

Abstract
Kidney epithelial cells have very high energy requirements, which are largely met by fatty acid oxidation. Complex changes in lipid metabolism are observed in patients with kidney disease. Defects in fatty acid oxidation and increased lipid uptake, especially in the context of hyperlipidemia and proteinuria, contribute to this excess lipid build-up and exacerbate kidney disease development. Recent studies have also highlighted the role of increased de novo lipogenesis in kidney fibrosis. The defect in fatty acid oxidation will cause energy starvation. Increased lipid uptake, synthesis and lower fatty acid oxidation can cause toxic lipid build-up, reactive oxygen species generation, and mitochondrial damage. Better understanding of these metabolic processes may open new treatment avenues for kidney diseases by targeting lipid metabolism.
Keywords: Acute kidney injury (AKI), chronic kidney disease (CKD), metabolism, lipid, fatty acid oxidation, de novo lipogenesis
INTRODUCTION
Several nephron segments have very high energy requirement. The proximal tubule (PT) of the nephron reabsorbs nearly 70% of the primary glomerular filtrate, which encompasses essential nutrients such as glucose and amino acids, electrolytes, and water.1 Similarly, the distal convoluted tubule cells (DCT) and principal cells transport sodium against their electrochemical gradient. The high energy demand of the reabsorptive and secretive processes necessitate a high mitochondrial density in PT and DCT cells2 and their reliance on mitochondrial oxidative phosphorylation (OxPhos) for ATP generation.
Proximal tubule cells preferentially metabolize fatty acids (FAs) to generate energy through fatty acid oxidation (FAO).3 This process provides tricarboxylic acid (TCA) cycle intermediates and produces more ATP per carbon atom, compared to glycolysis.
While PT cells reabsorb and transport hundreds of grams of glucose each day, PT cells do not typically use glucose as their energy source.4 PT cells likely avoid glucose utilization so PT cellular metabolism is uncoupled from the fluctuating luminal glucose exposure which is determined by the glomerular filtration rate (GFR) and serum glucose.5 To partition glucose reabsorption and cellular metabolism, proximal tubules seem to lack hexokinase which is essential for glucose phosphorylation (the first step for glucose metabolism). The healthy urine is glucose free by the time it reaches the distal segment, so distal tubule segments lack apical glucose transporters and utilize fatty acids as their energy substrates.
Changes in lipid uptake, utilization, lipid accumulation and cholesterol metabolism in podocytes was recently reviewed.6 In this review, we discuss changes in lipid metabolism in tubule cells and investigate the potential link between alterations in lipid metabolism, lipid accumulation, and the development of kidney disease.
I. Lipid accumulation in kidney proximal tubules is a common feature of acute and chronic kidney disease.
Kimmelstiel was one of the first pathologists to notice lipid accumulation in kidneys of patients with diabetic kidney disease (DKD).7 Lipid accumulation in kidney tubule cells is now described in a wide range of conditions, including renal ischemia-reperfusion, diabetes, and nephrotic syndrome.8–10 Multiple studies suggest that kidney lipid content correlate with disease severity. However, a key question remains whether lipid accumulation is the cause or the consequence of disease.11
Accumulating triglycerides (TG) in PT cells give rise to lipid droplets (LDs). LD formation and accumulation has been observed in both human kidney diseases and animal models.12–14 These LDs consist of free fatty acids (FFAs) that have been esterified with glycerol. Once formed, LDs undergo fusion, ripening, coalescence, or lipophagy (autophagic LD degradation), reflecting their dynamic nature. The morphology and behavior of LDs—varying in number, size, localization, and composition—mirror the cellular metabolic status and the oscillation between nutrient availability and energy demand. Furthermore, LDs interact with other cellular compartments like the endoplasmic reticulum, peroxisomes, mitochondria, and lysosomes, and integral for their life cycle and functions. Beyond the core roles in lipid storage, transport, synthesis, and hydrolysis, recent analyses reveal LDs’ involvement in membrane trafficking, protein storage and degradation, signal transduction, detoxification, and nucleic acid handling. Coating of LDs with perilipin 2 facilitates TG export.
A notable challenge is the comprehensive elucidation of lipid species that accumulate in disease states, including the diverse array of fatty acid side chains. A recent comprehensive characterization of the kidney lipidome in diabetic rats indicated distinctive lipid signature characterized by variations in side-chain compositions and unsaturation levels.15 Particularly, neutral lipids largely constituted this altered lipidome, with a higher degree of unsaturation and side chains of linoleic acid. On the other hand, phospholipids, and sphingolipids showed substantial alterations which may bear deleterious impacts on the renal function. Importantly, not all lipids appear to be damaging for kidney tubule cells. Polyunsaturated fatty acids (PUFAs)– like eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA)– have been noted to have renoprotective effects, while palmitate is deleterious.16
Recently an NMR-based metabolite analysis was performed on ~91,000 participants in the UK Biobank study and followed a median of 13 years to test the association with lipid metabolomic alterations and the incidence of CKD.17 There are 142 lipid biomarkers out of which 90 biomarkers belonging to 14 subclasses of lipoproteins were significantly associated with incidence of CKD. Consistent with previous findings, the study showed a positive association with VLDL, cholesteryl esters, free fatty acids and TG were concentrations with greater risk of CKD, whereas HDL, PUFA, and DHA concentrations were associated with lower risk of CKD. Furthermore, the study also identified concentrations of apolipoprotein A1 and apolipoprotein B to apolipoprotein A1 ratio were significantly associated with the risk of CKD. But apolipoprotein B concentration alone did not correlate with CKD risk.17 The role of these lipid species need to be carefully investigated for better understanding of CKD mechanisms. Future studies shall analyze changes in lipid species in kidney disease and understand the effect of such lipid species on tubule epithelial health and function.
II. Changes in lipid metabolism in CKD
Increased lipid uptake
The mechanism of lipid uptake by PT cells in normal and disease states need better characterization. At basal state, PT cells acquire lipids from the circulation through their basolateral membrane. This basolateral uptake of fatty acids (FAs) is facilitated mostly by fatty acid transport protein 2 (FATP2). CD36; another long chain fatty acid transporter, is mostly expressed by endothelial cells at basal conditions and are likely to be essential for delivering circulating lipids into PT cells.18 Basal lipid uptake might be sensitive to fluctuations of circulating lipid levels and systemic hyperlipidemia.
Tubule cell lipid uptake is far more complex in glomerular proteinuric state. Albumin is the primary carrier of circulating lipids.19 In the setting of glomerular proteinuria, FA-bound albumin is endocytosed via megalin. Albumin and FAs then dissociate, and lipids enter tubule cells by transporter-mediated mechanisms or diffusion.20 Increased CD36 expression is observed in patients DKD.21 In the heart, increased uptake via CD36 was associated with lipotoxicity,22 but this may not be the case in the kidney. Overexpression of CD36 in kidney tubule cells caused increased lipid uptake, but also enhanced fatty acid oxidation (FAO) without causing overt phenotypic changes.23 FATP2 appears to be the dominant fatty acid transporter in PT cells, as genetic deletion of FATP2 ameliorated lipid uptake in models of glomerular disease development in mice19 and tubule lipotoxicity.24 Additionally, small molecular FATP2 inhibitors have been developed, which have protected mice from kidney disease and tubule toxicity.24 Injured proximal tubule cells express KIM1 (Havcr1), which has been reported to mediate the uptake of oxidized lipids.25 Overall, it is possible that basolateral lipid uptake is the main mechanism for nutrient uptake in healthy kidney tubule cells, while apical lipid uptake occurs especially in the setting of glomerular injury.
Changes in fatty acid oxidation
Fatty acid oxidation (FAO) is the primary source of energy for kidney PT cells. A complex network of transcription factors that are mostly nuclear receptors control the expression of genes involved in FAO (Figure 1a). These transcription factors include the PPARα, liver X receptor (LXR), farnesoid X receptor (FXR), and estrogen-related receptor alpha (ESRRα), all which can directly control FAO genes. Transcription coactivators, such as peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1a), also regulate FAO by influencing mitochondrial biogenesis.26 These critical metabolic regulators work together with HNF1B and HNF4A transcription factors to establish cell identity and usually co-bind to genes involved in PT metabolism,27 ensuring PT cell metabolism and energy supply is linked to PT cell gene expression. As the most abundant genes expressed by PT cells are energy-requiring solute carriers, this mechanism essentially links PT cell metabolism to PT cell gene expression, likely ensuring that the generated energy is utilized.
