

Abstract
Background:
GLI1 (12q13.3) amplification is identified in a subset of mesenchymal neoplasms with a distinct nested round cell/epithelioid phenotype. MDM2 and CDK4 genes are situated along the oncogenic 12q13–15 segment, amplification of which defines well-differentiated (WDLPS)/dedifferentiated liposarcoma (DDLPS). The 12q amplicon can occasionally include GLI1 – a gene in close proximity to CDK4. We hereby describe the first cohort of GLI1/MDM2/CDK4 co-amplified WD/DDLPS.
Materials and Methods:
The departmental database was queried retrospectively for all cases of WD/DDLPS having undergone next generation (IMPACT) sequencing with confirmed MDM2, CDK4, and GLI1 co-amplification. Clinicopathologic data was obtained from review of the medical chart and available histologic material.
Results:
486 WD/DDLPS underwent DNA sequencing, 92 (19%) of which harbored amplification of the GLI1 locus in addition to MDM2 and CDK4. These included primary tumors (n=60), local recurrences (n=29), and metastases (n=3). Primary tumors were most frequently retroperitoneal (47/60,78%) mediastinal (4/60,7%), and paratesticular (3/60, 5%). Average age was 63 years with a male: female ratio of 3:2.
The cohort was comprised by DDLPS (86/92 [93%], 6 of which were comprised by WDLPS with early dedifferentiation), and WDLPS without any longitudinal evidence of dedifferentiation (6/92, 7%%). A fifth (13/86,17%) of DDLPS cases showed no evidence of a well-differentiated component in any of the primary, recurrent, or metastatic specimens. Dedifferentiated areas mostly showed high-grade undifferentiated pleomorphic sarcoma-like (26/86, 30%),) and high-grade myxofibrosarcoma-like (13/86, 16%)) morphology. A disproportionately increased incidence of meningothelial whorls with/without osseous metaplasia was observed as the predominant pattern in 16/86 (19%), and GLI1-altered morphology as described was identified in a total of 10/86 (12%) tumors.
JUN (1p32.1), also implicated in the pathogenesis of WD/DDLPS, was co-amplified with all three of MDM2, CDK4, and GLI1 in 7/91 (8%) cases. Additional loci along chromosomal arms 1p and 6q, including TNFAIP3, LATS1 and ESR1, were also amplified in a subset of cases.
Conclusions:
In this large-scale cohort of GLI1 co-amplified WD/DDLPS, we elucidate uniquely recurrent features including meningothelial whorls and GLI-altered morphology in dedifferentiated areas. Assessment of tumor location (retroperitoneal or mediastinal), identification of a well-differentiated liposarcoma component, and co-amplification of other spatially discrete genomic segments (1p, 6q) might aid in distinction from tumors with true driver GLI1 alterations.
Keywords: GLI1, MDM2, CDK4, dedifferentiated liposarcoma, well-differentiated liposarcoma, 12q, amplification
Introduction
With the ongoing molecular interrogation of mesenchymal tumors, our understanding of neoplasms with GLI1 aberrations has evolved significantly over the past decade. Originally conceptualized as group of pericyte-derived proliferations with underlying GLI1::ACTB fusions (1), this umbrella of ‘GLI1-altered mesenchymal tumors’ now encompasses an array of entities with not only distinct clinicopathologic attributes but also different mechanisms of genomic tumorigenesis. Pathogenic fusions between GLI1 and MALAT1, for example, give rise to both benign (plexiform fibromyxoma) and aggressive to malignant (gastroblastoma/’GNET’) lesions of the gastric wall (2–4). Other fusion partners including PTCH1, NCOR2, NEAT1, SYT, DDIT3, and AARS are recently described in multiple case series, sourcing tumors with heterogenous demographics but a conspicuously recurrent histologic motif: multilobulated proliferations of monotonous round to epithelioid cells, arranged in nests with intervening rich arborescent capillary vasculature (5–10).
From a structural perspective, the GLI1 gene is situated along an oncogenic expanse on chromosome 12q13.3 that is implicated in various soft tissue tumors, including MDM2 (12q15), CDK4 (12q14.1), DDIT3 (12q13.3), HMGA2 (12q14.3) and STAT6 (12q13.3). In particular, amplification of MDM2 and/or CDK4 define the spectrum of atypical lipomatous tumor/well-differentiated (WDLPS)/dedifferentiated liposarcoma (DDLPS) in appropriate contexts. Due to their conglomerate spatial proximity, the driver MDM2 and/or CDK4 amplicon is associated with co-amplifications of these neighboring genes in a subset of cases – a phenomenon which to date is not well-characterized in either clinicopathologic or genomic frameworks (11–14).
Prior investigation into an array of GLI1-amplified neoplasms revealed a considerable subset of DDLPS with evidence of concomitant GLI1 (along with axiomatic MDM2 and CDK4) amplification, comprising approximately a fifth of those archived in our institutional database. Remarkably, a minor proportion of these displayed an organoid arrangement of homogenous round/epithelioid cells evocative of underlying GLI1 alteration within the dedifferentiated component, implying a potentially shared morphologic contribution of this specific molecular signature (15). In our practice, we also recently encountered a few enigmatic tumors challenging to characterize as DDLPS or GLI1-altered mesenchymal neoplasm, prompting further investigation into these specific lesions.
In this study we characterize the first cohort of GLI1/MDM2/CDK4 co-amplified WD/DDLPS to elucidate any characteristic clinical, morphologic, or genomic patterns therein. The differential diagnoses and clinicopathologic implications of this subset of tumors are discussed.
Materials and Methods
Cohort selection
The cBioPortal platform at Memorial Sloan Kettering Cancer Center (a sequencing database including biopsies and resections from primary, recurrent, and metastatic in-house and consult specimens) was queried retrospectively for all cases of ‘well-differentiated liposarcoma,’ ‘dedifferentiated liposarcoma,’ and ‘sarcoma, not otherwise specified’ having undergone MSK-IMPACT from 2015 to the present (16, 17). Specimens with an initial diagnosis of well/dedifferentiated liposarcoma or nonspecific sarcoma without confirmatory MDM2 and/or CDK4 amplification were excluded. The resulting population was filtered for all cases with evidence of concurrent GLI1, MDM2, and CDK4 amplification (including those with only one of either MDM2 or CDK4 amplification). Within the MSK-IMPACT platform, amplification is defined as a fold change of ≥2 for any gene; specimens under this threshold were excluded.
Clinicopathologic analysis
Demographic information (age, sex, tumor size, sites of primary/recurrent/metastatic tumor) and survival parameters (date of diagnosis and primary resections, interval to recurrence, interval to death, cause of death) were tabulated from the pathology database and electronic medical record.
At our institution, dedifferentiated liposarcoma or ‘dedifferentiation’ of a well-differentiated liposarcoma is defined per the Weiss et al. criteria of at least a single low-power field lacking evidence of lipogenic differentiation, regardless of mitotic activity and in some cases regardless of cellularity (18, 19). Therein, ‘high-grade’ and ‘low-grade’ dedifferentiation are qualified by an integrated assessment of degree of cellularity, cytonuclear atypia/pleomorphism, and mitotic activity - similar to the conventional two-tiered appraisal of other soft tissue sarcomas. Well-differentiated liposarcoma with ‘early dedifferentiation’ is defined as well-differentiated liposarcoma with foci of cellular non-lipogenic/non-myxoid tissue comprising <5% of the overall appropriately sampled tumor volume (20, 21). As per prior publications, so-called ‘GLI1-altered morphology’ features a proliferation of monomorphic round to epithelioid cells with scant pale cytoplasm, partitioned into symmetric small nests/clusters by an anastomotic capillary network (5–10, 15).
Histologic features were recorded upon review of all available digital slides. These included the presence or absence of WDLPS, and the histologic subtypes represented therein (sclerosing, lipoma-like, inflammatory). Prevailing and secondary morphologies of the dedifferentiated component (spindle cell sarcoma, high-grade or low-grade myxofibrosarcoma-like, high-grade undifferentiated pleomorphic sarcoma-like, meningothelial whorls with or without osseous metaplasia, heterologous e.g., osteosarcomatous or rhabdomyosarcomatous differentiation, GLI1-altered morphology) were qualitatively tabulated. Non-GLI1-amplified DDLPS sequenced during the same interval served as an internal control cohort for the morphologic analysis.
