

Abstract
Background
Infantile spasms (West's Syndrome) is a syndrome that includes a peculiar type of epileptic seizure—the spasms—and an electroencephalographic (EEG) abnormality often called hypsarrhythmia. Psychomotor retardation is frequently found at follow‐up. Approximately two‐thirds of affected infants will have a detectable underlying neurological abnormality, but still little is known about the pathophysiological basis for infantile spasms, and treatment remains problematic.
Objectives
To compare the effects of single pharmaceutical therapies used to treat infantile spasms in terms of control of the spasms, resolution of the EEG, relapse rates, psychomotor development, subsequent epilepsy, side effects, and mortality.
Search methods
To identify published data, we searched the Cochrane Epilepsy Group Specialised Register (October 2012), CENTRAL (The Cochrane Library 2012, Issue 9), MEDLINE (1946 to September Week 4, 2012), EMBASE (1980 to March 2003), and the reference lists of all retrieved articles. To identify unpublished data, we searched the ISRCTN Register (www.controlled‐trials.com), corresponded with colleagues and drug companies, and made requests at international conferences.
Selection criteria
All randomised controlled trials (RCTs) of the administration of drug therapy to patients with infantile spasms.
Data collection and analysis
Data collection from all relevant publications was independently undertaken by three review authors (before 2010) or by two review authors using a standard proforma. Analysis included assessment of study quality and a search for sources of heterogeneity.
Main results
We found 16 small RCTs (fewer than 100 patients enrolled) and 2 larger RCTs (more than 100 patients enrolled). These 18 studies looked at a total of 916 patients treated with a total of 12 different pharmaceutical agents. Overall methodology of the studies was poor, in part because of ethical dilemmas such as giving placebo injections to children. Two studies showed that placebo was not as good as active treatment in resolving the spasms. The strongest evidence suggested that hormonal treatment (prednisolone or tetracosactide depot) leads to resolution of spasms faster and in more infants than does vigabatrin. Responses without subsequent relapse may be no different. The same study suggests that hormonal treatments might improve the long‐term developmental outcome compared with vigabatrin in infants not found to have an underlying cause for their infantile spasms.
Authors' conclusions
To date, few well‐designed RCTs have considered the treatment of infantile spasms, and the numbers of patients enrolled have been small. In the majority, methodology has been poor, hence it is not clear which treatment is optimal in the treatment of this epilepsy syndrome. Hormonal treatment resolves spasms in more infants than vigabatrin, but this may or may not translate into better long‐term outcomes. If prednisolone or vigabatrin is used, high dosage is recommended. Vigabatrin may be the treatment of choice in tuberous sclerosis. Resolution of the EEG features may be important, but this has not been proven. Further research using large studies with robust methodology is required.
Keywords: Humans; Infant; Anticonvulsants; Anticonvulsants/adverse effects; Anticonvulsants/therapeutic use; Cosyntropin; Cosyntropin/therapeutic use; Hormones; Hormones/therapeutic use; Prednisolone; Prednisolone/therapeutic use; Psychomotor Performance; Randomized Controlled Trials as Topic; Spasms, Infantile; Spasms, Infantile/complications; Spasms, Infantile/drug therapy; Vigabatrin; Vigabatrin/therapeutic use
Plain language summary
Treatment of infantile spasms
The optimum treatment for infantile spasms has yet to be proven with confidence, in part because of the different aims of existing studies. However, some useful conclusions can be drawn from current evidence.
Infantile spasms is a rare seizure disorder commonly associated with severe learning difficulties. Many different treatments are currently used worldwide in the treatment of this disorder, and many more have been tried in the past, often with little success. Not all treatments are licensed for use in all countries. Most treatments are associated with significant adverse effects. Additional research is needed to explore the long‐term benefits of different therapies for seizure control and for neurodevelopment. Two studies have shown that a placebo is not as good as an active treatment in resolving the spasms. The strongest evidence suggests that hormonal treatment (prednisolone or tetracosactide depot) leads to resolution of spasms faster and in more infants than does vigabatrin. Responses without subsequent relapse may be no different, but one study suggested that hormonal treatment (prednisolone or tetracosactide) might improve long‐term neurodevelopmental outcomes in infants and young children for whom no underlying cause for their infantile spasms has been identified. This makes hormonal treatment more attractive, at least for this group of infants. More information and further research are needed to compare currently available therapies.
Background
Infantile spasms, also known as West's syndrome or salaam spasms, were first described by Dr West in a letter he wrote to The Lancet in 1841. It is a syndrome that includes a peculiar type of seizure, high risk of psychomotor retardation, and usually a characteristic electroencephalographic (EEG) pattern known as hypsarrhythmia (ILAE 1989; Roger 1985). It is relatively uncommon, with an estimated incidence of 0.16 to 0.42 per 1000 live births (Cowan 1991). An association with several disorders has been noted, some of which are known at disease onset (e.g. cerebral palsy, Down syndrome), although others (e.g. tuberous sclerosis, neuronal migration disorders) are discovered on investigation after the onset of spasms. However, in a significant minority of cases, the aetiology remains unknown (Aicardi 1994). Despite awareness of the condition for over 150 years, little progress has been made in our understanding of the pathophysiology of the condition, and treatment of the disorder remained largely empirical for a long time.
Onset of seizures usually occurs within the first year of life, with a peak age of onset of three to five months. The seizures usually consist of sudden, generally bilateral, and symmetrical contractions of the neck, trunk, and extremities that are associated with a brief loss of consciousness. Less commonly, they may consist of an extensor spasm of the legs and spine, or simple head nodding. Rarely they are asymmetrical. Seizures often occur in clusters or runs; commonly 20 or so but as many as 100 spasms can occur in a single cluster, with each individual spasm lasting 1 to 2 seconds only. These clusters frequently occur as the infant is waking from sleep and are commonly associated with a cry. In most cases, they resolve by the age of three, although rarely they can persist up to 10 to 15 years of age (Aicardi 1994).
Hypsarrhythmia is used to describe an EEG pattern that is characterized by random, high‐voltage spikes and slow waves. The most striking features of hypsarrhythmia are high‐voltage slow waves with variable amplitude; spikes and waves from many foci, varying with time; and lack of synchrony, with a generally 'chaotic' appearance. The typical appearance is more likely to be noted in earlier stages of infantile spasms and when onset occurs at a younger age. The hypsarrhythmic pattern may disappear during rapid‐eye‐movement (REM) sleep, but it may be observed with greater sensitivity in some other stages of sleep (Lux 2004b).
The seizures are refractory to treatment with most conventional antiepileptic drugs. Although the spasms resolve with time, the long‐term prognosis is poor. Many children develop other forms of severe epilepsy, and most (80% to 90%) have psychomotor retardation (Riikonen 1996). Some children have delayed development before the onset of their seizures as part of a predisposing condition, for example, Down syndrome. Nevertheless, even in these patients, further regression of development is often seen after the onset of spasms. The degree of psychomotor delay is severe in approximately 70% of children, placing a great burden on both caregivers and the health system. Although few studies have been undertaken to look at long‐term outcomes for these patients, it is a widely accepted view that earlier diagnosis, along with quicker control of the spasms, might improve the prognosis. Evidence now supports this, in that a longer lead time to treatment (time from onset to start of treatment) leads to a worse developmental outcome (O'Callaghan 2011).
In the past, numerous clinical trials have investigated different treatment regimens. The main problem is that few of these trials were randomised controlled trials, and the drug under investigation has frequently been used long after the onset of seizures and in addition to, or after, other anticonvulsants. Wide variation in the drug dosages used and in the duration of their use has been reported. As a result, it is confusing when looking at the literature to know which is the best treatment for this disorder, with the consequence that many different treatment modalities have been used. Most trials have considered infantile spasms as a single entity rather than considering the underlying aetiology, but it is possible that infantile spasms resulting from one particular disorder (e.g. Down syndrome, tuberous sclerosis) might respond better to one form of treatment than another. Other factors than treatment that might affect the outcome also need to be considered, for example, the belief that the sooner the spasms are brought under control (i.e. the shorter the lead time), the better the outcome. Likewise, if infants entered into a trial have been previously treated for infantile spasms, this might also affect outcome. Recently, it has been suggested that age at onset affects developmental outcome, with improvement noted for each increase in age of one month (O'Callaghan 2011).
Infantile spasms are an unusual epilepsy syndrome. The interictal EEG is very abnormal in these infants, but occasionally it is abnormal only during sleep. The ictal EEG is variable, often with suppression or fast activity, but the ictal event is frequently (but not always) typical. Agreement on the exact definition of the EEG abnormality required to make the diagnosis has not been achieved, but fortunately, the combination of the unusual ictal event, the clinical semiology, and the very abnormal EEG makes any other diagnosis unlikely, even when either the EEG or the ictal event is not typical.
The fact that ictal episodes are individually very brief (lasting between one and two seconds), their occurrence in clusters or batches (many of which can occur in a day), and the frequency of the ictal episodes during a cluster (often many per cluster) are features also contributing to the fact that this is an unusual epilepsy. Absence of ictal activity can, therefore, be detected with some accuracy (depending on the accuracy of the witness) on a daily basis by direct observation. Cessation of all ictal activity is generally accompanied by resolution of the features usually seen on the EEG, but many infants will not then have a normal EEG because of other seizure types or underlying structural brain abnormalities. It is not practical to record the EEG on a daily basis; therefore, the relationship between resolution of ictal activity and improvement of the EEG is not clearly established. Therefore, disagreement continues over the importance of continuing treatment if the EEG remains severely abnormal in the absence of clinical evidence of continuing infantile spasms. Some clinicians believe that persistence of the EEG abnormality must indicate continuation of the epileptic encephalopathy, and that the EEG findings should, therefore, be treated even in the absence of observed ictal episodes.
Another unusual feature of this condition is the developmental regression that so often accompanies, or quickly follows, the onset of seizures. This, along with the severity of the EEG abnormality, has led many to the conclusion that infantile spasms are an epileptic encephalopathy. This theory lends support to the hypothesis that it may be important to continue treatment until the EEG abnormalities have improved. Most treatments are believed to work if a response occurs within a short time of the commencement of treatment, usually within 14 days. Such claims need to be backed up with evidence, especially since because the tendency in the literature is for claims of rapid response not to be supported by the evidence. However, the practical outcome of the developmental regression and the usual rapid response (viz other epilepsy syndromes) mean that continuation of spasms beyond 14 days of treatment is usually an indication that one should try an alternative treatment.