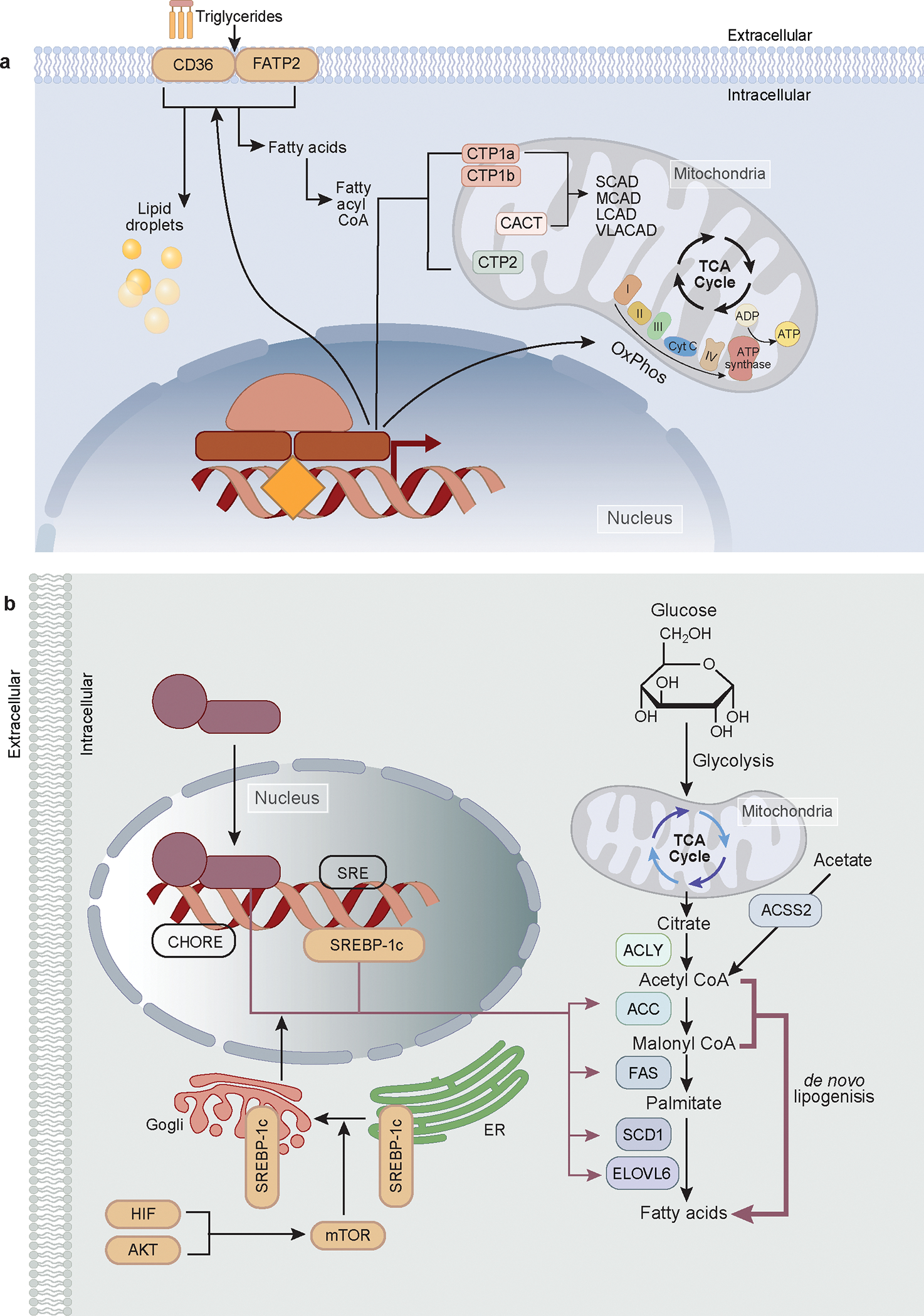
Figure 1. Schematic representation of fatty acid oxidation and de novo lipogenesis.
a. The fatty acids are transported by CD36 and FATP2 into the cells, where they either stored as lipid droplets or undergo mitochondrial oxidation. Prior to mitochondrial oxidation, fatty acids are converted into fatty acyl CoA. Two key molecules for transporting FA to the mitochondria; Carnitine palmiotyltransferases-1 and 2 (CPT1, CPT2), are highlighted. Also depicted are the central transcriptional regulators of fatty acid oxidation and mitochondrial biogenesis, namely PGC1a, PPARα, FXR, LXR, and ESRRα.
b. The insulin/AKT/PI3K pathway is the primary mechanism allowing cells to store excess nutrients. The phosphatidylinositol 3 phosphate kinase (P13K)/AKT / mechanistic target of rapamycin (mTOR) pathways control de Novo Lipogenesis (DNL). Mix and ChREBP dimerize and translocate into the nucleus, where they bind to the CHORE promoter, initiating transcription of target genes (ACC, FAS, SCD1, and ELOVL6) crucial for the final stages of fatty acid synthesis. HIF/AKT/mTOR pathways induce SREBP-1c, a transcription factor central to the biosynthesis of lipid, TG, FA, and cholesterol metabolism. SREBP-1c binds to the SRE promoter, promoting the transcription of the same target genes (ACC, FAS, SCD1, and ELOVL6). Both transcription factors increase the transcription of ACLY and ACSS, which convert citrate and acetate, respectively, into Acetyl CoA. In renal Proximal Tubule (PT) cells, acetate is converted into acetyl CoA via ACSS2 to promote de novo lipogenesis, is also illustrated, along with the utilization of cytosolic malonyl CoA. The final conversion of Acetyl-CoA to Malonyl-CoA to palmitate, carried out by ACC, FAS, SCD1, and ELOVL6 in the cytosol.
Once free fatty acids (FFAs) enter the cell, they are conjugated by acyl-CoA synthetase (ACS) into fatty acyl-CoA.28 Due to the impermeability of the inner mitochondrial membrane, fatty acyl-CoA cannot directly pass into the mitochondrial matrix. Instead, it is converted into fatty acyl-carnitine by the enzyme carnitine palmitoyl transferase I (CPT1). Once inside the mitochondria, the fatty acyl-carnitine is converted back into fatty acyl-CoA by carnitine palmitoyltransferase II (CPT2). In the mitochondria, fatty acyl-CoA is broken down to produce acetyl-CoA via a reaction that includes oxidation, hydration and thiolytic cleavage within mitochondria. The acetyl-CoA is then used in the tricarboxylic acid (TCA) cycle to generate FADH2 and NADH, which are then used in oxidative phosphorylation to produce ATP. Excess acetyl-CoA can be transported out of the mitochondria by CACT (carnitine-acylcarnitine translocase) and used to synthesize new FAs. Very long chain fatty acids (VLFAs) are initially oxidized in the peroxisome, releasing acetyl-CoA until they are shortened to eight carbons and can then be transported to the mitochondria to complete the oxidation.
