

Our airways and gut are home to a diverse array of microbes, which bring both potential benefits and dangers (1–3). These mucosal surfaces must guard against invading pathogens while remaining receptive to forming symbiotic relationships with beneficial microbes. Reconciling these demands is the primary function of the immune system (4–6). Establishing the appropriate immunological set-point at each mucosal surface therefore requires careful calibration. This is driven by local assimilation of information on microbe type, burden, proximity, and metabolic activity by each mucosa (4, 5, 7, 8). It is thought that this information, coupled with the physiological constraints imposed by nutrient extraction in the gut and gaseous exchange in the airway, provides the blueprint to program immune defenses at each of these sites. The work of Röwekamp et al. (9) is part of a growing body of work that is re-evaluating this localized view of host–microbe interactions. Their study shows that microbes at one mucosal surface—the gut—can shape immunity at distal mucosal surfaces, in this case coordinating lower airway defenses. The implications of this are significant: host resistance to pneumococcal pneumonia is defined by the complement of microbes colonizing the gut. It is part of an emerging area of research which suggests that the outcome of infectious disease is governed by the collective influence of local and distal microbial communities on immune defenses (10).
Despite being isolated in 1880, Streptococcus pneumoniae (the pneumococcus) remains the leading cause of bacterial lower airway infections (11). Understanding how the pneumococcus causes disease and how the host resist it are therefore important biomedical questions. Beyond its immediate clinical significance, the study of the pneumococcus has also been a rich source of fundamental biological insights. It was through investigations on the pneumococcus that it was demonstrated that DNA encodes genetic information and the importance of surface capsular polysaccharides in host–microbe interactions was cemented (12, 13). The study of Röwekamp et al. (9) continues this tradition, uncovering new fundamental insights into host-microbe interactions by dissecting the pathogenesis of this clinically important pathogen.
This study started with a simple question: Do alarmins play a role in pneumococcal pneumonia? Alarmins are endogenous molecules released by cells in response to tissue damage, infection, or inflammation (14). They act as danger signals, alerting the immune system to the presence of stress and initiating a protective immune response. To investigate the role of alarmins in pneumonia, the authors first tested whether the pneumococcus induces the secretion of the major alarmins, uric acid, adenosine triphosphate (ATP), and interleukin (IL)-33. They found that the pneumococcus triggers the release of these molecules from macrophages, lung epithelial cells, and lung tissue explants. This hinted at a potential role for alarmins in controlling pneumococcal pneumonia. To test this hypothesis, they used various pharmacological and genetic tools to inhibit their function in vivo. They found that the activity of uric acid and ATP had little impact on pneumonia progression. While in the absence of IL-33 or its receptor, ST2, the ability to control the early progress of pneumonia was altered. Specifically, they found that without IL-33, mice were more resistant to pneumonia. IL-33 is a nuclear cytokine from the IL-1 family which is particularly abundant at barrier tissues including the skin, lungs, and gut (15). Upon tissue damage, it is released into the extracellular environment where it activates immune cells, in particular, type 2 innate lymphoid cells (ILC2s). Their data suggested that IL-33 activity somehow stymies resistance to pneumococcal pneumonia. To explore this further and link their discoveries derived from mouse models with pneumococcal pneumonia in humans, the authors analyzed single nucleotide polymorphisms (SNPs) in components of the IL-33 pathway in patients with pneumonia and age- and sex-matched controls. Supporting their findings from mouse models, SNP alleles linked to lower levels of the soluble decoy receptor of IL-33, sST2, and thus indicative of greater IL-33 activity, were more common in pneumonia patients.
The elegant work of Röwekamp et al. underscores the extensive connections between gut microbes, the immune system, and respiratory pathogens.
The next phase of work focused on elucidating the mechanistic basis for how IL-33 influences resistance to pneumonia. To do this, the authors undertook comprehensive characterization of the early pulmonary immune response to the pneumococcus. Initially, this appeared unfruitful. They found that the absence of IL-33 had little impact on the production of an extensive panel of cytokines and chemokines involved in coordinating pulmonary immunity. The number of alveolar macrophages, recruited neutrophils, and inflammatory monocytes in the lung during pneumonia were also unaffected by IL-33. Similarly, the absence of ILC2 cells, or the cytokines IL-4 and IL-13 which, like ILC2s, are also components of the type 2 immune response, had little impact on pneumococcal pneumonia. Their exhaustive analysis of the pulmonary immune system did, however, reveal that without IL-33 there was significantly more IL-22 produced during pneumonia and this was likely derived from ILC3s. IL-22 is a key cytokine for managing microbes at the mucosa. It promotes the induction of antimicrobial peptides and recruitment of immune cells from the bloodstream and orchestrates the restoration of mucosal homeostasis by controlling barrier repair (16). By using mice deficient in both IL-22 and IL-33, they confirmed that without IL-22, loss of IL-33 no longer enhanced resistance to pneumonia. Collectively, these data revealed that the activity of IL-33 renders the host susceptible to pneumococcal pneumonia by tempering the activity of IL-22.