These issues, definitions, and outcomes have been subject to review by an independent group using Delphi methodology. This review concluded that clinical evidence of cessation of spasms should be reported independently of cessation of spasms combined with resolution of the EEG features of the condition. It was agreed that subsequent developmental progress was an important outcome, and that this might be a more important outcome than cessation of spasms. It is proposed that the protocol for the present review will need to be reviewed before the next review, if one is to take into account this internationally agreed consensus.
Additional difficulties have arisen from the use of terms such as 'high dose' and 'low dose' in publications. In some studies, the 'high dose' has been lower than the dose used in other studies as the 'low dose.' In addition, lack of study by pharmaceutical companies, a common problem in paediatric practice, means that the most effective dose often is not known. Some studies have corrected for body size through an adjustment for body weight; others have adjusted for surface area. However, such adjustment is not usually justified, and the scientific basis for or against adjustment is not as robust as it could be.
Therefore, it is clear that resolution of the clinical evidence of spasms is one outcome, and that this may occur quickly, in contrast to most epilepsy syndromes. Resolution of the EEG features also needs to be reported, and subsequent developmental progress is an important later outcome. Other seizure types occur before the spasms in many infants and may follow spasms, even when these resolve and other seizure types did not occur before the onset of spasms; therefore, epilepsy outcomes for all seizure types need to be reported.
Significant conventional reviews of the literature on the treatment of infantile spasms include Haines 1994, Riikonen 1996, Mackay 2004, and Go 2012.
Objectives
To compare the effects of single pharmaceutical therapies used to treat infantile spasms in terms of control of spasms, resolution of hypsarrhythmia, long‐term psychomotor development, and subsequent epilepsy rates.
The following hypotheses were tested.
Therapy versus placebo
(1) Therapy A* is more effective than placebo treatment in controlling infantile spasms (in terms of spasm cessation, reduction in total number of spasms, and relapse). . (2) Therapy A* is more effective than placebo treatment in resolving the EEG appearance. (3) Therapy A* leads to improved long‐term psychomotor development compared with placebo treatment. (4) Treatment of infantile spasms with therapy A* reduces subsequent epilepsy rates compared with placebo treatment.
Therapy versus no treatment
(1) Therapy A* is more effective than no treatment in controlling infantile spasms (in terms of spasm cessation, reduction in total number of spasms, and relapse). (2) Therapy A* is more effective than no treatment in resolving the EEG appearance. (3) Therapy A* leads to improved long‐term psychomotor development compared with no treatment. (4) Treatment of infantile spasms with therapy A* reduces subsequent epilepsy rates compared with no treatment.
Comparisons between therapies
(1) Therapy A* is more effective in controlling infantile spasms (in terms of spasm cessation, reduction in total number of spasms, and relapse) compared with any other single pharmaceutical therapy. (2) Therapy A* is more effective in resolving the EEG appearance compared with any other single pharmaceutical therapy. (3) Therapy A* leads to improved long‐term psychomotor development compared with any other single pharmaceutical therapy. (4) Treatment of infantile spasms with therapy A* reduces subsequent epilepsy rates compared with any other single pharmaceutical therapy.
*Therapy A = ACTH** or hydrocortisone or prednisone or prednisolone or carbamazepine or ethosuximide or gabapentin or lamotrigine or phenobarbitone or phenytoin or topiramate or vigabatrin or valproate or clonazepam or diazepam or nitrazepam or pyridoxine (vitamin B6), or methysergide, or methylparatyrosine, or sulthiame, or any other single therapeutic agent studied in the literature.
**ACTH – at the time this review was undertaken, two ACTH preparations were in widespread use: ACTH (adrenocorticotrophin hormone) and tetracosactide. ACTH is naturally occurring, and the therapeutic product is derived from a bovine or porcine source and is administered as an intramuscular injection. However in the UK, with existing concerns surrounding bovine spongiform encephalopathy (BSE), ACTH has been withdrawn from the market. Tetracosactide is a synthetic alternative to ACTH and consists of the first 24 amino acids occurring in ACTH. It displays the same physiological properties as ACTH. It can be provided in depot and non‐depot preparations' the Depot preparation is usually given on alternate days.
Methods
Criteria for considering studies for this review
Types of studies
All randomised controlled trials (RCTs) of the administration of drug therapy to infants and children with infantile spasms were included in this review, including trials that compared a therapy with no treatment or placebo, and trials that compared one drug with another or with different doses of the same drug.
Definition of an RCT: trials in which participants are prospectively allocated to treatment groups by a random (e.g. random number generation, coin flips) or a quasi random (e.g. by date of birth) process.
If the study was not an RCT, it was not included in the review. The existence of such studies was documented.
We considered studies looking at drug therapy as second‐line therapy, as well as studies looking at drug therapy as first‐line therapy.
Types of participants
Any infant or child treated for infantile spasms regardless of whether or not EEGs were performed, or whether therapy had been given before trial entry.
Most published studies on infantile spasms did not give the definition of infantile spasms used for their participants; therefore, for the purpose of this review, we assumed that a clinical diagnosis had been made for any participant that was entered into a trial. We documented any definitions that were given and considered whether differences in definition might account for variation between studies.
Types of interventions
(1) Any trial that compared at least one therapy against placebo treatment. (2) Any trial that compared at least one therapy against no therapy. (3) Any trial that compared at least one therapy against another therapy or a different dose of the same therapy.
Therapies included: (1) 'Steroids': ACTH, tetracosactide, hydrocortisone, prednisone or prednisolone. (2) Antiepileptic drugs: carbamazepine, clonazepam, diazepam, ethosuximide, flunarizine, gabapentin, ganaxolone lamotrigine, methysergide, methylparatyrosine, nitrazepam, phenobarbitone, phenytoin, pyridoxine, sodium valproate, sulthiame, topiramate, vigabatrin. (3) Any other single therapeutic agent studied in the literature.
Any dose regimen of the above therapies was included.
Types of outcome measures
(1) Cessation of spasms
This was defined as total cessation of spasms for at least 48 hours after the start of therapy but occurring within a month of commencement of therapy. It was measured as a dichotomous variable (i.e. ceased or continuing). Time taken from commencement of therapy to cessation of spasms was also to be measured as a continuous variable (measured in days).
(2) Quantitative reduction of spasms
This was measured as the number of spasms occurring before treatment was commenced compared with the number occurring after treatment and was to be measured as a dichotomous variable (i.e. greater than 50% reduction or less than 50% reduction in the number of spasms occurring per day over the seven‐day period before trial entry and over a seven‐day period at one month after the start of therapy).
(3) Resolution of EEG abnormality
Participants were divided into those in whom the EEG abnormality remained and those in whom there was resolution. This was measured as a dichotomous variable (i.e. resolved or unresolved).
(4) Relapse rates of spasms
A single spasm occurring by one year of age or within the study period but after cessation of spasms constituted a relapse. It was measured as a dichotomous variable (i.e. relapse occurred or no relapse occurred). Time taken from cessation of spasms to relapse was also to be measured as a continuous variable (measured in days) in those patients in whom spasms had ceased.
(5) Long‐term psychomotor development
This was to be analysed in months as a measure of deviation from the chronological age. It was to be measured at three months post entry into the trial and, where possible, at five years of age. It was measured as a continuous variable.
(6) Subsequent epilepsy rates
The diagnosis of any epileptic seizure type other than infantile spasms after commencement of therapy constituted subsequent epilepsy. This was to be measured at three months post entry into the trial and, where possible, at five years of age. Any seizure type plus each seizure type (e.g. primary generalised, partial) was to be measured as a dichotomous variable (i.e. present or absent) over the previous one month at three months post entry, and over the previous six months at five years of age. We used the original authors' diagnosis because of the difficulty involved in defining epileptic seizures in children.
(7) Adverse effects
Only side effects that were considered severe enough to warrant discontinuation of the test treatment were measured. They were measured as a dichotomous variable (i.e. therapy stopped vs therapy not stopped). They were also qualitatively summarised.
(8) Deaths
All deaths were measured as a dichotomous variable (i.e. alive or deceased).
Search methods for identification of studies
We searched the Epilepsy Group Specialised Register (5 October 2012). This register contains reports of trials identified from regular searches of the Cochrane Central Register of Controlled Trials (CENTRAL) and of MEDLINE. Relevant reports were also identified by handsearching selected journals and conference proceedings.
In addition, we carried out searching as follows.
Electronic searches
We searched the following databases. No language restrictions were applied.
(a) CENTRAL (The Cochrane Library 2012, Issue 9) was searched using the strategy outlined in Appendix 1. (b) MEDLINE (Ovid) (1946 to September Week 4, 2012) was searched using the strategy outlined in Appendix 2. (c) EMBASE (1980 to March 2003) was searched in a similar manner to MEDLINE. We no longer have direct access to that database. However, a project to identify reports of trials in EMBASE is being carried out by the UK Cochrane Centre. This search is updated annually and these records are published in CENTRAL. These records are therefore available to us via our searches of CENTRAL. (d) ISRCTN Register (www.controlled‐trials.com) was searched using the term "infantile spasms" (4 October 2012).
Searching other resources
The reference lists of the RCTs and of rejected articles were scanned (by ECH) to identify possible articles missed by the computerised search. We contacted pharmaceutical companies and colleagues, and made appeals at international conferences to try to identify unpublished data and studies not published in English.
Data collection and analysis
Relevant publications were reviewed independently by at least two review authors (ECH or SE and JO) and before 2010 by three review authors (ECH, JO, and Professor Milner or SE). Discrepancies were resolved by discussion. There was no blinding of authorship or results. All RCTs were considered, as were all non‐English studies.
Assessment of methodological quality
Selection bias: the studies were assessed as to whether allocation concealment was adequate, unclear, or inadequate.
Performance bias: the studies were assessed as to whether recipients and those measuring outcome were unaware of the assigned therapy (however, we accepted that most of the studies in this review would not be double‐blinded trials, except where blind assessment of outcomes was possible).
Attrition bias: assessed as to whether patients were lost to follow‐up in the studies.
Using these criteria, studies were then divided into: (1) those with a low risk of bias; (2) those with a moderate risk of bias; and (3) those with a high risk of bias (see the Cochrane Handbook for Systematic Reviews of Interventions, Section 8.5) (Higgins 2005).
Exclusion criteria
Any trial that was not a RCT was excluded from analysis but was documented.
If a clinical definition for infantile spasms was given, any trial in which there was doubt about the clinical diagnosis was excluded from analysis but was documented.
In studies in which it is unclear if the above criteria have been met, the review authors are endeavouring to obtain additional information by contacting the first author on up to three occasions. This is documented.
The following data were originally extracted independently by three review authors (ECH, JO, and Professor Milner or SE), and discrepancies were resolved by discussion. For this update, only two authors were used (ECH, JO).
