

Keywords: caspases, cell death, gasdermins, inflammasomes, pyroptosis
Abstract
The field of cell death has witnessed significant advancements since the initial discovery of apoptosis in the 1970s. This review delves into the intricacies of pyroptosis, a more recently identified form of regulated, lytic cell death, and explores the roles of pyroptotic effector molecules, with a strong emphasis on their mechanisms and relevance in various diseases. Pyroptosis, characterized by its proinflammatory nature, is driven by the accumulation of large plasma membrane pores comprised of gasdermin family protein subunits. In different contexts of cellular homeostatic perturbations, infections, and tissue damage, proteases, such as caspase-1 and caspase-4/5, play pivotal roles in pyroptosis by cleaving gasdermins. Gasdermin-D (GSDMD), the most extensively studied member of the gasdermin protein family, is expressed in various immune cells and certain epithelial cells. Upon cleavage by caspases, GSDMD oligomerizes and forms transmembrane pores in the cell membrane, leading to the release of proinflammatory cytokines. GSDMD-N, the NH2-terminal fragment, displays an affinity for specific lipids, contributing to its role in pore formation in pyroptosis. While GSDMD is the primary focus, other gasdermin family members are also discussed in detail. These proteins exhibit distinct tissue-specific functions and contribute to different facets of cell death regulation. Additionally, genetic variations in some gasdermins have been linked to diseases, underscoring their clinical relevance. Furthermore, the interplay between GSDM pores and the activation of other effectors, such as ninjurin-1, is elucidated, providing insights into the complexity of pyroptosis regulation. The findings underscore the molecular mechanisms that govern pyroptosis and its implications for various physiological and pathological processes.
INTRODUCTION
Regulated cell death is a crucial part of a cell’s life cycle and a significant event that contributes to physiologically essential processes in multicellular organisms, including the removal of unwanted cells, formation of organs, and maturation of immune cells. The first description of regulated, in other words programmed, cell death dates back to the 1970s, when the shrinkage and blebbing of cells were observed and the term apoptosis (from the Greek apo for off/away and ptosis for falling) was given to this type of cell demise (1). Initially, the characterization of cell death was merely restricted to a simple apoptosis/necrosis distinction, in which necrosis was identical to all types of lytic cell and tissue destruction following a physiologically intolerable event. In contrast, apoptosis was considered a cell death modality that followed a regulated, genetically programmed process. Apoptosis was distinct in its morphological appearance, showing a decrease in cell volume, condensation of the nucleus, nuclear fragmentation, and preservation of cytoplasm membrane intactness until the late stages of the process. All these distinctive traits of apoptosis enable the immunologically silent removal of compromised cells from the organism (2). During the early 1990s, the primary regulatory components responsible for this form of cell death were identified and coined with the term caspase, derived from the condensation of two words: cysteine and aspartase. Caspases exist in zymogen (inactive) forms within the cells and become activated in a cascade-like manner upon an apoptotic stimulus (3). In the early 2000s, a novel form of regulated cell death was identified in Shigella-infected macrophages, which shared features with necrosis in that it exhibited early cytoplasm membrane damage accompanied by osmotic imbalances and release of proinflammatory content to the extracellular space. The newly identified cell death process was given the name pyroptosis (4) by combining the terms pyro (from the Greek for fire) and ptosis, reflecting its highly proinflammatory nature. Pyroptosis was first associated with inflammatory caspase activation, the release of proinflammatory cytokines, and cell death. This general pathway was first defined by studies of inflammasomes, which are high molecular protein complexes. In response to stimuli, inflammatory caspases, such as caspase-1, caspase-4/5 in humans, and caspase-11 in mice, are recruited in inflammasomes (5). Inflammasomes are pathogen and stress sensors that recognize damage-associated molecular patterns (DAMPs), pathogen-associated molecular patterns (PAMPs), and metabolomic perturbations (6). Inflammatory caspases and initiator apoptotic caspases consist of a large prodomain, which facilitates their recruitment to active inflammasomes. Caspase-1 activation in the inflammasome results in the processing of prointerleukin-1β (IL-1β) and proIL-18 (7). Activation of both caspase-1 and caspase-4/5 (caspase-11 in mice) can result in the cleavage of gasdermin-D (GSDMD), one of the key effector proteins of pyroptosis (8). The NH2-terminal region of GSDMD is inserted into the lipid bilayer of the cytoplasm membrane and forms oligomeric pores (9), leading to the release of mature ILs into the extracellular space (10) and pyroptotic cell death. In light of recent findings, the field now widely recognizes pyroptosis as a lytic mode of proinflammatory cell death that is mediated by various members of the gasdermin family, including gasdermin-A, -B, -C, -D, and -E, and that inflammasome activation is not the only way to initiate gasdermin pore formation. In this review, the current knowledge of the cellular mechanisms that initiate, regulate, and execute the process of pyroptosis is discussed with a strong emphasis on the effector mechanisms and their relevance in diseases.
INITIATION OF PYROPTOSIS BY INFLAMMASOME ACTIVATION
Activation of large protein complexes called inflammasomes serves as a pivotal component of innate immune signaling, resulting in both inflammation and cell death (11). Various pathogens and danger signals trigger sensor proteins of inflammasomes, leading to the assembly of active inflammasome complexes, yet the mere presence of genes responsible for encoding inflammasome components in a cell does not necessarily imply the formation or activation of an inflammasome. In humans and mice, inflammasome components are expressed in various cell types, including both immune (12–20) and nonimmune cells (21–23). Several cell types from humans and mice exhibit inflammasome activation, including monocytes (12), macrophages (13, 18), neutrophils (14), peripheral blood mononuclear cells (15), T lymphocytes (16), and dendritic cells (17, 19). Intestinal epithelial cells (21), airway epithelial cells (22), and keratinocytes (23) also assemble active inflammasome complexes.
Different cell types and host species can assemble distinct inflammasome complexes and the activation of inflammasomes in these cell types can result in diverse biological outcomes and consequences. Specific inflammasome complexes are formed depending on which inflammasome sensor is activated. Typically, the sensor components of inflammasomes are the member of the nucleotide-binding domain (NBD), leucine-rich repeat (LRR)-containing protein (NLR) family with a pyrin domain (NLRP) (24, 25), including NLRP1, NLRP3, NLRP6, but other members, such as absent in melanoma 2 (AIM2), pyrin, caspase-4/5 (caspase-11 in mice), NLR containing caspase recruitment domain-containing protein (CARD) domain 4 (NLRC4), and caspase recruitment domain-containing protein 8 (CARD8) (18, 26–35) form also inflammasomes. When activated, most inflammasome sensors recruit an adaptor protein known as apoptosis-associated speck-like protein containing a CARD [ASC or PYCARD], and the protease enzyme caspase-1. Canonical inflammasomes consist of activated inflammasome sensor, ASC, and caspase-1, which create a functional inflammasome complex (25). Within this complex, caspase-1 catalyzes the proteolytic cleavage of proinflammatory cytokines and GSDMD (discussed later in detail).
The first identified inflammasome sensor, human NLRP1 (hNLRP1), becomes active in response to certain viral proteases like the 3 C protease of human coronavirus (26) and double-stranded RNA (dsRNA) and in the presence of the peptidase inhibitor Val-boroPro (VbP) (27). Highly expressed in immune cells, NLRP3 becomes active in response to several PAMPs or DAMPs, which initiate changes in ion homeostasis and organelle dysfunction (28, 29). NLRP6, an inflammasome component highly expressed in the intestinal epithelium, is triggered by the lipoteichoic acid of bacteria and other microbial byproducts (30). Pyrin was first identified as the genetic factor of familial Mediterranean fever. Pyrin becomes active when bacterial toxins inhibit Rho GTPases (18). Absent in melanoma 2 (AIM2), an intracellular DNA sensor, initially identified as a tumor suppressor, recognizes double-stranded DNA (dsDNA) derived from pathogens, including human papillomavirus (31). Neuronal apoptosis inhibitory proteins (NAIPs) can detect bacterial flagellin and proteins found in the bacterial type III secretion system (T3SS) apparatus. An example of this is Salmonella T3SS. The detection of these bacterial proteins leads to the activation of NLRC4 inflammasome (32, 33). CARD8 is activated by HIV protease activity through the dimerization of the HIV Gag-Pol polyprotein and the presence of the inhibitor VbP (34). Caspases, including caspase-4/5 in humans and caspase-11 in mice, recognize the bacterial cell wall component lipopolysaccharides (LPS) (35) and serve as noncanonical inflammasomes which require neither NLRs as initiators nor ASC as an adapter protein for activation.
This section attempted to give a brief overview of the various forms of inflammasomes and their way of stimulation. The activation of all these pathways converges in inflammatory caspase activation, rendering the induction of downstream pyroptotic effectors. Recent reviews dedicated to inflammasomes (25, 36, 37) offer more detailed insights into various inflammasome complexes and advances in the field of inflammasome biology. In the next sections of this review, the primary emphasis is placed on the comprehensive discussion of the effector mechanisms of pyroptosis.