Omega oxidation, (ω-oxidation), is an alternative pathway of fatty acid metabolism that takes place primarily in the endoplasmic reticulum. Omega oxidation is typically a minor pathway, except for some specific fatty acids.29 Omega oxidation starts at the omega carbon, which is the carbon furthest from the carboxyl group. The initial step is the addition of an oxygen atom by cytochrome P450 to the omega carbon, forming a fatty alcohol. The fatty alcohol is then oxidized to a fatty aldehyde by an alcohol dehydrogenase, and then to a fatty acid by an aldehyde dehydrogenase. The result is a dicarboxylic acid, which has carboxyl groups at both ends of the molecule. Dicarboxylic acids can then undergo beta-oxidation, starting from the carboxyl end. This results in shorter chain dicarboxylic acids and, eventually, in succinates that can enter the citric acid cycle to be further broken down for energy.30
In human kidney tissue samples, expression of genes playing role in FAO has shown a strong correlation with kidney function and fibrosis.31 Using microarray and RNA sequencing techniques, lower expression of PPARα, ESRRα, PGC1α, and CPT1A has been observed in patients with kidney disease and animal models of AKI and CKD31–35 indicating an overall decrease in FAO in diseased kidneys (Figures 2–3). Gene expression analysis performed in mouse kidney disease models indicated a consistently lower expression of genes associated with FAO.36 Single cell expression studies of mice AKI and CKD samples have further confirmed reduced expression of key FAO genes (PPARα, ESRRα) in PT cells.23,31,37 Elevated serum acyl carnitines observed in hospitalized patients indicating a potential blockage of metabolism further support the likely lower FAO in kidney injury.38,39 Finally, direct measurement of kidney FAO in vivo and in vitro in cultured cells showed consistent results with gene expression changes in animal models of kidney disease.40
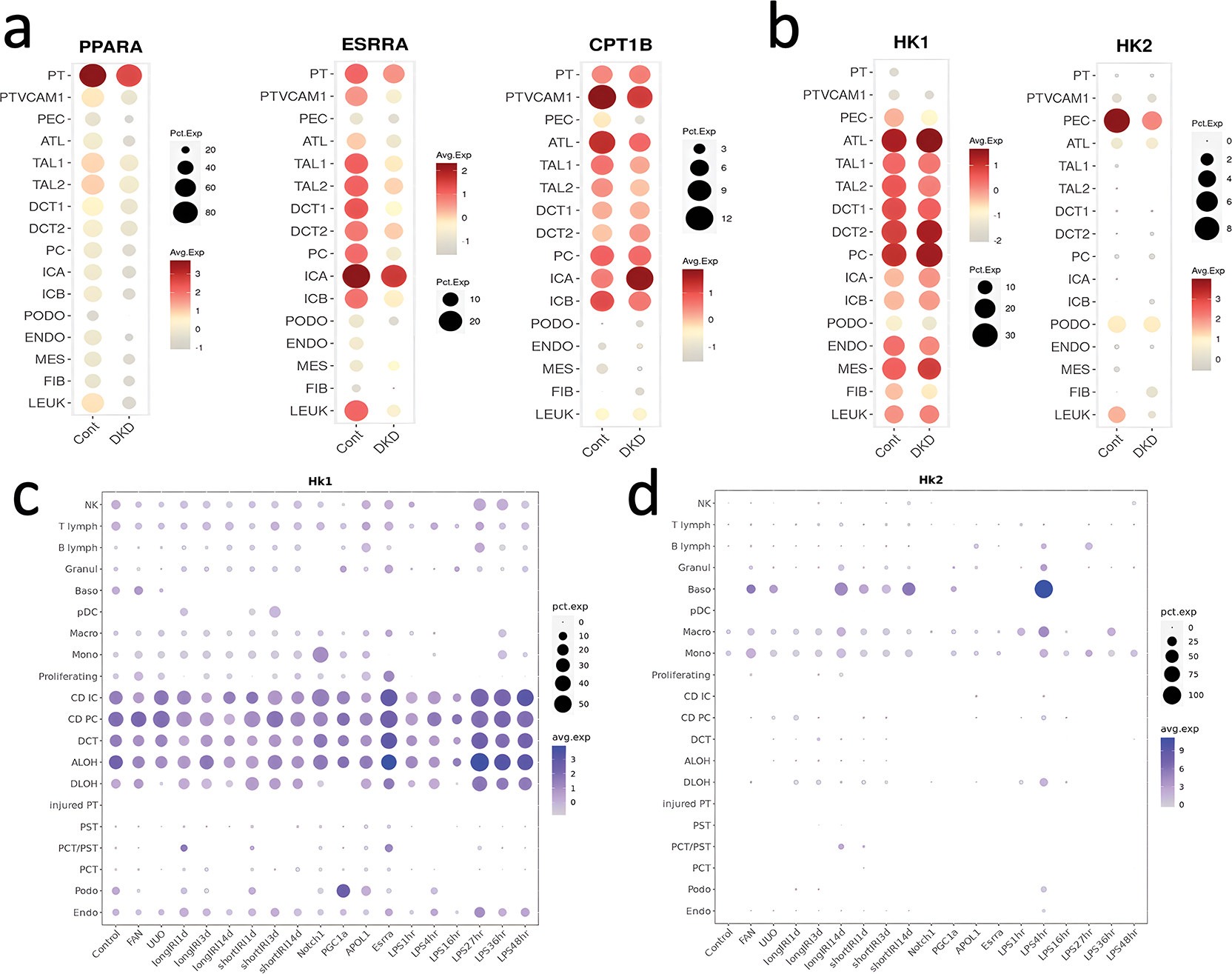
Figure 2. Expression of key metabolic regulators in healthy and diseased human and mouse kidneys at single cell level.
Single cell gene expression of FAO genes such as PPARA, ESRRA, and CPT1B and other metabolic genes hexokinase 1 (HK1) and 2 (HK2 in healthy controls and DKD patient kidneys.35
Single cell gene expression of metabolic regulators in various mouse kidney disease models5 including chronic kidney disease (CKD) and acute kidney injury (AKI) models. UUO (unilateral ureteral obstruction, a surgical CKD model), FAN (folic acid nephropathy, a crystal precipitation-induced CKD model), ischemia-reperfusion (IRI), Notch1 transgenic mice (Notch), Peroxisome proliferator-activated receptor γ-coactivator 1 alpha transgenic mice (PGC-1α), Apolipoprotein L1 transgenic mice (APOL1), Estrogen related receptor alpha (Esrrα) knockout mice, and bacterial lipopolysaccharide induced AKI (LPS). The size of the dot indicates the percent positive cells, and the intensity of the color indicates the transcript amount. The size of the circle indicates the percent of cells expressing the target gene, while the color intensity indicates the level of gene expression.
Figure 3. Expression of key genes in fatty acid oxidation (FAO) and de novo lipogenesis (DNL) in bulk mouse and human kidney samples.
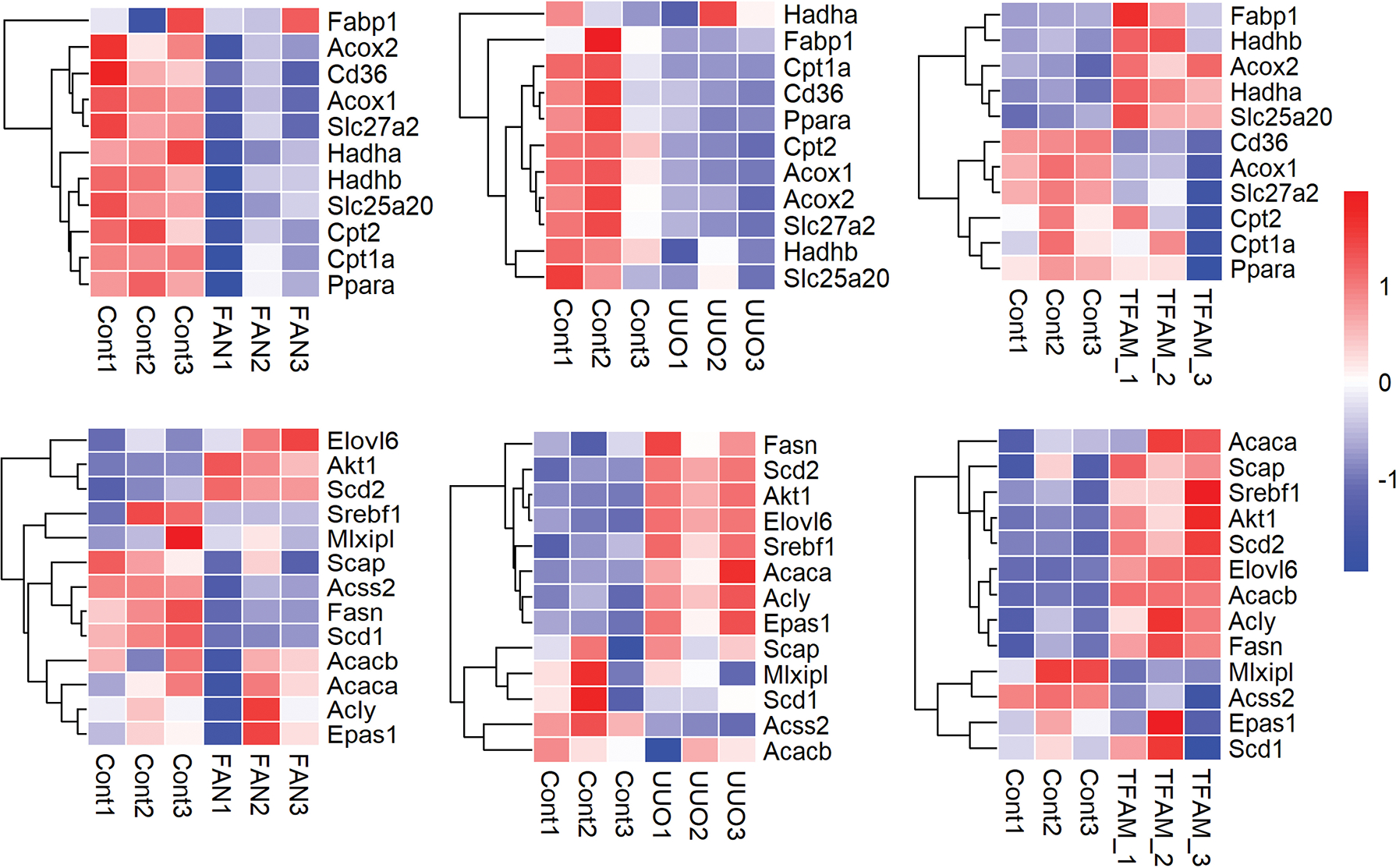
Top Row (FAO): Expression of major FAO genes in FAN and UUO kidney injury models and mice with genetic deletion of Tfam in kidney tubule cells compared to control kidneys. Genes: Acox1, Acox 2, Cd36, Cpt1a, Cpt2, Fabp1, Hadha, Hadhb, Ppara, Scl25a20, Slc27a2).
