

Abstract
Δ1‐pyrroline‐5‐carboxylate reductase isoform 1 (PYCR1) is the last enzyme of proline biosynthesis and catalyzes the NAD(P)H‐dependent reduction of Δ1‐pyrroline‐5‐carboxylate to l‐proline. High PYCR1 gene expression is observed in many cancers and linked to poor patient outcomes and tumor aggressiveness. The knockdown of the PYCR1 gene or the inhibition of PYCR1 enzyme has been shown to inhibit tumorigenesis in cancer cells and animal models of cancer, motivating inhibitor discovery. We screened a library of 71 low molecular weight compounds (average MW of 131 Da) against PYCR1 using an enzyme activity assay. Hit compounds were validated with X‐ray crystallography and kinetic assays to determine affinity parameters. The library was counter‐screened against human Δ1‐pyrroline‐5‐carboxylate reductase isoform 3 and proline dehydrogenase (PRODH) to assess specificity/promiscuity. Twelve PYCR1 and one PRODH inhibitor crystal structures were determined. Three compounds inhibit PYCR1 with competitive inhibition parameter of 100 μM or lower. Among these, (S)‐tetrahydro‐2H‐pyran‐2‐carboxylic acid (70 μM) has higher affinity than the current best tool compound N‐formyl‐l‐proline, is 30 times more specific for PYCR1 over human Δ1‐pyrroline‐5‐carboxylate reductase isoform 3, and negligibly inhibits PRODH. Structure‐affinity relationships suggest that hydrogen bonding of the heteroatom of this compound is important for binding to PYCR1. The structures of PYCR1 and PRODH complexed with 1‐hydroxyethane‐1‐sulfonate demonstrate that the sulfonate group is a suitable replacement for the carboxylate anchor. This result suggests that the exploration of carboxylic acid isosteres may be a promising strategy for discovering new classes of PYCR1 and PRODH inhibitors. The structure of PYCR1 complexed with l‐pipecolate and NADH supports the hypothesis that PYCR1 has an alternative function in lysine metabolism.
Keywords: enzyme inhibition, inhibitor screening, lysine metabolism, proline metabolism, X‐ray crystallography
1. INTRODUCTION
Proline metabolism has gained attention because of its involvement in the unique metabolism of cancer cells (Bogner et al., 2021; Tanner et al., 2018). The proline biosynthetic enzyme Δ1‐pyrroline‐5‐carboxylate reductase isoform 1 (PYCR1) is especially notable as the most highly expressed proline metabolism enzyme in tumor cells compared to non‐cancerous cells (Bogner et al., 2021). PYCRs catalyze the last step of proline biosynthesis, the NAD(P)H‐dependent reduction of Δ1‐pyrroline‐5‐carboxylate (P5C) to l‐proline (Figure 1). Among the three major isoforms of PYCR, PYCR1 is most closely associated with tumorigenesis and metastasis (Bogner et al., 2021; Hu, 2021; Li et al., 2021). PYCR1 forms a metabolic hub, known as the proline cycle, with the proline catabolic enzyme proline dehydrogenase (PRODH), which catalyzes the FAD‐dependent oxidation of l‐proline to P5C (Figure 1) (Phang, 2021; Phang et al., 2012). The proline cycle supports ATP production, protein and nucleotide synthesis, anaplerosis, and redox homeostasis in cancer cells, and both PYCR1 and PRODH are the targets of chemical probe development. (Tanner et al., 2018).
FIGURE 1.

Reactions catalyzed by the proline cycle enzymes Δ1‐pyrroline‐5‐carboxylate reductase isoform 1 (PYCR1) and proline dehydrogenase (PRODH).
Numerous correlative and mechanistic links of PYCR1 to cancer progression and metastasis have been reported. Many studies have shown that high PYCR1 expression is associated with tumor aggressiveness and poor patient outcomes (Alaqbi et al., 2022; Ding et al., 2017, 2020; Du et al., 2021; Li, Xu, et al., 2022; Li, Liu, et al., 2022; Liu et al., 2021; Oudaert et al., 2022; Shao et al., 2022; Shenoy et al., 2020; Weijin et al., 2019; Xiao et al., 2020; Xu et al., 2021; Zhang et al., 2021), and the knockdown of the PYCR1 gene or the inhibition of PYCR1 enzyme has been shown to inhibit tumorigenesis in cancer cell lines and animal models of cancer (Alaqbi et al., 2022; Cai et al., 2018; Christensen et al., 2020; Ding et al., 2020; Du et al., 2021; Elia et al., 2017; Gao et al., 2020; Li et al., 2023; Li, Liu, et al., 2022; Li, Xu, et al., 2022; Sang et al., 2019; Shenoy et al., 2020; Xiao et al., 2020; Xu et al., 2021; Yan et al., 2019; Zeng et al., 2017; Zhuang et al., 2019). The production of proline via PYCR1 may be an indicator of cancer stem‐like cells, which can survive chemotherapy treatments and lead to relapse in cancer patients (Sharif et al., 2019). PYCR1 is overexpressed in breast cancer cells and associated with tumor growth and invasiveness (Ding et al., 2017). PYCR1 maintains triple‐negative breast cancer stem cell‐like properties by producing proline, which activates the cGMP‐PKG signaling pathway in psychologically stressed conditions. Activation of the cGMP‐PKG pathway is seen in triple‐negative breast cancer patients and often leads to poor outcomes (Cui et al., 2023). PYCR1 is important for tumor growth under hypoxia. IGF1R increases PYCR1 expression in hypoxic environments to overcome effects of cisplatin chemotherapy treatments, which is commonly used to treat patients with esophageal squamous cell carcinoma (Fang et al., 2023). Mechanistically, PYCR1 supports tumor cells in hypoxic environments by generating NAD+ in the mitochondria, which is used to drive the TCA cycle (Westbrook et al., 2022). Traditionally considered to be a mitochondrial enzyme, new research has revealed a role for phosphorylated PYCR1 (at Tyr135) binding to the transcription factor ELK4 in the nucleus to promote colorectal cancer tumor growth under hypoxia (Zheng et al., 2023).
A motivation for the current study is to identify small molecule inhibitors that can be used as chemical probes to further elucidate the roles of PYCR1 in cancer. Currently, the best validated inhibitor is N‐formyl‐l‐proline (NFLP), which was identified in an in crystallo screen of 27 proline analogs (Christensen et al., 2020). NFLP has an inhibition constant (K i) of 100 μM against the purified enzyme and exhibits cellular activity by inhibiting proline biosynthesis and spheroid growth in MCF10A H‐RASV12 breast cancer cells. All the inhibitors identified in that initial screen were close proline analogs, featuring a five‐membered ring and carboxylate group. The crystal structures of these compounds showed how the carboxylate group anchors proline analogs in the αK–αL loop of the P5C binding site. The higher affinity of NFLP compared to other proline analogs is attributed to additional hydrogen bonding of the formyl group with the αK–αL loop.
