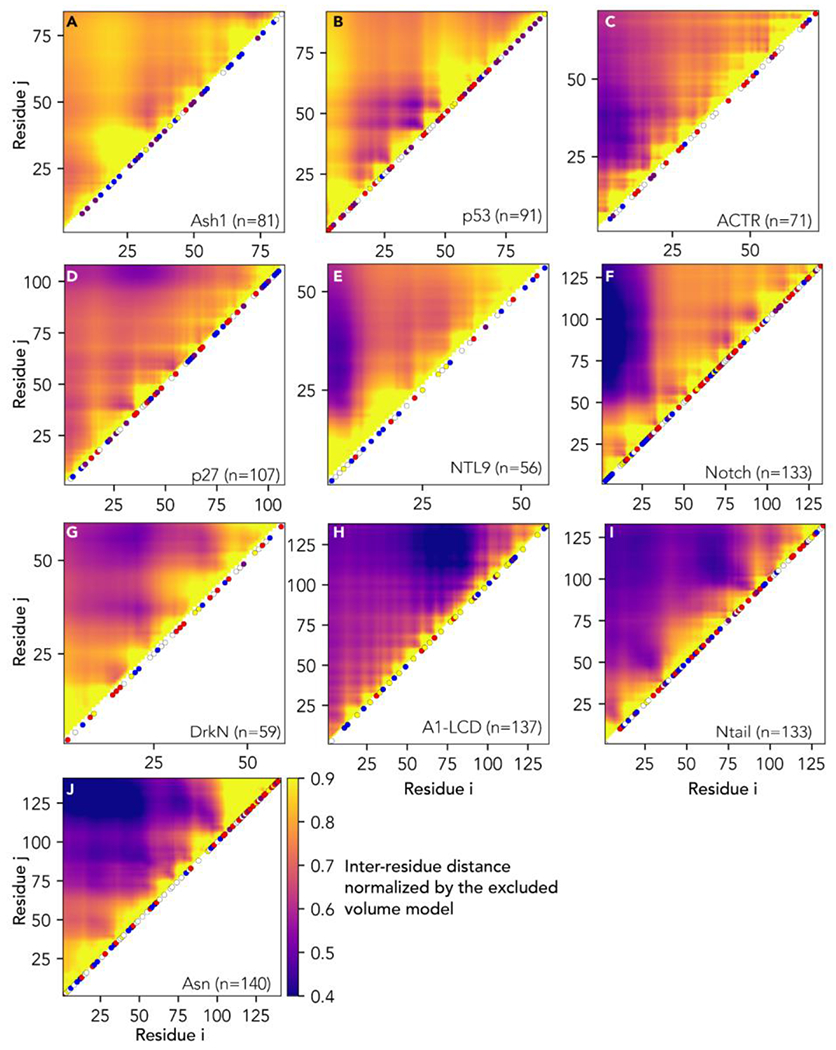
Preferential attraction and repulsion quantified via scaling maps that report the normalized distance between every pair of residues in the protein. (A-J) Individual plots of analysis for each protein ensemble as introduced in Figure 2. Normalized distances are calculated by dividing ensemble-average inter-residue distance by the distance obtained for the EV model. Attractive interactions emerge as darker colors, while repulsive interactions are lighter. Along the diagonal, subsets of residues are colored using the same color scheme used in Fig. 3.
Our analysis here across the set of simulations confirmed prior observations made by many groups: that charged, aromatic, and proline residues emerge as key determinants of IDR local and global interactions irrespective of the forcefield or simulations approach being used (Fig. 3, Fig. 4).
While our analysis is necessarily retrospective and correlative, it is in line with prior experimental work57,79–82. To explore this observation further, we performed all-atom simulations using the ABSINTH implicit solvent model of the p531–91 with three phosphomimetic mutations (S15E, T18E, S20E) and compared the result to previous simulations of the wildtype sequence (Fig. 5A)71. While glutamic acid is an imperfect analog for the phosphate group, the results revealed that relatively modest changes in linear charge density can cause local and long-range changes in the conformational ensemble. Despite substantial local conformational rearrangement, this leads only to a modest change of 0.5 Å in the mean radius of gyration (Fig. 5B). Charge effects leading to seemingly minor changes in global dimensions while altering local networks of intramolecular interactions mirrors prior work on the multi-phosphorylated proteins Ash1, Sic1, and a region of the RNA polymerase CTD 58,78,83,84. Taken together, these results suggest that while local changes in charge density can induce local conformational changes in ensemble behavior, compensatory changes in attractive (and repulsive) interactions that act on different or overlapping length scales can mask the effects of large-scale changes when global chain dimensions are examined.
Figure 5:
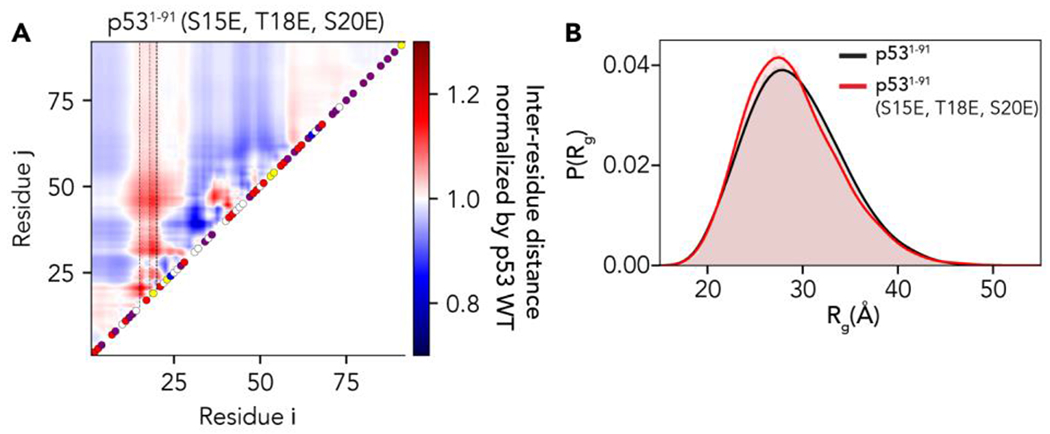
Comparison of changes in local and global dimensions for wildtype vs. phosphomimetic versions of p53. (A) Scaling maps where inter-residue distances for the phosphomimetic version of p53 N-terminal domain (p531–91) are normalized by distances for the wild-type protein. Despite differing by only three residues in the N-terminal quarter of the protein, the phosphomimetic version of p53 shows substantial differences in long-range and local dimensions, as shown by the emergence of both attractive (blue) and repulsive (red) interactions. (B) Despite these rearrangements, a relatively small change in overall global dimensions is observed. While the wildtype ensemble-average Rg is 29.4 Å, the phosphomimetic variant is 29.1 Å, a difference below the statistical detection limits for most experimental techniques.