Bottom Row (DNL): Expression of some of the core DNL genes in FAN,31 UUO kidney injury models,36 and mice with genetic deletion of Tfam in kidney tubule cells compared to control kidneys.36
Genes: Acaca, Acacb, Acly, Acss2, Elovl6, Epas1, Fasn, MIxipl, Scap, Scd1, Scd2, Srebf1.
UUO (unilateral ureteral obstruction, a surgical CKD model), FAN (folic acid nephropathy, a crystal precipitation-induced CKD model), TFAM (kidney tubule specific knockout of mitochondrial transcription factor A). Each row is one gene, each column is one animal, red indicates higher gene expression while blue indicates lower gene expression.
Genetic manipulation of genes and pathways associated with FAO indicate that defects in fatty acid oxidation cause lipid accumulation and downstream kidney disease development. In mice, deleting the mitochondrial transcription factorA (Tfam) caused lipid accumulation, fibrosis, and kidney dysfunction.41 While deleting Pparα, Pgc1α, or Esrrα in mice did not induce kidney disease at baseline, but these knockout animals were more susceptible to both acute and chronic injury.31–33,42 Consistently, transgenic expression of Ppparα or Pgc1α in tubule cells has been found to protect mice against AKI and CKD.23,43 These findings suggest that mitochondrial metabolism is critical for maintaining kidney homeostasis and preventing kidney disease.31,44
The rate-limiting step of beta oxidation is the carbon entry into the mitochondria, this step is regulated by the enzyme CPT1, which is located on the outer mitochondrial membrane. Lower CPT1 expression has been observed in later stages of CKD.23 Transgenic mice with CPT1A overexpression in kidney tubule cells are protected from lipid accumulation and kidney fibrosis.34 Pharmacological activation of CPT1 has also shown a beneficial effect in mouse kidney disease models.23 Recent single cell transcriptomic profiling of early human diabetic nephropathy samples has indicted a modest increase in PT CPT1A expression,45 which potentially suggests an early compensation mechanism in response to diabetes or elevated lipid levels.37 In summary, a pronounced and consistent defect in PT FAO has been observed both in kidneys of mice and patient with disease.
Changes in de novo lipogenesis
De novo lipogenesis (DNL) is the process by which new lipids are synthesized from carbohydrates (Figure 1b). The major site of DNL is the liver,46 storing excess nutrients in an efficient manner, but DNL could also take place in adipose tissue and kidneys (Figure 1b).47,48 The initial step of DNL is driven by the conversion of acetyl CoA to malonyl CoA by acetyl CoA carboxylase (ACC). Intracellular acetyl-CoA sources include acetate (generated via ACSS2), citrate (generated via ACLY), and pyruvate (generated via PHD).49 The accumulation of malonyl CoA will inhibit CPT1, thus negatively regulate FAO.50 Seven molecules of malonyl-CoA are used to synthesize one molecule of palmitate (C16:0) through a cycle of condensation, reduction, and dehydration with the enzyme fatty acid synthase (FAS). Converting acetyl-CoA to palmitate requires 7 ATP and 14 NADPH molecules, making it an energetically demanding process.51 PT cells have low FAS expression at baseline, which increases in disease state.40
It is important to note that DNL is linked to FAO. For example, fatty acyl-CoA can enter the mitochondria for catabolism, producing the energy required by homeostatic kidney function and transport. Excess fatty acyl-CoA enters the anabolic pathway and generates TG for storage. In addition, acetyl-CoA generated by FAs via beta-oxidation can also be transported out by CACT and converted to malonyl-CoA by ACC, which re-synthesizes new fatty acids.52 AMPK (AMP-activated protein kinase) is an enzyme that plays a key role in cellular energy homeostasis. AMPK can inhibit ACC activity, thereby reducing malonyl-CoA levels, and increasing CPT1 and PGC-1α activity to promote fatty acid oxidation.53,54
The key transcriptional regulators of DNL are SREBP and ChREBP. Together they promote ACC and FAS expression to increase fatty acid synthesis.55,56 ChREBP (carbohydrate response element binding protein) and SREBP-1c (sterol regulatory element binding protein 1c) are the key transcription factors regulating DNL in the liver.57–59 ChREBP is a mondo family member of transcription factors.60,61 It functions by dimerizing with MIx, to regulate the expression of glucose-responsive genes and anabolic pathways.62 Transcription of the SREBP-1c gene is controlled by several signaling pathways. One of the primary regulators of SREBP-1c transcription is insulin, which acts through the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) pathway63,64 and another one is called the Liver X Receptor (LXR) pathway. LXRs are activated by oxysterols and other cholesterol derivatives, and they bind to specific response elements on the SREBP-1c gene, promoting its transcription.65,66 SREBP1 activates lipogenic genes, while SREBP2 is more specific for cholesterologenic genes.67,68
The contribution of DNL to kidney disease development is not well defined. Cell culture studies have indicated that hyperglycemia can increase SREBP1 expression,69 which subsequently leads to increased accumulation of triglycerides.70 Similarly, treatment of diabetic rats with insulin has been shown to prevent SREBP-1 overexpression and kidney triglycerides accumulation.70 Genetic deletion of SREBP1c protected against the accumulation of triglycerides in the kidney,71 whereas SREBP1a transgenic mice have shown signs lipid accumulation, albuminuria and fibrosis.72 These studies indicate the important role of SREBP1 in driving kidney pathology in vivo and in vitro. Additionally, our recent studies using mouse genetic models and cell culture experiments indicate the key role of ACSS2 and DNL in lipid accumulation and kidney disease development.40 ACSS2 has been identified as one of kidney disease risk genes in genome wide association analysis.73 Furthermore, consistent changes in genes associated with DNL were observed human CKD and mouse models of kidney disease (Figure 3). Mice with genetic deletion of ACSS2 or tubule specific deletion of fatty acid synthase (FAS) were protected from kidney disease and fibrosis.40 In summary, new results suggest that increased synthesis of lipids in the kidney is an important factor in the progression of CKD.
III. From fatty kidneys to kidney disease
Careful analysis of patient samples and animal models indicate a relationship between lipid accumulation and disease development (Figure 2).14,23,74 It is unclear whether lipid accumulation itself is toxic to cells or if disease development is linked to upstream molecular defects (for example, decreased FAO and increased DNL) which induces downstream lipid accumulation and resulting kidney disease development. Lipid accumulation alone may not be harmful, but rather serve as a response to ensure that nutrients (lipids) are stored within the cell. Indeed, renal lipid accumulation occurs during overnight fasting, which is considered physiologic.18 For example, tubule specific CD36 expression led to lipid accumulation with no observable change in kidney function or fibrosis.23
It is exceedingly difficult to dissect the contribution of FAO defect and lipid accumulation to kidney disease development. Inefficient beta-oxidation has been suggested to generate toxic lipid intermediates.75 These partially oxidized lipid products are typically directed towards mitochondrial complex II, reducing the electron transport chain, inducing a back pressure on complex I,76 and resulting in excess production of superoxide and ROS. Not only decreased FAO but also higher DNL can create a more oxidate environment. DNL requires large amounts of NADH to build fatty acids.40,77 Utilization of NADH will cause the build-up of NAD+ to increase cellular ROS content likely exacerbating the injury.78,79
On the other hand, several (mostly) in vitro studies indicate that lipid accumulation cause lipotoxicity, cellular dysfunction, cell death, and fibroinflammation,11,80,81 but the molecular pathways linking them to disease are not well understood. Accumulation of toxic metabolites like acylcarnitines and ceramides,82,83 reactive oxygen species,84 and mitochondrial defects are proposed to play a significant role.85 Uptake of palmitate is associated with intracellular reactive oxygen species (ROS) release.24,37,86,87 Recently a novel cell death mechanism called ferroptosis have been described in renal tubule cells.88 The key component of ferroptosis is iron mediated lipid peroxidation.88,89 Ferroptosis has been described in multiple animal models of kidney disease and in patient samples. Lowering ferroptosis protected cells and mice from acute and chronic kidney disease development, indicating the key role of the pathway in kidney injury.89,90
In addition, multiple studies indicate that defects in FAO plays a primary role in kidney disease development, while lipid accumulation only happened secondarily.85 For example, the decline in FAO in kidney tubules will cause ATP starvation (Figure 2).23,31,91 While tubule cells tolerated some reduction of ATP levels, the decrease in energy was associated with lowering of expression of important membrane transporters, a process we call dedifferentiation. Extreme ATP loss amplified tubule cell death.23 Overall, these studies indicate that lower FAO will induce energy deficiency in PT cells, and thereby kidney disease development.