Herein, we further explore the chemical space of PYCR1 probes to identify new interactions that can be leveraged in the design of higher affinity inhibitors. We screened a library of 71 low molecular weight compounds (131 ± 24 Da) having various anchor groups intended to dock in the αK–αL loop, including carboxylate, hydroxyl, sulfonate, amide, and ester (Figure 2). Initial screening was based on activity measurements, and compounds with at least 50% inhibition of PYCR1 activity were validated with X‐ray crystallography. Twelve PYCR1 inhibitor complex structures were determined. To assess inhibitor specificity/promiscuity, the library was counter‐screened against human Δ1‐pyrroline‐5‐carboxylate reductase isoform 3 (PYCR3) and PRODH. (S)‐tetrahydro‐2H‐pyran‐2‐carboxylic acid (10) is revealed as a promising inhibitor. 10 has a lower K i than our best tool compound NFLP, is 30 times more specific for PYCR1 over PYCR3, and negligibly inhibits PRODH. The crystal structures of PYCR1 and PRODH complexed with 1‐hydroxyethane‐1‐sulfonate (43) show that the sulfonate group is a suitable carboxylic acid isostere anchor for targeting both enzymes of the proline cycle. The structures reported here could be used in template‐based docking to identify larger inhibitors with higher affinity and specificity (Meeks et al., 2024).
FIGURE 2.

Chemical structures of the screened compounds. Boxed compounds were characterized by inhibition kinetics and X‐ray crystallography. Note that some compounds were used as racemic mixtures (e.g., 10, 23, and 43).
2. RESULTS
2.1. Description of the library
Seventy‐one compounds were screened against PYCR1 (Figure 2). The names, vendor information, SMILES strings, and PubChem IDs of the compounds are provided in a spreadsheet in Supporting Information. The compounds were chosen based on our knowledge of PYCR1 inhibitor recognition elements, including the presence of an anchor group for binding to the αK–αL loop and small functional groups or heteroatoms that could potentially form additional hydrogen bonds to the αK–αL loop, analogous to those formed by NFLP. Anchoring groups include carboxylate, hydroxyl, sulfonate, amide, and ester (Figure 2). Thirty compounds are cyclic, including five with an aromatic ring.
The molecular weight range of the library is 86.1–207.7 Da, with an average of 131 ± 24 Da, and the number of heavy atoms is 8.6 ± 1.6. On average, these compounds are smaller than those typically used in fragment‐based inhibitor discovery (Erlanson, 2006; Rees et al., 2004; Scott et al., 2012) and slightly larger than the class of inhibitors known as ultra‐low molecular weight fragments, such as “MiniFrags” (94 ± 16 Da, 5–8 heavy atoms) (O'Reilly et al., 2019) and “MicroFrags” (102 ± 25 Da, 5–9 heavy atoms) (Whitehouse et al., 2023). The physicochemical descriptors of the library calculated with SwissADME are listed in Table S1 (Daina et al., 2017).
2.2. Primary screening of the library by enzyme activity
The library was screened by measuring the relative enzyme activity of PYCR1 in the presence of 5 mM of compound (Figure 3). NFLP was used as a positive control and lowers the activity of PYCR1 to 7%. 22 was the only compound that outperformed NFLP in the primary screen (4% relative activity).
FIGURE 3.

Initial Screening of Δ1‐pyrroline‐5‐carboxylate reductase isoform 1 (PYCR1). The bars represent the initial rate with l‐P5C at 250 μM, NADH at 175 μM, and the compound at 5 mM. The error bars represent the standard deviation from three technical replicates. The data are normalized to the rate of PYCR1 in the absence of an inhibitor (labeled “0”). The positive control (N‐formyl‐l‐proline) is denoted “+.”
Any compound that lowered activity by at least 50% was considered a hit and attempts were then made to obtain a crystal structure with PYCR1. Thirteen compounds met this criterion (Figure 3) (6, 10, 11, 22, 23, 28, 36, 38, 39, 43, 60, 66, 70). In addition, 7, 27, and 41 were carried forward to crystallization trials for various reasons. 7 had the highest inhibition of any of the dicarboxylate compounds in the library and was previously shown to be a weak inhibitor of PRODH (K i = 1.5 mM) (Bogner & Tanner, 2022). 27 (55% relative activity) is an acyclic analog of hit compound 23. 41 (l‐pipecolic acid) is of interest because of the proposed involvement of PYCR1 in lysine catabolism by catalyzing the conversion of Δ1‐piperideine‐6‐carboxylate (P6C) to l‐pipecolic acid (Struys et al., 2014).
2.3. Structures of PYCR1 complexed with hit compounds
Co‐crystallization was attempted with 16 compounds, including 13 that lowered enzyme activity to below 50% relative to control, and three additional compounds as described in the preceding section. Eleven PYCR1‐inhibitor structures were determined at high resolution limits of 1.64–2.35 Å (7, 10, 22, 23, 27, 36, 39, 41, 43, 66, 70) (Table S2). All but two of the structures have resolution better than 2.0 Å (27, 43). All except 36 were determined as ternary complexes with NADH. Co‐crystallization with 6, 11, 28, 38, and 60 was unsuccessful, resulting in no crystals, poor diffraction, or weak ligand electron density.
All the compounds are clearly defined by strong electron density in the P5C site (Figure 4). 7, 10, 22, 23, 36, 39, 41, and 43 were modeled into all five chains in the asymmetric unit. 27, 66, and 70 were modeled into four, three, and two chains, respectively. The refined ligand occupancies range from 0.72 to 1.0 (Table S2). NADH occupancy is similarly high, ranging from 0.8 to 1.0 (Table S2). Two conformations of 70 were modeled to satisfy the spherical shape of the density for the ring (Figure 4k). The refined occupancies of the two conformations are 0.49/0.51 in one chain and 0.44/0.56 in the other chain.
FIGURE 4.

Structures of Δ1‐pyrroline‐5‐carboxylate reductase isoform 1 (PYCR1) complexed with (a) 7, (b) 10, (c) 22, (d) 23, (e) 27, (f) 36, (g) 39, (h) 41, (i) 43, (j) 66, and (k) 70 and NADH. Note: The complex with 36 does not include NADH. Electron density for compounds is a polder omit map at 4σ.
All the compounds bind in the αK–αL loop through a carboxylate or sulfonate anchoring group (Figure 5). Superposition of all the structures shows the low variability in the orientation of the anchor (Figure S1). Interactions with the anchors include main chain hydrogen bonds with Val231, Ser233, Ala237, and Thr238. In some structures, the side chains of Ser233 and Thr238 also engage the anchor. All the anchors hydrogen bond with a conserved water molecule known as the “in‐loop water” described previously (Christensen et al., 2020). Several of the compounds also engage another water molecule in the active site. Considering only those interactions observed in all instances of the inhibitor in the asymmetric unit, the number of anchoring interactions varies from two (41) to six (7). Four is the most common number of anchoring interactions (10, 22, 43, 66, 70). The observed networks of interactions with the anchors resemble those of previous PYCR1‐inhibitor structures (Christensen et al., 2017, 2020; Meeks et al., 2024).
FIGURE 5.

Schematic diagrams of the interactions of hit compounds with Δ1‐pyrroline‐5‐carboxylate reductase isoform 1 (PYCR1). Dashed lines represent electrostatic interactions observed in all instances of the inhibitor in the asymmetric unit. Dotted lines represent electrostatic interactions observed in at least one but not all instances of the inhibitor. Compound numbers are listed in bold.