EFFECTOR MECHANISMS OF PYROPTOSIS
GSDMD as the main effector of pyroptosis was first identified by two research groups simultaneously by employing genome-wide genetic screening and by using forward genetic approaches in mouse macrophages in 2015 (8, 38). These two studies showed that the linker region between the COOH- and NH2-terminal domains of GSDMD was cleaved by inflammatory caspases, resulting in pyroptotic activity. GSDMD is a member of the gasdermin (GSDM) protein family (Fig. 1). Even though GSDMD is the most widely studied gasdermin, there is accumulating evidence showing that other members of this protein family contribute to cell death processes, such as pyroptosis and apoptosis. In humans, the gasdermin protein family consists of 6 members including GSDMA, B, C, D, E, and F (commonly known as pejvakin) (41). In general, GSDMs consist of an NH2-terminal and a COOH-terminal structured domain connected with a linker region. The only exception is pejvakin, which lacks the linker region between the two domains, and its pore formation potential is still debated (42). The NH2-terminal domain of gasdermins is responsible for oligomerization and subsequent pore formation in phospholipid bilayer, whereas the COOH-terminal part serves as an inhibitory structure. Consequently, full-length gasdermins, only with very few exceptions (see gasdermin-A3 below), are unable to oligomerize and form transmembrane pore structures. In most cases, protease cleavage in the linker region is necessary to free the NH2-terminal gasdermin fragment and initiate the process of membrane binding and oligomerization (8, 9, 38, 43). Expanding upon the shared mechanisms applicable to the GSDM family, individual members differ in specific proteases that cleave the linker region, in their binding affinity to specific membrane phospholipids, and in localization to certain subcellular compartments. They also participate in cell death regulation in various extents and in a context and tissue-specific fashion.
Figure 1.

Pore formation mechanisms of the gasdermin (GSDM) protein family. A: GSDMA is cleaved by the bacterial protease SpeB, leading to the free NH2-terminal (N) fragment. C, COOH terminal. Consequently, this results in membrane insertion in mitochondria and, with delayed kinetics, in cytoplasmic membrane pore formation by oligomerization. B: GSDMB is cleaved by granzyme A secreted by natural killer (NK) cells. The NH2-terminal fragment can be ubiquitinated, leading to its proteasomal degradation. Free NH2-terminal fragments insert into the cytoplasmic membrane and form pores. Apoptotic effector caspase-3 leads to GSDMB inactivation. C: GSDMC is cleaved by the apoptotic initiator caspase-8, resulting in a pore-forming NH2-terminal fragment. D: GSDMD is cleaved by inflammatory caspases, caspase-8, neutrophil N-elastase (39), and lysosomal enzyme cathepsin G. The NH2-terminal fragment is modified by reactive oxygen species (ROS), ubiquitination, and fumarate (40), rendering its membrane insertion and pore formation. Apoptotic effector caspase-3 leads to GSDMD inactivation. E: GSDME is cleaved by apoptotic effector caspase-3 and granzyme-B to form the active NH2-terminal fragment.
Gasdermin-A, -B, and -C
Gasdermin-A (GSDMA) is largely expressed in the gastrointestinal tract, lower esophagus, stomach, and skin epithelium, and it is frequently silenced in gastric cancers (44, 45). Based on recent studies, GSDMA is cleaved by the bacterial protease SpeB (Fig. 1A) produced by a major skin pathogen Streptococcus pyogenes (46). When GSDMA is cleaved into the NH2-terminal fragment, it can bind membrane phosphoinositides and cardiolipin resulting in oligomerization, pore formation, and membrane leakage (47). Given the cardiolipin binding capacity of GSDMA, it preferentially binds to the cardiolipin-rich mitochondrial membrane, and, accordingly, it accumulates only with delayed kinetic at the plasma membrane. Consequently, NH2-terminal GSDMA results in mitochondrial dysfunction and elevated reactive oxygen species (ROS) production (48, 49). The function of GSDMA seems to be also essential in epidermal cornification during normal skin regeneration processes (50). Earlier studies of murine gasdermin-A3 based on point mutations that caused hair loss in mice determined that the mutations disrupted the stability of the inhibitory interface between the NH2- and COOH-terminal lobes of the full-length gasdermin-A3. These mutations resulted in accumulation of GSDMA3 pores in mitochondria and consequently mitochondrial ROS-dependent cell death without any cleavage event (48, 51).
Gasdermin-B (GSDMB) is closely related to GSDMA, but it was shown to be expressed in intensively proliferating cells (44). Polymorphism in both GSDMA and GSDMB genes is associated with childhood asthma susceptibility. Additionally, several studies conclude GSDMDB gene association with asthma onset (52, 53). GSDMB is also overexpressed in metastatic breast cancer cells and promotes invasion and metastasis (54). Unlike other gasdermins, even intact GSDMB is capable of binding membrane phosphoinositides and, uniquely, sulphatides, the components of the apical membrane of epithelial cells. The crystal structure of the GSDMB COOH-terminal domain reveals that amino acids encoded by single nucleotide polymorphisms that are linked to asthma and inflammatory bowel disease result in a less flexible configuration and higher positive surface charge compared to the GSDMB from healthy individuals. GSDMB is not cleaved by caspase-1, -4, and -5, but it is cleaved by apoptotic caspase-3, -6, and -7 (Fig. 1B). Intriguingly, caspases cleave GSDMB inside the NH2-terminal domain and not in the linker region, resulting in a fragment unable to function in pore formation and pyroptosis (55). Supporting this, recent data confirm that cleavage of GDSMB by caspase-7 leads to pyroptosis inhibition instead of activation (56). GSDMB is also cleaved by granzyme-A, a protease produced by natural killer cells and cytotoxic T lymphocytes. This cleavage takes place within the linker region between the NH2- and COOH-terminal domains, which consequently results in pore formation and cancer cell pyroptosis (57). GSDMB is ubiquitinated and thus targeted for degradation by Ipah7.8 a protein secreted by the intracellular human enteropathogenic Shigella to block bactericidal activity (58, 59). Full-length GSDMB by itself does not induce cell death, but it facilitates caspase-4 activity and GSDMD cleavage in leukocytes (60). GSDMB also has pyroptosis-independent functions, for instance, in epithelial barrier repair (61). The conflicting data on GDSMB regarding pyroptosis susceptibility and oncogenic activity can be explained by the observation that GSDMB has several splicing variants that differ in their sequence of linker regions. This attribute defines which GSDMB isoform exhibits pyroptotic potential (62, 63). A recent study demonstrates that isoform 1 and 2 expressing tumor cells are susceptible to apoptosis only, isoform 3 containing cells exhibit pyroptosis susceptibility, and isoform 4 shows a mixed apoptosis/pyroptosis susceptibility. These isoforms likely differ in their membrane binding potential (63).
Gasdermin-C (GSDMC) has been implicated in the inflammatory response to parasitic infections, and genetic variation in GSDMC is associated with lower back pain (64). GSMDC triggers pyroptosis in human embryonal kidney cells. Immune response to parasitic worm infection initiates overexpression of GSDMC in mouse intestine, leading to lytic cell death, which may account for villus atrophy (65). Similarly, helminth infection leads to overexpression of GSDMC and results in pore formation and noncanonical release of IL-33, which is indispensable for anti-helminth immunity. GDSMC is cleaved to an NH2-terminal fragment (GSDMC-N) upon infection (66). GSDMC is cleaved by the apoptotic initiator caspase-8. Transcription of GSDMC is facilitated under hypoxia, and TNF-α stimulation initiates caspase-8 activation-driven GSDMC cleavage into GSDMC-N and pyroptosis in cancer cells (67). In concert with this, GSDMC was shown to be recruited to the internalized death receptor complex and cleaved by caspase-8 (Fig. 1C; Ref. 68).
Gasdermin-D
GSDMD stands out as the inaugural member of the GSDM family to be discovered in connection with pyroptosis. (8, 38) and the most widely studied gasdermin to date. GSDMD is highly expressed in immune cells, including macrophages, dendritic cells, and lymphocytes, and is present in some epithelial cell types of the gastrointestinal tract and endothelium (41).
GSDMD possesses a linker region between the NH2- and COOH- terminal domains, which is cleaved by active inflammatory caspase-1 and -4/5 (caspase-11 in mice; Fig. 1D). Caspase-1 is a common effector activated in inflammasomes in response to danger signals, whereas caspase-4/5 can be directly activated by LPS binding (35). Additionally, GSDMD is also cleaved by apoptotic caspase-8 upon Yersinia infection (69). The role of caspase-8 is further confirmed by the observation that cellular FLICE-like inhibitory protein (cFLIP) protects macrophages from LPS-induced pyroptosis via inhibition of caspase-8 containing complex formation (70). Finally, the lysosomal protease cathepsin-G can also cleave GSDMD. Supporting the role of cathepsin-G, serpins, endogenous inhibitors of cathepsins, negatively regulate pyroptosis in macrophages. (71). GSDMD is also cleaved by the SARS-CoV2 protease 3CL (26). Unlike apoptotic initiator caspase-8, the apoptotic effector caspase-3 cleaves GSDMD within the NH2-terminal domain, thereby leading to its inactivation (72). In conclusion, caspase-8 and -3 play opposite roles in GSDMD activation. In canonical apoptosis, caspase-8 activation is followed by caspase-3 activation, hence GSDMD-N fragments produced by caspase-8 can be quickly inactivated by the subsequent caspase-3 activity. If we hypothesize that the GSDMD-N fragments already inserted in membranes are not accessible by active caspase-3, this mechanism might serve as a molecular timer to control the number of GSDMD pores by limiting their formation for the period between the activation of caspase-8 and the action of caspase-3. Caspase-3 inhibits GSDMD pore formation but promotes pannexin-1 channel formation during extrinsic apoptosis. More studies conclude that Impaired GSDMD pore formation or absence of GSDMD does not guarantee protection from cell death; instead, it directs the cells to the apoptotic caspase-8 and -3/-7 axis (73–75).
