

Abstract
BACKGROUND:
Vascular Ehlers-Danlos syndrome (vEDS) is a rare connective tissue disorder with a high risk for arterial, bowel, and uterine rupture, caused by heterozygous pathogenic variants in COL3A1. The aim of this cohort study is to provide further insights into the natural history of vEDS and describe genotype-phenotype correlations in a Dutch multicenter cohort to optimize patient care and increase awareness of the disease.
METHODS:
Individuals with vEDS throughout the Netherlands were included. The phenotype was charted by retrospective analysis of molecular and clinical data, combined with a one-time physical examination.
RESULTS:
A total of 142 individuals (50% female) participated the study, including 46 index patients (32%). The overall median age at genetic diagnosis was 41.0 years. More than half of the index patients (54.3%) and relatives (53.1%) had a physical appearance highly suggestive of vEDS. In these individuals, major events were not more frequent (P=0.90), but occurred at a younger age (P=0.01). A major event occurred more often and at a younger age in men compared with women (P<0.001 and P=0.004, respectively). Aortic aneurysms (P=0.003) and pneumothoraces (P=0.029) were more frequent in men. Aortic dissection was more frequent in individuals with a COL3A1 variant in the first quarter of the collagen helical domain (P=0.03).
CONCLUSIONS:
Male sex, type and location of the COL3A1 variant, and physical appearance highly suggestive of vEDS are risk factors for the occurrence and early age of onset of major events. This national multicenter cohort study of Dutch individuals with vEDS provides a valuable basis for improving guidelines for the diagnosing, follow-up, and treatment of individuals with vEDS.
Keywords: aortic aneurysm; connective tissue; collagen; Ehlers-Danlos syndrome, type IV; uterine rupture
Heterozygous pathogenic variants in the COL3A1 gene (OMIM 120180), encoding for type III collagen, cause the heritable connective tissue disorder vascular Ehlers-Danlos syndrome (vEDS; OMIM 1300050, ORPHA 286)1 with an estimated prevalence range from 1:50 000 to 1:200 000 in the United States.2 Spontaneous arterial dissections, bowel rupture, and uterine rupture are the most characteristic and life-threatening complications of vEDS.3,4 At age 40 to 43 years, a first vascular, gastrointestinal, or obstetric complication has occurred in 80% to 85% of the individuals.1,4 Initially, the median survival for individuals with vEDS was 48 to 51 years, with a higher number of deaths due to arterial dissection or rupture in adolescent males.4,5 Patients with a phenotype classically associated with the disease are reported to have characteristic appearance/features including prominent (sunken) eyes, narrow nose, gingival recession/fragility, lobeless ears, acrogeria, easy bruising, translucent skin, and early-onset varicose veins.6 Revised suggestive major and minor criteria for vEDS have been published in 2017.7 These revised criteria were created to indicate when genetic testing is recommended. The presence of one of the following major criteria was proposed as an indication for genetic testing: (1) positive family history of vEDS, (2) arterial rupture or dissection under 40 years of age, (3) spontaneous sigmoid colon perforation, (4) unexplained uterine rupture in the third trimester of pregnancy, or (5) spontaneous carotid-cavernous sinus fistula formation. Genetic testing should also be considered in the presence of a combination of the following minor criteria: characteristic appearance as mentioned above, spontaneous pneumothorax, talipes equinovarus, congenital hip dislocation, hypermobility, keratoconus, and tendon and muscle rupture.7
COL3A1 is located on chromosome 2q32.2 and consists of 51 coding exons that encode a 1466 amino acid protein.8 More than half of the known pathogenic variants in COL3A1 are missense substitutions of a glycine in the repeating (Gly-X-Y) sequence within the triple helical region of type III collagen. Together with splice variants resulting in exon skipping and in-frame insertions, deletions, and duplications, these variants with a dominant negative effect comprise the majority of the pathogenic variants in COL3A1.1,4 Variants leading to haploinsufficiency are more rare and may result in a milder phenotype with reduced penetrance compared with dominant negative variants.1,5,9 Bi-allelic COL3A1 variants have been found in <1% of all vEDS individuals.10,11
Rarely specific heterozygous arginine-to-cysteine substitutions in COL1A1 cause a vEDS-like phenotype.12
Surveillance in vEDS is very difficult due to the unpredictability of the occurrence of arterial dissections, often without a prior aneurysm,2,4 and the potential complexity of surgical interventions.13 Although recommendations for patient management have been proposed,3,14,15 these are often based on observations in case reports or small cohorts due to the rarity of the disease. It is therefore essential to gain further insight into the natural history of the disorder by detailed studies in larger patient cohorts. To this end, we describe a detailed overview of the Dutch cohort of 142 patients with vEDS.
METHODS
The data that support the findings of this study are available from the corresponding author upon reasonable request. The full methods of this retrospective study are available in Supplemental Data, including Tables S1 and S2. This study was approved by the Medical and Ethics Review Committee of the Amsterdam University Medical Centre (MEC 2019-0662) and all participating centers. The alive participating individuals provided written informed consent before inclusion in the study.
RESULTS
Clinical Characteristics of the Study Population
In total, 142 individuals (50% female) from 70 families were included (Table 1). Forty-six of the participants (32.4%) were index patients. Fifteen individuals were deceased, including 10 index patients. The median age at death was 43.0 years (interquartile range [IQR], 23.75–56.50) for index patients, compared with 65.0 years (IQR, 51.5–72.5) for relatives. Six index patients and 2 relatives died due to vascular complications of vEDS. The cause of death was unknown for 3 individuals, including 2 index patients (40 and 61 years) and 1 relative (61 years). All 3 individuals with an unknown cause of death suffered from a major event earlier in life. Six individuals were deceased before vEDS was diagnosed. The median age at genetic diagnosis was 41.0 years (IQR, 28.0–59.5) and 40.0 years (IQR, 29.75–54.5) for index patients. The median time in follow-up at the time of inclusion in the study was 3.0 years (IQR, 1.0–6.0). The main reason for referral for index patients was an aneurysm or dissection (65.2%), and 7 index patients (14.9%) had a facial appearance or other features consistent with vEDS. A total of 34/96 (35.4%) of the relatives with a pathogenic/likely pathogenic (P/LP) variant in COL3A1 had a major event.
Table 1.
Clinical Characteristics

2017 Suggestive Criteria
Almost two-thirds (31/46) of the index patients met the 2017 suggestive criteria for vEDS; 17 of them had at least 1 major criterion, while 14 had a combination of minor criteria. Arterial rupture or dissection was the most common major suggestive criterion in index patients (76.5%). At least 1 major suggestive criterion was present in 75.4% of all individuals, including 37% of the index patients. Another major suggestive criterion besides the “positive family history for vEDS” criterion was present in 9 of 90 relatives (10%), including an arterial rupture or dissection <40 years in 4, a colon rupture in 4, and spontaneous carotid-cavernous sinus fistula formation in 1. Six index patients did not meet any of the major or minor suggestive criteria, including 3 with an isolated aortic aneurysm, 2 diagnosed due to an incidental finding while performing DNA analysis for another condition (age at inclusion, 15 and 61 years), and 1 with an isolated iliac artery dissection at 41 years of age. The individuals with isolated aortic aneurysms were a 63-year-old male with a 45-mm ascending aortic aneurysm (height 184 cm, weight 84 kg), a 56-year-old female who underwent a personalized external aortic root support procedure for a 53-mm aortic root aneurysm (Z-score 8.44), and a 69-year-old male who underwent an endovascular aortic repair procedure because of a 64-mm abdominal aortic aneurysm (height 171 cm, weight 77.8 kg). In total, 101 of 124 examined individuals (81.5%) had at least 1 characteristic facial feature, including 76 individuals with >4 facial features.
