

Abstract
Basal cell carcinoma (BCC), the most common cancer in humans, appears macroscopically and microscopically similar to many other skin lesions, which makes differential diagnosis difficult. We are developing an approach for quantitative molecular imaging of BerEP4, a transmembrane biomarker for BCC, with the goal of increasing the precision and accuracy of diagnosis. This pilot study was conducted to assess the affinity and selectivity of BerEp4 antibody and assess its possible use in designing theranostic probes for BCC. We provide evidence that our photon-counting fluorescence macrodetection system can recover specific signal increases from a film/pellet phantom. Additionally, we show that a two-photon excited fluorescence /backscatter confocal microscopy system can image BerEP4 antibody/antigen complex on the surface of BerEP4-expressing cancer cells in three dimensions. Based on the initial results, BerEP4 seems to be a promising biomarker for molecular imaging of BCC. To prepare BerEP4 for eventual theranostic use, we examined the feasibility of a combined macro-/micro-optical approach to imaging BCC with various histologies. These optical methods, endowed with the ability to monitor treatment in real time, may open an opportunity for noninvasive diagnosis, treatments, and follow-up.
Summary
We have examined applying a novel multiscale, macro/micro BerEP4 imaging system with an eye toward BCC. We anticipate that the same principle can be applied to molecular probes for other skin pathologies. The efficacy of treatment can be assessed by follow-up imaging. Quantitative fluorescence may provide a noninvasive, rapid approach to diagnosis, treatment, and follow-up of BCC while reducing the morbidity associated with surgical approaches. Given the high incidence, extraordinarily slow growth, and extremely rare mortality of BCC, noninvasive diagnostics seem both justified and prudent.
BASAL CELL CARCINOMA (BCC) is the most common cancer in humans. The annual incidence has increased exponentially over the past decades and has now reached ≈ 2.8 million new cases in the United States alone.1 The direct costs of diagnosis and treatment of nonmelanoma skin cancer in the United States were reported to be $1.4 billion in 2004, when the annual incidence was only 0.5 million.2 A trend toward a younger age at initial diagnosis has made the early detection and treatment of BCC and other skin cancers an increasingly important public health issue.
Benign solitary skin lesions, such as actinic keratosis (a premalignant skin lesion) and seborrheic keratosis (a benign skin lesion), have a higher prevalence than skin cancer. Actinic keratosis and seborrheic keratosis can present with clinically similar morphology to BCC, especially at early stages. Differentiating benign from cancerous lesions is a major challenge, especially since the clinical practice of diagnosing skin cancer has changed little in the past 100 years. BCC and other skin cancers are diagnosed by skin biopsy followed by histologic evaluation of the specimen and surgical treatment, which has considerable associated cost and morbidity.
Currently available clinical tools such as dermoscopy, which are used to aid early detection of skin cancer, improve clinical diagnosis above simple visual inspection.3,4 However, their utility is limited by their subjective nature and the limitations of visual assessment. In a recent retrospective review of 2,000 specimens submitted by a mixed group of academicians and private practitioners to the Pinkus Dermatopathology Laboratories (Monroe, MI) to rule out BCC, we found that > 70% were benign lesions.5
BCC can present with a variety of histologic morphologies at various depths and tumor cell collection sizes. Micronodular, infiltrative, and morpheaform variants penetrate more deeply. They also exhibit a markedly smaller collection of malignant cells, which warrant a modality that can image sufficiently deep while retaining highly sensitive signal detection.6 The visual perception of BCC and other skin cancers can be misleading because of concurrent secondary phenomena, including inflammation. Therefore, a large field of view is necessary to minimize sampling error. The need for deeper imaging with a wider field of view makes ‘‘macro-optics’’ an essential element for a hybrid multimodal system.
Given the microscopic profile of skin structures and cutaneous pathologies, most of the work done in the area of primary skin optical imaging has been in the microscopic domain.7–12 The small field of view has been one of the limiting steps in imaging the whole lesion. This results in a bottleneck effect in the workflow of a dermatology clinic. Additionally, given the microscopic nature of skin pathologies and their histopathologic similarities, a targeted and quantified method is needed to reach the diagnostic accuracy and precision that are expected. We have applied time-resolved fluorescent imaging in the diffuse regimen as the approach of choice.
Our ultimate goal is bedside application. Quantification of molecular signals with time-resolved macrofluorescent imaging is needed to objectively monitor treatment. Once the macrosystem is coupled with a two-photon excited fluorescence (TPEF)/confocal multimodal microscopy system, localized subcellular resolution with three-dimensional tomographic image reconstruction may also be achieved. The compatibility of near-infrared (NIR) lasers used with both macro- and micro-optical systems makes NIR the preferred spectrum for this purpose.13–15
NIR fluorophore–labeled antibody (Ab) is a well-established method for in vivo optical imaging.16–18 NIR fluorophores conjugated with BerEP4 Ab have been applied to imaging BerEP4-expressing carcinomas.17 BerEP4 or EpCAM, a human epithelial cell adhesion molecule, located on chromosome 4q, is a 40 kDa type 1 transmembrane glycoprotein with an extracellular domain.19 This biomarker is expressed on the basolateral surface of simple, pseudostratified, and transitional epithelium. Its in vivo expression is believed to be related to epithelial proliferation and dedifferentiation.20,21 BerEP4 is known to be overexpressed on carcinomas of various origins,22 including all histologic variants of BCC23 and colorectal carcinoma,24 making this molecule an attractive pancarcinoma biomarker with potential diagnostic and therapeutic (theranostic) application.25–27
BerEP4 is expressed on 90 to 95% of BCC tumors.23 BerEP4 is also expressed on some eccrine/apocrine glands as well as lesions of follicular origin, such as trichofolliculoma.28 However, these lesions represent a negligible fraction of the biopsies performed to rule out BCC. Most important, BerEP4 is not expressed on stratified epithelium of normal skin or on those benign lesions that are often biopsied to rule out BCC due to clinical morphologic similarities at early stages.23 Therefore, BerEP4 is a desirable biomarker to target for molecular imaging of BCC.