Molecular accessibility is context dependent in IDRs
It is often convenient to imagine IDRs as being uniformly solvent accessible.. While appealingly simple, given the complex conformational behavior observed in our analyses here and elsewhere, it may not be a given that every residue is equally accessible58,75,85–88. To examine this idea further, we computed local accessibility across an eight-residue sliding window for each IDR using a 10 Å spherical probe ([get_regional_SASA()] Fig. 6). Solvent accessibility here was calculated using the Shake Rupley algorithm89. We chose eight residues here to provide information on local conformational accessibility only (i.e., slightly larger than the blob size), although the window size used can be varied as an input parameter to the analysis function. This analysis allows us to assess how accessibility varies as a function of local sequence position.
Figure 6:
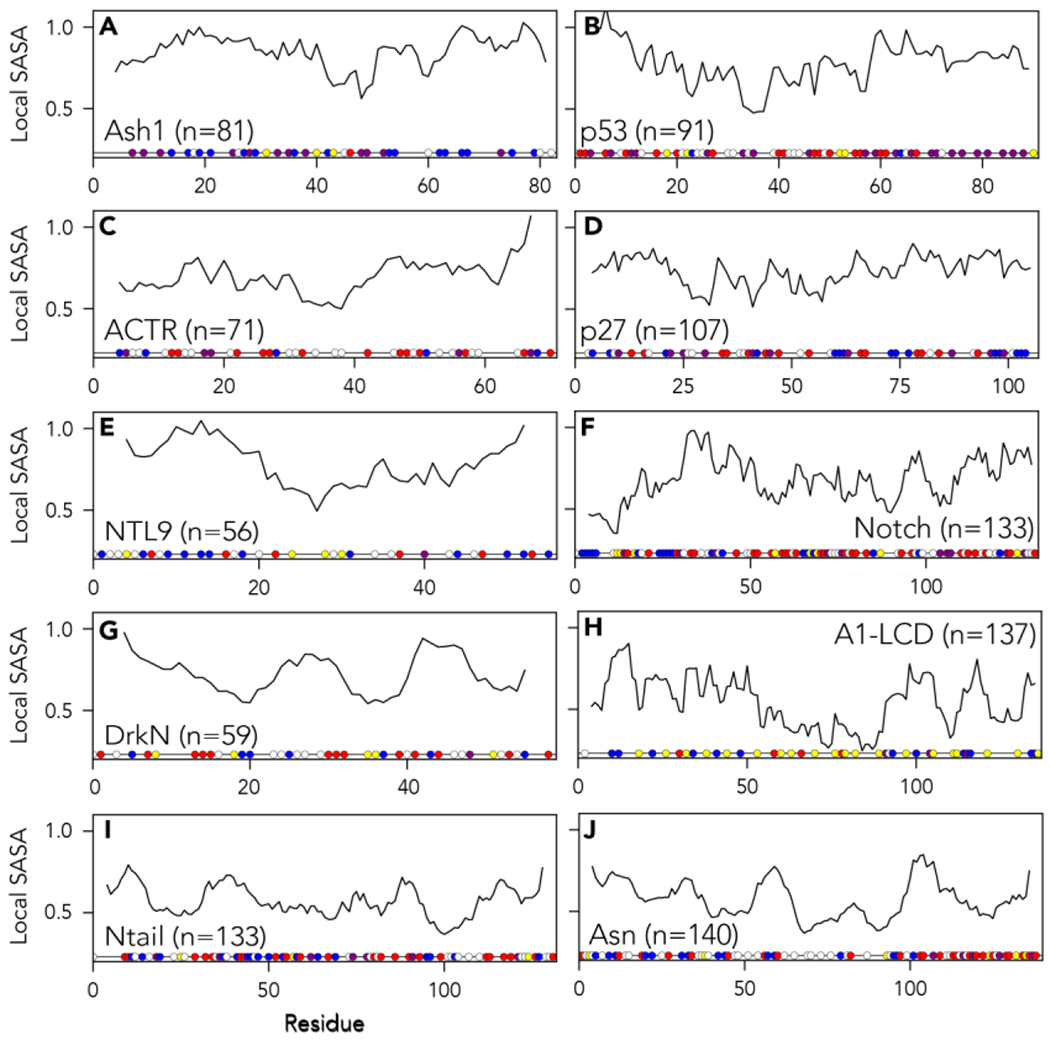
Normalized local solvent-accessible surface area (SASA) using an eight-residue sliding window and a 10 Å probe size. Normalization is done using excluded volume (EV) reference simulations to account for side-chain-dependent differences in solvent accessibility. Amino acid residues are colored as in Fig 3. Distinct patterns of accessibility are observed across different proteins, indicating long- and short-range intramolecular interactions can influence the accessibility of local binding sites.
Our analysis reveals substantial variation in molecular accessibility, suggesting that two residues of the same type may be differentially accessible depending on their broader sequence context (Fig. 6). Clearly, the local sequence environment offers a mechanism to control the effective concentration of a local binding motif. The importance of local sequence context on molecular interactions can be further expanded if sequence-encoded chemistry provides partner-specific attractive and repulsive interactions. Taken together, despite the lack of a fixed 3D structure, it seems reasonable to speculate that the binding of motifs from IDRs should be considered both in terms of molecular sterics and shape complementarity (as is the conventional view for rigid-body molecular recognition) but also in terms of if and how the local chain context influences their accessibility and chemical context 90,91.
