

Abstract
Mutations in the promoter of the telomerase reverse transcriptase (TERT) gene are the paradigm of a cross-cancer alteration in a noncoding region. TERT promoter mutations (TPM) are biomarkers of poor prognosis in cancer, including thyroid tumors. TPMs enhance TERT transcription, which is otherwise silenced in adult tissues, thus reactivating a bona fide oncoprotein. To study TERT deregulation and its downstream consequences, we generated a Tert mutant promoter mouse model via CRISPR/Cas9 engineering of the murine equivalent locus (Tert−123C>T) and crossed it with thyroid-specific BrafV600E-mutant mice. We also employed an alternative model of Tert overexpression (K5-Tert). Whereas all BrafV600E animals developed well-differentiated papillary thyroid tumors, 29% and 36% of BrafV600E+Tert−123C>T and BrafV600E+K5-Tert mice progressed to poorly differentiated cancers at week 20, respectively. Tert-upregulated tumors showed increased mitosis and necrosis in areas of solid growth, and older animals displayed anaplastic-like features, that is, spindle cells and macrophage infiltration. Murine TPM increased Tert transcription in vitro and in vivo, but temporal and intratumoral heterogeneity was observed. RNA-sequencing of thyroid tumor cells showed that processes other than the canonical Tert-mediated telomere maintenance role operate in these specimens. Pathway analysis showed that MAPK and PI3K/AKT signaling, as well as processes not previously associated with this tumor etiology, involving cytokine, and chemokine signaling, were overactivated. These models constitute useful preclinical tools to understand the cell-autonomous and microenvironment-related consequences of Tert-mediated progression in advanced thyroid cancers and other aggressive tumors carrying TPMs.
Introduction
Hotspot mutations in the proximal promoter of the telomerase reverse transcriptase gene (TERT) are a prototype of a noncoding genetic alteration present in multiple cancers. After the initial discovery of TERT promoter mutations (TPM) in melanomas (1, 2), they were also identified as frequent events in various tumor types, such as gliomas, hepatocellular, urothelial, and thyroid carcinomas (3–5). Early pan-cancer studies assessing the noncoding genome identified TERT as the top altered gene (6, 7). The former was subsequently confirmed by the Pan Cancer Analysis of Whole Genomes (PCAWG), which reported the TERT promoter as the most frequently mutated noncoding driver in 2,658 cancer genomes (8). Of note, TPMs correlate with metastatic and aggressive forms in most cancer lineages and have thus emerged as biomarkers of poor prognosis (1, 3, 9, 10).
TPMs occur in two hotspots at c.−124C>T and c.−146C>T and are mutually exclusive, suggesting a common effect. Tumors harboring TPMs reactivate TERT transcription, which is otherwise repressed in adult normal cells. The discovery of TPMs has restored interest in TERT as a bona fide potential cancer target. TERT is the catalytic component of the telomerase complex and is responsible of extending chromosome ends (i.e., telomeres) by adding hexanucleotide tandem repeats, thus preventing telomere erosion and avoiding replicative senescence. In recent years, there is growing evidence of extra-telomeric roles of TERT in cancer cells, suggesting that it enhances other pro-neoplastic features (11, 12).
Thyroid cancers are genetically simple tumors, typically driven by oncogenic mutations in BRAFV600E, RAS genes or receptor tyrosine kinase fusions, which result in the constitutive activation of the MAPK pathway. The presence of BRAF or RAS mutations in well-differentiated tumors, that is, papillary thyroid cancers (PTC), as well as in advanced forms, such as poorly differentiated (PDTC), and anaplastic thyroid cancers (ATC), strongly suggests a continuum in disease progression via the accumulation of key additional genetic defects. Indeed, we and others reported a stepwise increase in TPM frequency along the spectrum of thyroid cancer progression: 9% in PTCs, 40% in PDTCs, and 73% in ATCs. Interestingly, TPMs are subclonal in the few PTCs that harbor them, whereas they are clonal in PDTCs and ATCs, pointing to selection during tumor evolution (13–16).
So far, TERT biology in cancer has been studied in either transgenic mouse models, which do not recapitulate the endogenous levels of Tert expression, or in immortalized cell systems, which do not represent biologically accurate settings for telomerase biology. Early attempts to assess telomerase upregulation in vivo targeting Tert overexpression to various tissues showed an increased incidence of neoplastic transformation without evidence of critical telomere deregulation (17–19). Of note, Tert overexpression under the control of the keratin 5 promoter (K5-Tert) increased the frequency of pre-neoplastic lesions of the thyroid gland of aging mice, suggesting that the thyroid follicular cell lineage, even in the absence of MAPK constitutive signaling, might be particularly susceptible to telomerase upregulation (20).
In this study, we used CRISPR/Cas9 to generate a Tert mutant promoter mouse model and studied these animals in the context of thyroid-specific BrafV600E activation. We demonstrate that BrafV600E+Tert−123C>T mice increase Tert expression to levels comparable with those observed in human tumors harboring TPMs (e.g., BRAFV600E+TERT−124C>T), and that they induce thyroid cancer progression, mimicking the phenotypes observed in a model in which Tert overexpression was targeted to epithelial cells (BrafV600E+K5-Tert). Thyroid cells with an engineered TPM did not uniformly re-express telomerase, suggesting that additional steps are required for the enhanced Tert expression to manifest. Consistent with the fact that mice have longer telomeres than humans and show less restricted expression of telomerase in adult tissues (21, 22), our data indicate that events unrelated to telomere attrition operate in these tumors. Instead, transcriptomic analysis of telomerase-upregulated tumors showed that overactivation of MAPK and PI3K pathways, as well as immune-related signaling, probably in response to changes in the tumor microenvironment, likely play a role in disease progression.
Materials and Methods
Sequence analysis
To determine the degree of mouse:human interspecies conservation in the region surrounding the human TERT c.−124C>T and c.−146C>T mutation hotspots, we performed sequence alignment between human TERT (chr5: 1,295,347–1,294,746) and mouse Tert (chr13: 73,626,701–73,627,273) covering sequences around the transcription start site using the European Bioinformatics Institute (EMBL-EBI) pairwise sequence alignment tools (https://www.ebi.ac.uk/Tools/psa/). Sequence alignments were retrieved and manually inspected for similarities.
Luciferase assays
The 226-nucleotide sequence upstream of mouse Tert gene, containing the −123C wild-type allele, was synthesized at Genscript and cloned into the pGL4.20[luc2/Puro] reporter vector (Promega). A mutant version of this plasmid, containing the Tert −123T allele, was created by site-directed mutagenesis. Tert c.−123C and Tert c.−123T alleles were confirmed by direct sequencing. Reporter experiments were performed using the Dual-Luciferase Reporter Assays (Promega) following the manufacturer’s protocol and using Renilla (pGL4.73[hRluc/SV40]) as a control. Tert promoter-driven luciferase expression was tested in NIH-3T3 (mouse fibroblasts) and in two cell lines (B92 and B16509E) derived from murine BrafV600E-mutant thyroid tumors. Cells were grown in DMEM (NIH-3T3) or F12-Coon’s media (B92 and B16509E) media supplemented with 10% FBS. Luciferase results are expressed in relative units and represent the average of three independent experiments with each condition ran in quadruplicate.
Gene editing and Tert mutant promoter mouse generation
Two guide RNAs (gRNA) targeting the mouse Tert c.−123C locus were designed. Two rounds of Cas9/CRISPR injections using the selected gRNA and donor templates were performed on mouse zygotes, followed by embryo transfer, which resulted in 84 pups. The B6 hybrid (B6CBAF1) background was used. Gene editing was performed at the MSKCC Mouse Genetics Core Facility. Animals were first screened by a restriction fragment length polymorphism (RFLP) approach, using the Mnl1 enzyme. Agarose gel band patterns compatible with the presence of Tert c.−123C>T mutation were amplified and Sanger-sequenced using the following primers: Ter_123_2F: 5′-CATGCACCAGCATTGTGACCA-3′ and Tert_123_4R: 5′- CAACGAGGAGCGCGGGTCATTGT-3′. To evaluate off-target effects, six founder animals with confirmed Tert −123C>T mutation were subjected to targeted next-generation sequencing (NGS) and analyzed using CRISPResso (23). NGS showed founders with up to 20% of reads displaying the desired Tert c.−123C>T mutation and no off-target effects in cis. Three animals were used to generate the Tert mutant promoter mouse line. Off-target alterations were bred-out in subsequent generations, as assessed by Sanger sequencing. Animals were born at the expected Mendelian frequency. The offspring of a selected founder was used to establish the Tert−123C>T mouse line.
Mouse models and breeding strategies
Animal care and all experimental procedures were approved by the MSKCC and BWH Animal Care and Use Committees. The Tert−123C>T mouse line was studied in the context of thyroid-specific activation of BrafV600E oncoprotein by crossing these animals with LSL-BrafV600E/TPO-Cre/eYFP mice (“BrafV600E”), which express endogenous levels of Braf oncoprotein in thyroid follicular cells at E14.5, when Cre recombinase is expressed downstream of the thyroid peroxidase (Tpo) gene promoter (24). This BrafV600E model is also engineered to express yellow fluorescent protein (YEP; Jackson Lab stock, #007903) in thyroid cells, as previously described (25). The transgenic Tg-K5-Tert mouse line, in which Tert expression is targeted to epithelial tissue via the keratin 5 (K5) promoter (17, 20), was a generous gift from Dr. María Blasco at the Spanish National Cancer Research Centre. We crossed K5-Tert animals with our BrafV600E mice to generate an alternative model in which expression of BrafV600E oncoprotein and overexpression of Tert were present in thyrocytes.
Our breeding scheme was designed to minimize housing differences between the groups to be compared, ensuring that BrafV600E animals, and either BrafV600E+ Tert−123C>T or BrafV600E+K5-Tert mice were littermates. Tert c.−123C>T was studied in heterozygosis. BrafV600E alone was used as our baseline model of well-differentiated thyroid cancer for all comparisons. Mouse genotyping was primarily achieved by allele-specific assays designed and performed by Transnetyx.
Histology and IHC
Thyroid tissues were fixed in 4% paraformaldehyde, embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E). Histologic diagnosis was performed by thyroid pathologists (R.G. and B.X.) blinded to the genotype and the treatment status of each animal. IHC was performed on the Leica Bond III automated staining platform using the Leica Biosystems Refine Detection Kit. The following antibodies were used for IHC: phospho-Erk [Cell Signaling Technology (CST), catalog no. 4370), Ki-67 (CST, catalog no. 12202), CD11b (Abcam, catalog no. ab133357), F4/80 (CST, catalog no. 70076), Arg1 (Abcam, catalog no. ab 93668), and Pax8 (Proteintech, catalog no. 10336). The secondary antibodies were part of the Leica Bond Polymer Refine Detection Kit (catalog no. DS9800). QuPath (version 0.3.0; ref. 26) was used for H/E slide visualization and the quantification of 3,3′-diaminobenzidine (DAB) staining on IHC slides. Tumor boundaries were delineated, and the positive cell detection command was applied to the tumor area, applying default settings and a single intensity threshold of 0.2 to quantify the percentage of positive DAB-stained cells.