The purposes of this study were the following: (1) to develop a BerEP4 microtumor phantom to conduct ex vivo experiments, (2) to assess the sensitivity of our macro-optics system to detect and characterize positive phantoms, (3) to assess the affinity and selectivity of BerEp4 antibody, and (4) to consider needed specifications of a combined optical system to image BCC of various histologies while monitoring treatment.
We found that BerEP4 Ab can selectively detect and distinguish BerEP4-expressing cells from non–BerEP4-expressing cells on microtumor phantoms. Additionally, we show that time-resolved macro- and TPEF/backscatter confocal microscopy systems can image BerEP4 antibody/antigen complex on the surface of BerEP4-expressing cancer cells in three dimensions. Based on preliminary results, BerEP4 seems to be a promising biomarker for molecular imaging of BCC.
Materials and Methods
Sample Preparation
The human cell lines UACC257 (skin melanoma) and SW620 (colorectal adenocarcinoma) were obtained from the National Cancer Institute Drug Screen Cell Bank. Both were grown in RPMI medium incubating with 10% fetal bovine serum at 37°C in 5% CO2 atmosphere. Notably, the SW620 and UACC257 cell lines were selected from a list of candidates as positive and negative cell lines, respectively, for BerEP4 expression according to flow cytometry results. A BCC cell line was not selected because of its lengthy growth time and propensity to differentiate without a proper substrate in ex vivo cell culture while also losing BerEP4 receptors at their surface.
For micronodular tumor phantom experiments, cells were grown on CytoDex-3 microcarrier beads (Sigma-Aldrich, St. Louis, MO). These beads are cross-linked dextran coated with denatured porcine skin collagen. We rehydrated beads in phosphate-buffered saline (PBS) and then sterilized them by soaking in 70% v/v ethanol for several hours and washing with PBS, followed by repeatedly washing with RPMI medium. Beads were stored as a 50% v/v slurry in RPMI. For cell attachment, cells were trypsinized, removed from their growth flasks, and resuspended in RPMI medium. Cells were mixed with the beads in T25 growth flasks held just off vertical, so the cells and beads were joined in the corner of the flask. About 100 μL of the bead slurry was mixed with 1 mL of RPMI medium containing ≈ 105 cells. The flask was rocked occasionally to remix the cells and beads. After an hour of incubation, 3 mL of RPMI medium was gently added to the flask. Following overnight incubation, the cell-coated beads measuring 60 to 200 μm in diameter were used in experiments.
For antibody labeling, cell-coated beads were labeled with EpCAM mouse monoclonal antibody (Abcam, Cambridge, MA). Cell-coated beads were removed from their growth flask to small microcentrifuge tubes and allowed to settle. Excess RPMI medium was removed and replaced by 100 μL of anti-Ep-CAM antibody diluted 1:100 in RPMI complete medium. Microcentrifuge tubes were gently stirred and incubated in the 37°C incubator for 30 minutes with occasional mixing. Following this, the beads were allowed to settle, medium was removed, and beads were washed twice by resuspending them in RPMI medium, allowing them to settle, and removing the supernatant medium. Beads were then stained with secondary goat antimouse IgG (Sigma-Aldrich #M6898), conjugated to fluorophores DyLight 594 NHS (Thermo, Rockform, IL) or Alexa Fluor 750 carboxylic acid, succinimidyl ester (Life Technologies, Grand Island, NY), according to the manufacturer’s protocols. Labeled antibody was diluted 1:200 in RPMI medium from a 1 mg/mL solution, added to the cell-coated beads, stained, and washed as above for the primary antibody. Control beads were incubated with the secondary antibody alone.
Macroscopic Live Cell Time-Resolved Fluorescence Imaging of Labeled Micronodular Tumor Phantoms
Our in vivo time-resolved fluorescence small-animal imaging system, which consisted of a tunable pulsed laser (Newport Spectra-Physics model Tsunami self-mode-locked Ti:Sapphire laser pumped by a Millenia V green continous wave laser) with a pulse width of ≈ 100 fs and a repetition rate of 80 MHz, was described previously in detail.18 Briefly, the laser was tuned to an excitation wavelength of 750 nm. The light pulses delivered by an optical fiber scanned the target in a raster pattern through a scanning head in a noncontact fashion. The target phantom consisted of a droplet of Alexa Fluor750–labeled cell pellet in PBS buffer deposited on a glass microscope slide. The emittted fluorescence was filtered by a 780 nm long-pass glass emission filter. The detected photons were captured by a sensitive GaAs photomultiplier tube (Hamamatsu Corp., model H7422–50), and their arrival times were registered by a time-correlated single-photon counting board (Becker & Hickl, model SPC-830). Initialization, scanning, and acquisition were controlled by our LabVIEW-based software. The final data were processed by software written in Matlab.