4. DISCUSSION
Here we introduce SOURSOP, an integrative Python-based software package for the analysis of all-atom ensembles extracted from simulations of intrinsically disordered proteins. SOURSOP is easy to install and use and is accompanied by extensive documentation and unit tests. Here we have shown how SOURSOP can be applied to analyze all-atom ensembles extracted from two types of simulations (Monte Carlo simulations and molecular dynamic simulations) of different IDRs. SOURSOP contains a range of additional routines not explored in this work, but have been applied to various systems under a range of contexts, including local residual structure, intra-residue contacts, and the interaction between folded and disordered regions (Fig. S1) 57,58,92–94.
SOURSOP as a stand-alone package
SOURSOP was developed as a stand-alone analysis package built on the existing general-purpose simulation analysis package MDTraj41. The decision to develop SOURSOP as an independent package, as opposed to expanding the functionality of MDTraj, was motivated by several factors.
First, many of the analysis routines built into SOURSOP are of limited value for the analysis of well-folded proteins. At this juncture, MDTraj is a stable and mature software package that functions as the backend to a range of tools associated with molecular simulations 44,95–99. To add features into MDTraj would unavoidably lead to additional technical debt - more features to keep track of, manage, and test for. Technical debt adds viscosity, risks the introduction of new bugs, and can hamper future development if several coding styles are combined100. Accordingly, the drawbacks of integrating the analysis routines into MDTraj were judged to be substantially greater than the possible benefits.
Second, our goal is for SOURSOP to provide a general platform where novel analysis routines appropriate for disordered proteins can be implemented by the burgeoning community of labs performing simulations of disordered proteins. This requires our ability to maintain control over a consistent programmatic interface, which can be achieved via an interface layer between MDTraj and SOURSOP but becomes challenging if analysis routines are implemented directly inside of MDTraj. For this reason, providing SOURSOP as a loosely-coupled software component that works with MDTraj, as opposed to within MDTraj, enables the best of both worlds.
Finally, applying principles from polymer physics to analyze disordered proteins is not new. Several of the analysis routines provided by SOURSOP are also available in extant software, notably in the simulation engine CAMPARI (http://campari.sourceforge.net/) 6,30,60,61,101. SOURSOP provides a lightweight toolkit that is simple to install, simple to use, and interoperable with MDTraj and the collection of existing analysis tools therein. Therefore, while some overlap exists, we do not see SOURSOP as replacing the analysis routines in MDTraj or CAMPARI. Instead, SOURSOP is a complement to extant routines and packages. Furthermore, it makes it relatively straightforward for groups to publish scripts or Jupyter notebooks that enable full reproduction of their analysis workflow.
SOURSOP in the broader ecosystem of simulation software
Molecular simulations enables the characterization of biophysical properties that may be inaccessible to direct experimental measurement. Simulations typically involve either Molecular Dynamics (MD) or Monte Carlo (MC)-based approaches. In addition, the underlying molecular system must be represented in some way, be that at all-atom resolution or some degree of coarse-graining. All-atom models represent every atom in a system explicitly. In contrast, coarse-grain models combine two or more atoms into groups, and may included united-atom models (where typically 2-4 atoms are combined together) or lower resolution models in which residues or whole domains are represented as single beads.
As model resolution decreases, longer and larger simulations become increasingly accessible, yet assumptions and caveats made to justify the loss of details become increasingly broad. While it is tempting to expect all-atom models to be the gold standard, if simulations are unable to adequately sample a phenomenon of interest they may be inappropriate for a given question. In contrast, while coarse-grained simulations may seem appealing given their comparative simplicity when compared to all-atom simulations, the many assumptions that underlie their simplicitly can be, inadvertently, misleading. As such, the appropriate model depends entirely on the question of interest.
The emergence and refinement of molecular simulations has coincided with significant advancements in computational resources in the past thirty years. As a result, simulations are now commonly used in hypothesis generation, to aid in the interpretation of experimental data, and to build molecular models of complex biological processes. In parallel, as molecular simulations have become more common-place, there has been a veritable Cambrian explosion of APIs, toolkits, plugins, and frameworks for analyzing molecular simulations (see Table S1 for a survey of ~100 analysis packages). These tools help make complex, integrative analyses routine, and lower the barrier for scientific discovery.
SOURSOP analysis of coarse-grained simulations
While SOURSOP was built to analyze all-atom simulations, many of the routines provided can be directly applied when working with coarse-grained simulations. If an input topology (PDB) file defines beads as ‘CA’ (alpha carbon) as their atom type, then SOURSOP will correctly parse individual chains into individual residues, and the majority of the analyses described here are applicable for the analysis of coarse-grained simulations. It is important to emphasize that while we have used SOURSOP extensively to analyze coarse-grained simulations, certain functionality may not make sense or may fail (e.g., secondary structure analysis, sidechain bond vector analysis etc.)102–104. As such, while SOURSOP does not officially support the analysis of coarse-grained simulations at this juncture, unofficially, it works relatively well in this capacity.
SOURSOP is an extendable platform for novel analysis routines
Analyzing IDR ensembles to reveal clear and interpretable conclusions remains challenging. Absent a native reference state, it can be difficult to generate informative and visually coherent representations that fully capture the inherent high dimensionality of an IDR ensemble. While various ‘standard’ analyses have emerged for folded proteins (e.g., contact maps, per-residue RMSF, the fraction of native contacts), there is less consensus on what the standard analyses should be when assessing IDR ensembles.
Rather than a problem, this raises an opportunity, whereby novel analysis and visualization approaches are needed. With this in mind, we hope new analysis routines can be integrated into SOURSOP, facilitating distribution and packaging. Considering this objective, SOURSOP includes a well-defined style guide for new analysis routines and a collection of utility functions that provide automatic sanity checking and defensive programming for input data. We also provide documentation on how best to introduce a new routine and how to integrate it into the main codebase. These features, combined with the broad reach of the Python programming language, will lower the barrier to open-source and community-driven scientific development.
5. CONCLUSION
SOURSOP is an open-source Python toolkit for the general analysis of ensembles of disordered proteins. In addition to analyzing disordered protein ensembles, SOURSOP can also be used to analyze folded protein trajectories or individual PDB files. As such, SOURSOP offers a general interface for calculating molecular properties, polymeric parameters, and the development of new IDR-centric analysis routines.