Molecular analysis
All samples were selected per the above parameters after having undergone MSK-IMPACT sequencing for either diagnostic or therapeutic purposes (22). (Of note, for technical reasons this platform does not appraise copy number changes in the DDIT3 locus.) Amplification fold changes were recorded for each of MDM2, CDK4, GLI1, and JUN. Copy number alterations and pathogenic mutations in accessory loci were recorded if present, in particular those involving the 1p or 6q chromosomal arms due to their documentation as spatially discrete segments of co-amplification in DDLPS (23–26).
Statistical analysis
Recurrence-free survival (RFS) as an event was defined as the interval of time between primary resection and either radiologic or histologic recurrence/metastasis; death from disease was censored. (27) Disease-specific survival (DSS) was defined as the interval of time between primary resection and date of death due to WD/DDLPS; death from other causes were not censored. Statistical comparisons and survival analyses were performed using Graphpad Prism® and XLMiner.
Results
Demographic features
Clinicopathologic and genomic features of the entire cohort are summarized in Table 1. A total of 486 WD/DDLPS underwent sequencing via MSK-IMPACT between the years 2015 to the current time. 92 of these tumors (19%) harbored amplification of the GLI1 locus in addition to MDM2 and/or CDK4. This core cohort was comprised by 55 males and 37 females (male to female ratio of 3:2) with an average age of 63 years (median 64, range 31–86). Individual specimens from these patients previously selected for MSK-IMPACT were represented by primary tumors (n=60), local recurrences (n=29), and metastases (n=3). Primary tumors were most frequently designated as retroperitoneal (47/60, 78%) but also mediastinal (4/60, 7%), paratesticular (3/60, 5%), neck (1/57, 2%), duodenal (1/57. 2%), groin (1/57, 2%) and musculoskeletal tissues of the trunk or extremities (3/57, 5%). Sites of sequenced locally recurrent tumors were also predominated by the retroperitoneum (23/29, 80%), followed by the abdominal cavity (4/29, 10%) and mediastinum (2/29, 7%). All sequenced metastatic sites sourced from the lung (3/3), primary sites for which were the retroperitoneum (1/3), soft tissues of posterior neck (1/3), and paratesticular region (1/3).
Table 1:
Clinicopathologic and molecular features of the cohort of GLI1 co-amplified well-differentiated/dedifferentiated liposarcomas.
| Total patients | 92 |
| Age (years) | 63 (range 31–86) |
| Gender | |
| Male | 55/92 (60%) |
| Female | 37/92 (40%) |
| Site of primary tumor | |
| Retroperitoneum | 71/92 (77%) |
| Mediastinum | 6/92 (7%) |
| Paratesticular/spermatic cord | 4/92 (4%) |
| Abdominal cavity | 5/92 (6%) |
| Trunk/extremities | 6/92 (7%) |
| Diagnosis (sequenced tumors) | |
| Well-differentiated liposarcoma (without any dedifferentiation) | 6/92 (7%) |
| Well-differentiated liposarcoma with early dedifferentiation | 6/92 (7%) |
| Dedifferentiated liposarcoma… | 86/92 (94%) |
| ...with accompanying WDLPS* | 73/86 (94%) |
| ...without accompanying WDLPS* | 13/86 (17%) |
| Dedifferentiated morphology (predominant pattern) | 86 # |
| High-grade undifferentiated pleomorphic sarcoma | 26/86 (30%) |
| Meningothelial whorls with/without osseous metaplasia | 16/86 (19%) |
| High-grade spindle cell sarcoma, not otherwise specified | 14/86 (17%) |
| High-grade myxofibrosarcoma | 13/86 (16%) |
| Low-grade myxofibrosarcoma | 8/86 (8%) |
| GLI1 morphology | 5/86 (6%) |
| Low-grade spindle cell sarcoma, not otherwise specified | 1/86 (1%) |
| Osteosarcomatous differentiation | 1/86 (1%) |
| Rhabdomyoblastic differentiation | 1/86 (1%) |
| Inflammatory myofibroblastic tumor | 1/86 (1%) |
| GLI1 morphology (any proportion) | 10/86 (12%) |
| Meningothelial whorls with/without osseous metaplasia (any proportion) | 19/86 (86%) |
| Follow-up available | 83/92 (90%) |
| Local recurrence | 56/83 (67%) |
| Distant metastasis | 10/83 (13%) |
| Alive with no evidence of disease | 31/83 (37%) |
| Alive with disease | 27/83 (33%) |
| Dead of disease | 25/83 (31%) |
| Average MDM2 fold change | 13.2 (range 2.3–33.7) |
| Average MDM2:CDK4 fold change ratio | 1.4 (range 0.07–3.8) |
| Average CDK4 fold change | 11 (range 2.6–32.1) |
| Average GLI1 fold change | 6.7 (range 2–21.2) |
| Average GLI1:MDM2 fold change ratio | 0.67 (range 0.07–2.2) |
| Average GLI1:CDK4 fold change ratio | 0.7 (range 0.3–5.5) |
| 1p co-amplification | |
| JUN | 7/91 (8%) |
| NOTCH2 | 1/91 (1%) |
| 6p co-amplification | |
| TNFAIP3 | 11/92 (12%) |
| LATS1 | 5/92 (5%) |
| ESR1 | 5/92 (6%) |
| IFNG1 | 2/92 (2%) |
| FYN | 2/92 (2%) |
| ARID1B | 2/92 (2%) |
WDLPS: Well-differentiated liposarcoma;
86 cases with dedifferentiation includes cases a) with early dedifferentiation (6), b) with associated WDLPS (73), and c) without associated WDLPS (13).
Morphologic features
The cohort of sequenced tumors (primary, recurrent, and metastatic) was comprised by dedifferentiated liposarcoma (86/92, 94%, 6 of which were comprised by WDLS with early dedifferentiation) and well-differentiated liposarcoma without any longitudinal evidence of dedifferentiation (6/92, 7%). Of the sequenced DDLPS, 69/86 (80%) were associated with a WDLPS component in the same specimen. In 4/86 (5%) cases, a well-differentiated component was only appreciable in the subsequent recurrent specimen. A portion of the dedifferentiated samples (13/86, 15%) showed no evidence of a well-differentiated component in any of the primary, recurrent, or metastatic specimens.
Of the sequenced WDLPS, 6/9 (67%) never recurred and 3/9 (33%) had evidence of dedifferentiation in the subsequent recurrence (as above rendering 6 cases without any longitudinal evidence of dedifferentiation). Of the sequenced WDLPS with early dedifferentiation, 3/6 (50%) recurred with only 1 of 3 with evidence of dedifferentiation in the recurrence.
The tumors displayed a wide morphologic spectrum in regard to both well-differentiated and dedifferentiated components, frequently with multiple (2–3) discrete patterns within the same lesion. Of the sequenced tumors with an appreciable well-differentiated component with or without association with a dedifferentiated component (n=75), 10/75 showed a scant amount of well-differentiated tumor not amenable to further classification. The prevailing histology of WDLPS in the remaining tumors by proportion of tumor volume was lipoma-like (55/65, 85%), followed by the sclerosing subtype (9/65, 14%, Figure 1). Secondary and tertiary morphologies were comprised by the sclerosing subtype (26/65, 40% - frequently in tumors with a predominant lipoma-like morphology), and the inflammatory variant (3/65, 5%).
Figure 1.