TPEF and Confocal Microscopy of Labeled Micronodular Tumor Phantoms
Cancer cells grown on collagen microspheres as described above were placed on 0.17 mm glass-bottomed eight-well culture plates (#1 Lab-Tek chambered glass) and studied with an Olympus 60× oil immersion 1.35 N.A. UPLSAPO microscope objective. Confocal imaging was performed on an Olympus IX81 inverted microscope stand equipped with a mercury lamp/Texas Red filter cube, Melles Griot 561 nm DPSS laser for excitation, and an Olympus Fluoview 1000 laser scanner/detection head. Reflected (scattered) and DyLight 594 fluorescence light were detected through an 80/20 neutral density beamsplitter, 560 nm dichroic, and empty/580 to 660 emission filters correspondingly. TPEF of DyLight 594 imaging was performed as described previously. Briefly, excitation light from a self-mode-locked Ti:Sapphire laser (Newport Spectra-Physics model DeepSee Maitai) tuned to 790 nm (for DyLight 594) entered the IR-port of Fluoview 1000 and was filtered out from fluorescence through a Chroma 680 nm dichroic mirror. TPEF was directed to the nondescanned detection pathway and split with Semrock 500 nm dichroic mirror in two channels employing red-sensitive side-on photomultiplier tube assemblies from LSM Technologies (Stewartstown, PA). Fluorescence was filtered with emission filters from Oriel (for DyLight 594) and signal digitized with an Olympus Fluoview 1000 expansion board. Both confocal and multiphoton tomographic image stacks were acquired with 0.25 μm steps in z-direction.
Results
Design of an Ex Vivo Microtumor Phantom
A BerEP4-expressing SW620 (colon adenocarcinoma) cell line was cultured on collagen microspheres measuring ≈ 60 to 200 microns in diameter, and these were used as BerEP4 microtumor phantoms. UACC257 (melanoma) cells lacking BerEP4 antigen (Ag) expression (based on flow cytometry data) were used as BerEP4-negative microtumor phantoms.
BerEP4 Ag was detected with a primary mouse monoclonal anti-BerEP4 and a secondary goat antimouse IgG, which was fluorescently labeled with dyes having excitation at either 594 nm or 750 nm (see Materials and Methods for details). We adopted the approach for this proof of principle study so that we could test more than one primary antibody easily and in a timely fashion. For future in vivo studies, we plan to use a primary Ab that is directly labeled with fluorescent dye with no need for secondary antibody.
Primary Ab plus secondary Ab labeled with 594 or 750 nm fluorophores is referred to as anti-BerEP4(594) and anti-BerEP4(750), respectively. Secondary Ab alone labeled with 594 or 750 nm fluorophores is referred to as secondary Ab(594) and secondary Ab(750).
BerEP4 Molecular Imaging of Microtumor Phantoms Using a TPEF/Confocal Microscope System
Images of BerEP4-positive microtumor phantoms labeled with anti-BerEP4(594), obtained in both two-photon (2p) and confocal (1p) microtomographic modes, showed bright signals from the cytoplasmic membrane of SW620 cells. Notably, the signals coming from the cytoplasmic membrane of SW620 cells were visually and qualitatively stronger than, and distinct from, the collagen microsphere background. This background is presumed to originate from nonspecific binding of anti-BerEP4(594) to the collagen of microbeads on which the cells were cultured. Maximum intensity