Supplementary Material
ACKNOWLEDGMENTS
We thank all members of the Pappu and Holehouse labs for extensive testing, error reporting, and feature requests in SOURSOP. The development of SOURSOP was financially and intellectually supported by the Molecular Sciences Software Institute (MOLSSI), and A.S.H. received an NSF-sponsored MOLSSI postdoctoral fellowship (NSF-1547580 (subaward 479590)). This work was also supported by NSF grant no. 2128068 to ASH, and we thank members of the Water and Life Interface Institute (WALII), supported by NSF DBI grant #2213983, for helpful discussions. J.M.L. is a data scientist and scientific programmer in the Center for Biomolecular Condensates at Washington University in St. Louis. Work by members of the Pappu lab was supported by a grant from the Air Force Office of Scientific Research (FA9550-20-1-0241) and the US National Institutes of Health (R01NS121114).
SOURSOP was previously called CTraj and CAMPARITraj. We renamed the package SOURSOP to avoid confusion and the incorrect implication that this is a CAMPARI-specific analysis package.
Footnotes
Supporting Information Available: The supporting information includes details on the simulations performed for this manuscript, along with detailed description of the modern ecosystem of simulation analysis packages and several supplementary figures.
The Supporting Information is available free of charge on the ACS Publications website.
REFERENCES
- (1).van der Lee R; Buljan M; Lang B; Weatheritt RJ; Daughdrill GW; Dunker AK; Fuxreiter M; Gough J; Gsponer J; Jones DT; Kim PM; Kriwacki RW; Oldfield CJ; Pappu RV; Tompa P; Uversky VN; Wright PE; Babu MM Classification of Intrinsically Disordered Regions and Proteins. Chem. Rev 2014, 114 (13), 6589–6631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Sigler PB Acid Blobs & Negative Noodles. Nature 1988, 333, 210–212. [DOI] [PubMed] [Google Scholar]
- (3).Ptitsyn OB; Uversky VN The Molten Globule Is a Third Thermodynamical State of Protein Molecules. FEBS Lett. 1994, 341 (1), 15–18. [DOI] [PubMed] [Google Scholar]
- (4).Wright PE; Dyson HJ Intrinsically Unstructured Proteins: Re-Assessing the Protein Structure-Function Paradigm. J. Mol. Biol 1999, 293 (2), 321–331. [DOI] [PubMed] [Google Scholar]
- (5).Babu MM; Kriwacki RW; Pappu RV Structural Biology. Versatility from Protein Disorder. Science 2012, 337 (6101), 1460–1461. [DOI] [PubMed] [Google Scholar]
- (6).Mao AH; Lyle N; Pappu RV Describing Sequence-Ensemble Relationships for Intrinsically Disordered Proteins. Biochem. J 2013, 449 (2), 307–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Dyson HJ; Wright PE Unfolded Proteins and Protein Folding Studied by NMR. Chem. Rev 2004, 104 (8), 3607–3622. [DOI] [PubMed] [Google Scholar]
- (8).Mittag T; Forman-Kay JD Atomic-Level Characterization of Disordered Protein Ensembles. Curr. Opin. Struct. Biol 2007, 17 (1), 3–14. [DOI] [PubMed] [Google Scholar]
- (9).Das RK; Ruff KM; Pappu RV Relating Sequence Encoded Information to Form and Function of Intrinsically Disordered Proteins. Curr. Opin. Struct. Biol 2015, 32 (0), 102–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Martin EW; Holehouse AS Intrinsically Disordered Protein Regions and Phase Separation: Sequence Determinants of Assembly or Lack Thereof. Emerg Top Life Sci 2020, 4 (3), 307–329. [DOI] [PubMed] [Google Scholar]
- (11).Schuler B; Soranno A; Hofmann H; Nettels D Single-Molecule FRET Spectroscopy and the Polymer Physics of Unfolded and Intrinsically Disordered Proteins. Annu. Rev. Biophys 2016, 45, 207–231. [DOI] [PubMed] [Google Scholar]
- (12).Kikhney AG; Svergun DI A Practical Guide to Small Angle X-ray Scattering (SAXS) of Flexible and Intrinsically Disordered Proteins. FEBS Lett. 2015. [DOI] [PubMed] [Google Scholar]
- (13).Chemes LB; Alonso LG; Noval MG; de Prat-Gay G Circular Dichroism Techniques for the Analysis of Intrinsically Disordered Proteins and Domains. Methods Mol. Biol 2012, 895, 387–404. [DOI] [PubMed] [Google Scholar]
- (14).Gast K; Fiedler C Dynamic and Static Light Scattering of Intrinsically Disordered Proteins. Methods Mol. Biol 2012, 896, 137–161. [DOI] [PubMed] [Google Scholar]
- (15).Jensen MR; Zweckstetter M; Huang J-R; Blackledge M Exploring Free-Energy Landscapes of Intrinsically Disordered Proteins at Atomic Resolution Using NMR Spectroscopy. Chem. Rev 2014, 114 (13), 6632–6660. [DOI] [PubMed] [Google Scholar]
- (16).Gibbs EB; Cook EC; Showalter SA Application of NMR to Studies of Intrinsically Disordered Proteins. Arch. Biochem. Biophys 2017, 628, 57–70. [DOI] [PubMed] [Google Scholar]
- (17).Best RB Computational and Theoretical Advances in Studies of Intrinsically Disordered Proteins. Curr. Opin. Struct. Biol 2017, 42, 147–154. [DOI] [PubMed] [Google Scholar]
- (18).Wang X; Vitalis A; Wyczalkowski MA; Pappu RV Characterizing the Conformational Ensemble of Monomeric Polyglutamine. Proteins 2006, 63 (2), 297–311. [DOI] [PubMed] [Google Scholar]
- (19).Shea J-E; Best RB; Mittal J Physics-Based Computational and Theoretical Approaches to Intrinsically Disordered Proteins. Curr. Opin. Struct. Biol 2021, 67, 219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Alston JJ; Soranno A; Holehouse AS Integrating Single-Molecule Spectroscopy and Simulations for the Study of Intrinsically Disordered Proteins. Methods 2021, 193, 116–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Palazzesi F; Prakash MK; Bonomi M; Barducci A Accuracy of Current All-Atom Force-Fields in Modeling Protein Disordered States. J. Chem. Theory Comput 2015, 11 (1), 2–7. [DOI] [PubMed] [Google Scholar]