Participants (i.e. those characteristics of the population that may affect outcome regardless of treatment):
Age at spasm onset, at diagnosis, and at start of treatment (mean or median and range measured to the nearest completed week of age).
Time from onset of spasms to initiation of treatment (mean or median and range measured to the nearest completed week).
Sex (male, female).
Previous treatment for infantile spasms (e.g. prednisolone, ACTH, vigabatrin, valproate) – but not previous treatment for other seizure types.
Interventions:
Type of pharmaceutical agent used (e.g. prednisolone, ACTH, vigabatrin, valproate).
Dose (measured in internationally accepted units (e.g. mg for prednisolone, IU for ACTH).
Frequency (measured as the number of times the pharmaceutical agent was given in a 24 hour period).
Route of administration (i.e. oral, intramuscular, intravenous).
Treatment length (measured to the nearest completed day).
Outcome measures:
Cessation of spasms and the time taken to cessation of spasms.
Resolution of EEG and time taken for resolution.
Reduction in spasms.
Relapse rates and time taken to relapse.
Psychomotor development.
Subsequent epilepsy rates.
Side effects.
Deaths.
Analysis plan
Study quality. This was done with a table of met or unmet criteria for selection, performance, and attrition bias.
Dichotomous data. For each item of data requiring dichotomous analysis, the following were recorded: number of patients who experienced the event (or outcome) in each group for each comparison and the total number in each group. These data were analysed using Peto odds ratios (ORs) in the Cochrane RevMan software.
Continuous data. For each item of data requiring continuous data analysis (except psychomotor development), the following were recorded: number of patients in each group, mean or median value for the outcome in each group, and standard deviation. These values were analysed using weighted means in the Cochrane RevMan software.
When data for the same outcome were presented in some studies as dichotomous and in others as continuous data, we endeavoured to obtain continuous data from the investigators. If it was not possible to obtain continuous data, for example, because they had not been recorded, data were analysed either as dichotomous data with a 'cut‐off' point agreed by the three review authors, or as a mixture of dichotomous and continuous data, using two separate tables.
We looked for sources of heterogeneity between trials: methodological and clinical differences, including previous treatment for spasms, age at trial entry, and single underlying cause. We also looked for subgroup analysis of clinical features, for example, developmental delay before onset of spasms, differences in drug dosages, timing and length of treatment, and specific underlying diagnosis, such as tuberous sclerosis.
Results
Description of studies
The literature search identified 96 studies to be evaluated for inclusion in this review. Some studies were identified from more than one source. Review of the studies eliminated 64 studies because they were not RCTs and a further six because the participants did not suffer from infantile spasms; one paper was excluded because the data referred to the effects of hormonal treatment on the brain when imaged using computerised tomography and did not report any other outcome measure. This left a total of 27 papers reporting 18 different RCTs conducted to look at the treatment of infantile spasms, yielding a total of 858 patients. These 18 trials looked at 12 different pharmacological agents: vigabatrin; ACTH (nine different treatment regimens and different preparations); prednisolone; prednisone; hydrocortisone; magnesium sulphate; nitrazepam; sodium valproate; sulthiame; flunarizine; ganaxolone methysergide; and alpha‐methylparatyrosine (see table Characteristics of included studies and Table 1, Table 2, Table 3, Table 4). We have been careful to amalgamate for meta‐analysis only those treatments for which a rationale for combining treatments was provided.
1. Methodological quality of included studies.
| Study ID | No. patients | Placebo RCT | Allocation blinded | Recipients blinded | Outcome blinded | Loss of follow‐up | Multi‐centre | Risk of bias |
| Appleton 1999 | 40 | yes | yes | yes | yes | no | yes | low |
| Askalan 2003 | 9 | no | not clear | no | some | no | no | high |
| Baram 1996 | 29 | no | yes | no | yes | none | no | high |
| Chiron 1997 | 22 | no | not clear | no | no | none | yes | high |
| Debus 2004 | 51 | yes | yes | yes | no | no | yes | low |
| Dreifuss 1986 | 52 | no | not clear | no | yes | yes | yes | high |
| Dyken 1985 | 17 | yes | not clear | yes | yes | yes | no | high |
| Elterman 2010 | 221 | no | not clear | no | no | yes | yes | high |
| Hrachovy 1983 | 24 | no | not clear | yes | not clear | none | unclear | high |
| Hrachovy 1989 | 24 | no | not clear | not clear | not clear | none | unclear | high |
| Hrachovy 1994 | 59 | no | not clear | no | yes | yes | unclear | high |
| Lux 2004 | 107 | no | yes | no | no except developmental outcome | yes | yes | high |
| Bitton 2012 | 68 | yes | not clear | yes | yes | yes | yes | low |
| Shu 2009 | 30 | no | not clear | not clear | not clear | not clear | not clear | high |
| Tsai 2009 | 56 | yes | not clear | yes | yes | not clear | not clear | high |
| Vigevano 1997 | 42 | no | not clear | no | not clear | none | no | high |
| Yanagaki 1999 | 26 | no | not clear | not clear | not clear | yes | no | high |
| Zou 2010 | 38 | no | not clear | no | no | no | no | unclear |
2. Participants.
| Study ID | Intervention | Male:Female | Age at onset | Age at diagnosis | Age at trial entry | Delay to treatment | Previous treatment |
| Appleton 1999 | Vigabatrin | 11:9 | 30 weeks | not available | 35 weeks | 6 weeks | no |
| Placebo | 8:12 | 26 weeks | not available | 35 weeks | 7 weeks | no | |
| Askalan 2003 | ACTH | 1:2 | not available | not available | not available | not available | no |
| Vigabatrin | 3:3 | not available | not available | not available | not available | no | |
| Baram 1996 | ACTH | 4:11 | not available | not available | 22 weeks | not available | not available |
| Prednisone | 8:6 | not available | not available | 32 weeks | not available | not available | |
| Chiron 1997 | Vigabatrin | 5:6 | 25 weeks | not available | 29 weeks | 3 weeks | not available |
| Hydrocortisone | 5:6 | 26 weeks | not available | 34 weeks | 8 weeks | not available | |
| Debus 2004 | Sulthiame | 14:11 (3 unclear) | 31 weeks | 31 weeks | 35 weeks | 4 weeks | yes |
| Placebo | 7:12 (4 unclear) | 29 weeks | 29 weeks | 33 weeks | 4 weeks | yes | |
| Dreifuss 1986 | Nitrazepam | 14:13 | not available | not available | 37 weeks | not available | not available |
| ACTH | 15:10 | not available | not available | 35 weeks | not available | not available | |
| Dyken 1985 | Valproate | not available | not available | not available | not available | not available | yes |
| Placebo | not available | not available | not available | not available | not available | yes | |
| Elterman 2010 | Vigabatrin (low dose) | 42:33 | not available | 29.5 weeks | 32.9 weeks | not available | yes |
| Vigabatrin (high dose) | 29:38 | not available | 28.1 weeks | 31.6 weeks | not available | yes | |
| Hrachovy 1983 | ACTH | not available | not available | not available | not available | not available | not available |
| Prednisone | not available | not available | not available | not available | not available | not available | |
| Hrachovy 1989 | Methysergide | not available | not available | not available | not available | not available | not available |
| Methylparatyrosine | not available | not available | not available | not available | not available | not available | |
| Hrachovy 1994 | ACTH (high‐dose) | not available | not available | not available | not available | not available | not available |
| ACTH (low‐dose) | not available | not available | not available | not available | not available | not available | |
| Lux 2004 | Vigabatrin | 32:20 | 22 weeks | 26 weeks | 26 weeks | 4 weeks | no |
| Hormonal | 32:23 | 22 weeks | 26 weeks | 26 weeks | 4 weeks | no | |
| Bitton 2012 | Flunarizine | 25:9 | Not available | 28 weeks | 28weeks | Not available | no |
| Placebo | 22:12 | Not available | 31 weeks | 31 weeks | Not available | no | |
| Shu 2009 | ACTH (high ‐ dose) | Not available | Not available | Not available | Not available | Not available | Not available |
| ACTH (low dose) | Not available | Not available | Not available | Not available | Not available | Not available | |
| Tsai 2009 | Ganaxolone | Not available | Not available | Not available | 17.3 to 104 weeks | Not available | Not available |
| Placebo | Not available | Not available | Not available | 17.3 to 104 weeks | Not available | Not available | |
| Vigevano 1997 | Vigabatrin | 14:9 | 25 weeks | not available | 27 weeks | not available | no |
| ACTH | 7:12 | 26 weeks | not available | not available | not available | no | |
| Yanagaki 1999 | ACTH (high‐dose) | 8:5 | 20 weeks | not available | 28 weeks | 7 weeks | yes |
| ACTH (low‐dose) | 7:5 | 23 weeks | not available | 41 weeks | 16 weeks | yes | |
| Zou 2010 | ACTH | 14:5 | 6.6 months | not available | unclear | not available | yes, some |
| ACTH + magnesium sulphate | 9:10 | 5.5 months | not available | unclear | not available | yes, some |
