

Summary
Proximity is a fundamental concept in chemistry and biology, referring to the convergence of molecules to facilitate new molecular interactions or reactions. Hybrid biopolymers like GPI-anchored proteins, ubiquitinated proteins, glycosylated RNAs, and RNAylated proteins exemplify this by covalent bonding of moieties that are often orthogonally active. Hybrid molecules like glycoRNAs are localized to new physical spaces, generating new interfaces for biological functions. To fully investigate the compositional and spatial features of molecules like glycoRNAs, flexible genetic and chemical tools that encompass different encoding and targeting biopolymers are required. Here we discuss concepts of molecular proximity and explore newer proximity labeling technologies that facilitate applications in RNA biology, cell surface biology, and the interface therein with a particular focus on glycoRNA biology. We review the advantages and disadvantages of methods pertaining to cell-surface RNA identification and provide insights into the vast opportunities for method development in this area.
Graphical Abstract

eTOC
Kageler et al discuss concepts of molecular proximity and explore proximity labeling technologies that facilitate applications in RNA biology, cell surface biology and their interface. Technologies that employ genetic and chemical strategies are highlighted that will enable discovery of the compositional and organizational features of molecules like glycoRNAs.
Introduction
Across the scales of life, physical proximity governs many aspects of behavior and results in specific and predictable outcomes. In biology, and at the level of a single cell, molecular interactions can occur in 3D space unless constrained in some way. Cellular membranes, particularly the plasma membrane of mammalian cells, provide a physical container for intracellular biochemistry, information storage, and a platform upon which biochemistry occurs and is gated (e.g. membrane-spanning protein channels). Many cellular functions are regulated by molecular interactions between proteins, RNA, DNA, or glycans; and the dysregulation of these interactions has been tightly linked to human diseases. The composition, organization, and consequent molecular interactions on the cell surface are particularly important as they dictate how cells interact with their surroundings and how they initiate intracellular processes, including cell signaling, adhesion, recognition, and nutrient/waste transport. The cell surface is not a static structure but dynamic and constantly changing with cell state. A detailed understanding of the cell surface, especially in different disease states, is critical for the advancement of drug development efforts. Notably, 66% of approved drug candidates identified in the DrugBank database target proteins that are annotated as cell surface proteins1. Understanding the cell surface presentation of other biopolymers, including the newly discovered glycoRNA, may provide important insight for novel therapeutic development.
Induced proximity can occur biologically – through non-covalent interactions, for example between proteins and RNAs to form complexes with novel functions (e.g. the ribosome), or through covalent interactions between different molecules to form hybrid biopolymers (e.g. glycoRNA2,3, RNAylated proteins4) (Figure 1). Additionally, researchers have used proximity labeling techniques to characterize biological insights and have harnessed proximity concepts to develop novel therapeutic platforms.
Figure 1. Biologically induced proximity, chemically induced proximity, and hybrid conjugation of biopolymers.
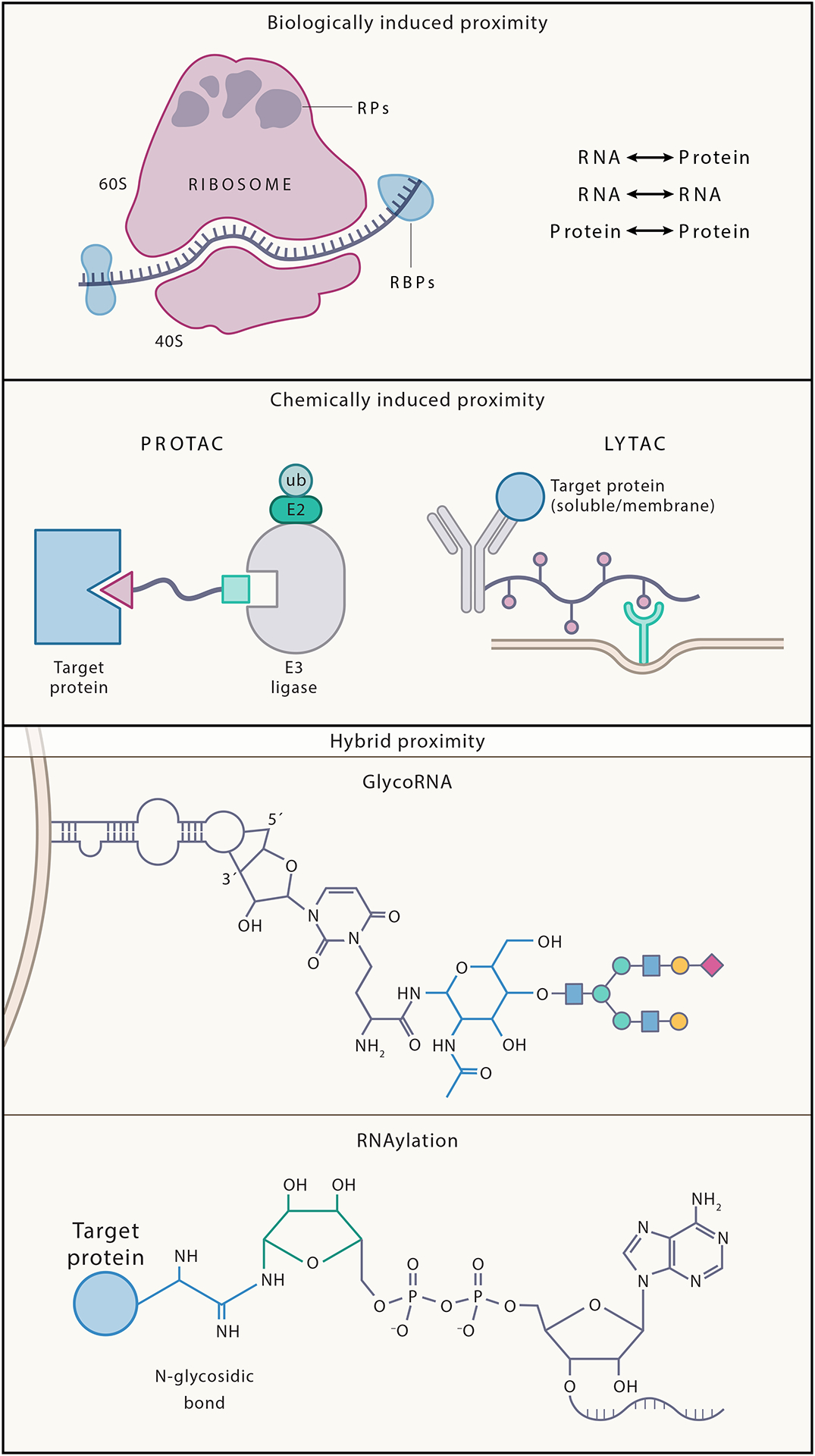
Examples of proximity include (A) Biologically induced proximity such as ribosomal RNA and ribosomal proteins that interact non-covalently to maintain the structure and function of the ribosome complex, which is essential for protein synthesis, (B) chemically induced proximity such as PROTACs and LYTACs. PROTACs are heterobifunctional molecules that degrade target proteins by hijacking the ubiquitin–proteasome system. They are composed of two active domains (ligands) and a linker: one ligand recruits and binds a protein of interest (POI) while the other recruits and binds an E3 ubiquitin ligase, bringing the ligase in proximity with the protein which causes subsequent degradation of the POI by the ubiquitin-proteasome system. LYTACs work by recruiting membrane-bound and extracellular proteins to lysosome-shuttling receptors, causing the lysosomal degradation of the POI. Both technologies present examples of how we can chemically induce proximity to cause novel outcomes. Finally (C) represents induced proximity between different biomolecules that can subsequently generate novel biopolymers with putatively different functions such as glycoRNAs and RNAylated proteins. GlycoRNAs represent a novel class of lectin (glycan binding protein) ligands and a novel class of cell surface molecules; both classifications present putatively important biological functions. RNAylated proteins represent a direct connection between RNA modification and post-translational protein modification, the first such characterization of post-translational protein modification, with identified roles in infection and pathogenicity.
Technologies such as proteolysis-targeting chimeras (PROTACs, often targeting intracellular proteins), bispecific T cell engagers (BiTEs, targeting cell surface antigens) and lysosome targeting chimeras (LYTACs, degrading cell surface or extracellular proteins) directly induce proximity between two molecules to perform a novel molecular action (Figure 1),5–7. Additionally, the extensive use of monoclonal antibodies and chimeric antigen receptor T-cell targeting have contributed to a better understanding of cell surface features including the presence, conformation, and organization of cell surface molecules in both health and disease.
Most chemical biology tools with applications on the cell surface were developed to study proteins, lipids or glycans. However, our discovery of glycosylated RNAs (glycoRNA) presented on the cell surface2, gives rise to new questions in cell-surface proximity and composition. In this initial work, we used proximity-based tools to assay the cell surface, including lectin bound peroxidase enzymes to label cell surface RNA species. However, the widespread use of highly selective (often for protein targets) tools means that unassayed features, such as cell surface glycoRNA biology, may be missed. In this review, we will discuss tools in proximity labeling that are amenable to RNA and cell-surface detection. We review the advantages and disadvantages of methods as it pertains to cell-surface RNA identification; and provide insights into the vast opportunities for method development in this area.