Decreased FAO might also cause a metabolic shift to glucose oxidation as indicated by some studies.92–94 A switching from FA utilization to glycolysis was described in DKD and ischemia-reperfusion injury. This metabolic change was attributed to the increased tubule hypoxia and expression of HIF-1α.95,96 The glycolytic shift was accompanied by alterations in mitochondrial number and morphology,97 limiting other nutrient flux such as pyruvate.98 The contribution of the glycolytic switch to kidney disease remains an open area of research.
Mitochondrial damage and the release of mitochondrial ribonucleotides plays a major role in kidney fibroinflammation and CKD development.44 Genetic deletion of the mitochondrial transcription factor; Tfam or acquired loss of Tfam in kidney disease resulted in a severe mitochondrial injury, causing mitochondrial DNA release into the cytosol.36 Cytosolic RNA and DNA are then sensed by cytosolic nucleotide sensing pathways; cGAS senses mitochondrial DNA and RIG-I senses mitochondrial RNA.99–102 Both proteins activate transcription factors such as IRF3/7 and NFKB to mediate the transcription of a variety of cytokines.103 A similar activation of nucleotide sensors was observed in cisplatin or ischemia reperfusion AKI injury in mice and in kidneys of patients with diabetic and hypertensive CKD.36,99,102
IV. Conclusion
Kidney disease is characterized by a complex dysregulation of lipid metabolism. Lipid accumulation has been noted in kidneys in various disease states and they often correlate with disease severity. At the same time, a defect in fatty acid oxidation, increase in kidney lipid synthesis, and lipid update are observed.23 Detailed genetic and molecular studies will be essential to dissect these pathways in the kidney. Defect in FAO is a key driver of lipid accumulation and kidney disease in animal models. Activation of PPARα, ESRRα, and CPT1A (key regulators of FAO) has been shown to improve kidney function and reduce fibrosis in animal models of acute and chronic kidney disease.34,104,105 The effect of fibrates (PPARα agonists) has been more complex in patients.106 Fenofibrate interferes with creatinine secretion, making it difficult to ascertain its effect on eGFRcre.107,108 The ACCORD study has examined the effect of fenofibrate in kidney function decline and renal outcomes.109,110 After the initial drop in eGFRcrea (which was likely due to change creatinine secretion), patients in the fenofibrate group had significantly lower eGFR decline than that of in the control group.111 The selective PPARα activator; pemafibrate, have showed an eGFR improvement in a clinical trial.112 These studies showed no difference in cardiovascular events and hypertriglyceridemia,113 indicating that these drugs might have a renal-specific mechanisms of action, consistent with animal model studies. In addition, the beneficial effects of SGLT2i could also be mediated via improving tubule lipid metabolism. The sodium mediated glucose reabsorption is one of the highest energy-requiring tasks of proximal tubule cells. SGLT2 inhibitors decrease the energy need and FAO of PT cells, thereby improving their metabolism and gene expression profiles further, indicating the key role of tubule metabolism in kidney disease development.
In conclusion, consistent changes in lipid metabolism including lipid update, oxidation, and synthesis have been observed in human, animal models, and in vitro studies, with potential therapeutic targets already emerging from studies via improving fatty acid oxidation and limiting de novo lipogenesis.
Sources of support:
NIDDK R01 DK076077, R01 DK087635, R01 DK105821, Regeneron, Gilead, Novo Nordisk, Genentech, Novartis Boehringer Ingelheim, Bayer, Variant Bio, Maze
Work in the Susztak laboratory is also supported by Regeneron, Gilead, Novo Nordisk, Genentech, Astra Zeneca, KKC, Novartis, Boehringer Ingelheim, Calico, ONO Pharma, and Maze. L.L, T.D., and D.M. have nothing to disclose.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Lauren E Lee, Renal, Electrolyte, and Hypertension Division, Department of Medicine, University of Pennsylvania, Perelman School of Medicine, Philadelphia, PA 19104, USA
Tomohito Doke, Institute for Diabetes, Obesity, and Metabolism, University of Pennsylvania, Perelman School of Medicine, Philadelphia, PA 19104, USA
Dhanunjay Mukhi, Department of Genetics, University of Pennsylvania, Perelman School of Medicine, Philadelphia, PA 19104, USA
Katalin Susztak, Penn-CHOP Kidney Innovation Center, University of Pennsylvania, Perelman School of Medicine, Philadelphia, PA 19104, USA
References
- 1.Balzer MS, Rohacs T, Susztak K. How Many Cell Types Are in the Kidney and What Do They Do? Annu Rev Physiol. Feb 10 2022;84:507–531. doi: 10.1146/annurev-physiol-052521-121841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhargava P, Schnellmann RG. Mitochondrial energetics in the kidney. Nat Rev Nephrol. Oct 2017;13(10):629–646. doi: 10.1038/nrneph.2017.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schaub JA, Venkatachalam MA, Weinberg JM. Proximal Tubular Oxidative Metabolism in Acute Kidney Injury and the Transition to CKD. Kidney360. Feb 25 2021;2(2):355–364. doi: 10.34067/KID.0004772020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kruger C, Nguyen TT, Breaux C, et al. Proximal Tubular Cell-Specific Ablation of Carnitine Acetyltransferase Causes Tubular Disease and Secondary Glomerulosclerosis. Diabetes Apr 2019;68(4):819–831. doi: 10.2337/db18-0090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou J, Abedini A, Balzer M, et al. Unified Mouse and Human Kidney Single-Cell Expression Atlas Reveal Commonalities and Differences in Disease States. J Am Soc Nephrol. Aug 28 2023;doi: 10.1681/ASN.0000000000000217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitrofanova A, Merscher S, Fornoni A. Kidney lipid dysmetabolism and lipid droplet accumulation in chronic kidney disease. Nat Rev Nephrol. Oct 2023;19(10):629–645. doi: 10.1038/s41581-023-00741-w [DOI] [PubMed] [Google Scholar]
- 7.Kimmelstiel P, Wilson C. Intercapillary Lesions in the Glomeruli of the Kidney. Am J Pathol. Jan 1936;12(1):83–98 7. [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Z, Jiang T, Li J, et al. Regulation of renal lipid metabolism, lipid accumulation, and glomerulosclerosis in FVBdb/db mice with type 2 diabetes. Diabetes. Aug 2005;54(8):2328–35. doi: 10.2337/diabetes.54.8.2328 [DOI] [PubMed] [Google Scholar]
- 9.Rinaldi A, Lazareth H, Poindessous V, et al. Impaired fatty acid metabolism perpetuates lipotoxicity along the transition to chronic kidney injury. JCI Insight. Sep 22 2022;7(18)doi: 10.1172/jci.insight.161783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Agrawal S, Zaritsky JJ, Fornoni A, Smoyer WE. Dyslipidaemia in nephrotic syndrome: mechanisms and treatment. Nat Rev Nephrol. Jan 2018;14(1):57–70. doi: 10.1038/nrneph.2017.155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bobulescu IA. Renal lipid metabolism and lipotoxicity. Curr Opin Nephrol Hypertens. Jul 2010;19(4):393–402. doi: 10.1097/MNH.0b013e32833aa4ac [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schonefeld M, Noble S, Bertorello AM, Mandel LJ, Creer MH, Portilla D. Hypoxia-induced amphiphiles inhibit renal Na+, K(+)-ATPase. Kidney Int. May 1996;49(5):1289–96. doi: 10.1038/ki.1996.184 [DOI] [PubMed] [Google Scholar]
- 13.Herman-Edelstein M, Scherzer P, Tobar A, Levi M, Gafter U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J Lipid Res. Mar 2014;55(3):561–72. doi: 10.1194/jlr.P040501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu H, Gonzalez Villalobos R, Yao X, et al. Mapping the single-cell transcriptomic response of murine diabetic kidney disease to therapies. Cell Metab. Jul 5 2022;34(7):1064–1078 e6. doi: 10.1016/j.cmet.2022.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hou B, He P, Ma P, et al. Comprehensive Lipidome Profiling of the Kidney in Early-Stage Diabetic Nephropathy. Front Endocrinol (Lausanne). 2020;11:359. doi: 10.3389/fendo.2020.00359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wieder N, Fried JC, Kim C, et al. FALCON systematically interrogates free fatty acid biology and identifies a novel mediator of lipotoxicity. Cell Metab. May 2 2023;35(5):887–905 e11. doi: 10.1016/j.cmet.2023.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geng TT, Chen JX, Lu Q, et al. Nuclear Magnetic Resonance-Based Metabolomics and Risk of CKD. Am J Kidney Dis. Jan 2024;83(1):9–17. doi: 10.1053/j.ajkd.2023.05.014 [DOI] [PubMed] [Google Scholar]