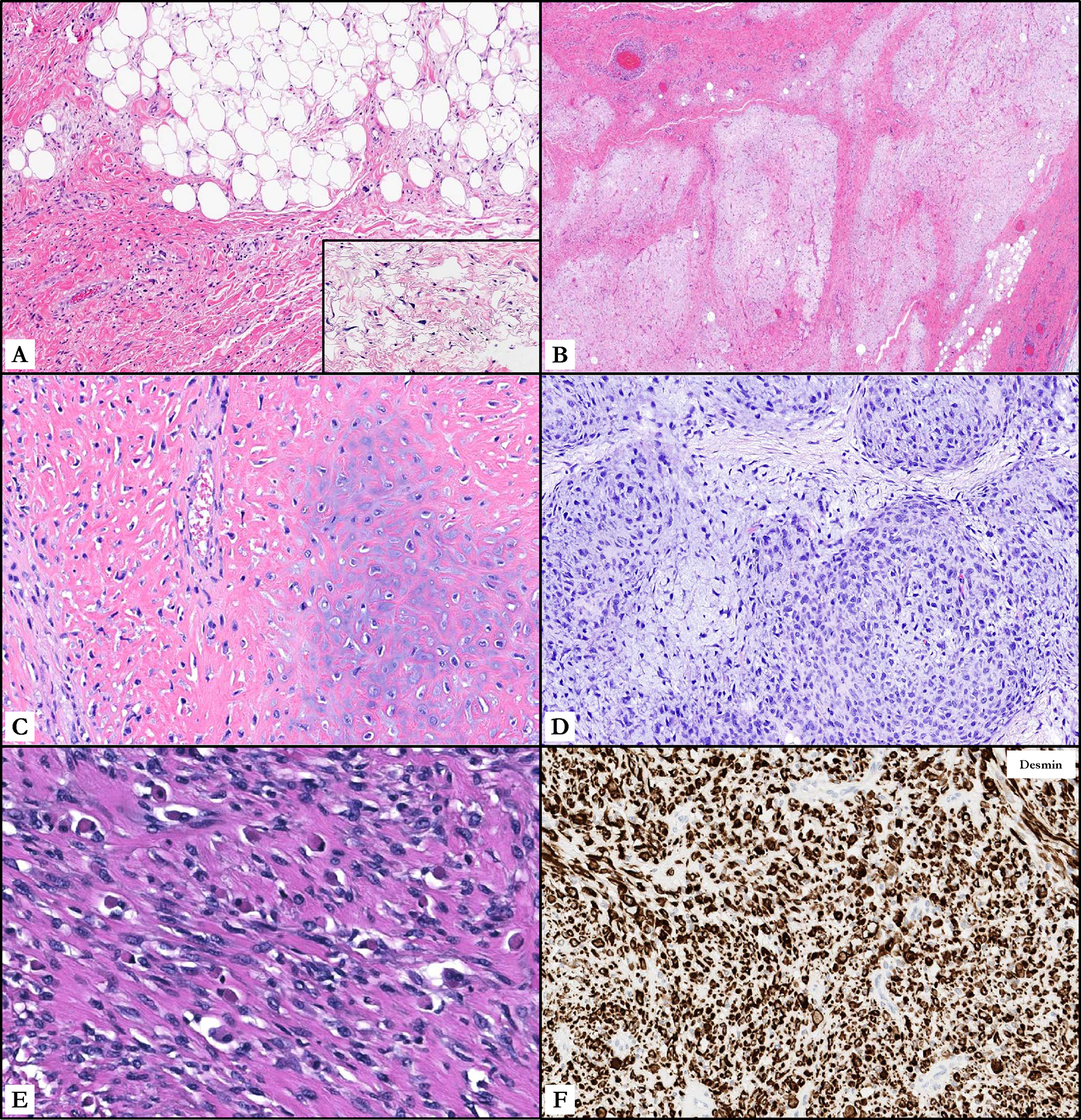
Histomorphologic features of GLI1-amplified well-differentiated/dedifferentiated liposarcomas. A. Lipoma-like and sclerosing subtypes comprised the most common variants of well-differentiated liposarcoma, identification of which is crucial in the context of a GLI1 co-amplified tumor. B. Area of ‘early dedifferentiation’ showing a predominantly non-lipogenic low-grade myxofibrosarcoma-like expanse. C. Heterogeneous morphology of dedifferentiated liposarcoma with malignant osteocartilaginous differentiation. D. Dedifferentiated liposarcoma with high-grade myxofibrosarcoma-like morphology. E. Dedifferentiated liposarcoma with heterologous rhabdomyoblastic differentiation. F. Immunohistochemical stain for desmin, which is diffusely positive in areas of rhabdomyosarcomatous differentiation.
Dedifferentiated areas (analyzed across all sequenced cases with evidence of dedifferentiation in either the primary, recurrent or metastatic lesion; n=86) were predominated by high-grade undifferentiated pleomorphic sarcoma-like (26/86, 30%) and high-grade myxofibrosarcoma-like (13/86, 15%) histologies, both frequently present in tandem (Figure 1). Other salient patterns included meningothelial whorls with or without osseous metaplasia (12/86 [14%] and 4/86 [5%], respectively), high-grade spindle cell sarcoma not otherwise specified (14/86 [16%], 2/14 of which showed storiform morphology), low-grade myxofibrosarcoma-like (8/86, 8%), low-grade spindle cell sarcoma not otherwise specified (1/86, 1%), inflammatory myofibroblastic tumor-like (1/86, 1%), heterologous osteosarcomatous differentiation (1/86, 1%), and heterologous rhabdomyosarcomatous differentiation (1/86, 1%). (Figures 1 and 2). GLI1-altered morphology, as described, was identified as the prevailing pattern in 5/86 (6%), as the secondary pattern in 3/86 (4%), and as the tertiary pattern in 2/86 (2%) tumors. Secondary morphologies by proportion of tumor volume were again predominated by high-grade myxofibrosarcoma-like morphology (14/86, 16%), low-grade spindle cell sarcoma not otherwise specified (7/86, 8%), high-grade undifferentiated pleomorphic sarcoma-like (4/86, 5%), low-grade myxofibrosarcoma-like (2/86, 2%), round cell sarcoma not otherwise specified (1/86, 1%), meningothelial whorls (3/86, 3%), low-grade myxoid liposarcoma-like (1/86, 1%), metaplastic ossification (1/86, 1%), desmoid fibromatosis-like (1/86, 1%), heterologous osteosarcomatous differentiation (1/86, 1%), and heterologous rhabdomyosarcomatous differentiation (1/86, 1%).
Figure 2.

Rare morphologic patterns of dedifferentiation observed in a subset of GLI1 co-amplified well-differentiated/dedifferentiated liposarcomas. A. Osseous metaplasia (left) accompanied by a meningothelial-like whorl (right); a rare dyad observed in the context of dedifferentiated liposarcoma. B. Multiple meningothelial-like nodules of swirling spindle cells. C. Dedifferentiated component composed of a low-grade spindle cell sarcoma with sweeping fascicles reminiscent of desmoid-type fibromatosis. D. ‘GLI1-altered morphology’ showing a distinctly nested architecture of monotonous round cells as a morphologic pattern of dedifferentiation. E. Characteristic GLI1-altered morphology of epithelioid cells in a vaguely organoid pattern imparted by a delicate arborizing capillary network. F. A richly vascularized and nested proliferation of monotonous undifferentiated round cells should raise the differential diagnosis of a GLI1-altered tumor or a GLI1 co-amplified dedifferentiated liposarcoma, the latter corroborated by an MDM2 immunostain (inset).
A control group of 255 cases of non-GLI1 co-amplified DDLPS was assessed by pathology reports for morphologic cross-comparison. 182/255 cases (71%) showed undifferentiated pleomorphic sarcoma-like, 62/255 (24%) showed myxofibrosarcoma-like, and 11/255 (5%) showed divergent osteosarcomatous (8) or rhabdomyosarcomatous (3) morphologies. No cases with meningothelial whorls with or without osseous metaplasia or GLI1-altered morphology were identified in this group.
Molecular features
All tumors showed co-amplification of MDM2, CDK4, and GLI1, save one which showed only CDK4 and GLI1 co-amplification (i.e., all 92 cases were CDK4 co-amplified, and 91 cases were MDM2 co-amplified). Average fold change for MDM2, CDK4, and GLI1 was 13.2 (median 12.6, range 2.3–33.7), 11.0 (median 10.0, range 2.6–32.1), and 6.7 (median 5.8, range 2–21.2), respectively. As absolute comparison of copy number alterations across different tumors is unreliable due to individual disparities in tumor purity and volume, GLI1:MDM2 and GLI1:CDK4 ratios within each individual tumor were instead assessed (Table 1). Average MDM2:CDK4 ratio was 1.4 (median 1.1, range 0.07–3.8), average GLI1:MDM2 ratio was 0.67 (median 0.5, range 0.07–2.2), and average GLI1:CDK4 ratio was 0.7 (median 0.68, range 0.3–5.5). One-way ANOVA with post-hoc Tukey test demonstrated significant differences between the average fold changes of MDM2 and GLI1 as well as between CDK4 and GLI1, but not between those of MDM2 and CDK4.