Generation of mouse thyroid cancer cell lines
To generate mouse cancer cell lines, thyroid tumors from age-matched 10-week animals with BrafV600E, BrafV600E+Tert−123C>T, and BrafV600E+K5-Tert genotypes were dissected from surrounding tissues and collected on minimum essential media (MEM). Specimens were subsequently minced using sterile razorblades, spun down by centrifugation at 500 × g for 5 minutes and resuspended in 10 mL of digestion medium (MEM containing 112 U/mL type I collagenase; Worthington, catalog no. CLS-1), 1.2 U/mL dispase (Gibco; catalog no. 17105–041), penicillin (50 U/mL), and streptomycin (50 mg/mL). Cells were incubated at 37°C for 60 minutes with vigorous shaking, after which cells were spun down and resuspended in Coon modified F12 medium with penicillin/streptomycin/L-glutamine (P/S/G; Gemini; #400–110) and 0.5% bovine brain extract (BBE, Hammond Cell Tech, catalog no. 2007-NZ), plated into CellBind plates (Corning Inc.) for 2 weeks and then switched to Coon modified F12 medium with P/S/G containing 5% FBS for routine culturing and subsequent experiments. Cell lines were maintained at 37°C and 5% CO2 in humidified atmosphere. For the experiments described here, we employed low passages of these cell lines, and passages were always synchronized across comparison groups.
Flow cytometry
To isolate YFP+ thyroid cells for subsequent RNA sequencing (RNA-seq), tumor and normal thyroids were harvested in cold digestion buffer [Hank’s Balanced Salt Solution (HBSS)] supplemented with 5% FBS and 1.5 mg/mL Collagenase A, and thoroughly minced with razorblades. Tissue suspensions were transferred to 15 mL tubes and incubated at 37°C for 1 hour with intermittent vortexing every 10 minutes. Dissociated thyroid cells were passed through a 70 μm cell strainer, pelleted by centrifugation at 500 × g for 5 minutes, washed twice with cold PBS and resuspended in cold sorting buffer (F-12 Coon media containing 2% FBS). A YFP+ pure thyroid cell population was sorted using a BD FACSAria flow cytometer into TRizol (Invitrogen, catalog no. 15596018). Each mouse tumor from BrafV600E, BrafV600E+Tert−123C>T, and BrafV600E+K5-Tert mice, was processed individually, and target cell yield was established at 20,000 YFP+ cells. For non-Braf specimens which did not generate tumors, we pooled several normal-looking thyroid glands from identical genotypes, to collect enough YFP+ cells, as follows: wildtype (WT, n = 25), Tert−123C>T only (n = 17), and K5-Tert only (n = 14).
RNA-seq
Total RNA was isolated from up to 20,000 YFP+ cells sorted into TRizol (range = 9,000–20,000 cells). Phase separation in cells lysed in TRizol reagent was achieved with chloroform. RNA was precipitated with isopropanol, washed with 75% ethanol, and resuspended in RNase-free water following the manufacturer’s protocol. RNA-seq was performed at the Dana-Farber Cancer Institute Molecular Biology Core Facility. RNA libraries were prepared following a low input mRNAseq protocol. RNA samples were fragmented at 94°C for 8 min with 14 cycles of PCR post-adapter ligation, according to manufacturer’s recommendation. The finished dsDNA libraries were quantified by Qubit fluorometer and Agilent TapeStation 2200. Libraries were pooled in equimolar ratios. Final sequencing was performed on an Illumina NovaSeq with paired-end 100 bp reads.
Standard analysis and visualization of RNA-seq data were performed using VIPER (27). These include the generation of heatmaps and volcano plots, as well as pathway and functional group analysis employing Gene Ontology (GO; 05/2021), Gene Set Enrichment Analysis (GSEA; version 4.1, 07/2020), and the Kyoto Encyclopedia of Genes and Genomes (KEGG; release 98, 04/2021) pathway database. Expression of specific genes was evaluated from RNA-seq-derived normalized counts and represented as median ± IQR)of gene transcripts for each animal. Specific pairwise comparisons from RNA-seq data were performed by comparing medians of normalized counts from defined groups, as described, and P values were calculated using unpaired two-tailed Mann–Whitney U tests, unless otherwise noted.
qRT-PCR
RNA was preserved in TRizol (Invitrogen) and isolated as described above. One microgram of total RNA per sample was reverse-transcribed into cDNA using SuperScript III Reverse Transcriptase (Invitrogen). qPCR was carried out in triplicates using Power SYBR Green PCR Master Mix (Applied Biosystems). Gene-specific, intron-spanning primer pairs for qPCR were designed for each target, and β-actin gene was used as housekeeping control for data normalization and qPCR analysis, using the delta-delta Ct method. Primer list is available in Supplementary Table S1.
Western blotting
Thyroid tumors were surgically resected under the microscope to ensure the removal of surrounding tissues and immediately placed in liquid nitrogen for preservation until protein extraction. Cultured cells were harvested with 0.05% trypsin/0.02% EDTA solution, and cell pellets were washed with cold PBS. Proteins were extracted using RIPA buffer (EMD Millipore) supplemented with protease and phosphatase inhibitors as per the manufacturer’s instructions. Protein concentrations were estimated by the BCA Kit (Thermo Fisher Scientific) on a microplate reader (SpectraMax M5). Comparable amounts of proteins were subjected to SDS-PAGE using NuPAGE 4% to 12% Bis–Tris gradient gels (Invitrogen) and were transferred to PVDF membranes. Following overnight incubation with the primary antibody at 4°C, membranes were incubated with goat anti-rabbit or goat anti-mouse secondary antibodies coupled to horseradish peroxidase (HRP) for 1 hour at room temperature. Chemoluminescence was detected using Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific) and visualized on a ChemiDoc equipment. The following primary antibodies were used: phospho-Erk (CST, catalog no. 4370), total Erk (CST, catalog no. 4695), phospho-Akt (at Ser473, CST, catalog no. 4060), total Akt (CST, catalog no. 4691), PI3 Kinase p85 (CST, catalog no. 4292), phospho-NF-κB p65 (at Ser536, CST, catalog no. 3033), and vinculin (CST, catalog no.13901). Quantification of Western blot bands was done using ImageJ Fiji (28) and normalizing phosphoproteins to loading controls.
In situ hybridization of single RNA molecules by RNAscope
In situ identification of messenger RNA (mRNA) molecules at single-cell resolution on mouse thyroid specimens was performed in conjunction with Harvard Medical School Neurobiology Imaging Facility using RNAscope. We evaluated a total of 20 murine thyroid specimens harvested from 20-week animals representing the following genotypes: wild-type (n = 2), BrafV600E (n = 5), BrafV600E+Tert−123C>T (n = 8), and BrafV600E+K5-Tert (n = 5). These thyroid specimens were FFPE-embedded and mounted on slides, ensuring that different genotypes were included on the same slide, and subsequently processed for RNAscope. We followed the standard protocol for RNAscope Multiplex Fluorescent Reagent Kit v2 assay (catalog no. 323100, ACD-BioTechne). Tert mRNA was detected using the specific RNAscope fluorescent probe catalog no. 313441. Images were acquired on an Olympus VS120 Whole Slide Scanner with Hamamatsu Orca R4 camera and 40×0.95NA objective. RNAscope image analysis was performed using QuPath V 0.3.2 (26). Image files in *.ets format were uploaded using the Bio-Formats builder. Tumor boundaries were outlined manually, and individual cells were detected via DAPI staining and counted using the automated cell detection algorithm. Subcellular detection was performed to identify RNA transcripts in the Cy5 or TRITC channels. The thresholds <1,000 and <8,000 were input for Cy5 and TRITC, respectively, and the size parameters of 4.5 μm2 average area per spot was input. Quantification of Tert mRNAs across specimens was performed by a single operator (CEMT) who was blinded to the tumor genotypes. Qupath-generated data on the number of RNAscope spots per cell was exported into Excel and genotypes were only unblinded for comparison between genotypes. Each specimen was mounted in duplicate on each slide and percentages of cells expressing Tert transcripts were averaged. Data were presented as the percentage of all cells within each specimen expressing 1, 2, 3, 4, and 5+ (range 5–10) fluorescent spots, representing single-cell level Tert mRNAs.
Thyroid ultrasonic imaging
Mice were anesthetized by inhalation of isoflurane. Thyroid tumors were imaged using Vevo 770 High-Resolution In Vivo Micro-Imaging System (VisualSonics). Aqueous ultrasonic gel was applied to the denuded skin overlying the thyroid gland prior to placement of the ultrasonic transducer. Volume was calculated by manually tracing the margin of the tumor every 250 μmol/L using the instrument software. Genotypes were blinded for volume analysis. Volume calculation for all tumors was performed by the same person (IL) to avoid interoperator bias.
Drug administration in vivo
A cohort of 24 animals, including eight BrafV600E, eight BrafV600E+Tert−123C>T, and eight BrafV600E+K5-Tert mice were ages for 20 weeks and randomized into two balanced groups within each genotype: “vehicle” and “treated” (n = 4 per genotype and condition). The treated group was administered a combination of the Raf inhibitor dabrafenib (GSK2118436, Selleckchem) at 30 mg/kg plus the Mek inhibitor trametinib (GSK1120212, Selleckchem) at 3 mg/kg. Drugs were prepared in 0.5% HPMC, 0.2% Tween80, in H2O pH 8.0, and administered daily by oral gavage. The vehicle cohort was given the drug solvent by oral gavage under the same conditions. Animals were treated daily for 12 days, with the weekend off drugs. Thyroid volumes were monitored by ultrasound on days 1 and 12. Mouse weight was recorded on days 2, 5, 8, 10, and 12. All animals were sacrificed on day 12, within 2 hours from the last dose. Thyroid glands were collected under the microscope to ensure the removal of surrounding tissues. Tumors were fixed and paraffin-embedded for histologic and IHC analyses.
Telomere length measurement
Telomere length from mouse thyroid primary tumors was measured via quantitative telomere FISH (Q-FISH). Briefly, paraffin-embedded tumor sections from 20-week-old animals with various genotypes were deparaffinized and fixed with 4% formaldehyde, followed by digestion with pepsin/HCl and a second fixation with 4% formaldehyde. Slides were dehydrated with increasing concentrations of ethanol (70%, 90%, 100%) and incubated with the telomeric peptide nucleic acid (PNA) probe (TTAGGG) labeled with Cy3 (Panagene) at 85°C for 3 minutes followed by 2 hours at room temperature in a wet chamber. The slides were extensively washed with 50% formamide and 0.08% TBS-Tween 20. After washing, slides were stained with DAPI (0.2 μg/mL) and dehydrated in a 70% to 90% to 100% ethanol series. Dried samples were finally mounted with VECTASHIELD mounting media (Vector Laboratories). Telomere length analysis is based on the specific and stable hybridization of the PNA with the telomeric region; the intensity of this PNA is directly related to telomere length allowing the measurement of telomeres at each individual chromosome end. Immunofluorescence images were obtained using a confocal laser-scanning microscope (Leica TSC SP8) using a Plan Apo 63Å-1.40 NA oil immersion objective (HCX). Maximal projection of z-stack images was generated using advanced fluorescence LAS AF 2.7.3.9723 software and analyzed with HALO image analysis platform (Indica Labs). The DAPI images were used to detect telomeric signals inside each nucleus. For each sample evaluation, four representative areas from each tumor were imaged for an unbiased study of telomere length. Telomeric length was calculated as mean intensity per nucleus, adjusted by the total of telomeric foci (to account for potential biases in nuclei segmentation), and expressed in arbitrary fluorescence units (auf).