- 18.Scerbo D, Son NH, Sirwi A, et al. Kidney triglyceride accumulation in the fasted mouse is dependent upon serum free fatty acids. J Lipid Res. Jun 2017;58(6):1132–1142. doi: 10.1194/jlr.M074427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khan S, Gaivin R, Abramovich C, Boylan M, Calles J, Schelling JR. Fatty acid transport protein-2 regulates glycemic control and diabetic kidney disease progression. JCI Insight. Aug 6 2020;5(15)doi: 10.1172/jci.insight.136845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trimble ME. Renal palmitate transport: possible sites for interaction with a plasma membrane fatty acid-binding protein. Mol Cell Biochem. Oct 15-Nov 8 1990;98(1–2):201–7. doi: 10.1007/BF00231385 [DOI] [PubMed] [Google Scholar]
- 21.Susztak K, Ciccone E, McCue P, Sharma K, Bottinger EP. Multiple metabolic hits converge on CD36 as novel mediator of tubular epithelial apoptosis in diabetic nephropathy. PLoS Med. Feb 2005;2(2):e45. doi: 10.1371/journal.pmed.0020045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiu HC, Kovacs A, Blanton RM, et al. Transgenic expression of fatty acid transport protein 1 in the heart causes lipotoxic cardiomyopathy. Circ Res. Feb 4 2005;96(2):225–33. doi: 10.1161/01.RES.0000154079.20681.B9 [DOI] [PubMed] [Google Scholar]
- 23.Kang HM, Ahn SH, Choi P, et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat Med. Jan 2015;21(1):37–46. doi: 10.1038/nm.3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen Y, Yan Q, Lv M, et al. Involvement of FATP2-mediated tubular lipid metabolic reprogramming in renal fibrogenesis. Cell Death Dis. Nov 20 2020;11(11):994. doi: 10.1038/s41419-020-03199-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mori Y, Ajay AK, Chang JH, et al. KIM-1 mediates fatty acid uptake by renal tubular cells to promote progressive diabetic kidney disease. Cell Metab. May 4 2021;33(5):1042–1061 e7. doi: 10.1016/j.cmet.2021.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scarpulla RC, Vega RB, Kelly DP. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab. Sep 2012;23(9):459–66. doi: 10.1016/j.tem.2012.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miao Z, Balzer MS, Ma Z, et al. Single cell regulatory landscape of the mouse kidney highlights cellular differentiation programs and disease targets. Nat Commun. Apr 15 2021;12(1):2277. doi: 10.1038/s41467-021-22266-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nowinski SM, Solmonson A, Rusin SF, et al. Mitochondrial fatty acid synthesis coordinates oxidative metabolism in mammalian mitochondria. Elife. Aug 17 2020;9doi: 10.7554/eLife.58041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wanders RJ, Komen J, Kemp S. Fatty acid omega-oxidation as a rescue pathway for fatty acid oxidation disorders in humans. FEBS J Jan 2011;278(2):182–94. doi: 10.1111/j.1742-4658.2010.07947.x [DOI] [PubMed] [Google Scholar]
- 30.Barbosa ACS, Pfister KE, Chiba T, et al. Dicarboxylic Acid Dietary Supplementation Protects Against Acute Kidney Injury. J Am Soc Nephrol. Dec 4 2023;doi: 10.1681/ASN.0000000000000266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dhillon P, Park J, Hurtado Del Pozo C, et al. The Nuclear Receptor ESRRA Protects from Kidney Disease by Coupling Metabolism and Differentiation. Cell Metab. Feb 2 2021;33(2):379–394 e8. doi: 10.1016/j.cmet.2020.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li SY, Susztak K. The Role of Peroxisome Proliferator-Activated Receptor gamma Coactivator 1alpha (PGC-1alpha) in Kidney Disease. Semin Nephrol. Mar 2018;38(2):121–126. doi: 10.1016/j.semnephrol.2018.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han SH, Wu MY, Nam BY, et al. PGC-1alpha Protects from Notch-Induced Kidney Fibrosis Development. J Am Soc Nephrol. Nov 2017;28(11):3312–3322. doi: 10.1681/ASN.2017020130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miguel V, Tituana J, Herrero JI, et al. Renal tubule Cpt1a overexpression protects from kidney fibrosis by restoring mitochondrial homeostasis. J Clin Invest. Mar 1 2021;131(5)doi: 10.1172/JCI140695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wilson PC, Muto Y, Wu H, Karihaloo A, Waikar SS, Humphreys BD. Multimodal single cell sequencing implicates chromatin accessibility and genetic background in diabetic kidney disease progression. Nat Commun. Sep 6 2022;13(1):5253. doi: 10.1038/s41467-022-32972-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chung KW, Dhillon P, Huang S, et al. Mitochondrial Damage and Activation of the STING Pathway Lead to Renal Inflammation and Fibrosis. Cell Metab. Oct 1 2019;30(4):784–799 e5. doi: 10.1016/j.cmet.2019.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H, Dixon EE, Wu H, Humphreys BD. Comprehensive single-cell transcriptional profiling defines shared and unique epithelial injury responses during kidney fibrosis. Cell Metab. Dec 6 2022;34(12):1977–1998 e9. doi: 10.1016/j.cmet.2022.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun J, Shannon M, Ando Y, et al. Serum metabolomic profiles from patients with acute kidney injury: a pilot study. J Chromatogr B Analyt Technol Biomed Life Sci. Apr 15 2012;893–894:107–13. doi: 10.1016/j.jchromb.2012.02.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Portilla D Energy metabolism and cytotoxicity. Semin Nephrol. Sep 2003;23(5):432–8. doi: 10.1016/s0270-9295(03)00088-3 [DOI] [PubMed] [Google Scholar]
- 40.Mukhi D, Li L, Liu H, et al. ACSS2 gene variants determine kidney disease risk by controlling de novo lipogenesis in kidney tubules. J Clin Invest. Dec 5 2023;doi: 10.1172/JCI172963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ishii K, Kobayashi H, Taguchi K, et al. Kidney epithelial targeted mitochondrial transcription factor A deficiency results in progressive mitochondrial depletion associated with severe cystic disease. Kidney Int. Mar 2021;99(3):657–670. doi: 10.1016/j.kint.2020.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tran M, Tam D, Bardia A, et al. PGC-1alpha promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Invest. Oct 2011;121(10):4003–14. doi: 10.1172/JCI58662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li S, Nagothu KK, Desai V, et al. Transgenic expression of proximal tubule peroxisome proliferator-activated receptor-alpha in mice confers protection during acute kidney injury. Kidney Int. Nov 2009;76(10):1049–62. doi: 10.1038/ki.2009.330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mohandes S, Doke T, Hu H, Mukhi D, Dhillon P, Susztak K. Molecular pathways that drive diabetic kidney disease. J Clin Invest. Feb 15 2023;133(4)doi: 10.1172/JCI165654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wilson PC, Wu H, Kirita Y, et al. The single-cell transcriptomic landscape of early human diabetic nephropathy. Proc Natl Acad Sci U S A. Sep 24 2019;116(39):19619–19625. doi: 10.1073/pnas.1908706116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Titchenell PM, Quinn WJ, Lu M, et al. Direct Hepatocyte Insulin Signaling Is Required for Lipogenesis but Is Dispensable for the Suppression of Glucose Production. Cell Metab. Jun 14 2016;23(6):1154–1166. doi: 10.1016/j.cmet.2016.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yang M, Geng CA, Liu X, Guan M. Lipid Disorders in NAFLD and Chronic Kidney Disease. Biomedicines. Oct 6 2021;9(10)doi: 10.3390/biomedicines9101405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mooli RGR, Mukhi D, Pasupulati AK, et al. Intestinal HIF-2alpha Regulates GLP-1 Secretion via Lipid Sensing in L-Cells. Cell Mol Gastroenterol Hepatol. 