projections of a Z-stack of tomographic images obtained with 2p and 1p microscopy confirmed that SW620 cells on the surface of collagen microbeads yield a signal stronger than (and distinct from) background signals emanating from the surface of the microbeads (Figure 3). When the above experiment was done using secondary Ab(594) only, neither 2p nor 1p images yielded significant emission from SW620 cells on collagen microspheres (Figure 4). The microbead itself could be visualized only because of random binding of the secondary Ab to the collagen on its surface. This implies that the BerEP4 Ab used in this experiment specifically binds to the surface of cultured BerEP4-expressing cells (SW620) on microtumor phantoms, making this antibody a candidate for in vivo imaging. To confirm the presence of SW620 cells labeled with secondary (nonspecific) Ab, which were not seen on 2p or 1p images, the same phantom was imaged with backscatter mode confocal microscopy (see Figure 3, B, E, F). Backscatter confocal images confirmed the presence of SW620 cells on the phantom despite the fact that they were not seen in fluorescent imaging as they lacked binding to secondary (nonspecific) Ab. This demonstrates the added value of backscatter confocal microscopy to fluorescent confocal/2p microscopy to show the histoanatomy of background tissue as well as nontargeted lesions.
Figure 3.
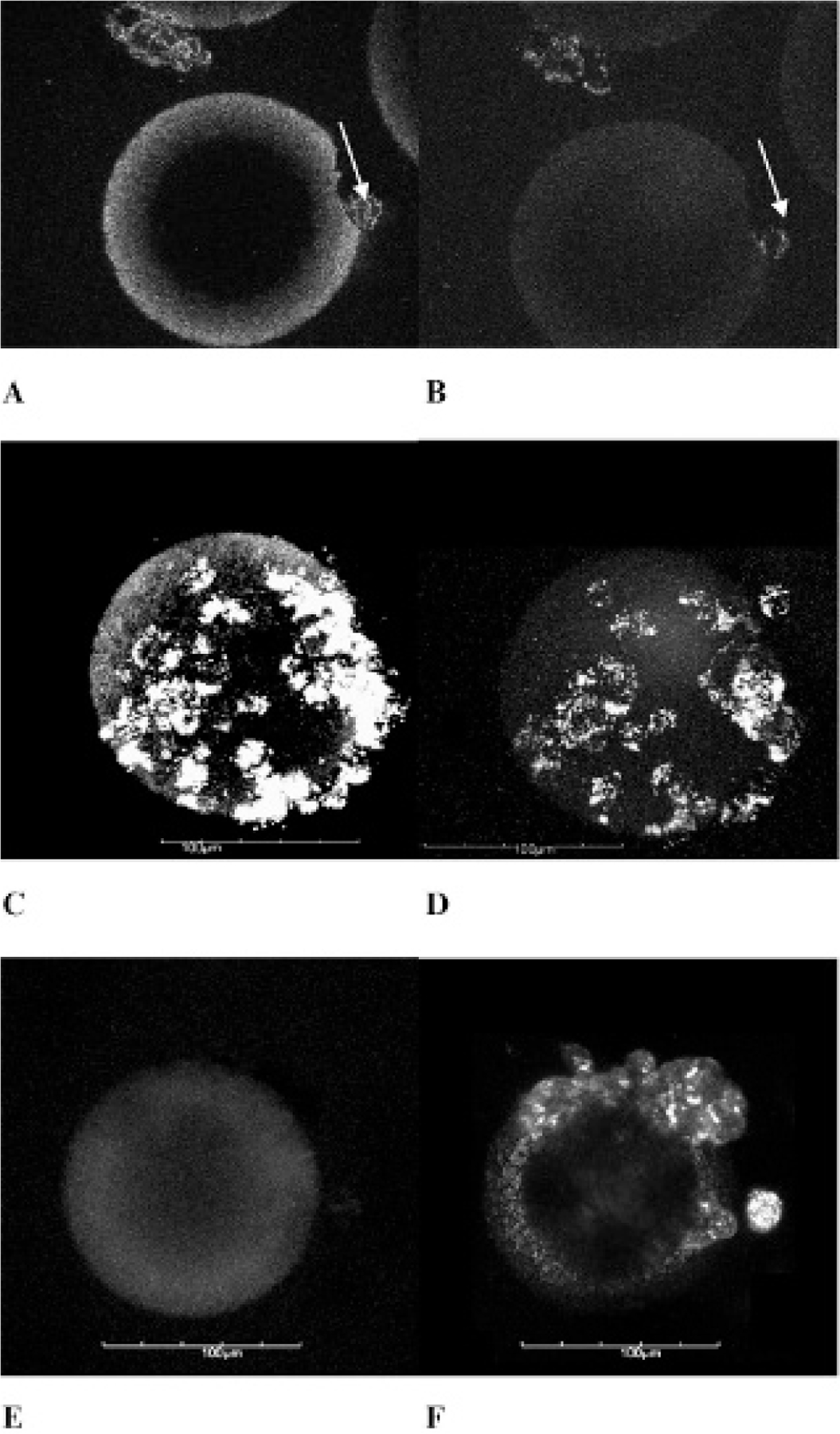
A, Colon cancer - SW620 – BerEP4-positive microtumor phantom labeled with anti-BerEP4(594) and imaged with 2p microtomography showing signals from the cell membrane (white arrow). B, 1p confocal image of the same tomographic section of the microtumor phantom shown in A. The 1p signals from the cell membranes (white arrow), although visually less intense than the 2p signals, are clearly distinct from background of the microbead. C, Maximum intensity projection of a z-stack of 2p images (sectioned at 0.5 μm steps) of the phantom shown in A as a single slice. Once again, 2p signals from the cell membrane show higher intensity compared to the background from collagen on microbeads. D, Maximum intensity projection of an image z-stack of the same phantom shown in B. E, 1p microscopy of colon cancer - SW620 – BerEP4-positive microtumor phantom labeled with secondary Ab(594) lacking signal from cells grown on a collagen microsphere. The microsphere itself is seen because of nonspecific binding of Ab to the collagen on the surface of the microsphere. F, Backscatter confocal image of the same phantom as in E showing that SW620 cells are present on the microsphere despite lack of signal on 1p images, confirming negative predictive value of selectivity of BerEP4 Ab for BerEP4 Ag. Scale bar 5 100 microns.
Figure 4.
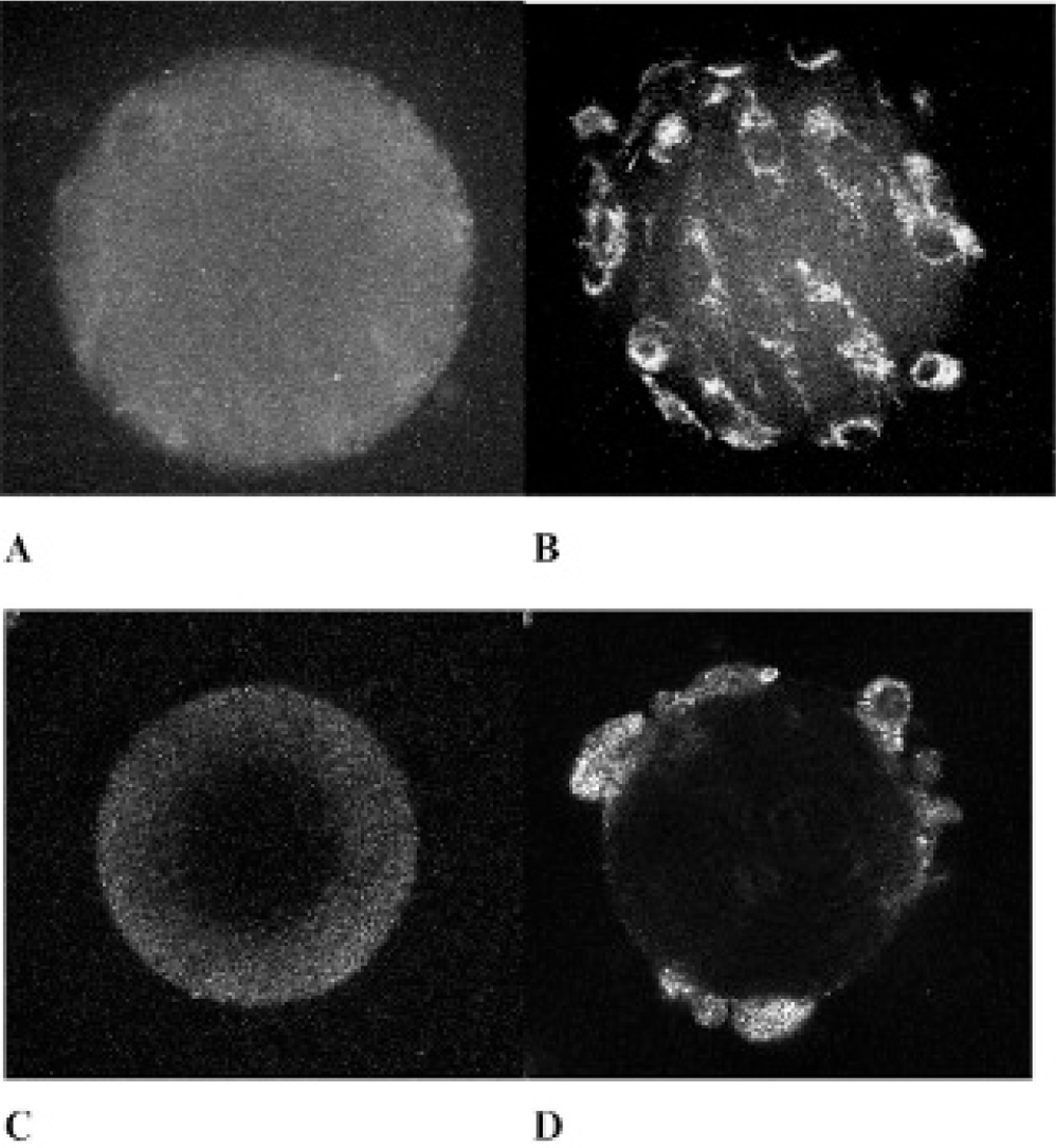
A, Maximum intensity projection of a 2p microtomo-graphic image z-stack of BerEP4-negative (UACC257 melanoma) microtumor phantom lacking signal from cells on microsphere labeled with anti-BerEP4(594). The microbead is seen because of nonspecific binding of fluorescent Ab to the collagen on the surface of the microsphere. B, Backscatter mode z-stack projection of the same phantom shown on A confirming the presence of UACC257cells on microbeads, once again confirming the negative predictive value of BerEP4 Ab as a molecular imaging probe. C, Tomographic image (section 50 at 0.5-micron section tomography) of A. D, Tomographic image (section 50 at 0.5-micron section tomography) of B. Scale bar 5 100 microns.