Hypertension
One of the risk factors for arterial aneurysms and dissections is hypertension. Data on blood pressure were known for 94 individuals (66.2%), and 27 of 94 (28.7%) had hypertension.
Antihypertensive drugs were prescribed in 74 of 142 (52.1%) individuals, including the 27 individuals with hypertension and 47 without hypertension who received antihypertensive drugs because of the diagnosis of vEDS. Twenty-two individuals used >1 antihypertensive drug. A beta blocker was prescribed in 61 of 74 individuals (82.4%), including celiprolol in 37 of 74 individuals (50.0%) and other beta blockers in 24 of 74 individuals (32.4%). Thirteen individuals (17.6%) received other antihypertensive drugs: calcium channel blockers in 7, angiotensin-converting enzyme (ACE) inhibitors in 3, angiotensin receptor blockers (ARB) in 2, and an ACE inhibitor plus a calcium channel blocker in 1.
Children
In total, 8 children (age <18; 6%) were included, of whom 3 were index patients (age range, 12–16 years). The median age at genetic diagnosis and inclusion was 4.5 years (IQR, 0.25–9.00) and 8.0 years (IQR, 3.50–12.75), respectively. Five children underwent genetic testing because 1 of their parents was diagnosed with vEDS, 2 because of facial appearance or features consistent with vEDS, and 1 underwent a genetic test for intellectual disability (a de novo deletion of the complete COL3A1 gene). None of these children were diagnosed with a major event (age range at inclusion, 1–16 years). In the total cohort, 2 individuals had a major event under the age of 18 years. Most children (5/8) received yearly follow-up by a pediatric cardiologist using different imaging modalities.
Molecular Studies
A total of 56 different P/LP COL3A1 variants were detected, including 29 glycine substitutions, 10 splice variants, 8 frameshift variants, 2 deletion-insertion (delins) variants, 2 nonsense variants, 3 in-frame deletions, 1 glutamate substitution, and 1 copy number variation of 14.5 Mb encompassing the entire COL3A1 gene, among others (Figure 1; Table S3). In total, 43 of 56 (76.8%) of the detected variants in COL3A1 are predicted to have a dominant negative effect. Forty-three variants were novel. More than half of the individuals were heterozygous for a glycine substitution, including 63% of the index patients. Nine variants (27%) were found in >1 family, and 9 variants (27%) were proven de novo. The origin of 23 variants was unknown as parental testing could not be performed. Segregation information was available for 39 families, and in total, 94 of 243 (38.7%) first-degree relatives were screened for the familial COL3A1 variant, revealing 54 heterozygotes, including 23 with a major event in their medical history.
Figure 1.

Schematic overview of the COL3A1 gene, with the location of pathogenic and likely pathogenic variants found in our cohort. The boxes represent the coding exons and the colors represent protein domains. Blue boxes represent N and C terminal domains, gray represents the transitional domain, and yellow represents the collagen helical domain. Glycine substitutions are displayed on the left. Each * is an individual with an aortic dissection. Variants in bold are proven de novo variants.
First Major Event
In total, 74 individuals (52%) experienced a major event, including 44 individuals with a glycine substitution, 11 with a splice/delins/del variant, and 19 with a predicted haploinsufficient variant. The first major event was lethal in 5 individuals with an aortic dissection, including 4 index patients (2 type A, 1 type B dissection, and 1 abdominal aortic dissection) and 1 relative (unknown origin of dissection). In all, 5 vEDS were diagnosed postmortem. The overall median age at the first major event was 41.0 years (IQR, 31.0–55.0). Significantly more men compared with women suffered from a major event (48 versus 26; odds ratio [OR], 3.61 [95% CI, 1.82–7.14]; P<0.001) and the Kaplan-Meier analysis showed a significantly younger age at the time of the first major event in men compared with women (P=0.004; Figure 2A).
Figure 2.

Kaplan-Meier curves for occurrence of major events. A, Sex and age at occurrence of the first major event. Age at the first major complication correlated with sex in 142 individuals with vEDS. B, Facial features highly suggestive of vEDS and age at occurrence of the first major event. Age at the first major complication correlated with the presence of ≥5 suggestive features of vEDS upon physical examination in 142 individuals with vEDS. C, Age at occurrence of the first major event in the total cohort. Age at the first major complication correlated with the type of P/LP COL3A1 variant in 142 individuals with vEDS. D, Age at occurrence of the first major event in relatives. Age at the first major complication correlated with the type of P/LP COL3A1 variant in 96 relatives with vEDS. P/LP indicates pathogenic/likely pathogenic; splice/delins/del, splice-site/deletion insertion/deletion variant; and vEDS, vascular Ehlers-Danlos syndrome.
The youngest individual with a major event was a male who suffered from a myocardial infarction at the age of 14 years due to an aneurysm combined with narrowing up to 70% of the left anterior descending artery. He was 24 years old at the time of inclusion and heterozygous for a pathogenic splice variant in COL3A1 with a predicted dominant negative effect. A major event was present in 17 of 27 individuals (63.0%) with hypertension compared with 27 of 63 (42.9%) without hypertension (OR, 2.27 [95% CI, 0.98–2.21]; P=0.08).
Major Event and Appearance Highly Suggestive of vEDS
An appearance highly suggestive of vEDS was defined as the presence of 5 or more suggestive features of vEDS upon physical examination and was seen in 76 individuals (53.5%), equally divided in index patients (54.3%) and relatives (53.1%; Table 1). Individuals with an appearance highly suggestive of vEDS did not suffer from a major event more frequently than those without (OR, 1.05 [95% CI, 0.54–2.02]; P=0.89). However, the median age at the first major event was significantly lower in individuals with an appearance highly suggestive of vEDS compared with those without: 39.5 years (IQR, 29.0–45.75) versus 48.5 years (IQR, 37.5–60.0), respectively (P=0.01). There was no confounding relationship between an appearance highly suggestive of vEDS and type of variant in COL3A1 (P=0.14). Also, the Kaplan-Meier analysis showed a significantly younger age at the first event in individuals with an appearance highly suggestive of vEDS compared with those without (P=0.045; Figure 2B).
Vascular Complications
Sixty-eight individuals were diagnosed with an arterial aneurysm or dissection at a median age of 44.0 years (IQR, 37–55), including 39 index patients (57.4%; Table 2). Twenty-eight individuals (41.2%) had >1 aneurysm or dissection; the majority (22/28) were index patients. Aortic aneurysms were found in 32 of 68 individuals, including 8 individuals with aneurysms/dissections elsewhere and 13 individuals with an ascending aortic aneurysm (Table S4). Aortic aneurysms were significantly more frequent in men than in women (24 versus 8; OR, 3.73 [95% CI, 1.52–9.17]; P=0.003). The youngest individual with an aortic aneurysm was 19 years old. No significant difference in the frequency of aortic dissections was seen between men (n=15) and women (n=8; OR, 1.94 [95% CI, 0.76–5.00]; P=0.16). The youngest age at which an aortic dissection occurred was 23 years. More than half of the aneurysms in middle-sized arteries (MSAs) were observed in the common and external iliac arteries (61%). Dissections occurred most frequently in the carotid arteries and the common and external iliac arteries (both 37%). The youngest age at which a dissection of a MSA occurred was 16 years. Five individuals (3 male and 2 female) had a spontaneous coronary artery dissection at a mean age of 39.0 years (±3.5). It was the first major event in 3 of them. Three of these individuals had vascular complications elsewhere as well. One individual with a spontaneous coronary artery dissection was an index patient without other phenotypic characteristics highly suggestive of vEDS, as reported by Bos et al.16
Table 2.