- (22).Zerze GH; Zheng W; Best RB; Mittal J Evolution of All-Atom Protein Force Fields to Improve Local and Global Properties. J. Phys. Chem. Lett 2019, 10 (9), 2227–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ruff KM; Pappu RV; Holehouse AS Conformational Preferences and Phase Behavior of Intrinsically Disordered Low Complexity Sequences: Insights from Multiscale Simulations. Curr. Opin. Struct. Biol 2019, 56, 1–10. [DOI] [PubMed] [Google Scholar]
- (24).Karplus M; Petsko GA Molecular Dynamics Simulations in Biology. Nature 1990, 347 (6294), 631–639. [DOI] [PubMed] [Google Scholar]
- (25).Bottaro S; Lindorff-Larsen K Biophysical Experiments and Biomolecular Simulations: A Perfect Match? Science 2018, 361 (6400), 355–360. [DOI] [PubMed] [Google Scholar]
- (26).Robustelli P; Piana S; Shaw DE Developing a Molecular Dynamics Force Field for Both Folded and Disordered Protein States. Proc. Natl. Acad. Sci. U. S. A 2018, 115 (21), E4758–E4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Piana S; Robustelli P; Tan D; Chen S; Shaw DE Development of a Force Field for the Simulation of Single-Chain Proteins and Protein-Protein Complexes. J. Chem. Theory Comput 2020, 16 (4), 2494–2507. [DOI] [PubMed] [Google Scholar]
- (28).Best RB; Mittal J Protein Simulations with an Optimized Water Model: Cooperative Helix Formation and Temperature-Induced Unfolded State Collapse. J. Phys. Chem. B 2010, 114 (46), 14916–14923. [DOI] [PubMed] [Google Scholar]
- (29).Best RB; Zheng W; Mittal J Balanced Protein-Water Interactions Improve Properties of Disordered Proteins and Non-Specific Protein Association. J. Chem. Theory Comput 2014, 10 (11), 5113–5124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Vitalis A; Pappu RV ABSINTH: A New Continuum Solvation Model for Simulations of Polypeptides in Aqueous Solutions. J. Comput. Chem 2009, 30 (5), 673–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Huang J; Rauscher S; Nawrocki G; Ran T; Feig M; de Groot BL; Grubmüller H; MacKerell AD Jr. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14 (1), 71–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Piana S; Donchev AG; Robustelli P; Shaw DE Water Dispersion Interactions Strongly Influence Simulated Structural Properties of Disordered Protein States. J. Phys. Chem. B 2015, 119 (16), 5113–5123. [DOI] [PubMed] [Google Scholar]
- (33).Appadurai R; Nagesh J; Srivastava A High Resolution Ensemble Description of Metamorphic and Intrinsically Disordered Proteins Using an Efficient Hybrid Parallel Tempering Scheme. Nat. Commun 2021, 12 (1), 958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Salomon-Ferrer R; Case DA; Walker RC An Overview of the Amber Biomolecular Simulation Package. Wiley Interdiscip. Rev. Comput. Mol. Sci 2013, 3 (2), 198–210. [Google Scholar]
- (35).Brooks BR; Brooks CL 3rd; Mackerell AD Jr; Nilsson L; Petrella RJ; Roux B; Won Y; Archontis G; Bartels C; Boresch S; Caflisch A; Caves L; Cui Q; Dinner AR; Feig M; Fischer S; Gao J; Hodoscek M; Im W; Kuczera K; Lazaridis T; Ma J; Ovchinnikov V; Paci E; Pastor RW; Post CB; Pu JZ; Schaefer M; Tidor B; Venable RM; Woodcock HL; Wu X; Yang W; York DM; Karplus M CHARMM: The Biomolecular Simulation Program. J. Comput. Chem 2009, 30 (10), 1545–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Abraham MJ; Murtola T; Schulz R; Páll S; Smith JC; Hess B; Lindahl E GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015/September, 1–2, 19–25. [Google Scholar]
- (37).Eastman P; Swails J; Chodera JD; McGibbon RT; Zhao Y; Beauchamp KA; Wang L-P; Simmonett AC; Harrigan MP; Stern CD; Wiewiora RP; Brooks BR; Pande VS OpenMM 7: Rapid Development of High Performance Algorithms for Molecular Dynamics. PLoS Comput. Biol 2017, 13 (7), e1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Thompson AP; Aktulga HM; Berger R; Bolintineanu DS; Brown WM; Crozier PS; in ‘t Veld PJ; Kohlmeyer A; Moore SG; Nguyen TD; Shan R; Stevens MJ; Tranchida J; Trott C; Plimpton SJ LAMMPS - a Flexible Simulation Tool for Particle-Based Materials Modeling at the Atomic, Meso, and Continuum Scales. Comput. Phys. Commun 2022, 271, 108171. [Google Scholar]
- (39).Bowers KJ; Chow E; Xu H; Dror RO; Eastwood MP; Gregersen BA; Klepeis JL; Kolossvary I; Moraes MA; Sacerdoti FD; Salmon JK; Shan Y; Shaw DE Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the 2006 ACM/IEEE conference on Supercomputing; SC ‘06; Association for Computing Machinery: New York, NY, USA, 2006; p 84 – es. [Google Scholar]
- (40).Phillips JC; Hardy DJ; Maia JDC; Stone JE; Ribeiro JV; Bernardi RC; Buch R; Fiorin G; Hénin J; Jiang W; McGreevy R; Melo MCR; Radak BK; Skeel RD; Singharoy A; Wang Y; Roux B; Aksimentiev A; Luthey-Schulten Z; Kalé LV; Schulten K; Chipot C; Tajkhorshid E Scalable Molecular Dynamics on CPU and GPU Architectures with NAMD. J. Chem. Phys 2020, 153 (4), 044130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).McGibbon RT; Beauchamp KA; Harrigan MP; Klein C; Swails JM; Hernández CX; Schwantes CR; Wang L-P; Lane TJ; Pande VS MDTraj: A Modern, Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J 2015, 109 (8), 1528–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Michaud-Agrawal N; Denning EJ; Woolf TB; Beckstein O MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem 2011, 32 (10), 2319–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Romo TD; Leioatts N; Grossfield A Lightweight Object Oriented Structure Analysis: Tools for Building Tools to Analyze Molecular Dynamics Simulations. J. Comput. Chem 2014, 35 (32), 2305–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Porter JR; Zimmerman MI; Bowman GR Enspara: Modelin4g Molecular Ensembles with Scalable Data Structures and Parallel Computing. J. Chem. Phys 2019, 150 (4), 044108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Scherer MK; Trendelkamp-Schroer B; Paul F; Pérez-Hernández G; Hoffmann M; Plattner N; Wehmeyer C; Prinz J-H; Noé F PyEMMA 2: A Software Package for Estimation, Validation, and Analysis of Markov Models. J. Chem. Theory Comput 2015, 11 (11), 5525–5542. [DOI] [PubMed] [Google Scholar]