Interactions beyond the anchor are also observed with some of the compounds. The heteroatoms of the rings of 10, 41, and 70 form hydrogen bonds with the hydroxyl of Thr238; the amino group of 41 also donates a hydrogen bond to the carbonyl of Val231. Similarly, the hydroxyl groups of the l‐lactate analogs, 36 and 39, also engage Val231 and Thr238.
The inhibitors also form interactions with the coenzyme. All but compound 36 were co‐crystallized as the ternary complex with NADH. The rings of 10, 22, 23, 41, 66, and 70 pack tightly against the nicotinamide ring of NADH, forming van der Waals contacts with the coenzyme (3.8 Å cutoff). Nonpolar contacts with the nicotinamide are also observed with the side chains of the acyclic compounds 27 and 39. Compound 2 is unusual in having a second carboxylate, which hydrogen bonds with the NADH amide group. Superposition of all the structures shows the packing of the inhibitors against NADH (Figure S1).
2.4. Structure of the ternary complex of PYCR1 with NADH and NFLP
The structures of the ternary complexes of PYCR1 with NADH and a low molecular weight compound demonstrate that the presence of the latter does not sterically impede the binding of the former. We wondered whether this is also true for NFLP, which was crystallized previously as a binary complex with PYCR1 (PDB 6XP0). The ternary complex structure was determined at 1.83 Å resolution (Table S2). Electron density for NFLP and NADH is strong, enabling both ligands to be modeled at occupancy of 1.0 in all five chains in the asymmetric unit (Figure 6a).
FIGURE 6.

Structure of the ternary complex of Δ1‐pyrroline‐5‐carboxylate reductase isoform 1 with N‐formyl‐l‐proline (NFLP) and NADH. (a) Electron density for the ligands (polder omit map at 4σ). (b) Comparison of the binary (salmon, PDB 6XP0) and ternary (white, PDB 8VRE) complexes of NFLP.
The pose and interactions of NFLP are similar to those of the binary complex; however, the presence of NADH causes NFLP to shift about 1 Å (Figure 6b). As a result, NFLP in the ternary complex forms fewer hydrogen bonds with the αK–αL loop. In particular, the formyl group forms only one hydrogen bond with Ser233 in the ternary complex, compared to two in the binary complex. Another difference is that the 230s helix is shifted by 1 Å and Val231 adopts a different rotamer. It is unclear whether these conformational differences are caused by the presence of NADH or reflect differences in crystal packing, since the binary PYCR1‐NFLP complex was determined in a different space group (P21212). Nevertheless, the ternary structure shows that NFLP binding does not sterically preclude the binding of NADH.
2.5. Validation of crystal structure hits with enzyme kinetic assays
The 11 compounds with electron density supporting binding to PYCR1 were further tested for inhibition of enzymatic activity. All but 70 were tested in assays with varying l‐P5C concentration, and the data were fit globally to the standard competitive model (Figure 7). Table 1 lists the competitive inhibition K i values. Inhibition data for 70 exhibited a poor fit to the standard inhibition models (e.g., competitive, mixed) and was instead analyzed with a concentration‐response approach (Figure 7k).
FIGURE 7.
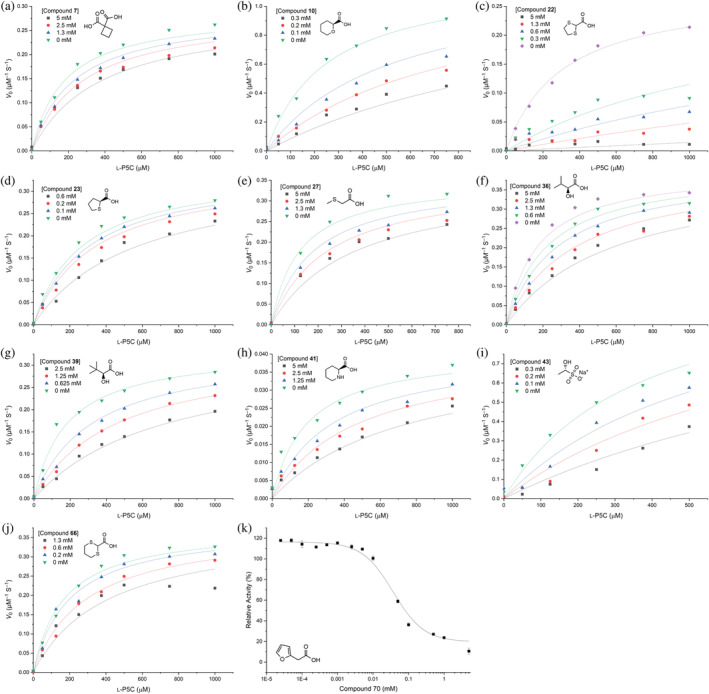
Kinetics of inhibition of Δ1‐pyrroline‐5‐carboxylate reductase isoform 1 (PYCR1) by (a) 7, (b) 10, (c) 22, (d) 23, (e) 27, (f) 36, (g) 39, (h) 41, (i) 43, and (j) 66 with l‐P5C as the variable substrate and NADH fixed at 175 μM. The lines are global fits to the competitive inhibition model. (k) Concentration–response curve for 70.
TABLE 1.
Competitive inhibition parameters for Δ1‐pyrroline‐5‐carboxylate reductase isoform 1 a .
| Compound | K i (mM) |
|---|---|
| 7 | 4.5 ± 0.7 |
| 10 b | 0.07 ± 0.01 |
| 22 | 0.04 ± 0.01 |
| 23 b | 0.5 ± 0.1 |
| 27 | 2.5 ± 0.5 |
| 36 | 2.0 ± 0.2 |
| 39 | 0.8 ± 0.1 |
| 41 | 1.7 ± 0.3 |
| 43 b | 0.1 ± 0.02 |
| 66 | 0.9 ± 0.2 |
K i values were obtained from global fitting to the competitive inhibition model.
A racemic mixture was used in activity assays and crystallization, yet only one stereoisomer was observed in the crystal structure. Thus, the inhibitor concentration used in curve fitting was decreased twofold.
1,3‐Dithiolane‐2‐carboxylic acid (22) exhibited the lowest K i among the competitive inhibitors, at 40 μM. Compounds 23, 27, and 66 are similar to 22 in that they have an S atom adjacent to the α‐carbon (Figure 2); however, they are much weaker inhibitors. Replacing one of the S atoms of 22 by a methylene to generate 23 increased K i 10‐fold. Increasing the ring size of 22 from 5 to 6, as in 66, increased K i about 20‐fold. The acyclic analog 27 has a K i of 2.5 mM.
Compound 43 (1‐hydroxyethane‐1‐sulfonate) is a novel PYCR1 inhibitor in that it has a sulfonate anchor instead of carboxylate. The K i of 43 is the second‐best in this study, at 100 μM, which is comparable to that of NFLP, a compound that phenocopies the PYCR1 gene knockdown in MCF10A H‐RASV12 breast cancer cells (Christensen et al., 2020).
The isostructural pair 10/41 differ only in the hydrogen bonding capacity of the heteroatom (O in 10 vs. NH in 41). The tetrahydropyran compound (10) has K i of 70 μM, whereas the piperidine compound (41) has a much weaker affinity of 1.7 mM. This result suggests that having a hydrogen bond acceptor at this location is preferred over a donor. We note the K i of 41 is similar to that of the product l‐proline, which also has a hydrogen bond donor in the ring (Christensen et al., 2020).