Major Events in Individuals With Vascular Ehlers-Danlos Syndrome
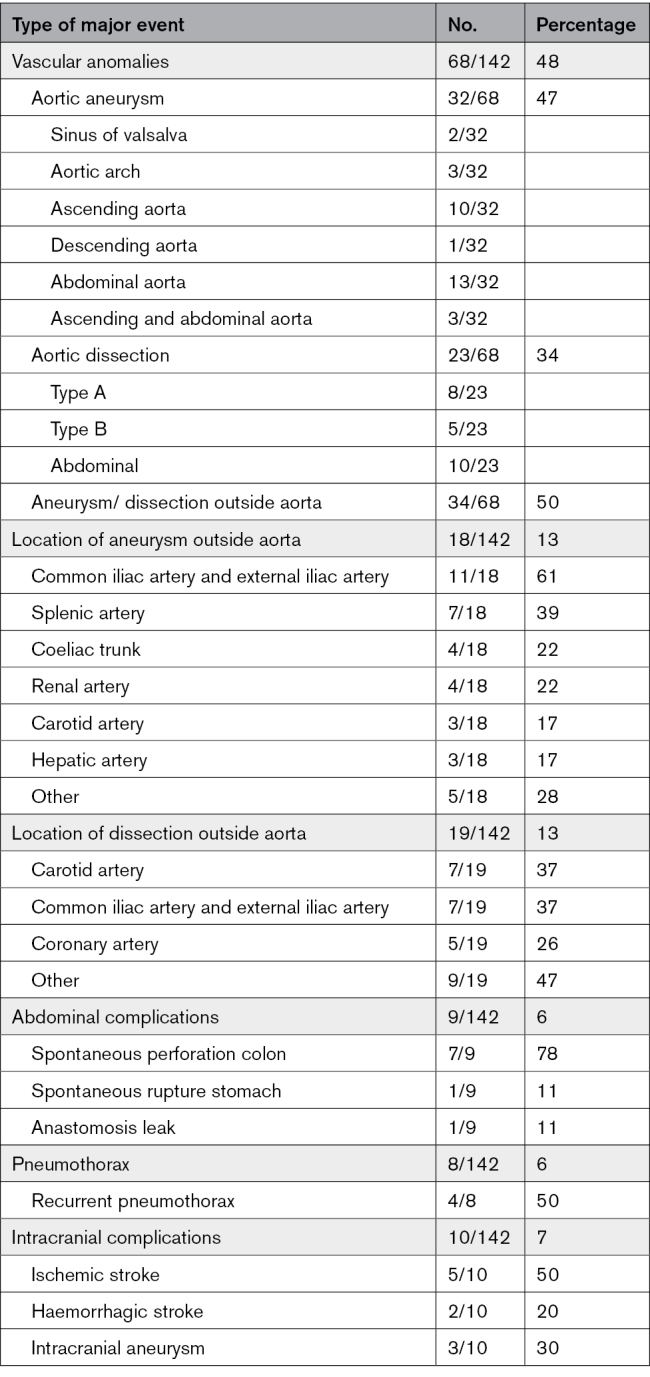
Forty-five surgical interventions were performed for vascular complications in 33 individuals (48.5%; Table S5). Severe complications were noted in 4 individuals, including a sternum infection in 1, a burst abdomen with enterocuteneous fistulas in 1, a type 3 endoleak resulting in an abdominal aortic aneurysm rupture leading to a serosa defect with ileocecal resection in 1, and an arteria hepatica rupture after coiling in 1.
Abdominal and Urogenital Complications
Major abdominal complications occurred in 9 individuals (7 male and 2 female), including spontaneous colon perforation in 7 (median age 32.0 years [IQR, 30.0–44.0]), 1 spontaneous stomach rupture (age unknown; male), and 1 anastomosis leak after colectomy (age 74 years; male; Table 2). The 2 women with spontaneous colon perforation were 31 years old (splice variant) and 27 years old (glycine substitution). Both had highly suggestive features for vEDS upon physical examination. Five of the spontaneous colon perforations (71.4%) were located in the sigmoid or rectum. Because of the spontaneous colon perforations, hemicolectomy or partial colectomy was performed in 5. Complications due to the surgery occurred in 2, namely an anastomosis leak plus an enterocutaneous fistula in 1 and a rupture of the spleen in the other individual.
In total, 129 pregnancies were noted, and no uterine ruptures were observed in our cohort.
Spontaneous Pneumothorax
Eight individuals suffered from spontaneous pneumothorax, including recurrent spontaneous pneumothorax in half of them (Table 2). Pneumothorax occurred significantly more frequently in men compared with women, namely 7 versus 1 (OR, 7.69 [95% CI, 0.92–62.5]; P=0.029). The median age at first spontaneous pneumothorax was 21.5 years (IQR, 17.25–29.0), and the youngest was 16 years. The diagnosis of vEDS was highly suspected in 2 individuals due to the presence of suggestive features of vEDS upon physical examination and vascular abnormalities in the MSAs.
Spontaneous pneumothorax was the first presentation of vEDS in 4, whereas vascular complications occurred later between the ages of 23 and 50 years. Two individuals (age 42 and 62 years) had recurrent pneumothorax as adolescents without other phenotypic characteristics for vEDS.
Cerebrovascular Complications
In total, 7 individuals (4 male and 3 female) endured a spontaneous stroke at a median age of 49.0 years (IQR, 45.0–58.0; 5 ischemic and 2 hemorrhagic strokes; Table 2). The youngest individual was a 22-year-old male with a spontaneous subdural hematoma. Ischemic stroke occurred once due to bilateral internal carotid artery dissection and once due to a vertebral artery dissection and of unknown etiology in the other 3.
Presymptomatic imaging of the cerebral arteries was performed in 58 of 142 individuals with vEDS, and 3 (5.2%) were diagnosed with an intracranial aneurysm. Carotid-cavernous fistula was diagnosed in 2 individuals (3.4%). Furthermore, 4 ischemic strokes occurred as complications during/after vascular surgery.
Genotype-Phenotype Correlation
Glycine Substitutions
The median age at the first major event for individuals with a glycine substitution and individuals with a splice/delins/del variant was significantly lower compared with individuals with a suspected haploinsufficient variant, namely, 40.5 years (IQR, 31.0–53.25) and 30.0 years (IQR, 21.0–45.0) versus 53.0 years (IQR, 43.0–58.0; P=0.027 and P=0.002).
The Kaplan-Meier analysis showed a significantly lower age at the first major event in individuals with a glycine substitution in COL3A1 compared with individuals with a suspected haploinsufficient variant (P=0.044; Figure 2C). Individuals with a splice/delins/del variant in COL3A1 were slightly younger at the time of their first major event compared with individuals with a suspected haploinsufficient variant (P=0.06; Figure 2C). Surprisingly, when taking only relatives into account, no difference in age at the first major event was seen between the different variant categories (P=0.62; Figure 2D).
Aortic dissections seem to be more present in individuals with a glycine substitution (23.0%) compared with suspected haploinsufficient variants (10.3%) or splice/delins/del variants (6.9%; P=0.09; Table 3). There was no difference in the frequency of aortic aneurysms, aneurysms, or dissections outside the aorta between the variant categories (P=0.60 and P=0.370, respectively). However, 4 of 5 individuals with spontaneous coronary artery dissection were heterozygous for a glycine substitution, including 2 individuals from the same family. About half of the individuals with a colon perforation (55.6%) had a glycine substitution in COL3A1. Only 1 individual had a variant predicted to result in haploinsufficiency, namely a 43-year-old male with a spontaneous sigmoid perforation. More than half of the individuals with pneumothorax (62.5%) or cerebral complications (57.1%) were heterozygous for a glycine substitution.
Table 3.