What links IL-33 and IL-22? The next part of the study revealed an unexpected and poorly understood connection between IL-33 and pulmonary IL-22: the gut microbiota. As will be familiar to many users of animal models, during their study the authors had to move between different vivaria for their work and this changed the phenotypes of their model systems. Rather than being a frustrating impediment to progress, these differences provided the first clue into the potential connection between IL-33 and the microbiota and how this was influencing pulmonary defenses. In “cleaner” vivaria, wild-type and IL-33-deficient mice had similar resistance to pneumonia, it was only in the “dirtier” vivaria that the difference between wild-type and IL-33-deficient mice became apparent. The authors then undertook an impressive set of experiments involving microbiota depletion, microbiota transfer by cohousing, and microbiota transfer by oral gavage, to test the importance of the gut microbiota in determining the outcome of pneumonia. These experiments confirmed that rather than acting directly to limit IL-22 during pulmonary infection, IL-33 acts indirectly, controlling the composition of the gut microbiota. These alterations in the gut microbiota then, somehow, curb pulmonary IL-22 production during pneumococcal pneumonia increasing susceptibility to disease. By complementing these mechanistic experiments with analysis of gut microbiota composition, the authors speculate that an increase in abundance of Lactobacillus spp., or reduction in Saccharicrinis fermentans and Peptococcus niger, due to IL-33 deficiency is what stimulates pulmonary IL-22 (Fig. 1). This idea aligns with other studies indicating that IL-33 activity helps define the composition of the microbiota. For example, IL-33 has been shown to indirectly influence the induction of airway hyperresponsiveness to ozone through its control of gut microbiota composition (17). Similarly, IL-33-deficient mice are more prone to colitis and colitis-associated cancer (18). In these disease models, increased abundance of the gut microbes, segmented filamentous bacteria, and Akkermansia muciniphila, coupled with decreased production of mucosal IgA, due to IL-33 ablation increased susceptibility to colitis (18). Thus, understanding the mechanistic basis for how IL-33 selects gut microbes could have important implications for not only pneumonia but a range of immunological disorders.
Fig. 1.
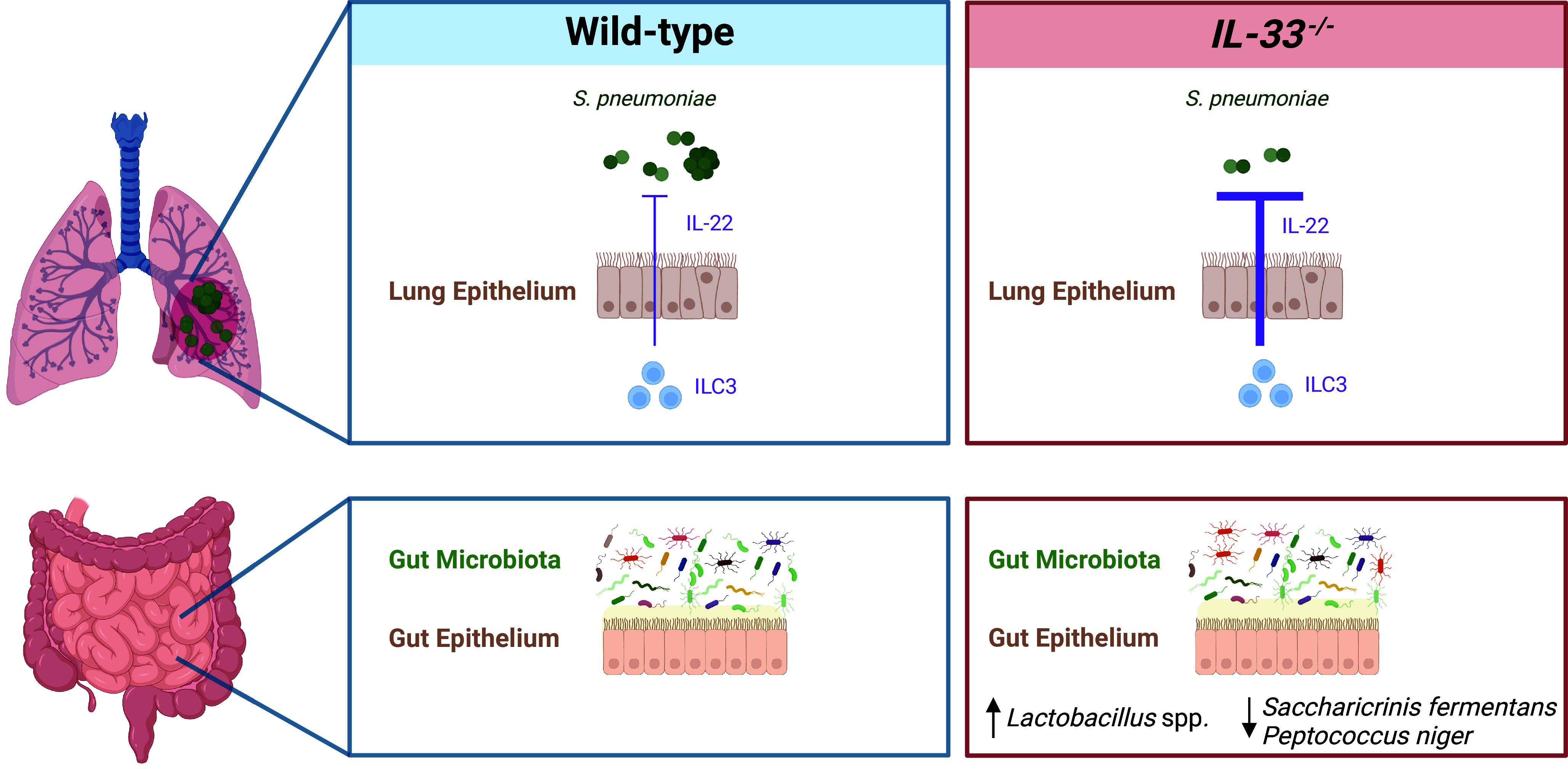
The coordination of defences against pneumococcal pneumonia by IL-33, IL-22, and the gut microbiota. IL-33 activity controls gut microbiota composition and this restrains pulmonary IL-22 during pneumococcal pneumonia leaving the host susceptible to disease. The proposed source of IL-22 in the lung are ILC3s.