- (46).Beauchamp KA; Bowman GR; Lane TJ; Maibaum L; Haque IS; Pande VS MSMBuilder2: Modeling Conformational Dynamics at the Picosecond to Millisecond Scale. J. Chem. Theory Comput 2011, 7 (10), 3412–3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Levine ZA; Shea J-E Simulations of Disordered Proteins and Systems with Conformational Heterogeneity. Curr. Opin. Struct. Biol 2017, 43, 95–103. [DOI] [PubMed] [Google Scholar]
- (48).Pappu RV; Wang X; Vitalis A; Crick SL A Polymer Physics Perspective on Driving Forces and Mechanisms for Protein Aggregation - Highlight Issue: Protein Folding. Arch. Biochem. Biophys 2008, 469 (1), 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Hofmann H; Soranno A; Borgia A; Gast K; Nettels D; Schuler B Polymer Scaling Laws of Unfolded and Intrinsically Disordered Proteins Quantified with Single-Molecule Spectroscopy. Proc. Natl. Acad. Sci. U. S. A 2012, 109 (40), 16155–16160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Cubuk J; Soranno A Macromolecular Crowding and Intrinsically Disordered Proteins: A Polymer Physics Perspective. ChemSystemsChem 2022. 10.1002/syst.202100051. [DOI] [Google Scholar]
- (51).Sørensen CS; Kjaergaard M Effective Concentrations Enforced by Intrinsically Disordered Linkers Are Governed by Polymer Physics. Proc. Natl. Acad. Sci. U. S. A 2019, 116 (46), 23124–23131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Cormen TH; Leiserson CE; Rivest RL; Stein C Introduction to Algorithms, Fourth Edition; MIT Press, 2022. [Google Scholar]
- (53).Virtanen P; Gommers R; Oliphant TE; Haberland M; Reddy T; Cournapeau D; Burovski E; Peterson P; Weckesser W; Bright J; van der Walt SJ; Brett M; Wilson J; Millman KJ; Mayorov N; Nelson ARJ; Jones E; Kern R; Larson E; Carey CJ; Polat İ; Feng Y; Moore EW; VanderPlas J; Laxalde D; Perktold J; Cimrman R; Henriksen I; Quintero EA; Harris CR; Archibald AM; Ribeiro AH; Pedregosa F; van Mulbregt P; SciPy 1.0 Contributors. SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 2020, 17 (3), 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).van der Walt S; Colbert SC; Varoquaux G The NumPy Array: A Structure for Efficient Numerical Computation. Computing in Science Engineering 2011, 13 (2), 22–30. [Google Scholar]
- (55).McKinney W DAta STructures for STatistical COmputing in PYthon. In Proceedings of the 9th Python in Science Conference; SciPy, 2010. 10.25080/majora-92bf1922-00a. [DOI] [Google Scholar]
- (56).Behnel S; Bradshaw R; Citro C; Dalcin L; Seljebotn DS; Smith K Cython: The Best of Both Worlds. Computing in Science Engineering 2011, 13 (2), 31–39. [Google Scholar]
- (57).Martin EW; Holehouse AS; Peran I; Farag M; Incicco JJ; Bremer A; Grace CR; Soranno A; Pappu RV; Mittag T Valence and Patterning of Aromatic Residues Determine the Phase Behavior of Prion-like Domains. Science 2020, 367 (6478), 694–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Martin EW; Holehouse AS; Grace CR; Hughes A; Pappu RV; Mittag T Sequence Determinants of the Conformational Properties of an Intrinsically Disordered Protein Prior to and upon Multisite Phosphorylation. J. Am. Chem. Soc 2016, 138 (47), 15323–15335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Crick SL; Pappu RV Thermodynamic and Kinetic Models for Aggregation of Intrinsically Disordered Proteins. Protein and Peptide Folding, Misfolding, and Non-Folding 2012, 413–440. [Google Scholar]
- (60).Vitalis A; Pappu RV Chapter 3 Methods for Monte Carlo Simulations of Biomacromolecules. In Annual Reports in Computational Chemistry; Wheeler RA, Ed.; Elsevier, 2009; Vol. 5, pp 49–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Vitalis A; Wang X; Pappu RV Quantitative Characterization of Intrinsic Disorder in Polyglutamine: Insights from Analysis Based on Polymer Theories. Biophys. J 2007, 93 (6), 1923–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Holehouse AS; Garai K; Lyle N; Vitalis A; Pappu RV Quantitative Assessments of the Distinct Contributions of Polypeptide Backbone Amides versus Side Chain Groups to Chain Expansion via Chemical Denaturation. J. Am. Chem. Soc 2015, 137 (8), 2984–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Zerze GH; Best RB; Mittal J Sequence- and Temperature-Dependent Properties of Unfolded and Disordered Proteins from Atomistic Simulations. J. Phys. Chem. B 2015, 119 (46), 14622–14630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Steinhauser MO A Molecular Dynamics Study on Universal Properties of Polymer Chains in Different Solvent Qualities. Part I. A Review of Linear Chain Properties. J. Chem. Phys 2005, 122 (9), 094901. [DOI] [PubMed] [Google Scholar]
- (65).Vitalis A Probing the Early Stages of Polyglutamine Aggregation with Computational Methods, Washington University in St. Louis, 2009. http://pappulab.wustl.edu/blog/wp-content/uploads/2013/06/Vitalis_thesis.pdf. [Google Scholar]