Vascular Complications and Genotype
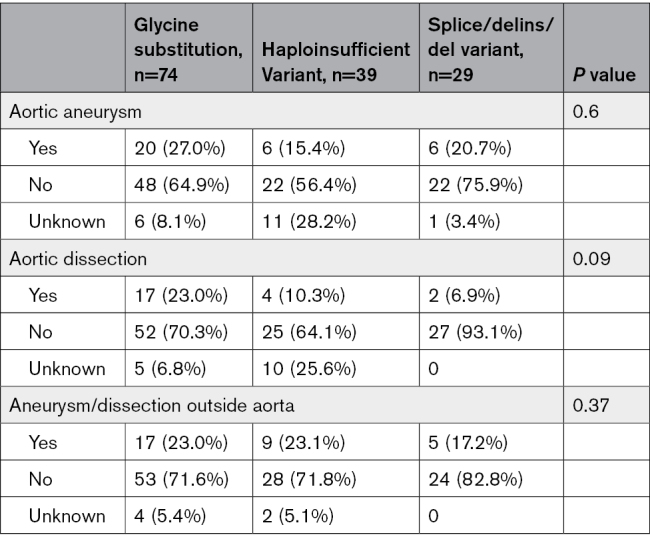
Localization
Interestingly, we found that individuals with an aortic dissection had significantly more often a P/LP variant located in the first quarter of the collagen helical domain of COL3A1 than in the other parts of the collagen helical domain, namely 13 of 20 (65.0%) versus 7 of 20 (35.0%; P=0.03) for unrelated individuals and 16 of 23 (69.6%) versus 7 of 23 (30.4%; P=0.01) when taking all individuals with an aortic dissection into account (Figure 1). There was no correlation between bowel complications, pneumothorax, or strokes and the type of location of the variant in the collagen helical domain in COL3A1.
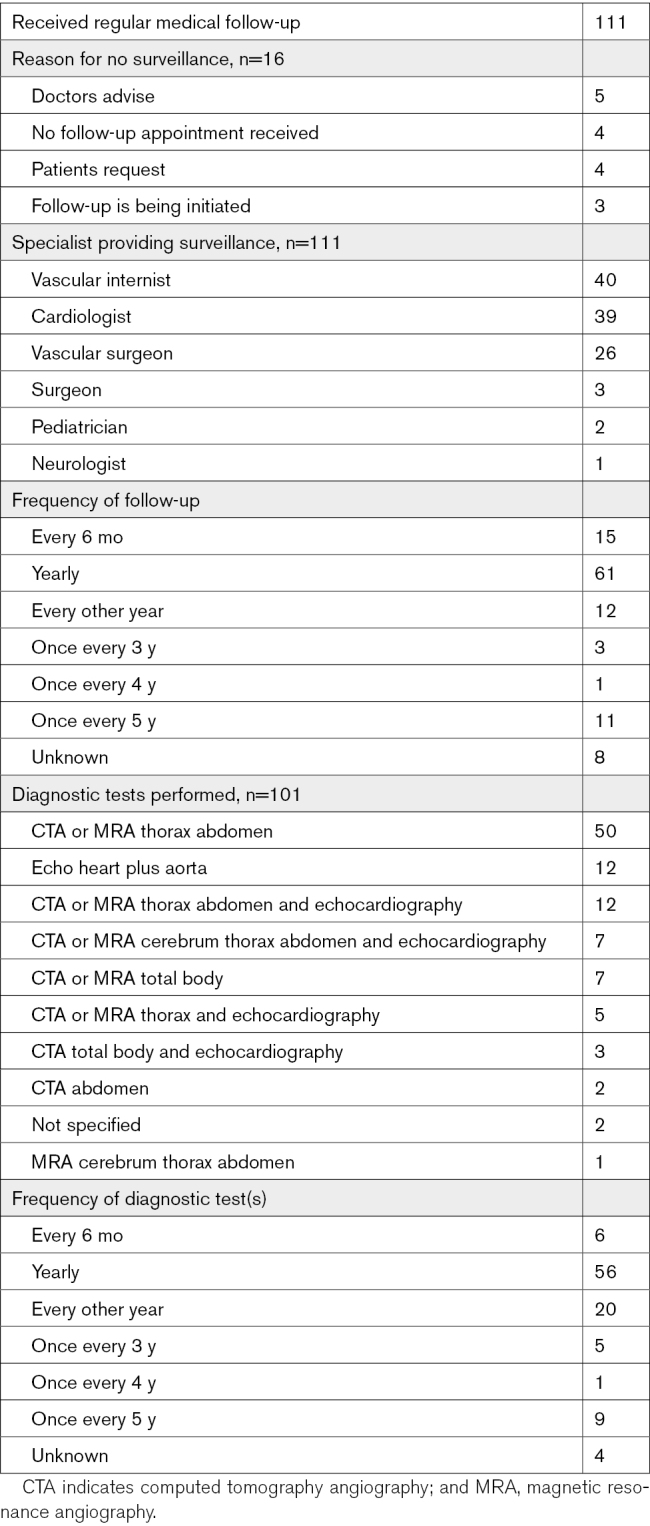
Surveillance
At the time of inclusion, 111 of 127 living individuals (87.4%) received periodic medical follow*up, including annual medical follow-up in more than half (55%). Medical follow-up was mainly provided by the (vascular) internist, cardiologist, or vascular surgeon (Table 4). The majority (73.9%) had arterial imaging at least every other year. A high degree of variability was seen in the imaging modalities used, including magnetic resonance angiography, computed tomography angiography, and echocardiography (Table 4). Sixteen individuals did not receive follow-up for several reasons (Table 4).
Table 4.
Surveillance of Individuals With Vascular Ehlers-Danlos Syndrome
DISCUSSION
In this Dutch national multicenter natural history study, we provide a detailed overview of the phenotype and genotype of 142 individuals from 70 families, harboring 56 different P/LP variants in COL3A1. Genetic examination revealed a glycine substitution in the COL3A1 gene in almost two-thirds (63%) of the index patients, which corresponds with the current literature1,5 and the Ehlers-Danlos Syndrome Variant Database (https://databases.lovd.nl/shared/variants/COL3A1). In index patients, the diagnosis of vEDS is most of the time made after a complication of vEDS. In this study, we describe the natural history of vEDS until diagnosis and the clinical evolution of the disease afterwards.
More than half of the individuals suffered from one or more major events. Male sex (P<0.001) and glycine substitutions (P=0.045) seem to give a higher risk for major events. Moreover, individuals with a glycine substitution were significantly younger at the time of the first major event compared with those with a suspected haploinsufficient variant (P=0.044), which is in line with several previous reports.1,5,9,17 Interestingly, when solely looking at the relatives in our cohort, this difference in age at the first major event between the different variant categories is not observed (P=0.62). We speculate that this might be due to the fact that relatives start with arterial screening and management of their blood pressure at a younger age or due to the presence of unknown genetic modifiers. In addition, vEDS individuals with hypertension seem to be younger at the time of the first major event compared with vEDS individuals without hypertension (P=0.08); therefore, adequate management of blood pressure is highly recommended.
Furthermore, major events were not more frequent in individuals with an appearance highly suggestive of vEDS (P=0.90); however, they did have a significantly lower median age at the time of their first major event compared with those without an appearance highly suggestive of vEDS (P=0.01). An appearance highly suggestive of vEDS was defined as the presence of more than half of the 9 mentioned features, so 5 or more. There were no differences in the predictive ability of individual features. Additionally, how obvious these features were varied between individuals, which can be challenging for the caregiver. The correlation between the incidence of major events and an appearance highly suggestive of vEDS has not been described before in vEDS. A correlation between the severity of craniofacial features and their cardiovascular outcome has also been described in individuals diagnosed with Loeys-Dietz syndrome.18,19 Caregivers should be aware of this increased risk in these individuals.
