

Abstract
Neurodevelopmental disorders (NDDs) are polygenic in nature and copy number variants (CNVs) are ideal candidates to study the nature of this polygenic risk. The disruption of striatal circuits is considered a central mechanism in NDDs. The 16p11.2 hemi-deletion (16p11.2 del) is one of the most common CNVs associated with NDD, and 16p11.2 del/+ mice show sex-specific striatum-related behavioral phenotypes. However, the critical genes among the 27 genes in the 16p11.2 region that underlie these phenotypes remain unknown. Previously, we applied a novel strategy to identify candidate genes associated with the sex-specific phenotypes of 16p11.2 del/+ mice and highlighted three significant genes within the deleted region: thousand and one amino acid protein kinase 2 (Taok2), seizure-related 6 homolog-like 2 (Sez6l2), and major vault protein (Mvp). Using the CRISPR/Cas9 technique, we generated mice carrying null mutations inTaok2, Sez6l2, and Mvp (3 gene hemi-deletion (3g del/+)). Hemi-deletion of these 3 genes recapitulates sex-specific behavioral alterations in striatum-dependent behavioral tasks observed in 16p11.2 del/+ mice, specifically male-specific hyperactivity and impaired motivation for reward seeking. Moreover, RNAseq analysis revealed that 3g del/+ mice exhibit gene expression changes in the striatum similar to 16p11.2 del/+ mice, but exclusively in males. Subsequent analysis identified translation dysregulation and/or extracellular signal-regulated kinase signaling as plausible molecular mechanisms underlying male-specific, striatum-dependent behavioral alterations. Interestingly, ribosomal profiling supported the notion of translation dysregulation in both 3g del/+ and 16p11.2 del/+ male mice. However, mice carrying a 4-gene deletion (with an additional deletion of Mapk3) exhibited fewer phenotypic similarities with 16p11.2 del/+ mice. Together, the mutation of 3 genes within the 16p11.2 region phenocopies striatal sex-specific phenotypes of 16p11.2 del/+ mice. These results support the importance of a polygenic approach to study NDDs and underscore that the effects of the large genetic deletions result from complex interactions between multiple candidate genes.
INTRODUCTION
Neurodevelopmental disorders (NDDs) are typically multi-factorial in origin and have heritable polygenic traits1–3. Although various studies have identified genetic variants associated with NDDs, it is still unclear how sets of genes interact with each other to impact the function of distinct neuronal circuits, ultimately resulting in behavioral phenotypes. Copy number variants (CNVs), which exhibit high effect sizes and contain multiple genes4–6, are ideal candidates to explore the polygenic mechanisms underlying NDDs. Although it is possible to study a single genetic variant at a time, currently there is no established strategy to identify and examine the effects of a group of candidate genes on distinct phenotypes.
16p11.2 hemi-deletion (16p11.2 del), one of the most common CNVs associated with NDDs7, 8, mediates several NDD-relevant phenotypes in mice, including reward processing and motor activity via alterations in striatal circuits8–13. Striatal dysfunction has also been observed in patients across various NDDs such as autism spectrum disorders (ASD) and attention deficit hyperactivity disorder (ADHD)14–25. In human carriers, 16p11.2 hemi-deletion has shown to be associated with ASD, ADHD and DCD, which, in turn, affect males disproportionately more than females8, 26–30. Consistent with this, female sex is a protective factor for 16p11.2 del carriers13. Noteworthy, mice modeling 16p11.2 hemi-deletion (16p11.2 del/+ mice) show several sex-specific phenotypes9, 11, 31, including male-specific deficits in behaviors dependent on striatal circuits, such as reward processing and motivation10. Male-specific alterations in peristriatal fiber tracts found in 16p11.2 del/+ mice could be a neuroanatomical correlate of these behavioral changes32. Thus, 16p11.2 del/+ mice provide a promising model to study male-specific striatal vulnerability; however, little is known about the genetic mechanisms underlying the sex-specific male vulnerability or female resilience of 16p11.2 del mice.
One particularly puzzling riddle is which of the 27 genes in the deletion contribute to these distinct sex-specific phenotypes. Single gene studies have partially replicated 16p11.2 del phenotypes33–39. However, these approaches are unable to examine synergistic combinatorial effects of multiple genes. Recent work underscores the idea that multiple genes within the 16p11.2 region may exhibit significant polygenic influences more broadly in ASD, thus establishing the 16p11.2 region as a source of both common and rare genetic variation40. One major obstacle for the identification of synergistic polygenic effects is the lack of established strategies to identify and select candidate genes. Here, we build upon a previous study from our lab that used a novel strategy to identify candidate genes based on their spatial expression profiles that were linked to structural alterations in 16p11.2 del/+ mice32. We selected a set of genes whose expression correlated with male-specific structural changes in striatal circuits in 16p11.2 del/+ mice to determine if haploinsufficiency of this set of genes might drive the striatum-related sex-specific behavioral phenotypes of 16p11.2 del/+ mice. These genes are thousand and one amino acid protein kinase 2 (Taok2), seizure-related 6 homolog-like 2 (Sez6l2), and major vault protein (Mvp). Previous studies have indicated the relevance of each of these three genes for neuronal development34, 35, 41 42–45, however, sex-specific behavioral or molecular phenotypes have not been reported in mice carrying deletions of any of these single candidate genes34, 36, 39, 46.
Considering the polygenic nature of NDDs, we hypothesized that the combination of mutations in these three candidate genes would give rise to the sex-specific phenotypes observed in 16p11.2 del/+ mice. Mice carrying mutations in all three genes cannot be produced by crossing existing single gene KO mice because of the close proximity of these genes in the genome. Therefore, we used the CRISPR/Cas9 system to develop a novel mouse model with the simultaneous hemi-deletion of these 3 candidate genes and found that the 3 gene mutations led to sex-specific changes in behavior, specifically hyperactivity and reduced motivation for reward in males. We also found that these mutations led to similar changes in gene expression in the striatum as 16p11.2 del/+ mice in males. We also tested whether Mapk3 plays an important role in male specific phenotypes with 3 genes, however, interestingly 4 gene hemi-deletion (Taok2, Sez6l2, Mvp, and Mapk3) displays fewer phenotypic similarities to 16p11.2 del/+ mice than 3g del/+ mice. These results suggest that the ‘combinatorial effects of genes’ are different from the ‘sum of single gene effects’. Together, this study emphasizes the significance of a polygenic approach in studying NDDs and offers fresh insights into the effects of gene combinations on NDD-relevant behavior.
METHODS AND MATERIALS
Animals
All procedures were approved by the University of Iowa Institutional Animal Care and Use Committees (IACUC), and we followed policies set forth by the National Institutes of Health Guide for the care and use of laboratory animals. Mice with a selective hemi-deletion (3g del/+ mice) were generated at the Genome Editing Core at University of Iowa using CRISPR/Cas9-induced gene modifications. 16p11.2 del/+ mice were purchased from The Jackson Lab (#013128). For 4g del/+ mice, we crossed 3g del/+ mice with Mapk3 hemi-deletion (Erk1 +/−) mice (bred from ERK1 KO mice (B6.129(Cg)-Mapk3tm1Gela/J), Jackson Lab #019113). Male and female littermate animals (10–14-week-old) during the same time period were used in all experiments unless otherwise specified.
Detailed methods and materials involving animals, behavioral tests, MRI imaging, biochemical experiments, and analyses are provided in supplementary materials.
Generation of 3g del/+ mice
C57BL/6J male mice were bred with super-ovulated females to produce zygotes for electroporation. Female ICR mice were used as recipients for embryo transfer. Cas9 ribonucleoprotein complexes (RNPs) and the electroporation mix were prepared. Pronuclear-stage embryos were collected using methods previously described47.
Behavioral tasks
Activity monitoring
Mice locomotor activity was measured by activity monitoring using an infrared beam-break system (Opto M3, Columbus Instruments) as previously described9, 48.
Operant task
Mice were allowed to acclimate to a 0900–2100 hours reversed light cycle one week prior to obtain food-restricted weights of 85–90% of free-feeding weights. Chocolate flavored Ensure Original Nutrition Shake (Abbott) diluted to 50% with water was used as a reinforcer. Fixed ratio (FR) responses per day for each animal and progressive ratio (PR) responses were recorded and analyzed using a repeated measures ANOVA and unpaired t-tests, as previously described10.
RNA quantification in striatal samples
RNA isolation was conducted as previously described10. The striatum was dissected bilaterally using a mouse brain matrix. The tissue was stored in RNAlater, then RNA was extracted using Qiagen RNeasy kit (Qiagen).
RNA-seq
Library preparation
RNA quality was assessed and samples with RNA integrity number (RIN) >8 were used to perform RNA-seq. RNA library preparation from WT mice (4 males and 4 females) and 3g del/+ mice (4 males and 4 females) for 3g del/+ mice and WT mice (4 males and 4 females) and 16p11.2 del/+ mice (4 males and 4 females) for 16p11.2 del/+ mice were prepared using the Illumina TruSeq Stranded Total RNA with Ribo-Zero gold sample preparation kit (Illumina, Inc., San Diego, CA). Pooled libraries were sequenced on Illumina NovaSeq 6000 sequencers with 100-bp Paired-End chemistry (Illumina). The dataset is available in the NCBI’s Gene Expression Omnibus repository, GEO Series accession GSE224750.
RNA-seq analysis
Sequencing data was processed with the bcbio-nextgen pipeline (https://github.com/bcbio/bcbio-nextgen). All further analyses were performed using R (version 4.1.2). Analysis code available through Github at https://github.com/YannVRB/16p11.2-del-and-3gKO-bulk-RNA-seq.git
Acquisition and analysis of diffusion-weighed MRI data sets
Image acquisition
Male and female mice at an age of PD 42–47 (6-week-old), 70-day-old (10-week-old), or 114–118-day-old (16-week-old) were imaged using a GE/Agilent Discovery 901 7-Telsa pre-clinical scanner. MRI imaging acquisition consisted of a 32-direction diffusion tensor imaging (DTI) scan.