GSDMD-N fragments efficiently bind phosphatidylinositol phosphates, and they have an affinity to phosphatidylserine (PS), which is restricted to the cell membrane inner leaflet. Mycobacterium tuberculosis PtpB dephosphorylates phosphatidylinositol-4-monophosphate and phosphatidylinositol-(4, 5)-bisphosphate in the host cell membrane, thereby inhibiting the membrane localization of cleaved GSDMD in macrophages (76). GSDMD-N also binds cardiolipin, which is present in mitochondria and bacterial membranes. Because of its selective lipid-binding affinity, GSDMD-N kills from inside the cell but does not affect neighboring mammalian cells when it is released during pyroptosis (9). Experiments employing atomic force microscopy indicate that the pore size of GSDMD can be largely variable with an average of ∼20–25 nm consisting of 33 monomers (77). The crystal structure of GSDMD reveals that disruption of the lipid-binding and oligomerization surface reduces pore-forming activity. The COOH-terminal GSDMD interacts with the NH2-terminal GSDMD, provides autoinhibition by blocking the lipid-binding surface of the GSDMD-N, and thus prevents pore formation. Cysteine 39 and cysteine 191/192 residues of neighboring GSDMD-Ns have been suggested to interact to form oligomers (9, 78). Along with this, pharmacological inhibitors, such as necrosulfonamide and disulfiram, that inhibit the reactive cysteine residue 191, disrupt GSDMD oligomerization efficiently (79, 80). NH2-acetylcysteine can abolish this effect, confirming the significance of reactivity toward thiol groups. Conversely, mutation in cysteine 191/192 has only a partial effect on pyroptosis. It suggests that these large chemical inhibitors might have additional effects, which can act at a larger interface than only one residue, resulting in complete oligomerization inhibition. LDC7559 is an additional GSDMD-N inhibitor, which blocks pyroptosis efficiently. The mechanism of its inhibitory effect is not entirely known, which suggests the interplay of multiple factors and larger interfaces in the process of efficient GSDMD oligomerization (81, 82). GSDMD cleavage is necessary but not sufficient to promote pore formation. Additional factors are needed to promote membrane insertion and oligomerization of the GSDMD-N monomers (Fig. 1D). Elevated mitochondrial reactive oxygen species (ROS) generation is one of the major additional components that lead to GSDMD-N activation (83) via the modification of cysteine 192 residue (84). Besides, in response to mitochondrial perturbation, mitochondrial ROS production directs GSDMD into the mitochondrial membrane (85). Very recent findings demonstrate that ROS-dependent palmitoylation of cysteine residues is essential to achieve membrane translocation and pore formation (86). Additionally, ubiquitination of GSDMD seems to be a significant regulatory mechanism. Based on site specificity, it may facilitate either oligomerization and pore formation (87) or promote degradation of GSDMD-N, such as it happens by the action of the Shigella protein IpaH7.8 (88).
Tissue-Specific Functions of Gasdermin-D Pores
GSDMD pores in living macrophages preferentially release positively charged and neutral cargoes and favor the passage of mature IL-1β and IL-18 over proIL forms. Acidic residues of GSDMD pores are responsible for the electrostatic filtering mechanism of the cargo (89). Macrophages tend to form GSDMD pores, which facilitate the release of IL-1β and IL-18 and act as conduits for ions (89), yet GSDMD pore formation is not restricted to macrophages. IL-33 is secreted by epithelial cells and has a significant role in initiating airway inflammatory disease. GSDMD pore formation is a key mechanism of IL-33 secretion upon allergen exposure to murine epithelial cells (90). Unlike IL-1β, IL-1α is not a substrate of caspase-1, but GSDMD mediates the maturation and release of IL-1α by initiating calcium influx and calpain activation (91). Plasma membrane pore formation by GSDMD is not a universal mechanism across all the immune cell types and there might be species-specific differences too. In human and mouse neutrophils, even though GSDMD is cleaved, it does not form plasma membrane pores, and this is mechanistically linked to marked trafficking of NH2-GSDMD to the very abundant azurophilic granules within neutrophils (92). Conversely, another study demonstrates that mouse neutrophils exhibit GSDMD-dependent plasma membrane permeabilization and IL-1β secretion in response to canonical inflammasome stimulus but not when cell death is stimulated with phorbol myristate acetate (93), highlighting the stimulus-specific complexity of pyroptotic signaling in neutrophils. Besides, GSDMD-N can create pores in the nuclear envelope of neutrophils. Extrusion of web-like structures called neutrophil extracellular traps (NETs) and concomitant cell death of neutrophils (NETosis) provides host defense against extracellular pathogens, whereas macrophage death by pyroptosis defends against intracellular pathogens. LPS stimulation of caspase-4/11 results in GSDMD cleavage. As a result, GSDMD contributes to NET formation by forming pores on the nuclear envelope and facilitates DNA expansion (94). Of note, the significance of GSDMD nuclear pores in NET formation requires further investigation because GSDMD-deficient mouse neutrophils are as competent as wild-type mouse neutrophils in producing NETs (95). In endothelial cells, LPS activates gasdermin-D, which forms mitochondrial pores and induces mitochondrial DNA (mtDNA) release into the cytosol of endothelial cells. The cytosolic mtDNA is recognized by the DNA sensor cyclic GMP-AMP synthase (cGAS) and generates the second messenger cGAMP, which suppresses endothelial cell proliferation (96).
Gasdermin-E
Gasdermin-E (GSDME) is another member of the same protein family that has NH2-terminal pore-forming domain. GSDME is not cleaved by caspase-1; instead, it is cleaved by apoptotic effector caspase-3 (97) and by granzyme-B (Fig. 1E) secreted by cytotoxic T cells and natural killer cells (98). GSDME leads to pyroptosis in cancer cells expressing GSDME treated with chemotherapy (97). GSDME is downregulated in chemotherapy-resistant cancer cells, including colon and breast cancer, and melanoma (99–101). Apoptosis induction via intrinsic or extrinsic pathway leads to caspase-3 activation. As a result, caspase-3 cleaves a variety of substrates that play a pivotal role in orchestrating apoptotic cell death. Unlike lytic cell death modalities, such as pyroptosis and necroptosis, apoptosis maintains the integrity of the cell membrane until the late stages of the process. At late stages, apoptotic cell membranes also rupture, which is known as secondary necrosis. In this process, GSDME pores take center stage (102). In the canonical apoptotic pathway, caspase-3 cleaves GSDMD-N (72) and thus inhibits pyroptosis and facilitates apoptosis instead. However, at later stages, GSDME-dependent secondary necrosis still occurs (102). Primarily, endogenous levels of GSDME act as a conduit for IL-1β release (103) and, in human T-helper cells, for IL-1α release (104) independent of cell killing potential. It seems GSDME acts as a backup mechanism to release IL-1β in GSDMD-depleted cells (105) that express moderate levels of GSDME. Contrarily, GSDME leads to pyroptosis in cells that have a high level of GSDME (97). GSDME also targets mitochondrial membranes. The mitochondrial GSDME pore formation promotes cytochrome c release, which then results in apoptosome-driven caspase-9 activation and augments caspase-3 activity (106).
Consequences of Gasdermin Pore Formation-Elicited Ion Perturbations
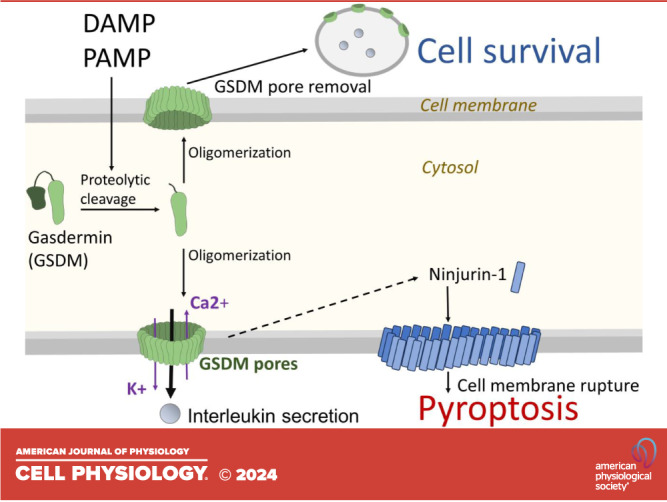
In living cells, an intact cell membrane, transmembrane ion channels, and ion pumps maintain high intracellular potassium (K+) and low calcium (Ca2+) levels (Fig. 2). This situation changes dramatically by the formation of GSDM pores. GSDM pores act as a conduit for ions, resulting in K+ efflux and Ca2+ entry into the cytoplasm (107). Increased Ca2+ and decreased K+ levels have consequences on cellular processes that impact the fate of the cells. For instance, the Ca2+ dependent phospholipid scramblase and ion channel transmembrane protein 16 F (TMEM16F) is activated (108), leading to translocation of, the normally inner membrane leaflet component, phosphatidyl serine (PS) to the outer leaflet of the plasma membrane (109). PS externalization was demonstrated to facilitate blood coagulation (109) and to play a vital role in recognition by macrophages and in triggering phagocytosis of apoptotic cell remnants (2) (Fig. 2). This indicates that pyroptosis shares common characteristics with apoptosis, not only in the activation of caspases but also in certain ultimate physiological outcomes. Ca2+ concentration increase has a major impact on membrane dynamics, including cell division and exosome formation. During GSDMD pore formation, Ca2+ influx triggers endosomal sorting complexes required for transport (ESCRT) that assist in removing GSDMD pore-containing membranes in the form of exosomes (110) (Fig. 2). This mechanism might be responsible for controlling the number and duration of pores in a calcium-dependent manner to maintain IL secretion but prevent pyroptosis. It is also demonstrated that the ESCRT complex helps to secrete IL-1β into exosomes (111). K+ efflux is one of the triggers of NLRP-3 inflammasome activation, thus GSDMD pore formation has an augmentation effect on NLRP3 inflammasome activity (28). Conversely, K+ efflux inhibits cGAS-dependent interferon-β response upon stimulus by cytosolic DNA (112), which might serve as a negative feedback loop to limit the extent of inflammatory response unleashed by GSDMD pore formation.
Figure 2.

Multifaceted impact of gasdermin-D (GSDMD) pores triggered Ca2+ influx. A: Ca2+ influx activates the endosomal sorting complexes required for transport (ESCRT) complex, leading to the removal of GSDMD pores and promoting cell survival. B: Ca2+ activates transmembrane protein 16 F (TMEM16F), resulting in phosphatidylserine (PS) externalization. Additionally, Ca2+ activates calpains, leading to cytoskeleton disruption. Ninjurin-1 (NINJ1) monomers form oligomers in the membrane. One possible mechanism of activation involves changes in membrane constitution, including PS externalization. All these events converge in cell membrane rupture.