We can only speculate on the factors involved in this correlation, since no proposed mechanism has been reported to date. Various features of the appearance of individuals with vEDS are likely to be explained by the expression of COL3A1 in the following tissues: (1) smooth muscle cells (varicose veins), (2) skin (easy bruising, translucent skin, gingival regression/fragility, lobeless ears, and acrogeria) and (3) bone marrow plus connective tissue (prominent [sunken] eyes and narrow nose; https://www.proteinatlas.org/ENSG00000168542-COL3A1/tissue). The presence of the abovementioned features might give an indication of the impact of the COL3A1 variant on an individual. When more features are present, the COL3A1 variant might have a greater impact on that individual, including a more severe effect on the hollow organs and blood vessels. Additionally, the great phenotypic variability even within families might implicate a role for cofounders. These cofounders likely differ per individual and might result in a different impact of the COL3A1 variant. Furthermore, the type of variant does not explain this correlation, since an appearance highly suggestive of vEDS was equally present in individuals with a dominant negative variant (55/103, 53.4%) and a haploinsufficient variant (21/39, 53.8%).
None of the 8 children in our cohort have had a major event, and only 2 individuals in the total cohort suffered from a major event as a child. Major events have repeatedly been described in childhood.1,5,20 The low number of children in our cohort causes a bias in the described age at the first major event and the prevalence of major events in children. An explanation for the low number of children is the restraint in presymptomatic genetic testing in children in the Netherlands. Genetic testing for vEDS is offered to children with an affected first-degree relative, but the majority of parents refrain from genetic testing in childhood. Dutch parents and clinicians generally attach great importance to the child’s autonomy and therefore prefer presymptomatic genetic testing at an age when their child can make his or her own decision.
Aortic dissections occurred mostly in individuals with a P/LP variant in the first quarter of the collagen helical domain of the COL3A1 gene. The extracellular matrix of the aortic wall consists primarily of elastin and collagen to provide strength to the aortic wall, to withstand the forces of blood pressure.21–23 The arterial collagen is mainly types I and III collagen.24 Previous studies in the skin biopsies of patients vEDS revealed an association between collagen fibril diameter and the location of the P/LP variant in COL3A1.25,26 P/LP variants near the N terminus were associated with larger collagen fibril diameters, whereas mutations near the C terminus of the COL3A1 gene were associated with smaller collagen fibril diameters. Lapiere et al27 showed a higher collagen I/III ratio in fibrils with larger collagen fibril thickness, which suggests reduced presence/incorporation of collagen type III when the P/LP variant is located near the N terminus (the start of the gene). We observed that vEDS individuals with aortic dissection mostly have a P/LP variant in the first quarter of the COL3A1 collagen helical domain, which is near the N terminus; thus, a shift in the collagen I/III ratio is expected to cause a more severe vascular phenotype. To the best of our knowledge, this genotype-phenotype correlation has not been described previously.
Interestingly, COL3A1-deficient, haploinsufficient, and glycine-mutant vEDS mouse models have been reported to show similar features as vEDS patients.28–32 Overexpression of COL3A1 with a variant at the N terminus showed reduced collagen in the aortic adventitia with fibrils highly variable and increased in diameter, similar to those found in the deficiency models. Upon fibril analysis, a higher collagen I/III ratio in fibrils with larger collagen fibril thickness was found in the skin of these COL3A1 mice.
To date, MSAs are described as the main location of vascular complications of vEDS.1,3,4 However, in our cohort, aneurysms or dissections occurred more frequently in the thoracic and abdominal aortas as compared with the MSA (55 versus 31, respectively). This is likely partially due to ascertainment bias, since the COL3A1 gene is present in the gene panel for thoracic aortic aneurysms and dissections and analyzed in all individuals with thoracic aortic aneurysms and dissections (n≥1000) who underwent genetic testing. Nevertheless, when taking only relatives into account, the aorta was still more prone to aneurysms or dissections compared with the MSA (23 versus 12).
However, not all individuals (101/127, 79.5%) received imaging at the time of inclusion in the study, and imaging of the abdominal arteries was not performed in 17 of 101 individuals (16.8%). Since vascular complications of the MSA are most commonly present in the abdominal arteries and not all individuals have received imaging of this section (yet), this might explain the underrepresentation of vascular aneurysms in the MSA in our cohort.
In our cohort, 33% of the index patients did not meet the revised criteria for vEDS, which is lower than the 45% previously reported by Ritelli et al,33 which suggests that the revised criteria might be too strict.33 The wide availability of genetic testing allows for a relatively low threshold for genetic testing. It is suggested to offer genetic analysis including the COL3A1 gene in all individuals with a dissection or unexplained aneurysm under the age of 50 years, or 60 years in the absence of hypertension.34 A previous study on the diagnostic yield of the thoracic aortic aneurysms and dissections genes showed a P/LP COL3A1 variant in 6 of 810 individuals (0.7%), including 2 individuals in the age range of 50 to 60 years.35
Spontaneous pneumothorax and abdominal complications were less frequently present than observed previously (12.5%–17% and 21%–22.6%, respectively).1,14,36,37 These complications occurred at a younger age than vascular complications and can be an early manifestation of vEDS.37 The median age at the time of a colon rupture or perforation in our cohort was higher (32.0 years [IQR, 30.0–44.0]) than described before by Frank et al1 (23.0 years [range, 19–34]). We recommend health care providers to be aware of vEDS in young individuals with spontaneous pneumothorax or abdominal complications, even in the absence of other vEDS features, and at least perform a physical examination and evaluate the family history for features suggestive of vEDS, but also Marfan syndrome and Birt-Hogg-Dubé syndrome, which are also associated with an increased risk for primary spontaneous pneumothorax.38,39 Genetic testing should be considered if additional physical or other features or a positive family history are present.
Most of the major abdominal complications (7/9) affected the colon and occurred in individuals with a glycine substitution or splice/delins/del variant in COL3A1.1 However, we also documented an abdominal complication in an individual heterozygous for a predicted haploinsufficient variant in COL3A1, which has only been reported once.17
Twelve individuals (8.5%) were diagnosed with intracranial vascular anomalies, including 2 individuals with an ischemic stroke due to the dissection of an internal carotid or vertebral artery, which is in line with the prevalence of intracranial vascular complications described by Adham et al.40 The prevalence of carotid-cavernous fistula in our cohort (2.4%) is lower than previously reported (9.8%).40,41 Despite the occurrence of intracranial vascular complications in 8.5% of vEDS individuals in our cohort, only 70% of the alive individuals received cerebrovascular imaging. We recommend to consider cerebrovascular screening at least once for all adult individuals with vEDS.
The described phenotypic spectrum of vEDS and genotype-phenotype correlations are based on the data of 142 individuals with vEDS. Since not all individuals with vEDS agreed to participate in this study and some were diagnosed after the closure of the study, the number of known individuals with vEDS in the Netherlands is currently estimated at ≈200. Given the total of 17.4 million inhabitants of the Netherlands, the minimal estimated prevalence of vEDS in the Netherlands is around 1.1:100 000, assuming all patients with vEDS have currently been diagnosed. This estimated prevalence is in line with the previously estimated prevalence range of 1:50 000 to 1:200 000 in the United States.2 However, it is likely that prevalence is even higher since the diagnosis can be missed due to the absence of the suggestive criteria for vEDS or unawareness of the suggestive criteria with the health professional.
Assessment of the type and frequency of surveillance revealed a great variability in surveillance strategies of caregivers. Around 70% of the 127 living individuals received surveillance including imaging at least every other year. Different imaging modalities are used, but magnetic resonance angiography or computed tomography angiography of the thorax and abdomen were the most common. This is in line with the observation that arterial monitoring of adult clinically silent patients with vEDS is standard clinical practice in European expert centers of the MSA working group of the European Reference Network For Rare Vascular Diseases.42 At this moment, there are no international guidelines for surveillance in vEDS. The objective of surveillance is the prevention of complications by performing elective procedures when abnormalities are present. However, no criteria for elective interventions in vEDS have been clearly established, probably due to the high risk of complications.1,2,14,17 Byers et al3 recommended to provide a care team for each vEDS patient with a primary physician to coordinate the management and surveillance. Previous studies examined the effect of celiprolol on the prevention of vascular complications and improving survival.1,43 Both studies showed a decrease in vascular complications and a higher survival rate in individuals who received celiprolol.
