

Abstract
Current methods to detect post-translational modifications of proteins, such as phosphate groups, cannot measure single molecules or differentiate between closely spaced phosphorylation sites. We detect post-translational modifications at the single-molecule level on immunopeptide sequences with cancer-associated phosphate variants by controllably drawing the peptide through the sensing region of a nanopore. We discriminate peptide sequences with one or two closely spaced phosphates with 95% accuracy for individual reads of single molecules.
Post-translational modifications (PTMs) play crucial roles in protein function and cell fate. Most PTMs involve attachment of a small chemical group (phosphoryl, acetyl, glycosyl and so on) to amino acids, which greatly expands the proteome. Mass spectrometry is the principal technique to detect PTMs, but this method requires substantial sample input (typically >109 copies) and often struggles to identify the correct position of a PTM between multiple candidate sites1. Improved detection of protein phosphorylation, the most frequent PTM2, is of particular interest, as dysregulation of phosphorylation pathways is linked to many diseases including cancers, Parkinson’s, Alzheimer’s and heart disease3. Specifically, certain phosphorylation patterns on immunopeptides, which are naturally digested protein products on the cell surface for immune cell recognition, have been directly linked to cancer cells, making these immunopeptide variants promising neoantigens (cancer-specific antigens) for targeted immunotherapy or cancer screening4. Nanopore techniques, where the change in ion current is measured as a single molecule passes through a nanopore in a membrane, have shown promise for PTM detection5–13. However, these approaches, which measure brief transient blockades, have so far lacked high accuracy in variant identification for single molecules.
In this Brief Communication, we apply a recently introduced nanopore single-peptide scanning method14–16 to PTM detection and demonstrate its capabilities to detect and discriminate single phosphate groups within individual peptides. In this approach14, a peptide of interest (up to ~25 amino acids) is chemically linked to a DNA oligonucleotide, creating a peptide–oligonucleotide conjugate (POC) that is slowly translocated in a stepwise manner through a nanopore (MspA17) using a DNA motor enzyme (Hel308 helicase18), as in nanopore DNA sequencing19–22. Previously14, individual amino acid substitutions on single peptides were discriminated with high accuracy, but the peptide sequence tested was atypical, with a near-uniform negatively charged chain of aspartate and glutamate residues to induce electrophoretic insertion of the POC into the nanopore. To test biologically relevant peptides with various charges, we chemically linked a second DNA oligo (the ‘threading DNA’) to the other end of the peptide16 (Fig. 1a). This DNA electrophoretically threads the POC into and through the nanopore where it is subsequently pulled back out of the pore in ~0.3 nm steps by the helicase, slowly scanning the peptide across the narrow sensing constriction of the pore (Fig. 1b). Figure 1c depicts a typical ion current trace from a single translocation event of a POC containing a 10-amino-acid peptide. The first part of the trace reads the template DNA section that corresponds well with the predicted pattern from nanopore DNA sequencing22 (Fig. 1d), whereas the second part contains the linker and peptide signal of interest.