Additional Effectors Essential for Pyroptosis
The previous examples illustrate that the formation and action of GSDM pores are regulated on multiple levels. In alignment with this, many studies agree that GSDM pores are important for releasing cytokines and transporting ions throughout the membrane, yet GSDM pore formation alone in the cytoplasm membrane does not always lead to cell death (113). The large molecule, nuclear protein high-mobility group box 1 (HMGB-1) is a chromatin-associated transcription factor. HMGB-1 normally is in the nucleus, but it can be released into the extracellular space in response to many inflammatory stimuli, where HMGB-1 acts as a DAMP. The released HMGB-1 binds to Toll-like receptors (TLRs) and triggers tissue injury by facilitating cytokine secretion of macrophages (114, 115). HMGB-1 is released during pyroptosis, but inflammasome activation alone is not sufficient to trigger HMGB-1 release. GSDMD knockout can prevent IL-1β and HMGB-1 release in vitro, suggesting that GSDMD pores are needed for both processes. However, glycine blocks pyroptosis-induced HMGB1 release without affecting IL-1β release in vitro. It suggests that HMGB-1 release is dependent on GSDMD, but GSDMD pores are not the direct route of HMGB-1 exit. This implies that pyroptosis triggers a larger pore, or other exit mechanism, downstream of GSDMD pores. On the other hand, in vivo endotoxemia-induced IL1-β appearance in the serum is completely blocked in GSDMD knockout mice, but HMGB-1 release is not. This supports the in vitro findings in that HMGB-1 does not exit through GSDMD pores but also suggests that this release is entirely independent of the presence of GSDMD in response to inflammatory cell death (116). Furthermore, GSDM pores are not large enough to release lactate dehydrogenase (LDH; 140 kDa) (117), another common marker of lytic cell death, and other macromolecules, including ASC specks (118). The release of these macromolecules and macromolecular complexes was thought to be dependent on passive cell rupture as a direct consequence of the GSDMD pore formation; however, the action of GSDM pores alone is not always sufficient to conduct cell lysis. The conventional notion that plasma membrane rupture occurs passively due to osmotic imbalances is not applicable in the context of pyroptosis. Therefore, what are the additional factors or effectors in pyroptosis that actively facilitate cell rupture?
Ninjurin-1 (NINJ1), a cell adhesion protein (119) first identified in nerve injury, plays a key role in active plasma membrane rupture (120). In normal conditions, NINJ1 is in 16-kDa monomer form embedded in the membrane. During pyroptosis, the two extracellular alpha helices of NINJ1 monomers translocate into the membrane and connect the monomers to form dense fence-like structures (up to 900 kDa in size) resulting in large pores and slits (121). These structures are responsible for the release of large macromolecules associated with lytic cell death (Fig. 2), such as LDH and HMGB-1 (120). Mutagenesis of positively charged NINJ1 residues to the noncharged glutamine unveils a mutation at lysine 45 that blocks NINJ1 cytotoxicity. However, what triggers the conformational changes of NINJ1 is largely unknown. GSDMD-driven PS externalization is one of the possible candidate mechanisms. Of note, NINJ1 mediates plasma membrane rupture universally across pyroptosis, necrosis, and apoptosis (120). Therefore, a common event such as an increase in cell volume or cytoskeleton collapse may trigger activation of NINJ1. Supporting this notion, calpain, a Ca2+-dependent protease, drives pyroptotic vimentin cleavage and intermediate filament loss, which ultimately ends with cell rupture (122). A very recent study highlights the crucial role of cell swelling in NINJ1 activation during necrosis (123). It is plausible that the interplay of large membrane pores, cell swelling, and the destruction of the cytoskeleton are all necessary to achieve fully completed cell rupture. Additionally, a further effector mechanism might be hidden in the restraining of membrane repair. Ca2+ influx stimulates ESCRT-dependent membrane repair in the form of exosomes (110), and active caspase-7 cleaves acid sphingomyelinase (ASM), leading to GSDMD pore internalization (124). These guarding mechanisms may keep the number and duration of pores at a sublytic level effectively; however, the inactivation of these repair functions might be sufficient to execute membrane rupture either alone or in combination with cytoskeleton disruption and NINJ1 (Fig. 2).
CONCLUDING REMARKS
Recent investigations have underscored the pivotal role of pyroptosis and its associated effector molecules across a diverse spectrum of pathological conditions. The activation of inflammasomes and the ensuing pyroptosis response have been extensively studied in response to viral and bacterial infections, with over 1,000 studies supporting this phenomenon. The recent revelation of the participation of gasdermin protein family in pyroptosis has opened novel avenues for research, offering promising prospects for therapeutic interventions aimed at mitigating inflammatory disorders. The significance of pyroptosis extends beyond infectious disease contexts; it has emerged as a pertinent factor in antitumor immunity (125) and is implicated in various neurological disorders, including depression (126). In the context of bone biology, the absence of GSDMD and GSDME in mice compromises fracture healing due to reduced interleukin release (127). Of note, GSDMD gene exhibits considerable genetic variation within the human population. These genetic polymorphisms can result in diverse functional alterations, ranging from the preservation of normal pyroptotic function to the inhibition of caspase cleavage and interference with oligomerization and pore formation. Consequently, structural polymorphisms in GSDMD contribute to phenotypic diversity in pyroptotic cell death susceptibility among individuals (128), highlighting its impact on individual differences in inflammatory responses, antitumor immunity, or even bone regeneration. Despite significant progress made in recent years to elucidate the mechanisms underlying pyroptosis, our understanding remains incomplete, leaving several intriguing questions unanswered. One notable question pertains to the realization that gasdermin pores, in many instances, do not suffice to trigger membrane rupture. This prompts the inquiry into the additional factors orchestrating membrane ruptures, with a proposed effector being NINJ1 (120). Antibodies specifically targeting NINJ1 completely inhibit cell death and release of LDH, HMGB-1, and other DAMPs in an in vivo model of hepatotoxicity in mice (129). However, the activation mechanisms upstream of NINJ1 remain unclear. Additionally, the absence of NINJ1 does not entirely inhibit membrane rupture in some cell types during apoptosis and necroptosis (123). This raises the question of identifying other regulators involved in this process, further underscoring the complexity of pyroptosis and its associated cellular events.
DATA AVAILABILITY
Data will be made available upon reasonable request.
GRANTS
This work was supported by the National Institute of General Medical Sciences Grant P20GM135008. This work is also supported by the National Institute of Food and Agriculture, U.S. Department of Agriculture, Accession No. 7002758, Project No. SD00H768-22.
DISCLAIMERS
The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
G.I. prepared figures; G.I. drafted manuscript; G.I edited and revised manuscript; G.I. approved final version of manuscript.