36 and 39 are isopropyl and tert‐butyl analogs of l‐lactic acid (40), respectively. Both compounds are rather weak inhibitors (millimolar K i), but the tert‐butyl group yields slightly better (two‐fold) inhibition. Although the isobutyl analog (38) seemed promising in the initial screen by lowering relative activity to 40% (Figure 3), its electron density was very weak, and the estimated K i was 3.5 ± 0.5 mM (data not shown).
The dicarboxylic acid (7) showed the weakest inhibition of the compounds tested, with K i >4 mM. We note this compound is also a very weak inhibitor of PRODH (1.5 mM) (Bogner & Tanner, 2022).
Finally, the kinetic inhibition pattern of 70 could not be fit to any of the standard models. 70 appears to affect V max, suggesting the inhibition is not entirely competitive with P5C, although it binds in the P5C site (Figure 4k). The concentration‐response behavior of 70 yields an IC50 of 39 ± 8 μM.
2.6. Selectivity of inhibition
The 71‐compound library was screened against PYCR3 and the proline cycle enzyme PRODH. For each enzyme, the relative activity was measured in the presence of 5 mM of the compound (Figure S2). A threshold of 50% relative activity was used for hit identification. Scatter plots of the activity data were used to qualitatively assess specificity (Figure 8a,b). In these plots, compounds in quadrants II and IV show preferential inhibition of one of the two enzymes being compared, whereas compounds in quadrant III potentially inhibit both enzymes, and those in quadrant I inhibit neither enzyme. Most of the compounds fall into quadrant I; that is, they inhibit neither PYCR1, PYCR3, nor PRODH (based on 50% threshold). Twenty‐nine compounds inhibited at least one of the three enzymes by 50% or more. The enzyme preferences of these compounds are represented in a Venn diagram (Figure 8c).
FIGURE 8.

Selectivity/promiscuity of inhibition. (a) Scatter plot of the relative activities of Δ1‐pyrroline‐5‐carboxylate reductase isoform 1 (PYCR1) and proline dehydrogenase (PRODH) at 5 mM of the compound. (b) Scatter plot of the relative activities of PYCR1 and human Δ1‐pyrroline‐5‐carboxylate reductase isoform 3 (PYCR3) at 5 mM of the compound. (c) Scatter plot of the relative activities of PYCR3 and PRODH at 5 mM of the compound. (d) Venn diagram of compounds that inhibit either PYCR1, PYCR3, or PRODH by 50% or more.
Five compounds show a preference for PYCR1 over PYCR3 and PRODH: 10, 11, 28, 36, and 60 (Figure 8c). Two have been validated with a crystal structure (10, 36). Attempts to crystallize 11, 28, and 60 with PYCR1 were unsuccessful, resulting in poor ligand density or the crystals were unsuitable for structure determination. 10 is notable among this group because its K i for PYCR1 of 70 μM is comparable to that of our best tool compound NFLP (100 μM) and negligibly inhibits PRODH (Figure S2A). The K i for 10 against PYCR3 was estimated to be 2.1 ± 0.3 mM (Figure S3). Thus, 10 is 30 times more specific for PYCR1 over PYCR3.
Three compounds are promiscuous, inhibiting PYCR1, PYCR3, and PRODH by 50% or more: 43, 66, and 70. 66 is likely a photoinduced covalent inactivator of PRODH based on its structural similarity to the previously characterized photoinduced covalent inactivators 22 and 23 (Campbell et al., 2021). 70 is also a known inhibitor of PRODH (Bogner et al., 2022). The structure of PRODH complexed with 70 shows the inhibitor occupies the proline substrate site, similar to proline analog inhibitors, and a mixed inhibition pattern was observed with a competitive K i of about 200 μM (Bogner et al., 2022). Note that 70 exhibited a similar level of inhibition against PYCR1 (IC50 of 39 μM).
The promiscuous inhibitor 43 is the only compound in the library with a sulfonate anchor. Being the first sulfonate‐anchored PRODH inhibitor, we determined the structure of a PRODH domain (SmPutAΔα2) complexed with 43. The inhibitor binds against the si face of the FAD, contacting the middle and pyrimidine rings of the isoalloxazine (Figure 9a). This is the usual location for proline analog inhibitors, such as S‐(−)‐tetrahydro‐2‐furoic acid (THFA) (Figure 9b). The sulfonate group is stabilized by three positively charged residues, Lys265, Arg488, and Arg489, and a conserved active site water molecule (Figure 9a). The inhibitor hydroxyl forms a hydrogen bond with the FAD 2'–OH group. The pose of 43 may be compared to that of the best proline analog inhibitor of PRODH, THFA (Figure 9b). The conformations of the two inhibited active sites are virtually identical, and in both structures, the anionic anchor is engaged by three positively charged side chains (Figure 9c). The structures differ in the role of the conserved water molecule, which hydrogen bonds to the sulfonate of 43 (Figure 9a) and the heteroatom of THFA (Figure 9b). Structurally, it appears that the one of the sulfonate O atoms has taken the place of the heteroatom of THFA.
FIGURE 9.

Structure of the proline dehydrogenase (PRODH) domain SmPutAΔα2 complexed with 43. (a) Interactions and electron density for 43 (violet) bound to SmPutAΔα2 (polder omit map at 4σ). The FAD is colored yellow. (b) Pose and interactions of S‐(−)‐tetrahydro‐2‐furoic acid (THFA) (cyan) bound to PRODH (PDB 8DKP). The FAD is colored yellow. (c) Superposition of SmPutAΔα2 complexed with 43 (violet) and THFA (cyan, PDB 8DKP).
3. DISCUSSION
Screening of a knowledge‐based library of small molecular weight compounds revealed new inhibitors of PYCR1. 10 is promising because it has a lower K i (70 μM) than our best inhibitor NFLP (100 μM), a compound that phenocopies the PYCR1 gene knockdown in MCF10A H‐RASV12 breast cancer cells (Christensen et al., 2020). Furthermore, 10 is 30 times more specific for PYCR1 over PYCR3 and negligibly inhibits PRODH. The results for the structurally similar compounds 41 and 66 provide insight into the origins of the affinity of 10 for PYCR1. 41 and 66 are also six‐membered‐ring carboxylic acids but are much weaker inhibitors with mM K i values. 10 differs from these compounds by having a hydrogen bond acceptor in the ring that interacts with the side chain of Thr238. Neither 41 nor 66 exhibit this feature. The amino group of 41 is a hydrogen bond donor, whereas the S atoms of 66 cannot form hydrogen bonds. Apparently, the hydrogen bonding of the heteroatom of 10 is important for affinity.
The discovery of 43 as an inhibitor of PYCR1 and PRODH is a significant outcome because it is the first inhibitor of either proline cycle enzyme featuring a sulfonate anchor in place of carboxylate. Although hundreds of drugs on the market contain a free carboxylic acid group, its presence can lead to undesirable effects such as limited permeability across biological membranes, metabolic instability, and toxicity (Kalgutkar & Daniels, 2010). Thus, it is sometimes desirable to replace the carboxylic acid group with an isostere that avoids these problems while maintaining binding to the target. Many carboxylic acid isosteres are available, including the sulfonate (Ballatore et al., 2013; Lassalas et al., 2016). Here we showed that sulfonate may be substituted for carboxylate in PYCR1 and PRODH inhibitors. Note that 43 is the sulfonate analog of l‐lactate (40 in our library), which is a weak inhibitor of PYCR1 and PRODH. In fact, l‐lactate was one of the first PRODH inhibitors discovered (Kowaloff et al., 1977). Replacement of the carboxylate of l‐lactate apparently increases the affinity to both enzymes. This result shows that the carboxylate group is not essential for inhibitor binding to PYCR1 or PRODH and suggests that testing compounds with carboxylic acid isosteres might be a fruitful line of inquiry. In particular, the sulfonate analogs of the larger molecular weight (>200 Da) PYCR1 inhibitors that we reported recently could be promising (Meeks et al., 2024).