Fig. 1 |. Nanopore PTM detection experimental schematic and data workflow.
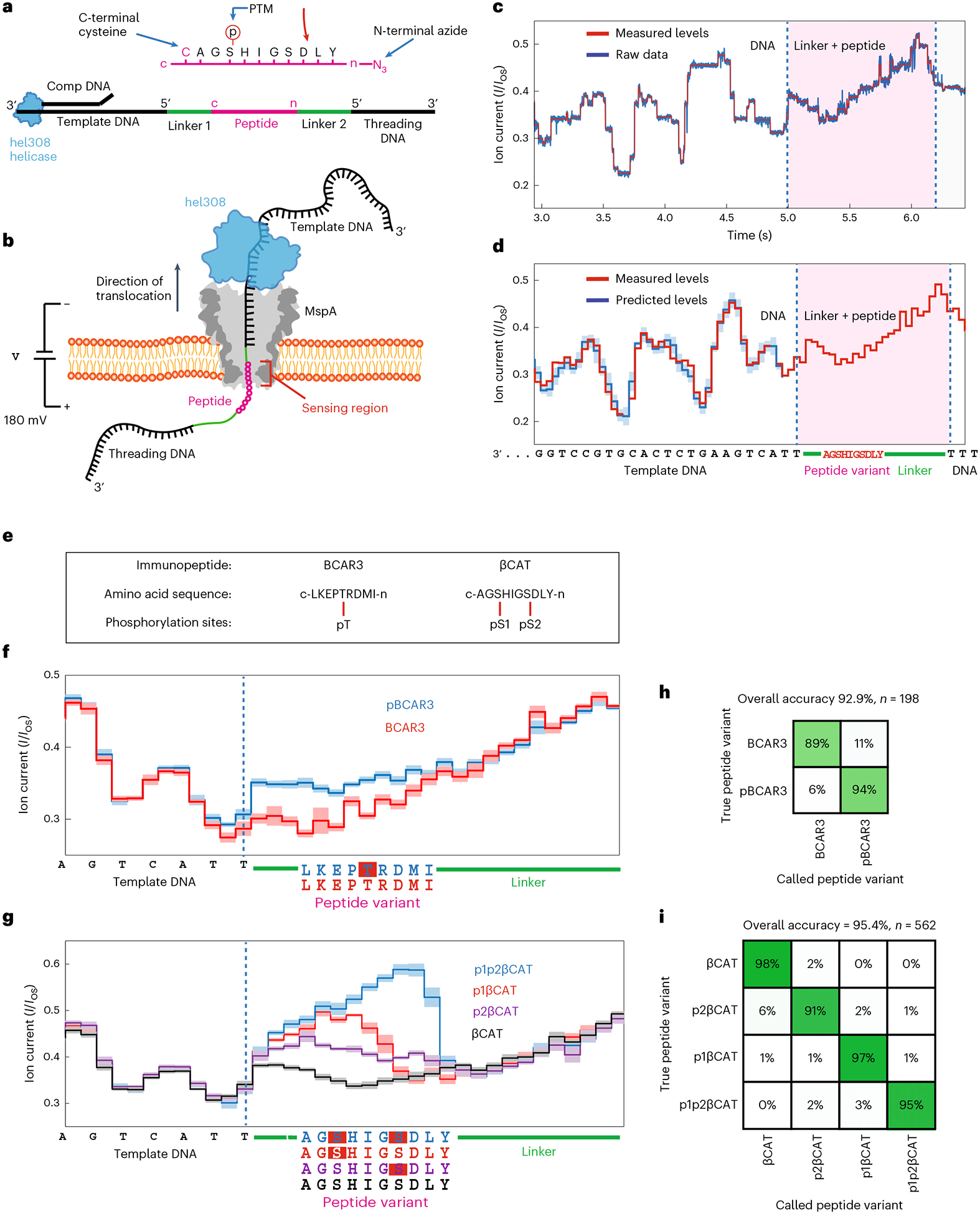
a, Schematic of the POC. A (phosphorylated) immunopeptide (pink) is linked by its C-terminus to the 5′ end of a DNA template (linker 1, cysteine–maleimide bond), while its N-terminus is linked to the 5′ end of the threading DNA (linker 2, azide-DBCO bond). Hel308 helicase loads onto the single-stranded DNA/double-stranded DNA junction made by a complementary oligo (comp DNA) that is annealed to the template DNA. b, Schematic of POC reading. An MspA nanopore (gray) is embedded in a lipid bilayer. Applied voltage (180 mV) drives a current of K+ and Cl− ions through the nanopore. The threading DNA is electrophoretically driven into and through the nanopore, translocating the POC, stripping off the comp DNA and docking the Hel308 onto the rim of MspA. As Hel308 steps along the template DNA, the POC is pulled up through the pore in ~0.33 nm increments, thereby pulling residues through the narrowest portion of MspA (sensing region) where they modulate the ion current. c, Ion current trace of a typical POC reading event for βCAT. Ion currents (I) are normalized to the unblocked open-state pore current (IOS). Measured levels (red) are determined using a data segmentation algorithm. After reading the template DNA, linker 1 enters the sensing region (at 5 s), followed by peptide, linker 2, and the start of the threading DNA. d, Consensus sequence of ion current steps (red), which in the DNA section is closely matched by the ion current levels predicted by the DNA sequence (blue). Error bars in the measured ion current levels are errors in the mean value, often too small to see. Error bars in the prediction are standard deviations of the ion current levels that were used to build the predictive map25. e, Immunopeptides with amino acid sequences and phosphorylation sites. BCAR3 contains a single phosphorylation site at a threonine residue (pT). βCAT contains two serine phosphorylation sites (termed pS1 and pS2) separated by three amino acids. Phosphopeptide variants studied were BCAR3, pBCAR3 (with pT), βCAT, p1βCAT (with pS1), p2βCAT (with pS2) and p1p2βCAT (with both pS1 and pS2). f, Consensus ion current patterns for BCAR3 and for the PTM variant pBCAR3 (data are the mean value with standard deviation for N = 40 reads for each trace). Dashed line marks the end of the template DNA in the sensing region. g, Consensus ion current patterns for βCAT and its phosphopeptide variants (data are the mean value with standard deviation for N = 40 reads for each trace). h, Single-read blinded-variant-calling matrix for BCAR3 variants yielding an overall variant-calling accuracy of 93%. i, Same for βCAT variants, yielding an overall variant-calling accuracy of 95%.