However, both studies had multiple limitations in study design and inclusion criteria, and therefore, no clear conclusions about the effect of celiprolol can be made at this moment.44
Given the high variability in type and frequency of surveillance and even the absence of medical follow-up in some vEDS patients, it is important to create an international consensus statement for the management and surveillance of individuals with vEDS. The collaboration of vEDS expert teams is essential in the creation of such a consensus statement, which should be created by using the merged genotypic and phenotypic data of different vEDS cohorts from all over the world.
CONCLUSIONS
Male sex, the phenotypic appearance, and the type and location of the variant are risk factors for the occurrence and age of onset of a major event. The type of COL3A1 variant does not influence the age at the first major event in relatives. Caregivers should be aware of the potential diagnosis of vEDS in patients with an isolated major event, even in the absence of characteristic features highly suggestive of vEDS given the phenotypic variability. Because of the great variability in type and frequency of surveillance in current daily practice, it is important to create guidelines for the management and surveillance of individuals with vEDS.
Limitations
This Dutch cohort of individuals with vEDS is relatively large given the rarity of vEDS. However, the number of included individuals is still small to interpret the results and draw conclusions. Therefore, it would be very valuable for further research to merge all the described vEDS cohorts and perform a meta-analysis. In addition, estimation of the penetrance of rare genetic disorders in retrospective cohort studies has several limitations, including missing data and the likelihood of preferential testing in individuals and relatives affected by complications (eg, inclusion bias). It is therefore not surprising that the penetrance of pathogenic variants in population databases is lower than that observed in index patients and their relatives opting for genetic testing, as recently underlined by Forrest et al.45 However, since these observations are representative of families opting for genetic testing (eg, the population reported in our current study), they are highly relevant. Nevertheless, it is important to consider these limitations.
Another common limitation of retrospective cohort studies for such rare diseases is the inclusion of family members, which might pose a risk for dependent data. We assumed that the variability in the vEDS phenotype is mutually independent, even within families.
Only a few children are included in this study due to the reluctance of presymptomatic genetic testing in children in the Netherlands. The low number of children results in a higher median age of the studied cohort. Additionally, this cohort study is limited by the lack of a unified strategy for imaging and follow-up of individuals with vEDS. For example, vascular complications in the MSA were underrepresented in our cohort. This may be explained by the fact that asymptomatic vascular complications of the MSA are most commonly present in the abdominal arteries, and imaging of the abdominal MSA was not performed in all individuals.
ARTICLE INFORMATION
Acknowledgments
The authors thank all the research participants, physicians, genetic counsellors, and investigators who have included individuals in this study and collected clinical data.
Sources of Funding
None.
Disclosures
None.
Supplemental Material
Supplemental Methods
Tables S1–S5
Supplementary Material
Nonstandard Abbreviations and Acronyms
- ACE
- angiotensin-converting enzyme
- ARB
- angiotensin receptor blockers
- Delins
- deletion-insertion
- MSA
- middle-sized artery
- P/LP
- pathogenic/likely pathogenic
- vEDS
- vascular Ehlers-Danlos syndrome
S. Demirdas and L.M. van den Bersselaar are joint first authors.
I.M.B.H. van de Laar and A.C. Houweling are joint last authors.
For Sources of Funding and Disclosures, see page 235.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/CIRCGEN.122.003978.
Contributor Information
Rosan Lechner, Email: r.lechner@erasmusmc.nl.
Jessica Bos, Email: jessicabos10@hotmail.com.
Annette F. Baas, Email: a.f.baas@umcutrecht.nl.
Özlem Baysal, Email: Ozlem.Baysal@radboudumc.nl.
Saskia N. van der Crabben, Email: s.n.vandercrabben@amsterdamumc.nl.
Eelco Dulfer, Email: dulfere@umcg.nl.
Yvonne Hilhorst-Hofstee, Email: Y.Hilhorst-Hofstee@lumc.nl.
Bart Loeys, Email: bart.loeys@uantwerp.be.
Eline Overwater, Email: e.overwater@umcg.nl.
Marsha Voorendt, Email: Marsha.Voorendt@radboudumc.nl.
Alessandra Maugeri, Email: a.maugeri@amsterdamumc.nl.
REFERENCES
- 1.Frank M, Albuisson J, Ranque B, Golmard L, Mazzella JM, Bal-Theoleyre L, Fauret AL, Mirault T, Denarie N, Mousseaux E, et al. The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular Ehlers-Danlos syndrome. Eur J Hum Genet. 2015;23:1657–1664. doi: 10.1038/ejhg.2015.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Byers PH. Vascular Ehlers-Danlos syndrome. Gene Reviews. 2019 [Google Scholar]
- 3.Byers PH, Belmont J, Black J, De Backer J, Frank M, Jeunemaitre X, Johnson D, Pepin M, Robert L, Sanders L, et al. Diagnosis, natural history, and management in vascular Ehlers-Danlos syndrome. Am J Med Genet C Semin Med Genet. 2017;175:40–47. doi: 10.1002/ajmg.c.31553 [DOI] [PubMed] [Google Scholar]
- 4.Pepin M, Schwarze U, Superti-Furga A, Byers PH. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med. 2000;342:673–680. doi: 10.1056/NEJM200003093421001 [DOI] [PubMed] [Google Scholar]
- 5.Pepin MG, Schwarze U, Rice KM, Liu M, Leistritz D, Byers PH. Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV). Genet Med. 2014;16:881–888. doi: 10.1038/gim.2014.72 [DOI] [PubMed] [Google Scholar]
- 6.Pope FM, Narcisi P, Nicholls AC, Germaine D, Pals G, Richards AJ. COL3A1 mutations cause variable clinical phenotypes including acrogeria and vascular rupture. Br J Dermatol. 1996;135:163–181. doi: 10.1111/j.1365-2133.1996.tb01143.x [PubMed] [Google Scholar]
- 7.Malfait F, Francomano C, Byers P, Belmont J, Berglund B, Black J, Bloom L, Bowen JM, Brady AF, Burrows NP, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175:8–26. doi: 10.1002/ajmg.c.31552 [DOI] [PubMed] [Google Scholar]
- 8.Kuivaniemi H, Tromp G. Type III collagen (COL3A1): gene and protein structure, tissue distribution, and associated diseases. Gene. 2019;707:151–171. doi: 10.1016/j.gene.2019.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leistritz DF, Pepin MG, Schwarze U, Byers PH. COL3A1 haploinsufficiency results in a variety of Ehlers-Danlos syndrome type IV with delayed onset of complications and longer life expectancy. Genet Med. 2011;13:717–722. doi: 10.1097/GIM.0b013e3182180c89 [DOI] [PubMed] [Google Scholar]