We used an alternative qPCR-based method to measure telomere length from an independent series of mouse thyroid primary tumors and tumor-derived cell lines as described previously (29). Briefly, thyroid tumors from each genotype from 20-week-old animals were flash-frozen. Frozen tumors were mechanically homogenized in DNeasy (Qiagen) Extraction Kit buffer, and genomic DNA (gDNA) was isolated following the manufacturer instructions. For cell line experiments, cells derived from age-matched animals were grown until a similar passage, and gDNA was isolated as explained above. Up to 30 ng of gDNAs were used in equal amounts for qPCR-based measurement of telomeres, using primers for mouse telomeric region and 36b4 as the housekeeping gene, as reported previously (29). Reactions were run in quadruplicate wells, and results were normalized across two independent experiments.
Statistical analysis and data graphing
GraphPad Prism software (version 9.3.1) was used for statistical analyses and graphics generation. Data from qPCR, ultrasonic imaging, quantification of IHC, and RNAscope images are represented as either mean ± SD, or median ± IQR, depending on whether distributions passed normality tests. Consequently, P values for pairwise comparisons were calculated using either unpaired two-tailed student t tests or Mann-Whitney U tests, as noted. Three-group comparisons were performed by either ANOVA or Kruskal–Wallis tests. Conceptual figures describing experimental approaches were created with BioRender.com.
Data availability
The RNA-seq data generated in this study are publicly available in Gene Expression Omnibus (GEO) at GSE227539.
Results
Mutation at mouse tert c.−123C>T is conserved and mimics the transcriptional effects of human TERT c.−124C>T
We first checked whether the TERT proximal promoter region, where hotspot mutations occur in human tumors, was conserved in the mouse. Pairwise sequence alignments showed that the human TERT site around −124C>T mutation, which accounts for ~80% of TPMs in human tumors (3), was conserved in murine sequences. We identified a mouse promoter residue at −123C (from the ATG codon), whose flanking nucleotides align well with the human ortholog, and which upon a C>T substitution create an identical core binding sequence for ETS factors, as defined in both species (Fig 1A; Supplementary Fig S1; ref. 30). We then tested if a mouse Tert promoter construct harboring the −123C>T mutation led to increased Tert expression in luciferase assays performed in NIH-3T3 (mouse fibroblasts) and two cell lines derived from murine Braf V600E-mutant thyroid tumors. All three mouse cell lines showed a two- to three-fold increase in Tert promoter activity for the mutant promoter (“mTert −123T”) compared with wild-type (“mTert −123C”) constructs (Fig 1B). This is consistent with the effect seen in luciferase assays for human TERT −124C>T/−146C>T constructs (31), as well as with TERT expression in patients’ tumors carrying TPMs (32, 33).
Figure 1.
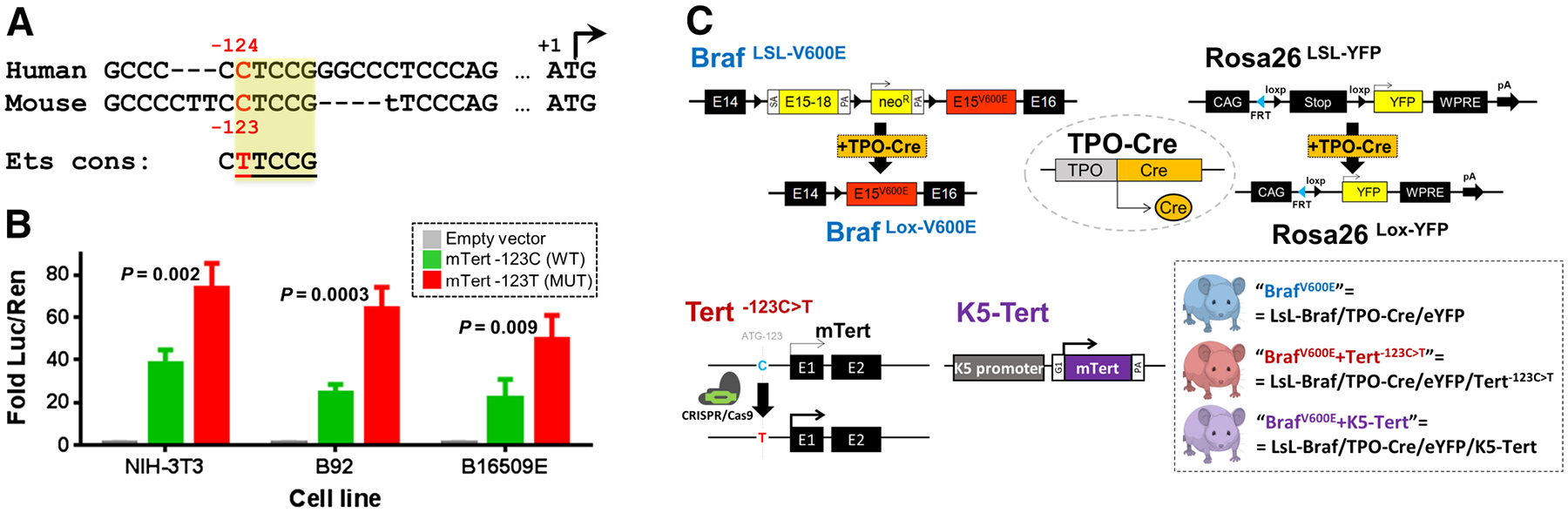
Modeling of Tert upregulation in genetically engineered mouse models with endogenous BrafV600E expression. A, Schematic representation of the sequence alignment of human TERT (top) and mouse Tert promoter (bottom) sequences. A single C>T transition at the conserved human c.−124C and mouse c.−123C loci generates a consensus binding motif for Ets transcription factors (“Ets cons”). B, Tert promoter-driven luciferase expression, normalized by renilla (“Luc/Ren”), in mouse cell lines using Tert wild-type (mTert −123C, green) and mutant (mTert −123T, red) constructs. C, Genetic schema of the mouse models: Top, Thyroid-specific expression of Cre recombinase [driven by thyroid peroxidase (TPO) in TPO-Cre constructs] substitutes exon 15 of WT Braf by V600E mutant allele, resulting in endogenous expression of Braf oncoprotein (66). Cre-mediated excision of stop cassette also enables YFP expression in thyroid cells; Bottom: Tert−123C>T is knocked-in in the germline via CRISPR/Cas9 editing of mouse zygotes, as described (left); mouse Tert is driven by the keratin 5 (K5) promoter (middle; ref. 17); breeding of LsL-Braf, TPO-Cre/eYFP, and Tert alleles generates the three genotype combinations used in this study (right).
Generation of a Tert mutant promoter mouse model
To investigate the role of TPMs in vivo, we generated a Tert c.−123C>T mouse model by Cas9/CRISPR gene editing (“Tert−123C>T” from now on). We successfully targeted the region in mouse zygotes and obtained 10/84 F0 animals carrying the desired −123C>T mutation. Targeted NGS around the Tert promoter locus on six F0 animals confirmed the knock-in of Tert c.−123C>T mutation without off-target effects in cis in 1% to 20% of the sequencing reads (Supplementary Table S2). Off-target mutations typically consisted of single-nucleotide changes or indels around the desired residue and were bred-out in the subsequent generations. A mouse Tert c.−123C>T line without any off-target alterations was established and confirmed by direct sequencing of the offspring. We subsequently crossed Tert−123C>T animals with LSL-BrafV600E/TPO-Cre/eYFP mice (“BrafV600E”), which express endogenous levels of the Braf oncoprotein and YFP in thyroid follicular cells. In parallel, we crossed mice in which Tert expression was targeted to epithelial tissues via the keratin 5 promoter (“K5-Tert”) with the same thyroid-specific BrafV600E model (Fig. 1C). All animals were viable and born at the expected Mendelian rates.
Mice carrying BrafV600E+Tert−123C>T alleles develop advanced thyroid cancers
We monitored tumor progression in BrafV600E versus BrafV600E+Tert−123C>T animals and employed the BrafV600E+K5-Tert mice as positive controls of sustained activation of Braf and Tert oncoproteins in thyroid follicular cells. BrafV600E mice developed PTCs at 4 to 6 weeks with nearly 100% penetrance, as reported previously (24), but they hardly ever progressed to advanced disease. We first sacrificed a cohort of ~10-week-old BrafV600E and BrafV600E+Tert−123C>T animals (Fig. 2A). All Braf-mutant animals developed PTCs, characterized by the presence of papillae and specific nuclear features (Supplementary Fig S2A), and so did most of the BrafV600E+Tert−123C>T mice, except 1/12 BrafV600E+Tert−123C>T animals, which displayed a PDTC phenotype (Table 1). We then monitored a larger cohort of mice for an extended period and confirmed that a subset of BrafV600E+Tert−123C>T animals progressed to histologically more aggressive tumors at around 20 weeks. As shown in Table 1 and Fig. 2B and C, none of the BrafV600E animals in this age range developed PDTCs (0/12), whereas 7/24 (29.2%) BrafV600E+Tert−123C>T mice did (chi-squared P-value = 0.0371). BrafV600E+Tert−123C>T tumors typically preserved a PTC component and displayed areas of PDTC (Fig. 2B). A subset of these BrafV600E+Tert−123C>T tumors showed characteristics compatible with the Turin definition of PDTC (Fig. 2B, second panel), whereas most fulfilled the criteria for the “differentiated high-grade thyroid carcinoma” (DHGTC) category (Fig. 2B, third panel; refs. 34–36). Interestingly, a similar phenotype was observed in an age-matched cohort of the Tert overexpression transgenic model (BrafV600E+K5-Tert): 4 of 11 (36.4%) developed PDTCs (Fig. 2B, bottom, and C). We note dother histologic features in these tumors. A nonsignificant trend in muscle invasion was observed in 25.0%, 37.5%, and 45.5% of BrafV600E, BrafV600E+Tert−123C>T, and BrafV600E+K5-Tert animals, respectively. In Tert-upregulated tumors, areas of solid growth correlated with proliferating cells and the presence of mitotic figures (Supplementary Fig 2B). These animals tended to have larger tumors and diminished survival, but differences did not reach statistical significance (Supplementary Figs. S2C and S2D). The presence of inflammation and calcifications were observed only in 20-week BrafV600E+Tert−123C>T and BrafV600E+K5-Tert groups.
Figure 2.