We found that this approach allows extremely sensitive measurements that can clearly distinguish peptides with or without a single PTM. We measured POCs containing the immunopeptide BCAR3 (Fig. 1e), a promising neoantigen for immunotherapy4. We compared BCAR3 (with sequence LKEPTRDMI, written C to N terminus) and its phosphate-PTM-containing variant pBCAR3 where a single threonine residue was phosphorylated (LKEP[pT]RDMI). Consensus ion current patterns were determined by aligning and averaging n = 40 reads of each variant (Fig. 1f). The addition of phosphothreonine (pT), a single small PTM of only five atoms, produced a dramatic change to the current pattern. Specifically, the pattern for the phosphate-containing variant was consistent with unphosphorylated BCAR3 until pT entered the sensing region, whereupon the current increased significantly by up to 25% for 13 steps, until the current returned to match for the rest of the remaining steps. These data clearly show that a single PTM can be well distinguished with even one nanopore read of a single molecule.
We next demonstrated the sensitivity of this method to discriminate between closely spaced PTMs along a peptide. We repeated the procedure to analyze another clinically relevant immunopeptide4, βCAT (AGSHIGSDLY), that contains two phosphorylation sites, one at each serine (termed pS1 and pS2), at positions separated by three amino acids (Fig. 1e). We determined the current patterns for the unphosphorylated variant, both single-phosphoserine (pS) variants (p1βCAT containing pS1, AG[pS]HIGSDLY; and p2βcat containing pS2, AGSHIG[pS] DLY), and the double pS variant (p1p2βCAT containing both pS1 and pS2, AG[pS]HIG[pS]DLY) (Fig. 1g). All four βCAT variants produced a distinct ion current pattern that could clearly be discriminated from that of the other variants. Just like for pT (Fig. 1f), the addition of pS had the consistent effect of increasing the current. Notably, the magnitude of the increase and the number of steps that were affected varied between the two single phosphorylation sites (9 steps for p1βCAT and 12 steps for p2βCAT). For the double phosphopeptide (p1p2βCAT), the two phosphoserines combined to increase the current even more than with the two single variants, reaching large current values that exceeded the nonphosphorylated variant by up to 64% for 12 steps.
These differences in ion current patterns can be used to accurately identify the correct variant for individual reads of these immunopeptides—as can be quantified in a so-called confusion matrix. For 198 single reads of BCAR3 and its variant, we blindly determined the correct variant using a hidden Markov model with an accuracy of 93% (Fig. 1h). For 562 reads of βCAT and its variants, we determined the correct variant with 95% accuracy, while individual variant-calling accuracies ranged between 91% (βCAT) and 98% (p2βCAT) (Fig. 1i). Overall, the single-read variant-calling accuracy was 95% for all of the measured phosphopeptides, highlighting the capabilities of this technique to reliably determine the correct PTM location on single molecules.
The heterogeneous charge profile of these peptides leads to variations in the POC polymer’s stretching as it is stepped through the pore. The constant k-mer reading frame19 that is commonly used in models of nanopore DNA sequencing is therefore inadequate to describe the influence of amino acid sequence on ion current patterns. We developed a physical model to better understand this behavior. For each of the four βCAT variants, we performed a Markov-chain Monte Carlo (MCMC) calculation (Methods), where the POC was modeled as a freely jointed chain with units of varying charge (Fig. 2a), anchored at the top of the MspA pore by Hel308, and subject to ion-screened Coulomb forces between charges as well as to the applied electrostatic potential (Fig. 2b). By performing the MCMC calculation with each βCAT variant at 30 consecutive Hel308 steps, we simulated the movement of the POCs through the nanopore. Figure 2c depicts typical configurations found for p2βCAT at a selection of Hel308 steps, while Fig. 2d plots the corresponding mean z-location (vertical axis along the pore) of the pS2 PTM, calculated for every step. We find that, after the template DNA is stepped through the sensing region, the linker/peptide polymer bunches within the pore, until the large negative charge (pS2) is held just below the nanopore constriction by the voltage drop. As stepping continues, the slack is gradually pulled out of the polymer and the phosphate is slowly pulled up into the pore constriction, reaching a critical point at which the charged phosphate quickly pops up into the pore vestibule and the polymer returns to a bunched slack configuration. This is illustrated in the trace of Fig. 2d where the pS2 PTM stalls at z ~ 8 nm, until it suddenly jumps up at step 19. While residing in the stalling position just below the pore, the negative phosphate group probably promotes the transit of K+ ions, thus increasing the nanopore current, as seen in the experimental data (Fig. 1g). The stalling and jumping behavior was consistently observed for all PTMs in all βCAT peptides (Fig. 2e).
Fig. 2 |. MCMC calculations of phosphate-containing peptides.