2022;13(4):1057–1072. doi: 10.1016/j.jcmgh.2021.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhao S, Jang C, Liu J, et al. Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature. Mar 2020;579(7800):586–591. doi: 10.1038/s41586-020-2101-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Foster DW. Malonyl-CoA: the regulator of fatty acid synthesis and oxidation. J Clin Invest. Jun 2012;122(6):1958–9. doi: 10.1172/jci63967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Batchuluun B, Pinkosky SL, Steinberg GR. Lipogenesis inhibitors: therapeutic opportunities and challenges. Nat Rev Drug Discov. Apr 2022;21(4):283–305. doi: 10.1038/s41573-021-00367-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Felix JB, Cox AR, Hartig SM. Acetyl-CoA and Metabolite Fluxes Regulate White Adipose Tissue Expansion. Trends Endocrinol Metab. May 2021;32(5):320–332. doi: 10.1016/j.tem.2021.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lee M, Katerelos M, Gleich K, et al. Phosphorylation of Acetyl-CoA Carboxylase by AMPK Reduces Renal Fibrosis and Is Essential for the Anti-Fibrotic Effect of Metformin. J Am Soc Nephrol. Sep 2018;29(9):2326–2336. doi: 10.1681/ASN.2018010050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Han SH, Malaga-Dieguez L, Chinga F, et al. Deletion of Lkb1 in Renal Tubular Epithelial Cells Leads to CKD by Altering Metabolism. J Am Soc Nephrol. Feb 2016;27(2):439–53. doi: 10.1681/ASN.2014121181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu Y, Maguire TG, Alwine JC. ChREBP, a glucose-responsive transcriptional factor, enhances glucose metabolism to support biosynthesis in human cytomegalovirus-infected cells. Proc Natl Acad Sci U S A. Feb 4 2014;111(5):1951–6. doi: 10.1073/pnas.1310779111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stoeckman AK, Ma L, Towle HC. Mlx is the functional heteromeric partner of the carbohydrate response element-binding protein in glucose regulation of lipogenic enzyme genes. J Biol Chem. Apr 9 2004;279(15):15662–9. doi: 10.1074/jbc.M311301200 [DOI] [PubMed] [Google Scholar]
- 57.Witte N, Muenzner M, Rietscher J, et al. The Glucose Sensor ChREBP Links De Novo Lipogenesis to PPARgamma Activity and Adipocyte Differentiation. Endocrinology. Nov 2015;156(11):4008–19. doi: 10.1210/EN.2015-1209 [DOI] [PubMed] [Google Scholar]
- 58.Herman MA, Peroni OD, Villoria J, et al. A novel ChREBP isoform in adipose tissue regulates systemic glucose metabolism. Nature. Apr 19 2012;484(7394):333–8. doi: 10.1038/nature10986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yokoyama A, Suzuki S, Okamoto K, Sugawara A. The physiological and pathophysiological roles of carbohydrate response element binding protein in the kidney. Endocr J. Jun 28 2022;69(6):605–612. doi: 10.1507/endocrj.EJ22-0083 [DOI] [PubMed] [Google Scholar]
- 60.Katz LS, Baumel-Alterzon S, Scott DK, Herman MA. Adaptive and maladaptive roles for ChREBP in the liver and pancreatic islets. J Biol Chem. Jan-Jun 2021;296:100623. doi: 10.1016/j.jbc.2021.100623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Steinberg P, Storkel S, Oesch F, Thoenes W. Carbohydrate metabolism in human renal clear cell carcinomas. Lab Invest. Oct 1992;67(4):506–11. [PubMed] [Google Scholar]
- 62.Xu X, So JS, Park JG, Lee AH. Transcriptional control of hepatic lipid metabolism by SREBP and ChREBP. Semin Liver Dis. Nov 2013;33(4):301–11. doi: 10.1055/s-0033-1358523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Repa JJ, Liang G, Ou J, et al. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. Nov 15 2000;14(22):2819–30. doi: 10.1101/gad.844900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yoshikawa T, Shimano H, Yahagi N, et al. Polyunsaturated fatty acids suppress sterol regulatory element-binding protein 1c promoter activity by inhibition of liver X receptor (LXR) binding to LXR response elements. J Biol Chem. Jan 18 2002;277(3):1705–11. doi: 10.1074/jbc.M105711200 [DOI] [PubMed] [Google Scholar]
- 65.Watanabe M, Houten SM, Wang L, et al. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J Clin Invest. May 2004;113(10):1408–18. doi: 10.1172/JCI21025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Calkin AC, Tontonoz P. Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat Rev Mol Cell Biol. Mar 14 2012;13(4):213–24. doi: 10.1038/nrm3312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Crewe C, Zhu Y, Paschoal VA, et al. SREBP-regulated adipocyte lipogenesis is dependent on substrate availability and redox modulation of mTORC1. JCI Insight. Jul 16 2019;5(15)doi: 10.1172/jci.insight.129397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Horton JD, Goldstein JL, Brown MS. SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest. May 2002;109(9):1125–31. doi: 10.1172/JCI15593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shimomura I, Bashmakov Y, Ikemoto S, Horton JD, Brown MS, Goldstein JL. Insulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-induced diabetes. Proc Natl Acad Sci U S A. Nov 23 1999;96(24):13656–61. doi: 10.1073/pnas.96.24.13656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sun L, Halaihel N, Zhang W, Rogers T, Levi M. Role of sterol regulatory element-binding protein 1 in regulation of renal lipid metabolism and glomerulosclerosis in diabetes mellitus. J Biol Chem. May 24 2002;277(21):18919–27. doi: 10.1074/jbc.M110650200 [DOI] [PubMed] [Google Scholar]
- 71.Jiang T, Wang Z, Proctor G, et al. Diet-induced obesity in C57BL/6J mice causes increased renal lipid accumulation and glomerulosclerosis via a sterol regulatory element-binding protein-1c-dependent pathway. J Biol Chem. Sep 16 2005;280(37):32317–25. doi: 10.1074/jbc.M500801200 [DOI] [PubMed] [Google Scholar]
- 72.Ishigaki N, Yamamoto T, Shimizu Y, et al. Involvement of glomerular SREBP-1c in diabetic nephropathy. Biochem Biophys Res Commun. Dec 21 2007;364(3):502–8. doi: 10.1016/j.bbrc.2007.10.038 [DOI] [PubMed] [Google Scholar]
- 73.Liu H, Doke T, Guo D, et al. Epigenomic and transcriptomic analyses define core cell types, genes and targetable mechanisms for kidney disease. Nat Genet. Jul 2022;54(7):950–962. doi: 10.1038/s41588-022-01097-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Abedini A, Ma Z, Frederick J, et al. Spatially resolved human kidney multi-omics single cell atlas highlights the key role of the fibrotic microenvironment in kidney disease progression. bioRxiv. 2022:2022.10.24.513598. doi: 10.1101/2022.10.24.513598 [DOI] [Google Scholar]
- 75.Afshinnia F, Nair V, Lin J, et al. Increased lipogenesis and impaired beta-oxidation predict type 2 diabetic kidney disease progression in American Indians. JCI Insight. Nov 1 2019;4(21)doi: 10.1172/jci.insight.130317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pagliarini DJ, Calvo SE, Chang B, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. Jul 11 2008;134(1):112–23. doi: 10.1016/j.cell.2008.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bertolio R, Napoletano F, Mano M, et al. Sterol regulatory element binding protein 1 couples mechanical cues and lipid metabolism. Nat Commun. Mar 22 2019;10(1):1326. doi: 10.1038/s41467-019-09152-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bidault G, Virtue S, Petkevicius K, et al. SREBP1-induced fatty acid synthesis depletes macrophages antioxidant defences to promote their alternative activation. Nat Metab. Sep 2021;3(9):1150–1162. doi: 10.1038/s42255-021-00440-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mooli RGR, Mukhi D, Ramakrishnan SK. Oxidative Stress and Redox Signaling in the Pathophysiology of Liver Diseases. Compr Physiol. Mar 29 2022;12(2):3167–3192. doi: 10.1002/cphy.c200021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Moorhead JF, Chan MK, El-Nahas M, Varghese Z. Lipid nephrotoxicity in chronic progressive glomerular and tubulo-interstitial disease. Lancet. Dec 11 1982;2(8311):1309–11. doi: 10.1016/s0140-6736(82)91513-6 [DOI] [PubMed] [Google Scholar]