REFERENCES
- 1. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26: 239–257, 1972. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol 148: 2207–2216, 1992. doi: 10.4049/jimmunol.148.7.2207. [DOI] [PubMed] [Google Scholar]
- 3. Yuan J, Shaham S, Ledoux S, Ellis HM, Horvitz HR. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell 75: 641–652, 1993. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- 4. Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol 9: 113–114, 2001. doi: 10.1016/s0966-842x(00)01936-3. [DOI] [PubMed] [Google Scholar]
- 5. van Opdenbosch N, Lamkanfi M. Caspases in cell death, inflammation, and disease. Immunity 50: 1352–1364, 2019. doi: 10.1016/j.immuni.2019.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Janeway CA Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol 20: 197–216, 2002. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 7. Black RA, Kronheim SR, Merriam JE, March CJ, Hopp TP. A pre-aspartate-specific protease from human leukocytes that cleaves pro-interleukin-1 beta. J Biol Chem 264: 5323–5326, 1989. doi: 10.1016/S0021-9258(18)83546-3. [DOI] [PubMed] [Google Scholar]
- 8. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT, Liu PS, Lill JR, Li H, Wu J, Kummerfeld S, Zhang J, Lee WP, Snipas SJ, Salvesen GS, Morris LX, Fitzgerald L, Zhang Y, Bertram EM, Goodnow CC, Dixit VM. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526: 666–671, 2015. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 9. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535: 153–158, 2016. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, Yang ZH, Zhong CQ, Han J. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res 25: 1285–1298, 2015. doi: 10.1038/cr.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vanaja SK, Rathinam VA, Fitzgerald KA. Mechanisms of inflammasome activation: recent advances and novel insights. Trends Cell Biol 25: 308–315, 2015. doi: 10.1016/j.tcb.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gaidt MM, Ebert TS, Chauhan D, Ramshorn K, Pinci F, Zuber S, O'Duill F, Schmid-Burgk JL, Hoss F, Buhmann R, Wittmann G, Latz E, Subklewe M, Hornung V. The DNA inflammasome in human myeloid cells is initiated by a STING-cell death program upstream of NLRP3. Cell 171: 1110–1124.e18, 2017. doi: 10.1016/j.cell.2017.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Baker PJ, Boucher D, Bierschenk D, Tebartz C, Whitney PG, D'Silva DB, Tanzer MC, Monteleone M, Robertson AA, Cooper MA, Alvarez‐Diaz S, Herold MJ, Bedoui S, Schroder K, Masters SL. NLRP3 inflammasome activation downstream of cytoplasmic LPS recognition by both caspase-4 and caspase-5. Eur J Immunol 45: 2918–2926, 2015. doi: 10.1002/eji.201545655. [DOI] [PubMed] [Google Scholar]
- 14. Chen KW, Groß CJ, Sotomayor FV, Stacey KJ, Tschopp J, Sweet MJ, Schroder K. The neutrophil NLRC4 inflammasome selectively promotes IL-1beta maturation without pyroptosis during acute Salmonella challenge. Cell Rep 8: 570–582, 2014. doi: 10.1016/j.celrep.2014.06.028. [DOI] [PubMed] [Google Scholar]
- 15. Zhu S, Ding S, Wang P, Wei Z, Pan W, Palm NW, Yang Y, Yu H, Li HB, Wang G, Lei X, de Zoete MR, Zhao J, Zheng Y, Chen H, Zhao Y, Jurado KA, Feng N, Shan L, Kluger Y, Lu J, Abraham C, Fikrig E, Greenberg HB, Flavell RA. Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature 546: 667–670, 2017. doi: 10.1038/nature22967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Linder A. CARD8 inflammasome activation triggers pyroptosis in human T cells. EMBO J 39: e105071, 2020. doi: 10.15252/embj.2020105071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hatscher L, Lehmann CH, Purbojo A, Onderka C, Liang C, Hartmann A, Cesnjevar R, Bruns H, Gross O, Nimmerjahn F, Ivanović-Burmazović I, Kunz M, Heger L, Dudziak D. Select hyperactivating NLRP3 ligands enhance the TH1- and TH17-inducing potential of human type 2 conventional dendritic cells. Sci Signal 14: eabe1757, 2021. doi: 10.1126/scisignal.abe1757. [DOI] [PubMed] [Google Scholar]
- 18. Xu H, Yang J, Gao W, Li L, Li P, Zhang L, Gong YN, Peng X, Xi JJ, Chen S, Wang F, Shao F. Innate immune sensing of bacterial modifications of Rho GTPases by the pyrin inflammasome. Nature 513: 237–241, 2014. doi: 10.1038/nature13449. [DOI] [PubMed] [Google Scholar]
- 19. Koch KN, Hartung ML, Urban S, Kyburz A, Bahlmann AS, Lind J, Backert S, Taube C, Müller A. Helicobacter urease-induced activation of the TLR2/NLRP3/IL-18 axis protects against asthma. J Clin Invest 125: 3297–3302, 2015. doi: 10.1172/JCI79337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Johnson DC, Okondo MC, Orth EL, Rao SD, Huang HC, Ball DP, Bachovchin DA. DPP8/9 inhibitors activate the CARD8 inflammasome in resting lymphocytes. Cell Death Dis 11: 628, 2020. doi: 10.1038/s41419-020-02865-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Knodler LA, Crowley SM, Sham HP, Yang H, Wrande M, Ma C, Ernst RK, Steele-Mortimer O, Celli J, Vallance BA. Noncanonical inflammasome activation of caspase-4/caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host Microbe 16: 249–256, 2014. doi: 10.1016/j.chom.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Robinson KS, Teo DE, Tan KS, Toh GA, Ong HH, Lim CK, Lay K, Au BV, Lew TS, Chu JJ, Chow VT, Wang DY, Zhong FL, Reversade B. Enteroviral 3C protease activates the human NLRP1 inflammasome in airway epithelia. Science 370: eaay2002, 2020. doi: 10.1126/science.aay2002. [DOI] [PubMed] [Google Scholar]
- 23. Zhong FL, Mamaï O, Sborgi L, Boussofara L, Hopkins R, Robinson K, et al. Germline NLRP1 mutations cause skin inflammatory and cancer susceptibility syndromes via inflammasome activation. Cell 167: 187–202.e17, 2016. doi: 10.1016/j.cell.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 24. Poyet JL, Srinivasula SM, Tnani M, Razmara M, Fernandes-Alnemri T, Alnemri ES. Identification of Ipaf, a human caspase-1-activating protein related to Apaf-1. J Biol Chem 276: 28309–28313, 2001. doi: 10.1074/jbc.C100250200. [DOI] [PubMed] [Google Scholar]
- 25. Barnett KC, Li S, Liang K, Ting JP. A 360° view of the inflammasome: mechanisms of activation, cell death, and diseases. Cell 186: 2288–2312, 2023. doi: 10.1016/j.cell.2023.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Planès R, Pinilla M, Santoni K, Hessel A, Passemar C, Lay K, et al. Human NLRP1 is a sensor of pathogenic coronavirus 3CL proteases in lung epithelial cells. Mol Cell 82: 2385–2400.e9, 2022. doi: 10.1016/j.molcel.2022.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhong FL, Robinson K, Teo DE, Tan KY, Lim C, Harapas CR, Yu CH, Xie WH, Sobota RM, Au VB, Hopkins R, D'Osualdo A, Reed JC, Connolly JE, Masters SL, Reversade B. Human DPP9 represses NLRP1 inflammasome and protects against autoinflammatory diseases via both peptidase activity and FIIND domain binding. J Biol Chem 293: 18864–18878, 2018. doi: 10.1074/jbc.RA118.004350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gong T, Yang Y, Jin T, Jiang W, Zhou R. Orchestration of NLRP3 inflammasome activation by ion fluxes. Trends Immunol 39: 393–406, 2018. doi: 10.1016/j.it.2018.01.009. [DOI] [PubMed] [Google Scholar]
- 29. Seoane PI, Lee B, Hoyle C, Yu S, Lopez-Castejon G, Lowe M, Brough D. The NLRP3–inflammasome as a sensor of organelle dysfunction. J Cell Biol 219: e202006194, 2020. doi: 10.1083/jcb.202006194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hara H, Seregin SS, Yang D, Fukase K, Chamaillard M, Alnemri ES, Inohara N, Chen GY, Núñez G. The NLRP6 inflammasome recognizes lipoteichoic acid and regulates Gram-positive pathogen infection. Cell 175: 1651–1664.e14, 2018. doi: 10.1016/j.cell.2018.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Reinholz M, Kawakami Y, Salzer S, Kreuter A, Dombrowski Y, Koglin S, Kresse S, Ruzicka T, Schauber J. HPV16 activates the AIM2 inflammasome in keratinocytes. Arch Dermatol Res 305: 723–732, 2013. doi: 10.1007/s00403-013-1375-0. [DOI] [PubMed] [Google Scholar]
- 32. Miao EA, Alpuche-Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, Aderem A. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1β via Ipaf. Nat Immunol 7: 569–575, 2006. doi: 10.1038/ni1344. [DOI] [PubMed] [Google Scholar]
- 33. Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, Liu L, Shao F. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477: 596–600, 2011. doi: 10.1038/nature10510. [DOI] [PubMed] [Google Scholar]
- 34. Wang Q, Gao H, Clark KM, Mugisha CS, Davis K, Tang JP, Harlan GH, DeSelm CJ, Presti RM, Kutluay SB, Shan L. CARD8 is an inflammasome sensor for HIV-1 protease activity. Science 371: eabe1707, 2021. doi: 10.1126/science.abe1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, Hu L, Shao F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514: 187–192, 2014. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- 36. Pandey A, Shen C, Feng S, Man SM. Cell biology of inflammasome activation. Trends Cell Biol 31: 924–939, 2021. doi: 10.1016/j.tcb.2021.06.010. [DOI] [PubMed] [Google Scholar]