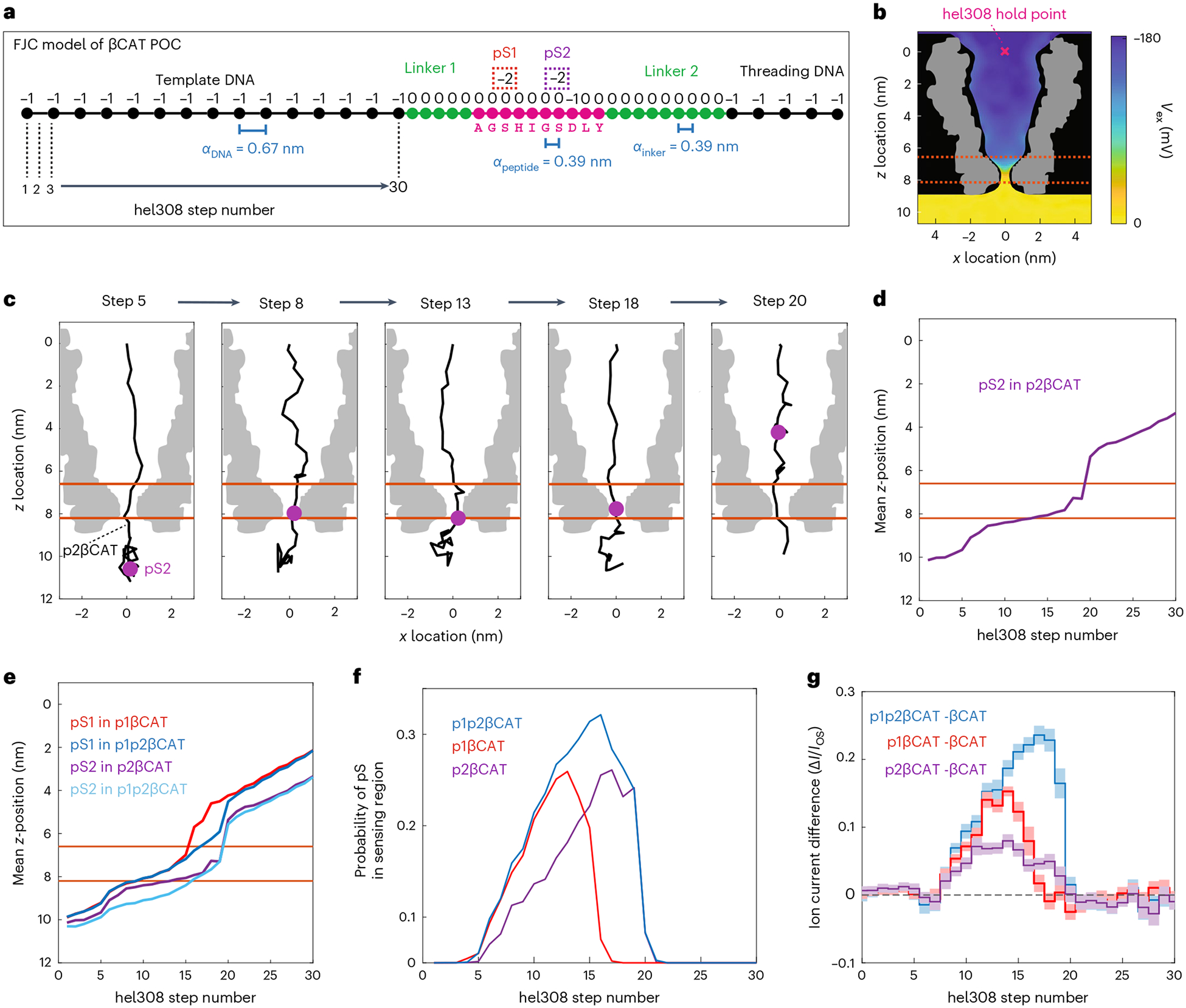
a, Freely jointed chain (FJC) model of a POC-containing peptide βCAT. Each unit in the polymer has an electrical charge and a typical distance α between residues. Phosphoserine PTMs pS1 and pS2 add a charge of −2 to that unit. b, Electric potential profile (color gradient) in the MspA pore26. The POC was confined within the physical boundaries of MspA (black) and anchored at the Hel308 hold point (pink x). The volumetric map of the MspA cross-section is shown in gray. c, Snapshots of the polymer configuration within MspA from MCMC calculations for the p2βCAT POC at five Hel308 steps. The phosphoserine residue (pS2) (purple) is observed to move through the pore in a nonlinear fashion. Note that the POC polymer gets stretched towards Hel308 step 19, after which the PTM moves into the pore lumen and the polymer relaxes. Orange lines indicate the sensing region of MspA. d, Mean z-location of the pS2 PTM versus Hel308 step number. e, Same for all PTMs in the peptide variants. f, Probability that a pS occupied the sensing region for various βCAT PTM variants versus Hel308 step. g, Experimentally measured ion currents for βCAT phosphopeptide variants where the ion current measured for the non-PTM βCAT was subtracted (from data in Fig. 1g). Shaded error bar is the standard deviation.