More recently, our understanding of cell surface composition has been expanding and thus a reevaluation of the biopolymers that are functionally in proximity to one another on the cell surface may be necessary and biologically informative. In particular, our discovery of glycosylated RNAs (glycoRNA) organized on the cell surface of many cell types2 and our more recent identification of a direct attachment site of N-glycans in RNA3 represent new concepts in induced proximity. GlycoRNAs are templated on small noncoding RNAs and attached to sialic acid and fucose containing N-glycans, which were traditionally only thought to modify proteins or lipids. Therefore, by definition, a glycoRNA is a hybrid biopolymer that represents a novel class of putative ligands for glycan-binding proteins (lectins). In fact, we have shown that glycoRNAs can interact with Siglec receptors (sialic-acid-binding immunoglobulin-like lectins), which are widely expressed on immune cells and play critical roles in immune cell signaling8. Beyond this and more generally, purposeful presentation of glycoRNAs now places RNAs proximal to cell surface proteins and lipids, expanding the potential for unknown functional regulation and interaction of these molecules.
The growing set of examples of cell surface glycoRNAs3,9–13 and or topologically luminal RNA biology14,15 highlights the need for a better understanding of the context in which this biology occurs. Specifically, the characterization of the glycoRNA hybrid biopolymers themselves as well as their proximity to and interactions with other well-studied aspects of cell surface biology will be pertinent to further understanding their functional roles. Additionally, there has been little exploration of non-sialic acid containing glycoRNAs and the direct characterization of the role of glycans on an RNA has not yet been defined.
To date, we have adapted specific chemical tools for glycoRNA labeling that enabled the initial discovery and characterization of glycoRNAs on the cell surface, including proximity labeling, rPAL labeling and flow cytometry3. Other groups have recently developed multiple in situ labeling techniques by taking advantage of the hybrid RNA-glycan nature of these species10,11 (Figure 2). These recent efforts highlight the potential opportunities for method development in this new space and the importance of adapting current techniques to study lesser-known biology. In reviewing concepts and methods that have been important for elucidating aspects of glycoRNA biology (Figure 3) and the limitations that currently exist for examining this biopolymer, we will use a framework of methods focused on (A) detecting proximity directly with fluorophores-based techniques, (B) detecting proximity through oligonucleotide ligation, and (C) detecting proximity through catalyst-based labeling. These methods have traditionally focused on proteins as target biopolymers and intracellular biology as the topological space, but we will highlight more recent work to expand towards RNA and cell surface characterization.
Figure 2. Proximity Ligation Assays with RNA biology.

Types of PLA shown including (A) Intermolecular indirect PLA between proteins in close proximity, (B) Intermolecular hybrid PLA between RNA and an RNA-binding proteins. Here, a RISH probe targets the RNA, while the RBP is indirectly immunolabeled for PLA as in42. And (C) Intramolecular PLA of glycoRNA (ARPLA) as shown in11. Sialic acid of the glycan is targeted with a DNA aptamer, while the RNA is targeted with a RISH probe, enabling ligation and readout. For all PLA methods shown, DNA connectors are ligated, amplified by RCA, bound by reporter oligos, and imaged to detect proximity events. Antibodies in all PLA methods may also be substituted with suitable DNA aptamers.
Figure 3. Proximity Labeling as a versatile tool for studying cell surface glycoRNA biology.

Tools to study the cell surface in its native context are important for our understanding of cell biology and therapeutic drug design. (A) Shown here, glycoRNAs and cell surface localized RBPs form clusters together that are physically distinct from other surface receptors like MHC class I (MHC-I). (B) To investigate these cell surface clusters we have used HRP-mediated proximity labeling, with biotin-phenol and biotin-aniline substrates, as it is a flexible and suitable tool for cell surface applications. Compared to biotin ligase-mediated proximity labeling and light-activated proximity labeling, HRP has a larger labeling radius. (C) Light-activated proximity labeling has seen major developments recently and can be used to label various distances from the target molecule on the cell surface, including very short distances. For example, using a singular photocatalyst, i.e. Eosin Y shown here, coupled to (in independent experiments) various biotinylated reagents, one can generate carbene, nitrene, and phenoxyl radicals (in order of smallest to largest labeling radius) dictated by the stability of each species.
Detecting proximity through Immunofluorescent colocalization, BiFC, FCS, and FRET
Directly or indirectly immunolabeling proteins on live cells enables selective labeling of cell surface proteins due to the impermeability of the plasma membrane to antibodies. Thus, labeling the surfaces of live cells for multiple proteins with distinct fluorophores facilitates both qualitative comparison of cell surface antigen positions and enables quantitation of their colocalization by calculating the Pearson correlation between objects in these channels16. However, this approach is susceptible to correlating signals not associated with the objects of interest. This pitfall may be overcome through segmentation of the image and object-based detection for determining distances between labeled objects17. This method requires either identification of objects by hand or systematic classification by software. Regardless of the method of analysis, all efforts to assess the colocalization of signal with conventional confocal microscopy are limited to Abbe’s diffraction limit of light–generally around 250 nm depending on the objective and wavelength of emitted light (Table 1). To overcome this, multiple methods including bimolecular fluorescence complementation (BiFC)18, Fluorescence Correlation Spectroscopy (FCS)19, and Forster Resonance Energy Transfer (FRET)20,21 have been developed.
Table 1.
Ligation- and fluorophore-based means for assessing proximity
| Method | Resolution | Advantages | Limitations | Applicability to glycoRNA |
|---|---|---|---|---|
| Immuno-fluorescent co-labeling and colocalization analysis 94 |
250 nm | Widely used for protein detection; can be used on formalin-fixed paraffin-embedded or fresh tissues; multiplexing capability; routinely used in diagnostic pathology. | Relies on indirect visualization; requires high-fidelity affinity reagents against targets of interest; limited ability to study dynamic interactions; colocalization does not necessarily indicate interaction | Development of novel antibodies specific to glycoRNAs holds the potential to easily and quickly visualize glycoRNAs on the cell surface and intracellularly. |
| PLA 37 |
<40 nm | Detects proteins, protein-protein interactions, and post-translational modifications; sensitive detection even in minute amounts; applicable in serum and tissue sections.95 | Requires high-fidelity affinity reagents against targets of interest; complex conjugation workflows; not easily amenable to monitoring live-cell dynamics | Methods like ARPLA capitalize on oligo-conjugated probes that either uniquely identify the RNA sequence of interest and a carbohydrate residue expected to be in the glycan to detect glycoRNAs on the cell surface. |
| FRET 27,28 |
<10 nm | High temporal sensitivity; simultaneously observation of multiple interactions; highly sensitive signal detection | Susceptible to photobleaching; complex experimental setup; spectral overlap must be deconvolved | Hybrid probes akin to those used in PLA could be employed in FRET; such a technology would enable monitoring of conformational changes in the glycoRNA itself or with the glycoRNA’s intermolecular interactors. |
| BiFC 22 |
20 nm | Direct visualization of protein-protein interactions in vivo; sensitive and proportional to interaction strength; reflects intrinsic interactions | Slow maturation of BiFC complex; not suitable for real-time observation of transient interactions; enhanced background at high expression levels. | Not recommended due to long maturation of fluorophore and difficulty of conjugating glycan/RNA/glycoRNA with a fluorescent protein fragment. |
For high-fidelity labeling of colocalization of distinct proteins on the cell surface, researchers have exploited bimolecular fluorescence complementation (BiFC). In this technique, a gene coding a fluorescence protein is split into two coding sequences that produce individually non-photoactivatable proteins that only fluoresce when reconstituted. Reconstitution events–indicating colocalization–may then be measured by fluorescence microscopy. For cell-surface targeting of this technique, Nickerson et al. generated Ras and Raf fusion proteins with complementary halves of PAmCherry122; the authors then visualized Ras-Raf interactions by detecting them with super-resolution photoactivated localization microscopy, yielding 18 nm resolution. This approach enabled visualization of clustering and diffusion of Ras-Raf complexes in real time. BiFC techniques such as that employed by Nickerson et al. offer advantages like stable signal and limited spectral crosstalk but suffer from slow maturation, non-specific reconstitution of BiFC fragments, influencing fusion protein function, and slow maturation of reconstituted BiFCs; split eGFP requires at least four hours to reconstitute and emit fluorescence23. The requirement of conjugating fragments of fluorescent proteins to analytes of interest BiFC also limits its use in understanding proximity in RNA-centric pathways.
To better study the dimerization of RNAs themselves, techniques such as FCS may be employed24. FCS involves the excitation of fluorescently tagged molecules in solution, detection of changes in fluorescence as molecules travel in and out of the path of illumination, and determination of an autocorrelation function, which can be used to indirectly assess the diffusion coefficients of molecules, concentrations, and rates of molecular binding or unbinding. While fluorescently tagged complementary RNAs of the same size may be leveraged to simply measure RNA dimerization events, FCS may also be used to disentangle mRNA composition. Recently, Regué et al. used FCS to measure GFP-tagged RNA binding protein (RBP) stoichiometry in mRNPs and further cross-correlation of dynamics with a different RFP-tagged RBP to understand how mRNA composition changes in the presence or absence of RNA-binding25. This enabled researchers to discover that the RBP YBX1’s interaction with RBP IMP1 depends on IMP1’s RNA-binding capability. However, FCS assays are generally limited to in vitro applications and only indirectly detects colocalization or oligomerization events of molecules through mathematical correlation of particle dynamics. An alternative with superior spatial resolution, live-cell applicability, and directness of analysis lies in Forster Resonance Energy Transfer (FRET).