In addition to amplification, 3 tumors harbored additional structural and sequence abnormalities in the GLI1 locus. Two demonstrated gene rearrangements – one a GLI1::YEATS4 fusion of indeterminate functional significance, which histologically appeared as a lipoma-like WDLPS with early dedifferentiation into low-grade myxofibrosarcoma-like morphology. The other harbored a GLI1::ZFAND3 fusion, also of indeterminate significance and possibly structurally related to the concomitant GLI1 amplification. All specimens from this patient lacked evidence of a juxtaposed well-differentiated component, and histologically were composed entirely of low-grade spindle cell sarcoma-like DDLPS. The third showed a pathogenic GLI1 mutation (exon12 p.P869A [c.2605C>G]); all specimens from this patient also lacked evidence of a juxtaposed WDLPS, and histologically appeared as a low-grade spindle cell sarcoma and desmoid fibromatosis-like DDLPS. GLI1 morphology was conspicuously absent from any specimen in these three patients.
The JUN gene (located on 1p32.1) was co-amplified with all three of MDM2, CDK4, and GLI1 in 7/91 cases (8%, not including the MDM2 non-amplified), with an average fold change of 11.3 (median 9, range 2.5–30). Genes along chromosomal arms 1p and 6q were also amplified in a fraction of all tumors, including most commonly TNFAIP3 (11/92, 11%), followed by LATS1 (5/92, 5%), ESR1 (5/92, 5%), IFNGR1 (2/92, 2%), FYN (2/92, 2%), ARID1B (2/92, 2%), and NOTCH2 (1/92, 1%).
Analysis of MDM2, CDK4 and GLI1 fold changes in the 8 tumors displaying any proportion of ‘GLI1 morphology’ revealed no correlation between magnitude of amplification nor ratio of fold change between each of GLI:MDM2 and GLI1:CDK4.
Survival metrics
Follow-up was available for most patients (83/92, 90%), an advantage of longitudinal treatment and surveillance at a dedicated referral cancer center (Table 1). Average length of follow-up was 65 months (median 52, range 1–240), during which 67% (56/83) of patients experienced a local recurrence or distant metastasis within an average of 37 months (range 4–156), and 30% (25/83) of patients died of disease within an average of 82 months (range 4–132). 33% (27/83) of patients remained alive with persistent disease at the time of last follow-up, and 39% (31/80) patients are alive without evidence of disease at last follow-up. 12% (10/83) of patients developed distant metastatic disease, occasionally multiple sites of which included liver (3/10), lung (5/10), thoracic vertebrae (2/10), humeral bone (1/10), and soft tissues of the fourth metacarpal ray (1/10).
Kaplan-Meier curves for the core cohort are provided in Figures 3A (recurrence-free survival, RFS) and 3B (disease-specific survival, DSS). Median RFS was 36 months, with 3-year recurrence-free survival at 48%, and 5-year recurrence-free survival at 30%. Median DSS was 141 months, with 5-year DSS at 80% and 10-year DSS at 60%.
Figure 3.

Kaplan-Meier curves for the core cohort: A. shows recurrence-free survival (RFS). Median RFS was 36 months, with 3-year recurrence-free survival at 48%, and 5-year recurrence-free survival at 30%. B. shows disease-specific survival (DSS). Median DSS was 141 months, with 5-year DSS at 80% and 10-year DSS at 60%.
Subset analysis of GLI1 co-amplified dedifferentiated liposarcoma with no well-differentiated component.
In as much as one diagnostic hinge of DDLPS in the setting of GLI1 co-amplification is the identification of a well-differentiated component, the clinicopathologic and molecular features of tumors in our cohort lacking the latter are also summarized briefly. A total of 13 DDLPS without evidence of a well-differentiated component in any of the primary, recurrent, or metastatic tumors were identified within the core cohort, as above. These were composed of 7 males and 6 females (male to female ratio of 1:1) with an average age of 59 years (median 61, range 40–86). Anatomic sites were represented by retroperitoneal soft tissues (7/13, 54%), mediastinum (4/13, 31%), gluteus muscle (1/13, 8%), and abdominal cavity (1/13, 8%). In contrast to the core cohort, tumors in this particular subdivision were more likely to feature only a single homogeneous pattern of dedifferentiated morphology (observed in 12/13, 92%): high-grade undifferentiated pleomorphic sarcoma (4/13, 30%), high-grade spindle cell sarcoma not otherwise specified (2/13, 15%), meningothelial whorls with osseous metaplasia (3/13, 2%), high-grade myxofibrosarcoma-like (1/13, 8%), and low-grade spindle cell sarcoma not otherwise specified (1/13, 8%). In a proportion similar to that of the overall cohort, GLI1-altered morphology was observed as the dominant component by overall tumor volume in 2/13 (15%) cases and the minor component in 1/13 (8%, as above).
Average fold change for MDM2, CDK4, and GLI1 was 9.9 (median 12.7, range 3.2–16.4), 10.3 (median 10.2, range 4.4–22.8), and 9.1 (median 8, range 4–21.2). JUN was co-amplified with all three of MDM2, CDK4, and GLI1 in 2/13 (15%) of cases, with fold changes of 14.1 and 9.1. Genes along 1p and 6q were additionally amplified in a subset of tumors, including most commonly TNFAIP3 (1/13, 8%), followed by LATS1 (1/13, 8%), and ESR1 (1/13, 8%).
Discussion
As with any tumor harboring a recurrent molecular signature, the morphologic heterogeneity of the WD/ DDLPS spectrum stands in contrast to its relatively invariable underlying genetic driver (28–37). Expansion of our technical capabilities to an unprecedented degree of genomic granularity, along with the ongoing classification of other molecularly defined mesenchymal entities has refined our understanding of their histology and clinical trajectory. An essentially relentless locally recurrent disease with a risk of metastatic potential much lower than that of undifferentiated pleomorphic sarcoma, high-grade myxofibrosarcoma, and pleomorphic liposarcoma, this subtype of liposarcoma is typically a surgical disease with repetitive radical debulking as a primary modality of treatment (38–41). Ongoing clinical and basket trials with targeted MDM2 and CDK4 inhibitors have shown promise in adjuvant and advanced-stage settings, but their efficacy and impact on survival are still to be established (42–46).
The topography of chromosome 12q, with coding segments for not only MDM2 and CDK4 but also oncogenes such as DDIT3 and STAT6 in close juxtaposition, renders its derivative tumors susceptible to copy alteration ‘carryover’ (i.e., secondary co-amplification likely ascribed to this structural coincidence). The ensuing morphologic and immunohistochemical overlap can be misleading, especially when a diagnostic body of evidence credibly aligns. For example, pathogenic fusions involving DDIT3 and STAT6 are definitional of myxoid/round cell liposarcoma and solitary fibrous tumor, respectively; nuclear upregulation of translated protein products can be exploited immunohistochemically towards their diagnosis. Indeed, early studies assessing the evidently excellent specificity of the STAT6 antibody demonstrated its exceptionally rare expression in WD/DDLPS as a potent pitfall, alluding perhaps inadvertently to this particular genomic phenomenon (14, 47). Similarly, myxoid portions of a WDLPS or DDLPS can show DDIT3 immunoreactivity so to misconstrue myxoid liposarcoma, which represents a diagnostic challenge in plausible clinicopathologic circumstances (12, 33).
The GLI1 locus is positioned within the oncogenic 12q segment and hence also geographically involved in this event. An abbreviation of ‘glioma-associated oncogene,’ it encodes a zinc finger transcription factor activated by the canonical sonic hedgehog signaling cascade (Shh), dyscrasias of which are fundamental to certain malignancies of the brain, breast, and melanocytes. (48–53) Unlike the DDIT3 and STAT6 genes, which appear relatively specific in their tumorigenic potential, GLI1 has demonstrated remarkable and categorical plasticity. Whether by rearrangement or amplification (occasionally concurrently), GLI1 aberrations are documented to source tumors of as yet ambiguous lineage – hypothesized over recent years to represent the gamut of myoepithelial, neural crest, myopericytic, and/or fibroblastic/myofibroblastic derivation due to their inconsistent and noncontextual expression of SMA, S100, and cytokeratin cocktails. Histologic features of GLI1-altered tumors are also commensurate with their genomic diversity, characterized by dichotomy even within the same location. In the stomach, a single pathogenic MALAT1::GLI1 fusion propagates two diametrically different tumors in young patients (identical structural and functional fusion properties across both) – one a benign plexiform growth comprised by bland spindle cells in richly vascularized myxoid matrix (plexiform fibromyxoma), and the other a primitive and highly aggressive biphasic spindle and epithelial/epithelioid neoplasm (gastroblastoma/’GNET’) (2, 4). In the submucosa of the small bowel, another biphasic but putatively indolent neoplasm with tubular epithelial differentiation, diffuse cytokeratin expression, and consistent negativity for S100, has also just emerged (52). In the head and neck with a peculiar predilection for the tongue, these neoplasms range from SMA-positive spindled perivasculocentric proliferations (so-called ‘t[7:12] pericytoma’), to S100-positive monomorphic round/ovoid-appearing cells partitioned into nests by a rich arborizing vasculature. The latter morphology was subsequently recapitulated in three larger series of GLI1-altered tumors accompanied by tentative labels of ‘GLI-altered mesenchymal neoplasm,’ and ‘nested glomoid neoplasm,’ - an appearance which is now a recognized suggestion of underlying GLI1 influence (6, 8, 15) (54, 55).