3. Outcomes.
| Study ID | No. participants | Intervention | Development | Seizures | Spasms stopped | Spasms reduced | Relapse rates | Resolution of EEG |
| Appleton 1999 | 20 | Vigabatrin | not reported | not reported | 35% | 40% reduction | 57% | 71% |
| 20 | Placebo | not reported | not reported | 10% | 15% reduction | 100% | 50% | |
| Askalan 2003 | 3 | ACTH | not reported | not reported | 100% | not reported | not reported | 100% |
| 6 | Vigabatrin | not reported | not reported | 100% | not reported | not reported | 100% | |
| Baram 1996 | 15 | ACTH | not reported | 50% (2‐48 month F.U.) | 87% | not reported | 14% | 87% |
| 14 | Prednisone | not reported | 50% (2‐46 month F.U.) | 29% | not reported | 29% | 29% | |
| Chiron 1997 | 11 | Vigabatrin | not reported | not reported | 100% | not reported | 9% | not reported |
| 11 | Hydrocortisone | not reported | not reported | 45% | not reported | not reported | not reported | |
| Debus 2004 | 20 | Sulthiame | not reported | not reported | 29% | not reported | none | 29% |
| 17 | Placebo | not reported | not reported | <1% | not reported | none | <1% | |
| Dreifuss 1986 | 27 | Nitrazepam | not reported | not reported | not reported | 66% | not reported | not reported |
| 25 | ACTH | not reported | not reported | not reported | 50% | not reported | not reported | |
| Dyken 1985 | 13 (/17) | Valproate | not reported | not reported | not reported | p<0.04 | not reported | not reported |
| 17 (/17) | Placebo | not reported | not reported | not reported | p<0.04 | not reported | not reported | |
| Elterman 2010 | 114 | Vigabatrin (low‐dose) | not reported | not reported | 11% | not reported | not reported | 11% |
| 107 | Vigabatrin (high‐dose) | not reported | not reported | 36% | not reported | not reported | 36% | |
| Hrachovy 1983 | 12 | ACTH | not reported | not reported | 42% | not reported | 60% | not reported |
| 12 | Prednisone | not reported | not reported | 33% | not reported | 25% | not reported | |
| Hrachovy 1989 | 12 | Methysergide | no significant difference | not reported | 8% | 25% | 100% | not reported |
| 12 | Methylparatyrosine | no significant difference | not reported | 16% | 17% | 50% | not reported | |
| Hrachovy 1994 | 30 | ACTH (high‐dose) | not reported | not reported | 43% | not reported | 15% | 23% |
| 29 | ACTH (low‐dose) | not reported | not reported | 48% | not reported | 21% | 21% | |
| Lux 2004 | 52 | Vigabatrin | see text | see text | 54% | not reported | 56% | |
| 55 | Hormonal | see text | see text | 72% | not reported | 88% | ||
| Bitton 2012 | 34 | Flunarazine | no significant improvement | not reported | 50% | not reported | not reported | not reported |
| 34 | Placebo | no significant improvement | not reported | 62% | not reported | not reported | not reported | |
| Shu 2009 | 30 | ACTH (high‐dose) | not reported | not reported | not reported | not reported | not reported | not reported |
| ACTH (low‐dose) | not reported | not reported | not reported | not reported | not reported | not reported | ||
| Tsai 2009 | 56 | Ganaxolone | not reported | not reported | not reported | not reported | not reported | not reported |
| Placebo | not reported | not reported | not reported | not reported | not reported | not reported | ||
| Vigevano 1997 | 23 | Vigabatrin | no significance difference | 25% (9‐44 months F.U.) | 48% | not reported | not reported | 36% |
| 19 | ACTH | no significance difference | 25% (9‐44 months F.U.) | 74% | not reported | not reported | 79% | |
| Yanagaki 1999 | 13 | ACTH (high‐dose) | no significance difference | not reported | 84% | not reported | 27% | 61% |
| 13 | ACTH (low‐dose) | no significance difference | not reported | 69% | not reported | 33% | 33% | |
| Zou 2010 | 19 | ACTH alone | not clear | not reported | 42% | 7 of 11 remaining | none | 26% |
| 19 | ACTH + magnesium sulphate | not clear | not reported | 63% | 3 of 7 remaining | 17% | 47% |
4. Adverse effects and deaths.
| Study ID | Intervention | No. adverse effects | Types of side effects | No. deaths | Cause of death |
| Appleton 1999 | Vigabatrin | none | none | ||
| Placebo | none | none | |||
| Askalan 2003 | ACTH | not reported | not reported | ||
| Vigabatrin | not reported | not reported | |||
| Baram 1996 | ACTH | none | |||
| Prednisone | not reported | none | |||
| Chiron 1997 | Vigabatrin | none | none | ||
| Hydrocortisone | 1 | not given | none | ||
| Debus 2004 | Sulthiame | 1 | somnolence | none | |
| Placebo | 0 | none | |||
| Dreifuss 1986 | Nitrazepam | none | none | ||
| ACTH | 6 | 1 = meleana, 1 = hypertension, 4 = not given | 1 | unknown | |
| Dyken 1985 | Valproate | not reported | not reported | ||
| Placebo | not reported | not reported | |||
| Elterman 2010 | Low‐dose vigabatrin | 9, unclear which group | not given | 2 deaths occurred, unclear which group | one unknown, one due to pulmonary haemorrhage |
| High‐dose vigabatrin | 9, unclear which group | not given | 2 deaths occurred, unclear which group | one unknown, one due to pulmonary haemorrhage | |
| Hrachovy 1983 | ACTH | not reported | none | ||
| Prednisone | not reported | none | |||
| Hrachovy 1989 | Methysergide | none | none | ||
| Methylparatyrosine | none | none | |||
| Hrachovy 1994 | High‐dose ACTH | not reported | not available | ||
| Low‐dose ACTH | not reported | not available | |||
| Lux 2004 | Vigabatrin | 2 | drowsiness and vomiting | 3 | |
| Hormonal | 2 | rash and irritability, vomiting, and high BP | 2 | ||
| Bitton 2012 | Flunarizine | not reported | not reported | 3 | not reported |
| Placebo | not reported | not reported | 3 | not reported | |
| Shu 2009 | High‐dose ACTH | 93% | not reported | not reported | not reported |
| High‐dose ACTH | 20% | not reported | not reported | not reported | |
| Tsai 2009 | Ganaxolone | more frequent | somnolence, lethargy, and irritability | not reported | not reported |
| Placebo | less frequent | not reported | not reported | ||
| Vigevano 1997 | Vigabatrin | 1 | excessive irritability and agitation | 1 death occurred, unclear which group | unknown |
| ACTH | 1 | not given | 1 death occurred, unclear which group | unknown | |
| Yanagaki 1999 | High‐dose ACTH | none | none | ||
| Low‐dose ACTH | none | none | |||
| Zou 2010 | ACTH | 16 | pyrexia, URTI, diarrhoea, anorexia, hypertension, insomnia, irritability, decreased heart rate with prolonged PR interval. | none | |
| ACTH + magnesium sulphate | 10 | pyrexia, URTI, diarrhoea, anorexia, vomiting | none |
Appleton 1999 This was a short‐term, randomised, double‐blind, placebo‐controlled multicentre trial of 40 participants. Inclusion criteria included participants between 1 and 20 months of age with newly diagnosed and previously untreated infantile spasms in whom the EEG demonstrated classical or modified hypsarrhythmia. A spasm was defined as a sudden, generally bilateral and symmetrical contraction of the muscles of the neck, trunk, and extremities (flexor, extensor, or mixed). The principal exclusion criterion was the use of any medication, including steroids, that could be considered to be an anti‐epileptic drug within a two‐month period before entry into the trial. The trial consisted of a five‐day double‐blind phase, during which patients received vigabatrin or placebo. Twenty participants received vigabatrin and 20 received placebo. The initial starting dose was 50 mg/kg/day for 24 hours; if spasms did not cease completely, the dose was increased to 100 mg/kg/day, and this dose was maintained for a further 48 hours. The dose could then be increased further to 150 mg/kg/day. Randomisation was pre‐determined by a code held by the pharmacy department for each participating hospital. Both recipients and assessors were blinded. Outcomes reported were cessation of spasms, reduction in spasms, resolution of hypsarrhythmia, and relapse rates. Some patients were lost to follow‐up, but not during the double‐blind phase.
Askalan 2003 This was an open‐label, randomised, single‐centred trial of nine participants. Inclusion criteria included participants between 3 and 16 months of age who presented with infantile spasms. The diagnosis of infantile spasms was confirmed by the presence of myoclonic flexor spasms, extensor spasms, or both, with video‐EEG showing hypsarrhythmia or modified hypsarrhythmia. Individuals were excluded if they had previously received vigabatrin or corticosteroids; had a known visual disturbance; or had a known medical condition for which corticosteroids would be contraindicated. The trial consisted of three participants receiving 150 IU/m2/day ACTH intramuscularly in two divided doses for one week, followed by 75 IU/m2/day ACTH for one week; and six participants receiving 100 mg/kg/day vigabatrin orally in two divided doses for two days, then increasing to 150 mg/kg/day for the remainder of the two‐week period. After the initial two‐week period, responders remained on their initial trial treatment and completed either a 12‐week tapering protocol for ACTH or 18 months of vigabatrin. Non‐responders were crossed over to the alternate trial drug while being tapered off the original study drug over one week. The method of randomisation was not stated. Assessors interpreting the EEGs and psychologists were blinded. Outcomes reported were cessation of spasms, resolution of hypsarrhythmia, short‐term cognitive outcomes, language scores, and the evolution of epilepsy and autism.
Baram 1996 This was a randomised, single‐blinded, single‐centred trial of 29 participants. Infants with clinical infantile spasms were considered for the study and underwent 24‐hour video‐EEG to ascertain hypsarrhythmia and clinical spasms. The trial consisted of a two‐week period during which 15 participants were treated with ACTH 150 units/m2/day intramuscularly in two divided doses; and 14 participants were treated with prednisone 2 mg/kg/day given orally in two divided doses. A computer‐generated random number list determined treatment, and allocation was concealed (as confirmed by Dr Baram, direct correspondence). Recipients were not blinded, but blinded assessment of outcomes was performed. Outcomes reported were cessation of spasms, resolution of hypsarrhythmia, relapse rates, and development of other seizure types within the follow‐up period.
Chiron 1997 This was a randomised, multicentre trial of 22 participants with infantile spasms, all of whom had tuberous sclerosis. Inclusion criteria included epileptic spasms recorded on EEG or seen by an experienced clinician, diffuse interictal paroxysmal activity, and age ranging from one month to two years. Participants were excluded if they had been previously treated with steroids or vigabatrin but not with other anticonvulsant medications. The trial spanned a one‐month period, during which 11 infants received vigabatrin at a dose of 150 mg/kg/day, and 11 infants received hydrocortisone at a dose of 15 mg/kg/day. The method of randomisation was not stated. Neither recipients nor assessors were blinded. Outcomes reported were cessation of spasms, resolution of hypsarrhythmia, and relapse rates.
Debus 2004 This was a randomised, double‐blind, multicentre trial of 51 participants. Inclusion criteria included children between 3 and 18 months of age with newly diagnosed infantile spasms. The diagnosis was confirmed by the presence of hypsarrhythmia or hemi‐hypsarrhythmia on an EEG that had to contain periods of sleep and wakefulness. Participants were excluded if they had a history of epilepsy before the development of infantile spasms, unless they were not treated or had been treated with phenytoin or phenobarbitone; or if they had other ongoing or acute diseases. The trial consisted of a nine‐day (days one to nine) treatment period with pyridoxine for all participants. On days one to three, all participants received just pyridoxine. From day four of the trial, 28 participants received sulthiame at 5 mg/kg/day in two divided doses, and 23 participants received placebo. If no response was seen after three days, the dose of the trial drug was doubled for three days. Randomisation was concealed by sealed envelopes (personal communication). The recipients were blinded, but the assessors were not blinded to treatment allocation. Outcomes reported were cessation of spasms, resolution of hypsarrhythmia, and relapse rates.