FRET uses donor and acceptor chromophores, and upon colocalization and excitation of the donor chromophore, the emission of the donor chromophore excites the acceptor, resulting in emission from the acceptor (Table 1). In 2012, Yaffe et al. discovered that the junction protein occludin (Ocln) self-associates through its MARVEL domain for basolateral membrane localization by using a YFP-Ocln/CFP-Ocln FRET pair21. Crucially, FRET enabled real-time monitoring of basolateral translocation, demonstrating the method’s temporal resolution. This feature of FRET also allows the method to measure the dynamics between an RBP and its RNA binding partner. For example, FRET enabled Niaki et al. to observe how different mutations in the RBP FUS affected the dynamics of its interactions with RNA, genetically fusing GFP to FUS and using NHS labeling chemistry to bind a Cy5 acceptor to RNA26. Similar methods could be applied to study the dynamics within the clusters of cell surface RBPs (csRBPs) and glycoRNAs that we have recently described12. However, new methods for labeling only the cell surface pools of the RNA and RBPs while the cells are alive may be required in this context.
FRET exhibits extremely high spatial resolution. Because the energy transferred from the donor to the acceptor chromophore varies inversely with the sixth power of the distance between donor and acceptor chromophores, FRET achieves sub-10 nm resolution27. The extremely high resolution of FRET makes the technique useful for detecting proximal components of biomolecules, including intramolecular features of cell surface molecules. Accordingly, FRET has been leveraged to detect the presence of glycans on cell surface proteins. Through metabolic labeling with an alkyne-modified sialic acid precursor, Lin et al. used click chemistry to couple azide-AlexaFluor 647 acceptor fluorophore to sialoglycans and a genetically encoded LplA acceptor peptide (LAP) to enable site-specific conjugation of the AlexaFluor 488 donor to the protein of interest28. Because this acceptor-donor pair requires a distance less than 10 nm for FRET, intramolecular interactions are greatly favored for FRET over intermolecular interactions. This method facilitated visualization of the glycosylation of EGFR, TGFBR1, and ITGB2; leading to the observation that ITGB2 sialylation is required for its activation28. Future intramolecular analysis of hybrid molecules such as glycoproteins and glycoRNAs may be enhanced by simultaneously detecting more than two features, as FRET allows concurrent use of one donor with multiple acceptors such as a Cy3 donor with Cy5 and Cy5.5 acceptors29. Such a technology would enable both visualization of interactions between cell surface proteins and glycoRNAs and how those interactions alter proximity between the RNA- and glyco-probes, potentially shedding light on how protein binding affects glycoRNA secondary structure. However, in designing a FRET experiment such as this, one should consider the differing dynamic ranges of different FRET pairs; different FRET pairs exhibit different Förster radii—the distance between donor and acceptor at which 50% FRET occurs. Below the lower bound of a FRET pair’s dynamic range (1 nm for a CFP-YFP FRET pair), energy transfer from donor to acceptor becomes saturated and decreases in separation of acceptor and donor fluorophores may no longer be detected. Above the upper bound of the FRET pair’s dynamic range (10 nm for a CFP-YFP FRET pair), energy transfer between donor and acceptor becomes so weak as to be indistinguishable from background signal30. Thus, conjugation strategies of the FRET donors and acceptors to biomolecules of interest and choice of FRET pairs is extremely important for successful analysis of the interaction of interest.
Detecting proximity through PLA
Leveraging the power of affinity reagents and PCR, the proximity ligation assay (PLA) was developed to detect and quantify interactions between two known biomolecules of interest (Figure 2). PLA canonically involves the immunolabeling of two targets with affinity reagents, often antibodies but sometimes DNA aptamers; affinity reagent is conjugated to sequence-specific DNA oligos that each bind one gap of two fragments of circular DNA. If oligos from both affinity reagents are in sufficient proximity to each bind the fragments of the circular DNA, these can be ligated, enabling rolling circle amplification of DNA. Then, fluorophore-conjugated oligos can be used to bind the amplified DNA, resulting in detection of a colocalization event. A now widely used method, PLA was first developed in 2002 leveraging DNA aptamers that targeted platelet-derived growth factor (PDGF)31. This method provided the key insight that simultaneous binding of the aptamers to PDGF would enable covalent ligation to one another followed by PCR amplification and quantification of protein levels. This was essentially an intramolecular detection of proximity where the stoichiometric binding of the aptamers would enable quantification of abundance.
Advancing the method, PLA was expanded to antibodies tagged with oligos which expanded the array of protein quantification measurements (Figure 2A). These developments facilitated detection of changes in Myc and Max heterodimer formation in response to interferon-gamma, demonstrating that PLA could be used to detect biomarkers for pancreatic and ovarian cancer from patient serum32–34. Considering the known physical size of antibodies and the 20 to 60 nucleotides that comprise the conjugated oligos, the maximum distance between oligo-conjugated antibodies that still allowed ligation and subsequent signal detection is estimated to be 40 nm35. Within the domains of proteins, PLA has also been shown to have applicability to post-translational modifications. For example, PLA was implemented to quantify levels of cell surface receptor PDGFRβ phosphorylation in situ, providing higher fidelity insight than PLA in homogenous solutions and revealing the spatial distribution of PDGFRβ activation during wound healing36. Subsequently, by using secondary antibodies conjugated with DNA oligonucleotides, the utility of PLA was dramatically expanded to include the wide array of epitopes targeted by primary unconjugated antibodies. However, this “indirect PLA” suffers a loss in spatial resolution (while remaining below 40 nm) due to the size associated with the addition of a secondary antibody37. Antibody-based PLA ultimately presents a high-resolution, high-sensitivity method for measuring abundance of protein, detecting colocalization events of distinct proteins, and quantifying levels of post-translational modification of a given protein.
In the context of nucleic acids, antibody-based PLA methods can be challenging as many antibodies are poorly optimized for selectively binding specific nucleic acids and exhibit great variability in their specificity38–41. This gap in the field motivated the development of hybrid techniques that require labeling methods uniquely amenable to the detection of each type of biomolecule in the complex (“hybrid PLA”, Figure 2B). In the case of RNA, one half of the PLA pair can be a DNA probe complementary to a target RNA for RNA in situ hybridization (RISH) often paired with a protein-targeting antibody42. For example, this strategy was used to characterize the interaction of the U1 and U2 small nuclear RNAs with Sm proteins. This work validated that the La protein engages the viral EBER2 RNA and provided insight into protein pathways exploited by viruses42. In the same year, Roussis et al. developed an alternative approach in which the RISH probe exhibited biotin rather than a primer site43. Here, a biotinylated hybridization probe and a protein of interest targeted with a primary antibody was detected with indirect PLA. This study confirmed the interaction between the Xenopus Staufen1 protein and the Vg1 RNA, allowing detection of heterogeneity in Vg1-Staufen1 interactions across oocytes43. While this method boasts simpler RISH probe design, the labeling strategy is notably more complex than that used by Zhang et al. PLA methods also facilitate detection of RNA-RNA interactions. Recently, Basavappa et al. employed two hybridizing RNA probes that bind distinct fragments of the nucleic acid reporter that, when ligated, can be amplified by PCR44. This strategy enabled the discovery that the endogenous lnRNA ALPHA binds genomic chikungunya virus RNA, attenuating viral reproduction and consequently, infection. Researchers have also recently developed RNA-centric PLA assays to detect and characterize the spatial distribution of mRNA translation. In this method termed RIBOmap, Zeng et al. designed one probe complementary to rRNA and a pair of padlock and primer probes that hybridize to mRNAs45. Upon binding of the rRNA splint probe and the padlock/primer pair probes to a ribosome and an mRNA engaged by that ribosome, respectively, the padlock probe is ligated, enabling rolling circle amplification, binding of reporter probes, and detection of active sites of translation of specific mRNAs. This method facilitated visualization of the translation of over 5000 genes in situ in intact murine brain tissues, enabling cell-type-specific and tissue-specific translational profiles. These and other examples demonstrate the utility of PLA in the context of RNA biology, however they have been focused on intracellular processes.
Moving to the cell surface, where RNA biology has been less commonly studied, a PLA-based technology has been developed that combines the idea of characterizing post-synthesis modifications with the study of RNA biology (Figure 2C). Specifically, a sialic acid aptamer and RNA in situ hybridization-mediated proximity ligation assay (ARPLA) enabled the first evaluation of specific glycoRNA on a mammalian cell11. This technology leveraged two pieces of knowledge: (A) that many glycoRNAs contain glycans with sialic acids and (B) the sequence of specific RNA templates that were modified with glycans2. To target (A), the authors used a selective DNA aptamer with binding specificity for N-acetylneuraminic acid (Neu5Ac), and to target (B) the authors optimized various complementary probes for sequence-specific hybridization. The authors first fixed cells to preserve features of interest and blocked nonspecific binding. After binding of the on-target RISH probe and an empirically optimized incubation time, the authors washed the cells and bound the aptamer in an optimized buffer to enhance both aptamer binding and specificity. DNA Ligation followed by the addition of connectors enabled rolling circle amplification and visualization of glycoRNA localization by binding fluorophore-tagged DNA oligo reporters. To ensure the accuracy of their method, the authors also demonstrated that removal of any probe, connector, ligation step, or reporter resulted in the loss of effectively all ARPLA signal. Moreover, digestion of cell surface RNA with RNase A or cleavage of the sialic acid with neuraminidase was sufficient to destroy ARPLA signal, further supporting the conclusion that ARPLA enables detection of glycoRNA. Crucially, because RISH probes exhibit specificity to the RNA to which they are complementary, this method facilitates the quantification and detection of individual known glycoRNAs of interest. In this study, the authors successfully visualized the U1, U35, and Y5 glycoRNAs on single cells and characterized their association with lipid rafts as well as SNARE proteins, suggesting a pathway for possible trafficking of glycoRNAs to or from the cell surface. Future efforts to quantify glycoRNA expression may consider performing super-resolution techniques to more accurately assess the spatial distribution of glycoRNA. Additionally, multiplexing strategies could be used to detect more than one cell surface glycoRNA transcript on a single cell.