BerEP4-negative (UACC257 melanoma cell line) microtumor phantoms incubated with either anti-BerEP4(594) or secondary Ab(594) yielded only dim fluorescence from UACC257 cells (see Figure 4). This affirms the specificity and affinity of BerEP4 Ab for BerEP4 Ag. Once the same BerEP4-negative phantom was imaged in backscatter mode by confocal microscopy, the anatomic presence of UACC25 cells on the surface of the phantom was evident. The above finding bodes well for the specificity and affinity of our BerEP4 Ab to detect BerEP4 expression on the surface of cultured BerEP4-expressing cells
BerEP4 Molecular Imaging of Microtumor Phantoms Using a Photon-Counting Fluorescence Macrosystem
To investigate the ability of a time-resolved fluorescent macrosystem to detect signal from microtumor phantoms, we imaged two samples of 20 μL of SW620 cell pellet labeled with anti-BerEP4(750) and secondary Ab(750), respectively. The same experiment was repeated with a UACC257 cell pellet labeled with anti-BerEP4(750) as a BerEP4-negative control. All three samples were imaged with the time-resolved macrofluorescence imaging system. Compared to control experiments in Figure 5C, SW620 cell pellet labeled with secondary Ab(750), or Figure 5D, UACC257 cell pellet labeled with anti-BerEP4(750), the photon counts detected from SW620 cell pellet labeled with anti-BerEP4(750) (Figure 5B) in 2-second acquisition times were higher. The above results were reproduced using two different primary Abs (see Materials and Methods) on two separate occasions.
Figure 5.

A, Picture of 20 μL pellet of cells in PBS buffer deposited on a glass microscope slide. B, SW620 pellet labeled with anti-BerEP4(750). C, SW620 pellet labeled with secondary Ab(750). D, UACC257 pellet labeled with anti-BerEP4(750). Data in panels B to D were obtained with time-resolved fluorescence macroscopy (2-second signal acquisition time). The increase in fluorescence photon count rates detected by the macrosystem from sample B compared to that from samples C and D is clearly remarkable. Each color pixel represents an area of 2 × 2 mm2.
Discussion
This study is a proof of principle test for a specific biomarker, BerEP4, as a target for molecular imaging of BCC. We explored the feasibility of a macro-/micro-optical system to image the expression of BerEP4 on cultured cancer cells. (SW620, a BerEP4-expressing cell line, was used as a surrogate.) Our results imply that such a multiscale imaging system, providing the high resolution of micro-optics and the wide field advantages of macro–optics, will be well suited for molecular-based diagnosis of various histologic types of BCC. Furthermore, it may add an ability to monitor treatment.
Although there are other known BCC biomarkers, such as extracellular domain of Sonic hedgehog and PTCH receptor, they are reported to be expressed on only 10 to 60% of sporadic BCC.6 Based on the results herein, we recommend BerEP4 as a biomarker for molecular imaging of BCC. The relative lack of BerEP4 expression on skin-stratified squamous epithelium (keratinocytes), which should lead to a less troublesome background, also makes the BerEP4 an attractive molecular target for BCC.