Image preprocessing and TBSS analysis
Diffusion-weighed images were converted from DICOM to NIFTI format using DCM2NIIX and controlled for quality. We generated fractional anisotropy (FA) data set. Voxel-wise statistical analysis was performed on the FA data using a version of Tract-Based Spatial Statistics (TBSS), as previously described32.
RESULTS
Generation of 3 gene hemi-deletion mice
We generated mice with a simultaneous hemi-mutation in each of the 3 candidate genes (Taok2, Sez6l2, and Mvp) using CRISPR/Cas9-induced gene modifications to introduce a premature stop codon in the coding region of each gene (Figure 1A and B). The targeting modification was designed to activate nonsense-mediated mRNA decay (NMD) for specific degradation of transcripts of each of the genes. For Taok2, a 10 bp deletion and inversion of part of the 9 bp deleted sequence were introduced in exon 2, creating a premature stop codon in exon 4 (Figure 1C and S1A). We introduced a 17 bp deletion in exon 2 of Sez6l2 to generate a frame shift mutation causing a premature stop codon in exon 3 (Figure 1D and S1B). We made a 1 bp insertion in exon 2 of the Mvp gene to generate a premature stop codon in exon 2 (Figure 1E and S1C). Sanger DNA sequencing was performed to confirm the modification in several brain regions (cortex, cerebellum, and striatum) and peripheral tissues (ear and tails). The results verified a successful modification of the candidate genes. To validate the impact of these modifications on expression levels, we analyzed mRNA levels of the target genes (Taok2, Sez6l2, and Mvp) and nearby genes (Hirip3, Tmem219, Asphd1, Cdipt, and Prrt2) in the striatum using qPCR. In addition, TAOK2, SEZ6L2, and MVP protein expression levels were tested. These results confirmed that the gene modifications decreased mRNA levels and protein levels of each of the 3 target genes but did not alter the expression of nearby genes (Figure 1F–K and S2). No significant deaths of 3g del/+ pups were found (Figure S3A). Interestingly, we found a reduction in body weight in male 3g del/+ mice compared to sex- and age-matched wildtype (wt) mice at the age of 7 weeks (Figure S3B), resembling 16p11.2 del/+ male mice that show decreased body weight compared to wt mice11, 49, 50. However, the weight change was not found in female 3g del/+ mice (Figure S3B), unlike 16p11.2 del/+ female mice, which have lower body weight than wt female mice11.
Figure 1.
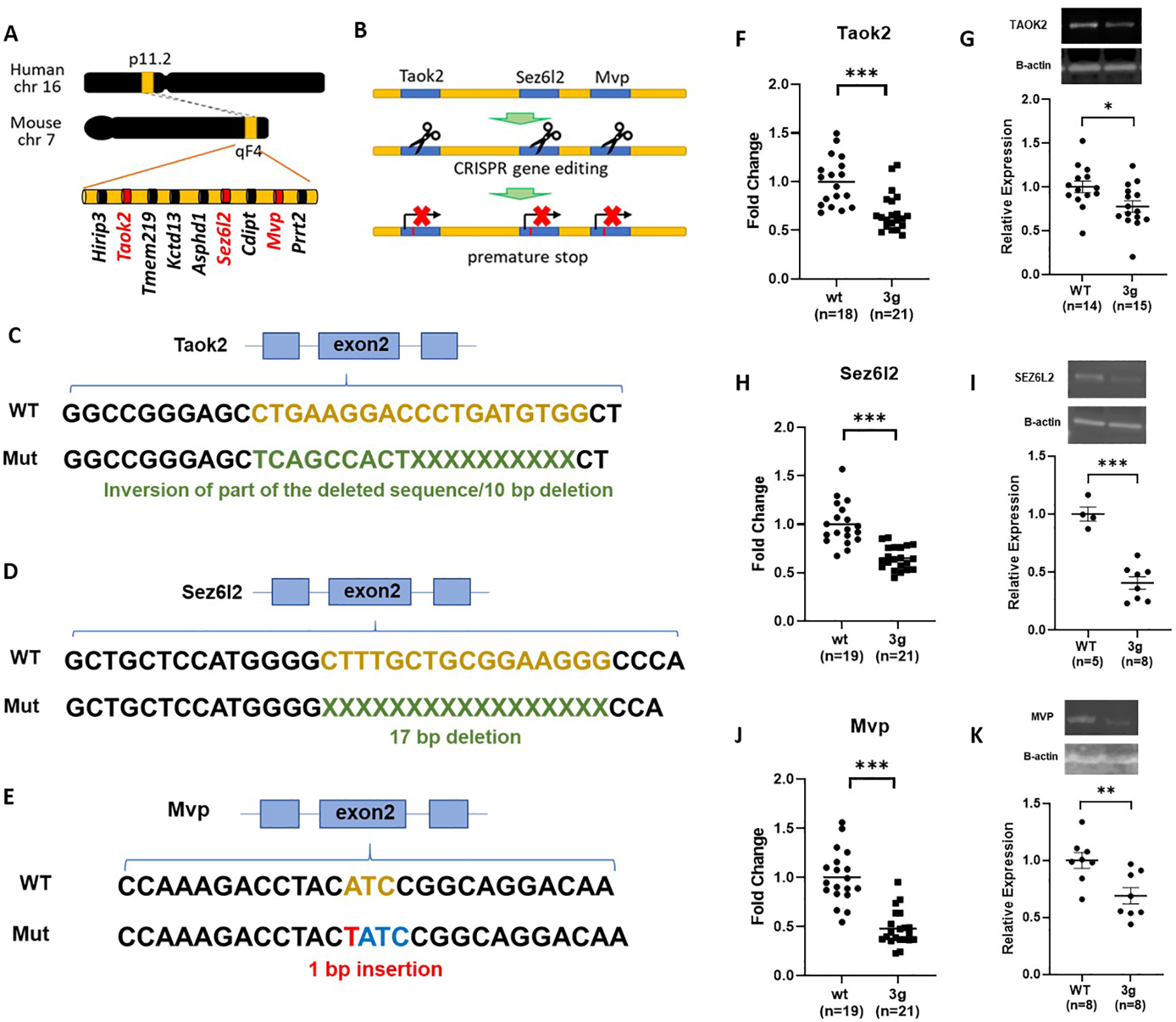
Generation of 3g del/+ mice. (A) Schematic diagram shows the target sites of mutations, mouse chromosome 7qF4. (B) A schematic presentation of the generation of 3 gene hemi-deletion mice by CRIPSR/Cas9. (C-E) Schematic presentations indicate the strategies of modifications. (F-K) qPCR (F,H,J) and western blot (G,I,K) results demonstrated decreased gene expressions and protein expressions of the 3 genes in the mutant mice (two-tailed Student t test, * p < 0.05, ** p < 0.01, *** p < 0.001).
3 gene hemi-deletion mice recapitulate male-specific striatum-dependent behavioral alterations observed in 16p11.2 del/+ mice
We first focused on behaviors mediated by striatal circuits in the 3g del/+ mouse model, as the three candidate genes were chosen based on the overlap between their expression patterns and structural alterations in striatal circuits of 16p11.2 del/+ mice32. We investigated whether 3g del/+ mice exhibit hyperactive behavior and deficits in reward learning and motivation as observed in 16p11.2 del/+ mice9, 10. First, we examined locomotor behavior in 3g del/+ mice using activity monitoring in the home-cage across the 24-hour cycle, as in previous experiments with 16p11.2 del/+ mice9. Interestingly, male 3g del/+ mice displayed increased locomotor activity (Figure 2A), but female mice did not (Figure 2B). When activity was analyzed across the light/dark cycle, we found significantly increased activity in the dark (active) phase (Figure 2C) for male 3g del/+ mice, but not female 3g del/+ mice (Figure 2D). Although female 3g del/+ mice show a trend of hyperactive behavior with a significant behavioral difference during certain time periods, they did not fully recapitulate the hyperactivity observed in 16p11.2 del/+ females9. These results point to the significance of the 3 candidate genes in the regulation of locomotor behavior regulation in male mice, phenocopying hyperactive behaviors of male 16p11.2 del/+ mice. In addition, motor coordination and balance of 3g del/+ mice was evaluated in the rotarod test in which 16p11.2 del/+ mice exhibit enhanced learning. Both male and female 3g del/+ mice showed similar performance to wt mice (Figure S4), suggesting that hyperactivity in male 3g del/+ mice is not linked to alterations in motor performance, coordination, or learning.
Figure 2.