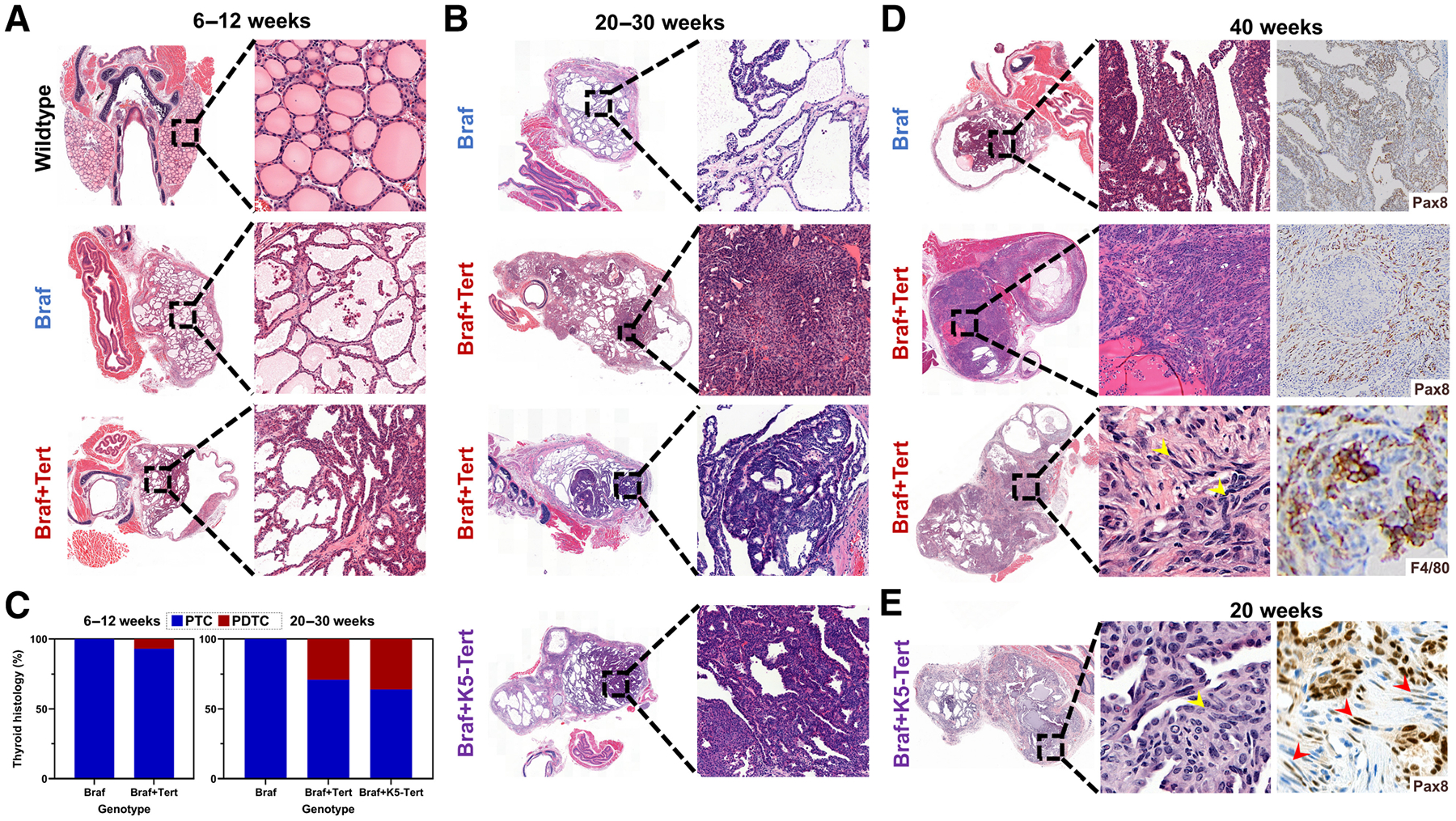
Histologic analysis of the Braf+Tert mouse models. Representative H&E-stained thyroid sections of BrafV600E, BrafV600E+Tert−123C>T, and BrafV600E+K5-Tert mice at (A) 6 to 12 weeks (all displaying PTC phenotypes); and (B) 20–30 weeks. In B, the second panel represents a PDTC fulfilling the Turin criteria (solid growth and high mitotic index), whereas the third and fourth panels show tumors within the newly defined DHGTC category. C, Percentage of animals with the indicated genotypes showing PTC or PDTC/DHGTC at 6 to 12 weeks (left) and 20 to 30 weeks (right). D, Examples of 40-week BrafV600E (top) and BrafV600E+Tert−123C>T tumors (middle and bottom). Some BrafV600E animals at this age develop signs of PDTC transformation (top) and typically retain Pax8 expression (top, right). Only BrafV600E+Tert−123C>T animals of this age developed tumors with ATC-like features within a PTC component (middle and bottom), overall loss of Pax8 with spindle cells retaining Pax8 positivity (middle, right), spindle cells (bottom, center, yellow arrows), and positivity for F4/80 stain, denoting macrophage infiltration (bottom, right). E, Example of a 20-week BrafV600E+K5-Tert tumor with similar features than tumors from E, that is, ATC-like areas, spindle cells (yellow arrowhead) with focal positivity for Pax8 (spindle cells with red arrowheads). “Braf+Tert” and “Braf+K5-Tert” labels denote “BrafV600E+Tert−123C>T” and “BrafV600E+keratin 5-driven Tert” genotypes, respectively.
Table 1.
Histologic features of mouse thyroid tumors.
| 6–12 weeks | 20–30 weeks | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Genotype | Age, avg weeks | PTC, n (%) | PDTC, n (%) | P value | Total, n | Age, avg weeks | PTC, n (%) | PDTC, n (%) | P value | Total, n |
| BrafV600E | 9.9 | 8 (100.0) | 0 (0.0) | 0.4215 | 8 | 22.2 | 12 (100.0) | 0 (0.0) | — | 12 |
| BrafV600E+Tert−123C>T | 10.2 | 12 (93.3) | 1 (7.7) | 13 | 22.0 | 17 (70.8) | 7 (29.2) | 0.0371 | 24 | |
| BrafV600E+K5-Tert | N/A | N/A | N/A | N/A | N/A | 23.7 | 7 (63.6) | 4 (36.4) | 0.0765 | 11 |
Note: P values are derived from two-sided chi-square tests for the indicated groups, employing BrafV600E animals as controls.
We then followed a small cohort of BrafV600E and BrafV600E+Tert−123C>T animals up to 40 weeks. At that time point, a subset of mice from both groups showed more advanced phenotypes; however, only BrafV600E+Tert−123C>T mice displayed ATC-like features, including pleomorphic or spindle nuclei, loss of positivity for Pax8, and macrophage infiltration (Fig 2D, middle and bottom). These same features were also observed in some 20-week-old Braf+K5-Tert mice, suggesting that high level expression of Tert might cooperate with oncogenic Braf to promote PTC-to-ATC transformation (Fig 2E). Tumor infiltration by myeloid cells was observed in 40-week-old BrafV600E and BrafV600E+Tert−123C>T animals; however, a subset of BrafV600E+Tert−123C>T tumors, displayed the highest proportion of cells staining positive for markers of M2-like macrophages, although these observations did not reach statistical significance. They suggest, however, that telomerase upregulation in murine tumors mimics a hallmark of human ATCs (Supplementary Figs. S2E and S2F).
Overall, Tert upregulation accelerated Braf-driven PTC progression towards more advanced forms, as observed in patients’ tumors carrying this genetic combination. Our results suggested that 20 weeks was an optimal window to evaluate Tert-mediated changes on thyroid cancer progression, so we used this time point for all subsequent experiments.
BrafV600E+Tert−123C>T tumors ultimately acquire the ability to increase Tert transcription, but Tert upregulation displays temporal and intratumoral heterogeneity
We then tested whether the engineering of a Tert−123C>T mutation led to an increase in Tert transcription in vivo. Tert mRNA levels of YFP-sorted cells from thyroid tumors of 10-week BrafV600E+Tert−123C>T animals were similar from those from BrafV600E mice, which, unlike human tissues, retain baseline Tert expression (21). However, at 20 weeks, Tert mRNA levels of a subset of BrafV600E+Tert−123C>T increased and were higher than 10-week-old specimens with the same genotype (Fig. 3A, P = 0.006). These suggest that Tert enhanced expression tracks with progression from PTC to PDTC, probably because cells with higher Tert levels are clonally selected over time. To further dissect the heterogeneity of Tert expression in animals engineered for the promoter mutation, we evaluated the levels of Tert transcripts in situ at single-cell resolution via RNAscope analysis of 20-week thyroid tumors from each genotype. A subset of thyroid cells from wild-type and BrafV600E mice maintained some Tert expression, as expected. Not surprisingly, Braf+K5-Tert animals showed sustained, overexpression of Tert mRNAs in about half of thyroid cells (Fig. 3B). Interestingly, the average number of Tert transcripts per cell showed a nonsignificant trend towards higher values in BrafV600E+Tert−123C>T tumors compared with BrafV600E for cells displaying high Tert transcription (Fig 3C, see “3”, “4”, and “+ ” transcript categories). In addition, the distribution of Tert transcripts in BrafV600E+Tert−123C>T animals showed an intratumoral heterogeneity, which was not observed in BrafV600 tumors. Crucially, areas of solid growth identified as PDTC regions typically correlated with a much higher number of fluorescent Tert mRNAs (Supplementary Figs. S3A–S3L; compare D vs. F).
Figure 3.
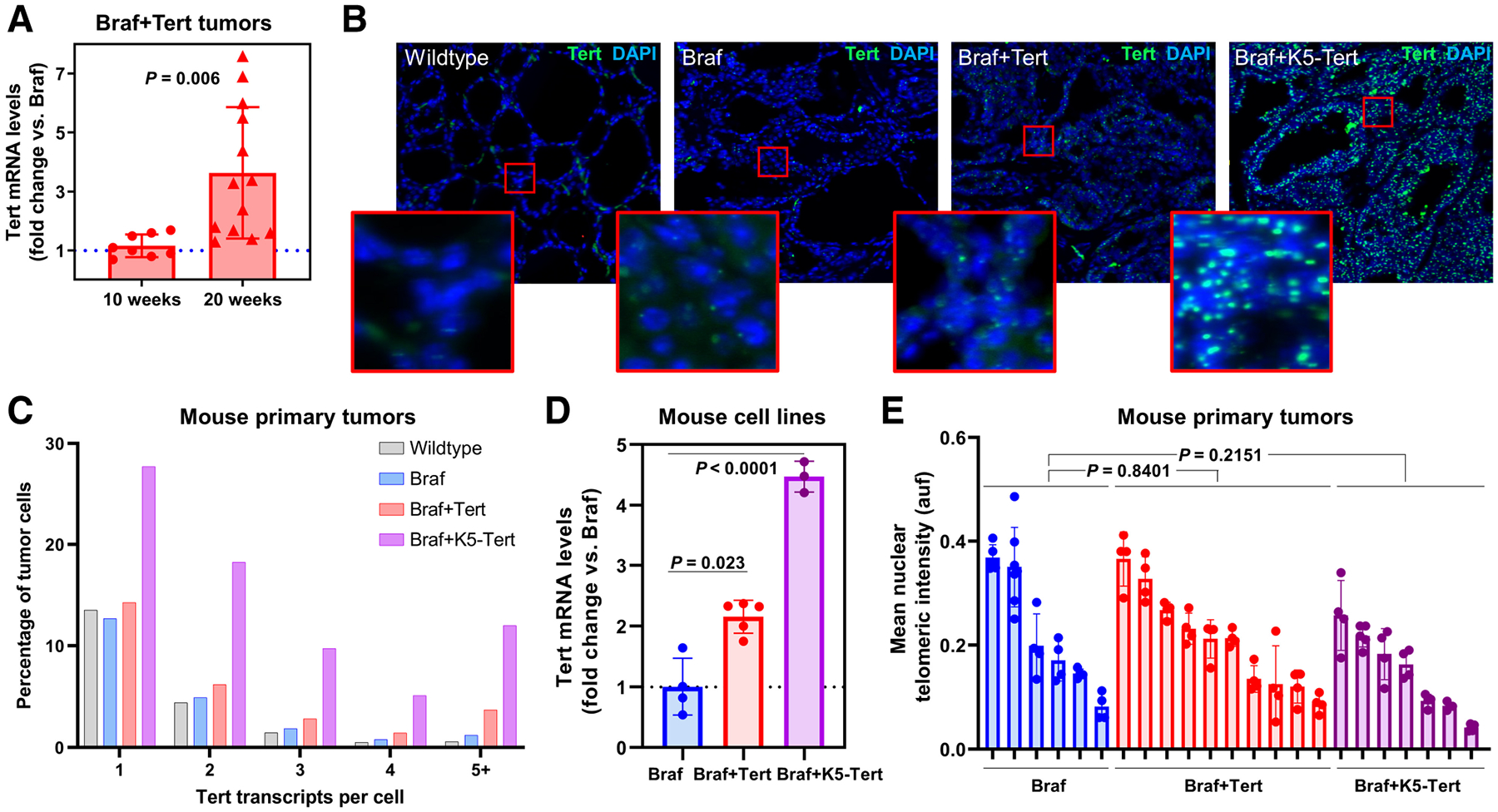
Characteristics of Tert re-expression in telomerase-upregulated thyroid cancers. A, Relative levels of Tert transcription in YFP-sorted cells isolated from BrafV600E+Tert−123C>T mouse tumors, showing an increase in Tert mRNA levels for specimens collected at 20 weeks versus 10 weeks. Each red point represents a tumor collected from a different animal at the indicated age. Results are expressed as fold change compared with the Tert baseline expression of BrafV600E tumors from the same ages (dotted blue line). B, Representative examples of thyroid specimens from 20-week animals with the indicated genotypes subjected to RNAscope to detect Tert single mRNA molecules (green dots). DAPI (blue) is used for contrast. Red squares denote areas for which higher magnifications are provided. C, Quantification of number of Tert transcripts at single-cell resolution from RNAscope data on mouse tumors with the indicated genotypes. Data are represented as percentage of cells within each tumor expressing 1, 2, 3, 4, or 5+ transcripts. Tumor boundaries were defined manually and cell detection and quantification of green fluorescent dots was performed employing the built-in automated analysis tools on QuPath, using the same detection parameters across specimens. D, Relative mRNA levels in cell lines derived from BrafV600E (n = 4), BrafV600E+Tert−123C>T (n = 5), and from BrafV600E+K5-Tert (n = 3) mouse tumors, showing that genotype-dependent increases in Tert transcription are maintained in vitro. E, Mean nuclear telomeric intensity, measured in arbitrary fluorescence units (auf) via Q-FISH (quantitative FISH) in BrafV600E (n = 6), BrafV600E+Tert−123C>T (n = 10) and BrafV600E+K5-Tert (n = 7) mouse primary tumors derived from 20-week animals. “Braf+Tert” and “Braf+K5-Tert” labels denote “BrafV600E+Tert−123C>T” and “BrafV600E+keratin 5-driven Tert” genotypes, respectively.