- 37. Tweedell RE, Kanneganti TD. Advances in inflammasome research: recent breakthroughs and future hurdles. Trends Mol Med 26: 969–971, 2020. doi: 10.1016/j.molmed.2020.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526: 660–665, 2015. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 39. Kambara H, Liu F, Zhang X, Liu P, Bajrami B, Teng Y, Zhao L, Zhou S, Yu H, Zhou W, Silberstein LE, Cheng T, Han M, Xu Y, Luo HR. Gasdermin D exerts anti-inflammatory effects by promoting neutrophil death. Cell Rep 22: 2924–2936, 2018. doi: 10.1016/j.celrep.2018.02.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Humphries F, Shmuel-Galia L, Ketelut-Carneiro N, Li S, Wang B, Nemmara VV, Wilson R, Jiang Z, Khalighinejad F, Muneeruddin K, Shaffer SA, Dutta R, Ionete C, Pesiridis S, Yang S, Thompson PR, Fitzgerald KA. Succination inactivates gasdermin D and blocks pyroptosis. Science 369: 1633–1637, 2020. doi: 10.1126/science.abb9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Huston HC, Anderson MJ, Fink SL. Pyroptosis and the cellular consequences of gasdermin pores. Semin Immunol 69: 101803, 2023. doi: 10.1016/j.smim.2023.101803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Domínguez-Ruiz M, Rodríguez-Ballesteros M, Gandía M, Gómez-Rosas E, Villamar M, Scimemi P, Mancini P, Rendtorff ND, Moreno-Pelayo MA, Tranebjaerg L, Medà C, Santarelli R, Del Castillo I. Novel pathogenic variants in PJVK, the gene encoding pejvakin, in subjects with autosomal recessive non-syndromic hearing impairment and auditory neuropathy spectrum disorder. Genes 13: 149, 2022. doi: 10.3390/genes13010149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vasudevan SO, Behl B, Rathinam VA. Pyroptosis-induced inflammation and tissue damage. Semin Immunol 69: 101781, 2023. doi: 10.1016/j.smim.2023.101781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tamura M, Tanaka S, Fujii T, Aoki A, Komiyama H, Ezawa K, Sumiyama K, Sagai T, Shiroishi T. Members of a novel gene family, Gsdm, are expressed exclusively in the epithelium of the skin and gastrointestinal tract in a highly tissue-specific manner. Genomics 89: 618–629, 2007. doi: 10.1016/j.ygeno.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 45. Saeki N, Usui T, Aoyagi K, Kim DH, Sato M, Mabuchi T, Yanagihara K, Ogawa K, Sakamoto H, Yoshida T, Sasaki H. Distinctive expression and function of four GSDM family genes (GSDMA-D) in normal and malignant upper gastrointestinal epithelium. Genes Chromosomes Cancer 48: 261–271, 2009. doi: 10.1002/gcc.20636. [DOI] [PubMed] [Google Scholar]
- 46. Deng W, Bai Y, Deng F, Pan Y, Mei S, Zheng Z, Min R, Wu Z, Li W, Miao R, Zhang Z, Kupper TS, Lieberman J, Liu X. Streptococcal pyrogenic exotoxin B cleaves GSDMA and triggers pyroptosis. Nature 602: 496–502, 2022. doi: 10.1038/s41586-021-04384-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, Sun H, Wang DC, Shao F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535: 111–116, 2016. doi: 10.1038/nature18590. [DOI] [PubMed] [Google Scholar]
- 48. Lin PH, Lin HY, Kuo CC, Yang LT. N-terminal functional domain of Gasdermin A3 regulates mitochondrial homeostasis via mitochondrial targeting. J Biomed Sci 22: 44, 2015. doi: 10.1186/s12929-015-0152-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kondolf HC, D'Orlando DA, Dubyak GR, Abbott DW. Protein engineering reveals that gasdermin A preferentially targets mitochondrial membranes over the plasma membrane during pyroptosis. J Biol Chem 299: 102908, 2023. doi: 10.1016/j.jbc.2023.102908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Huang LY, Li ST, Lin SC, Kao CH, Hong CH, Lee CH, Yang LT. Gasdermin A is required for epidermal cornification during skin barrier regeneration and in an atopic dermatitis-like model. J Invest Dermatol 143: 1735–1745.e11, 2023. doi: 10.1016/j.jid.2023.03.1657. [DOI] [PubMed] [Google Scholar]
- 51. Shi P, Tang A, Xian L, Hou S, Zou D, Lv Y, Huang Z, Wang Q, Song A, Lin Z, Gao X. Loss of conserved Gsdma3 self-regulation causes autophagy and cell death. Biochem J 468: 325–336, 2015. doi: 10.1042/BJ20150204. [DOI] [PubMed] [Google Scholar]
- 52. Yu J, Kang MJ, Kim BJ, Kwon JW, Song YH, Choi WA, Shin YJ, Hong SJ. Polymorphisms in GSDMA and GSDMB are associated with asthma susceptibility, atopy and BHR. Pediatr Pulmonol 46: 701–708, 2011. doi: 10.1002/ppul.21424. [DOI] [PubMed] [Google Scholar]
- 53. Verlaan DJ, Berlivet S, Hunninghake GM, Madore AM, Larivière M, Moussette S, Grundberg E, Kwan T, Ouimet M, Ge B, Hoberman R, Swiatek M, Dias J, Lam KC, Koka V, Harmsen E, Soto-Quiros M, Avila L, Celedón JC, Weiss ST, Dewar K, Sinnett D, Laprise C, Raby BA, Pastinen T, Naumova AK. Allele-specific chromatin remodeling in the ZPBP2/GSDMB/ORMDL3 locus associated with the risk of asthma and autoimmune disease. Am J Hum Genet 85: 377–393, 2009. doi: 10.1016/j.ajhg.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hergueta-Redondo M, Sarrió D, Molina-Crespo Á, Megias D, Mota A, Rojo-Sebastian A, García-Sanz P, Morales S, Abril S, Cano A, Peinado H, Moreno-Bueno G. Gasdermin-B promotes invasion and metastasis in breast cancer cells. PLoS One 9: e90099, 2014. doi: 10.1371/journal.pone.0090099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chao KL, Kulakova L, Herzberg O. Gene polymorphism linked to increased asthma and IBD risk alters gasdermin-B structure, a sulfatide and phosphoinositide binding protein. Proc Natl Acad Sci U S A 114: E1128–E1137, 2017. doi: 10.1073/pnas.1616783114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li X, Zhang T, Kang L, Xin R, Sun M, Chen Q, Pei J, Chen Q, Gao X, Lin Z. Apoptotic caspase-7 activation inhibits non-canonical pyroptosis by GSDMB cleavage. Cell Death Differ 30: 2120–2134, 2023. doi: 10.1038/s41418-023-01211-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, Wang Y, Li D, Liu W, Zhang Y, Shen L, Han W, Shen L, Ding J, Shao F. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 368: eaaz7548, 2020. doi: 10.1126/science.aaz7548. [DOI] [PubMed] [Google Scholar]
- 58. Hansen JM, de Jong MF, Wu Q, Zhang LS, Heisler DB, Alto LT, Alto NM. Pathogenic ubiquitination of GSDMB inhibits NK cell bactericidal functions. Cell 184: 3178–3191.e18, 2021. doi: 10.1016/j.cell.2021.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang C, Shivcharan S, Tian T, Wright S, Ma D, Chang J, Li K, Song K, Xu C, Rathinam VA, Ruan J. Structural basis for GSDMB pore formation and its targeting by IpaH7.8. Nature 616: 590–597, 2023. doi: 10.1038/s41586-023-05832-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen Q, Shi P, Wang Y, Zou D, Wu X, Wang D, Hu Q, Zou Y, Huang Z, Ren J, Lin Z, Gao X. GSDMB promotes non-canonical pyroptosis by enhancing caspase-4 activity. J Mol Cell Biol 11: 496–508, 2019. doi: 10.1093/jmcb/mjy056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Rana N, Privitera G, Kondolf HC, Bulek K, Lechuga S, De Salvo C, Corridoni D, Antanaviciute A, Maywald RL, Hurtado AM, Zhao J, Huang EH, Li X, Chan ER, Simmons A, Bamias G, Abbott DW, Heaney JD, Ivanov AI, Pizarro TT. GSDMB is increased in IBD and regulates epithelial restitution/repair independent of pyroptosis. Cell 185: 283–298.e17, 2022. doi: 10.1016/j.cell.2021.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhong X, Zeng H, Zhou Z, Su Y, Cheng H, Hou Y, She Y, Feng N, Wang J, Shao F, Ding J. Structural mechanisms for regulation of GSDMB pore-forming activity. Nature 616: 598–605, 2023. doi: 10.1038/s41586-023-05872-5. [DOI] [PubMed] [Google Scholar]
- 63. Kong Q, Xia S, Pan X, Ye K, Li Z, Li H, Tang X, Sahni N, Yi SS, Liu X, Wu H, Elowitz MB, Lieberman J, Zhang Z. Alternative splicing of GSDMB modulates killer lymphocyte-triggered pyroptosis. Sci Immunol 8: eadg3196, 2023. doi: 10.1126/sciimmunol.adg3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lie MU, Pedersen LM, Heuch I, Winsvold B, Gjerstad J, Hasvik E, Nygaard ØP, Grotle M, Matre D, Zwart JA, Nilsen KB. Low back pain with persistent radiculopathy; the clinical role of genetic variants in the genes SOX5, CCDC26/GSDMC and DCC. Front Genet 12: 757632, 2021. doi: 10.3389/fgene.2021.757632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Xi R, Montague J, Lin X, Lu C, Lei W, Tanaka K, Zhang YV, Xu X, Zheng X, Zhou X, Urban JF Jr, Iwatsuki K, Margolskee RF, Matsumoto I, Tizzano M, Li J, Jiang P. Up-regulation of gasdermin C in mouse small intestine is associated with lytic cell death in enterocytes in worm-induced type 2 immunity. Proc Natl Acad Sci USA 118: e2026307118, 2021. doi: 10.1073/pnas.2026307118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zhao M, Ren K, Xiong X, Xin Y, Zou Y, Maynard JC, Kim A, Battist AP, Koneripalli N, Wang Y, Chen Q, Xin R, Yang C, Huang R, Yu J, Huang Z, Zhang Z, Wang H, Wang D, Xiao Y, Salgado OC, Jarjour NN, Hogquist KA, Revelo XS, Burlingame AL, Gao X, von Moltke J, Lin Z, Ruan HB. Epithelial STAT6 O-GlcNAcylation drives a concerted anti-helminth alarmin response dependent on tuft cell hyperplasia and gasdermin C. Immunity 55: 623–638.e5, 2022. doi: 10.1016/j.immuni.2022.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hou J, Zhao R, Xia W, Chang CW, You Y, Hsu JM, Nie L, Chen Y, Wang YC, Liu C, Wang WJ, Wu Y, Ke B, Hsu JL, Huang K, Ye Z, Yang Y, Xia X, Li Y, Li CW, Shao B, Tainer JA, Hung MC. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat Cell Biol 22: 1264–1275, 2020. doi: 10.1038/s41556-020-0575-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhang JY, Zhou B, Sun RY, Ai YL, Cheng K, Li FN, Wang BR, Liu FJ, Jiang ZH, Wang WJ, Zhou D, Chen HZ, Wu Q. The metabolite α-KG induces GSDMC-dependent pyroptosis through death receptor 6-activated caspase-8. Cell Res 31: 980–997, 2021. doi: 10.1038/s41422-021-00506-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, Brooks A, Xia S, Wu H, Kelliher MA, Berger SB, Gough PJ, Bertin J, Proulx MM, Goguen JD, Kayagaki N, Fitzgerald KA, Lien E. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 362: 1064–1069, 2018. doi: 10.1126/science.aau2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Muendlein HI, Jetton D, Connolly WM, Eidell KP, Magri Z, Smirnova I, Poltorak A. cFLIPL protects macrophages from LPS-induced pyroptosis via inhibition of complex II formation. Science 367: 1379–1384, 2020. doi: 10.1126/science.aay3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Burgener SS, Leborgne NG, Snipas SJ, Salvesen GS, Bird PI, Benarafa C. Cathepsin G inhibition by Serpinb1 and Serpinb6 prevents programmed necrosis in neutrophils and monocytes and reduces GSDMD-driven inflammation. Cell Rep 27: 3646–3656.e5, 2019. doi: 10.1016/j.celrep.2019.05.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Chen KW, Demarco B, Heilig R, Shkarina K, Boettcher A, Farady CJ, Pelczar P, Broz P. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J 38: e101638, 2019. doi: 10.15252/embj.2019101638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tsuchiya K, Nakajima S, Hosojima S, Thi Nguyen D, Hattori T, Manh Le T, Hori O, Mahib MR, Yamaguchi Y, Miura M, Kinoshita T, Kushiyama H, Sakurai M, Shiroishi T, Suda T. Caspase-1 initiates apoptosis in the absence of gasdermin D. Nat Commun 10: 2091, 2019. doi: 10.1038/s41467-019-09753-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mascarenhas DP, Cerqueira DM, Pereira MS, Castanheira FV, Fernandes TD, Manin GZ, Cunha LD, Zamboni DS. Inhibition of caspase-1 or gasdermin-D enable caspase-8 activation in the Naip5/NLRC4/ASC inflammasome. PLoS Pathog 13: e1006502, 2017. doi: 10.1371/journal.ppat.1006502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Schneider KS, Groß CJ, Dreier RF, Saller BS, Mishra R, Gorka O, Heilig R, Meunier E, Dick MS, Ćiković T, Sodenkamp J, Médard G, Naumann R, Ruland J, Kuster B, Broz P, Groß O. The inflammasome drives GSDMD-independent secondary pyroptosis and IL-1 release in the absence of caspase-1 protease activity. Cell Rep 21: 3846–3859, 2017. doi: 10.1016/j.celrep.2017.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chai Q, Yu S, Zhong Y, Lu Z, Qiu C, Yu Y, Zhang X, Zhang Y, Lei Z, Qiang L, Li BX, Pang Y, Qiu XB, Wang J, Liu CH. A bacterial phospholipid phosphatase inhibits host pyroptosis by hijacking ubiquitin. Science 378: eabq0132, 2022. doi: 10.1126/science.abq0132. [DOI] [PubMed] [Google Scholar]
- 77. Sborgi L, Rühl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, Farady CJ, Müller DJ, Broz P, Hiller S. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J 35: 1766–1778, 2016. doi: 10.15252/embj.201694696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Liu Z, Wang C, Yang J, Zhou B, Yang R, Ramachandran R, Abbott DW, Xiao TS. Crystal structures of the full-length murine and human gasdermin d reveal mechanisms of autoinhibition, lipid binding, and oligomerization. Immunity 51: 43–49.e4, 2019. doi: 10.1016/j.immuni.2019.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Rathkey JK, Zhao J, Liu Z, Chen Y, Yang J, Kondolf HC, Benson BL, Chirieleison SM, Huang AY, Dubyak GR, Xiao TS, Li X, Abbott DW. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci Immunol 3: eaat2738, 2018. doi: 10.1126/sciimmunol.aat2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, Ruan J, Luo X, Lou X, Bai Y, Wang J, Hollingsworth LR, Magupalli VG, Zhao L, Luo HR, Kim J, Lieberman J, Wu H. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat Immunol 21: 736–745, 2020. doi: 10.1038/s41590-020-0669-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, Menninger S, Eickhoff J, Nussbaumer P, Klebl B, Krüger R, Herzig A, Zychlinsky A. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol 3: eaar6689, 2018. doi: 10.1126/sciimmunol.aar6689. [DOI] [PubMed] [Google Scholar]
- 82. Cai W, Wu Z, Lai J, Yao J, Zeng Y, Fang Z, Lin W, Chen J, Xu C, Chen X. LDC7559 inhibits microglial activation and GSDMD-dependent pyroptosis after subarachnoid hemorrhage. Front Immunol 14: 1117310, 2023. doi: 10.3389/fimmu.2023.1117310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Evavold CL, Hafner-Bratkovič I, Devant P, D'Andrea JM, Ngwa EM, Boršić E, Doench JG, LaFleur MW, Sharpe AH, Thiagarajah JR, Kagan JC. Control of gasdermin D oligomerization and pyroptosis by the Ragulator-Rag-mTORC1 pathway. Cell 184: 4495–4511.e19, 2021. doi: 10.1016/j.cell.2021.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Devant P, Boršić E, Ngwa EM, Xiao H, Chouchani ET, Thiagarajah JR, Hafner-Bratkovič I, Evavold CL, Kagan JC. Gasdermin D pore-forming activity is redox-sensitive. Cell Rep 42: 112008, 2023. doi: 10.1016/j.celrep.2023.112008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Weindel CG, Martinez EL, Zhao X, Mabry CJ, Bell SL, Vail KJ, Coleman AK, VanPortfliet JJ, Zhao B, Wagner AR, Azam S, Scott HM, Li P, West AP, Karpac J, Patrick KL, Watson RO. Mitochondrial ROS promotes susceptibility to infection via gasdermin D-mediated necroptosis. Cell 185: 3214–3231.e23, 2022. doi: 10.1016/j.cell.2022.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Zhuang Z, Gu J, Li BO, Yang L. Inhibition of gasdermin D palmitoylation by disulfiram is crucial for the treatment of myocardial infarction. Transl Res 264: 66–75, 2023. doi: 10.1016/j.trsl.2023.09.007. [DOI] [PubMed] [Google Scholar]
- 87. Shi Y, Yang Y, Xu W, Shi D, Xu W, Fu X, Lv Q, Xia J, Shi F. E3 ubiquitin ligase SYVN1 is a key positive regulator for GSDMD-mediated pyroptosis. Cell Death Dis 13: 106, 2022. doi: 10.1038/s41419-022-04553-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Luchetti G, Roncaioli JL, Chavez RA, Schubert AF, Kofoed EM, Reja R, Cheung TK, Liang Y, Webster JD, Lehoux I, Skippington E, Reeder J, Haley B, Tan MW, Rose CM, Newton K, Kayagaki N, Vance RE, Dixit VM. Shigella ubiquitin ligase IpaH7.8 targets gasdermin D for degradation to prevent pyroptosis and enable infection. Cell Host Microbe 29: 1521–1530.e10, 2021. doi: 10.1016/j.chom.2021.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Xia S, Zhang Z, Magupalli VG, Pablo JL, Dong Y, Vora SM, Wang L, Fu TM, Jacobson MP, Greka A, Lieberman J, Ruan J, Wu H. Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature 593: 607–611, 2021. doi: 10.1038/s41586-021-03478-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Chen W, Chen S, Yan C, Zhang Y, Zhang R, Chen M, Zhong S, Fan W, Zhu S, Zhang D, Lu X, Zhang J, Huang Y, Zhu L, Li X, Lv D, Fu Y, Iv H, Ling Z, Ma L, Jiang H, Long G, Zhu J, Wu D, Wu B, Sun B. Allergen protease-activated stress granule assembly and gasdermin D fragmentation control interleukin-33 secretion. Nat Immunol 23: 1021–1030, 2022. doi: 10.1038/s41590-022-01255-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Tsuchiya K, Hosojima S, Hara H, Kushiyama H, Mahib MR, Kinoshita T, Suda T. Gasdermin D mediates the maturation and release of IL-1α downstream of inflammasomes. Cell Rep 34: 108887, 2021. doi: 10.1016/j.celrep.2021.108887. [DOI] [PubMed] [Google Scholar]
- 92. Karmakar M, Minns M, Greenberg EN, Diaz-Aponte J, Pestonjamasp K, Johnson JL, Rathkey JK, Abbott DW, Wang K, Shao F, Catz SD, Dubyak GR, Pearlman E. N-GSDMD trafficking to neutrophil organelles facilitates IL-1β release independently of plasma membrane pores and pyroptosis. Nat Commun 11: 2212, 2020. doi: 10.1038/s41467-020-16043-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Chauhan D, Demon D, Vande Walle L, Paerewijck O, Zecchin A, Bosseler L, Santoni K, Planès R, Ribo S, Fossoul A, Gonçalves A, Van Gorp H, Van Opdenbosch N, Van Hauwermeiren F, Meunier E, Wullaert A, Lamkanfi M. GSDMD drives canonical inflammasome-induced neutrophil pyroptosis and is dispensable for NETosis. EMBO Rep 23: e54277, 2022. doi: 10.15252/embr.202154277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Chen KW, Monteleone M, Boucher D, Sollberger G, Ramnath D, Condon ND, von Pein JB, Broz P, Sweet MJ, Schroder K. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci Immunol 3: eaar6676, 2018. doi: 10.1126/sciimmunol.aar6676. [DOI] [PubMed] [Google Scholar]
- 95. Stojkov D, Claus MJ, Kozlowski E, Oberson K, Schären OP, Benarafa C, Yousefi S, Simon HU. NET formation is independent of gasdermin D and pyroptotic cell death. Sci Signal 16: eabm0517, 2023. doi: 10.1126/scisignal.abm0517. [DOI] [PubMed] [Google Scholar]
- 96. Huang LS, Hong Z, Wu W, Xiong S, Zhong M, Gao X, Rehman J, Malik AB. mtDNA activates cGAS signaling and suppresses the YAP-mediated endothelial cell proliferation program to promote inflammatory injury. Immunity 52: 475–486.e5, 2020. doi: 10.1016/j.immuni.2020.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Wang Y, Gao W, Shi X, Ding J, Liu W, He H, Wang K, Shao F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547: 99–103, 2017. doi: 10.1038/nature22393. [DOI] [PubMed] [Google Scholar]