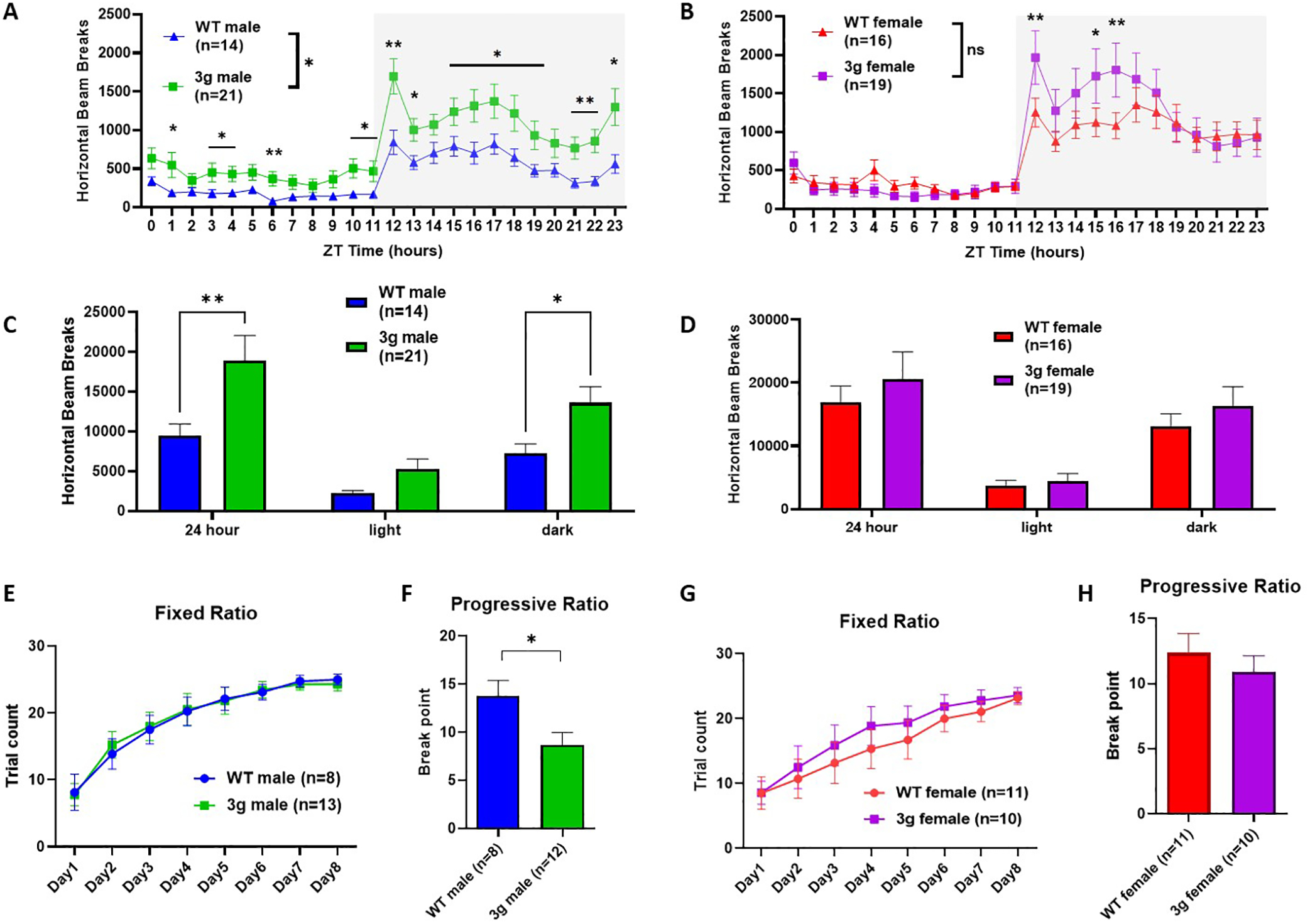
3g del/+ mice reproduced sex-specific changes in striatal behavior. (A, B) Infrared beam breaks in the horizontal axis are plotted for male and female 3g del/+ mice and wt mice across the 24-hr day with 1-hr bin. Gray box indicates the dark cycle. Male 3g mice have significantly greater activity relative to wt littermates (main effect of genotype, F (1, 33) = 5.373, p = 0.0268; main effect of time, F (4.368, 144.2) = 29.41, p < 0.001; genotype x time interaction, F (23, 759) = 2.249, p < 0.001), but not female 3g del/+ mice (main effect of genotype, F (1, 33) = 0.2537, p = 0.6178; main effect of time, F (23, 759) = 36.19, p < 0.001; genotype x time interaction, F (23, 759) = 2.791, p < 0.001). (C, D) Infrared beam breaks are plotted by light/dark cycle. Male 3g mice show hyperactive behavior relative to wt littermates in dark cycle (main effect of genotype, F (1, 99) = 13.60, p = 0.0004; post hoc, p = 0.034). Female 3g del/+ mice have no differences in horizontal activity (main effect of genotype, F (1, 99) = 1.256, p = 0.2650). (E, G) Both male and female 3g del/+ mice acquire a nosepoke response under a fixed ratio schedule of reinforcement indistinguishable from wt mice (male, main effect of genotype, F (1, 19) = 0.002, p = 0.968; female, main effect of genotype, F (1, 19) = 0.3530, p = 0.559). (F, H) In a progressive ratio test of motivation, male 3g del/+ mice responses after significantly fewer trials than wt males (t(18) = 2.43, p = 0.026), while female 3g del/+ mice show no different responds than wt mice (t(19) = 0.750, p = 0.463). Values are displayed as mean (±SEM) and significance values are set at *p<0.05 and **p<0.01.
Next, we investigated the performance of 3g del/+ mice in an operant reward learning task. After mild food restriction, 3g del/+ and wt mice were trained on a fixed-ratio task (FR) in mouse operant chambers. The 3g del/+ mice and wt mice showed similar weight changes after food restriction regardless of sex (Figure S5). In the FR task, both male and female 3g del/+ mice did not show differences in reward learning compared to wt mice (Figure 2E and G), suggesting that the learning of reward-stimuli association is unaltered. A week after FR training, we tested motivation to obtain a reward using a progressive ratio schedule (PR). Interestingly, male 3g del/+ mice showed a decreased break point for the PR compared to male wt mice (Figure 2F), suggesting diminished motivation to work for reinforcement. However, female 3g del/+ mice showed no difference compared to wt females (Figure 2H). These results reveal that only male 3g del/+ mice exhibit reduced motivation to work for reinforcement. This finding is consistent with 16p11.2 del/+ mice (Figure S6), which show a male-specific impairment in motivation10. In sum, the data from our behavioral characterization suggest that the selective 3 gene hemi-deletion phenocopies distinct male-specific behavioral phenotypes that critically depend on striatal circuits. We examined other behavioral phenotypes of the 3g del/+ mice including the open field task and social approach task, revealing male-specific reduced anxiety-like behavior and normal sociability of 3g del/+ mice (Figure S7).
3 gene hemi-deletion males engage transcriptional patterns in the striatum consistent with 16p11.2 del/+ males
Patterns of gene expression shape neuronal structure and function, which eventually affect the activity of neural circuits that mediate complex behaviors, including cognition, affective processing, and addiction51–53. To understand the mechanisms underlying the behavioral phenotypes, we compared gene expression between male and female 3g del/+ and wt mice using RNA-seq. Our analysis revealed 817 genes differentially expressed between 3g del/+ and wt males in the striatum, and 346 genes differentially expressed between 3g del/+ and wt females in the striatum (Figure 3A, Table S1 and S2). Only 36 differentially expressed genes (DEGs) were shared between male and female 3g del/+ mice (Figure 3A, Table S5), suggesting that distinct transcriptomic changes in the striatum between male and female 3g del/+ mice may underlie the sex-specific alterations in behaviors mediated by striatal circuits. Next, we compared the transcriptomic signatures of the 3g hemi-deletion mice to the16p11.2 deletion. We performed RNA-seq from the striatum of 16p11.2 del/+ mice and littermate controls. We found 881 DEGs between male 16p11.2 del/+ mice, and 127 DEGs in 16p11.2 females with 42 overlapping DEGs, both compared to littermate controls (Figure 3B, Table S3, S4, and S6). We compared DEGs between 3g del/+ mice and 16p11.2 del/+ mice for each sex. Remarkably, there are 248 overlapping DEGs in the striatum of male 3g del/+ mice and male 16p11.2 del/+ mice (Figure 3C, Table S7, p < 1.890e-112), but only 6 overlapping DEGs between female groups (Figure 3D, Table S8, p < 0.093), supporting the hypothesis that the hemi-deletion of the 3 candidate genes plays an important role in male-specific striatal transcriptomic alterations and behavioral phenotypes observed in 16p11.2 del. Moreover, the quadrant plot of 248 DEGs indicates that all DEGs in common between 3g del/+ and 16p11.2 del/+ males change in the same direction without a single exception (Figure 3E). These results strongly support the hypothesis that the 3 gene hemi-deletion largely shares its transcriptomic fingerprint with 16p11.2 deletion in the striatum of male mice. Next, we performed a pathway analysis to identify the molecular functions of the 248 overlapping male DEGs (Figure 3F) to provide insights into the potential mechanisms underlying male-specific, striatum-dependent behavioral alterations. The pathway analysis revealed significant dysregulation of DEGs within ribosome-related pathways, with a total of 24 ribosomal genes downregulated in the male striatum of both 3g del/+ and 16p11.2 del/+ mice (Figure S8A). Further, an overrepresentation test in PANTHER displayed significant alterations in a translation-related pathway (FDR < 6.73E-17) in the striatum of male 3g del/+ and 16p11.2 del/+ mice (Figure 3G). Transcription Factor Enrichment Analysis (TFEA) implicated 19 transcription factors involved in regulating expression of the 248 DEGs (Figure 3H). Protein-Protein interaction (PPI) using STRING database between 19 transcription factors and 27 genes within the 16p11.2 region indicated that SPN and MAPK3 (ERK1) interact with 10 transcription factors (Figure 3I).
Figure 3.