The structures PYCR1 and PRODH complexed with 43 provide an example of how two different receptors can bind the same ligand using very different molecular recognition strategies. PYCR1 exclusively uses hydrogen bonding to bind the negatively charged sulfonate (Figure 5i), whereas PRODH uses primarily ionic interactions with Arg and Lys side chains (Figure 9a). The hydroxyl of 43 hydrogen bonds to the protein in PYCR1 and to the FAD cofactor in PRODH. A commonality is that both enzymes provide conserved water molecules that bridge the sulfonate to the enzyme.
In addition to chemical probe discovery, our results provide insight into the proposed role of PYCR1 in lysine metabolism. Struys et al. (2014) reported that PYCR1 can catalyze the NAD(P)H‐dependent reduction of P6C to l‐pipecolic acid (41 in our library). This alternative enzymatic activity was also reported for a plant P5C reductase (Fujii et al., 2002). P6C is the 6‐membered ring analog of P5C and the substrate for l‐pipecolate oxidase in the pipecolate lysine catabolic pathway (Hallen et al., 2013). The nonenzymatic hydrolysis of P6C produces α‐aminoadipate semialdehyde, the substrate for the lysine catabolic enzyme ALDH7A1 (Korasick & Tanner, 2021), which appears in both the saccharopine and pipecolate pathways of lysine catabolism. The alternative activity of PYCR1 in the reduction of P6C to l‐pipecolic would impact lysine metabolism and potentially contribute to the metabolic mechanisms of inherited metabolic disorders of lysine metabolism.
The structure of PYCR1 complexed with 41 and NADH is consistent with the reported P6C reductase activity. The structure demonstrates that the active site can simultaneously bind 41 and the coenzyme NADH. Since 41 is isostructural to P6C, the structure is consistent with formation of the ternary PYCR1‐P6C‐coenzyme complex, a necessary requirement for catalysis. Further, the pose of 41 in relation to NADH is consistent with the hydride transfer mechanism of PYCR1. The C6 of 41, which represents the hydride acceptor of P6C, is positioned near the hydride donor of NADH (C4). Among the atoms in the ring of 41, C6 is the closest to NADH C4 (3.5–3.7 Å). Thus, the structure provides a model for the PYCR1‐P6C‐coenzyme ternary complex and supports the proposed role of PYCR1 in lysine metabolism.
4. MATERIALS AND METHODS
4.1. PYCR1 enzyme activity assays
Recombinant PYCR1 with a truncated C‐terminus to aid crystallization (residues 1–294) was expressed and purified as described previously (Meeks et al., 2024). Enzyme activity measurements were performed using a BioTek Epoch 2 microplate spectrophotometer in Corning 96‐well plates at 25°C. The assay buffer contained 50 mM HEPES pH 7.5 and 1 mM EDTA, and the total assay volume was 200 μL. The decrease in absorbance at 340 nm was monitored and corresponds to the oxidation of NADH in the reaction. A stock solution of d,l‐P5C (6–20 mM) was diluted with 1M Tris pH 8.5 and neutralized to pH 7–9 by dropwise addition of 6M NaOH. The concentration of l‐P5C was assumed to be one‐half of the d,l‐P5C stock.
The primary screen measured the relative activity of PYCR1 in the presence of 5 mM of a library compound, with l‐P5C and NADH present at 250 and 175 μM, respectively. Stock solutions of each compound were prepared at 100 mM by dissolving (for solids) or diluting (for liquids) the compound in 1M Tris pH 8.5 until the pH of the solution was neutral. 6M NaOH was added in some cases. Once neutral pH was reached, the volume was adjusted with water to achieve the desired concentration of 100 mM. Ten microliter of the 100 mM compound solutions were added to the plate. Then, 40 μL of 1250 μM neutralized l‐P5C was added to each well. The reaction was initiated by the addition of 150 μL of mastermix containing PYCR1, NADH, and buffer (50 mM HEPES pH 7.5 and 1 mM EDTA) to each well via multichannel pipette. The final concentration of PYCR1 in the assay was 10 nM. Each compound was tested in triplicate. A negative control (no inhibitor) was included in each plate in which H2O was used in place of the compound. A positive control using the known inhibitor NFLP was also included in each plate. The library was screened in batches of 8–24 compounds per plate. The positive and negative controls were included in every plate.
Data from the primary screen were analyzed as follows. Initial velocities were determined by linear regression with up to 15 min of absorbance data. The initial rates were converted to units of μM s−1 using the negative value of the slope and the extinction coefficient of NADH at 340 nM of 6220 M−1 cm−1. The negative control reactions (no inhibitor) were averaged and used to normalize the rates of reactions in presence of a compound. The percent relative activity of each compound was averaged after dividing by the rate of the negative control.
Selected compounds showing inhibition in the primary screen were validated using kinetic measurements with l‐P5C as the variable substrate and NADH fixed at 175 μM. Compound stock solutions were prepared as described above. Ten microliter of the hit compound (0–200 mM) were added to each well. Then, 40 μL of neutralized l‐P5C (0–5000 μM) was added to each well. The mastermix (150 μL) containing PYCR1, NADH, and assay buffer was added via multichannel pipette to start the reaction. The final concentration of PYCR1 was 10 nM. Initial velocities were calculated as described above for the primary screen. The initial rate data for each inhibitor were fitted globally to the competitive model of inhibition using Origin software to obtain the inhibition constant, K i. Concentration–response data for 70 were fit to the generalized dose–response function for enzyme inhibition (equation 8.25 of Copeland, 2000).
4.2. Co‐crystallization of PYCR1‐inhibitor complexes
Protein solutions used for co‐crystallization included 12 mg mL−1 PYCR1 and 10–30 mM of inhibitor. Table S4 lists the inhibitor concentrations used in co‐crystallization. In most cases, NADH (2 mM) was also used during crystallization. Crystals were grown in sitting drops using a reservoir solution of 0.34–0.4M Li2SO4, 0.1M HEPES pH 7.5, and 17–22% (w/v) PEG 3350. Sitting drops were made by combining 2 μL of reservoir solution with 2 μL of protein‐ligand solution. Drops were seed‐streaked with unliganded PYCR1 microcrystals. Crystals were prepared for cryogenic data collection by soaking in a solution containing 0–0.04M Li2SO4, 0.1M HEPES pH 7.5, 22% (w/v) PEG 3350, and 20% PEG 200, plus additional inhibitor at 25–125 mM and then flash‐cooled in liquid nitrogen. Table S4 lists the inhibitor concentrations used in cryo‐protection. This method produced the C2 crystal form described previously (Meeks et al., 2024).