Dreifuss 1986 This was a randomised multicentre trial of 52 participants. Inclusion criteria included participants between 1 and 24 months of age with infantile spasms documented by a hypsarrhythmic or a modified hypsarrhythmic pattern on EEG. None of the participants had received steroids or nitrazepam before entry into the trial. The trial consisted of a four‐week period during which 27 participants received nitrazepam, and 25 participants ACTH. Nitrazepam was started at 0.2 mg/kg/day in two divided doses or at 1 mg twice daily, whichever was greater, and was adjusted twice weekly by increments of 0.3 to 0.4 mg/kg/day. Maintenance dosage ranged from 4.8 to 9 mg/day. ACTH was given as a once‐daily intramuscular injection of 40 units. A computer‐generated randomisation code determined treatment, but concealment of allocation was not clearly stated. Recipients were not blinded, but blinded assessment of outcome was performed. The only outcome reported was reduction in spasm frequency. A discrepancy between the review authors was noted over the assessment of how many participants were lost to follow‐up in this trial; this discrepancy arose because the authors were unclear in their paper as to how many participants were lost to follow‐up. We have been unable to contact the author to clarify this point, so the number of participants lost to follow‐up remains unclear.
Dyken 1985 This was a randomised, placebo‐controlled, cross‐over, single‐centre trial of 17 participants. Inclusion criteria included participants between 8 and 83 months of age with a clinical diagnosis of infantile spasms and hypsarrhythmia on EEG, who had previously failed to respond to steroids. Many participants had received other anticonvulsant medications. The trial compared sodium valproate against placebo, but the length of the trial and the dosages used were not given. A computer‐generated random numbers table was used to allocate participants, but concealment of allocation was not clearly stated. Recipients were blinded, and blinded assessment of outcome was performed. Only reduction in spasm frequency was reported as an outcome. Four participants were lost to follow‐up.
Elterman 2010 This was a randomised, multicentre trial of 221 participants. Inclusion criteria included participants younger than two years of age but weighing at least 3.5 kg. Diagnostic criteria included infantile spasms and an EEG pattern typical of hypsarrhythmia or modified hypsarrhythmia, or multifocal spike wave discharges, or a video‐EEG recording capturing an event confirming the diagnosis of infantile spasms electroencephalographically. Participants were receiving stable doses of antiepilepsy medication, if applicable, and were unresponsive to a 50‐mg bolus of pyridoxine, when appropriate. Participants were excluded if they had received an investigational drug within 30 days of enrolment (one‐week washout for ganaxolone): and had been or were currently taking corticosteroids (brief exposure to low‐dose corticosteroids for inflammatory conditions or replacement therapy was acceptable but required a one‐week washout), valproic acid; or felbamate within 60 days of enrolment; or were receiving more than two standard antiepilepsy drugs to treat non–infantile spasm seizures. Participants were also excluded if they had a treatable or progressive cause of seizures; had current active medical disorders; had a caregiver who was unable to reliably record seizures or recall adverse events; or had showed poor compliance with medication. The trial consisted of a two‐week period during which participants received low‐dose vigabatrin 18 to 36 mg/kg/day or high‐dose vigabatrin 100 to 148 mg/kg/day. The dose was based on the participant's weight and, in the high‐dose group, was titrated over the first seven days of the study. Medication was given twice daily unless the total dose was less than 250 mg per day, in which case daily dosing was permitted. Participants in either group who continued to have infantile spasms after seven days of the full dose of vigabatrin specified for their weight could have the dose increased weekly by 25 to 50 mg/kg/day to a maximum dose of 200 mg/kg/day. The method of randomisation was not stated. The authors stated that this was a single‐blinded study, and that the recipients were blinded but the assessors were not. However, the recipients were not blinded to the dose of vigabatrin given (it was supplied as 500‐mg tablets in open‐labelled bottles), and caregivers were simply 'masked' as to whether their children were assigned to the high‐dose or the low‐dose group. Outcomes reported were cessation of spasms;resolution of hypsarrhythmia;time to cessation of spasms; and relapse rates. Safety data were also published. A total of 221 participants were randomly assigned: all were followed up as intention to treat, none were lost to follow‐up.
Hrachovy 1983 This was a randomised single‐centre trial (using a double‐dummy technique) of 24 participants. Inclusion criteria included participants with infantile spasms and hypsarrhythmic EEG patterns on serial 24‐hour video and polygraphic monitoring. No participant had been previously treated with steroids, but some had received other anticonvulsant medications. Twelve participants received ACTH 20 to 30 units/day for two weeks, which was then tapered over one week if a response was seen. If a response was not seen, participants continued a further four weeks before the dose was tapered. These participants also received a prednisone placebo. Twelve participants received prednisone, 2 mg/kg/day for two weeks, which was then tapered over one week if a response was seen. If a response was not seen, they continued a further four weeks before the dose was tapered. These participants also received an ACTH gel placebo. The method of randomisation was not given. Recipients were not blinded, but it was unclear if blinded assessment of outcome was performed. Outcomes reported were complete cessation of spasms and relapse rates.
Hrachovy 1989 This was a randomised trial of 24 participants. Inclusion criteria included newly diagnosed participants with infantile spasms and hypsarrhythmic EEGs. The trial consisted of a three‐week period during which 12 participants were treated with methysergide 2 mg/m2/day for the first seven days, then 5 mg/m2/day for the next 14 days, and 12 participants were treated with alpha‐methylparatyrosine 500 mg/m2/day, increasing by 150 mg/m2/day on day two and thereafter until a maximum dosage of 1250 mg/m2/day was reached. The method of randomisation was not given. It was not clear whether recipients and assessors were blinded. Authors reported cessation of spasms, a reduction in the number of spasms, and developmental status at three weeks as outcome measures.
Hrachovy 1994 This was a randomised controlled trial of 59 participants. Inclusion criteria included infants in whom a diagnosis of infantile spasms had recently been made, who had hypsarrhythmic EEG findings. They were excluded if they had previously received steroids. Thirty participants received 150 units/m2 of ACTH per day for three weeks, with a tapering dose over a further nine weeks, and 29 participants received 20 to 30 units/day of ACTH for two to six weeks, tapering over one week. The method of randomisation was not stated. Recipients were not blinded, but blinded assessment of outcomes was performed. Outcomes reported were complete cessation of spasms, resolution of hypsarrhythmia, and relapse rates. Nine participants were lost to follow‐up.
Lux 2004 This was an open, randomised, controlled, multicentre trial of 107 participants. Inclusion criteria were participants between 2 and 12 months of age with a clinical diagnosis of infantile spasms and a hypsarrhythmic or similar EEG. Exclusion criteria included a diagnosis or high risk of tuberous sclerosis; previous treatment with or a contraindication to vigabatrin or hormonal treatments; a lethal or potentially lethal disorder other than infantile spasms; inability of parents or guardians to give informed, signed consent or to know when spasms stop; leaving the UK unexpectedly within one month of randomisation; enrolment in a concurrent trial that either used treatment that might affect the outcome measures or was labour‐intensive for participants, guardians, or medical practitioners. Fifty‐two participants received vigabatrin given orally in two divided doses: 50 mg/kg/day for the first two doses, increasing to 100 mg/kg/day and then to 150 mg/kg/day at 96 hours if no response was seen. Twenty‐five participants received tetracosactide depot given intramuscularly, 0.5 mg (40 IU) on alternate days for two weeks, and increased to 0.75 mg (60 IU) on alternate days if no response was seen after one week. Thirty participants received prednisolone given orally, 10 mg four times daily for two weeks, increasing to 20 mg three times a day after one week if no response was seen. Randomisation was concealed by sealed envelopes. Neither recipients nor assessors were blinded. Outcomes reported were cessation of spasms and resolution of hypsarrhythmia, relapse rates, development at 14 months of age, seizure rates at 14 months of age, development at 4 years of age, and seizure rates at 4 years of age. One participant was lost to developmental follow‐up at 14 months of age, and 30 participants were lost to follow‐up at 4 years of age.
This was a study in which flunarizine or placebo was administered as an add‐on drug to vigabatrin to 68 infants aged 3 to 18 months, with new onset of infantile spasms in a double‐blind, randomised, controlled trial. Infants were included if they had 'de novo' onset of infantile spasms and an EEG with hypsarrhythmia or modified hypsarrhythmia Children were excluded if they had a known neurometabolic or neurodegenerative disease at study entry or a pre‐existing medical condition contraindicating steroid treatment, if they could not obtain the neuropsychological evaluation before enrolment, or if families were unable to comply with follow‐up visits.All children received vigabatrin 100 mg/kg/day for 3 days followed by 150 mg/kg/day when spasms persisted, as first‐line treatment. Responders continued to take vigabatrin for 6 months. If after 2 weeks, vigabatrin failed to arrest clinical spasms and abolish hypsarrhythmia, children were switched to synthetic adrenocorticotropic hormone (sACTH) given intramuscularly, and vigabatrin was tapered over a week. The dose of sACTH given every 2 days was calculated to be equivalent to 150 IU of natural ACTH/m2/day for 2 weeks, followed by a 10‐week tapering schedule. If clinical spasms and/or hypsarrhythmia persisted after 2 weeks of treatment with sACTH,the children were switched to topiramate (10 mg/kg/day).Outcome was reported as development at 24 months of age using the Vineland Adaptive Behaviour Scale and the Bayley Scales of Infant Development. Some participants were lost to follow‐up.
This was a prospective, randomised, controlled trial of 30 children with West's syndrome. Participants received either ACTH 50 IU/day for two weeks, then tapered to zero over two weeks, or 0.4 IU/kg for two weeks, increased to 1 IU/ kg for a further two weeks if no reponse was seen, then tapered to zero over two weeks. The method of randomisation is not clear. It is not clear whether both participants and assessors were blinded. Outcomes reported were response rates, relapse rates, time to relapse, long‐term outcome at 12 months, and adverse effects.
Thisdouble‐blind, placebo‐controlled randomised trial of 56 participants with infantile spasms compared ganaxolone with placebo. Infants were randomly assigned to receive either ganaxolone or placebo for one week, then all infants received ganaxolone while still blinded to the treatment of the previous week. Subjects were allowed two concomitant antiepileptic drugs (AEDs). The method of randomisation is not clear. Outcomes reported were the reduction of spasm cluster frequency in the first week, spasm frequency after two weeks of treatment, investigator/caregiver global assessment, and responder rates (defined as greater than 50% reduction in spasm frequency). Side effects were reported. It is not clear whether any subjects were lost to follow‐up.
Vigevano 1997 This was a randomised (response‐mediated, cross‐over) single‐centre trial of 42 participants. Inclusion criteria included participants with newly diagnosed and previously untreated infantile spasms diagnosed according to the International League Against Epilepsy (ILAE) classification. Twenty‐three participants received vigabatrin at 100 to 150 mg/kg/day, and 19 received ACTH at 10 units/day. The method of randomisation was not stated, but direct correspondence with the authors revealed a quasi‐random method (i.e. alternate allocation of therapy). Recipients were not blinded, and it was not clear whether assessors of outcomes were blinded. Outcomes reported were cessation of spasms, resolution of hypsarrhythmia, and short‐term effects on subsequent epilepsy rates and cognitive function.