As a proxy for the ion current patterns, we extracted the percentage of time that a phosphate PTM was present in the sensing region (defined as 6.6 nm < z < 8.2 nm, Methods) at each step in the simulations (Fig. 2f). The results display an excellent correspondence with the experimental ion current differences (Fig. 2g), capturing the wider region of influence for pS2 in p2βCAT compared to pS1 in p1βCAT (12 steps versus 9 steps). In addition, the combined effect of pS1 and pS2 in p1p2βCAT influencing the same region as pS2 on its own in p2βCAT (12 total steps for both, Fig. 2g) is well represented by the model (Fig. 2f). Overall, this model provides a starting point for understanding how the charge distribution along peptides relates to ion current traces. In addition, this type of modeling provides a future pathway towards accurate PTM mapping, where the expected region of influence can be used to identify the amino acid location of a PTM along the peptide.
Our data provide demonstration of a technique that can accurately differentiate between single-molecule phosphopeptide variants by controllably drawing the peptide through the sensing region of a nanopore. The technique can clearly distinguish phosphopeptides with phosphates that are separated by only a few amino acids (three in our example), where mass spectrometry faces particular difficulties, and it does not require chemical labeling of the PTM as in other single-molecule proteomics methods23. Notably, nanopores were previously used to distinguish peptide variants with PTMs during free translocation5, but the method developed presently, which analyzes the changes in current patterns over many subsequent steps (Fig. 1), presents a greatly improved sensitivity and versatility, and is capable of detection of both PTMs and amino acid substitutions14.
While the accuracy that we realized was already very high (95%) in single reads, it can, if desired, be further improved upon using the rereading capability14 of our nanopore scanning approach, as is illustrated in Supplementary Figs. 11–13 and described in detail in Supplementary Text 2. This analysis revealed a residual variant-calling error due a finite synthesis purity of our test samples. The difficulty in assessing low error rates such as these (comparable to the percent impurity of the sample) also underscores the need for high-quality peptide and PTM standards as single-molecule peptide analysis tools become more accurate.
Detection of other PTM types (acetylation, hydroxylation, methylation and so on) can probably be achieved with an identical approach, as long as the PTM is not too bulky to translocate through MspA. It is of interest to note that the charge profile of the peptide sequences tested here was heterogeneous, and mixed-charge peptides did not impede the generation of well-reproducible current traces upon scanning these peptides through the pore. In addition to the anionic sequences tested in our previous work14, this demonstrates the versatility of this method for PTM or amino acid composition analysis on a wide variety of peptide sequences and charges. Highly cationic peptides are thus far untested and may pose additional challenges, but such sequences are rare within the proteome24.
Further method developments may involve increasing throughput using arrays of nanopores in parallel, developing robust methods to attach DNA to the N- and C-termini of peptides without the a priori modifications used here, and using this to move from using synthetic peptides to natural peptides collected from a biological sample. Engineering improvements could also be implemented to reduce the read-head size and stretch the peptide, using nanopore protein engineering to increase the pore height and minimize the sensing region, or adding charged residues to increase the electroosmotic forces within the pore25. Even before such next steps, this demonstration of single PTM detection within individual peptides presents a tool for phosphorylation investigation, enabling measurements currently unachievable with other proteomics tools.
Methods
Construction of POCs
Peptides (sequences in Supplementary Table 1) were purchased from Life Technologies and diluted to 10 μM in degassed PBS buffer. Phosphorylated amino acids were incorporated during solid-phase peptide synthesis. DNA oligos (sequences in Supplementary Table 2) were purchased from Biomers and diluted to 100 μM in degassed PBS buffer. Orthogonal click-chemistry reactions were used to attach a C-terminal cysteine on the peptide to a 5′ maleimide on the template DNA, and to attach a N-terminal azide on the peptide to a 5′ dibenzocyclooctyne group (DBCO) on the threading DNA.
The cysteine–maleimide reaction and the DBCO-azide copper-free click reaction were performed in one pot. Peptides and DNAs were mixed at a ratio of 1:2:6 (peptide:threading DNA:template DNA) at a concentration of 7 μM peptide in PBS and were incubated for 20 h at +4 °C under argon gas (50 μl reaction volume). Excess DNA was used to ensure the majority of peptide was conjugated. Supplementary Fig. 1 depicts the chemical structure of the full POC. The mixture was purified using DynaBeads strep-biotin polyA cleanup, ensuring that only constructs containing threading DNA (poly T) remained. The resulting product was estimated to be at a final concentration of 12.5 μM (20 μl elution volume) based on the maximum binding capacity of the beads. Comp DNA was added at a concentration of 15 μM and annealed to template DNA. The construction of the entire POC was verified using the nanopore measurements. Approximately 90% of the reads measured contained the entire POC construct. The other ~10% contained only template DNA, indicating that some template DNA without peptide remained after purification.