A different crystallization approach was used for compound 43. The above crystallization recipe contains sulfate ion, which can compete with the inhibitor for the P5C/proline binding site (Meeks et al., 2024). Because of the similarity of sulfate ion and the sulfonate group of 43, we anticipated challenges in distinguishing 43 from sulfate ion based on electron density. Therefore, we used an alternative crystallization reservoir containing 0.01M MgCl2, 0.1M HEPES pH 7.5, and 24% poly(acrylic acid sodium salt) 5100, which had been discovered during crystal screening trials. The protein solution contained 10 mM of 43 and 2 mM NADH. The crystal was cryoprotected with 0.04M MgCl2, 0.1M HEPES pH 7.5, 25% poly(acrylic acid sodium salt) 5100, and 20% PEG 200, plus 50 mM of 43, and then flash‐cooled in liquid nitrogen. Despite the different crystallization solution, the crystal form is the same as that of the PEG 3350 recipe.
4.3. X‐ray crystal structure determination of PYCR1 complexes
Shutterless X‐ray diffraction data were collected at the Advanced Photon Source using NECAT beamlines 24‐ID‐C and 24‐ID‐E. The data were processed with XDS (Kabsch, 2010) and Aimless (Evans & Murshudov, 2013) through the automated pipeline of the beamline. The space group is C2 with five molecules in the asymmetric unit arranged as one‐half of a pentamer‐of‐dimers decamer. Data collection statistics are listed in Table S2.
Initial phases were calculated by Fourier synthesis using a model derived from PDB entry 6XP3. Iterative cycles of refinement and model building were performed with Phenix (Afonine et al., 2012) and Coot (Emsley et al., 2010). The B‐factor model consisted of one TLS group per protein chain and isotropic B‐factors for all non‐hydrogen atoms. Phenix eLBOW was used to generate restraints and initial coordinates of ligands from the three‐digit PDB residue name (if available) or the SMILES string in the case of ligands that were not in the PDB (Moriarty et al., 2009). The occupancies of NADH and inhibitors were refined, starting from an initial value of ~0.7; the range of occupancies in the final structures are listed in Table S2. NADH in one of the chains of the complex with 39 was modeled in two conformations, which differ in the orientation of the amide group. The structures were validated using MolProbity (Chen et al., 2010) and the wwPDB validation service (Gore et al., 2017). Modeling of ligands was validated with polder omit maps (Liebschner et al., 2017). Refinement statistics are listed in Table S2. The coordinates and structure factor amplitudes of the PYCR1 structures have been deposited in the PDB under the following accession codes, which are also listed in Table S2: 8TD2 (7); 8TD3 (10), 8TD4 (22), 8TD5 (23), 8TD6 (27), 8TD7 (36), 8TD8 (39), 8TD9 (41), 8TDB (43), 8TDC (66), 8TDD (70), 8VRE (NFLP).
4.4. PYCR3 enzyme activity assays
Recombinant PYCR3 was expressed as a SUMO fusion and purified as described previously (Meeks & Tanner, 2023). PYCR3 activity was assayed with the same experimental setup as PYCR1. The final l‐P5C and NADH concentration were 300–400 and 175 μM, respectively. The protein concentration was 1 nM.
4.5. Enzyme assays and X‐ray crystallography of PRODH
A bacterial PRODH domain construct engineered for inhibitor discovery (SmPutAΔα2) was used for enzyme activity assays and X‐ray crystallography. SmPutAΔα2 was expressed and purified as described previously (Bogner et al., 2022). The PRODH activity assay uses l‐proline as the substrate and measures the production of the product P5C trapped as a covalent adduct with ortho‐aminobenzaldehyde at 443 nm. Menadione is included as the electron acceptor for the reduced FAD to enable catalytic cycling. For screening assays, l‐proline, ortho‐aminobenzaldehyde, and menadione were fixed at 25, 2.5, and 0.15 mM, respectively. The final protein concentration of SmPutAΔα2 was 200 nM. Each compound in the library was tested at 5 mM. The percent relative activity was calculated as described above for PYCR1 assays. Compounds 1–4, 12, 18–20, and 22–34, and 36–71 were tested for inhibition in this study; data for 5–11, 13–17, 21, and 35 were published previously (Bogner & Tanner, 2022).
The crystal structure of SmPutAΔα2 complexed with 43 was determined. Crystals were grown in sitting drops using conditions reported previously (Bogner et al., 2022). The drops were formed by combining equal volumes of the enzyme‐inhibitor solution (10 mg mL−1 SmPutAΔα2, 20 mM 43) and reservoir (0.2M sodium formate, 20% (w/v) PEG 3350). The crystal was prepared for low‐temperature data collection by soaking in cryobuffer containing 20% PEG 200 and 20 mM of mM 43 prior to flash‐cooling in liquid nitrogen. The starting coordinates for structure refinement were obtained from PDB entry 8DKP. Data collection and refinement statistics are summarized in Table S3. The coordinates and structure factor amplitudes of the SmPutAΔα2‐43 structure have been deposited in the PDB under the accession code 8W0K.
AUTHOR CONTRIBUTIONS
Kaylen R. Meeks: Conceptualization; methodology; investigation; writing – original draft; writing – review and editing; visualization. Alexandra N. Bogner: Conceptualization; methodology; investigation; writing – review and editing. John J. Tanner: Conceptualization; methodology; supervision; project administration; visualization; funding acquisition; writing – original draft; writing – review and editing.
FUNDING INFORMATION
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R01GM132640.
Supporting information
Data S1. Supporting information.
ACKNOWLEDGMENTS
We thank Jonathan Schuerman and Igor Kourinov for help with X‐ray diffraction data collection and processing. We thank Oseeyi Daudu from the Becker lab at University of Nebraska for supplying d,l‐P5C. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The Eiger 16M detector on 24‐ID‐E is funded by a NIH‐ORIP HEI grant (S10OD021527). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE‐AC02‐06CH11357.
Meeks KR, Bogner AN, Tanner JJ. Screening a knowledge‐based library of low molecular weight compounds against the proline biosynthetic enzyme 1‐pyrroline‐5‐carboxylate 1 (PYCR1) . Protein Science. 2024;33(7):e5072. 10.1002/pro.5072
Review Editor: John Kuriyan
DATA AVAILABILITY STATEMENT
Crystallographic data for the structures reported in this article have been deposited at the Protein Data Bank with the accession numbers 8TD2 (PYCR1‐7), 8TD3 (PYCR1‐10), 8TD4 (PYCR1‐22), 8TD5 (PYCR1‐23), 8TD6 (PYCR1‐27), 8TD7 (PYCR1‐36), 8TD8 (PYCR1‐39), 8TD9 (PYCR1‐41), 8TDB (PYCR1‐43), 8TDC (PYCR1‐66), 8TDD (PYCR1‐70), 8VRE (PYCR1‐NFLP), and 8W0K (SmPutAΔα2‐43).