- (66).Alston JJ; Ginell GM; Soranno A; Holehouse AS The Analytical Flory Random Coil Is a Simple-to-Use Reference Model for Unfolded and Disordered Proteins. J. Phys. Chem. B 2023, 127 (21), 4746–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Tesei G; Trolle AI; Jonsson N; Betz J; Pesce F; Johansson KE; Lindorff-Larsen K Conformational Ensembles of the Human Intrinsically Disordered Proteome: Bridging Chain Compaction with Function and Sequence Conservation. bioRxiv, 2023, 2023.05.08.539815. 10.1101/2023.05.08.539815. [DOI] [PubMed] [Google Scholar]
- (68).Peran I; Holehouse AS; Carrico IS; Pappu RV; Bilsel O; Raleigh DP Unfolded States under Folding Conditions Accommodate Sequence-Specific Conformational Preferences with Random Coil-like Dimensions. Proc. Natl. Acad. Sci. U. S. A 2019, 116 (25), 12301–12310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).González-Foutel NS; Glavina J; Borcherds WM; Safranchik M; Barrera-Vilarmau S; Sagar A; Estaña A; Barozet A; Garrone NA; Fernandez-Ballester G; Blanes-Mira C; Sánchez IE; de Prat-Gay G; Cortés J; Bernadó P; Pappu RV; Holehouse AS; Daughdrill GW; Chemes LB Conformational Buffering Underlies Functional Selection in Intrinsically Disordered Protein Regions. Nat. Struct. Mol. Biol 2022, 29 (8), 781–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Sherry KP; Das RK; Pappu RV; Barrick D Control of Transcriptional Activity by Design of Charge Patterning in the Intrinsically Disordered RAM Region of the Notch Receptor. Proc. Natl. Acad. Sci. U. S. A 2017, 114 (44), E9243–E9252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Holehouse AS; Sukenik S Controlling Structural Bias in Intrinsically Disordered Proteins Using Solution Space Scanning. J. Chem. Theory Comput 2020, 16 (3), 1794–1805. [DOI] [PubMed] [Google Scholar]
- (72).Das RK; Huang Y; Phillips AH; Kriwacki RW; Pappu RV Cryptic Sequence Features within the Disordered Protein p27Kip1 Regulate Cell Cycle Signaling. Proc. Natl. Acad. Sci. U. S. A 2016, 113 (20), 5616–5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Moses D; Yu F; Ginell GM; Shamoon NM; Koenig PS; Holehouse AS; Sukenik S Revealing the Hidden Sensitivity of Intrinsically Disordered Proteins to Their Chemical Environment. J. Phys. Chem. Lett 2020, 11 (23), 10131–10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Song J; Gomes G-N; Shi T; Gradinaru CC; Chan HS Conformational Heterogeneity and FRET Data Interpretation for Dimensions of Unfolded Proteins. Biophys. J 2017, 113 (5), 1012–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Fuertes G; Banterle N; Ruff KM; Chowdhury A; Mercadante D; Koehler C; Kachala M; Estrada Girona G; Milles S; Mishra A; Onck PR; Gräter F; Esteban-Martín S; Pappu RV; Svergun DI; Lemke EA Decoupling of Size and Shape Fluctuations in Heteropolymeric Sequences Reconciles Discrepancies in SAXS vs. FRET Measurements. Proc. Natl. Acad. Sci. U. S. A 2017, 114 (31), E6342–E6351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Ruff KM; Holehouse AS SAXS versus FRET: A Matter of Heterogeneity? Biophys. J 2017, 113 (5), 971–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Das RK; Pappu RV Conformations of Intrinsically Disordered Proteins Are Influenced by Linear Sequence Distributions of Oppositely Charged Residues. Proc. Natl. Acad. Sci. U. S. A 2013, 110 (33), 13392–13397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Gomes G-NW; Krzeminski M; Namini A; Martin EW; Mittag T; Head-Gordon T; Forman-Kay JD; Gradinaru CC Conformational Ensembles of an Intrinsically Disordered Protein Consistent with NMR, SAXS, and Single-Molecule FRET. J. Am. Chem. Soc 2020, 142 (37), 15697–15710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Marsh JA; Forman-Kay JD Sequence Determinants of Compaction in Intrinsically Disordered Proteins. Biophys. J 2010, 98 (10), 2383–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Mao AH; Crick SL; Vitalis A; Chicoine CL; Pappu RV Net Charge per Residue Modulates Conformational Ensembles of Intrinsically Disordered Proteins. Proc. Natl. Acad. Sci. U. S. A 2010, 107 (18), 8183–8188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Müller-Späth S; Soranno A; Hirschfeld V; Hofmann H; Rüegger S; Reymond L; Nettels D; Schuler B Charge Interactions Can Dominate the Dimensions of Intrinsically Disordered Proteins. Proc. Natl. Acad. Sci. U. S. A 2010, 107 (33), 14609–14614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Basu S; Martínez-Cristóbal P; Pesarrodona M; Frigolé-Vivas M; Lewis M; Szulc E; Bañuelos CA; Sánchez-Zarzalejo C; Bielskutė S; Zhu J; Pombo-García K; Garcia-Cabau C; Batlle C; Mateos B; Biesaga M; Escobedo A; Bardia L; Verdaguer X; Ruffoni A; Mawji NR; Wang J; Tam T; Brun-Heath I; Ventura S; Meierhofer D; García J; Robustelli P; Stracker TH; Sadar MD; Riera A; Hnisz D; Salvatella X Rational Optimization of a Transcription Factor Activation Domain Inhibitor. bioRxiv, 2022. 