Detecting proximity by catalyst-based labeling
Proximity labeling (PL) has emerged as an indispensable tool in chemical biology, initially developed to study protein-protein interactions46,47. This technique, central to understanding molecular networks and their biological functions across cell types, has mainly been optimized to study intracellular proteins. However, its application extends further and has more recently been adapted to investigate nucleic acids, lipids, and glycans in some contexts48–52, with highlights in Table 2 (focusing here on methods that can assay cell surface RNAs directly). The ability to probe diverse biopolymer networks, especially on membranes and the cell surface, have the potential to transform our understanding of important molecular processes, so it is critical that these methods are robust for all biopolymers. There are many ways to consider catalyst-based proximity labeling – through a target-centric lens focused on how to label RNA, DNA, or protein, etc.; or through a technology-centric lens focused on comparing the pros and cons of different techniques as they pertain to different molecular species. Here we will use a technology-centric approach to describe the various catalyst-based proximity labeling methods, with a focus on their utility for studying RNA interactions, including glycoRNA. We also highlight the considerations and adaptations required for proximity labeling on the cell surface.
Table 2.
Examples of cell surface catalyst-based proximity labeling
| Method | Probe | Targets | Applications in Cell Surface Biology | Notes | Design |
|---|---|---|---|---|---|
| HRP |
|
|
|
|
Probe: > Available Enzyme: > Available |
| APEX/APEX2 |
|
|
|
|
Probe: > Available Enzyme: > Available |
| uMAP | photocatalyst and dexter energy transfer | Proteins | Microenvironments on the cell surface, with shorter, more precise labeling strategies, e.g. PD-L1 | Photocatalyst-antibody conjugate used to spatially localize carbene generation and selectively label protein-protein interactions on membranes, Geri et al.86 | Probe: > Available Catalyst: > Custom |
| MultiMap | Eosin Y and diazirine biotin, aryl-azide-biotin, phenol-biotin | Proteins | Maps protein networks at various distances – short, intermediate and long range - from the photocatalyst, e.g. EGFR neighborhoods | A single photocatalyst coupled with three photoprobes can generate three sets of protein network datasets at various distances from the POI, Lin et al.93 | Probe: > Available Catalyst: > Available |
Proximity labeling historically leverages enzymes, such as peroxidases and biotin ligases, that are genetically or chemically tethered to a protein of interest. Upon activation, the enzymes convert an inert small-molecule substrate into a short-lived reactive species that covalently tags proximal molecules limited by diffusion of the reactive species. Tagged molecules can then be imaged or enriched and subsequently identified by mass spectrometry (MS) or nucleic acid sequencing. Controlling variables such as temperature, time, and reaction conditions is critical to establish specificity in proximity labeling and has been extensively reviewed elsewhere53. The labeling radius is often a key consideration and tool-specific feature that is determined by both the half-life of the reactive species and the concentration of quenchers in the environment. The reported labeling radii for peroxidases (e.g. HRP – horseradish peroxidase, and APEX – engineered ascorbate peroxidase) and biotin ligases (e.g. BioID and TurboID) fall in the range of 1–20 nm in living cells47,53. Reactive species from both enzyme classes are membrane impermeant, so the labeling radius ends at membrane boundaries - a feature that can be exploited to restrict labeling to the cell surface or constrained to other membranous organelles54.
Biotin-ligase-dependent PL harnesses engineered promiscuous biotin ligases, such as mutated BirA, that are fused to a bait protein of interest and expressed in cells. The enzyme catalyzes the synthesis of a bioAMP radical from biotin and ATP to label proximal molecules. BioID, the first of its kind developed, has a labeling period of 15–18 hours, which generates a ‘history’ of candidate proximal proteins upon biotin supplementation, which can be further screened for novel substrates or binding partners to the bait46,55. BioID and BioID2, a smaller more specific ligase, have also been used to provide useful information on the constituency of subcellular structures such as lamins, nuclear envelope proteins, and nuclear transporters55,56. Since BioID is simple and non-toxic, only requiring the addition of biotin, it has been widely used to study protein interactions in mammalian cells, parasites, mice, yeast, and plants57. However, the slow kinetics of BioID make it less amenable to study dynamic processes occurring in minutes or even a few hours. This prompted the development of TurboID and miniTurbo, an evolved biotin ligase, optimized for rapid labeling in living cells. These enzymes have the advantage of working at near-physiological conditions with faster labeling times of 10 minutes, compared to the BioID and BioID2, with no issues in toxicity58. However, biotin-ligase-based methods initially had limited applicability to studying RNA-centric mechanisms because the bioAMP radical is reactive with lysine residues on protein not RNA moieties.
Adaptation of biotin-ligase methods to identify proteins that are proximal to RNA species of interest have occurred by designing the enzyme fused to proteins that are known to bind RNA sequences. RaPID (RNA-protein interaction detection) for example, uses a BoxB RNA stem loop that is tagged to an RNA sequence of interest to recruit a mutated biotin ligase fusion protein that biotinylates proximal proteins in the presence of biotin59. RaPID was used to identify host proteins that interact with Zika virus RNA. Similarly, the MS2 coat protein has been fused to BioID to recruit these PL enzymes to MS2-tagged RNAs, such as β-actin mRNA60. However, one major caveat is that these methods label proximity neighborhoods of exogenously expressed tagged RNA which may differ from their native counterparts. To circumvent this issue, RNA-directed CRISPR systems have recently been developed to identify proteins associated with a specific RNA sequence in its native context. These methods require a catalytically dead Cas protein, such as dCas9 or dCas13, fused to a PL enzyme, including BioID, BASU, TurboID or APEX, that is directed to the RNA by gRNAs61–64. This approach has identified novel interactors of human telomerase RNA, nuclear lncRNAs, and different genomic loci. The CRISPR-based methods have varying advantages and disadvantages, depending on the dCas used and its ability to bind to the target RNA as well as the features of the PL enzyme chosen – as both biotin-ligases and peroxidase enzymes can be used. In some contexts, these methods may have great utility, but the possibility that the Cas protein will sterically hinder RNA-protein binding and the need for genetic expression of the system necessitate development of improved RNA-based PL strategies. Notably, the aforementioned RNA-based PL techniques label protein proximal to specific RNA of interest and are not designed to discover novel RNAs that interact with targeted proteins.
While peroxidase-based labeling schemes are also employed in RNA-directed CRISPR methods, they are generally more amenable than biotin ligases to directly tag RNA. This has been best represented using APEX enzymes and biotin-phenol as a substrate. APEX was first designed to overcome the intracellular restrictions of HRP and then an improved version, APEX2, was evolved with better activity and sensitivity65,66. APEX and HRP can both generate biotin-phenoxyl radicals from biotin-phenol upon oxidation, which covalently attach to electron-rich amino acid side chains such as tyrosine on proteins. It was later reported, with APEX2, that biotin-phenoxyl radicals can also directly biotinylate guanosine in RNAs52. APEX-PL was then combined with RNA sequencing (APEX-seq) to determine the enrichment or depletion of RNA near key RNA-binding proteins and to determine subcellular transcriptomes67,68. Other protein-RNA PL strategies include APEX-RIP which uses APEX2-mediated proximity biotinylation of proteins with formaldehyde cross-linking to enrich endogenous RNAs in membrane-enclosed organelles69. Similarly, proximity-CLIP (crosslinking and immunoprecipitation) uses APEX2 followed by UV crosslinking to identify localized RNAs and proteins in subcellular compartments70. However, APEX-seq is the more direct approach, bypassing the need for crosslinking by using an APEX fusion protein to directly biotinylate proximal RNAs.
Peroxidase-based PL has been used widely for probing protein interactions since 2012, and only more recently for RNA interactions. Consequently, there have been various protein-centric method adaptations including the combination of PL strategies with crosslinking, phase separation or sequencing as mentioned above, as well as the optimization of the substrates71. Notably, biotin-phenol has relatively poor cellular permeability, which prompted the development of novel substrates including alkyne-phenol with improved membrane permeability. Following APEX-mediated labeling via the activated phenol radical, the alkyne handle is derivatized using click chemistry with biotin-conjugated azide for enrichment and readout72,73. This alkyne-phenol mediated PL boasts both improved membrane permeability and activity compared to its biotin counterpart. Although, the poor cellular permeability of biotin-phenol can be advantageous when targeting the cell surface or a subcellular compartment. Labeling can be restricted to a physical locale, such as the cell surface, by combining antibody-mediated74 or surface-targeted HRP75 with a membrane impermeant biotin-phenol variant BxxP76. These method developments have expanded the scope of peroxidase proximity labeling and highlight the vast potential for opportunities to develop similar adaptations for RNA-centric PL.