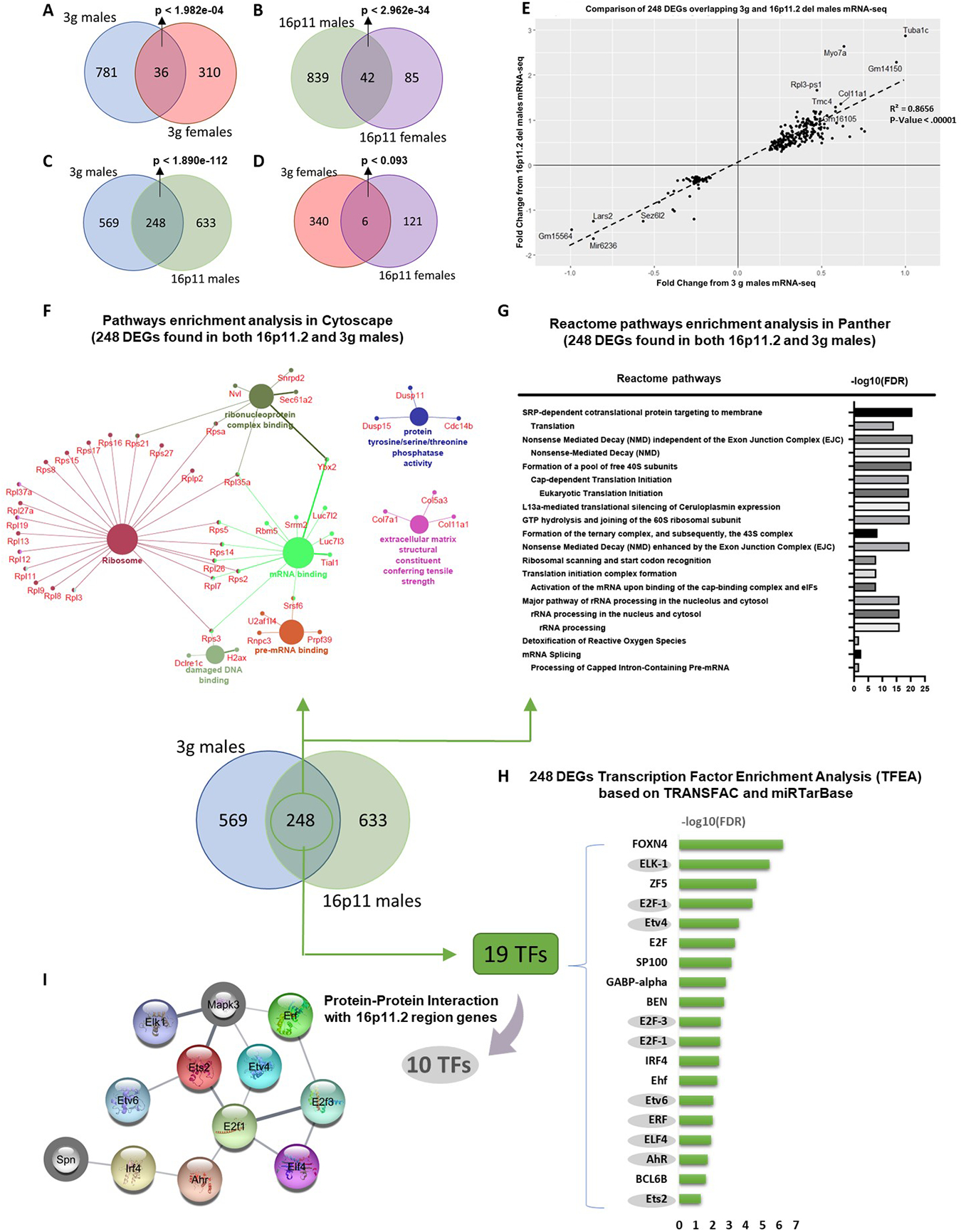
Gene expression changes in the striatum of male 3g del/+ mice are consistent with 16p11.2 del/+ males. (A, B) Venn diagram showing the number of overlapping DEGs that were altered in 3g del/+ mice (A) and 16p11.2 del/+ mice (B) (males and females separately). (C, D) Venn diagram representing the number of overlapping DEGs between 3g del/+ mice and 16p11.2 del/+ mice sex-specifically. (E) Quadrant plot based on comparison of genes regulated in 3g del/+ male mice and 16p11.2 del/+ male mice. Linear regression was applied that generated a coefficient of correlation which was used to calculate the p-value using Pearson (R) Calculator. The value of coefficient of determination (R2) is 0.8656 with significant p value (p<0.00001) in regression. (F) Pathway analysis using ClueGo (KEGG and GO: Molecular function terms) of 248 DEGs, the overlapping DEGs between male 3g del/+ mice with male 16p11.2 del/+ mice, shows functional groupings of network of enriched categories for the DEGs. Significant pathways are presented with a corrected p-value <0.05 (Bonferroni step down) (G) An overrepresentation test on 248 DEGs was performed in PANTHER using the reactome pathway annotation dataset. Only pathways with FDR <0.05 were displayed. (H) Transcription Factor Enrichment Analysis on 248 DEGs was performed based on TRANSFAC and miTRarBase. Only transcription factors with FDR < 0.05 were listed. (I) Protein-protin interaction (PPI) networks of 19 transcription factors and 27 genes within the 16p11.2 del region show closely connected proteins. The network of interaction score of 0.400 is shown. Genes from 16p11.2 del region are gray border.
These results indicate that changes of gene expression in ribosomal pathways linked to protein translation constitute the transcriptomic basis for striatal male vulnerability/female resilience to NDDs. To investigate translational dysregulation, we examined translational efficiency (TE) by performing ribosome profiling (RIBO-seq) in the striatum of 3g del/+ and 16p11.2 del/+ male mice (Figure S9A). TE is the ratio of ribosome occupancy over input mRNA, indicating changes in protein expression54. Interestingly, both 3g del/+ and 16p11.2 del/+ male mice showed TE changes in several genes (Figure S9B–E, Table S9, S10), demonstrating translation dysregulation in the striatum of both 3g del/+ and 16p del/+ male mice. We also examined whether these translation alterations are linked with activation of mammalian target of rapamycin (mTOR). 3g del/+ mice do not show mTOR signaling changes in the striatum (Figure S9F), suggesting that translational dysregulation in 3g del/+ male mice is mTOR independent.
We next investigated whether DEGs in the male 3g del/+ and 16p11.2 del/+ mice showed any overlap with a database of candidate genes relevant to ASD using the SFARI Gene database. We found that 32 DEGs in 3g del/+ male mice overlapped with ASD risk genes (Figure S8B, p < 9.838e-05), and 28 overlapping DEGs in 16p11.2 del/+ male mice overlapped with ASD risk genes (Figure S8B, p < 3.608e-07). Additional pathway analyses for DEGs for 3g del/+ males only (569 DEGs, Figure S10), 16p11.2 del/+ males only (633 DEGs, Figure S11), 3g del/+ females only (340 DEGs, Figure S12), and 16p11.2 del/+ females only (121 DEGs, Figure S13) are provided in the supplementary document.
Sex-specific impact of 3 gene hemi-deletion of brain structure
Previously we have shown sex-specific fiber tract changes in 16p11.2 del/+ mice using high-resolution MRI with diffusion tensor imaging (DTI)32. DTI scans enable the estimation of the fiber tract architecture of the brain55. Homologous changes in increased fractional anisotropy (FA) in medial white matter regions was reported in 16p11.2 hemi-deletion patients56. Therefore, we investigated brain structural changes of 6-week-old 3g del/+ mice using DTI as described previously32. To our surprise, we did not find any significant differences in FA between 3g del/+ mice and wt male mice (Figure 4A and S14A), in contrast to previous findings in 16p11.2 del/+ mice32. Female 3g del/+ mice showed significant FA decreases in a small region of striatal fiber tracts compared to wt female mice (Figure 4B and S14B). Because our previous 16p11.2 del/+ MRI study used older mice32, we subjected 6-week-old 16p11.2 del/+ mice to an identical DTI sequence to explore for age-related effects. In contrast to older mice, there was no significant FA difference between 16p11.2 del/+ male mice and wt mice (Figure 4C and S14C) at 6 weeks of age. However, we found widespread decreased FA in 16p11.2 del/+ females compared to wt females (Figure 4D and S14D), consistent with previous results in 10-week-old female mice32. As no FA change was detected in both 6-week-old male 3g mice and 16p11.2 del mice, we inspected 16-week-old male and female 3g mice and 10-week-old male 16p11.2 del mice to examine the age-dependent brain structural changes. Consistent with our previous study, FA changes are detected in male 10-week-old 16p11.2 del mice (Figure S14G). However, FA changes are not detected in male and female 16-week-old 3g mice at this age (Figure 4E,F and S14E,F). These findings suggest complex disruptions in the brain maturation process in mice with 16p11.2 del/+ and 3g del/+ genotypes, warranting further investigation.
Figure 4.
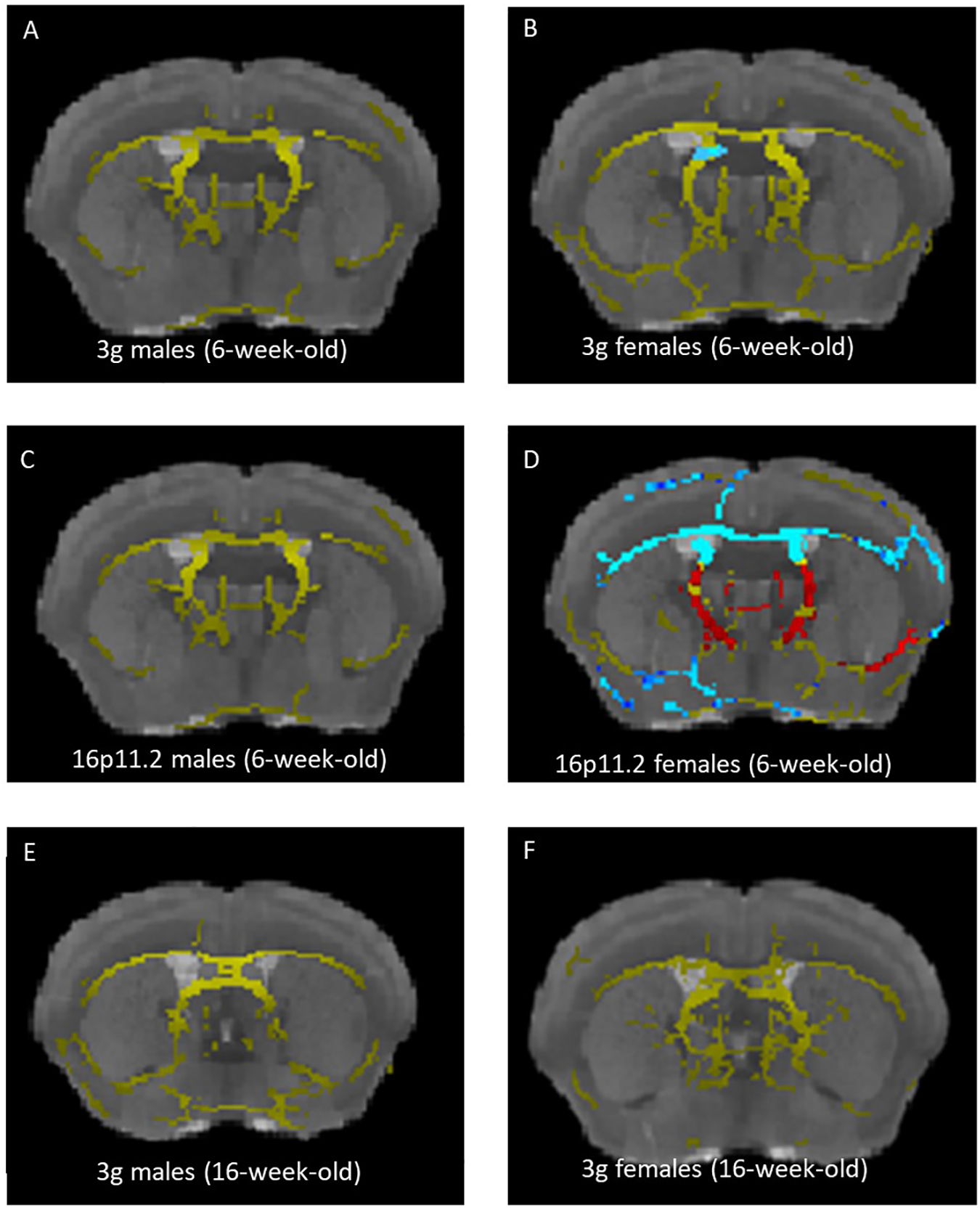
Sex-specific changes in white matter during development in 3g del/+ mice. (A-D) Increased fractional anisotropy (FA) in 3g del/+ or 16p11.2 del/+ mice compared to wt mice is represented in blue and decreased FA is displayed in red. The mean fiber tract skeleton is shown in yellow. FA alterations are not detected in 6-week-old 3g del/+ and 16p11.2 del/+ male mice (A, C), while increased FA in small region in 3g del/+ female mice (B). Widespread increased FA and pronounced FA decreases are displayed 16p11.2 del/+ female mice (D). FA alterations are not detected in 16-week-old 3g del/+ mice (E, F). Significant changes on the brain region are indicated on representative images (p<0.05).