Nanopore experimental methods
Nanopore measurements were conducted as in Brinkerhoff et al.14 and previous studies17–22 with a few notable differences. DPhPC lipids purchased from Avanti were used to paint bilayers on ~10 μM Teflon apertures in custom U-tube experimental devices. MspA mutant M2-NNN17 was purified by Genscript. All experiments were conducted at 37 ± 1 °C with 1 mM ATP, 400 mM KCl, 10 mM MgCl2, 10 mM HEPES pH 8.00 ± 0.05 in the cis well and 400 mM KCl, 10 mM HEPES pH 8.00 ± 0.05 in the trans well. Hel308 was added to the cis well to a final concentration of 50 nM. POCs were added to a final concentration of 5 nM. Hel308 used in this study is from Thermococcus gammatolerans (accession number WP_015858487.1) and was cloned into the pET-28b(+) vector plasmid at Ndel/NotI sites by Genscript. Ion current data were acquired at 50 kHz sampling frequency using an Axopatch 200B patch clamp amplifier and filtered with 10 kHz 4-pole Bessel filter. Applied voltage was set to 180 mV for all experiments and controlled by a National Instruments X series DAQ and operated with custom LabVIEW software. Using these methods, many ion current reads (termed ‘events’) were gathered for each of the six POCs used in this study.
Data analysis
All data analysis was performed in MATLAB. Custom MATLAB software as described in detail in Brinkerhoff et al.14, and briefly below, was used for data preprocessing, reduction, filtering, alignment and variant identification:
Event selection and filtering.
Data were further Bessel filtered and decimated to 5 kHz, and potential events were identified using a thresholding algorithm based on the unblocked pore current as in previous work14,17–22. Events were then selected by eye by discrete selection criteria for further analysis. Occasionally, Hel308 fell off of the DNA before the end of the template strand. Therefore, we selected events that included steps for both template DNA and the entire peptide region into the second linker (for example events that fit selection criteria, see Supplementary Figs. 2–7).
Level finding and filtering.
To determine the transition points between Hel308 steps in the data, we used a change point algorithm as described in previous work17 and originally developed in Wiggins et al.27. A sample of the typical behavior of this change point ‘level finder’ across an entire event is shown in Supplementary Fig. 8. These measured levels were further filtered, first by excluding any levels outside the bounds of expected current values (I/IOS < 0.15 or I/IOS > 0.7). Levels outside of these bounds correspond to noise spikes or mid-event gating of MspA pore. We next applied a recombination filter, as described in Noakes et al.28, which identifies helicase backsteps and eliminates repeated levels from the trace. We delineated each event by eye by noting the position of the end of the DNA template in the measured levels, creating a DNA section (before this position) and peptide section (after this position, including both linker 1 and peptide and linker 2) for each event.
Reread removal.
In our previous study14, it was determined that, at high Hel308 concentration, a string of multiple Hel308 enzymes can be loaded onto the DNA template strand during translocation. After the first Hel308 reaches the end of the DNA template and dissociates, the POC falls back through the pore until the next Hel308 sits on the rim of the pore and continues stepping. This produces a ‘reread’ of the polymer, where the reread usually includes the final ~16 steps (equal to the footprint of one Hel 308 enzyme, 8 DNA bases). In the experiments presented in the main text, the rereads did not include the variable region within the peptide but merely included levels corresponding to linker 2 within the pore (Supplementary Figs. 9 and 10). However, rereading of the relevant region was enabled by using a different linker design (Supplementary Text 1). In the current study, rereads were removed for subsequent analysis. Supplementary Fig. 9 depicts a typical rereading pattern and sections of removal.
DNA level prediction.
The current pattern for the template DNA sequence was predicted using an empirically derived 6-mer map29, where each six-base sequence was given two ion current states, corresponding to the two substeps of Hel308 helicase (‘pre’ and ‘post’ steps) per DNA base. The ion currents in the map are the mean of the set of ion currents assigned to each state, and the uncertainty in the ion current is the standard deviation in that set of ion currents. The prediction matched well with the experimentally measured DNA levels in this study (Fig. 1d).
Initial consensus generation.
For the peptide section of the measured ion current, the ion current patterns had to be experimentally determined for each POC variant, as no predictive map exists for peptides or other polymers that are not DNA or RNA. To determine the ion current patterns for the linker and peptide regions of each POC (Fig. 1), we determined an initial ‘best guess’ of the ion current pattern. A selection of typical reads (n = 6–10) of each construct was compared by eye to determine the unique ion current states and place them in the appropriate order. This process eliminated Hel308 backsteps, repeated levels and spurious states that are not representative of typical reads. These initial consensuses included the last 15 steps of the DNA template section and 28 steps after the DNA template (where the helicase typically fell off of the DNA).
Reads were then calibrated, applying a scaling factor m to the measured ion current to account for slight variations in buffer salt concentration due to evaporation. Determination of the scaling factor was done as in Brinkerhoff et al.14, where the maximum likelihood estimator for m that limited the error between reads was calculated for each read. After applying the scales, a mean and standard deviation value was calculated for each position in the consensus. Next, a second round of calibration was applied to the mean consensus values in order to ensure cross-calibration consistency between consensus of the different POC variants. We calculated a scale and offset factor by performing a single-polynomial fit of the first 15 steps of each initial consensus (corresponding to the DNA section) to the last 15 levels of the predicted ion current pattern for the DNA template sequence. This ensured that all of the consensus patterns were calibrated to the same reference.