While biotin- and alkyne-phenol can be used to label proximal RNA, the phenolic compounds preferentially react with proteins, limiting the scope of RNA PL applications. Therefore, to improve the RNA-specific capture of APEX-seq, Zhou et al. synthesized novel probes and reported biotin-aniline (Figure 3B) to be approximately 20-fold more reactive than biotin-phenol towards RNA52. The addition of biotin-aniline to the toolbox of proximity labeling substrates significantly expands the potential of peroxidase enzymes to define novel RNA interactions. For example, to validate the observation that glycoRNAs are tethered to the cell surface, we utilized biotin-aniline and a non-genetic HRP labeling strategy74. HRP, like APEX enzymes, can generate reactive radicals from both biotin-phenol and biotin-aniline substrates. We leveraged lectins as cell surface affinity tools to bind live cells, a strategy previously reported as a means to tether exogenous peroxidases to the cell surface for labeling77. We selected a lectin which was expected to bind glycoRNA directly and one that was not, finding that only the predicted glycoRNA-binding lectin was able to deposit biotin-aniline radicals on proximal glycoRNAs2. The versatility of HRP to be used non-genetically and to generate radicals that can label protein and RNA makes it a useful tool for researchers to study hybrid interactions of both biomolecules using the same enzyme. We continued to take advantage of these method features to better understand the glycoRNA cell surface neighborhoods by probing the proximal proteins and RNAs of newly defined csRBPs12. Using antibody-directed HRP labeling with biotin-aniline or -phenol, we were able to characterize glycoRNA-csRBP clusters on the cell surface (Figure 3C). We found that a large but specific suite of RBPs is presented on the external surface of cells and cluster together. These clusters form regular patterns reminiscent of a tessellated structure and contain glycoRNAs. Interestingly, these glycoRNA-csRBP clusters are sites where extracellular factors like cell penetrating peptides (e.g. TAT) can bind and internalize in a manner gated by the presence of cell surface RNA. The use of HRP in our study was critical to uncover this glycoRNA-csRBP biology. HRP, one of the earliest enzymes utilized for PL, has primarily been used to study cell surface and compartment (ER, secretory pathway) biology, offering a convenient, non-genetic and robust method to study cell surface biology, such as glycoRNA interactions.
The advent of RNA-centric PL adaptations is encouraging, but there remain many opportunities for method development including the design of clickable, smaller probes for RNA-specific, peroxidase-mediated labeling. Recently, Liang et al. reported a set of novel phenol derivatives with improved RNA labeling capabilities compared to biotin-phenol and alkyne-phenol78. They showed that a derivative, Ph_N3, with APEX2 can effectively label transcriptomes in the mitochondrial matrix, demonstrating how chemical innovation can deepen our ability to query RNA in hard-to-reach subcellular spaces.
While APEX/HRP-mediated PL has many benefits and opportunities to be adapted for RNA-centric, cell surface and membrane labeling in vitro, it has been more difficult to probe in vivo interactomes. Transgenic Drosophila and mouse lines with APEX or HRP-fused constructs have been used to identify cell-type specific proteomes75,79. These strategies have been useful in some contexts, but they do require the tissues to be dissected for labeling, and require exposure to H2O2 which is toxic to living cells80. On the other hand, the in vivo BioID and TurboID methods developed have more demonstrated success. However, they are not well suited for probing RNA interactomes, and there has been very little PL data using these methods on cell surfaces in live animals81–85. Altogether, there have been many exciting developments in the PL space and there remain many opportunities to adapt current methods for diverse biopolymer studies on the cell surface in vitro and in vivo.
Beyond the broadly employed enzymatic PL, researchers have developed alternative PL methods using photosensitizers, such as metal catalysts and chromophores, as the artificial enzyme. Such methods, μMAP, CAT-ex, PhoXCELL, and PhoTag, offer improved temporal responses strictly controlled by light exposure86–89. Targeting biomolecules, such as antibodies, are typically used to link the photosensitizer catalyst to the target of interest, and upon light exposure, the precursor molecules are converted to reactive species via electron transfer or energy transfer. The relatively large molecular size and charged nature of these compounds are beneficial when probing the cell surface and cell-cell interactions. While these methods have been powerful for mapping protein neighborhoods, novel methods for unbiased probing of RNA interactomes have been scarcer. Light-activated, RNA-specific targeting methods, such as CAP-seq and Halo-seq, have been developed that use genetically encoded photosensitizers, including singlet-oxygen, dibromofluorescein or miniSOG, to mediate nucleobase oxidation50,90. However, these methods rely on radical or reactive oxygen species (ROS)-reactivity, which can be less active towards RNA-like polymers50,91. Therefore, increased reagent concentrations and labeling times are required to overcome poor RNA reactivity, which could have unknown effects on the native cellular conformation and conditions. Therefore, there is space for optimization of light-activated, RNA-centric PL methods. Recently, Pani et al. reported a novel method, BAP-seq, for spatial analysis of RNA that combines a subcellular-targeted esterase from Bacillus subtilis (BS2) with a new class of masked enol-ester probes to generate highly reactive acylating agents that label nearby RNAs92. The highly reactive acid chloride acylating agent labels nearby nucleophiles with an alkyne that can be imaged or enriched with click chemistry. This method was combined with RNAseq to map mitochondrial, nuclear and nucleolar RNAs, and is an exciting addition to RNA-PL methods for membrane-bound and membrane-less organelles. Additionally, researchers developed “MultiMap” (Figure 3C), a method that uses Eosin Y as a singular photocatalyst coupled to (in independent experiments) various biotinylated reagents to generate carbene, nitrene, and phenoxyl radicals for labeling proximal molecules93. The relative stability of each of these light-generated species dictates the radius of labeling (with carbene, nitrene, and phenoxyl radicals in order of smallest to largest labeling radius) enabling a common Eosin Y tagged affinity tool to produce data on proximal neighborhoods of different sizes. Notably, it has not yet been directly tested how efficiently the carbene single oxygen species reacts with RNA.
Outlook
Many tools have been developed that leverage chemical biology principles to study induced proximity across a wide array of biological conditions. To date, the focus of most of these has been protein-centric, however as the methods become better distributed, they have been applied to glycobiology and RNA biology questions. Most recently, the use of ligation-based and enzymatic proximity labeling assays has enabled early investigations into glycoRNA biology on the cell surface. The ability to maintain topological specificity (on the extracellular side of the plasma membrane) and interrogate across different biopolymers (protein to RNA, RNA to glycan, etc.) are key features for accurate induced proximity assays. As more work leverages these tools, we propose a flexible naming scheme that indicates the proximity labeling concept (PL), the biopolymer targeted (RNA, protein, etc.), and a post-label that indicates the analysis method (MS/mass spec, seq/sequencing, etc.). For example, “pPL-MS” could be used to note proximity labeling of proteins with TurboID or APEX+biotin-phenol and subsequent proteomics analysis whereas “rPL-seq” could be used to denote proximity labeling of RNA with HRP+biotin-aniline and subsequent analysis by deep sequencing.
While we have access to this biology with the above-mentioned tools, improvements in key areas would deepen and expand our ability to understand how the cell surface is constructed and better define the role and organization of hybrid molecules like glycoRNAs. Specific features include (1) multiplexing, (2) chemical selectivity and efficiency, and (3) in vivo tools.
In the case of ligation-based tools like ARPLA, having the ability to assess multiple glycoRNAs on the cell surface at the same time would significantly increase our understanding of their spatial organization. For example, a currently unanswered question is: given a single cluster of glyco-Y5, are there other glycoRNAs within this physical domain, or does each cell surface domain only contain a singular transcript? Establishing orthogonal PLA pairs or detection sequences with a suitable protocol for this multiplexed labeling would enable examination of putative glycoRNA heterogeneity in situ. This approach could be expanded to RNA-protein interactions on the cell surface, providing additional motivation for the development of such a tool.
New chemical labeling schemes on the cell surface could also be developed. There has been a recent expansion of catalyst-based labeling tools. However, no concerted effort has evaluated their labeling properties using a common cell type, common anchoring point, and common conditions to detail how each performs on the cell surface while labeling RNA, proteins, and glycans. Additional developments could include methods that are purposely designed for cell surface studies using probes that are never cell permeable, perhaps reducing the off-target labeling of intracellular proteins.
In vitro models of biological systems are useful starting points. However, blotting and mass spectrometry tools are now sensitive enough to detect material from in vivo systems. Examples of in vivo ER lumen and extracellular protein labeling by TurboID82 and75 provide a starting point for expanding into cell surface glycoRNA biology. However, the in vivo study in the Drosophila brain used BxxP, of which an aniline version is not yet available, and the fly brains needed to be isolated by dissection first to allow for BxxP exposure. Whereas the TurboID work was feasible in living mice, this labeling scheme is not robustly compatible with RNA labeling. Optimizing efficient RNA-directed live animal labeling methods could enable more accurate capture of the cell surface RNA biology in the context of native or disease 3D tissues.
As we use and develop tools that enable us to peer deeper and with higher resolution into the organization of domains like the cell surface, we are sure to uncover a broader appreciation for the known, and likely new, ways that biology leverages induced proximity to establish precise structure and function across life.
Acknowledgements
This work was supported by grants from Burroughs Wellcome Fund Career Award for Medical Scientists (R.A.F.) and the National Institute of General Medical Sciences of the National Institutes of Health under award number GM151157 (R.A.F.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
R.A.F. is a cofounder and stockholder of GanNA Bio. R.A.F. is a board of directors member and stockholder of Chronus Health and Blue Planet Systems.