We also derived primary cultures from these tumors, which grew well over multiple passages. Interestingly, these in vitro models maintained their original genotype-dependent differences in Tert expression: BrafV600E+Tert−123C>T and Braf+K5-Tert showed a 2.1-fold (P = 0.023) and 4.5-fold (P < 0.0001) increase in Tert transcription, respectively, versus Braf cells (Fig. 3D). These suggest that BrafV600E+Tert−123C>T lines select for cells with increased Tert mRNA levels during in vitro immortalization.
Telomere length is not altered in tumors with Tert upregulation
We then explored whether engineered re-expression of Tert, either via c.−123C>T mutation or keratin 5 promoter-mediated transcription, affected telomere length in mouse primary tumors. Q-FISH on 20-week, age-matched tumors showed mean ± SD nuclear telomeric intensities of 0.22 ± 0.11, 0.21 ± 0.09, and 0.15 ± 0.08 in BrafV600E, BrafV600E+Tert−123C>T, BrafV600E+K5-Tert thyroid tumors (ANOVA P-value = 0.3415; Fig 3E). Compared with BrafV600E, neither BrafV600E+Tert−123C>T nor BrafV600E+K5-Tert showed differences in telomere length (Student t P-values = 0.8401 and 0.2151, respectively). These suggested that telomere attrition is likely not a critical factor in the observed murine Tert-driven thyroid cancer progression. Our findings are in line with previous reports in which telomerase overexpression induced cancers without critical changes in telomere length (18–20), as well as the known fact that mice have unusually long telomeres compared with humans (22).
We also evaluated telomere length in an independent group of flash-frozen, 20-week mouse tumors via qPCR methods. Relative median ± IQR telomere length in BrafV600E, BrafV600E+Tert−123C>T, and BrafV600E+K5-Tert thyroid tumors was 5.08 ± 1.78, 5.13 ± 1.05, and 4.78 ± 2.31, respectively (Kruskal–Wallis P-value = 0.8087; Supplementary Fig. S3M). In addition, we measured telomere lengths of immortalized cell lines that we derived from these murine tumors, confirming the lack of differences in specimens from each genotype evaluated at a similar passage: median ± IQR was 3.20 ± 0.40, 3.20 ± 2.75, and 2.40 ± 0.80, in BrafV600E, BrafV600E+Tert−123C>T, and BrafV600E+K5-Tert cell lines, respectively (Kruskal–Wallis P-value = 0.3929; Supplementary Fig. S3N). Overall, these findings prompted us to look for no-telomeric effects as the primary mechanisms driving the observed Tert-induced thyroid cancer progression in our models.
Thyroid tumors with telomerase upregulation display distinct transcriptomes
To evaluate whether telomerase enhanced expression impacted the transcriptome of Braf-driven cancers, we ran RNA-seq on YFP-sorted cells from primary thyroid tumors from unselected 20-week BrafV600E, BrafV600E+Tert−123C>T, and BrafV600E+K5-Tert animals (Fig. 4A). For comparison purposes, we also ran RNA-seq on YFP+thyroid cells from pooled thyroid glands from WT, Tert−123C>T, and K5-Tert animals (without oncogenic Braf activation), which, as expected, did not develop tumors. RNA-seq normalized counts for Tert were increased in pooled YFP+cells from Tert−123C>T and K5-Tert thyroids compared with WT (Fig. 4B). When comparing tumor-bearing animals, BrafV600E mice showed high baseline expression of Tert (compared with WT), suggesting that MAPK activation impacts wild-type Tert transcription. Tert constitutive upregulation was observed in BrafV600E+K5-Tert (P = 0.029, compared with BrafV600E), but not in BrafV600E+Tert−123C>T ((P 0.254; Fig 4C). The latter might be a consequence of our observation that only certain cells within the tumor engineered for Tert c.−123C>T mutation overexpress this gene (Fig. 3B and C; Supplementary Figs. S3A–S3L).
Figure 4.
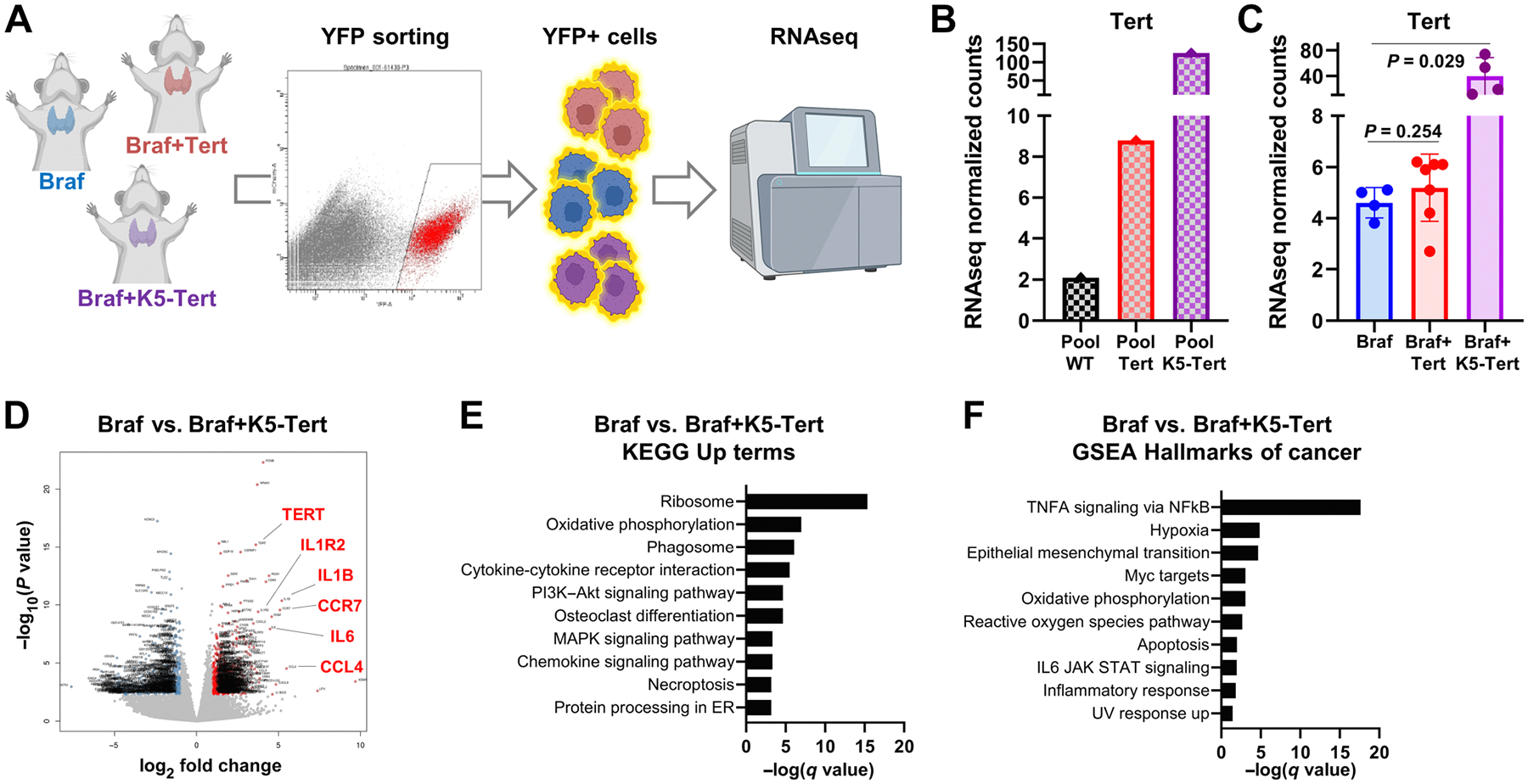
Transcriptomic characterization of telomerase-upregulated mouse thyroid tumors. A, Schematic representation of the isolation of thyroid tumor cells for RNA-seq. B, Comparison of RNA-seq normalized counts for Tert in pooled thyroid glands with the indicated genotypes. Values represent average expression levels for pooled WT (n = 25), Tert−123C>T (n = 17), and K5-Tert (n = 14) animals. C, Comparison of RNA-seq normalized counts for Tert in thyroid tumors from BrafV600E (n = 4), BrafV600E+Tert−123C>T (n = 7), and BrafV600E+K5-Tert (n = 4) animals. Each dot represents an individual mouse. D, Volcano plot showing significantly under- and overexpressed genes in BrafV600E+K5-Tert tumors compared with BrafV600E. Specific genes are indicated in red. E, Top 10 upregulated terms from the KEGG pathway database analysis for the BrafV600E versus BrafV600E+K5-Tert comparison. F, Top 10 upregulated terms from the GSEA hallmarks of cancer analysis for the BrafV600E versus BrafV600E+K5-Tert comparison.