Once a BerEP4-based diagnostic probe is developed and vetted, it might be further engineered for therapeutic application as a theranostic probe. One advantage of molecular probe–based approaches to dermatologic lesions is the ability to deliver the theranostic probe topically rather than systemic dosing, if it demonstrates optimal selectivity and clearance in vivo. Systemic application of such a theranostic probe can be further investigated for other BerEP4-expressing carcinomas, especially colorectal, pancreatic, and ovarian. When compared to keratinocytes, other epithelial tissues—such as columnar epithelium in colon—express BerEP4 but to a lesser degree than corresponding carcinomas.21
BCC cell lines are especially difficult to grow ex vivo. Even when achieved, the rate of growth for many lines is exceptionally slow, and they tend to differentiate, losing malignancy-related biomarkers. Because of its rapid rate of growth ex vivo, we selected the SW620 cell line as a BerEP4-expressing microtumor phantom. This allows for repeated experiments, which will help optimize our results while still being applicable to BCC. We based this assumption on our previously published study showing that all histologic variants of BCC highly express BerEP4. Moreover, we demonstrated that most skin lesions that come in the clinical differential diagnosis of BCC—especially in early stages—do not express BerEP4. It is important to note that for in vivo imaging, BerEP4 does not bind to the background normal epidermis and dermis.23 All of these findings, as well as the data presented in this article, speak to the likelihood that BerEP4 is a favorable target for in vivo molecular imaging of BCC, and the BerEP4 antibody presented here is a promising candidate to achieve this goal.
To assess the resolution and sensitivity of our imaging modalities, microspheres measuring from 60 to 200 microns in diameter were selected to simulate molecular signals from foci as small as micronodular BCC. In our initial attempt, when conventional microscopy with an NIR filter cube was applied, we observed that collective signals from background activity from nonspecific Ab binding to the collagen on the surface of microspheres obscured the signals emitted from the BerEP4 Ag/Ab complex (Figure 8). This suggested that a more sophisticated, tomographic system was needed to image molecular signals in BCC. When a 2p microscope is implemented in concert with total emission detection (TED) methodology, one can obtain stronger signals deeper than currently reported into skin and other scattering tissues.15,29 Additionally, both macro- and microsystems may rely on the same source of NIR excitation pulses. A multiscale optical system could enhance the precision and accuracy needed for diagnosing BCC and other skin cancers.
Based on our preliminary ex vivo phantom data, as detailed above, we recommend time-resolved macrofluorescent imaging and 2p-TED/confocal microscopy as systems of choice. We next plan to use BerEP4-expressing BCC mice models to reexamine the persistence of binding of BerEP4 Ab in vivo. These experiments will focus on discrimination of BerEP4 signals from background as washout progresses.
A unique opportunity in primary skin imaging is ease of access, permitting the design of a noninvasive, transcutaneous vehicle to deliver the theranostic probe. Skin has evolved to function as a barrier to protect us from loss of fluid and environmental harm. Nevertheless, our skin is susceptible to various permeation phenomena such as allergic contact dermatitis and viral diseases.30 We plan to exploit such phenomena to our advantage to investigate the preferred size, electrical charge, and chemistry of various nanovehicles for a noninvasive, transcutaneous delivery system. These would facilitate real-time diagnosis of BCC. Refined probes may allow simultaneous therapy and monitoring of treatment, thus improving the standard of care.
Looking beyond the skin, BerEP4 may be a screening and diagnostic molecular biomarker for carcinomas of other organs, especially the colon and pancreas. Given high specificity, systemic delivery of a BerEP4 Ab-based molecular probe may avoid the need for a vehicle for delivery.
Figure 1.
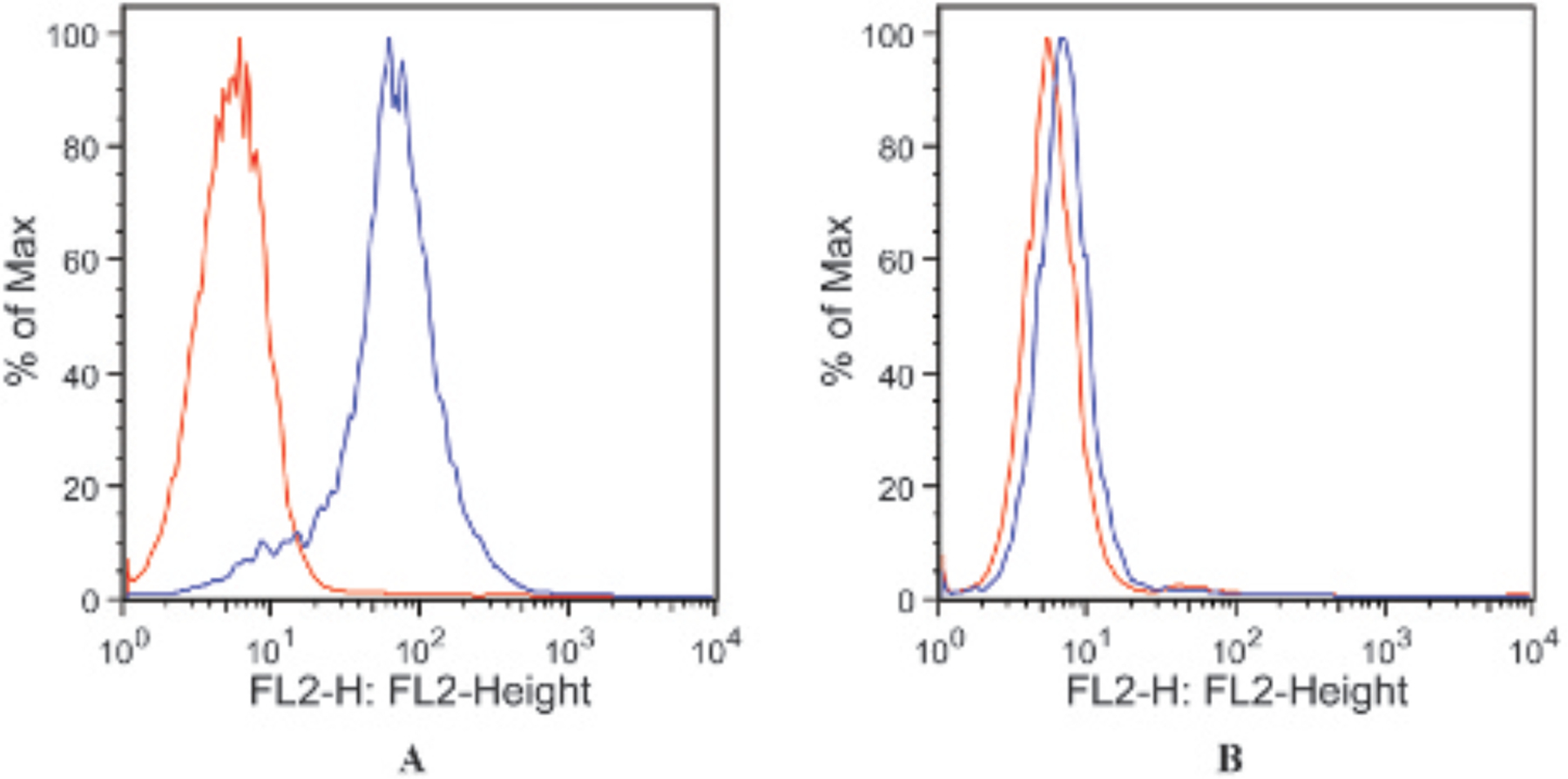
Flow cytometry results showing expression of BerEP4 antigen in the SW620 colon cancer cell line as positive control (A) and absence of BerEP4 antigen in the UACC257 melanoma cell line as the negative control (B). The blue line represents fluorophore-labeled BerEP4 antibody, and the red line represents background cell autofluorescence.