Final consensus generation.
These calibrated initial consensuses were then used as initial guesses for a customized Baum–Welch algorithm, a type of expectation maximization for the hidden Markov model. This algorithm was performed identically as in Brinkerhoff et al.14 and described fully in Noakes et al.28. We randomly chose 40 events of each POC variant for the EM algorithm. To calibrate these events, we performed a hidden-Markov-model alignment of the levels in the DNA section of each event to the template DNA prediction over a range of scale factors (m = 0.8 to 1.2 with increments of 0.01) and calculated a likelihood score for each m value. We chose the m value that produced the highest alignment score. We then applied this event specific scale factor to the associated measured levels from the peptide section of the same event. The expectation maximization algorithm was then used to generate a final consensus for each POC variant, using this set of calibrated events of each variant and the initial consensus as a seed for the algorithm. Using this procedure, we obtained six final consensus ion current patterns (one for each of the immunopeptide variants used in this study). Figure 1f,g depicts the final consensus ion current patterns for each immunopeptide variant.
Variant identification.
All filtered events that were not included in the initial or final consensus were used for variant identification. We calibrated each set of peptide levels using the DNA section alignment as previously. For each set of now calibrated peptide section levels, we performed a hidden Markov model alignment to the final consensus for each variant. Events containing βCAT and its associated variants were separated from BCAR3 and its associated variant and only aligned to the set of variants of the appropriate immunopeptide. The alignment producing the maximum alignment score was chosen as ‘called variant’ (Fig. 1h,i). Alignment accuracy for each variant was calculated as the percentage of correct calls compared to the total number of calls. The overall accuracy was calculated by calculating the percentage of correct calls for all variants divided by total calls.
Simulation methods
MCMC calculations26 were implemented in MATLAB. Degrees of freedom were encoded as polar and azimuthal angles between each polymer joint, with the first joint being fixed at the origin located at the top of MspA’s vestibule. Spacings between joints were chosen to be 0.67 nm for DNA and 0.39 nm for linker and peptide regions. Charges were assigned to each joint: +1e− for each K or R residue, −1e− for each D or E residue and for each DNA monomer, −2e− for each phosphorylated residue, and 0 for all other joints (Supplementary Fig. 14). The update distribution for both the polar and azimuthal angles was a normal distribution with mean 0 and standard deviation 0.05 radians.
The potential energy was calculated as the sum of (1) the interaction between the joint charges and a previously published electric potential map of MspA (Supplementary Fig. 15)30; (2) the Coulomb interaction between pairs of joint charges, screened with a Debye radius of 0.40 nm; and (3) a hard wall excluding any polymer joints from the wall of MspA, defined using a cylindrically symmetric spline derived from the electric potential map.
The MCMC calculation was performed at different ‘enzyme steps’ by removing monomers from the top part of the chain one by one, thereby shifting the entire sequence up through the constriction. At each step, the calculation started with a completely extended chain, with all polar and azimuthal angles set to 0, and the calculation was iterated 106 times. We discarded the first 104 samples at each step in order to allow for thermalization of the samples before inclusion in the calculated distributions (for a detailed description of the simulations, see Supplementary Text 2).
Figure 2f was produced by computing the fraction of samples in which a phosphorylation lay in the region where the z-component of the electric field along the z axis was greater than 1 kBT e−1 nm−1 per electron (6.6 nm < z < 8.2 nm, Supplementary Fig. 16) Figure 2d,e was produced by computing the mean position of the phosphorylated residue in the samples for each construct during each step.
Supplementary Material
Acknowledgements
We thank A. Laszlo for discussions on the MCMC calculations, J. van der Torre for help in troubleshooting POC construction, E. van der Sluis and A. Goutou for Hel308 purification, and A. Aksimentiev for discussions. The work was supported by funding from the Dutch Research Council (NWO) project NWO-I680 (SMPS) (C.D.); European Research Council Advanced Grant 883684 (C.D.); European Commission Marie Skłodowska-Curie Fellowship 897672 (H.B.); and NIH NHGRI project HG012544-01 (J.G. and C.D.).
Footnotes
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41587-023-01839-z.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Competing interests
H.B. and C.D. have filed a provisional patent for the nanopore peptide measurement method (NL patent N2024579 P1600131NL00). The remaining authors declare no competing interests.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41587-023-01839-z.
Data availability
The raw nanopore data for all of the reads used in this study are publicly available at https://doi.org/10.57760/sciencedb.0833831.