We subsequently assessed global gene expression changes in these specimens. Unsupervised clustering classified tumors primarily based on their genotypes (Supplementary Fig. S4A). Bulk RNA-seq identified only a small subset of genes differentially expressed in the BrafV600E+Tert−123C>T models compared with BrafV600E (Supplementary Table S3), as changes were likely diluted by the observed intratumoral heterogeneity of Tert upregulation (Fig. 3B and C). The amount and extent of differences were more pronounced for BrafV600E+K5-Tert than for BrafV600E+Tert−123C>T, so we primarily used keratin 5-mediated Tert overexpression models to inform our subsequent analyses (Fig 4D; Supplementary Table S4).
BrafV600E+K5-Tert tumors showed distinct transcriptomes, with multiple under- and overexpressed genes, compared with their BrafV600E counterparts (Fig. 4D). In addition to Tert, genes encoding several cytokines, chemokines, and some of their receptors were overexpressed in these specimens (see highlighted genes in Fig. 4D). Pathway analysis confirmed these observations: cytokine and chemokine signaling were among the top overexpressed terms in BrafV600E+K5-Tert tumors when employing the KEGG pathway database (Fig. 4E; Supplementary Table S5). In addition, the top two upregulated terms by GO analysis of this same dataset were “immune system process” (q-value <1E−60) and “inflammatory response” (q-value <1E−40). The paucity of transcriptomic changes observed in BrafV600E+Tert−123C>T by bulk RNA-seq prevented us from running in-depth pathway analysis for these specimens. However, it is worth noting that the top two KEGG upregulated terms (employing unadjusted P values) in BrafV600E+Tert−123C>T, compared with BrafV600E, were “chemokine signaling” and “cytokine-cytokine receptor interaction,” pointing to a similar signaling hub regardless of the mechanisms by which we induced Tert upregulation (Supplementary Fig. 4B; Supplementary Table S6). Seeking to more precisely pin down the signaling pathways by which these cytokines might be relevant in thyroid tumors, we applied the GSEA hallmarks of cancer database analysis to our RNA-seq dataset. As shown in Fig. 4F, genes belonging to the “tumor necrosis alpha (TNFA) signaling via NF-κB” category were the most significantly overexpressed in BrafV600E+K5-Tert tumors (full details in Supplementary Table S7). To confirm this result, we assessed the levels of phospho-p65, a key component of the TNFA-mediated canonical NF-κB signaling (37), in unselected thyroid tumors from BrafV600E, BrafV600E+Tert−123C>T, and BrafV600E+K5-Tert 20-week animals. Western blotting showed higher levels of phospho-p65 in telomerase-overexpressed extracts, particularly BrafV600E+Tert−123C>T, suggesting this pathway is also overactivated in these specimens (Fig. 5A; Supplementary Fig. 4C). Overall, RNA-seq unveiled that key mediators of inflammation, likely in crosstalk with the tumor immune infiltrate, may play a role in Tert-induced thyroid cancer progression.
Figure 5.
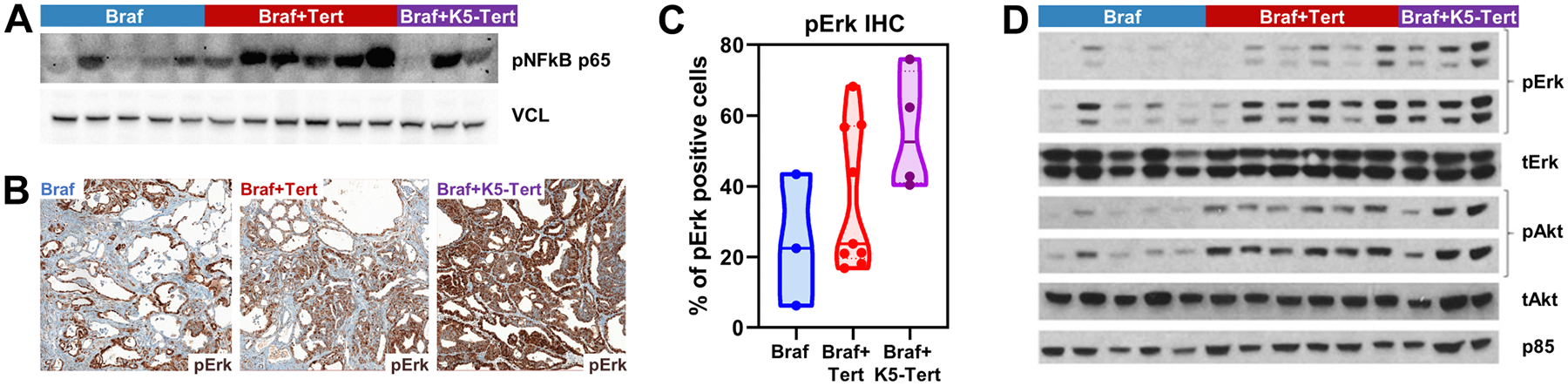
Validation of telomerase-upregulated pathways. A, Western blot analysis using protein extracts from thyroid tumors from 20-week-old animals with the indicated genotypes for phospho-NF-κB p65 and vinculin (VCL, loading control). B, Representative images from phospho-Erk (pErk) IHC performed in mouse tumors from 20-week-old animals with the indicated genotypes. C, Quantification of the percentage of tumor cells staining positive for pErk in mice with the indicated genotypes. D, Western blot analysis using protein extracts from thyroid tumors from 20-week-old animals with the indicated genotypes for phospho- and total Erk (MAPK pathway) and Akt (PI3K effector), as well as p85 (loading control; only protein that was reblotted on stripped membranes). “Braf+Tert” and “Braf+K5-Tert” labels denote “BrafV600E+Tert−123C>T” and “BrafV600E+keratin 5-driven Tert” genotypes, respectively.
In addition, pathway analysis showed that several bona fide signaling hubs in thyroid cancer pathogenesis, including the MAPK and PI3K/AKT pathways, ranked high as hyperactivated pathways in BrafV600E+K5-Tert tumors (Fig. 4F). Despite some inter-mouse variability, MAPK overactivation was confirmed via IHC analysis for phospho-Erk (pErk) of tumors from 20-week animals, showing that, compared with BrafV600E, BrafV600E+K5-Tert have higher proportion of tumor cells staining positive for pErk (P = 0.0673, Fig. 5B and C). Western blotting with protein extracts from 20-week mouse tumors from each genotype further showed increased phosphorylation levels for Erk and Akt in Tert-engineered tumors (Fig 5D; Supplementary Fig. S4D). Because MAPK pathway activation in human and mouse thyroid tumors inversely correlate with transcriptional programs of thyroid differentiation, we evaluated whether key thyroid transcription factors and iodine metabolism genes were suppressed across different genotypes in our RNA-seq data. In general, thyroid transcription factors Pax8, Nkx2-1, and Foxe1, and iodine metabolism genes Tshr, Slc5a5, and Tpo, tended to be reduced in BrafV600E+K5-Tert cells, which showed the highest MAPK overactivation, but not in BrafV600E+Tert−123C>T tumors (Supplementary Fig. S4E).
Because MAPK pathway overactivation was detected in tumors with high telomerase levels, we evaluated whether in vivo treatment with dabrafenib (Raf inhibitor) plus trametinib (Mek inhibitor) impacted tumor growth (Supplementary Fig. S5A). Sustained treatment for 12 days was well tolerated, and body weight of treated mice did not vary over time. Compared with vehicle, dabrafenib plus trametinib treatment reduced tumor volume in all three groups (BrafV600E, BrafV600E+Tert−123C>T, and BrafV600E+K5-Tert; Supplementary Figs. S5B and S5C). Overall, despite the observed higher MAPK output of telomerase-upregulated thyroid tumors, combination treatment with dabrafenib plus trametinib was effective at reducing tumor volume across all evaluated mouse models.
Discussion
Telomerase reactivation occurs in 90% of human tumors and is considered a hallmark of cancer (38, 39). The discovery of TPM occurring in multiple cancers, and the fact that TPMs are more prevalent in metastatic/advanced forms of the disease, have heightened interest in studying this oncogene. Accurate experimental models to mechanistically assess the biological consequences of telomerase upregulation in a neoplastic background are scarce. Here we studied how enhanced expression of telomerase contributes to thyroid cancer progression in BrafV600E-driven murine tumors. To this end, we edited a C>T change in the mouse Tert promoter (Tert−123C>T), which we believe is the orthologue nucleotide of human TERT−124C>T, and also employed a previously generated model of Tert overexpression (K5-Tert). Our experiments showed that Tert upregulation likely cooperates with oncogenic Braf to promote advanced thyroid cancers, which was associated with activation of oncogenic pathways unrelated to telomere maintenance. Tert−123C>T mice were born at Mendelian rates comparable with Tert wildtype littermates and displayed no cancer phenotypes, consistent with the observation that this promoter variant is found in families that have no abnormalities other than a higher frequency of metastatic melanoma (1).
We and others have previously reported that TPMs are enriched in advanced thyroid tumors, that is, PDTC and ATC (4, 5). Furthermore, in the context of constitutive MAPK pathway activation, they are predictors of poorer outcomes within each subtype. The presence of TPMs in combination with BRAFV600E is associated with more aggressive PTCs, including higher mortality rates (9, 40, 41). In advanced disease, TPMs correlate with the presence of distant metastases in PDTC and with diminished survival in patients with ATC (13). This is particularly relevant in thyroid cancers, which are genetically simple tumors that remain exquisitely dependent on MAPK activation, making them good models to study MAPK–TERT contributions to cancer biology. The models described here could thus be useful tools to determine the cellular processes by which Braf-driven clones, upon the upregulation of Tert, evolve to more aggressive and less differentiated cancers.
Our in vivo experiments suggested that Tert upregulation, regardless of its mechanism of overexpression, induced BrafV600E-driven PTCs to progress to PDTC/DHGTC. This phenomenon was observed starting at 20 weeks, when no BrafV600E animals, but around one-third of BrafV600E+Tert−123C>T and BrafV600E+K5-Tert mice showed advanced phenotypes. The variability in Tert-driven thyroid cancer progression observed in our models remains to be fully understood: it is unclear, at this point, whether thyroid dedifferentiation is a stochastic process or follows a defined pattern of events. In addition, a subset of older BrafV600E+Tert−123C>T, but none of the BrafV600E group, displayed ATC-like features. These included a strong infiltration of the tumor microenvironment by myeloid cells, particularly M2-macrophages and myeloid-derived suppressor cells (MDSC). This is relevant because human ATCs are known to be infiltrated by tumor-promoting macrophages and MDSCs (42–44). Given that BRAFV600E+TERT−124C>T is the most common genetic combination in ATC (13), our findings suggest that telomerase overexpression is a stepping stone towards the progressive immunosuppression that tracks with PTC-to-ATC transformation (45). In any case, some of the pathways that we identified to be activated in murine thyroid tumor cells upon Tert upregulation (see next paragraphs) suggest a continuous crosstalk between cancer cells and components of the tumor immune milieu.
Another feature of these mouse models that conveys potentially important translational consequences is the increased activation of BrafV600E-induced MAPK signaling, which we observed in telomerase-upregulated tumors. The MAPK pathway is a central signaling hub in both PTC and PDTC/ATC. The use of dabrafenib (RAF inhibitor) plus trametinib (MEK inhibitor) in ATCs harboring BRAFV600E mutations remains one of the few effective therapeutic strategies in these otherwise highly lethal cancers (46). Our in vivo treatment with dabrafenib plus trametinib showed comparable tumor volume reductions in telomerase-upregulated specimens. The relatively small number of animals per group and condition was insufficient for robust statistical association. Overall, future work using these and other models, combined with clinical trials with extensive genomic characterization, might ultimately help refine the specifics of MAPK blockade in patients with ATC, given that most of their tumors harbor TPMs. Of note, interactions between TERT and MAPK pathway effectors have been previously reported in other tumors (47).