Exploring the role Mapk3 in male-specific striatal phenotypes
Interestingly, the 3 candidate genes are all related to the regulation of ERK signaling. MVP has a well-established association with ERK signaling, serving as a scaffold protein for ERK57, 58. Taok2 has been shown to interact with Septin7 at the postsynaptic density, which can activate ERK signaling59, and Sez6l2 phosphorylates PKC42, which is a known activator of ERK. Therefore, we hypothesized that the connection between the 3 candidate genes and the ERK signaling pathway may provide an explanation for the molecular mechanisms of sex-specific striatal changes in 16p11.2 del/+ mice. Previously, we have observed reward-mediated ERK1 hyperphosphorylation in the dorsal striatum from 16p11.2 del/+ males, but not females, consistent with the male-specific reward learning deficit in 16p11.2 del/+ mice10, suggesting a role for ERK1 in striatum-dependent reward learning in males specifically. Therefore, we examined the effects of reward on ERK signaling in the striatum of 3g del/+ mice. After sucrose consumption, both 3g del/+ and wild type males showed increased phospho-ERK (Figure S15A), but no genotype-dependent differences as observed in the 16p11.2 del/+ males10. In addition, we did not observe altered levels of striatum-enriched protein-tyrosine phosphatase (StEP), a negative regulator of ERK phosphorylation, in 3g del/+ mice (Figure S15B), as has been described previously in 16p11.2 del/+ mice10. To further characterize the impact of multiple genes in the 16p11.2 region on striatal circuits and behaviors, we examined how the additional hemi-deletion of the Mapk3 gene affected striatum-dependent phenotypes and the transcriptome. The choice of Mapk3 was motivated by three factors: 1) it is one of the genes within the 16p11.2 deletion, 2) PPI analysis (Figure 3H) showed possible interactions between ERK1 and several transcription factors from TFEA analysis (Figure 3I), and 3) it codes for ERK1, which interacts with the gene products of the 3 candidate genes and has also been identified as a key molecular mediator of reward learning. We generated mice carrying a hemi-deletion of all four candidate genes (MVP, Sez6l2, Taok2 and Mapk3) by crossing 3g del/+ mice with Mapk3 hemi-deletion (Erk1 +/−) mice (Figure 5A). We validated decreased mRNA levels of the target genes (Mapk3, Taok2, Sez6l2, and Mvp) via qPCR (Figure 5B–E, S16A–D). We compared gene expression in the striatum between male and female 4g del/+ and wt mice using RNA sequencing and found 539 DEGs in 4g del/+ compared to wt males, and 493 DEGs in females (Figure 5F). Only 87 DEGs were shared between male and female 4g del/+ mice, pointing towards sex-specific transcriptomic changes. Strikingly, unlike in 3g del/+ mice, the transcriptomic fingerprint in the striatum of 4g del/+ males only showed minor overlap with gene expression changes in 16p11.2 del/+ males. We found 44 overlapping DEGs in the striatum of male 4g del/+ and male 16p11.2 del/+ mice. Moreover, 4g del/+ male mice do not show reward motivation deficits as16p11.2 del/+ or 3g del/+ male mice (Figure 5N), although 4g del/+ mice display hyperactive behavior similar to 3g del/+ mice (Figure 5I–L) and unaltered motor coordination and learning in rotarod (Figure S16F). We could not find significant reduction in body weight in male 4g del/+ mice compared to sex- and age-matched wt mice (Figure S16E), again showing a different phenotype as 3g del/+ or 16p11.2 del/+ mice. These findings – less pronounced phenotypes observed in the 4g del/+ mice – challenge our expectation of a central role for Mapk3 hemi-deletion in male-specific striatal phenotype.
Figure 5.
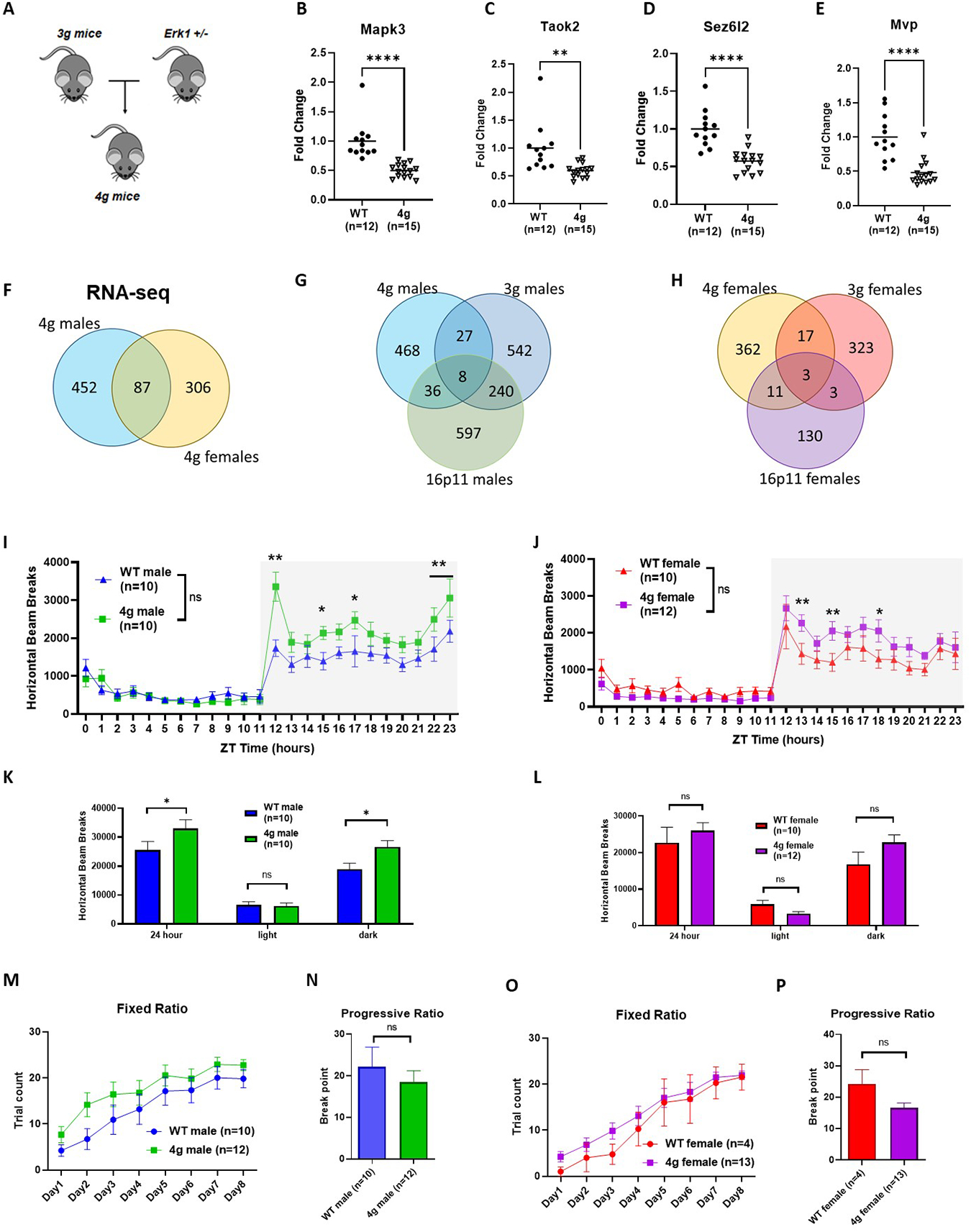
Additional hemi-deletion of Mapk3 does not increase molecular and behavioral alterations observed in 3g del/+ mice. (A) Schematic presentations indicate the breeding strategies for 4g del/+ mice. (B-E) qPCR results demonstrated decreased gene expressions of the 4 genes in the mutant mice (two-tailed Student t test, ** p < 0.05, **** p < 0.001). (F) Venn diagram showing the number of overlapping DEGs that were altered in male and female 4g del/+ mice. (G,H) Venn diagram showing the number of overlapping DEGs that were altered in 4g del/+, 3g del/+, and 16p11.2 del/+ male (G) and female (H) mice. (I,J) Male 4g del/+ mice have greater activity trend relative to wt littermates (main effect of genotype, F (1, 18) = 3.015, p = 0.0996; main effect of time, F (5.948, 107.1) = 39.10, p < 0.0001; genotype x time interaction, F (23, 414) = 3.434, p < 0.001), but not female 4g del/+ mice (main effect of genotype, F (1, 20) = 0.5448, p = 0.4690; main effect of time, F (23, 460) = 31.54, p < 0.001; genotype x time interaction, F (23, 460) = 2.657, p < 0.001). (K, L) Infrared beam breaks are plotted by light/dark cycle. Male 4g del/+ mice show hyperactive behavior relative to wt littermates in dark cycle (main effect of genotype, F (1, 54) = 7.465, p = 0.0085; post hoc, p = 0.017). Female 3g del/+ mice have no differences in horizontal activity (main effect of genotype, F (1, 60) = 1.231, p = 0.2717). (M, O) Both male and female 4g del/+ mice acquire a nosepoke response under a fixed ratio schedule of reinforcement indistinguishable from wt mice (male, main effect of genotype, F (1, 20) = 1.829, p = 0.1914; female, main effect of genotype, F (1, 15) = 0.6168, p = 0.4445). (F, H) In a progressive ratio test of motivation, male 4g del/+ mice (t(20) = 0.6847, p = 0.5014) and female 4g del/+ mice (t(15) = 2.018, p = 0.062) show no different responds than sex-matched wt mice. Values are displayed as mean (±SEM) and significance values are set at *p<0.05 and **p<0.01.