We recently examined ten retrospectively identified specimens unified by this nested round/ovoid cell morphology and evidence of GLI1 amplification confirmed by fluorescent in situ hybridization (FISH) (15). When queried for fold changes in these neighboring loci, a substantial proportion did show concurrent amplifications in DDIT3, HMGA2, STAT6 and – of particular relevance here – MDM2 and CDK4. A tangential inquiry into the incidence of GLI1 co-amplification in WD/DDLPS resulted in a substantial subset of patients from the MSK-IMPACT database by which to address the diagnostic corollaries of this observation. This is relevant from both histomorphologic and therapeutic standpoints insofar as evolving investigations into sonic hedgehog pathway inhibition (of which GLI1 is a member) might render another effective target in the trajectory of this primarily surgical disease. (56–59)
A brief overview of our data reveals that approximately a fifth of malignancies called as WD/DDLPS harbor underlying GLI1/MDM2/CDK4 co-amplification. The demographic profile of this subgroup was comparable to that of WD/DDLPS as documented in the literature, presenting in the retroperitoneal or mediastinal soft tissues of older adults. Importantly, many of the dedifferentiated tumors were associated with a well-differentiated component, either adjacent to or evident within the recurrence or metastasis. These two features, namely tumor location and association with a morphologically well-differentiated lipogenic sarcoma, are conceptually central to the classification of these GLI1 co-amplified tumors, especially given what we now understand about the extent of their overlap.
When evaluating GLI1/MDM2/CDK4 co-amplified neoplasms (especially referencing those with accompanying eponymous GLI1 morphology as in our cohort), the issue of parsing liposarcomas with driving MDM2/CDK4 amplification and incidental GLI1 amplification from tumors with true ‘primary’ oncogenic GLI1 alteration becomes especially germane. Indeed - MDM2 and CDK4 amplification is pathogenic of other sarcomas such as intimal sarcoma, low-grade central osteosarcoma, parosteal osteosarcoma, and rhabdomyosarcoma. It has even been reported incidentally in other soft tissue malignancies including malignant peripheral nerve sheath tumor, endometrial stromal sarcoma, and leiomyosarcoma, which are distinguished from liposarcoma via antecedent factors such as demography, site/radiology, histology, and immunoprofile. As such, assignment of a sarcomatoid or malignant epithelioid neoplasm as a DDLPS frequently and circumstantially relies on the detection of a proximal well-differentiated lipogenic sarcoma in addition to this characteristic molecular aberration – a common diagnostic scenario in which lineage immunohistochemistry is almost categorically inconclusive. It goes without saying that extensive gross sampling of any peripheral or intralesional fatty tissue is a simple but powerful tool applied to this conundrum. Careful histologic examination of these areas might establish adipocytic lineage, frequently indiscernible within swathes of completely dedifferentiated lesional material. Admittedly, absence of an unequivocal WDLPS does not exclude the diagnosis of DDLPS, as the latter may occur completely de novo or represent covertly unsampled tumor. It does provoke speculation that a few of the 13 tumors in our cohort lacking this feature in the setting of an oncogenic ‘hit’ at the GLI1 locus - might actually represent primary GLI1-altered neoplasms; however, this is unlikely given the classic clinical presentations and additional genomic co-amplifications characteristic of true DDLPS.
Primary tumor location is also directional towards this distinction, in that WD/DDLPS typically have epicenters in the retroperitoneum, mediastinum, or deep somatic soft tissues. In contrast, GLI1-altered neoplasms rarely derive from viscera (case reports in the stomach, small bowel, uterus, ovary, as above) or retroperitoneum/mediastinal soft tissues; they are encountered more often in the head and neck (particularly the tongue) and trunk, but also long and flat bones and both deep and superficial soft tissues of the extremities. (6–10) (54, 55, 60–62). For example, one patient in our cohort presented with a soft tissue lesion in the ring finger with classic GLI1 morphology; the possibility of DDLPS was, quite reasonably, not entertained given both the unconventional location and histology. Only after resection of a paratesticular mass which revealed a MDM2/CDK4/GLI1 co-amplified WD/DDLPS was a metastasis of the former to the finger postulated.
Morphologically, the dedifferentiated components in our cohort were usually variegated with a combination of high-grade undifferentiated pleomorphic sarcoma-like and high-grade myxofibrosarcoma-like features being common. Notably, we observed a salient proportion of the otherwise quite rare meningothelial whorls/osseous metaplasia pattern identified as a primary/secondary/tertiary element in 22% (19/86) of tumors with dedifferentiated morphology (32, 37, 63, 64). Ironically, this was recorded as nigh commensurate with the characteristic high-grade myxofibrosarcoma-like pattern (present in 30% [26/86] of cases), and more commonly than the nested GLI1-altered pattern (present in 12% [10/86] of cases). In contrast, review of a control group of 255 non-GLI1 coamplified DDLPS, there were no cases with meningothelial whorls/osseous metaplasia or GLI1-altered pattern - a novel finding meriting further investigation. Conceptually, recognizing either histologic variant – especially in an isolated core biopsy with minimal peripheral tissue for context – can be helpful to postulate a DDLPS when the appearances would otherwise prompt workup for a nonspecific small round cell tumor, carcinoma, extraskeletal osteosarcoma, follicular dendritic cell sarcoma, or even a true GLI1-altered tumor. Somewhat counterintuitively, the presence or absence of either GLI1 morphology or meningothelial whorls/osseous metaplasia did not correlate with the magnitude (either absolute or relative) of GLI1 amplification, nor were these patterns observed exclusively in tumors lacking a WDLPS.
From an objective genomic standpoint, a wide array of comparative fold changes in MDM2, CDK4, and GLI1 was observed between each individual specimen. Although the comparison of fold changes across tumors is technically unsound as their derivative calculations are multifactorial and contingent in part upon tumor quantity and purity - in any single tumor, MDM2 and CDK4 were duplicated in generally similar magnitudes (i.e., MDM2:CDK4 fold ratio of approximately ~1), whereas the degree of GLI1 amplification was about 60–70% of these two.
A subset of these GLI1-amplified liposarcomas also showed co-amplification of other regions previously described in DDLPS, namely those along the 1p and 6q chromosomal arms. In contrast to the structural contiguity of 12q, the precise mechanism of this phenomenon is not entirely clear but can be exploited as another ancillary tool towards this diagnosis - especially when clinicopathologic circumstances are equivocal and comprehensive sequencing data is available. For example, in a neoplasm that defies classification (due to either noncommittal histology, lack of a lipogenic component, and/or peculiar site/demographic as above) with documented underlying GLI1/MDM2/CDK4 co-amplification, the presence of concurrent copy number gains along these axes would align most closely with the molecular profile of a DDLPS. In our cohort, the presence of any of JUN, TNFAIP3, ESR1, and/or IFNGR1 co-amplifications (among others) was observed in 21/92 (23%) of tumors, and in 3/13 (24%) of tumors comprehensively lacking a well-differentiated liposarcomatous component. Admittedly the specificity of 1p/6q amplification is not well-established across the spectrum of soft tissue sarcomas, but in the background of GLI1/MDM2/CDK4 co-amplification is likely indicative of WD/DDLPS.