Yanagaki 1999 This was a randomised single‐centre trial of 26 participants. Inclusion criteria included participants with infantile spasms and hypsarrhythmia on video‐EEG. Individuals were excluded if they had received previous steroid or intravenous gamma globulin treatment. Thirteen participants received 1 unit/kg/day of synthetic ACTH for two weeks, tapering over a further two weeks; and 13 participants received 0.2 units/kg/day of synthetic ACTH for two weeks, tapering over a further two weeks. The method of randomisation was not stated. It was not clear whether recipients and assessors were blinded. Outcomes reported were cessation of spasms, resolution of hypsarrhythmia, relapse rates, and the short‐term effect on development. One participant was lost to follow‐up.
This was a single‐centre trial of 38 participants. Inclusion criteria included participants with infantile spasms and hypsarrhythmia. Exclusions included acute associated illness and previous treatment with ACTH, prednisolone, hydrocortisone, or magnesium sulphate. All participants received ACTH 25 units/day, and half received magnesium sulphate 0.25 g/kg/day for three weeks. Allocation was not concealed, but randomisation was applied. Recipients and outcomes were not blinded. Outcomes reported were cessation of spasms and resolution of hypsarrhythmia: some developmental outcome data were given, but no between group comparisons were done. No deaths occurred.
Risk of bias in included studies
Overall, methodological quality of the included studies was poor. All but two of the studies had small numbers of participants—the largest included 221 and the second largest 107 participants. All the other studies enrolled fewer than 70 participants, and subsequently the power of most of the studies was low. There were five placebo‐controlled trials (Appleton 1999; Debus 2004; Dyken 1985; Bitton 2012; Tsai 2009 ). One trial used a double‐dummy technique (Hrachovy 1983). Although all studies stated that they were RCTs, only six stated the method of randomisation, and only four indicated that concealment of allocation had been performed. Recipients were blinded in five studies; blinding did not take place in eight studies, and it was unclear whether blinding was performed in the remaining studies. Six studies clearly stated that those measuring outcomes were unaware of the assigned therapy. Six studies had losses to follow‐up. Six studies were multicentre trials, and six recruited participants from a single centre; it was unclear how many centres were involved in recruiting participants in the remaining studies. Of the 18 RCTs considered in this review, all but two had a high risk of bias (seeTable 1: Methodological quality of included studies).
Also of importance for interpretation of the meta‐analyses below, the results of longer‐term outcomes were complicated by the fact that infants initially randomly assigned to receive one therapy were likely to have gone on to receive the comparative or other therapies, or both, within the follow‐up period if they failed to respond to the first therapy.
Effects of interventions
There were characteristics of the population that may affect outcome regardless of the treatment (seeTable 2: Participants). The male to female ratio was given for 12 of the studies and was 1.1:1. The mean age at spasm onset was given in seven studies and ranged from 20 to 31 weeks. The mean age at which diagnosis of spasms was made was given for four studies and ranged from 26 to 31 weeks. The mean age at trial entry (i.e. the age at which the trial treatment was started) was given in 11 studies and ranged from 15.1 to 104 weeks. The time delay between onset of spasms and start of treatment was given in three studies and ranged from 3 to 16 weeks. In five studies, other treatments had been previously tried; in five studies, other treatments had not been used; and in eight studies, it was unclear whether previous treatment had been received.
We had also planned to look for heterogeneity according to different diagnostic groupings, for example, tuberous sclerosis and Down syndrome; and we planned to look at whether participants had demonstrated developmental delay before the onset of spasms. Unfortunately, for most of the studies selected, this information was not available and therefore could not be analysed.
Vigabatrin versus placebo
One study was included (Appleton 1999).
Effects on cessation of spasms: The Appleton 1999 study enrolled 40 participants and showed complete cessation of spasms in 7 of 20 (35%) participants treated with vigabatrin compared with 2 of 20 (10%) treated with placebo (Peto OR 4.1, 95% confidence interval (CI) 0.9 to 17.5). Effects on time taken to achieve cessation of spasms were not reported as an outcome in this study. Effects on reduction in the number of spasms: The Appleton 1999 study showed > 70% reduction in spasms in 40% of the group treated with vigabatrin compared with 15% in the group treated with placebo. However, it was not clear from the article what proportion of the two groups of participants was reflected in these figures, and whether the figures applied to the whole group, or just to those participants in whom complete cessation of spasms was not achieved. Effects on relapse rates: Four of seven participants who responded to vigabatrin relapsed, and all participants successfully treated with placebo relapsed. Overall, only three participants treated with vigabatrin and no participant treated with placebo remained spasm free within the four‐week study period (Peto OR 8.2, 95% CI 0.8 to 84). Effects on time taken to relapse were not reported as an outcome in this study. Effects on resolution of EEG: Five of the seven participants who were spasm free with vigabatrin showed resolution of EEG, compared with one of the two participants who had become spasm free while taking placebo (Peto OR 2.4, 95% CI 0.1 to 54.6). Effects on psychomotor development were not reported as an outcome in this study. Effects on subsequent seizure rates were not reported as an outcome in this study. No side effects severe enough to warrant stopping treatment were reported in this study. No deaths were reported.
Sodium valproate versus placebo
One study was included (Dyken 1985).
Effects on cessation of spasms were not reported as an outcome in this study. Effects on time taken to achieve cessation of spasms were not reported as an outcome in this study. Effects on reduction in the number of spasms: The Dyken 1985 study used a spasm index (SI) = (spasm frequency x spasm duration/total observation time) x 1000 to calculate the mean reduction in spasm frequency from baseline. At the end of the four‐week treatment period, valproate treatment was found to have a lower mean spasm index than placebo when valproate was administered first (P < 0.04). No significant difference between placebo and valproate was noted during the second level of treatment. It was not possible to calculate a Peto OR. Effects on relapse rates were not reported as an outcome in this study. Effects on time taken to relapse were not reported as an outcome in this study. Effects on resolution of EEG were not reported as an outcome in this study. Effects on psychomotor development were not reported as an outcome in this study. Effects on subsequent seizure rates were not reported as an outcome in this study. Side effects were not reported as an outcome in this study. Deaths were not reported as an outcome in this study.
Sulthiame versus placebo
One study was included (Debus 2004).
Effects on cessation of spasms:The Debus 2004 study found 8 of 28 participants treated with sulthiame to be spasm free as compared with 1 of the 23 participants given placebo (Peto OR 5.13, 95% CI 1.22 to 21.47). Effects on time taken to cessation of spasms were not reported as an outcome in this study. Effects on reduction in the number of spasms were not reported as an outcome in this study. Effects on relapse rates were not reported as an outcome in this study. Effects on time taken to relapse were not reported as an outcome in this study. Effects on resolution of EEG: The Debus 2004 study stated that 8 of 28 participants treated with sulthiame had resolution of their EEG as compared with 1 of 23 participants given placebo (Peto OR 5.13, 95% CI 1.22 to 21.47). Effects on psychomotor development were not reported as an outcome in this study. Effects on subsequent seizure rates were not reported as an outcome in this study. Side effects: Sulthaime had to be withdrawn in one patient because of somnolence. No deaths occurred in either group.
Low‐dose vigabatrin versus high‐dose vigabatrin
One study was included (Elterman 2010).
Effects on cessation of spasms: On the basis of primary criteria and an intention‐to‐treat approach (please see below), 17 of 107 participants treated with high‐dose vigabatrin became spasm free compared with 8 of 114 participants treated with low‐dose vigabatrin (Peto OR 0.41, 95% CI 0.18 to 0.95). Effects on time taken to achieve cessation of spasms within the initial two‐week study period were not reported as an outcome in this study. Effects on reduction in the number of spasms were not reported as an outcome in this study. Effects on relapse rates: Again on the basis of primary criteria, 2 of 17 responders in the high‐dose group subsequently relapsed compared with 2 of 8 participants in the low‐dose group. Effects on time taken to relapse: In the high‐dose group, the time taken to relapse was 162 days (range 53 to 270 days) compared with 45 days (range 31 to 58 days) in the low‐dose group (Peto OR 0.25, 95% CI 0.25 to 24.32). Effects on resolution of EEG: 8 of 75 participants treated with low‐dose vigabatrin had resolution of their EEG compared with 24 of 67 participants treated with high‐dose vigabatrin (Peto OR 0.24, 95% CI 0.11 to 0.52). Effects on psychomotor development were not reported as an outcome in this study. Effects on subsequent seizure rates were not reported as an outcome in this study. Side effects: In total, vigabatrin was stopped in five participants as a result of an adverse event directly attributed to the vigabatrin therapy; the reasons were not given, and the dose that had been given was not stated. Three deaths were reported in this study; all three were judged to be unrelated to vigabatrin.
However, it is important to note the following when assessing the results of this study.
This study was initially published in 2001. In the initial paper, many participants had been 'lost to follow‐up' because they were excluded from the analysis for violating the protocol. However, in the final article, published in 2010, the authors report the findings on an intention‐to‐treat basis (now more widely accepted as best practise). This explains why the numbers presented in this updated review are different from those presented previously, with a lower rate of participants lost to follow‐up.
In their original study, primary responders were defined as subjects who were spasm free and obtained a video‐EEG within 3 days of the seventh day of spasm freedom, which showed no indication of spasms or hypsarrhythmia. During the course of the study, it became apparent that obtaining the video‐EEG within 3 days of spasm cessation was not feasible for most subjects. A post hoc analysis was therefore performed, relaxing the timing of vidoe‐EEG to any subsequent visit. This analysis yielded response rates of 33/107 in the high‐dose group and 15/114 in the low‐dose group (P = 0.0014).
Vigabatrin versus hydrocortisone
One study (Chiron 1997) considered only participants with tuberous sclerosis.