DISCUSSION
We previously identified 3 candidate genes from the 16p11.2 region, Taok2, Sez6l2, and Mvp, using a novel strategy to identify genes-of-interest based on their spatial gene expression patterns32. Our hypothesis was that these 3 genes would be sufficient to mediate sex-specific behavioral phenotypes that depend on striatal circuits. Our newly generated 3g del/+ mouse model displays sex-specific striatal behavioral and transcriptomic characteristics, resembling the phenotypes of 16p11.2 del/+ mice. These results demonstrate that our novel approach to selecting genes of interest based on their spatial gene expression patterns in structurally altered brain regions is a valid path to develop data-driven hypotheses that can be later tested by generating model organisms.
The comparisons of genes differentially expressed in the striatum between male 3g del/+ mice and 16p11.2 del/+ mice revealed 248 genes. The pathway analysis of these 248 genes revealed that a ribosome-related pathway and translation regulation are significantly dysregulated, highlighting potential mechanisms that underlie the male-specific striatum-dependent behavioral alterations. Changes in translation efficiency changes are seen in both 3g del/+ and 16p11.2 del/+ male mice using ribosome profiling. A recent tissue-specific transcriptome profiling study on 16p11.2 del/+ mouse model reported significant GO-term enrichments related to translation, in the striatum and cerebellum60. Although they did not provide a sex-specific analysis, these results support the hypothesis that translational regulation is disrupted in 16p11.2 del/+ mice. The downregulation of ribosomal genes could affect translation efficiency leading to protein synthesis dysregulation and corresponding to the presentation of NDDs61–63. Recent studies of NDDs including Fragile X Syndrome and Tuberous Sclerosis have shown sex-specific differences in ribosomal function or translation regulation that may contribute to sex-specific phenotypes64–67. An interesting consideration here is the potential impact of heterogeneity in ribosome function and activity on the transcript-specific regulation of gene expression potentially resulting from the presence of specialized ribosomes in distinct cell types68. Therefore, cell type-specific approaches may provide better insights, considering the importance of the interplay between D1- and D2- receptor expressing spiny projection neurons that are required for striatal function. These cell type-specific investigations focused on transcriptomics and translational regulation promise to provide insights into the sex-specific pathological mechanisms underlying NDDs.
It has been reported that patients with 16p11.2 del syndrome show regional volumetric differences including in striatal nuclei such as the accumbens, pallidum, caudate, and putamen, along with increased FA in medial white matter12, 56, 69, 70. Similarly, 16p11.2 del/+ mice show increased volumes of several brain regions including the striatum, nucleus accumbens, and globus pallidus50, 71, 72. However, sex- and age-dependent brain structural changes in 16p11.2 deletion have not been fully investigated in either human subjects or rodent models. Previously, we have shown fiber tract changes in both male and female 16p11.2 del mice at 10 weeks of age32. Here, we found decreased FA in 16p11.2 del/+ females compared to wt females at 6 weeks of age, however, we did not observe differences in males. These results indicate that FA changes in 16p11.2 del/+ mice are sex- and age-dependent, suggesting structural changes may occur earlier in development in female 16p11.2 del/+ mice compared to male mice. In addition, these results imply that the 3 gene hemi-deletion may not affect brain structural changes in the same way as 16p11.2 del. A previous study indicates that Taok2 hemi-deletion mice show increased total brain volume compared with wt mice34, however, 16p11.2 del/+ mice display decreased total brain volume50, 71. These opposite brain structural changes between Taok2 hemi-deletion mice and 16p11.2 del/+ mice may provide a hint to understand these distinctive brain structural changes between 3g del/+ mice and 16p11.2 del/+ mice. Uncovering the genetic mechanism of brain structural changes across development will require further study.
ERK signaling has been suggested to underlie phenotypes observed in 16p11.2 del/+ mice and has been the focus of potential therapeutic approaches10, 73, 74. To explore this issue, we created 4g del/+ mice that carry the 3g deletion on one copy of chromosome 7 and a hemi deletion of Mapk3 on the other chromosome 7. We found that 4g del/+ mice exhibit fewer behavioral alterations than 3g del/+ mice, challenging our initial assumption about the crucial involvement of Mapk3 hemi-deletion in the specific striatal phenotypes observed in males. Although it has been shown that the ERK pathway plays an important role in development, learning, and synaptic plasticity75, 76 and ERK signaling is involved in behaviors dependent on striatal circuits38, 77–80, it also has been shown that deletion of both ERK1 and ERK2 is required due to functional redundancy between the isoforms38, 78. Therefore, subsequent studies will need to clarify the connection between ERK signaling and the 3 candidate genes as an underlying mechanism of sex-specific phenotypes. Despite this caveat, we would like to emphasize that our findings underscore the important idea that the ‘combinatorial effects of genes’ are more than just the ‘sum of single gene effects’. Our findings point towards complex non-linear, rather than simple additive, effects of risk genes, and, hence, further highlight the need for multigenic models to study NDDs. This idea is supported by a recently published study on neuroanatomical and behavioral phenotypes that identified MVP as the gene with the highest impact on neuroanatomy in the 16p11.2 region, but failed to detect any behavioral changes in animals with a hemi-deletion of MVP, strongly hinting at multigenic interactions underlying distinct phenotypes39. Previous other single gene studies have been shown that each gene (Taok2, Sez6l2, and Mvp) plays an important role in neuronal development as well as NDD34–36, 41–46, 81. However, sex-specific phenotypes have not been observed in these studies and few studies have examined hemi-deletion of these individual genes. These additional studies suggest that the 3 gene hemi-deletion mediates male-specific phenotypes via the combinatorial effect of 3 genes rather than just a summation of the individual effects of each gene. It is still possible that the combination of 2 genes may be sufficient to replicate certain phenotypes include male-specific phenotypes, so we cannot exclude the possibility that one out of the three genes is unnecessary to cause these effects without future experiments. Importantly, our findings demonstrate that mutation of a single copy of the 3 genes is sufficient to induce male-specific phenotypes. However, it is noteworthy that the necessity of these 3 genes for the observed phenotypes has not been examined.
Overall summary
This study demonstrates the validity of a novel approach to dissecting large chromosomal regions linked to disease to identify candidate sets of genes-of-interest. To accomplish this, we developed a novel experimental model carrying a selective hemi-deletion of three genes in the 16p11.2 region. 3g del/+ animals recapitulate several male-specific striatal phenotypes observed in 16p11.2 del/+ mice, indicating that these 3 candidate genes may serve as ‘core genes’ to increased male striatal vulnerability to neurodevelopmental disorders. Moreover, we highlight the importance of the combinatorial effect of genes, rather than merely additive effects. These findings could pave the way towards novel therapeutic and preventive strategies for NDDs.
Supplementary Material
PUBLICATION ACKNOWLEDGEMENT
This work was supported by The University of Iowa Hawkeye Intellectual and Developmental Disabilities Research Center (HAWK-IDDRC) P50 HD103556 (T.A. and Lane Strathearn, PI), the Roy J. Carver Chair in Neuroscience (T.A.), Interdisciplinary Graduate Program in Genetics at University of Iowa (Y.V.), NIH grant R01 MH 087463 (T.A.), Simons Foundation Autism Research Initiative (SFARI) grant 345034 (T.A.), NIH grants T32 GM067795 and F31 MH134542 (B.K.), Eagles Autism Challenge (T.N.-J.) and the Andrew H. Woods Professorship (T.N-J.).
Transgenic mice were generated at the University of Iowa Genome Editing Core Facility directed by William Paradee, PhD and supported in part by grants from the NIH and from the Roy J. and Lucille A. Carver College of Medicine. We wish to thank Norma Sinclair, Patricia Yarolem, Joanne Schwarting and Rongbin Guan for their technical expertise in generating transgenic mice.
The Neural Circuits and Behavior Core in the Iowa Neuroscience Institute provided equipment, facilities, and consultations services to support investigators in performing behavioral tasks. The Iowa Institute of Human Genetics provided Sanger DNA sequencing and RNA sequencing services. The Iowa Magnetic Resonance Research Facility provides access to a small animal MRI scanner as well as the necessary data processing equipment for animal MR imaging.
We thank TB-SEQ, Inc. (Palo Alto, CA) for Ribosome profiling and helpful discussion.
Footnotes
CONFLICT OF INTEREST
Dr. Ted Abel serves on the Scientific Advisory Board of EmbarkNeuro and is a scientific advisor to Aditum Bio and Radius Health.