References
- 1.Bausch-Fluck D, Goldmann U, Müller S, Oostrum M. van, Müller M, Schubert OT, and Wollscheid B (2018). The in silico human surfaceome. PNAS 115, E10988–E10997. 10.1073/pnas.1808790115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Flynn RA, Pedram K, Malaker SA, Batista PJ, Smith BAH, Johnson AG, George BM, Majzoub K, Villalta PW, Carette JE, et al. (2021). Small RNAs are modified with N-glycans and displayed on the surface of living cells. Cell 184, 3109–3124.e22. 10.1016/j.cell.2021.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xie Y, Hemberger H, Till NA, Chai P, Watkins C, Lebedenko CG, Caldwell RM, George B, Bertozzi CR, Garcia BA, et al. (2023). The modified RNA base acp3U is an attachment site for N-glycans in glycoRNA. Preprint at bioRxiv, 10.1101/2023.11.06.565735 10.1101/2023.11.06.565735. [DOI] [Google Scholar]
- 4.Wolfram-Schauerte M, Pozhydaieva N, Grawenhoff J, Welp LM, Silbern I, Wulf A, Billau FA, Glatter T, Urlaub H, Jäschke A, et al. (2023). A viral ADP-ribosyltransferase attaches RNA chains to host proteins. Nature 620, 1054–1062. 10.1038/s41586-023-06429-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banik SM, Pedram K, Wisnovsky S, Ahn G, Riley NM, and Bertozzi CR (2020). Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature. 10.1038/s41586-020-2545-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huehls AM, Coupet TA, and Sentman CL (2015). Bispecific T-cell engagers for cancer immunotherapy. Immunol Cell Biol 93, 290–296. 10.1038/icb.2014.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tender GS, and Bertozzi CR (2023). Bringing enzymes to the proximity party. RSC Chem Biol 4, 986–1002. 10.1039/d3cb00084b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crocker PR, Paulson JC, and Varki A (2007). Siglecs and their roles in the immune system. Nat Rev Immunol 7, 255–266. 10.1038/nri2056. [DOI] [PubMed] [Google Scholar]
- 9.Li J, Yue S, Gao Z, Hu W, Liu Z, Xu G, Wu Z, Zhang X, Zhang G, Qian F, et al. (2023). Novel Approach to Enriching Glycosylated RNAs: Specific Capture of GlycoRNAs via Solid-Phase Chemistry. Anal. Chem 95, 11969–11977. 10.1021/acs.analchem.3c01630. [DOI] [PubMed] [Google Scholar]
- 10.Liu H, Li X, Ren Y, Yang Y, Chen Y, and Ju H (2024). In Situ Visualization of RNA-Specific Sialylation on Living Cell Membranes to Explore N-Glycosylation Sites. J Am Chem Soc 146, 8780–8786. 10.1021/jacs.4c01826. [DOI] [PubMed] [Google Scholar]
- 11.Ma Y, Guo W, Mou Q, Shao X, Lyu M, Garcia V, Kong L, Lewis W, Ward C, Yang Z, et al. (2023). Spatial imaging of glycoRNA in single cells with ARPLA. Nat Biotechnol. 10.1038/s41587-023-01801-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perr J, Langen A, Almahayni K, Nestola G, Chai P, Lebedenko C, Volk R, Caldwell R, Spiekermann M, Hemberger H, et al. (2023). RNA binding proteins and glycoRNAs form domains on the cell surface for cell penetrating peptide entry. Preprint at bioRxiv, 10.1101/2023.09.04.556039 10.1101/2023.09.04.556039. [DOI] [Google Scholar]
- 13.Zhang N, Tang W, Torres L, Wang X, Ajaj Y, Zhu L, Luan Y, Zhou H, Wang Y, Zhang D, et al. (2024). Cell surface RNAs control neutrophil recruitment. Cell 187, 846–860.e17. 10.1016/j.cell.2023.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang N, Fan X, Zaleta-Rivera K, Nguyen TC, Zhou J, Luo Y, Gao J, Fang RH, Yan Z, Chen ZB, et al. (2020). Natural display of nuclear-encoded RNA on the cell surface and its impact on cell interaction. Genome Biol 21, 225. 10.1186/s13059-020-02145-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ren Z, Li R, Zhou X, Chen Y, Fang Y, and Zou P (2023). Enzyme-Mediated Proximity Labeling Identifies Small RNAs in the Endoplasmic Reticulum Lumen. Biochemistry. 10.1021/acs.biochem.3c00142. [DOI] [PubMed] [Google Scholar]
- 16.Dunn KW, Kamocka MM, and McDonald JH (2011). A practical guide to evaluating colocalization in biological microscopy. Am J Physiol Cell Physiol 300, C723–742. 10.1152/ajpcell.00462.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ryder PV, and Lerit DA (2020). Quantitative analysis of subcellular distributions with an open-source, object-based tool. Biol Open 9, bio055228. 10.1242/bio.055228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu C-D, Chinenov Y, and Kerppola TK (2002). Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol Cell 9, 789–798. 10.1016/s1097-2765(02)00496-3. [DOI] [PubMed] [Google Scholar]
- 19.Magde D, Elson E, and Webb WW (1972). Thermodynamic Fluctuations in a Reacting System---Measurement by Fluorescence Correlation Spectroscopy. Phys. Rev. Lett 29, 705–708. 10.1103/PhysRevLett.29.705. [DOI] [Google Scholar]
- 20.Förster T (2012). Energy migration and fluorescence. 1946. J Biomed Opt 17, 011002. 10.1117/1.JBO.17.1.011002. [DOI] [PubMed] [Google Scholar]
- 21.Yaffe Y, Shepshelovitch J, Nevo-Yassaf I, Yeheskel A, Shmerling H, Kwiatek JM, Gaus K, Pasmanik-Chor M, and Hirschberg K (2012). The MARVEL transmembrane motif of occludin mediates oligomerization and targeting to the basolateral surface in epithelia. J Cell Sci 125, 3545–3556. 10.1242/jcs.100289. [DOI] [PubMed] [Google Scholar]
- 22.Nickerson A, Huang T, Lin L-J, and Nan X (2014). Photoactivated localization microscopy with bimolecular fluorescence complementation (BiFC-PALM) for nanoscale imaging of protein-protein interactions in cells. PLoS One 9, e100589. 10.1371/journal.pone.0100589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ozawa T, Takeuchi TM, Kaihara A, Sato M, and Umezawa Y (2001). Protein splicing-based reconstitution of split green fluorescent protein for monitoring protein-protein interactions in bacteria: improved sensitivity and reduced screening time. Anal Chem 73, 5866–5874. 10.1021/ac010717k. [DOI] [PubMed] [Google Scholar]
- 24.Werner A, Skakun VV, Meyer C, and Hahn U (2011). RNA dimerization monitored by fluorescence correlation spectroscopy. Eur Biophys J 40, 907–921. 10.1007/s00249-011-0701-8. [DOI] [PubMed] [Google Scholar]
- 25.Mateu-Regué À, Christiansen J, Bagger FO, Hellriegel C, and Nielsen FC (2021). Unveiling mRNP composition by fluorescence correlation and cross-correlation spectroscopy using cell lysates. Nucleic Acids Res 49, e119. 10.1093/nar/gkab751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niaki AG, Sarkar J, Cai X, Rhine K, Vidaurre V, Guy B, Hurst M, Lee JC, Koh HR, Guo L, et al. (2020). Loss of Dynamic RNA Interaction and Aberrant Phase Separation Induced by Two Distinct Types of ALS/FTD-Linked FUS Mutations. Mol Cell 77, 82–94.e4. 10.1016/j.molcel.2019.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holden SJ, Uphoff S, Hohlbein J, Yadin D, Le Reste L, Britton OJ, and Kapanidis AN (2010). Defining the limits of single-molecule FRET resolution in TIRF microscopy. Biophys J 99, 3102–3111. 10.1016/j.bpj.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lin W, Du Y, Zhu Y, and Chen X (2014). A cis-membrane FRET-based method for protein-specific imaging of cell-surface glycans. J Am Chem Soc 136, 679–687. 10.1021/ja410086d. [DOI] [PubMed] [Google Scholar]
- 29.Hohng S, Joo C, and Ha T (2004). Single-molecule three-color FRET. Biophys J 87, 1328–1337. 10.1529/biophysj.104.043935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bajar BT, Wang ES, Zhang S, Lin MZ, and Chu J (2016). A Guide to Fluorescent Protein FRET Pairs. Sensors (Basel) 16, 1488. 10.3390/s16091488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fredriksson S, Gullberg M, Jarvius J, Olsson C, Pietras K, Gústafsdóttir SM, Ostman A, and Landegren U (2002). Protein detection using proximity-dependent DNA ligation assays. Nat Biotechnol 20, 473–477. 10.1038/nbt0502-473. [DOI] [PubMed] [Google Scholar]
- 32.Fredriksson S, Horecka J, Brustugun OT, Schlingemann J, Koong AC, Tibshirani R, and Davis RW (2008). Multiplexed proximity ligation assays to profile putative plasma biomarkers relevant to pancreatic and ovarian cancer. Clin Chem 54, 582–589. 10.1373/clinchem.2007.093195. [DOI] [PubMed] [Google Scholar]
- 33.Gullberg M, Gústafsdóttir SM, Schallmeiner E, Jarvius J, Bjarnegård M, Betsholtz C, Landegren U, and Fredriksson S (2004). Cytokine detection by antibody-based proximity ligation. Proc Natl Acad Sci U S A 101, 8420–8424. 10.1073/pnas.0400552101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Söderberg O, Gullberg M, Jarvius M, Ridderstråle K, Leuchowius K-J, Jarvius J, Wester K, Hydbring P, Bahram F, Larsson L-G, et al. (2006). Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods 3, 995–1000. 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