A recent investigation illustrated the utility of a GLI1 antibody in the distinction of ‘primary’ GLI1-altered (both rearranged and amplified) neoplasms from histologic analogs but also its positivity in a subset of tumors with molecularly confirmed, but presumed ‘secondary,’ GLI1 amplification (2/5 DDLPS, 1/1 alveolar rhabdomyosarcoma, and 1/2 unclassifiable uterine sarcomas). (65) While GLI1 positivity by immunohistochemistry may be helpful, it should also be deployed in an appropriate clinicopathologic context, and thereafter assessed with caution rather than serve as a diagnostic endpoint. As the literature and our findings imply, GLI1 morphology is identified in tumors with both primary and secondary underlying GLI1 abnormality - as would translate to GLI1 immunoreactivity in most instances - but the presence of either should consequently prompt both additional molecular and histologic evaluation to clarify its active or passive role in oncogenesis. Similarly, the DDIT3 antibody in these circumstances can function not only as a diagnostic aid but also red herring, as the DDIT3 and GLI1 loci are more closely apposed than any other paired coding regions of relevance along this 12q13–15 stretch. Immunohistochemical or select molecular techniques exploiting amplification or protein product upregulation of either gene will also – inadvertently or otherwise - straddle the other. (5, 66–68) Employing already established DDIT3 immunohistochemistry as an inexpensive predictive screening test in a neoplasm with standard GLI1 morphology (wherein positivity would extrapolate to potential GLI1 alteration) could adjudicate further molecular workup (but should not be misinterpreted as evidence of a high-grade round cell myxoid liposarcoma - an unlikely but sometimes credible morphologic mimic). This concept requires validation within a molecular platform that can definitively characterize the DDIT3 locus, which is another limitation of this study.
It may be that details innate to the cohort we enumerate above become academic in the overarching context of longitudinal tumor behavior. Unfortunately, the fact that most basket survival analyses of retroperitoneal and somatic soft tissue sarcomas neglect to perform sub-stratification of only WD/DDLPS, along with widely discrepant use of survival metrics and definitions, renders comparison to prior studies quite challenging (69–73). In the future, statistical cross-comparison to an internal group of GLI1-normal ‘control’ DDLPS will be necessary to elucidate any outcome metric discrepancies.
In conclusion, we present the seminal large-scale characterization of GLI1 co-amplified WD/DDLPS in a cohort benefitted by longitudinal oncologic follow-up from a single cancer center. Distinguishing these neoplasms from morphologic mimics – including true primary GLI1-altered neoplasms – is important towards treatment and prognostication. This entails careful examination for a well-differentiated lipogenic component, assessment of primary tumor location, and integration of any coincident genomic co-amplifications, in particular those along chromosomal arms 1p/6q. While many clinicopathologic aspects mirror those of conventional DDLPS, certain attributes including a predilection for variant meningothelial whorls/osseous metaplastic and eponymous nested round cell GLI1 morphology can alert the practicing pathologist to this important consideration.
Funding Statement
Supported in part by: P30 CA008748 (NA)
Footnotes
Conflicts of Interest:
The authors have disclosed that they have no significant relationships with, or financial interest in, any commercial companies pertaining to this article.
Ethics Approval / Consent to Participate
This study was approved by the Institutional Review Board at Memorial Sloan Kettering Cancer Center.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.
References
- 1.Dahlen A, Fletcher CD, Mertens F, Fletcher JA, Perez-Atayde AR, Hicks MJ, et al. Activation of the GLI oncogene through fusion with the beta-actin gene (ACTB) in a group of distinctive pericytic neoplasms: pericytoma with t(7;12). Am J Pathol. 2004. May;164(5):1645–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spans L, Fletcher CD, Antonescu CR, Rouquette A, Coindre JM, Sciot R, Debiec-Rychter M. Recurrent MALAT1-GLI1 oncogenic fusion and GLI1 up-regulation define a subset of plexiform fibromyxoma. J Pathol. 2016. Jul;239(3):335–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arslan ME, Li H, Fu Z, Jennings TA, Lee H. Plexiform fibromyxoma: Review of rare mesenchymal gastric neoplasm and its differential diagnosis. World J Gastrointest Oncol. 2021. May 15;13(5):409–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Graham RP, Nair AA, Davila JI, Jin L, Jen J, Sukov WR, et al. Gastroblastoma harbors a recurrent somatic MALAT1-GLI1 fusion gene. Mod Pathol. 2017. Oct;30(10):1443–52. [DOI] [PubMed] [Google Scholar]
- 5.Palsgrove DN, Rooper LM, Stevens TM, Shin C, Damm DD, Gagan J, et al. GLI1-Altered Soft Tissue Tumors of the Head and Neck: Frequent Oropharyngeal Involvement, p16 Immunoreactivity, and Detectable Alterations by DDIT3 Break Apart FISH. Head Neck Pathol. 2022. Dec;16(4):1146–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu J, Mao R, Lao IW, Yu L, Bai Q, Zhou X, Wang J. GLI1-altered mesenchymal tumor: a clinicopathological and molecular analysis of ten additional cases of an emerging entity. Virchows Arch. 2022. May;480(5):1087–99. [DOI] [PubMed] [Google Scholar]
- 7.Xu B, Chang K, Folpe AL, Kao YC, Wey SL, Huang HY, et al. Head and Neck Mesenchymal Neoplasms With GLI1 Gene Alterations: A Pathologic Entity With Distinct Histologic Features and Potential for Distant Metastasis. Am J Surg Pathol. 2020. Jun;44(6):729–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Papke DJ Jr., Dickson BC, Oliveira AM, Sholl LM, Fletcher CDM. Distinctive Nested Glomoid Neoplasm: Clinicopathologic Analysis of 20 Cases of a Mesenchymal Neoplasm With Frequent GLI1 Alterations and Indolent Behavior. Am J Surg Pathol. 2023. Jan 1;47(1):12–24. [DOI] [PubMed] [Google Scholar]
- 9.Antonescu CR, Agaram NP, Sung YS, Zhang L, Swanson D, Dickson BC. A Distinct Malignant Epithelioid Neoplasm With GLI1 Gene Rearrangements, Frequent S100 Protein Expression, and Metastatic Potential: Expanding the Spectrum of Pathologic Entities With ACTB/MALAT1/PTCH1-GLI1 Fusions. Am J Surg Pathol. 2018. Apr;42(4):553–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kerr DA, Pinto A, Subhawong TK, Wilky BA, Schlumbrecht MP, Antonescu CR, et al. Pericytoma With t(7;12) and ACTB-GLI1 Fusion: Reevaluation of an Unusual Entity and its Relationship to the Spectrum of GLI1 Fusion-related Neoplasms. Am J Surg Pathol. 2019. Dec;43(12):1682–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cancer Genome Atlas Research Network. Electronic address edsc, Cancer Genome Atlas Research N. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell. 2017. Nov 2;171(4):950–65 e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mantilla JG, Ricciotti RW, Chen EY, Liu YJ, Hoch BL. Amplification of DNA damage-inducible transcript 3 (DDIT3) is associated with myxoid liposarcoma-like morphology and homologous lipoblastic differentiation in dedifferentiated liposarcoma. Mod Pathol. 2019. Apr;32(4):585–92. [DOI] [PubMed] [Google Scholar]
- 13.Saada-Bouzid E, Burel-Vandenbos F, Ranchere-Vince D, Birtwisle-Peyrottes I, Chetaille B, Bouvier C, et al. Prognostic value of HMGA2, CDK4, and JUN amplification in well-differentiated and dedifferentiated liposarcomas. Mod Pathol. 2015. Nov;28(11):1404–14. [DOI] [PubMed] [Google Scholar]