Effects on cessation of spasms: The Chiron 1997 study compared vigabatrin (150 mg/kg/day) and hydrocortisone (15 mg/kg/day) in 22 infants with infantile spasms due to tuberous sclerosis and found in the initial phase that 11 of the 11 participants (100%) treated with vigabatrin were spasm free as compared with 5 of 11 participants (45%) treated with hydrocortisone (Peto OR 13.8, 95% CI 2.21 to 86.35). Effects on time taken to achieve cessation of spasms: On average, the 11 responders to vigabatrin took 4 days (range 0.5 to 14 days, median 2 days) to achieve complete cessation of spasms, and the 5 responders to hydrocortisone took an average of 13 days (range 3 to 30 days, median 23.5 days) (weighted mean difference (WMD) ‐8.8, 95% CI ‐19.2 to 1.6). Effects on reduction in the number of spasms were not reported as an outcome in this study. Effects on relapse rates: 10 of the 11 participants who responded to vigabatrin remained spasm free; this information was not given for the five responders to hydrocortisone. Effects on time taken to relapse were not reported as an outcome in this study. Effects on resolution of EEG were not reported as an outcome in this study. Effects on psychomotor development were not reported as an outcome in this study. Effects on subsequent seizure rates were not reported as an outcome in this study. Side effects: Hydrocortisone had to be stopped in one patient; no reason was given. No deaths were reported in this study.
Vigabatrin versus hormonal treatment (ACTH, tetracosactide, or high‐dose prednisolone), excluding hydrocortisone
Three studies were included (Askalan 2003, Lux 2004, and Vigevano 1997).
Effects on cessation of spasms: The Askalan 2003 trial of nine participants showed that all six of the participants randomly assigned to vigabatrin had cessation of spasms, as did the three participants randomly assigned to ACTH. The Lux 2004 trial showed that 28 of the 52 participants randomly assigned to receive vigabatrin had cessation of their spasms compared with 40 of the 55 participants randomly assigned to receive either tetracosactide or high‐dose prednisolone. The Vigevano 1997 trial of 42 participants showed cessation of spasms in 11 of the 23 participants randomly assigned to vigabatrin compared with 14 of the 19 participants randomly assigned to ACTH. Combining these three studies revealed that 45 of the 81 participants randomly assigned to vigabatrin had cessation of their spasms compared with 57 of the 77 randomly assigned to ACTH, tetracosactide, or high‐dose prednisolone (Peto OR 0.42, 95% CI 0.21 to 0.80).
Effects on time taken to achieve cessation of spasms: The Askalan 2003 study did not report on this outcome. The Lux 2004 study reported that the 28 responders to vigabatrin took a median of 11 and a half days to respond, but the 40 responders to tetracosactide or high‐dose prednisolone had a median response time of three days. In the Vigevano 1997 trial, the 11 responders to vigabatrin took between 1 and 14 days to achieve complete cessation of spasms, and the 14 responders to ACTH took between 2 and 12 days.
Effects on reduction in the number of spasms were not reported as an outcome in these three studies.
Effects on relapse rates were not reported as an outcome in the Askalan 2003 and Vigevano 1997 studies. The Lux 2004 study reported relapse in 9 of 28 responders on vigabatrin compared with 18 of 40 responders on tetracosactide or high‐dose prednisolone (Peto OR 0.59, 95% CI 0.22 to 1.57). Overall, 19 of the 52 participants randomly assigned to receive vigabatrin remained spasm free as compared with 22 of the 55 participants randomly assigned to receive tetracosactide or prednisolone.
Effects on time taken to first relapse were reported by Lux 2004 using actuarial curves. This outcome was not reported in two of the studies.
Effects on resolution of EEG: In the Askalan 2003 study, resolution occurred in all six responders to vigabatrin and in all three responders to ACTH. In the Lux 2004 investigation, resolution occurred in 20 of 28 responders to vigabatrin for whom it was measured, compared with 26 of 32 responders to tetracosactide or high‐dose prednisolone for whom it was measured. In the Vigevano 1997 study, resolution occurred in 4 of 11 participants responding to vigabatrin and in 11 of 14 participants responding to ACTH. Combining these three studies revealed that resolution occurred in 30 of 45 participants responding to vigabatrin and in 40 of 49 participants responding to ACTH (Peto OR 0.38, 95% CI 0.15 to 0.99).
Effects on psychomotor development:
Investigators in the Askalan 2003 study administered the Bayley Scales of Infant Development at trial entry and subsequently at 3, 12, and 24 months' follow‐up.
The Lux 2004 study reported that Vineland Adaptive Behaviour Scales (VABS) were obtained in 101 of 102 survivors (Figure 1) at 14 months of age. This study reported that no significant differences were noted in mean composite scores between tetracosactide and high‐dose prednisolone and vigabatrin (78.6 (SD 16.8) vs 77.5 (SD 12.7); difference 1.0, 95% CI ‐4.9 to 7.0; t99 = 0.35, P = 0.73). Mean composite scores were significantly higher in infants with no identified aetiology than in infants with a known aetiology (83.8 (SD 16.5) vs 73.3 (SD 11.4); difference 10.5, 95% CI 4.9 to 16.1; t97 = 3.74, P = 0.0003). Composite scores were also significantly higher in infants who had cessation of spasms than in those who did not (80.9 (SD 15.0) vs 72.7 (SD 13.4); difference 8.2, 95% CI 2.2 to 14.2; t99 = 2.70, P = 0.008). No significant differences in composite scores were noted between those with an interval between onset of spasms and treatment (lead time to treatment) of less than one month and those with an interval greater than one month (where this interval was known) for all infants (80.3 (SD 14.9) vs 76.1 (SD 15.1); difference 4.3, 95% CI ‐2.2 to 10.7; t89 = 1.32, P = 0.19) and for the subgroup with no identified underlying aetiology (87.4 (SD 16.2) vs 81.9 (SD 16.1); difference 5.5, 95% CI ‐5.1 to 16.1; t38 = 1.05, P = 0.30). Analysis of composite scores using ANOVA showed a significant interaction between treatment and underlying aetiology (F1,95 = 6.78, P = 0.011). For the subgroup of infants with no identified underlying aetiology, mean composite scores were higher in infants allocated hormone treatment than in those allocated vigabatrin (88.2 (SD 17.3) vs 78.9 (SD 14.3); difference 9.3, 95% CI 1.2 to 17.3; least significant difference test t95 = 2.28, P = 0.025). If the two infants whose aetiology was not fully investigated were added to the no identified aetiology group (the sensitivity analysis with the most extreme data against the detected effect), the interaction remained significant (F1,97 = 5.02, P = 0.027), but the difference between means was not significant (86.9 (SD 18.1) vs 79.5 (SD 14.3); difference 7.4, 95% CI ‐0.7 to 15.4; t97 = 1.82, P = 0.071). The mean composite scores in the group with an identified aetiology did not differ significantly between treatment groups (hormone 70.8 (SD 11.1) vs vigabatrin 75.9 (SD 11.3); difference 5.0, 95% CI ‐2.3 to 12.4; least significance difference test t95 = 1.36, P = 0.18).
The Lux study (Lux 2004) reported that VABS were obtained at four years of age in 77 infants of the original cohort. VABS scores for the 39 infants allocated hormonal treatment were 60 compared with 50 for the 38 infants allocated vigabatrin (Mann‐Whitney U = 575; P = .091; median difference 8 (95%CI ‐1 to 19). For those with no identified aetiology, VABS scores were 96 for the 21 infants allocated hormonal treatment compared with 63 for the 16 allocated vigabatrin (Mann Whitney U = 98.5; P = 0.033; median difference 14, 95% CI 1 to 42. VABS scores for those with proven aetiology were 45 for the 18 infants allocated hormonal treatment compared with 50 for the 21 allocated vigabatrin (Mann‐Whitney U = 179.5; P = 0.79; median difference ‐1, 95% CI ‐11 to 7).
The Vigevano report (Vigevano 1997) stated that there was no apparent difference in cognitive development between the two groups during follow‐up, which ranged from 9 to 44 months. They did not state how they assessed development and did not quantify it any further.
Effects on subsequent seizure rates:
The Askalan study (Askalan 2003) stated that the ACTH responder and two of the vigabatrin responders subsequently developed epilepsy during the two‐year follow‐up period; no further information was available. The Lux 2004 study (reported 2005) reported seizure rates at 14 months of age. They indicated that in the vigabatrin group, seven participants continued to have infantile spasms (no other seizures present) compared with six in the hormonal group. In the vigabatrin group, seven participants were free of spasms but had other seizures present, compared with 13 in the hormonal group; and five participants in the vigabatrin group had both infantile spasms and other seizures present compared with eight in the hormonal group. None of these comparisons were statistically significant. The Lux study (Lux 2004) also reported seizure rates at 4 years of age. They indicated that in the vigabatrin group, five participants continued to have infantile spasms, as did five participants in the hormonal group. All ten participants had seizures of other types. In addition, in the vigabatrin group, 16 participants were free of spasms but had other seizures present, compared with 12 in the hormonal group. None of these comparisons were statistically significant.
The Vigevano study (Vigevano 1997) found the incidence of other types of epileptic seizures to be 25% in both groups during follow‐up at 9 to 44 months. No study considered subsequent epilepsy rates at five years of age. Combining these studies revealed that 27 of 81 participants receiving vigabatrin had seizures at follow‐up compared with t33 of 77 participants receiving hormonal treatment, Peto OR ratio 0.69 (95% CI 0.36 to 1.32).
Side effects:
The Lux trial (Lux 2004) stopped vigabatrin in two participants because of drowsiness and vomiting; tetracosactide in one participant because of a rash; and high‐dose prednisolone in one participant because of irritability, vomiting, and high blood pressure. In the Vigevano trial (Vigevano 1997), investigators stopped vigabatrin in one participant because of excessive irritability, and ACTH in one participant with no reason given.
Deaths:
The Lux study (Lux 2004) reported nine deaths during follow‐up to four years of age; five occurred in the vigabatrin group and four in the hormonal group. One participant in the Vigevano 1997 study died, but the cause was not given, and it was unclear to which treatment group they had been randomly assigned.
ACTH versus high‐dose prednisolone
One study (Lux 2004) was included (nested within the comparison of vigabatrin with hormonal treatment).
Effects on cessation of spasms: 19 of 25 participants treated with ACTH (40 to 60 units/alternate days) had cessation of their spasms compared with 21 of 30 participants treated with prednisolone (40 to 60 mg/day) (Peto OR 1.36, 95% CI 0.41 to 4.53). Effects on time taken to achieve cessation were not reported as an outcome in this study. Effects on reduction in the number of spasms were not reported as an outcome in this study. Effects on relapse rates were not reported individually for these two treatments in this study. Effects on time taken to relapse were not reported as an outcome in this study. Effects on resolution of EEG: 16 of 18 ACTH responders, in whom it was measured, showed resolution of their EEG compared with 10 of 14 of the prednisolone responders (Peto OR 3.20, 95% CI 0.49 to 20.81). Effects on psychomotor development were not reported individually for these two treatments in this study. Effects on subsequent seizure rates were not reported individually for these two treatments in this study.
Side effects:
ACTH was withdrawn in one participant because of a rash, and prednisolone in one participant because of vomiting.