We observed heterogeneity in Tert re-expression in BrafV600E+Tert−123C>T tumors, in line with reports showing intermittent expression of TERT in human cancer cells (48). A much more granular evaluation of promoter binding preferences around the mouse Tert promoter would be needed to unveil these processes, which are likely related with chromatin accessibility, histone modifications and/or differential binding of transcriptions factors at the Tert locus. In this regard, Tert−123C>T created a consensus binding sequence for transcription factors of the ETS (“E26 transformation specific”) family which was identical to the one generated by TERT−124C>T in humans (Fig. 1A). The ETS family is a diverse group of proteins, and the specific factor(s) influencing transcription from the TERT mutant promoter in thyroid cancers remain to be fully elucidated (49–52). In addition, MAPK activation is also known to control TERT transcription (52–54). Overall, although these studies are beyond the scope of this paper, we believe that the BrafV600E+Tert−123C>T model can be leveraged for future work on Tert mutant promoter regulation. The ease at generating tumors and deriving cell lines from these animals should pave the way towards this goal, but it remains to be established which specific Ets proteins control expression from Tert c.−123C>T mutant promoter, and whether those Ets factors show the same promiscuity that was reported in TERT mutant human thyroid cancer cells (49–52). Of note, despite the intratumoral heterogeneity in Tert expression, cell lines derived from these tumors maintained a genotype-dependent increase in Tert transcription, which might reflect a selection process by which cells expressing higher levels of Tert are favored over time.
We acknowledge that mouse models are not ideal settings to study the canonical role of telomerase biology in cancer, that is, telomere maintenance. This limitation has been extensively reported in the literature and relies on two features of mouse cells which do not occur in humans: having longer telomeres and retaining some degree of telomerase expression in their adult tissues (21, 22). We do not rule out the possibility of telomere-related effects playing a role in thyroid cancer progression in human tumors, as recently suggested (33). However, here we demonstrate that Tert enhanced expression leads to thyroid cancer progression and upregulation of non-telomeric pathways. Indeed, the demonstration that telomerase upregulation causes disease progression in mice further substantiates the evidence that noncanonical effects of Tert play an important role in this process. Our RNA-seq-derived pathway analyses support a role for cytokines and chemokines produced by Tert-activated thyroid cancer cells, probably in response to signals from the tumor microenvironment. Signaling of these molecules via the canonical (i.e., TNFA-activated) NF-κB pathway, a key node linking inflammation, immune-related effects and cancer, was particularly enriched in our analysis. Inhibition of the NF-κB/TNFA signaling in human thyroid cancer cell lines, most of which are now known to harbor TPMs (55), has been shown to impact cell proliferation and invasion (56). Interestingly, our results in murine models recapitulate observations reported in human cancers, that is, telomerase controlling NF-κB-dependent transcriptional programs, and NF-κB in turn determining TERT nuclear localization and influencing transcription from the mutant TERT promoter, in what was described as a feed-forward mechanism (57–59). Our observations of telomerase-mediated increases in chemokine/cytokine-related pathways remain to be validated in human thyroid cancers. Of note, a re-analysis of TCGA RNA-seq data from human PTCs showed that chemokine and cytokine signaling ranked on top of the overexpressed pathways in BRAFV600E-mutant PTCs with TERT re-expression (vs. their BRAFV600E counterparts; ref. 51).
Tert−123C>T constitutes the first attempt at engineering a promoter mutation in a murine Tert noncoding locus. To our knowledge, there had not been in vivo studies of TPMs in genetically engineered mouse models. The closest effort is a recent publication in which human stem cells were engineered for TERT−124C>T mutation, differentiated into neural precursors and orthotopically injected into mice to study glioblastoma in the context of EGFR overexpression and loss of CDKN2A and PTEN (60). Our results showing that murine Tert upregulation promotes cancer progression in oncogenic Braf-driven thyroid tumors, open the door at devising equivalent studies in tumors from other lineages with high prevalence of TPMs. Although some of the effects reported here might pertain exclusively to thyroid tumors, we believe that the combination of the Tert−123C>T allele with, for instance, tissue-specific drivers of melanoma or glioblastoma, such as the Tyr:NRasQ61K/INK4a−/− (61, 62) or EGFRvIII (63) mouse models, respectively, can provide hints into the telomerase-dependent biology of those cancers. Of note, although the K5-Tert model of continuous Tert overexpression is less biologically relevant, in light of our results, we argue that employing BrafV600E+K5-Tert animals can be an informative setting to study telomerase-mediated thyroid cancer progression. This statement relies on the fact that the observed increase of Tert expression in BrafV600E+K5-Tert thyroid tumor cells was about 10-fold, in contrast to a much higher (and thus less biologically accurate) increases in other epithelial cells. It is also worth noting that other mechanisms of TERT upregulation, such as TERT copy-number gains and aberrant methylation of the TERT promoter, have been identified in advanced thyroid cancers (33, 64, 65). Nevertheless, we acknowledge that both BrafV600E+Tert−123C>T and BrafV600E+K5-Tert groups showing similar cancer phenotypes, but only the latter displaying strong differences in their transcriptomes (compared with those of BrafV600E tumors) is an observation that remains to be reconciled and might be related to the sequencing of different admixtures of PTC and PDTC cells.
In conclusion, here we show that the in vivo engineering of a non-coding alteration in the mouse Tert promoter induces thyroid cancer progression. We show that Tert upregulation induces changes in the transcriptome of fluorescence-sorted cancer cells isolated from murine thyroid tumors, and point to several cellular processes, not involved in telomere elongation, that are altered in Braf- plus Tert-driven advanced thyroid cancers. Our study can shed light into the multiple mechanistic underpinnings that telomerase overexpression bears in cancer.
Supplementary Material
Implications:
Telomerase-driven cancer progression activates pathways that can be dissected and perhaps therapeutically exploited.
Acknowledgments
This research was funded by the NIH grants 1R01CA249663-02 and 5R01CA050706-31 (to J.A. Fagin), the NCI Career Transition Award, grant no. 1K22CA230381-01A1 (to I. Landa), and the Brigham and Women’s Hospital, Department of Medicine, Innovation Evergreen Fund (to I. Landa). We would like to thank Talia Gebhard from the Landa lab, and Joe Giacalone from the MSKCC Mouse Genetics Core Facility, for technical assistance. I. Landa would like to thank Luis Javier Leandro-Garcia for his scientific input and logistical support. We thank the Harvard Medical School Neurobiology Imaging Facility for technical assistance, as well as the Dana-Farber/Harvard Cancer Center, for the use of the Specialized Histopathology Core, which provided histology and immunohistochemistry service. The MSKCC Mouse Genetics Core Facility is supported by the NCI Cancer Center Support Grant P30 CA008748. The Dana-Farber/Harvard Cancer Center was supported in part by an NCI Cancer Center Support Grant No. NIH 5 P30 CA06516.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authors’ Disclosures
R. Ghossein reports grants from NIH during the conduct of the study. J.A. Fagin reports grants from NIH during the conduct of the study. No disclosures were reported by the other authors.
Footnotes
Supplementary data for this article are available at Molecular Cancer Research Online (http://mcr.aacrjournals.org/).
References
- 1.Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, et al. TERT promoter mutations in familial and sporadic melanoma. Science 2013;339:959–61. [DOI] [PubMed] [Google Scholar]
- 2.Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science 2013;339: 957–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Killela PJ, Reitman ZJ, Jiao Y, Bettegowda C, Agrawal N, Diaz LA Jr., et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc Natl Acad Sci U S A 2013;110: 6021–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Landa I, Ganly I, Chan TA, Mitsutake N, Matsuse M, Ibrahimpasic T, et al. Frequent somatic TERT promoter mutations in thyroid cancer: higher prevalence in advanced forms of the disease. J Clin Endocrinol Metab 2013;98:E1562–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu X, Bishop J, Shan Y, Pai S, Liu D, Murugan AK, et al. Highly prevalent TERT promoter mutations in aggressive thyroid cancers. Endocr Relat Cancer 2013;20: 603–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weinhold N, Jacobsen A, Schultz N, Sander C, Lee W. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat Genet 2014;46:1160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fredriksson NJ, Ny L, Nilsson JA, Larsson E. Systematic analysis of noncoding somatic mutations and gene expression alterations across 14 tumor types. Nat Genet 2014;46:1258–63. [DOI] [PubMed] [Google Scholar]
- 8.Rheinbay E, Nielsen MM, Abascal F, Wala JA, Shapira O, Tiao G, et al. Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature 2020;578: 102–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Melo M, da Rocha AG, Vinagre J, Batista R, Peixoto J, Tavares C, et al. TERT promoter mutations are a major indicator of poor outcome in differentiated thyroid carcinomas. J Clin Endocrinol Metab 2014;99:E754–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Griewank KG, Murali R, Puig-Butille JA, Schilling B, Livingstone E, Potrony M, et al. TERT promoter mutation status as an independent prognostic factor in cutaneous melanoma. J Natl Cancer Inst 2014;106:dju246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blasco MA. Telomeres and human disease: ageing, cancer and beyond. Nat Rev Genet 2005;6:611–22. [DOI] [PubMed] [Google Scholar]
- 12.Martinez P, Blasco MA. Telomeric and extra-telomeric roles for telomerase and the telomere-binding proteins. Nat Rev Cancer 2011;11:161–76. [DOI] [PubMed] [Google Scholar]
- 13.Landa I, Ibrahimpasic T, Boucai L, Sinha R, Knauf JA, Shah RH, et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J Clin Invest 2016;126:1052–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pozdeyev N, Gay L, Sokol ES, Hartmaier RJ, Deaver KE, Davis SN, et al. Genetic analysis of 779 advanced differentiated and anaplastic thyroid cancers. Clin Cancer Res 2018;24:3059–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi X, Liu R, Qu S, Zhu G, Bishop J, Liu X, et al. Association of TERT promoter mutation 1,295,228 C>T with BRAF V600E mutation, older patient age, and distant metastasis in anaplastic thyroid cancer. J Clin Endocrinol Metab 2015; 100:E632–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014;159:676–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gonzalez-Suarez E, Samper E, Ramirez A, Flores JM, Martin-Caballero J, Jorcano JL, et al. Increased epidermal tumors and increased skin wound healing in transgenic mice overexpressing the catalytic subunit of telomerase, mTERT, in basal keratinocytes. EMBO J 2001;20:2619–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Artandi SE, Alson S, Tietze MK, Sharpless NE, Ye S, Greenberg RA, et al. Constitutive telomerase expression promotes mammary carcinomas in aging mice. Proc Natl Acad Sci U S A 2002;99:8191–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Canela A, Martin-Caballero J, Flores JM, Blasco MA. Constitutive expression of tert in thymocytes leads to increased incidence and dissemination of T-cell lymphoma in Lck-Tert mice. Mol Cell Biol 2004;24:4275–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gonzalez-Suarez E, Geserick C, Flores JM, Blasco MA. Antagonistic effects of telomerase on cancer and aging in K5-mTert transgenic mice. Oncogene 2005; 24:2256–70. [DOI] [PubMed] [Google Scholar]