- 14.Doyle LA, Tao D, Marino-Enriquez A. STAT6 is amplified in a subset of dedifferentiated liposarcoma. Mod Pathol. 2014. Sep;27(9):1231–7. [DOI] [PubMed] [Google Scholar]
- 15.Agaram NP, Zhang L, Sung YS, Singer S, Stevens T, Prieto-Granada CN, et al. GLI1-amplifications expand the spectrum of soft tissue neoplasms defined by GLI1 gene fusions. Mod Pathol. 2019. Nov;32(11):1617–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012. May;2(5):401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013. Apr 2;6(269):pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weiss SW, Rao VK. Well-differentiated liposarcoma (atypical lipoma) of deep soft tissue of the extremities, retroperitoneum, and miscellaneous sites. A follow-up study of 92 cases with analysis of the incidence of “dedifferentiation”. Am J Surg Pathol. 1992. Nov;16(11):1051–8. [DOI] [PubMed] [Google Scholar]
- 19.Henricks WH, Chu YC, Goldblum JR, Weiss SW. Dedifferentiated liposarcoma: a clinicopathological analysis of 155 cases with a proposal for an expanded definition of dedifferentiation. Am J Surg Pathol. 1997. Mar;21(3):271–81. [DOI] [PubMed] [Google Scholar]
- 20.Huang HY, Brennan MF, Singer S, Antonescu CR. Distant metastasis in retroperitoneal dedifferentiated liposarcoma is rare and rapidly fatal: a clinicopathological study with emphasis on the low-grade myxofibrosarcoma-like pattern as an early sign of dedifferentiation. Mod Pathol. 2005. Jul;18(7):976–84. [DOI] [PubMed] [Google Scholar]
- 21.Elgar F, Goldblum JR. Well-differentiated liposarcoma of the retroperitoneum: a clinicopathologic analysis of 20 cases, with particular attention to the extent of low-grade dedifferentiation. Mod Pathol. 1997. Feb;10(2):113–20. [PubMed] [Google Scholar]
- 22.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. The Journal of molecular diagnostics : JMD. 2015. May;17(3):251–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tap WD, Eilber FC, Ginther C, Dry SM, Reese N, Barzan-Smith K, et al. Evaluation of well-differentiated/de-differentiated liposarcomas by high-resolution oligonucleotide array-based comparative genomic hybridization. Genes Chromosomes Cancer. 2011. Feb;50(2):95–112. [DOI] [PubMed] [Google Scholar]
- 24.Mandahl N, Magnusson L, Nilsson J, Viklund B, Arbajian E, von Steyern FV, et al. Scattered genomic amplification in dedifferentiated liposarcoma. Mol Cytogenet. 2017;10:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nishio J, Nakayama S, Nabeshima K, Yamamoto T. Biology and Management of Dedifferentiated Liposarcoma: State of the Art and Perspectives. J Clin Med. 2021. Jul 22;10(15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Snyder EL, Sandstrom DJ, Law K, Fiore C, Sicinska E, Brito J, et al. c-Jun amplification and overexpression are oncogenic in liposarcoma but not always sufficient to inhibit the adipocytic differentiation programme. J Pathol. 2009. Jul;218(3):292–300. [DOI] [PubMed] [Google Scholar]
- 27.Kurtin SE, Taher R. Clinical Trial Design and Drug Approval in Oncology: A Primer for the Advanced Practitioner in Oncology. J Adv Pract Oncol. 2020. Sep-Oct;11(7):736–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McCormick D, Mentzel T, Beham A, Fletcher CD. Dedifferentiated liposarcoma. Clinicopathologic analysis of 32 cases suggesting a better prognostic subgroup among pleomorphic sarcomas. Am J Surg Pathol. 1994. Dec;18(12):1213–23. [DOI] [PubMed] [Google Scholar]
- 29.Horvai AE, DeVries S, Roy R, O’Donnell RJ, Waldman F. Similarity in genetic alterations between paired well-differentiated and dedifferentiated components of dedifferentiated liposarcoma. Mod Pathol. 2009. Nov;22(11):1477–88. Epub 20090904. [DOI] [PubMed] [Google Scholar]
- 30.Evans HL. Liposarcoma: a study of 55 cases with a reassessment of its classification. Am J Surg Pathol. 1979. Dec;3(6):507–23. [DOI] [PubMed] [Google Scholar]
- 31.Marino-Enriquez A, Fletcher CD, Dal Cin P, Hornick JL. Dedifferentiated liposarcoma with “homologous” lipoblastic (pleomorphic liposarcoma-like) differentiation: clinicopathologic and molecular analysis of a series suggesting revised diagnostic criteria. Am J Surg Pathol. 2010. Aug;34(8):1122–31. [DOI] [PubMed] [Google Scholar]
- 32.Nascimento AG, Kurtin PJ, Guillou L, Fletcher CD. Dedifferentiated liposarcoma: a report of nine cases with a peculiar neurallike whorling pattern associated with metaplastic bone formation. Am J Surg Pathol. 1998. Aug;22(8):945–55. [DOI] [PubMed] [Google Scholar]
- 33.Sioletic S, Dal Cin P, Fletcher CD, Hornick JL. Well-differentiated and dedifferentiated liposarcomas with prominent myxoid stroma: analysis of 56 cases. Histopathology. 2013. Jan;62(2):287–93. [DOI] [PubMed] [Google Scholar]
- 34.Folpe AL, Weiss SW. Lipoleiomyosarcoma (well-differentiated liposarcoma with leiomyosarcomatous differentiation): a clinicopathologic study of nine cases including one with dedifferentiation. Am J Surg Pathol. 2002. Jun;26(6):742–9. [DOI] [PubMed] [Google Scholar]
- 35.Agaimy A, Michal M, Hadravsky L, Michal M. Dedifferentiated liposarcoma composed predominantly of rhabdoid/epithelioid cells: a frequently misdiagnosed highly aggressive variant. Hum Pathol. 2018. Jul;77:20–7. Epub 20180105. [DOI] [PubMed] [Google Scholar]
- 36.Yamashita K, Kohashi K, Yamada Y, Ishii T, Nishida Y, Urakawa H, et al. Osteogenic differentiation in dedifferentiated liposarcoma: a study of 36 cases in comparison to the cases without ossification. Histopathology. 2018. Apr;72(5):729–38. Epub 20171222. [DOI] [PubMed] [Google Scholar]
- 37.Hasegawa T, Seki K, Hasegawa F, Matsuno Y, Shimodo T, Hirose T, et al. Dedifferentiated liposarcoma of retroperitoneum and mesentery: varied growth patterns and histological grades--a clinicopathologic study of 32 cases. Hum Pathol. 2000. Jun;31(6):717–27. [DOI] [PubMed] [Google Scholar]
- 38.Yu Z, Zhao X, Gao J, Zhou S, Li P, Liu N. Correlation Analysis Between Demographic, Surgical, and Pathological Characteristics with Local Recurrence-Free Survival for Surgical Resected Retroperitoneal Liposarcoma. World J Surg. 2023. Aug;47(8):1946–55. Epub 20230418. [DOI] [PubMed] [Google Scholar]
- 39.Muratori F, Frenos F, Bettini L, Matera D, Mondanelli N, Scorianz M, et al. Liposarcoma: Clinico-pathological analysis, prognostic factors and survival in a series of 307 patients treated at a single institution. J Orthop Sci. 2018. Nov;23(6):1038–44. Epub 20180711. [DOI] [PubMed] [Google Scholar]
- 40.Molina G, Hull MA, Chen YL, DeLaney TF, De Amorim Bernstein K, Choy E, et al. Preoperative radiation therapy combined with radical surgical resection is associated with a lower rate of local recurrence when treating unifocal, primary retroperitoneal liposarcoma. J Surg Oncol. 2016. Dec;114(7):814–20. Epub 20160916. [DOI] [PubMed] [Google Scholar]
- 41.Tseng WW, Madewell JE, Wei W, Somaiah N, Lazar AJ, Ghadimi MP, et al. Locoregional disease patterns in well-differentiated and dedifferentiated retroperitoneal liposarcoma: implications for the extent of resection? Annals of surgical oncology. 2014. Jul;21(7):2136–43. Epub 20140407. [DOI] [PubMed] [Google Scholar]
- 42.Stein EM, DeAngelo DJ, Chromik J, Chatterjee M, Bauer S, Lin CC, et al. Results from a First-in-Human Phase I Study of Siremadlin (HDM201) in Patients with Advanced Wild-Type TP53 Solid Tumors and Acute Leukemia. Clin Cancer Res. 2022. Mar 1;28(5):870–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gounder MM, Bauer TM, Schwartz GK, Weise AM, LoRusso P, Kumar P, et al. A First-in-Human Phase I Study of Milademetan, an MDM2 Inhibitor, in Patients With Advanced Liposarcoma, Solid Tumors, or Lymphomas. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2023. Mar 20;41(9):1714–24. Epub 20230120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takahashi S, Fujiwara Y, Nakano K, Shimizu T, Tomomatsu J, Koyama T, et al. Safety and pharmacokinetics of milademetan, a MDM2 inhibitor, in Japanese patients with solid tumors: A phase I study. Cancer Sci. 2021. Jun;112(6):2361–70. Epub 20210502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Assi T, Kattan J, Rassy E, Nassereddine H, Farhat F, Honore C, et al. Targeting CDK4 (cyclin-dependent kinase) amplification in liposarcoma: A comprehensive review. Crit Rev Oncol Hematol. 2020. Sep;153:103029. Epub 20200618. [DOI] [PubMed] [Google Scholar]