- 10.Plancke A, Holder-Espinasse M, Rigau V, Manouvrier S, Claustres M, Khau Van Kien P. Homozygosity for a null allele of COL3A1 results in recessive Ehlers-Danlos syndrome. Eur J Hum Genet. 2009;17:1411–1416. doi: 10.1038/ejhg.2009.76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jorgensen A, Fagerheim T, Rand-Hendriksen S, Lunde PI, Vorren TO, Pepin MG, Leistritz DF, Byers PH. Vascular Ehlers-Danlos Syndrome in siblings with biallelic COL3A1 sequence variants and marked clinical variability in the extended family. Eur J Hum Genet. 2015;23:796–802. doi: 10.1038/ejhg.2014.181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malfait F, Symoens S, De Backer J, Hermanns-Le T, Sakalihasan N, Lapiere CM, Coucke P, De Paepe A. Three arginine to cysteine substitutions in the pro-alpha (I)-collagen chain cause Ehlers-Danlos syndrome with a propensity to arterial rupture in early adulthood. Hum Mutat. 2007;28:387–395. doi: 10.1002/humu.20455 [DOI] [PubMed] [Google Scholar]
- 13.Elsisy MF, Pochettino A, Dearani JA, Bower TC, McBane RD, 2nd, Graham GC, Deyle DR, Bonnichsen CR, Stephens EH. Early and late outcomes of cardiovascular surgery in patients with Ehlers-Danlos Syndrome. World J Pediatr Congenit Heart Surg. 2021;12:773–777. doi: 10.1177/21501351211049253 [DOI] [PubMed] [Google Scholar]
- 14.Oderich GS, Panneton JM, Bower TC, Lindor NM, Cherry KJ, Noel AA, Kalra M, Sullivan T, Gloviczki P. The spectrum, management and clinical outcome of Ehlers-Danlos syndrome type IV: a 30-year experience. J Vasc Surg. 2005;42:98–106. doi: 10.1016/j.jvs.2005.03.053 [DOI] [PubMed] [Google Scholar]
- 15.Olubajo F, Kaliaperumal C, Choudhari KA. Vascular Ehlers-Danlos syndrome: literature review and surgical management of intracranial vascular complications. Clin Neurol Neurosurg. 2020;193:105775. doi: 10.1016/j.clineuro.2020.105775 [DOI] [PubMed] [Google Scholar]
- 16.Bos EO J, Dirksen MT, Simsek S, Demirdas S, Houweling AC. Spontaneous coronary artery dissection as presenting feature of vascular Ehlers-Danlos syndrome. Cardiogenetics. 2021;11:129–131. doi: 10.1161/CIRCGENETICS.117.001933 [Google Scholar]
- 17.Shalhub S, Black JH, 3rd, Cecchi AC, Xu Z, Griswold BF, Safi HJ, Milewicz DM, McDonnell NB. Molecular diagnosis in vascular Ehlers-Danlos syndrome predicts pattern of arterial involvement and outcomes. J Vasc Surg. 2014;60:160–169. doi: 10.1016/j.jvs.2014.01.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Teixido-Tura G, Franken R, Galuppo V, Gutierrez Garcia-Moreno L, Borregan M, Mulder BJ, Garcia-Dorado D, Evangelista A. Heterogeneity of aortic disease severity in patients with Loeys-Dietz syndrome. Heart. 2016;102:626–632. doi: 10.1136/heartjnl-2015-308535 [DOI] [PubMed] [Google Scholar]
- 19.Loeys BL, Schwarze U, Holm T, Callewaert BL, Thomas GH, Pannu H, De Backer JF, Oswald GL, Symoens S, Manouvrier S, et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med. 2006;355:788–798. doi: 10.1056/NEJMoa055695 [DOI] [PubMed] [Google Scholar]
- 20.Solyst S, Oksjoki R, Farholt S, Nielsen DG, Christensen AH, Fagerberg CR, Risom L, Gregersen PA, Christensen MB, Rasmussen TB, et al. Carriers of COL3A1 pathogenic variants in Denmark: interfamilial variability in severity and outcome of elective surgical procedures. Clin Genet. 2022;102:191–200. doi: 10.1111/cge.14176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nichols WW, O’Rourke MF. Properties of the arterial wall. 3rd ed. McDonald’s Blood Flow in Arteries. Lea and Febiger. 1990:77–124. [Google Scholar]
- 22.Dobrin PB. Mechanics of normal and diseased blood vessels. Ann Vasc Surg. 1988;2:283–294. doi: 10.1016/S0890-5096(07)60016-8 [DOI] [PubMed] [Google Scholar]
- 23.Sidawy AN, Sumpio BE, DePalma RG, Marty AT. The basic science of vascular disease. Chest. 1997;112:A36. doi: 10.1002/clc.4960210321 [Google Scholar]
- 24.Morton LF, Barnes MJ. Collagen polymorphism in the normal and diseased blood vessel wall. Investigation of collagens types I, III and V. Atherosclerosis. 1982;42:41–51. doi: 10.1016/0021-9150(82)90124-1 [DOI] [PubMed] [Google Scholar]
- 25.Hausser I, Anton-Lamprecht I. Differential ultrastructural aberrations of collagen fibrils in Ehlers-Danlos syndrome types I-IV as a means of diagnostics and classification. Hum Genet. 1994;93:394–407. doi: 10.1007/BF00201664 [DOI] [PubMed] [Google Scholar]
- 26.Smith LT, Schwarze U, Goldstein J, Byers PH. Mutations in the COL3A1 gene result in the Ehlers-Danlos syndrome type IV and alterations in the size and distribution of the major collagen fibrils of the dermis. J Invest Dermatol. 1997;108:241–247. doi: 10.1111/1523-1747.ep12286441 [DOI] [PubMed] [Google Scholar]
- 27.Lapiere CM, Nusgens B, Pierard GE. Interaction between collagen type I and type III in conditioning bundles organization. Connect Tissue Res. 1977;5:21–29. doi: 10.3109/03008207709152608 [DOI] [PubMed] [Google Scholar]
- 28.Liu X, Wu H, Byrne M, Krane S, Jaenisch R. Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development. Proc Natl Acad Sci USA. 1997;94:1852–1856. doi: 10.1073/pnas.94.5.1852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cooper TK, Zhong Q, Krawczyk M, Tae HJ, Muller GA, Schubert R, Myers LA, Dietz HC, Talan MI, Briest W. The haploinsufficient Col3a1 mouse as a model for vascular Ehlers-Danlos syndrome. Vet Pathol. 2010;47:1028–1039. doi: 10.1177/0300985810374842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith LB, Hadoke PW, Dyer E, Denvir MA, Brownstein D, Miller E, Nelson N, Wells S, Cheeseman M, Greenfield A. Haploinsufficiency of the murine Col3a1 locus causes aortic dissection: a novel model of the vascular type of Ehlers-Danlos syndrome. Cardiovasc Res. 2011;90:182–190. doi: 10.1093/cvr/cvq356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.D’Hondt S, Guillemyn B, Syx D, Symoens S, De Rycke R, Vanhoutte L, Toussaint W, Lambrecht BN, De Paepe A, Keene DR, et al. Type III collagen affects dermal and vascular collagen fibrillogenesis and tissue integrity in a mutant Col3a1 transgenic mouse model. Matrix Biol. 2018;70:72–83. doi: 10.1016/j.matbio.2018.03.008 [DOI] [PubMed] [Google Scholar]
- 32.Bowen CJ, Calderon Giadrosic JF, Burger Z, Rykiel G, Davis EC, Helmers MR, Benke K, Gallo MacFarlane E, Dietz HC. Targetable cellular signaling events mediate vascular pathology in vascular Ehlers-Danlos syndrome. J Clin Invest. 2020;130:686–698. doi: 10.1172/JCI130730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ritelli M, Rovati C, Venturini M, Chiarelli N, Cinquina V, Castori M, Colombi M. Application of the 2017 criteria for vascular Ehlers-Danlos syndrome in 50 patients ascertained according to the Villefranche nosology. Clin Genet. 2020;97:287–295. doi: 10.1111/cge.13653 [DOI] [PubMed] [Google Scholar]
- 34.Verhagen JMA, Kempers M, Cozijnsen L, Bouma BJ, Duijnhouwer AL, Post JG, Hilhorst-Hofstee Y, Bekkers SCAM, Kerstjens-Frederikse WS, van Brakel TJ, et al. ; National Working Group on BAV & TAA. Expert consensus recommendations on the cardiogenetic care for patients with thoracic aortic disease and their first-degree relatives. Int J Cardiol. 2018;258:243–248. doi: 10.1016/j.ijcard.2018.01.145 [DOI] [PubMed] [Google Scholar]