REFERENCES
- Afonine PV, Grosse‐Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr. 2012;68(Pt 4):352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alaqbi SS, Burke L, Guterman I, Green C, West K, Palacios‐Gallego R, et al. Increased mitochondrial proline metabolism sustains proliferation and survival of colorectal cancer cells. PLoS One. 2022;17(2):e0262364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatore C, Huryn DM, Smith AB 3rd. Carboxylic acid (bio)isosteres in drug design. ChemMedChem. 2013;8(3):385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogner AN, Ji J, Tanner JJ. Structure‐based engineering of minimal proline dehydrogenase domains for inhibitor discovery. Protein Eng Des Sel. 2022;35:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogner AN, Stiers KM, Tanner JJ. Structure, biochemistry, and gene expression patterns of the proline biosynthetic enzyme pyrroline‐5‐carboxylate reductase (PYCR), an emerging cancer therapy target. Amino Acids. 2021;53(12):1817–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogner AN, Tanner JJ. Structure‐affinity relationships of reversible proline analog inhibitors targeting proline dehydrogenase. Org Biomol Chem. 2022;20(4):895–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai F, Miao Y, Liu C, Wu T, Shen S, Su X, et al. Pyrroline‐5‐carboxylate reductase 1 promotes proliferation and inhibits apoptosis in non‐small cell lung cancer. Oncol Lett. 2018;15(1):731–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell AC, Prater AR, Bogner AN, Quinn TP, Gates KS, Becker DF, et al. Photoinduced covalent irreversible inactivation of proline dehydrogenase by S‐heterocycles. ACS Chem Biol. 2021;16(11):2268–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, et al. MolProbity: all‐atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;D66(Pt 1):12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen EM, Bogner AN, Vandekeere A, Tam GS, Patel SM, Becker DF, et al. In crystallo screening for proline analog inhibitors of the proline cycle enzyme PYCR1. J Biol Chem. 2020;295(52):18316–18327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen EM, Patel SM, Korasick DA, Campbell AC, Krause KL, Becker DF, et al. Resolving the cofactor‐binding site in the proline biosynthetic enzyme human pyrroline‐5‐carboxylate reductase 1. J Biol Chem. 2017;292(17):7233–7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland RA. Enzymes: a practical Introduction to structure, mechanism, and data analysis. New York, NY: Wiley‐VCH; 2000. [Google Scholar]
- Cui B, He B, Huang Y, Wang C, Luo H, Lu J, et al. Pyrroline‐5‐carboxylate reductase 1 reprograms proline metabolism to drive breast cancer stemness under psychological stress. Cell Death Dis. 2023;14(10):682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug‐likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Kuo ML, Su L, Xue L, Luh F, Zhang H, et al. Human mitochondrial pyrroline‐5‐carboxylate reductase 1 promotes invasiveness and impacts survival in breast cancers. Carcinogenesis. 2017;38(5):519–531. [DOI] [PubMed] [Google Scholar]
- Ding Z, Ericksen RE, Escande‐Beillard N, Lee QY, Loh A, Denil S, et al. Metabolic pathway analyses identify proline biosynthesis pathway as a promoter of liver tumorigenesis. J Hepatol. 2020;72:725–735. [DOI] [PubMed] [Google Scholar]
- Du S, Sui Y, Ren W, Zhou J, Du C. PYCR1 promotes bladder cancer by affecting the Akt/Wnt/beta‐catenin signaling. J Bioenerg Biomembr. 2021;53(2):247–258. [DOI] [PubMed] [Google Scholar]
- Elia I, Broekaert D, Christen S, Boon R, Radaelli E, Orth MF, et al. Proline metabolism supports metastasis formation and could be inhibited to selectively target metastasizing cancer cells. Nat Commun. 2017;8:15267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of coot. Acta Cryst D Biol Crystallogr. 2010;66(Pt 4):486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlanson DA. Fragment‐based lead discovery: a chemical update. Curr Opin Biotechnol. 2006;17(6):643–652. [DOI] [PubMed] [Google Scholar]
- Evans PR, Murshudov GN. How good are my data and what is the resolution? Acta Crystallogr D Biol Crystallogr. 2013;69(Pt 7):1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang K, Sun M, Leng Z, Chu Y, Zhao Z, Li Z, et al. Targeting IGF1R signaling enhances the sensitivity of cisplatin by inhibiting proline and arginine metabolism in oesophageal squamous cell carcinoma under hypoxia. J Exp Clin Cancer Res. 2023;42(1):73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii T, Mukaihara M, Agematu H, Tsunekawa H. Biotransformation of L‐lysine to L‐pipecolic acid catalyzed by L‐lysine 6‐aminotransferase and pyrroline‐5‐carboxylate reductase. Biosci Biotechnol Biochem. 2002;66(3):622–627. [DOI] [PubMed] [Google Scholar]
- Gao Y, Luo L, Xie Y, Zhao Y, Yao J, Liu X. PYCR1 knockdown inhibits the proliferation, migration, and invasion by affecting JAK/STAT signaling pathway in lung adenocarcinoma. Mol Carcinog. 2020;59(5):503–511. [DOI] [PubMed] [Google Scholar]
- Gore S, Sanz Garcia E, Hendrickx PMS, Gutmanas A, Westbrook JD, Yang H, et al. Validation of structures in the protein data Bank. Structure. 2017;25(12):1916–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallen A, Jamie JF, Cooper AJ. Lysine metabolism in mammalian brain: an update on the importance of recent discoveries. Amino Acids. 2013;45(6):1249–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu CA. Isozymes of P5C reductase (PYCR) in human diseases: focus on cancer. Amino Acids. 2021;53(12):1835–1840. [DOI] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66(Pt 2):125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalgutkar AS, Daniels JS. Carboxylic acids and their bioisosteres. In: Smith DA, editor. Metabolism, pharmacokinetics and toxicity of functional groups: impact of the building blocks of medicinal chemistry on ADMET. London: The Royal Society of Chemistry; 2010:99–167. [Google Scholar]
- Korasick DA, Tanner JJ. Impact of missense mutations in the ALDH7A1 gene on enzyme structure and catalytic function. Biochimie. 2021;183:49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowaloff EM, Phang JM, Granger AS, Downing SJ. Regulation of proline oxidase activity by lactate. Proc Natl Acad Sci U S A. 1977;74(12):5368–5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassalas P, Gay B, Lasfargeas C, James MJ, Tran V, Vijayendran KG, et al. Structure property relationships of carboxylic acid isosteres. J Med Chem. 2016;59(7):3183–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Bie J, Song C, Liu M, Luo J. PYCR, a key enzyme in proline metabolism, functions in tumorigenesis. Amino Acids. 2021;53(12):1841–1850. [DOI] [PubMed] [Google Scholar]