- 98. Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, Junqueira C, Meza-Sosa KF, Mok TM, Ansara J, Sengupta S, Yao Y, Wu H, Lieberman J. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 579: 415–420, 2020. doi: 10.1038/s41586-020-2071-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Yokomizo K, Harada Y, Kijima K, Shinmura K, Sakata M, Sakuraba K, Kitamura Y, Shirahata A, Goto T, Mizukami H, Saito M, Kigawa G, Nemoto H, Hibi K. Methylation of the DFNA5 gene is frequently detected in colorectal cancer. Anticancer Res 32: 1319–1322, 2012. [PubMed] [Google Scholar]
- 100. Kim MS, Lebron C, Nagpal JK, Chae YK, Chang X, Huang Y, Chuang T, Yamashita K, Trink B, Ratovitski EA, Califano JA, Sidransky D. Methylation of the DFNA5 increases risk of lymph node metastasis in human breast cancer. Biochem Biophys Res Commun 370: 38–43, 2008. doi: 10.1016/j.bbrc.2008.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Grottke C, Mantwill K, Dietel M, Schadendorf D, Lage H. Identification of differentially expressed genes in human melanoma cells with acquired resistance to various antineoplastic drugs. Int J Cancer 88: 535–546, 2000. doi:. [DOI] [PubMed] [Google Scholar]
- 102. Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun 8: 14128, 2017. doi: 10.1038/ncomms14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Zhou B, Abbott DW. Gasdermin E permits interleukin-1 beta release in distinct sublytic and pyroptotic phases. Cell Rep 35: 108998, 2021. doi: 10.1016/j.celrep.2021.108998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Chao YY, Puhach A, Frieser D, Arunkumar M, Lehner L, Seeholzer T, Garcia-Lopez A, van der Wal M, Fibi-Smetana S, Dietschmann A, Sommermann T, Ćiković T, Taher L, Gresnigt MS, Vastert SJ, van Wijk F, Panagiotou G, Krappmann D, Groß O, Zielinski CE. Human TH17 cells engage gasdermin E pores to release IL-1α on NLRP3 inflammasome activation. Nat Immunol 24: 295–308, 2023. doi: 10.1038/s41590-022-01386-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Wang C, Yang T, Xiao J, Xu C, Alippe Y, Sun K, Kanneganti TD, Monahan JB, Abu-Amer Y, Lieberman J, Mbalaviele G. NLRP3 inflammasome activation triggers gasdermin D-independent inflammation. Sci Immunol 6: eabj3859, 2021. doi: 10.1126/sciimmunol.abj3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun 10: 1689, 2019. doi: 10.1038/s41467-019-09397-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Chen X, He WT, Hu L, Li J, Fang Y, Wang X, Xu X, Wang Z, Huang K, Han J. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res 26: 1007–1020, 2016. doi: 10.1038/cr.2016.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Ousingsawat J, Wanitchakool P, Schreiber R, Kunzelmann K. Contribution of TMEM16F to pyroptotic cell death. Cell Death Dis 9: 300, 2018. doi: 10.1038/s41419-018-0373-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Yang X, Cheng X, Tang Y, Qiu X, Wang Y, Kang H, Wu J, Wang Z, Liu Y, Chen F, Xiao X, Mackman N, Billiar TR, Han J, Lu B. Bacterial endotoxin activates the coagulation cascade through gasdermin D-dependent phosphatidylserine exposure. Immunity 51: 983–996.e6, 2019. doi: 10.1016/j.immuni.2019.11.005. [DOI] [PubMed] [Google Scholar]
- 110. Rühl S, Shkarina K, Demarco B, Heilig R, Santos JC, Broz P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 362: 956–960, 2018. doi: 10.1126/science.aar7607. [DOI] [PubMed] [Google Scholar]
- 111. Liao Y, Chen X, Miller-Little W, Wang H, Willard B, Bulek K, Zhao J, Li X. The Ras GTPase-activating-like protein IQGAP1 bridges gasdermin D to the ESCRT system to promote IL-1β release via exosomes. EMBO J 42: e110780, 2023. doi: 10.15252/embj.2022110780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Banerjee I, Behl B, Mendonca M, Shrivastava G, Russo AJ, Menoret A, Ghosh A, Vella AT, Vanaja SK, Sarkar SN, Fitzgerald KA, Rathinam VA. Gasdermin D restrains type I interferon response to cytosolic DNA by disrupting ionic homeostasis. Immunity 49: 413–426.e5, 2018. doi: 10.1016/j.immuni.2018.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The pore-forming protein gasdermin D regulates interleukin-1 secretion from living macrophages. Immunity 48: 35–44.e6, 2018. doi: 10.1016/j.immuni.2017.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ. HMG-1 as a late mediator of endotoxin lethality in mice. Science 285: 248–251, 1999. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 115. Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, Lu B, Chavan S, Rosas-Ballina M, Al-Abed Y, Akira S, Bierhaus A, Erlandsson-Harris H, Andersson U, Tracey KJ. A critical cysteine is required for HMGB-1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci U S A 107: 11942–11947, 2010. doi: 10.1073/pnas.1003893107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Volchuk A, Ye A, Chi L, Steinberg BE, Goldenberg NM. Indirect regulation of HMGB1 release by gasdermin D. Nat Commun 11: 4561, 2020.doi: 10.1038/s41467-020-18443-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. den Hartigh AB, Fink SL. Pyroptosis induction and detection. Curr Protoc Immunol 122: e52, 2018. doi: 10.1002/cpim.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Franklin BS, Bossaller L, De Nardo D, Ratter JM, Stutz A, Engels G, Brenker C, Nordhoff M, Mirandola SR, Al-Amoudi A, Mangan MS, Zimmer S, Monks BG, Fricke M, Schmidt RE, Espevik T, Jones B, Jarnicki AG, Hansbro PM, Busto P, Marshak-Rothstein A, Hornemann S, Aguzzi A, Kastenmüller W, Latz E. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat Immunol 15: 727–737, 2014. doi: 10.1038/ni.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Araki T, Milbrandt J. Ninjurin, a novel adhesion molecule, is induced by nerve injury and promotes axonal growth. Neuron 17: 353–361, 1996. doi: 10.1016/S0896-6273(00)80166-X. [DOI] [PubMed] [Google Scholar]
- 120. Kayagaki N, Kornfeld OS, Lee BL, Stowe IB, O'Rourke K, Li Q, Sandoval W, Yan D, Kang J, Xu M, Zhang J, Lee WP, McKenzie BS, Ulas G, Payandeh J, Roose-Girma M, Modrusan Z, Reja R, Sagolla M, Webster JD, Cho V, Andrews TD, Morris LX, Miosge LA, Goodnow CC, Bertram EM, Dixit VM. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 591: 131–136, 2021. doi: 10.1038/s41586-021-03218-7. [DOI] [PubMed] [Google Scholar]
- 121. Degen M, Santos JC, Pluhackova K, Cebrero G, Ramos S, Jankevicius G, Hartenian E, Guillerm U, Mari SA, Kohl B, Müller DJ, Schanda P, Maier T, Perez C, Sieben C, Broz P, Hiller S. Structural basis of NINJ1-mediated plasma membrane rupture in cell death. Nature 618: 1065–1071, 2023. doi: 10.1038/s41586-023-05991-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Davis MA, Fairgrieve MR, Den Hartigh A, Yakovenko O, Duvvuri B, Lood C, Thomas WE, Fink SL, Gale M Jr.. Calpain drives pyroptotic vimentin cleavage, intermediate filament loss, and cell rupture that mediates immunostimulation. Proc Natl Acad Sci U S A 116: 5061–5070, 2019. doi: 10.1073/pnas.1818598116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Dondelinger Y, Priem D, Huyghe J, Delanghe T, Vandenabeele P, Bertrand MJ. NINJ1 is activated by cell swelling to regulate plasma membrane permeabilization during regulated necrosis. Cell Death Dis 14: 755, 2023. doi: 10.1038/s41419-023-06284-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Nozaki K, Maltez VI, Rayamajhi M, Tubbs AL, Mitchell JE, Lacey CA, Harvest CK, Li L, Nash WT, Larson HN, McGlaughon BD, Moorman NJ, Brown MG, Whitmire JK, Miao EA. Caspase-7 activates ASM to repair gasdermin and perforin pores. Nature 606: 960–967, 2022. doi: 10.1038/s41586-022-04825-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Wang Q, Wang Y, Ding J, Wang C, Zhou X, Gao W, Huang H, Shao F, Liu Z. A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature 579: 421–426, 2020. doi: 10.1038/s41586-020-2079-1. [DOI] [PubMed] [Google Scholar]
- 126. Li S, Sun Y, Song M, Song Y, Fang Y, Zhang Q, Li X, Song N, Ding J, Lu M, Hu G. NLRP3/caspase-1/GSDMD-mediated pyroptosis exerts a crucial role in astrocyte pathological injury in mouse model of depression. JCI Insight 6: e146852, 2021.doi: 10.1172/jci.insight.146852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Sun K, Wang C, Xiao J, Brodt MD, Yuan L, Yang T, Alippe Y, Hu H, Hao D, Abu-Amer Y, Silva MJ, Shen J, Mbalaviele G. Fracture healing is delayed in the absence of gasdermin-interleukin-1 signaling. eLife 11: e75753, 2022. doi: 10.7554/eLife.75753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Rathkey JK, Xiao TS, Abbott DW. Human polymorphisms in GSDMD alter the inflammatory response. J Biol Chem 295: 3228–3238, 2020. doi: 10.1074/jbc.RA119.010604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Kayagaki N, Stowe IB, Alegre K, Deshpande I, Wu S, Lin Z, Kornfeld OS, Lee BL, Zhang J, Liu J, Suto E, Lee WP, Schneider K, Lin WY, Seshasayee D, Bhangale T, Chalouni C, Johnson MC, Joshi P, Mossemann J, Zhao S, Ali D, Goldenberg NM, Sayed BA, Steinberg BE, Newton K, Webster JD, Kelly RL, Dixit VM. Inhibiting membrane rupture with NINJ1 antibodies limits tissue injury. Nature 618: 1072–1077, 2023. doi: 10.1038/s41586-023-06191-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Man SM. Inflammasomes in the gastrointestinal tract: infection, cancer and gut microbiota homeostasis. Nat Rev Gastroenterol Hepatol 15: 721–737, 2018. doi: 10.1038/s41575-018-0054-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available upon reasonable request.