How do gut microbes communicate with distal organs and how are these systemic influences controlled? By governing the development and operation of immune defenses throughout the host, gut microbes establish a critical barrier against infectious disease. Mechanistically, this long-range, interorgan communication can occur via an array of nonmutually exclusive mechanisms (10). Gut microbes produce various immunologically active molecules, including metabolic products and structural components of their cell walls, that spread from the gut to systemic sites through the bloodstream, priming immune responses in distal tissues (19). The activity of cytokine signaling networks is also critical for gut microbes to control systemic immunity. For example, previous work from investigators in this study demonstrated that gut microbes prime conventional dendritic cells (cDCs) in systemic tissues via type I interferon, and this enhances the immune response to subsequent microbial challenge (20). Immune cell migration from the gut to the periphery is a further mechanism that enables the transfer of microbial-derived signals to distal tissues (21). Collectively, these processes allow the immune system beyond the gut to integrate information from intestinal microbes and fortify its responses to infectious diseases. These organism-wide influences cannot operate unlicensed. Gut microbes exert their power through a range of different tissues, many of which are exquisitely sensitive to microbial signals. Without proper control, this can lead to systemic inflammation and other forms of over-exuberant immune responses. For example, the Firmicutes phylum is enriched with immunoregulatory gut microbes that enhance systemic innate immune defense to infection. However, without mechanisms to restrain the immunological activity of the Firmicutes cell wall molecules that coordinate systemic immunity, the beneficial influence of these microbes becomes pathological, driving inflammatory cachexia and organ damage (19). Similarly, the previous work from investigators in this study demonstrating that the microbiota controls cDCs in systemic tissues also comes with downsides. Specifically, mechanisms of peripheral tolerance, such as regulatory T cells, are needed to restrain the immune system because programming of cDCs by the microbiota primes CD8+ T cell responses to harmless antigens (20). Thus, while the systemic effect of gut microbes can lead to more robust responses to pathogens and their components, these responses must be appropriately moderated to prevent immunopathology. Setting the work of Röwekamp et al. (9) in this framework of balance perhaps provides a rationale for their observations. During homeostasis, IL-33 restricts the expansion of highly immunologically active gut microbes. Consequently, the host is protected against these organisms causing excessive inflammation, especially at the sites of colonization in the gut, but the trade-off is that priming of immune defenses to infections like pneumonia is reduced. While potentially appealing, this hypothesis remains to be tested. The elegant work of Röwekamp et al. (9) underscores the extensive connections between gut microbes, the immune system, and respiratory pathogens. As pneumonia remains a major cause of human mortality, defining the gut microbes that provide protection against it could reveal new tactics to battle this disease, once described as the “Captain of the Men of Death.”