- 81.Ruan XZ, Varghese Z, Moorhead JF. An update on the lipid nephrotoxicity hypothesis. Nat Rev Nephrol. Dec 2009;5(12):713–21. doi: 10.1038/nrneph.2009.184 [DOI] [PubMed] [Google Scholar]
- 82.Nicholson RJ, Pezzolesi MG, Summers SA. Rotten to the Cortex: Ceramide-Mediated Lipotoxicity in Diabetic Kidney Disease. Front Endocrinol (Lausanne). 2020;11:622692. doi: 10.3389/fendo.2020.622692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nicholson RJ, Holland WL, Summers SA. Ceramides and Acute Kidney Injury. Semin Nephrol. May 2022;42(3):151281. doi: 10.1016/j.semnephrol.2022.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Irazabal MV, Torres VE. Reactive Oxygen Species and Redox Signaling in Chronic Kidney Disease. Cells. May 28 2020;9(6)doi: 10.3390/cells9061342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Doke T, Susztak K. The multifaceted role of kidney tubule mitochondrial dysfunction in kidney disease development. Trends Cell Biol. Oct 2022;32(10):841–853. doi: 10.1016/j.tcb.2022.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Khan S, Cabral PD, Schilling WP, et al. Kidney Proximal Tubule Lipoapoptosis Is Regulated by Fatty Acid Transporter-2 (FATP2). J Am Soc Nephrol. Jan 2018;29(1):81–91. doi: 10.1681/ASN.2017030314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li LC, Yang JL, Lee WC, et al. Palmitate aggravates proteinuria-induced cell death and inflammation via CD36-inflammasome axis in the proximal tubular cells of obese mice. Am J Physiol Renal Physiol. Dec 1 2018;315(6):F1720–F1731. doi: 10.1152/ajprenal.00536.2017 [DOI] [PubMed] [Google Scholar]
- 88.Linkermann A, Skouta R, Himmerkus N, et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci U S A. Nov 25 2014;111(47):16836–41. doi: 10.1073/pnas.1415518111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Balzer MS, Doke T, Yang YW, et al. Single-cell analysis highlights differences in druggable pathways underlying adaptive or fibrotic kidney regeneration. Nat Commun. Jul 11 2022;13(1):4018. doi: 10.1038/s41467-022-31772-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Guan Y, Liang X, Ma Z, et al. A single genetic locus controls both expression of DPEP1/CHMP1A and kidney disease development via ferroptosis. Nat Commun. Aug 23 2021;12(1):5078. doi: 10.1038/s41467-021-25377-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bataille A, Galichon P, Chelghoum N, et al. Increased Fatty Acid Oxidation in Differentiated Proximal Tubular Cells Surviving a Reversible Episode of Acute Kidney Injury. Cell Physiol Biochem. 2018;47(4):1338–1351. doi: 10.1159/000490819 [DOI] [PubMed] [Google Scholar]
- 92.Tran DH, Wang ZV. Glucose Metabolism in Cardiac Hypertrophy and Heart Failure. J Am Heart Assoc. Jun 18 2019;8(12):e012673. doi: 10.1161/JAHA.119.012673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sas KM, Kayampilly P, Byun J, et al. Tissue-specific metabolic reprogramming drives nutrient flux in diabetic complications. JCI Insight. Sep 22 2016;1(15):e86976. doi: 10.1172/jci.insight.86976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Meyer C, Dostou J, Nadkarni V, Gerich J. Effects of physiological hyperinsulinemia on systemic, renal, and hepatic substrate metabolism. Am J Physiol. Dec 1998;275(6):F915–21. doi: 10.1152/ajprenal.1998.275.6.F915 [DOI] [PubMed] [Google Scholar]
- 95.Cai T, Ke Q, Fang Y, et al. Sodium-glucose cotransporter 2 inhibition suppresses HIF-1alpha-mediated metabolic switch from lipid oxidation to glycolysis in kidney tubule cells of diabetic mice. Cell Death Dis. May 22 2020;11(5):390. doi: 10.1038/s41419-020-2544-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Dickman KG, Mandel LJ. Differential effects of respiratory inhibitors on glycolysis in proximal tubules. Am J Physiol. Jun 1990;258(6 Pt 2):F1608–15. doi: 10.1152/ajprenal.1990.258.6.F1608 [DOI] [PubMed] [Google Scholar]
- 97.Lan R, Geng H, Singha PK, et al. Mitochondrial Pathology and Glycolytic Shift during Proximal Tubule Atrophy after Ischemic AKI. J Am Soc Nephrol. Nov 2016;27(11):3356–3367. doi: 10.1681/ASN.2015020177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zager RA, Johnson AC, Becker K. Renal cortical pyruvate depletion during AKI. J Am Soc Nephrol. May 2014;25(5):998–1012. doi: 10.1681/ASN.2013070791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Maekawa H, Inoue T, Ouchi H, et al. Mitochondrial Damage Causes Inflammation via cGAS-STING Signaling in Acute Kidney Injury. Cell Rep. Oct 29 2019;29(5):1261–1273 e6. doi: 10.1016/j.celrep.2019.09.050 [DOI] [PubMed] [Google Scholar]
- 100.West AP, Khoury-Hanold W, Staron M, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. Apr 23 2015;520(7548):553–7. doi: 10.1038/nature14156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tigano M, Vargas DC, Tremblay-Belzile S, Fu Y, Sfeir A. Nuclear sensing of breaks in mitochondrial DNA enhances immune surveillance. Nature. Mar 2021;591(7850):477–481. doi: 10.1038/s41586-021-03269-w [DOI] [PubMed] [Google Scholar]
- 102.Doke T, Mukherjee S, Mukhi D, et al. NAD(+) precursor supplementation prevents mtRNA/RIG-I-dependent inflammation during kidney injury. Nat Metab. Mar 2023;5(3):414–430. doi: 10.1038/s42255-023-00761-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wu J, Ma Z, Raman A, et al. APOL1 risk variants in individuals of African genetic ancestry drive endothelial cell defects that exacerbate sepsis. Immunity. Nov 9 2021;54(11):2632–2649 e6. doi: 10.1016/j.immuni.2021.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Park CW, Kim HW, Ko SH, et al. Accelerated diabetic nephropathy in mice lacking the peroxisome proliferator-activated receptor alpha. Diabetes. Apr 2006;55(4):885–93. doi: 10.2337/diabetes.55.04.06.db05-1329 [DOI] [PubMed] [Google Scholar]
- 105.Park CW, Zhang Y, Zhang X, et al. PPARalpha agonist fenofibrate improves diabetic nephropathy in db/db mice. Kidney Int. May 2006;69(9):1511–7. doi: 10.1038/sj.ki.5000209 [DOI] [PubMed] [Google Scholar]
- 106.Davis TM, Ting R, Best JD, et al. Effects of fenofibrate on renal function in patients with type 2 diabetes mellitus: the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) Study. Diabetologia. Feb 2011;54(2):280–90. doi: 10.1007/s00125-010-1951-1 [DOI] [PubMed] [Google Scholar]
- 107.Kostapanos MS, Florentin M, Elisaf MS. Fenofibrate and the kidney: an overview. Eur J Clin Invest. May 2013;43(5):522–31. doi: 10.1111/eci.12068 [DOI] [PubMed] [Google Scholar]
- 108.Broeders N, Knoop C, Antoine M, Tielemans C, Abramowicz D. Fibrate-induced increase in blood urea and creatinine: is gemfibrozil the only innocuous agent? Nephrol Dial Transplant. Dec 2000;15(12):1993–9. doi: 10.1093/ndt/15.12.1993 [DOI] [PubMed] [Google Scholar]
- 109.Mychaleckyj JC, Craven T, Nayak U, et al. Reversibility of fenofibrate therapy-induced renal function impairment in ACCORD type 2 diabetic participants. Diabetes Care. May 2012;35(5):1008–14. doi: 10.2337/dc11-1811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Group AS, Ginsberg HN, Elam MB, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. Apr 29 2010;362(17):1563–74. doi: 10.1056/NEJMoa1001282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Frazier R, Mehta R, Cai X, et al. Associations of Fenofibrate Therapy With Incidence and Progression of CKD in Patients With Type 2 Diabetes. Kidney Int Rep. Jan 2019;4(1):94–102. doi: 10.1016/j.ekir.2018.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Iwasaki M, Suzuki H, Umezawa Y, et al. Efficacy and safety of pemafibrate in patients with chronic kidney disease: A retrospective study. Medicine (Baltimore). Feb 17 2023;102(7):e32818. doi: 10.1097/MD.0000000000032818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Das Pradhan A, Glynn RJ, Fruchart JC, et al. Triglyceride Lowering with Pemafibrate to Reduce Cardiovascular Risk. N Engl J Med. Nov 24 2022;387(21):1923–1934. doi: 10.1056/NEJMoa2210645 [DOI] [PubMed] [Google Scholar]