- 21.Prowse KR, Greider CW. Developmental and tissue-specific regulation of mouse telomerase and telomere length. Proc Natl Acad Sci U S A 1995;92:4818–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kipling D, Cooke HJ. Hypervariable ultra-long telomeres in mice. Nature 1990; 347:400–2. [DOI] [PubMed] [Google Scholar]
- 23.Pinello L, Canver MC, Hoban MD, Orkin SH, Kohn DB, Bauer DE, et al. Analyzing CRISPR genome-editing experiments with CRISPResso. Nat Biotechnol 2016;34:695–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Franco AT, Malaguarnera R, Refetoff S, Liao XH, Lundsmith E, Kimura S, et al. Thyrotrophin receptor signaling dependence of Braf-induced thyroid tumor initiation in mice. Proc Natl Acad Sci U S A 2011;108:1615–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saqcena M, Leandro-Garcia LJ, Maag JLV, Tchekmedyian V, Krishnamoorthy GP, Tamarapu PP, et al. SWI/SNF complex mutations promote thyroid tumor progression and insensitivity to redifferentiation therapies. Cancer Discov 2021; 11:1158–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bankhead P, Loughrey MB, Fernandez JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: open source software for digital pathology image analysis. Sci Rep 2017;7:16878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cornwell M, Vangala M, Taing L, Herbert Z, Koster J, Li B, et al. VIPER: visualization pipeline for RNA-seq, a snakemake workflow for efficient and complete RNA-seq analysis. BMC Bioinf 2018;19:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schneider CA, Rasband WS, Eliceiri KW. NIH image to imageJ: 25 years of image analysis. Nat Methods 2012;9:671–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Callicott RJ, Womack JE. Real-time PCR assay for measurement of mouse telomeres. Comp Med 2006;56:17–22. [PubMed] [Google Scholar]
- 30.Wei GH, Badis G, Berger MF, Kivioja T, Palin K, Enge M, et al. Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J 2010;29: 2147–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bullock M, Ren Y, O’Neill C, Gill A, Aniss A, Sywak M, et al. TERT promoter mutations are a major indicator of recurrence and death due to papillary thyroid carcinomas. Clin Endocrinol 2016;85:283–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vinagre J, Almeida A, Populo H, Batista R, Lyra J, Pinto V, et al. Frequency of TERT promoter mutations in human cancers. Nat Commun 2013;4:2185. [DOI] [PubMed] [Google Scholar]
- 33.Montero-Conde C, Leandro-Garcia LJ, Martinez-Montes AM, Martinez P, Moya FJ, Leton R, et al. Comprehensive molecular analysis of immortalization hallmarks in thyroid cancer reveals new prognostic markers. Clin Transl Med 2022; 12:e1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hiltzik D, Carlson DL, Tuttle RM, Chuai S, Ishill N, Shaha A, et al. Poorly differentiated thyroid carcinomas defined on the basis of mitosis and necrosis: a clinicopathologic study of 58 patients. Cancer 2006;106:1286–95. [DOI] [PubMed] [Google Scholar]
- 35.Volante M, Collini P, Nikiforov YE, Sakamoto A, Kakudo K, Katoh R, et al. Poorly differentiated thyroid carcinoma: the Turin proposal for the use of uniform diagnostic criteria and an algorithmic diagnostic approach. Am J Surg Pathol 2007;31:1256–64. [DOI] [PubMed] [Google Scholar]
- 36.Baloch ZW, Asa SL, Barletta JA, Ghossein RA, Juhlin CC, Jung CK, et al. Overview of the 2022 WHO classification of thyroid neoplasms. Endocr Pathol 2022;33:27–63. [DOI] [PubMed] [Google Scholar]
- 37.Taniguchi K, Karin M. NF-kappaB, inflammation, immunity and cancer: coming of age. Nat Rev Immunol 2018;18:309–24. [DOI] [PubMed] [Google Scholar]
- 38.Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, et al. Specific association of human telomerase activity with immortal cells and cancer. Science 1994;266:2011–5. [DOI] [PubMed] [Google Scholar]
- 39.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011; 144:646–74. [DOI] [PubMed] [Google Scholar]
- 40.Liu R, Bishop J, Zhu G, Zhang T, Ladenson PW, Xing M. Mortality risk stratification by combining BRAF V600E and TERT promoter mutations in papillary thyroid cancer: genetic duet of BRAF and TERT promoter mutations in thyroid cancer mortality. JAMA Oncol 2017;3:202–8. [DOI] [PubMed] [Google Scholar]
- 41.Xing M, Liu R, Liu X, Murugan AK, Zhu G, Zeiger MA, et al. BRAF V600E and TERT promoter mutations cooperatively identify the most aggressive papillary thyroid cancer with highest recurrence. J Clin Oncol 2014;32:2718–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caillou B, Talbot M, Weyemi U, Pioche-Durieu C, Al Ghuzlan A, Bidart JM, et al. Tumor-associated macrophages (TAMs) form an interconnected cellular supportive network in anaplastic thyroid carcinoma. PLoS One 2011;6:e22567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ryder M, Ghossein RA, Ricarte-Filho JC, Knauf JA, Fagin JA. Increased density of tumor-associated macrophages is associated with decreased survival in advanced thyroid cancer. Endocr Relat Cancer 2008;15:1069–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Suzuki S, Shibata M, Gonda K, Kanke Y, Ashizawa M, Ujiie D, et al. Immunosuppression involving increased myeloid-derived suppressor cell levels, systemic inflammation and hypoalbuminemia are present in patients with anaplastic thyroid cancer. Mol Clin Oncol 2013;1:959–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Luo H, Xia X, Kim GD, Liu Y, Xue Z, Zhang L, et al. Characterizing dedifferentiation of thyroid cancer by integrated analysis. Sci Adv 2021;7:eabf3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Subbiah V, Kreitman RJ, Wainberg ZA, Cho JY, Schellens JHM, Soria JC, et al. Dabrafenib and trametinib treatment in patients with locally advanced or metastatic BRAF V600-mutant anaplastic thyroid cancer. J Clin Oncol 2018; 36:7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Z, Ivanov AA, Su R, Gonzalez-Pecchi V, Qi Q, Liu S, et al. The OncoPPi network of cancer-focused protein-protein interactions to inform biological insights and therapeutic strategies. Nat Commun 2017;8:14356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ravindranathan A, Cimini B, Diolaiti ME, Stohr BA. Preliminary development of an assay for detection of TERT expression, telomere length, and telomere elongation in single cells. PLoS One 2018;13:e0206525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu R, Zhang T, Zhu G, Xing M. Regulation of mutant TERT by BRAF V600E/MAP kinase pathway through FOS/GABP in human cancer. Nat Commun 2018; 9:579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bullock M, Lim G, Zhu Y, Aberg H, Kurdyukov S, Clifton-Bligh R. ETS factor ETV5 activates the mutant telomerase reverse transcriptase promoter in thyroid cancer. Thyroid 2019;29:1623–33. [DOI] [PubMed] [Google Scholar]
- 51.Song YS, Yoo SK, Kim HH, Jung G, Oh AR, Cha JY, et al. Interaction of BRAF-induced ETS factors with mutant TERT promoter in papillary thyroid cancer. Endocr Relat Cancer 2019;26:629–41. [DOI] [PubMed] [Google Scholar]
- 52.Thornton CEM, Hao J, Tamarapu PP, Landa I. Multiple ETS factors participate in the transcriptional control of TERT mutant promoter in thyroid cancers. Cancers 2022;14:357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Greenberg RA, O’Hagan RC, Deng H, Xiao Q, Hann SR, Adams RR, et al. Telomerase reverse transcriptase gene is a direct target of c-Myc but is not functionally equivalent in cellular transformation. Oncogene 1999;18:1219–26. [DOI] [PubMed] [Google Scholar]
- 54.Takakura M, Kyo S, Kanaya T, Hirano H, Takeda J, Yutsudo M, et al. Cloning of human telomerase catalytic subunit (hTERT) gene promoter and identification of proximal core promoter sequences essential for transcriptional activation in immortalized and cancer cells. Cancer Res 1999;59:551–7. [PubMed] [Google Scholar]
- 55.Landa I, Pozdeyev N, Korch C, Marlow LA, Smallridge RC, Copland JA, et al. Comprehensive genetic characterization of human thyroid cancer cell lines: a validated panel for preclinical studies. Clin Cancer Res 2019;25: 3141–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bauerle KT, Schweppe RE, Haugen BR. Inhibition of nuclear factor-kappa B differentially affects thyroid cancer cell growth, apoptosis, and invasion. Mol Cancer 2010;9:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li Y, Zhou QL, Sun W, Chandrasekharan P, Cheng HS, Ying Z, et al. Non-canonical NF-kappaB signalling and ETS1/2 cooperatively drive C250T mutant TERT promoter activation. Nat Cell Biol 2015;17:1327–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ghosh A, Saginc G, Leow SC, Khattar E, Shin EM, Yan TD, et al. Telomerase directly regulates NF-kappaB-dependent transcription. Nat Cell Biol 2012;14: 1270–81. [DOI] [PubMed] [Google Scholar]
- 59.Akiyama M, Hideshima T, Hayashi T, Tai YT, Mitsiades CS, Mitsiades N, et al. Nuclear factor-kappaB p65 mediates tumor necrosis factor alpha-induced nuclear translocation of telomerase reverse transcriptase protein. Cancer Res 2003;63:18–21. [PubMed] [Google Scholar]
- 60.Miki S, Koga T, McKinney AM, Parisian AD, Tadokoro T, Vadla R, et al. TERT promoter C228T mutation in neural progenitors confers growth advantage following telomere shortening in vivo. Neuro Oncol 2022;24:2063–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ackermann J, Frutschi M, Kaloulis K, McKee T, Trumpp A, Beermann F. Metastasizing melanoma formation caused by expression of activated N-RasQ61K on an INK4a-deficient background. Cancer Res 2005;65:4005–11. [DOI] [PubMed] [Google Scholar]
- 62.Sharpless NE, Kannan K, Xu J, Bosenberg MW, Chin L. Both products of the mouse Ink4a/Arf locus suppress melanoma formation in vivo. Oncogene 2003; 22:5055–9. [DOI] [PubMed] [Google Scholar]
- 63.Stockhausen MT, Broholm H, Villingshoj M, Kirchhoff M, Gerdes T, Kristoffersen K, et al. Maintenance of EGFR and EGFRvIII expressions in an in vivo and in vitro model of human glioblastoma multiforme. Exp Cell Res 2011;317:1513–26. [DOI] [PubMed] [Google Scholar]
- 64.McKelvey BA, Zeiger MA, Umbricht CB. Characterization of TERT and BRAF copy number variation in papillary thyroid carcinoma: an analysis of The Cancer Genome Atlas study. Genes Chromosomes Cancer 2021;60:403–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lee DD, Leao R, Komosa M, Gallo M, Zhang CH, Lipman T, et al. DNA hypermethylation within TERT promoter upregulates TERT expression in cancer. J Clin Invest 2019;129:223–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mercer K, Giblett S, Green S, Lloyd D, DaRocha Dias S, Plumb M, et al. Expression of endogenous oncogenic V600EB-raf induces proliferation and developmental defects in mice and transformation of primary fibroblasts. Cancer Res 2005;65:11493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq data generated in this study are publicly available in Gene Expression Omnibus (GEO) at GSE227539.