Acknowledgments
Author contributions
C.C. and T.B.C. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
See companion article, “IL-33 controls IL-22-dependent antibacterial defense by modulating the microbiota,” 10.1073/pnas.2310864121.
References
- 1.Cho I., Blaser M. J., The human microbiome: At the interface of health and disease. Nat. Rev. Genet. 13, 260–270 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegel S. J., Weiser J. N., Mechanisms of bacterial colonization of the respiratory tract. Annu. Rev. Microbiol. 69, 425–444 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gilbert J. A., et al. , Current understanding of the human microbiome. Nat. Med. 24, 392–400 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chu H., Mazmanian S. K., Innate immune recognition of the microbiota promotes host-microbial symbiosis. Nat. Immunol. 14, 668–675 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Donaldson G. P., Lee S. M., Mazmanian S. K., Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 14, 20–32 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belkaid Y., Harrison O. J., Homeostatic immunity and the microbiota. Immunity 46, 562–576 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kayama H., Okumura R., Takeda K., Interaction between the microbiota, epithelia, and immune cells in the intestine. Annu. Rev. Immunol. 38, 23–48 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Perdijk O., Azzoni R., Marsland B. J., The microbiome: An integral player in immune homeostasis and inflammation in the respiratory tract. Physiol. Rev. 104, 835–879 (2024). [DOI] [PubMed] [Google Scholar]
- 9.Röwekamp I., et al. , IL-33 controls IL-22-dependent antibacterial defense by modulating the microbiota. Proc. Natl. Acad. Sci. U.S.A. 121, e2310864121 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jordan C. K. I., Clarke T. B., How does the microbiota control systemic innate immunity? Trends Immunol. 45, 94–102 (2024). [DOI] [PubMed] [Google Scholar]
- 11.GBD 2019 Antimicrobial Resistance Collaborators, Global mortality associated with 33 bacterial pathogens in 2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 400, 2221–2248 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Austrian R., Pneumococcus: The first one hundred years. Rev. Infect. Dis. 3, 183–189 (1981). [DOI] [PubMed] [Google Scholar]
- 13.Austrian R., The pneumococcus at the millennium: Not down, not out. J. Infect. Dis. 179, S338–S341 (1999). [DOI] [PubMed] [Google Scholar]
- 14.Yang D., Han Z., Oppenheim J. J., Alarmins and immunity. Immunol. Rev. 280, 41–56 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liew F. Y., Girard J. P., Turnquist H. R., Interleukin-33 in health and disease. Nat. Rev. Immunol. 16, 676–689 (2016). [DOI] [PubMed] [Google Scholar]
- 16.Keir M., Yi Y., Lu T., Ghilardi N., The role of IL-22 in intestinal health and disease. J. Exp. Med. 217, e20192195 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kasahara D. I., et al. , The interleukin-33 receptor contributes to pulmonary responses to ozone in male mice: Role of the microbiome. Respir. Res. 20, 197 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malik A., et al. , IL-33 regulates the IgA-microbiota axis to restrain IL-1alpha-dependent colitis and tumorigenesis. J. Clin. Invest. 126, 4469–4481 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jordan C. K. I., et al. , Symbiotic Firmicutes establish mutualism with the host via innate tolerance and resistance to control systemic immunity. Cell Host Microbe 31, 1433–1449.e9 (2023). [DOI] [PubMed] [Google Scholar]
- 20.Schaupp L., et al. , Microbiota-induced type I interferons instruct a poised basal state of dendritic cells. Cell 181, 1080–1096.e19 (2020). [DOI] [PubMed] [Google Scholar]
- 21.Galvan-Pena S., Zhu Y., Hanna B. S., Mathis D., Benoist C., A dynamic atlas of immunocyte migration from the gut. Sci. Immunol. 9, eadi0672 (2024). [DOI] [PMC free article] [PubMed] [Google Scholar]