- Li Y, Xu J, Bao P, Wei Z, Pan L, Zhou J, et al. Survival and clinicopathological significance of PYCR1 expression in cancer: a meta‐analysis. Front Oncol. 2022;12:985613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Jiang Y, Liu J, Fu H, Yang Q, Song W, et al. Exosomes from PYCR1 knockdown bone marrow mesenchymal stem inhibits aerobic glycolysis and the growth of bladder cancer cells via regulation of the EGFR/PI3K/AKT pathway. Int J Oncol. 2023;63(1):84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Liu J, Fu H, Li Y, Liu Q, Song W, et al. SENP3 affects the expression of PYCR1 to promote bladder cancer proliferation and EMT transformation by deSUMOylation of STAT3. Aging (Albany NY). 2022;14(19):8032–8045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Moriarty NW, Poon BK, Sobolev OV, Terwilliger TC, et al. Polder maps: improving OMIT maps by excluding bulk solvent. Acta Crystallogr D Struct Biol. 2017;73(Pt 2):148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Sun T, Zhang Z, Bi J, Kong C. An 18‐gene signature based on glucose metabolism and DNA methylation improves prognostic prediction for urinary bladder cancer. Genomics. 2021;113(1 Pt 2):896–907. [DOI] [PubMed] [Google Scholar]
- Meeks KR, Ji J, Protopopov MV, Tarkhanova OO, Moroz YS, Tanner JJ. Novel fragment inhibitors of PYCR1 from docking‐guided X‐ray crystallography. J Chem Inf Model. 2024;64(5):1704–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeks KR, Tanner JJ. Expression and kinetic characterization of PYCR3. Arch Biochem Biophys. 2023;733:109468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriarty NW, Grosse‐Kunstleve RW, Adams PD. Electronic ligand builder and optimization workbench (eLBOW): a tool for ligand coordinate and restraint generation. Acta Crystallogr D Biol Crystallogr. 2009;65(Pt 10):1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly M, Cleasby A, Davies TG, Hall RJ, Ludlow RF, Murray CW, et al. Crystallographic screening using ultra‐low‐molecular‐weight ligands to guide drug design. Drug Discov Today. 2019;24(5):1081–1086. [DOI] [PubMed] [Google Scholar]
- Oudaert I, Satilmis H, Vlummens P, De Brouwer W, Maes A, Hose D, et al. Pyrroline‐5‐carboxylate reductase 1: a novel target for sensitizing multiple myeloma cells to bortezomib by inhibition of PRAS40‐mediated protein synthesis. J Exp Clin Cancer Res. 2022;41(1):45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phang JM. Perspectives, past, present and future: the proline cycle/proline‐collagen regulatory axis. Amino Acids. 2021;53(12):1967–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phang JM, Liu W, Hancock C, Christian KJ. The proline regulatory axis and cancer. Front Oncol. 2012;2:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees DC, Congreve M, Murray CW, Carr R. Fragment‐based lead discovery. Nat Rev Drug Discov. 2004;3(8):660–672. [DOI] [PubMed] [Google Scholar]
- Sang S, Zhang C, Shan J. Pyrroline‐5‐carboxylate reductase 1 accelerates the migration and invasion of nonsmall cell lung cancer in vitro. Cancer Biother Radiopharm. 2019;34(6):380–387. [DOI] [PubMed] [Google Scholar]
- Scott DE, Coyne AG, Hudson SA, Abell C. Fragment‐based approaches in drug discovery and chemical biology. Biochemistry. 2012;51(25):4990–5003. [DOI] [PubMed] [Google Scholar]
- Shao Z, Lu L, Cui Y, Deng L, Xu Q, Liang Q, et al. PYCR in kidney renal papillary cell carcinoma: expression, prognosis, gene regulation network, and regulation targets. Front Biosci (Landmark ed). 2022;27(12):336. [DOI] [PubMed] [Google Scholar]
- Sharif T, Dai C, Martell E, Ghassemi‐Rad MS, Hanes MR, Murphy PJ, et al. TAp73 modifies metabolism and positively regulates growth of cancer stem‐like cells in a redox‐sensitive manner. Clin Cancer Res. 2019;25(6):2001–2017. [DOI] [PubMed] [Google Scholar]
- Shenoy A, Belugali Nataraj N, Perry G, Loayza Puch F, Nagel R, Marin I, et al. Proteomic patterns associated with response to breast cancer neoadjuvant treatment. Mol Syst Biol. 2020;16(9):e9443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Struys EA, Jansen EE, Salomons GS. Human pyrroline‐5‐carboxylate reductase (PYCR1) acts on Delta(1)‐piperideine‐6‐carboxylate generating L‐pipecolic acid. J Inherit Metab Dis. 2014;37(3):327–332. [DOI] [PubMed] [Google Scholar]
- Tanner JJ, Fendt SM, Becker DF. The proline cycle as a potential cancer therapy target. Biochemistry. 2018;57(25):3433–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weijin F, Zhibin X, Shengfeng Z, Xiaoli Y, Qijian D, Jiayi L, et al. The clinical significance of PYCR1 expression in renal cell carcinoma. Medicine (Baltimore). 2019;98(28):e16384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westbrook RL, Bridges E, Roberts J, Escribano‐Gonzalez C, Eales KL, Vettore LA, et al. Proline synthesis through PYCR1 is required to support cancer cell proliferation and survival in oxygen‐limiting conditions. Cell Rep. 2022;38(5):110320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehouse RL, Alwan WS, Ilyichova OV, Taylor AJ, Chandrashekaran IR, Mohanty B, et al. Fragment screening libraries for the identification of protein hot spots and their minimal binding pharmacophores. RSC Med Chem. 2023;14(1):135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao S, Li S, Yuan Z, Zhou L. Pyrroline‐5‐carboxylate reductase 1 (PYCR1) upregulation contributes to gastric cancer progression and indicates poor survival outcome. Ann Transl Med. 2020;8(15):937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Zuo W, Wang X, Zhang Q, Gan X, Tan N, et al. Deciphering the effects of PYCR1 on cell function and its associated mechanism in hepatocellular carcinoma. Int J Biol Sci. 2021;17(9):2223–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan K, Xu X, Wu T, Li J, Cao G, Li Y, et al. Knockdown of PYCR1 inhibits proliferation, drug resistance and EMT in colorectal cancer cells by regulating STAT3‐mediated p38 MAPK and NF‐κB signalling pathway. Biochem Biophys Res Commun. 2019;520(2):486–491. [DOI] [PubMed] [Google Scholar]
- Zeng T, Zhu L, Liao M, Zhuo W, Yang S, Wu W, et al. Knockdown of PYCR1 inhibits cell proliferation and colony formation via cell cycle arrest and apoptosis in prostate cancer. Med Oncol. 2017;34(2):27. [DOI] [PubMed] [Google Scholar]
- Zhang T, Liu Y, Liu W, Li Q, Hou W, Huang Y, et al. Increased PYCR1 mRNA predicts poor prognosis in kidney adenocarcinoma: a study based on TCGA database. Medicine (Baltimore). 2021;100(38):e27145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng K, Sha N, Hou G, Leng Z, Zhao Q, Zhang L, et al. IGF1R‐phosphorylated PYCR1 facilitates ELK4 transcriptional activity and sustains tumor growth under hypoxia. Nat Commun. 2023;14(1):6117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang J, Song Y, Ye Y, He S, Ma X, Zhang M, et al. PYCR1 interference inhibits cell growth and survival via c‐Jun N‐terminal kinase/insulin receptor substrate 1 (JNK/IRS1) pathway in hepatocellular cancer. J Transl Med. 2019;17(1):343. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting information.
Data Availability Statement
Crystallographic data for the structures reported in this article have been deposited at the Protein Data Bank with the accession numbers 8TD2 (PYCR1‐7), 8TD3 (PYCR1‐10), 8TD4 (PYCR1‐22), 8TD5 (PYCR1‐23), 8TD6 (PYCR1‐27), 8TD7 (PYCR1‐36), 8TD8 (PYCR1‐39), 8TD9 (PYCR1‐41), 8TDB (PYCR1‐43), 8TDC (PYCR1‐66), 8TDD (PYCR1‐70), 8VRE (PYCR1‐NFLP), and 8W0K (SmPutAΔα2‐43).