DATA AVAILABILTY
The dataset supporting the conclusions of this article is available in the NCBI’s Gene Expression Omnibus repository, GEO Series accession GSE224750.
REFERENCES
- 1.Baxter AJ, Brugha TS, Erskine HE, Scheurer RW, Vos T, Scott JG. The epidemiology and global burden of autism spectrum disorders. Psychol Med 2015; 45(3): 601–613. [DOI] [PubMed] [Google Scholar]
- 2.Grove J, Ripke S, Als TD, Mattheisen M, Walters RK, Won H et al. Identification of common genetic risk variants for autism spectrum disorder. Nat Genet 2019; 51(3): 431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Faraone SV, Larsson H. Genetics of attention deficit hyperactivity disorder. Mol Psychiatry 2019; 24(4): 562–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sanders SJ, He X, Willsey AJ, Ercan-Sencicek AG, Samocha KE, Cicek AE et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron 2015; 87(6): 1215–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T et al. Strong association of de novo copy number mutations with autism. Science 2007; 316(5823): 445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fu JM, Satterstrom FK, Peng M, Brand H, Collins RL, Dong S et al. Rare coding variation provides insight into the genetic architecture and phenotypic context of autism. Nat Genet 2022; 54(9): 1320–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hanson E, Bernier R, Porche K, Jackson FI, Goin-Kochel RP, Snyder LG et al. The cognitive and behavioral phenotype of the 16p11.2 deletion in a clinically ascertained population. Biol Psychiatry 2015; 77(9): 785–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rein B, Yan Z. 16p11.2 Copy Number Variations and Neurodevelopmental Disorders. Trends Neurosci 2020; 43(11): 886–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Angelakos CC, Watson AJ, O’Brien WT, Krainock KS, Nickl-Jockschat T, Abel T. Hyperactivity and male-specific sleep deficits in the 16p11.2 deletion mouse model of autism. Autism Res 2017; 10(4): 572–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grissom NM, McKee SE, Schoch H, Bowman N, Havekes R, O’Brien WT et al. Male-specific deficits in natural reward learning in a mouse model of neurodevelopmental disorders. Mol Psychiatry 2018; 23(3): 544–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lynch JF 3rd, Ferri SL, Angelakos C, Schoch H, Nickl-Jockschat T, Gonzalez A et al. Comprehensive Behavioral Phenotyping of a 16p11.2 Del Mouse Model for Neurodevelopmental Disorders. Autism Res 2020; 13(10): 1670–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sonderby IE, Gustafsson O, Doan NT, Hibar DP, Martin-Brevet S, Abdellaoui A et al. Dose response of the 16p11.2 distal copy number variant on intracranial volume and basal ganglia. Mol Psychiatry 2020; 25(3): 584–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hudac CM, Bove J, Barber S, Duyzend M, Wallace A, Martin CL et al. Evaluating heterogeneity in ASD symptomatology, cognitive ability, and adaptive functioning among 16p11.2 CNV carriers. Autism Res 2020; 13(8): 1300–1310. [DOI] [PubMed] [Google Scholar]
- 14.Nickl-Jockschat T, Habel U, Michel TM, Manning J, Laird AR, Fox PT et al. Brain structure anomalies in autism spectrum disorder--a meta-analysis of VBM studies using anatomic likelihood estimation. Hum Brain Mapp 2012; 33(6): 1470–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Langen M, Bos D, Noordermeer SD, Nederveen H, van Engeland H, Durston S. Changes in the development of striatum are involved in repetitive behavior in autism. Biol Psychiatry 2014; 76(5): 405–411. [DOI] [PubMed] [Google Scholar]
- 16.Langen M, Schnack HG, Nederveen H, Bos D, Lahuis BE, de Jonge MV et al. Changes in the developmental trajectories of striatum in autism. Biol Psychiatry 2009; 66(4): 327–333. [DOI] [PubMed] [Google Scholar]
- 17.Haznedar MM, Buchsbaum MS, Hazlett EA, LiCalzi EM, Cartwright C, Hollander E. Volumetric analysis and three-dimensional glucose metabolic mapping of the striatum and thalamus in patients with autism spectrum disorders. Am J Psychiatry 2006; 163(7): 1252–1263. [DOI] [PubMed] [Google Scholar]
- 18.Di Martino A, Zuo XN, Kelly C, Grzadzinski R, Mennes M, Schvarcz A et al. Shared and distinct intrinsic functional network centrality in autism and attention-deficit/hyperactivity disorder. Biol Psychiatry 2013; 74(8): 623–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Langen M, Leemans A, Johnston P, Ecker C, Daly E, Murphy CM et al. Fronto-striatal circuitry and inhibitory control in autism: findings from diffusion tensor imaging tractography. Cortex 2012; 48(2): 183–193. [DOI] [PubMed] [Google Scholar]
- 20.Castellanos FX, Giedd JN, Eckburg P, Marsh WL, Vaituzis AC, Kaysen D et al. Quantitative morphology of the caudate nucleus in attention deficit hyperactivity disorder. Am J Psychiatry 1994; 151(12): 1791–1796. [DOI] [PubMed] [Google Scholar]
- 21.Semrud-Clikeman M, Pliszka SR, Bledsoe J, Lancaster J. Volumetric MRI differences in treatment naive and chronically treated adolescents with ADHD-combined type. J Atten Disord 2014; 18(6): 511–520. [DOI] [PubMed] [Google Scholar]
- 22.Badgaiyan RD, Sinha S, Sajjad M, Wack DS. Attenuated Tonic and Enhanced Phasic Release of Dopamine in Attention Deficit Hyperactivity Disorder. PLoS One 2015; 10(9): e0137326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fuccillo MV. Striatal Circuits as a Common Node for Autism Pathophysiology. Front Neurosci 2016; 10: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Janouschek H, Chase HW, Sharkey RJ, Peterson ZJ, Camilleri JA, Abel T et al. The functional neural architecture of dysfunctional reward processing in autism. Neuroimage Clin 2021; 31: 102700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chase HW, Loriemi P, Wensing T, Eickhoff SB, Nickl-Jockschat T. Meta-analytic evidence for altered mesolimbic responses to reward in schizophrenia. Hum Brain Mapp 2018; 39(7): 2917–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Faheem M, Akram W, Akram H, Khan MA, Siddiqui FA, Majeed I. Gender-based differences in prevalence and effects of ADHD in adults: A systematic review. Asian J Psychiatr 2022; 75: 103205. [DOI] [PubMed] [Google Scholar]
- 27.Santos S, Ferreira H, Martins J, Goncalves J, Castelo-Branco M. Male sex bias in early and late onset neurodevelopmental disorders: Shared aspects and differences in Autism Spectrum Disorder, Attention Deficit/hyperactivity Disorder, and Schizophrenia. Neurosci Biobehav Rev 2022; 135: 104577. [DOI] [PubMed] [Google Scholar]
- 28.Werling DM, Geschwind DH. Understanding sex bias in autism spectrum disorder. Proc Natl Acad Sci U S A 2013; 110(13): 4868–4869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferri SL, Abel T, Brodkin ES. Sex Differences in Autism Spectrum Disorder: a Review. Curr Psychiatry Rep 2018; 20(2): 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bolte S, Neufeld J, Marschik PB, Williams ZJ, Gallagher L, Lai MC. Sex and gender in neurodevelopmental conditions. Nat Rev Neurol 2023; 19(3): 136–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agarwalla S, Arroyo NS, Long NE, O’Brien WT, Abel T, Bandyopadhyay S. Male-specific alterations in structure of isolation call sequences of mouse pups with 16p11.2 deletion. Genes Brain Behav 2020; 19(7): e12681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kumar VJ, Grissom NM, McKee SE, Schoch H, Bowman N, Havekes R et al. Linking spatial gene expression patterns to sex-specific brain structural changes on a mouse model of 16p11.2 hemideletion. Transl Psychiatry 2018; 8(1): 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martin Lorenzo S, Nalesso V, Chevalier C, Birling MC, Herault Y. Targeting the RHOA pathway improves learning and memory in adult Kctd13 and 16p11.2 deletion mouse models. Mol Autism 2021; 12(1): 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richter M, Murtaza N, Scharrenberg R, White SH, Johanns O, Walker S et al. Altered TAOK2 activity causes autism-related neurodevelopmental and cognitive abnormalities through RhoA signaling. Mol Psychiatry 2019; 24(9): 1329–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Anda FC, Rosario AL, Durak O, Tran T, Graff J, Meletis K et al. Autism spectrum disorder susceptibility gene TAOK2 affects basal dendrite formation in the neocortex. Nat Neurosci 2012; 15(7): 1022–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ip JPK, Nagakura I, Petravicz J, Li K, Wiemer EAC, Sur M. Major Vault Protein, a Candidate Gene in 16p11.2 Microdeletion Syndrome, Is Required for the Homeostatic Regulation of Visual Cortical Plasticity. J Neurosci 2018; 38(16): 3890–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ferguson SM, Fasano S, Yang P, Brambilla R, Robinson TE. Knockout of ERK1 enhances cocaine-evoked immediate early gene expression and behavioral plasticity. Neuropsychopharmacology 2006; 31(12): 2660–2668. [DOI] [PubMed] [Google Scholar]
- 38.Mazzucchelli C, Vantaggiato C, Ciamei A, Fasano S, Pakhotin P, Krezel W et al. Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron 2002; 34(5): 807–820. [DOI] [PubMed] [Google Scholar]