References
- 1.Kim MS, Zhong J & Pandey A Common errors in mass spectrometry-based analysis of post-translational modifications. Proteomics 16, 700–714 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khoury GA, Baliban RC & Floudas CA Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database. Sci. Rep 1, 1–5 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu H et al. ‘PTMD: a database of human disease-associated post-translational modifications’. Genomics Proteomics Bioinformatics 16, 244–251 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Engelhard VH et al. MHC-restricted phosphopeptide antigens: preclinical validation and first-in-humans clinical trial in participants with high-risk melanoma. J. Immunother. Cancer 8, e000262 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rosen CB et al. Single-molecule site-specific detection of protein phosphorylation with a nanopore. Nat. Biotechnol 32, 179–181 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Restrepo-Pérez L, Wong CH, Maglia G, Dekker C & Joo C Label-free detection of post-translational modifications with a nanopore. Nano Lett. 19, 7957–7964 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huo MZ, Hu ZL, Ying YL & Long YT Enhanced identification of tau acetylation and phosphorylation with an engineered aerolysin nanopore. Proteomics 22, 2100041 (2022). [DOI] [PubMed] [Google Scholar]
- 8.Li S et al. T232K/K238Q aerolysin nanopore for mapping adjacent phosphorylation sites of a single tau peptide. Small Methods 4, 2000014 (2020). [Google Scholar]
- 9.Wloka C et al. Label-free and real-time detection of protein ubiquitination with a biological nanopore. ACS Nano 11, 4387–4394 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shorkey SA, Du J, Pham R, Strieter ER & Chen M Real-time and label-free measurement of deubiquitinase activity with a MspA nanopore. ChemBioChem 22, 2688–2692 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nir I, Huttner D & Meller A Direct sensing and discrimination among ubiquitin and ubiquitin chains using solid-state nanopores. Biophys. J 108, 2340–2349 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fahie MA & Chen M Electrostatic interactions between OmpG nanopore and analyte protein surface can distinguish between glycosylated isoforms. J. Phys. Chem. B 119, 10198–10206 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Versloot RCA et al. Quantification of protein glycosylation using nanopores. Nano Lett. 22, 5357–5364 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brinkerhoff H, Kang AS, Liu J, Aksimentiev A & Dekker C Multiple rereads of single proteins at single–amino acid resolution using nanopores. Science 374, 1509–1513 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan S et al. Single molecule ratcheting motion of peptides in a Mycobacterium smegmatis porin A (MspA) nanopore. Nano Lett. 21, 6703–6710 (2021). [DOI] [PubMed] [Google Scholar]
- 16.Chen Z et al. Controlled movement of ssDNA conjugated peptide through Mycobacterium smegmatis porin A (MspA) nanopore by a helicase motor for peptide sequencing application. Chem. Sci 12, 15750–15756 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Butler TZ, Pavlenok M, Derrington IM, Niederweis M & Gundlach JH Single-molecule DNA detection with an engineered MspA protein nanopore. Proc. Natl Acad. Sci. USA 105, 20647–20652 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Derrington IM et al. Subangstrom single-molecule measurements of motor proteins using a nanopore. Nat. Biotechnol 33, 1073–1075 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manrao EA et al. Reading DNA at single-nucleotide resolution with a mutant MspA nanopore and phi29 DNA polymerase. Nat. Biotechnol 30, 349–353 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cherf GM et al. Automated forward and reverse ratcheting of DNA in a nanopore at 5-Å precision. Nat. Biotechnol 30, 344–348 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laszlo AH et al. Detection and mapping of 5-methylcytosine and 5-hydroxymethylcytosine with nanopore MspA. Proc. Natl Acad. Sci. USA 110, 18904–18909 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laszlo AH et al. Decoding long nanopore sequencing reads of natural DNA. Nat. Biotechnol 32, 829–833 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swaminathan J et al. Highly parallel single-molecule identification of proteins in zeptomole-scale mixtures. Nat. Biotechnol 36, 1076–1082 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Requião RD et al. Protein charge distribution in proteomes and its impact on translation. PLoS Comput. Biol 13, e1005549 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang G et al. Electro-osmotic capture and ionic discrimination of peptide and protein biomarkers with FraC nanopores. Nat. Commun 8, 935 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Metropolis N, Rosenbluth AW, Rosenbluth MN, Teller AH & Teller E Equation of state calculations by fast computing machines. J. Chem. Phys 21, 1087–1092 (1953). [Google Scholar]
- 27.Wiggins PA An information-based approach to change-point analysis with applications to biophysics and cell biology. Biophys. J 109, 346–354 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Craig JM et al. Determining the effects of DNA sequence on Hel308 helicase translocation along single-stranded DNA using nanopore tweezers. Nucleic Acids Res. 47, 2506–2513 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Noakes MT et al. Increasing the accuracy of nanopore DNA sequencing using a time-varying cross membrane voltage. Nat. Biotechnol 37, 651–656 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bhattacharya S, Yoo J & Aksimentiev A Water mediates recognition of DNA sequence via ionic current blockade in a biological nanopore. ACS Nano 10, 4644–4651 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nova IC Nanopore data traces for PTM detection on peptides. Science Data Bank 10.57760/sciencedb.08338 (2023). [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw nanopore data for all of the reads used in this study are publicly available at https://doi.org/10.57760/sciencedb.0833831.