- 35.Zatloukal B, Kufferath I, Thueringer A, Landegren U, Zatloukal K, and Haybaeck J (2014). Sensitivity and specificity of in situ proximity ligation for protein interaction analysis in a model of steatohepatitis with Mallory-Denk bodies. PLoS One 9, e96690. 10.1371/journal.pone.0096690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jarvius M, Paulsson J, Weibrecht I, Leuchowius K-J, Andersson A-C, Wählby C, Gullberg M, Botling J, Sjöblom T, Markova B, et al. (2007). In situ detection of phosphorylated platelet-derived growth factor receptor beta using a generalized proximity ligation method. Mol Cell Proteomics 6, 1500–1509. 10.1074/mcp.M700166-MCP200. [DOI] [PubMed] [Google Scholar]
- 37.Alam MS (2018). Proximity Ligation Assay (PLA). Curr Protoc Immunol 123, e58. 10.1002/cpim.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ausserwöger H, Schneider MM, Herling TW, Arosio P, Invernizzi G, Knowles TPJ, and Lorenzen N (2022). Non-specificity as the sticky problem in therapeutic antibody development. Nat Rev Chem 6, 844–861. 10.1038/s41570-022-00438-x. [DOI] [PubMed] [Google Scholar]
- 39.Bonin M, OBERSTRAß J, Lukacs N, Ewert K, Oesterschulze E, Kassing R, and Nellen W (2000). Determination of preferential binding sites for anti-dsRNA antibodies on double-stranded RNA by scanning force microscopy. RNA 6, 563–570. 10.1017/S1355838200992318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bumbaca D, Wong A, Drake E, Reyes AE, Lin BC, Stephan J-P, Desnoyers L, Shen B-Q, and Dennis MS (2011). Highly specific off-target binding identified and eliminated during the humanization of an antibody against FGF receptor 4. MAbs 3, 376–386. 10.4161/mabs.3.4.15786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Loberg LI, Chhaya M, Ibraghimov A, Tarcsa E, Striebinger A, Popp A, Huang L, Oellien F, and Barghorn S (2021). Off-target binding of an anti-amyloid beta monoclonal antibody to platelet factor 4 causes acute and chronic toxicity in cynomolgus monkeys. MAbs 13, 1887628. 10.1080/19420862.2021.1887628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang W, Xie M, Shu M-D, Steitz JA, and DiMaio D (2016). A proximity-dependent assay for specific RNA-protein interactions in intact cells. RNA 22, 1785–1792. 10.1261/rna.058248.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roussis IM, Guille M, Myers FA, and Scarlett GP (2016). RNA Whole-Mount In situ Hybridisation Proximity Ligation Assay (rISH-PLA), an Assay for Detecting RNA-Protein Complexes in Intact Cells. PLoS One 11, e0147967. 10.1371/journal.pone.0147967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Basavappa MG, Ferretti M, Dittmar M, Stoute J, Sullivan MC, Whig K, Shen H, Liu KF, Schultz DC, Beiting DP, et al. (2022). The lncRNA ALPHA specifically targets chikungunya virus to control infection. Mol Cell 82, 3729–3744.e10. 10.1016/j.molcel.2022.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zeng H, Huang J, Ren J, Wang CK, Tang Z, Zhou H, Zhou Y, Shi H, Aditham A, Sui X, et al. (2023). Spatially resolved single-cell translatomics at molecular resolution. Science 380, eadd3067. 10.1126/science.add3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kim DI, and Roux KJ (2016). Filling the Void: Proximity-Based Labeling of Proteins in Living Cells. Trends Cell Biol 26, 804–817. 10.1016/j.tcb.2016.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Qin W, Cho KF, Cavanagh PE, and Ting AY (2021). Deciphering molecular interactions by proximity labeling. Nat Methods 18, 133–143. 10.1038/s41592-020-01010-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guo Y, Wang P, Jiang L, Deng C, Zheng L, Song C, and Jiao J (2023). Multifunctional Proximity Labeling Strategy for Lipid Raft-Specific Sialic Acid Tracking and Engineering. Bioconjug Chem 34, 1719–1726. 10.1021/acs.bioconjchem.3c00236. [DOI] [PubMed] [Google Scholar]
- 49.Reeves AE, and Huang ML (2023). Proximity labeling technologies to illuminate glycan-protein interactions. Curr Opin Chem Biol 72, 102233. 10.1016/j.cbpa.2022.102233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang P, Tang W, Li Z, Zou Z, Zhou Y, Li R, Xiong T, Wang J, and Zou P (2019). Mapping spatial transcriptome with light-activated proximity-dependent RNA labeling. Nat Chem Biol. 10.1038/s41589-019-0368-5. [DOI] [PubMed] [Google Scholar]
- 51.Wu G, Nagala M, and Crocker PR (2017). Identification of lectin counter-receptors on cell membranes by proximity labeling. Glycobiology 27, 800–805. 10.1093/glycob/cwx063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou Y, Wang G, Wang P, Li Z, Yue T, Wang J, and Zou P (2019). Expanding APEX2 Substrates for Proximity-Dependent Labeling of Nucleic Acids and Proteins in Living Cells. Angewandte Chemie 131, 11889–11893. 10.1002/ange.201905949. [DOI] [PubMed] [Google Scholar]
- 53.Bosch JA, Chen C-L, and Perrimon N (2021). Proximity-dependent labeling methods for proteomic profiling in living cells: An update. Wiley Interdiscip Rev Dev Biol 10, e392. 10.1002/wdev.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rhee H-W, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, and Ting AY (2013). Proteomic Mapping of Mitochondria in Living Cells via Spatially-Restricted Enzymatic Tagging. Science 339, 1328–1331. 10.1126/science.1230593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Roux KJ, Kim DI, Raida M, and Burke B (2012). A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. Journal of Cell Biology 196, 801–810. 10.1083/jcb.201112098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim DI, Jensen SC, Noble KA, Kc B, Roux KH, Motamedchaboki K, and Roux KJ (2016). An improved smaller biotin ligase for BioID proximity labeling. Mol Biol Cell 27, 1188–1196. 10.1091/mbc.E15-12-0844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sears RM, May DG, and Roux KJ (2019). BioID as a Tool for Protein-Proximity Labeling in Living Cells. Methods Mol Biol 2012, 299–313. 10.1007/978-1-4939-9546-2_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Branon TC, Bosch JA, Sanchez AD, Udeshi ND, Svinkina T, Carr SA, Feldman JL, Perrimon N, and Ting AY (2018). Efficient proximity labeling in living cells and organisms with TurboID. Nat Biotechnol 36, 880–887. 10.1038/nbt.4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ramanathan M, Majzoub K, Rao DS, Neela PH, Zarnegar BJ, Mondal S, Roth JG, Gai H, Kovalski JR, Siprashvili Z, et al. (2018). RNA-protein interaction detection in living cells. Nat Methods 15, 207–212. 10.1038/nmeth.4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mukherjee J, Hermesh O, Eliscovich C, Nalpas N, Franz-Wachtel M, Maček B, and Jansen R-P (2019). β-Actin mRNA interactome mapping by proximity biotinylation. Proc Natl Acad Sci U S A 116, 12863–12872. 10.1073/pnas.1820737116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Han S, Zhao BS, Myers SA, Carr SA, He C, and Ting AY (2020). RNA–protein interaction mapping via MS2- or Cas13-based APEX targeting. Proc Natl Acad Sci USA, 202006617. 10.1073/pnas.2006617117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li Y, Liu S, Cao L, Luo Y, Du H, Li S, Zhang Z, Guo X, Tian W, Wong CC, et al. (2021). CBRPP: a new RNA-centric method to study RNA-protein interactions. RNA Biol 18, 1608–1621. 10.1080/15476286.2021.1873620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Myers SA, Wright J, Peckner R, Kalish BT, Zhang F, and Carr SA (2018). Discovery of proteins associated with a predefined genomic locus via dCas9-APEX-mediated proximity labeling. Nat Methods 15, 437–439. 10.1038/s41592-018-0007-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yi W, Li J, Zhu X, Wang X, Fan L, Sun W, Liao L, Zhang J, Li X, Ye J, et al. (2020). CRISPR-assisted detection of RNA–protein interactions in living cells. Nat Methods. 10.1038/s41592-020-0866-0. [DOI] [PubMed] [Google Scholar]