- 46.Nassif EF, Cope B, Traweek R, Witt RG, Erstad DJ, Scally CP, et al. Real-world use of palbociclib monotherapy in retroperitoneal liposarcomas at a large volume sarcoma center. Int J Cancer. 2022. Jun 15;150(12):2012–24. Epub 20220218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Doyle LA, Vivero M, Fletcher CD, Mertens F, Hornick JL. Nuclear expression of STAT6 distinguishes solitary fibrous tumor from histologic mimics. Mod Pathol. 2014. Mar;27(3):390–5. Epub 20130913. [DOI] [PubMed] [Google Scholar]
- 48.Zhu H, Lo HW. The Human Glioma-Associated Oncogene Homolog 1 (GLI1) Family of Transcription Factors in Gene Regulation and Diseases. Curr Genomics. 2010. Jun;11(4):238–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bigner SH, Friedman HS, Vogelstein B, Oakes WJ, Bigner DD. Amplification of the c-myc gene in human medulloblastoma cell lines and xenografts. Cancer Res. 1990. Apr 15;50(8):2347–50. [PubMed] [Google Scholar]
- 50.Wang B, Yu T, Hu Y, Xiang M, Peng H, Lin Y, et al. Prognostic role of Gli1 expression in breast cancer: a meta-analysis. Oncotarget. 2017. Oct 6;8(46):81088–97. Epub 20170707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stecca B, Mas C, Clement V, Zbinden M, Correa R, Piguet V, et al. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc Natl Acad Sci U S A. 2007. Apr 3;104(14):5895–900. Epub 20070328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jessurun J, Orr C, McNulty SN, Hagen CE, Alnajar H, Wilkes D, et al. GLI1 -Rearranged Enteric Tumor : Expanding the Spectrum of Gastrointestinal Neoplasms With GLI1 Gene Fusions. Am J Surg Pathol. 2023. Jan 1;47(1):65–73. Epub 20220815. [DOI] [PubMed] [Google Scholar]
- 53.Tusa I, Gagliardi S, Tubita A, Pandolfi S, Menconi A, Lulli M, et al. The Hedgehog-GLI Pathway Regulates MEK5-ERK5 Expression and Activation in Melanoma Cells. Int J Mol Sci. 2021. Oct 19;22(20). Epub 20211019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Argani P, Boyraz B, Oliva E, Matoso A, Gross J, Fridman E, et al. GLI1 Gene Alterations in Neoplasms of the Genitourinary and Gynecologic Tract. Am J Surg Pathol. 2022. May 1;46(5):677–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Machado I, Hosler GA, Traves V, Claramunt R, Sanmartin O, Santonja C, et al. Superficial GLI1-amplified mesenchymal neoplasms: Expanding the spectrum of an emerging entity which reaches the realm of dermatopathology. Journal of cutaneous pathology. 2023. Jun;50(6):487–99. Epub 20221123. [DOI] [PubMed] [Google Scholar]
- 56.Avery JT, Zhang R, Boohaker RJ. GLI1: A Therapeutic Target for Cancer. Front Oncol. 2021;11:673154. Epub 20210525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peer E, Tesanovic S, Aberger F. Next-Generation Hedgehog/GLI Pathway Inhibitors for Cancer Therapy. Cancers (Basel). 2019. Apr 15;11(4). Epub 20190415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xie H, Paradise BD, Ma WW, Fernandez-Zapico ME. Recent Advances in the Clinical Targeting of Hedgehog/GLI Signaling in Cancer. Cells. 2019. Apr 29;8(5). Epub 20190429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schneider RK, Mullally A, Dugourd A, Peisker F, Hoogenboezem R, Van Strien PMH, et al. Gli1(+) Mesenchymal Stromal Cells Are a Key Driver of Bone Marrow Fibrosis and an Important Cellular Therapeutic Target. Cell Stem Cell. 2017. Jun 1;20(6):785–800 e8. Epub 20170427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Punjabi LS, Goh CHR, Sittampalam K. Expanding the spectrum of GLI1-altered mesenchymal tumors-A high-grade uterine sarcoma harboring a novel PAMR1::GLI1 fusion and literature review of GLI1-altered mesenchymal neoplasms of the gynecologic tract. Genes Chromosomes Cancer. 2023. Feb;62(2):107–14. Epub 20221026. [DOI] [PubMed] [Google Scholar]
- 61.Koh NWC, Seow WY, Lee YT, Lam JCM, Lian DWQ. Pericytoma With t(7;12): The First Ovarian Case Reported and a Review of the Literature. Int J Gynecol Pathol. 2019. Sep;38(5):479–84. [DOI] [PubMed] [Google Scholar]
- 62.Alwaqfi RR, Samuelson MI, Guseva NN, Ouyang M, Bossler AD, Ma D. PTCH1-GLI1 Fusion-Positive Ovarian Tumor: Report of a Unique Case With Response to Tyrosine Kinase Inhibitor Pazopanib. J Natl Compr Canc Netw. 2021. Sep 20;19(9):998–1004. [DOI] [PubMed] [Google Scholar]
- 63.Fanburg-Smith JC, Miettinen M. Liposarcoma with meningothelial-like whorls: a study of 17 cases of a distinctive histological pattern associated with dedifferentiated liposarcoma. Histopathology. 1998. Nov;33(5):414–24. [DOI] [PubMed] [Google Scholar]
- 64.Patton A, McKenney JK, Alruwaii FI, Angulo KA, Fuller LD, Calvaresi E, et al. Paratesticular Dedifferentiated Liposarcoma with Epithelioid Features: A Diagnostic Pitfall. International journal of surgical pathology. 2023. Aug;31(5):721–7. [DOI] [PubMed] [Google Scholar]
- 65.Parrack PH, Marino-Enriquez A, Fletcher CDM, Hornick JL, Papke DJ Jr. GLI1 Immunohistochemistry Distinguishes Mesenchymal Neoplasms With GLI1 Alterations From Morphologic Mimics. Am J Surg Pathol. 2023. Apr 1;47(4):453–60. [DOI] [PubMed] [Google Scholar]
- 66.Baranov E, Black MA, Fletcher CDM, Charville GW, Hornick JL. Nuclear expression of DDIT3 distinguishes high-grade myxoid liposarcoma from other round cell sarcomas. Mod Pathol. 2021. Jul;34(7):1367–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Scapa JV, Cloutier JM, Raghavan SS, Peters-Schulze G, Varma S, Charville GW. DDIT3 Immunohistochemistry Is a Useful Tool for the Diagnosis of Myxoid Liposarcoma. Am J Surg Pathol. 2021. Feb 1;45(2):230–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kuczkiewicz-Siemion O, Wisniewski P, Dansonka-Mieszkowska A, Grabowska-Kieryl M, Olszewska K, Goryn T, et al. The utility of fluorescence in situ hybridization (FISH) in determining DNA damage-inducible transcript 3 (DDIT3) amplification in dedifferentiated liposarcomas - an important diagnostic pitfall. Pathol Res Pract. 2021. Sep;225:153555. [DOI] [PubMed] [Google Scholar]
- 69.Yan Y, Xia S, Teng D, Hu S, Li S, Wang Y, et al. Resection outcomes for primary and local recurrent retroperitoneal liposarcoma patients. Ann Transl Med. 2020. Nov;8(21):1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ishii K, Yokoyama Y, Nishida Y, Koike H, Yamada S, Kodera Y, et al. Characteristics of primary and repeated recurrent retroperitoneal liposarcoma: outcomes after aggressive surgeries at a single institution. Jpn J Clin Oncol. 2020. Dec 16;50(12):1412–8. [DOI] [PubMed] [Google Scholar]
- 71.Homsy P, Heiskanen I, Sampo M, Ronty M, Tukiainen E, Blomqvist C. Single centre 30-year experience in treating retroperitoneal liposarcomas. J Surg Oncol. 2020. Nov;122(6):1163–72. [DOI] [PubMed] [Google Scholar]
- 72.Paik B, Seo CJ, Tan JW, Juan WKD, Soo KC, Ong CJ, et al. A systematic review of margin status in retroperitoneal liposarcomas: Does the R0 margin matter? Front Oncol. 2022;12:891710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Improta L, Pasquali S, Iadecola S, Barisella M, Fiore M, Radaelli S, et al. Organ Infiltration and Patient Risk After Multivisceral Surgery for Primary Retroperitoneal Liposarcomas. Annals of surgical oncology. 2023. Jul;30(7):4500–10. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.