10.1101/2022.08.18.504385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Gibbs EB; Lu F; Portz B; Fisher MJ; Medellin BP; Laremore TN; Zhang YJ; Gilmour DS; Showalter SA Phosphorylation Induces Sequence-Specific Conformational Switches in the RNA Polymerase II C-Terminal Domain. Nat. Commun 2017, 8 (1), 15233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Portz B; Lu F; Gibbs EB; Mayfield JE; Rachel Mehaffey M; Zhang YJ; Brodbelt JS; Showalter SA; Gilmour DS Structural Heterogeneity in the Intrinsically Disordered RNA Polymerase II C-Terminal Domain. Nat. Commun 2017, 8, 15231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Wicky BIM; Shammas SL; Clarke J Affinity of IDPs to Their Targets Is Modulated by Ion-Specific Changes in Kinetics and Residual Structure. Proc. Natl. Acad. Sci. U. S. A 2017, 114 (37), 9882–9887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Moses D; Guadalupe K; Yu F; Flores E; Perez A; McAnelly R; Shamoon NM; Cuevas-Zepeda E; Merg AD; Martin EW; Holehouse AS; Sukenik S Structural Biases in Disordered Proteins Are Prevalent in the Cell. bioRxiv, 2022, 2021.11.24.469609. 10.1101/2021.11.24.469609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Naudi-Fabra S; Tengo M; Jensen MR; Blackledge M; Milles S Quantitative Description of Intrinsically Disordered Proteins Using Single-Molecule FRET, NMR, and SAXS. J. Am. Chem. Soc 2021, 143 (48), 20109–20121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Guseva S; Milles S; Jensen MR; Salvi N; Kleman J-P; Maurin D; Ruigrok RWH; Blackledge M Measles Virus Nucleo- and Phosphoproteins Form Liquid-like Phase-Separated Compartments That Promote Nucleocapsid Assembly. Sci Adv 2020, 6 (14), eaaz7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Shrake A; Rupley JA Environment and Exposure to Solvent of Protein Atoms. Lysozyme and Insulin. J. Mol. Biol 1973, 79 (2), 351–371. [DOI] [PubMed] [Google Scholar]
- (90).Prestel A; Wichmann N; Martins JM; Marabini R; Kassem N; Broendum SS; Otterlei M; Nielsen O; Willemoës M; Ploug M; Boomsma W; Kragelund BB The PCNA Interaction Motifs Revisited: Thinking Outside the PIP-Box. Cell. Mol. Life Sci 2019. 10.1007/s00018-019-03150-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Bugge K; Brakti I; Fernandes CB; Dreier JE; Lundsgaard JE; Olsen JG; Skriver K; Kragelund BB Interactions by Disorder - A Matter of Context. Front Mol Biosci 2020, 7, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Cubuk J; Alston JJ; Incicco JJ; Singh S; Stuchell-Brereton MD; Ward MD; Zimmerman MI; Vithani N; Griffith D; Wagoner JA; Bowman GR; Hall KB; Soranno A; Holehouse AS The SARS-CoV-2 Nucleocapsid Protein Is Dynamic, Disordered, and Phase Separates with RNA. Nat. Commun 2021, 12 (1), 1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Sankaranarayanan M; Emenecker RJ; Wilby EL; Jahnel M; Trussina IREA; Wayland M; Alberti S; Holehouse AS; Weil TT Adaptable P Body Physical States Differentially Regulate Bicoid mRNA Storage during Early Drosophila Development. Dev. Cell 2021, 56 (20), 2886–2901.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Moses D; Guadalupe K; Yu F; Flores E; Perez A; McAnelly R; Shamoon NM; Cuevas-Zepeda E; Merg A; Martin EW; Holehouse AS; Sukenik S Hidden Structure in Disordered Proteins Is Adaptive to Intracellular Changes. bioRxiv, 2021, 2021.11.24.469609. 10.1101/2021.11.24.469609. [DOI] [Google Scholar]
- (95).Husic BE; Charron NE; Lemm D; Wang J; Pérez A; Majewski M; Krämer A; Chen Y; Olsson S; de Fabritiis G; Noé F; Clementi C Coarse Graining Molecular Dynamics with Graph Neural Networks. arXiv [physics.comp-ph], 2020. http://arxiv.org/abs/2007.11412. [DOI] [PMC free article] [PubMed]
- (96).Parton DL; Grinaway PB; Hanson SM; Beauchamp KA; Chodera JD Ensembler: Enabling High-Throughput Molecular Simulations at the Superfamily Scale. PLoS Comput. Biol 2016, 12 (6), e1004728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Singh S; Bowman GR Quantifying Allosteric Communication via Both Concerted Structural Changes and Conformational Disorder with CARDS. J. Chem. Theory Comput 2017, 13 (4), 1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Tubiana T; Carvaillo J-C; Boulard Y; Bressanelli S TTClust: A Versatile Molecular Simulation Trajectory Clustering Program with Graphical Summaries. J. Chem. Inf. Model 2018, 58 (11), 2178–2182. [DOI] [PubMed] [Google Scholar]
- (99).Lotz SD; Dickson A Wepy: A Flexible Software Framework for Simulating Rare Events with Weighted Ensemble Resampling. ACS Omega 2020, 5 (49), 31608–31623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Richard RM; Bertoni C; Boschen JS; Keipert K; Pritchard B; Valeev EF; Harrison RJ; de Jong WA; Windus TL Developing a Computational Chemistry Framework for the Exascale Era. Computing in Science Engineering 2019, 21 (2), 48–58. [Google Scholar]
- (101).Meng W; Lyle N; Luan B; Raleigh DP; Pappu RV Experiments and Simulations Show How Long-Range Contacts Can Form in Expanded Unfolded Proteins with Negligible Secondary Structure. Proc. Natl. Acad. Sci. U. S. A 2013, 110 (6), 2123–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Jing H; Yang X; Emenecker RJ; Feng J; Zhang J; Figueiredo M. R. A de; Chaisupa P; Wright RC; Holehouse AS; Strader LC; Zuo J Nitric Oxide-Mediated S-Nitrosylation of IAA17 Protein in Intrinsically Disordered Region Represses Auxin Signaling. J. Genet. Genomics 2023. 10.1016/j.jgg.2023.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Lotthammer JM; Ginell GM; Griffith D; Emenecker RJ; Holehouse AS Direct Prediction of Intrinsically Disordered Protein Conformational Properties from Sequence. bioRxiv, 2023, 2023.05.08.539824. 10.1101/2023.05.08.539824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Cubuk J; Alston JJ; Jeremías Incicco J; Holehouse AS; Hall KB; Stuchell-Brereton MD; Soranno A The Disordered N-Terminal Tail of SARS CoV-2 Nucleocapsid Protein Forms a Dynamic Complex with RNA. bioRxiv, 2023, 2023.02.10.527914. 10.1101/2023.02.10.527914. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.