- 35.Overwater E, Marsili L, Baars MJH, Baas AF, van de Beek I, Dulfer E, van Hagen JM, Hilhorst-Hofstee Y, Kempers M, Krapels IP, et al. Results of next-generation sequencing gene panel diagnostics including copy-number variation analysis in 810 patients suspected of heritable thoracic aortic disorders. Hum Mutat. 2018;39:1173–1192. doi: 10.1002/humu.23565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Boussouar S, Benattia A, Escudie JB, Gibault L, Capron F, Legrand A, Brillet PY, Jeunemaitre X, Grenier PA, Mousseaux E, et al. Vascular Ehlers-Danlos syndrome (vEDS): CT and histologic findings of pleural and lung parenchymal damage. Eur Radiol. 2021;31:6275–6285. doi: 10.1007/s00330-021-07710-6 [DOI] [PubMed] [Google Scholar]
- 37.Shalhub S, Neptune E, Sanchez DE, Dua A, Arellano N, McDonnell NB, Milewicz DM. Spontaneous pneumothorax and hemothorax frequently precede the arterial and intestinal complications of vascular Ehlers-Danlos syndrome. Am J Med Genet A. 2019;179:797–802. doi: 10.1002/ajmg.a.61094 [DOI] [PubMed] [Google Scholar]
- 38.Karpman C, Aughenbaugh GL, Ryu JH. Pneumothorax and bullae in Marfan syndrome. Respiration. 2011;82:219–224. doi: 10.1159/000322958 [DOI] [PubMed] [Google Scholar]
- 39.Johannesma PC, Houweling AC, van Waesberghe JH, van Moorselaar RJ, Starink TM, Menko FH, Postmus PE. The pathogenesis of pneumothorax in Birt-Hogg-Dube syndrome: a hypothesis. Respirology. 2014;19:1248–1250. doi: 10.1111/resp.12405 [DOI] [PubMed] [Google Scholar]
- 40.Adham S, Billon C, Legrand A, Domigo V, Denarie N, Charpentier E, Jeunemaitre X, Frank M. Spontaneous cervical artery dissection in vascular Ehlers-Danlos syndrome: a cohort study. Stroke. 2021;52:1628–1635. doi: 10.1161/STROKEAHA.120.032106 [DOI] [PubMed] [Google Scholar]
- 41.Adham S, Trystram D, Albuisson J, Domigo V, Legrand A, Jeunemaitre X, Frank M. Pathophysiology of carotid-cavernous fistulas in vascular Ehlers-Danlos syndrome: a retrospective cohort and comprehensive review. Orphanet J Rare Dis. 2018;13:100. doi: 10.1186/s13023-018-0842-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van de Laar I, Baas AF, De Backer J, Blankenstein JD, Dulfer E, Helderman-van den Enden A, Houweling AC, Kempers MJ, Loeys B, Malfait F, et al. Surveillance and monitoring in vascular Ehlers-Danlos syndrome in European Reference Network For Rare Vascular Diseases (VASCERN). Eur J Med Genet. 2022;65:104557. doi: 10.1016/j.ejmg.2022.104557 [DOI] [PubMed] [Google Scholar]
- 43.Ong KT, Perdu J, De Backer J, Bozec E, Collignon P, Emmerich J, Fauret AL, Fiessinger JN, Germain DP, Georgesco G, et al. Effect of celiprolol on prevention of cardiovascular events in vascular Ehlers-Danlos syndrome: a prospective randomised, open, blinded-endpoints trial. Lancet. 2010;376:1476–1484. doi: 10.1016/S0140-6736(10)60960-9 [DOI] [PubMed] [Google Scholar]
- 44.De Backer J, De Backer T. Vascular Ehlers-Danlos syndrome management: the Paris way, a step forward on a long road. J Am Coll Cardiol. 2019;73:1958–1960. doi: 10.1016/j.jacc.2019.02.025 [DOI] [PubMed] [Google Scholar]
- 45.Forrest IS, Chaudhary K, Vy HMT, Petrazzini BO, Bafna S, Jordan DM, Rocheleau G, Loos RJF, Nadkarni GN, Cho JH, et al. Population-based penetrance of deleterious clinical variants. JAMA. 2022;327:350–359. doi: 10.1001/jama.2021.23686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hirsch AT, Haskal ZJ, Hertzer NR, Bakal CW, Creager MA, Halperin JL, Hiratzka LF, Murphy WR, Olin JW, Puschett JB, et al. ; American Association for Vascular Surgery. ACC/AHA 2005 practice guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease): endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; Transatlantic Inter-Society Consensus; and Vascular Disease Foundation. Circulation. 2006;113:e463–e654. doi: 10.1161/CIRCULATIONAHA.106.174526 [DOI] [PubMed] [Google Scholar]
- 47.Thompson MM, Bell PR. ABC of arterial and venous disease. Arterial aneurysms. BMJ. 2000;320:1193–1196. doi: 10.1136/bmj.320.7243.1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Devereux RB, de Simone G, Arnett DK, Best LG, Boerwinkle E, Howard BV, Howard BV, Kitzman D, Lee ET, Mosley TH, et al. Normal limits in relation to age, body size and gender of two-dimensional echocardiographic aortic root dimensions in persons >/=15 years of age. Am J Cardiol. 2012;110:1189–1194. doi: 10.1016/j.amjcard.2012.05.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Du Bois DBE. A formula to estimate the approximate surface area if height and weight be known. 1916. Nutrition. 1989;5:303–311, discussion 312–313. doi: 10.1001/archinte.1916.00080130010002 [PubMed] [Google Scholar]
- 50.Williams B, Mancia G, Spiering W, Agabiti Rosei E, Azizi M, Burnier M, Clement DL, Coca A, de Simone G, Dominiczak A, et al. ; Authors/Task Force Members. 2018 ESC/ESH guidelines for the management of arterial hypertension: the task force for the management of arterial hypertension of the European Society of Cardiology and the European Society of Hypertension: the task force for the management of arterial hypertension of the European Society of Cardiology and the European Society of Hypertension. J Hypertens. 2018;36:1953–2041. doi: 10.1097/HJH.0000000000001940 [DOI] [PubMed] [Google Scholar]
- 51.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector A, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.ClinVar: National Library of Medicine, NCBI. doi: 10.1080/15360280801989377. https://www.ncbi.nlm.nih.gov/clinvar/ [DOI] [PubMed] [Google Scholar]
- 53.Schwarze U, Goldstein JA, Byers PH. Splicing defects in the COL3A1 gene: marked preference for 5’ (donor) spice-site mutations in patients with exon-skipping mutations and Ehlers-Danlos syndrome type IV. Am J Hum Genet. 1997;61:1276–1286. doi: 10.1086/301641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van de Luijtgaarden KM, Heijsman D, Maugeri A, Weiss MM, Verhagen HJ, IJpma A, Bruggenwirth HT, Majoor-Krakauer D. First genetic analysis of aneurysm genes in familial and sporadic abdominal aortic aneurysm. Hum Genet. 2015;134:881–893. doi: 10.1007/s00439-015-1567-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cho YKDGW, Changwon K, Lee BH, Seo GH, Byung-Chul L. A novel PS4 criterion approach based on symptoms of rare diseases and in-house frequency data in a Bayesian framework. BioRxiv. Preprint posted online July 2020. doi: 10.1101/2020.07.22.215426 [Google Scholar]
- 56.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. ; Exome Aggregation Consortium. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sian Ellard ELB, Alison C, Ian B, Natalie F, Clare T, Martina O, Diana ME, Stephen A, Richard S, Zandra C, et al. ACGS best practice guidelines for variant classification in rare disease 2020. Association for Clinical Genomic Science. 2020. https://www.acgs.uk.com/media/11631/uk-practice-guidelines-for-variant-classification-v4-01-2020.pdf [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.