- 65.Lam SS, Martell JD, Kamer KJ, Deerinck TJ, Ellisman MH, Mootha VK, and Ting AY (2015). Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat Methods 12, 51–54. 10.1038/nmeth.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Martell JD, Deerinck TJ, Sancak Y, Poulos TL, Mootha VK, Sosinsky GE, Ellisman MH, and Ting AY (2012). Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat Biotechnol 30, 1143–1148. 10.1038/nbt.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fazal FM, Han S, Parker KR, Kaewsapsak P, Xu J, Boettiger AN, Chang HY, and Ting AY (2019). Atlas of Subcellular RNA Localization Revealed by APEX-Seq. Cell, S0092867419305550. 10.1016/j.cell.2019.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Padrón A, Iwasaki S, and Ingolia NT (2019). Proximity RNA Labeling by APEX-Seq Reveals the Organization of Translation Initiation Complexes and Repressive RNA Granules. Mol Cell 75, 875–887.e5. 10.1016/j.molcel.2019.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kaewsapsak P, Shechner DM, Mallard W, Rinn JL, and Ting AY (2017). Live-cell mapping of organelle-associated RNAs via proximity biotinylation combined with protein-RNA crosslinking. eLife 6, 623. 10.7554/eLife.29224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Benhalevy D, Anastasakis DG, and Hafner M (2018). Proximity-CLIP provides a snapshot of protein-occupied RNA elements in subcellular compartments. Nature Methods 15, 1074–1082. 10.1038/s41592-018-0220-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Qin W, Myers SA, Carey DK, Carr SA, and Ting AY (2021). Spatiotemporally-resolved mapping of RNA binding proteins via functional proximity labeling reveals a mitochondrial mRNA anchor promoting stress recovery. Nat Commun 12, 4980. 10.1038/s41467-021-25259-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Li Y, Tian C, Liu K, Zhou Y, Yang J, and Zou P (2020). A Clickable APEX Probe for Proximity-Dependent Proteomic Profiling in Yeast. Cell Chem Biol 27, 858–865.e8. 10.1016/j.chembiol.2020.05.006. [DOI] [PubMed] [Google Scholar]
- 73.Qin W, Cheah JS, Xu C, Messing J, Freibaum BD, Boeynaems S, Taylor JP, Udeshi ND, Carr SA, and Ting AY (2023). Dynamic mapping of proteome trafficking within and between living cells by TransitID. Cell 186, 3307–3324.e30. 10.1016/j.cell.2023.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bar DZ, Atkatsh K, Tavarez U, Erdos MR, Gruenbaum Y, and Collins FS (2018). Biotinylation by antibody recognition—a method for proximity labeling. Nature Methods 15, 127–133. 10.1038/nmeth.4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li J, Han S, Li H, Udeshi ND, Svinkina T, Mani DR, Xu C, Guajardo R, Xie Q, Li T, et al. (2020). Cell-Surface Proteomic Profiling in the Fly Brain Uncovers Wiring Regulators. Cell 180, 373–386.e15. 10.1016/j.cell.2019.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Loh KH, Stawski PS, Draycott AS, Udeshi ND, Lehrman EK, Wilton DK, Svinkina T, Deerinck TJ, Ellisman MH, Stevens B, et al. (2016). Proteomic Analysis of Unbounded Cellular Compartments: Synaptic Clefts. Cell 166, 1295–1307.e21. 10.1016/j.cell.2016.07.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kirkemo LL, Elledge SK, Yang J, Byrnes J, Glasgow J, Blelloch R, and Wells JA (2021). Cell-surface tethered promiscuous biotinylators enable small-scale surface proteomics of human exosomes. Preprint at bioRxiv, 10.1101/2021.09.22.461393 10.1101/2021.09.22.461393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liang J, Han J, Gao X, Jia H, Li R, Tse ECM, and Li Y (2023). Clickable APEX2 Probes for Enhanced RNA Proximity Labeling in Live Cells. Anal Chem. 10.1021/acs.analchem.3c03614. [DOI] [PubMed] [Google Scholar]
- 79.Chen C-L, Hu Y, Udeshi ND, Lau TY, Wirtz-Peitz F, He L, Ting AY, Carr SA, and Perrimon N (2015). Proteomic mapping in live Drosophila tissues using an engineered ascorbate peroxidase. Proceedings of the National Academy of Sciences of the United States of America 112, 12093–12098. 10.1073/pnas.1515623112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yang X, Wen Z, Zhang D, Li Z, Li D, Nagalakshmi U, Dinesh-Kumar SP, and Zhang Y (2021). Proximity labeling: an emerging tool for probing in planta molecular interactions. Plant Commun 2, 100137. 10.1016/j.xplc.2020.100137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Droujinine IA, Meyer AS, Wang D, Udeshi ND, Hu Y, Rocco D, McMahon JA, Yang R, Guo J, Mu L, et al. (2021). Proteomics of protein trafficking by in vivo tissue-specific labeling. Nat Commun 12, 2382. 10.1038/s41467-021-22599-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim K-E, Park I, Kim J, Kang M-G, Choi WG, Shin H, Kim J-S, Rhee H-W, and Suh JM (2021). Dynamic tracking and identification of tissue-specific secretory proteins in the circulation of live mice. Nat Commun 12, 5204. 10.1038/s41467-021-25546-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Strack R (2021). Revealing the secretome. Nat Methods 18, 1273. 10.1038/s41592-021-01320-2. [DOI] [PubMed] [Google Scholar]
- 84.Long J Cell type-selective secretome profiling in vivo. 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yang R, Meyer AS, Droujinine IA, Udeshi ND, Hu Y, Guo J, McMahon JA, Carey DK, Xu C, Fang Q, et al. (2022). A genetic model for in vivo proximity labelling of the mammalian secretome. Open Biol 12, 220149. 10.1098/rsob.220149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Geri JB, Oakley JV, Reyes-Robles T, Wang T, McCarver SJ, White CH, Rodriguez-Rivera FP, Parker DL, Hett EC, Fadeyi OO, et al. (2020). Microenvironment mapping via Dexter energy transfer on immune cells. Science 367, 1091–1097. 10.1126/science.aay4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liu H, Luo H, Xue Q, Qin S, Qiu S, Liu S, Lin J, Li JP, and Chen PR (2022). Antigen-Specific T Cell Detection via Photocatalytic Proximity Cell Labeling (PhoXCELL). J Am Chem Soc 144, 5517–5526. 10.1021/jacs.2c00159. [DOI] [PubMed] [Google Scholar]
- 88.Liu Z, Xie X, Huang Z, Lin F, Liu S, Chen Z, Qin S, Fan X, and Chen PR (2022). Spatially resolved cell tagging and surfaceome labeling via targeted photocatalytic decaging. Chem 8, 2179–2191. 10.1016/j.chempr.2022.04.016. [DOI] [Google Scholar]
- 89.Rc O, T R-R, Ch W, Jh T, Ka C, Ep B, D C, Vm P, L L, S F, et al. (2022). Detection of cell-cell interactions via photocatalytic cell tagging. Nature chemical biology 18. 10.1038/s41589-022-01044-0. [DOI] [PubMed] [Google Scholar]
- 90.Li Y, Aggarwal MB, Nguyen K, Ke K, and Spitale RC (2017). Assaying RNA Localization in Situ with Spatially Restricted Nucleobase Oxidation. ACS Chem. Biol 12, 2709–2714. 10.1021/acschembio.7b00519. [DOI] [PubMed] [Google Scholar]
- 91.Lo H-YG, Engel KL, Goering R, Li Y, Spitale RC, and Taliaferro JM (2022). Halo-seq: An RNA Proximity Labeling Method for the Isolation and Analysis of Subcellular RNA Populations. Curr Protoc 2, e424. 10.1002/cpz1.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pani S, Qiu T, Kentala K, Azizi S-A, and Dickinson B (2023). Biorthogonal masked acylating agents for proximity-dependent RNA labeling. Preprint at ChemRxiv, 10.26434/chemrxiv-2023-vx3hm 10.26434/chemrxiv-2023-vx3hm. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lin Z, Schaefer K, Lui I, Yao Z, Fossati A, Swaney DL, Palar A, Sali A, and Wells JA (2023). Multi-scale photocatalytic proximity labeling reveals cell surface neighbors on and between cells. bioRxiv, 2023.10.28.564055. 10.1101/2023.10.28.564055. [DOI] [Google Scholar]
- 94.Moreno V, Smith EA, and Piña-Oviedo S (2022). Fluorescent Immunohistochemistry. Methods Mol Biol 2422, 131–146. 10.1007/978-1-0716-1948-3_9. [DOI] [PubMed] [Google Scholar]
- 95.Weibrecht I, Leuchowius K-J, Clausson C-M, Conze T, Jarvius M, Howell WM, Kamali-Moghaddam M, and Söderberg O (2010). Proximity ligation assays: a recent addition to the proteomics toolbox. Expert Rev Proteomics 7, 401–409. 10.1586/epr.10.10. [DOI] [PubMed] [Google Scholar]
- 96.Kotani N, Gu J, Isaji T, Udaka K, Taniguchi N, and Honke K (2008). Biochemical visualization of cell surface molecular clustering in living cells. Proc Natl Acad Sci U S A 105, 7405–7409. 10.1073/pnas.0710346105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ogawa Y, Lim BC, George S, Oses-Prieto JA, Rasband JM, Eshed-Eisenbach Y, Hamdan H, Nair S, Boato F, Peles E, et al. (2023). Antibody-directed extracellular proximity biotinylation reveals that Contactin-1 regulates axo-axonic innervation of axon initial segments. Nat Commun 14, 6797. 10.1038/s41467-023-42273-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Xie Y, Mao Y, Mao Z-W, and Xia W (2023). Identification of Substrates of Secreted Bacterial Protease by APEX2-Based Proximity Labeling. Methods Mol Biol 2674, 169–179. 10.1007/978-1-0716-3243-7_11. [DOI] [PubMed] [Google Scholar]
- 99.Tuomivaara ST, Teo CF, Jan YN, Jan LY, and Wiita AP (2023). SLAPSHOT reveals rapid dynamics of extracellularly exposed proteome in response to calcium-activated plasma membrane phospholipid scrambling. bioRxiv, 2023.03.26.534250. 10.1101/2023.03.26.534250. [DOI] [Google Scholar]
- 100.Hung V, Lam SS, Udeshi ND, Svinkina T, and Guzman G (2017). Proteomic mapping of cytosol-facing outer mitochondrial and ER membranes in living human cells by proximity biotinylation. eLife. 10.7554/eLife.24463.001. [DOI] [PMC free article] [PubMed] [Google Scholar]