- 39.Kretz PF, Wagner C, Mikhaleva A, Montillot C, Hugel S, Morella I et al. Dissecting the autism-associated 16p11.2 locus identifies multiple drivers in neuroanatomical phenotypes and unveils a male-specific role for the major vault protein. Genome Biol 2023; 24(1): 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weiner DJ, Ling E, Erdin S, Tai DJC, Yadav R, Grove J et al. Statistical and functional convergence of common and rare genetic influences on autism at chromosome 16p. Nat Genet 2022; 54(11): 1630–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yadav S, Oses-Prieto JA, Peters CJ, Zhou J, Pleasure SJ, Burlingame AL et al. TAOK2 Kinase Mediates PSD95 Stability and Dendritic Spine Maturation through Septin7 Phosphorylation. Neuron 2017; 93(2): 379–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miyazaki T, Hashimoto K, Uda A, Sakagami H, Nakamura Y, Saito SY et al. Disturbance of cerebellar synaptic maturation in mutant mice lacking BSRPs, a novel brain-specific receptor-like protein family. FEBS Lett 2006; 580(17): 4057–4064. [DOI] [PubMed] [Google Scholar]
- 43.Boonen M, Staudt C, Gilis F, Oorschot V, Klumperman J, Jadot M. Cathepsin D and its newly identified transport receptor SEZ6L2 can modulate neurite outgrowth. J Cell Sci 2016; 129(3): 557–568. [DOI] [PubMed] [Google Scholar]
- 44.Lotsch D, Steiner E, Holzmann K, Spiegl-Kreinecker S, Pirker C, Hlavaty J et al. Major vault protein supports glioblastoma survival and migration by upregulating the EGFR/PI3K signalling axis. Oncotarget 2013; 4(11): 1904–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steiner E, Holzmann K, Pirker C, Elbling L, Micksche M, Sutterluty H et al. The major vault protein is responsive to and interferes with interferon-gamma-mediated STAT1 signals. J Cell Sci 2006; 119(Pt 3): 459–469. [DOI] [PubMed] [Google Scholar]
- 46.Nash A, Aumann TD, Pigoni M, Lichtenthaler SF, Takeshima H, Munro KM et al. Lack of Sez6 Family Proteins Impairs Motor Functions, Short-Term Memory, and Cognitive Flexibility and Alters Dendritic Spine Properties. Cereb Cortex 2020; 30(4): 2167–2184. [DOI] [PubMed] [Google Scholar]
- 47.Pinkert CA, Pinkert CA. Transgenic animal technology : a laboratory handbook. 2nd edn. Academic Press: Amsterdam ; Boston, 2002. [Google Scholar]
- 48.Angelakos CC, Tudor JC, Ferri SL, Jongens TA, Abel T. Home-cage hypoactivity in mouse genetic models of autism spectrum disorder. Neurobiol Learn Mem 2019; 165: 107000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brunner D, Kabitzke P, He D, Cox K, Thiede L, Hanania T et al. Comprehensive Analysis of the 16p11.2 Deletion and Null Cntnap2 Mouse Models of Autism Spectrum Disorder. PLoS One 2015; 10(8): e0134572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Portmann T, Yang M, Mao R, Panagiotakos G, Ellegood J, Dolen G et al. Behavioral abnormalities and circuit defects in the basal ganglia of a mouse model of 16p11.2 deletion syndrome. Cell Rep 2014; 7(4): 1077–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bali P, Kenny PJ. Transcriptional mechanisms of drug addiction Dialogues Clin Neurosci 2019; 21(4): 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miyashita Y Cognitive memory: cellular and network machineries and their top-down control. Science 2004; 306(5695): 435–440. [DOI] [PubMed] [Google Scholar]
- 53.Engeln M, Fox ME, Chandra R, Choi EY, Nam H, Qadir H et al. Transcriptome profiling of the ventral pallidum reveals a role for pallido-thalamic neurons in cocaine reward. Mol Psychiatry 2022; 27(10): 3980–3991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Brar GA, Weissman JS. Ribosome profiling reveals the what, when, where and how of protein synthesis. Nat Rev Mol Cell Biol 2015; 16(11): 651–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meoded A, Huisman T. Diffusion Tensor Imaging of Brain Malformations: Exploring the Internal Architecture. Neuroimaging Clin N Am 2019; 29(3): 423–434. [DOI] [PubMed] [Google Scholar]
- 56.Maillard AM, Ruef A, Pizzagalli F, Migliavacca E, Hippolyte L, Adaszewski S et al. The 16p11.2 locus modulates brain structures common to autism, schizophrenia and obesity. Mol Psychiatry 2015; 20(1): 140–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kolli S, Zito CI, Mossink MH, Wiemer EA, Bennett AM. The major vault protein is a novel substrate for the tyrosine phosphatase SHP-2 and scaffold protein in epidermal growth factor signaling. J Biol Chem 2004; 279(28): 29374–29385. [DOI] [PubMed] [Google Scholar]
- 58.Kim E, Lee S, Mian MF, Yun SU, Song M, Yi KS et al. Crosstalk between Src and major vault protein in epidermal growth factor-dependent cell signalling. FEBS J 2006; 273(4): 793–804. [DOI] [PubMed] [Google Scholar]
- 59.Zhang N, Liu L, Fan N, Zhang Q, Wang W, Zheng M et al. The requirement of SEPT2 and SEPT7 for migration and invasion in human breast cancer via MEK/ERK activation. Oncotarget 2016; 7(38): 61587–61600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tai DJC, Razaz P, Erdin S, Gao D, Wang J, Nuttle X et al. Tissue- and cell-type-specific molecular and functional signatures of 16p11.2 reciprocal genomic disorder across mouse brain and human neuronal models. Am J Hum Genet 2022; 109(10): 1789–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hetman M, Slomnicki LP. Ribosomal biogenesis as an emerging target of neurodevelopmental pathologies. J Neurochem 2019; 148(3): 325–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Santini E, Klann E. Reciprocal signaling between translational control pathways and synaptic proteins in autism spectrum disorders. Sci Signal 2014; 7(349): re10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tee AR, Blenis J. mTOR, translational control and human disease. Semin Cell Dev Biol 2005; 16(1): 29–37. [DOI] [PubMed] [Google Scholar]
- 64.Ifrim MF, Williams KR, Bassell GJ. Single-Molecule Imaging of PSD-95 mRNA Translation in Dendrites and Its Dysregulation in a Mouse Model of Fragile X Syndrome. J Neurosci 2015; 35(18): 7116–7130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jishi A, Qi X, Miranda HC. Implications of mRNA translation dysregulation for neurological disorders. Semin Cell Dev Biol 2021; 114: 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ceman S, O’Donnell WT, Reed M, Patton S, Pohl J, Warren ST. Phosphorylation influences the translation state of FMRP-associated polyribosomes. Hum Mol Genet 2003; 12(24): 3295–3305. [DOI] [PubMed] [Google Scholar]
- 67.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev 2003; 17(15): 1829–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xue S, Barna M. Specialized ribosomes: a new frontier in gene regulation and organismal biology. Nat Rev Mol Cell Biol 2012; 13(6): 355–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Qureshi AY, Mueller S, Snyder AZ, Mukherjee P, Berman JI, Roberts TP et al. Opposing brain differences in 16p11.2 deletion and duplication carriers. J Neurosci 2014; 34(34): 11199–11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Martin-Brevet S, Rodriguez-Herreros B, Nielsen JA, Moreau C, Modenato C, Maillard AM et al. Quantifying the Effects of 16p11.2 Copy Number Variants on Brain Structure: A Multisite Genetic-First Study. Biol Psychiatry 2018; 84(4): 253–264. [DOI] [PubMed] [Google Scholar]
- 71.Horev G, Ellegood J, Lerch JP, Son YE, Muthuswamy L, Vogel H et al. Dosage-dependent phenotypes in models of 16p11.2 lesions found in autism. Proc Natl Acad Sci U S A 2011; 108(41): 17076–17081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ellegood J, Anagnostou E, Babineau BA, Crawley JN, Lin L, Genestine M et al. Clustering autism: using neuroanatomical differences in 26 mouse models to gain insight into the heterogeneity. Mol Psychiatry 2015; 20(1): 118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pucilowska J, Vithayathil J, Tavares EJ, Kelly C, Karlo JC, Landreth GE. The 16p11.2 deletion mouse model of autism exhibits altered cortical progenitor proliferation and brain cytoarchitecture linked to the ERK MAPK pathway. J Neurosci 2015; 35(7): 3190–3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pucilowska J, Vithayathil J, Pagani M, Kelly C, Karlo JC, Robol C et al. Pharmacological Inhibition of ERK Signaling Rescues Pathophysiology and Behavioral Phenotype Associated with 16p11.2 Chromosomal Deletion in Mice. J Neurosci 2018; 38(30): 6640–6652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sweatt JD. The neuronal MAP kinase cascade: a biochemical signal integration system subserving synaptic plasticity and memory. J Neurochem 2001; 76(1): 1–10. [DOI] [PubMed] [Google Scholar]
- 76.Kelleher RJ 3rd, Govindarajan A, Tonegawa S Translational regulatory mechanisms in persistent forms of synaptic plasticity. Neuron 2004; 44(1): 59–73. [DOI] [PubMed] [Google Scholar]
- 77.Shiflett MW, Balleine BW. Contributions of ERK signaling in the striatum to instrumental learning and performance. Behav Brain Res 2011; 218(1): 240–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Selcher JC, Nekrasova T, Paylor R, Landreth GE, Sweatt JD. Mice lacking the ERK1 isoform of MAP kinase are unimpaired in emotional learning. Learn Mem 2001; 8(1): 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Engel SR, Creson TK, Hao Y, Shen Y, Maeng S, Nekrasova T et al. The extracellular signal-regulated kinase pathway contributes to the control of behavioral excitement. Mol Psychiatry 2009; 14(4): 448–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Talley MJ, Nardini D, Qin S, Prada CE, Ehrman LA, Waclaw RR. A role for sustained MAPK activity in the mouse ventral telencephalon. Dev Biol 2021; 476: 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Scharrenberg R, Richter M, Johanns O, Meka DP, Rucker T, Murtaza N et al. TAOK2 rescues autism-linked developmental deficits in a 16p11.2 microdeletion mouse model. Mol Psychiatry 2022; 27(11): 4707–4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The dataset supporting the conclusions of this article is available in the NCBI’s Gene Expression Omnibus repository, GEO Series accession GSE224750.