Figure 2.

A, Collagen microspheres (scale bar 5 100 microns). B, Melanoma - UACC257 – BerEP 4-negative micro-tumor phantom. C, Colon cancer - SW620 – BerEP4-positive microtumor phantom.
Figure 6.

A, BerEP4-expressing (SW620 colon adenocarcinoma) cells on collagen microspheres simulating a BerEP4-positive microtumor phantom imaged with a wide-field microscope. B, The same phantoms in A labeled with anti-BerEP4(750) and imaged with NIR wide-field microscopy only showing fluorescent signals from the nonspecific binding of primary probe to the collaged background on microspheres. C, Overlay picture of A and B clearly showing the seemingly negative cells on microbeads. This phenomenon is the result of a higher number of signals detected from a markedly larger volume of microsphere background—although nonspecific binding to collagen—compared to the number of specific signals emitted from a small volume of BerEP4-expressing cells on the microbeads labeled with anti-BerEP4(750). This imaging method would result in false-negative results, especially for small-size tumors. This further supports the need for a tomographic 2p microsystem to detect specific signals from small-volume point of interest of an imaged sample and discriminate them from nonspecific signals from a larger volume background. Scale bar 5 100 microns.
Acknowledgments
We would like to thank the Drug Screen Cell Bank of the National Cancer Institute, National Institutes of Health (NIH), for donating cancer cell lines as a public service. Also, we express appreciation to the Light Microscopy Core of the National Heart, Lung, and Blood Institute (NHLBI), NIH, for the use of the Olympus Fluoview 1000 Experimental Microscope and facilities. We are also grateful to Joan Liebmann-Smith, PhD, for her help in editing the manuscript. Bahar Dasgeb would like to express special thanks to Jane and Jack Zahrobsly for their generosity in providing logistical support.
Financial disclosure of authors: Support in funding and facilities was provided by the Intramural Research Programs of the Eunice Kennedy Shriver National Institute of Child Health and Human Development and the NHLBI.
Footnotes
Financial disclosure of reviewers: None reported.
References
- 1.Skin Cancer Foundation. Skin cancer facts. Available at: http://www.skincancer.org/skin-cancer-information/skin-cancer-facts (accessed April 21, 2013). [Google Scholar]
- 2.The Lewin Group, Inc. The burden of skin diseases 2005. Prepared for The Society for Investigative Dermatology and The American Academy of Dermatology Association; 2005. [Google Scholar]
- 3.Vestergaard ME, Macaskill P, Holt PE, et al. Dermoscopy compared with naked eye examination for the diagnosis of primary melanoma: a meta-analysis of studies performed in a clinical setting. Br J Dermatol 2008;159:669–76. [DOI] [PubMed] [Google Scholar]
- 4.Braun RP, Rabinovitz H, Tzu JE, et al. Dermoscopy research—an update. Semin Cutan Med Surg 2009;28:165–71, doi: 10.1016/j.sder.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 5.Kolatch J New type of ultrasound might substitute for some biopsies. The Washington Post 2010. Mar 30. Health Section, Page E1, Science Column. [Google Scholar]
- 6.LeBoit PE, International Agency for Research on Cancer, World Health Organization, et al. Pathology and genetics of skin tumours. World Health Organization classification of tumours. Lyon: IARC Press; 2006. [Google Scholar]
- 7.Caspers PJ, Lucassen GW, Puppels GJ. Combined in vivo confocal Raman spectroscopy and confocal microscopy of human skin. Biophys J 2003;85:572–80, doi: 10.1016/S0006-3495(03)74501-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Giorgi V, Massi D, Sestini S, et al. Combined non-linear laser imaging (two-photon excitation fluorescence microscopy, fluorescence lifetime imaging microscopy, multispectral multiphoton microscopy) in cutaneous tumours: first experiences. J Eur Acad Dermatol Venereol 2009;23:314–6, doi: 10.1111/j.1468-3083.2008.03045.x. [DOI] [PubMed] [Google Scholar]
- 9.Galletly NP, McGinty J, Dunsby C, et al. Fluorescence lifetime imaging distinguishes basal cell carcinoma from surrounding uninvolved skin. Br J Dermatol 2008;159:152–61, doi: 10.1111/j.1365-2133.2008.08577.x. [DOI] [PubMed] [Google Scholar]
- 10.Manfredini M, Arginelli F, Dunsby C, et al. High-resolution imaging of basal cell carcinoma: a comparison between multiphoton microscopy with fluorescence lifetime imaging and reflectance confocal microscopy. Skin Res Technol 2013;19:194–204, doi: 10.1111/j.1600-0846.2012.00661.x. [DOI] [PubMed] [Google Scholar]
- 11.Seidenari S, Arginelli F, Dunsby C, et al. Multiphoton laser tomography and fluorescence lifetime imaging of basal cell carcinoma: morphologic features for non-invasive diagnostics. Exp Dermatol 2012;21:831–6, doi: 10.1371/journal.pone.0070682. [DOI] [PubMed] [Google Scholar]
- 12.Cicchi R, Sestini S, De Giorgi V, et al. Nonlinear laser imaging of skin lesions. J Biophoton 2008;1:62–73, doi: 10.1002/jbio.200710003. [DOI] [PubMed] [Google Scholar]
- 13.Hassan M, Riley J, Chernomordik V, et al. Fluorescence lifetime imaging system for in vivo studies. Mol Imaging 2007;6:229–36. [PMC free article] [PubMed] [Google Scholar]
- 14.Hassan M, Chernomordik V, Zielinski R, et al. In vivo method to monitor changes in HER2 expression using near-infrared fluorescence imaging. Mol Imaging 2012;11:177–86. [PMC free article] [PubMed] [Google Scholar]
- 15.Combs CA, Smirnov A, Chess D, et al. Optimizing multiphoton fluorescence microscopy light collection from living tissue by noncontact total emission detection (epiTED). J Microsc 2011;241: 153–61, doi: 10.1111/j.1365-2818.2010.03411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alford R, Ogawa M, Hassan M, et al. Fluorescence lifetime imaging of activatable target specific molecular probes. Contrast Media Mol Imaging 2010;5:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ardeshirpour Y, Chernomordik V, Capala J, et al. Using in-vivo fluorescence imaging in personalized cancer diagnostics and therapy, an image and treat paradigm. Technol Cancer Res Treat 2011;10:549–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ardeshirpour Y, Chernomordik V, Zielinski R, et al. In vivo fluorescence lifetime imaging monitors binding of specific probes to cancer biomarkers. PLoS One 2012;7:e31881, doi: 10.1371/journal.pone.0031881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Linnenbach AJ, Seng BA, Wu SA, et al. Retroposition in a family of carcinoma-associated antigen genes. Mol Cell Biol 1993;13: 1507–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Litvinov SV, van Driel W, van Rhijn CM, et al. Expression of Ep-CAM in cervical squamous epithelia correlates with an increased proliferation and the disappearance of markers for terminal differentiation. Am J Pathol 1996;148:865–75. [PMC free article] [PubMed] [Google Scholar]
- 21.Balzar M, Winter MJ, de Boer CJ, et al. The biology of the 17–1A antigen (Ep-CAM). J Mol Med (Berl) 1999;77:699–712, doi: 10.1007/s001099900038. [DOI] [PubMed] [Google Scholar]
- 22.Winter MJ, Nagelkerken B, Mertens AE, et al. Expression of Ep-CAM shifts the state of cadherin-mediated adhesions from strong to weak. Exp Cell Res 2003;285:50–8, doi: 10.1016/S0014-4827(02)00045-9. [DOI] [PubMed] [Google Scholar]
- 23.Dasgeb B, Mohammadi TM, Mehregan DR. BerEP4 and epithelial specific antigen to differentiate clinical simulators of basal cell carcinoma. Biomark Cancer 2013;5:7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herlyn M, Steplewski Z, Herlyn D, et al. Colorectal carcinoma-specific antigen: detection by means of monoclonal antibodies. Proc Natl Acad Sci U S A 1979;76:1438–42, doi: 10.1073/pnas.76.3.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Went PT, Lugli A, Meier S, et al. Frequent EpCam protein expression in human carcinomas. Hum Pathol 2004;35:122–8, doi: 10.1016/j.humpath.2003.08.026. [DOI] [PubMed] [Google Scholar]
- 26.Schwartzberg LS. Clinical experience with edrecolomab: a monoclonal antibody therapy for colorectal carcinoma. Crit Rev Oncol Hematol 2001;40:17–24, doi: 10.1016/S1040-8428(01)00131-7. [DOI] [PubMed] [Google Scholar]
- 27.Naundorf S, Preithner S, Mayer P, et al. In vitro and in vivo activity of MT201, a fully human monoclonal antibody for pancarcinoma treatment. Int J Cancer 2002;100:101–10, doi: 10.1002/ijc.10443. [DOI] [PubMed] [Google Scholar]
- 28.Swanson PE, Fitzpatrick MM, Ritter JH, et al. Immunohistologic differential diagnosis of basal cell carcinoma, squamous cell carcinoma, and trichoepithelioma in small cutaneous biopsy specimens. J Cutan Pathol 1998;25:153–9, doi: 10.1111/j.1600-0560.1998.tb01708.x. [DOI] [PubMed] [Google Scholar]
- 29.Combs CA, Smirnov AV, Riley JD, et al. Optimization of multiphoton excitation microscopy by total emission detection using a parabolic light reflector. J Microsc 2007;228:330–7, doi: 10.1111/j.1365-2818.2007.01851.x. [DOI] [PubMed] [Google Scholar]
- 30.Barry BW. Novel mechanisms and devices to enable successful transdermal drug delivery. Eur J Pharm Sci 2001;14:101–14, doi: 10.1016/S0928-0987(01)00167-1. [DOI] [PubMed] [Google Scholar]