Deaths:
Two deaths were reported during follow‐up to 14 months.
ACTH versus low‐dose prednisone
Two studies (Baram 1996; Hrachovy 1983) were included. However, it is known that prednisone requires metabolism to convert it to the active form, prednisolone. It is now known that at birth, the newborn infant has poor capacity of HSD11B1 to reduce prednisone to prednisolone, and that this ability only slowly increases over the first six months of life. Hence, prednisone and prednisolone should not be considered equivalent treatments for infants, and it is likely that prednisone will not be as effective as prednisolone in the treatment of infantile spasms.
Effects on cessation of spasms:
Baram (Baram 1996) compared 15 participants treated with natural ACTH (150 units/m2/day) with 14 treated with prednisone (2 mg/kg/day), and showed ACTH to be superior to prednisone, with cessation of spasms in 13 of 15 participants and 4 of 14 participants, respectively. Hrachovy (Hrachovy 1983) compared 12 participants treated with ACTH (20 to 30 units/day) with 12 participants treated with prednisone (2 mg/kg/day). In the initial phase of the trial, 5 of 12 participants treated with ACTH had complete cessation of spasms compared with 4 of 12 treated with prednisone. Combining these two studies revealed that ACTH stopped the spasms in 18 of 27 participants compared with prednisone in 8 of 26 participants (Peto OR 4.19, 95% CI 1.42 to 12.35). Effects on time taken to achieve cessation:
In the Barram study (Baram 1996), on average, the 13 responders to ACTH took 3.2 days (range 1 to 7 days, median 2 days) to achieve complete cessation of spasms, and the 4 responders to prednisone took an average of 4 days (range 2 to 7 days, median 3.5 days; WMD ‐0.8, 95% CI ‐3.3 to 1.7). Effects on reduction in the number of spasms were not reported as an outcome in these studies. Effects on relapse rates:
In the Barram study (Baram 1996), 2 of the 13 participants who responded to ACTH relapsed, and none of the four responders to prednisone relapsed. In the Hrachovy trial (Hrachovy 1983), three of the five participants who responded to ACTH relapsed, and one of the four responders to prednisone also relapsed.
Overall, for Barram (Baram 1996), 11 participants who responded to ACTH remained spasm free and the four responders to prednisone also remained spasm free. For Hrachovy (Hrachovy 1983), two participants successfully treated with ACTH remained spasm free, and three successfully treated with prednisone also remained spasm free within the study period. The combined Peto OR for these two studies was 2.6 (95% CI 0.8 to 8.1). Effects on time taken to relapse were not reported as an outcome in these studies. Effects on resolution of EEG:
For Baram (Baram 1996), this study showed resolution of their EEG in 13 of 15 participants treated with ACTH compared with 4 of 14 participants treated with prednisone (Peto OR 10.10, 95% CI 2.36 to 43.19). For Hrachovy (Hrachovy 1983), 5 of 12 participants treated with ACTH had resolution of their EEG, but this was not reported for the group treated with prednisone, and their results were not therefore included in the OR calculation.
Effects on psychomotor development were not reported as an outcome in these studies. Effects on subsequent seizure rates:
In the Baram study (Baram 1996), investigators compared ACTH with prednisone and found that seven participants in both groups developed other seizure types over the period of follow‐up, 2 to 48 months. They did not report subsequent epilepsy rates at five years of age. Side effects were not reported in these studies. Deaths were not reported in these studies.
High‐dose ACTH versus low‐dose ACTH
Three studies were included (Hrachovy 1994; Yanagaki 1999; Shu 2009 ). Hrachovy and Shu used natural ACTH, and Yanagaki used synthetic ACTH. This made direct comparison difficult. Yanagaki stated that the dose equivalent of 1 IU of natural ACTH is 0.025 IU of synthetic ACTH. The calculation of the dose to be given also varied: some doses were calculated on body weight, some on surface area, and others were given a standard dose regardless of weight, height, or age. The results must be interpreted with caution.
Effects on cessation of spasms: The Hrachovy study (Hrachovy 1994) randomly assigned 30 participants to high‐dose ACTH (150 units/m2/day for three weeks with a tapering dose over a further nine weeks), and 29 participants to low‐dose ACTH (20 to 30 units/day for two to six weeks, tapering over one week). Of 30 participants treated with high‐dose therapy, 13 had complete cessation of spasms, and of 29 participants treated with low‐dose therapy, 14 had complete cessation of spasms. The Yanagaki trial (Yanagaki 1999) randomly assigned 13 participants to receive 1 unit /kg/day (equivalent to 40 units/kg/day of natural ACTH) for two weeks, tapering over a further two weeks, and 13 participants to receive 0.2 units/kg/day (equivalent to 8 units/kg/day of natural ACTH) for two weeks, tapering over a further two weeks. Among those treated with high‐dose ACTH, spasm cessation occurred in 11 of 13 participants compared with 9 of 13 participants treated with low‐dose ACTH. Combining these two studies revealed that high‐dose ACTH stopped the spasms in 79.5% participants compared with 76.5% of participants with low‐dose ACTH (Peto OR 1.1, 95% CI 0.4 to 2.6). The Shu study (Shu 2009) randomly assigned 30 children to receive either conventional‐dose (high‐dose) ACTH of 50 IU/day for 2 weeks, then tapered off to zero over 2 weeks, or low‐dose ACTH of 0.4 IU/kg/day for 2 weeks, increased to 1 IU/kg /day if no response was seen, and then tapered to zero over 2 weeks. Investigators report that 53% of the high‐dose group had a "good initial response" compared with 60% of participants in the low‐dose group. Effects on time taken to achieve cessation of spasms were not reported as an outcome in these studies. Effects on reduction in the number of spasms were not considered as an outcome in these studies. Effects on relapse rates: In the Hrachovy study (Hrachovy 1994), 2 of the 13 participants who responded to high‐dose ACTH relapsed, and 2 of the 14 participants who responded to low‐dose ACTH also relapsed. In the Yanagaki study (Yanagaki 1999), 3 of the 11 participants who responded to high‐dose ACTH relapsed, and three of the nine responders to low‐dose ACTH relapsed. The Shu study (Shu 2009) reported no difference in relapse rates between the two groups. The Hrachovy study (Hrachovy 1994) reported that 11 participants who responded to high‐dose ACTH remained spasm free and 12 participants who responded to low‐dose ACTH also remained spasm free. In the Yanagaki study (Yanagaki 1999), eight participants who responded to high‐dose ACTH remained spasm free and six participants who responded to low‐dose ACTH remained spasm free. Combining these two studies yields a Peto OR of 0.8 (95% CI 0.3 to 1.9). Effects on time taken to relapse were not reported in these studies. Effects on resolution of the EEG: In the Hrachovy study (Hrachovy 1994), 3 of 13 responders to high‐dose ACTH showed resolution of their EEG, as did 3 of 14 responders to low‐dose ACTH. Yanagaki (Yanagaki 1999) reported that 8 of 13 treated with high‐dose ACTH showed resolution of their EEG, as did 7 of 12 treated with low‐dose ACTH. Combining these two studies yields a Peto OR for remaining spasm free participants of 1.9 (95% CI 0.6 to 6.2). The Shu study (Shu 2009) reported no significant differences between the two groups. Effects on psychomotor development: The Yanagaki study (Yanagaki 1999) compared high‐dose ACTH with low‐dose ACTH and used the Japanese Tumor Scale to evaluate developmental status at entry into the trial as well as in 17 responders who were followed up for longer than one year. Investigators found no significant differences in developmental quotients between the two groups at the end of the follow‐up period. However, they considered only psychomotor development in the responders, which gives rise to a biased analysis; this should be interpreted with caution. Investigators did not consider long‐term developmental status at five years of age. Effects on subsequent seizure rates were not reported as an outcome in these studies. Side effects were not reported as an outcome in the Hrachovy and Yanagaki studies (Hrachovy 1994, Yanagaki 1999). The Shu study (Shu 2009) reported that ACTH therapy–related adverse effects were seen in 20% of the conventional (high‐dose) group as compared with 93% of the low‐dose group. No deaths were reported in these studies.
Nitrazepam versus ACTH
One study was included (Dreifuss 1986).
Effects on cessation of spasms were not reported as an outcome in this study. Effects on time taken to achieve cessation of spasms were not reported as an outcome in this study. Effects on reduction in the number of spasms: The Dreifuss study (Dreifuss 1986) showed greater than 50% reduction in spasms in 66% of the group treated with nitrazepam compared with 50% in the group treated with ACTH (Peto OR 2.0, 95% CI 0.7 to 5.9). Effects on relapse rates were not reported as an outcome in this study. Effects on time taken to relapse were not reported as an outcome in this study. Effects on resolution of EEG were not reported as an outcome in this study. Effects on psychomotor development were not reported as an outcome in this study. Effects on subsequent seizure rates were not reported as an outcome in this study. Side effects: Investigators in the Dreifuss study (Dreifuss 1986) had to withdraw six patients who were receiving ACTH; hypertension and meleana were given as two causes for withdrawal. Deaths: One patient in the ACTH group died, but no cause was found at autopsy.
Methysergide versus alpha‐methylparatyrosine
One study was included (Hrachovy 1989).
Effects on cessation of spasms: The Hrachovy study (Hrachovy 1989) found that only one patient (8%) treated with methysergide and two (16%) treated with alpha‐methylparatyrosine responded to therapy (Peto OR 0.5, 95% CI 0.1 to 5.2). Effects on time taken to achieve cessation of spasms were not reported as an outcome in this study. Effects on reduction in the number of spasms: The Hrachovy study (Hrachovy 1989) showed a greater than 50% reduction in spasms in 25% of the group treated with methysergide compared with 17% in the group treated with alpha‐methylparatyrosine (Peto OR 1.6, 95% CI 0.2 to 11.2). Effects on relapse rates: No patient treated with methysergide remained spasm free, and only one of the two patients successfully treated with alpha‐methylparatyrosine remained spasm free (Peto OR 0.14, 95% CI 0.0 to 6.8). Effects on time taken to relapse were not reported as an outcome in this study. Effects on resolution of EEG were not reported as an outcome in this study. Effects on psychomotor development: Investigators in the Hrachovy study (Hrachovy 1989) used the Denver Developmental Screening Test to evaluate developmental status at entry into the trial and at three weeks post entry. They stated, in their results section, that two patients in each group showed developmental improvement, but they did not quantify this further. They did not report developmental status at three months post entry or at five years of age. Effects on subsequent seizure rates were not reported as an outcome in this study. No side effects severe enough to warrant stopping treatment were reported in this study. No deaths were reported in this study.
Ganaxolone versus placebo
One study was included (Tsai 2009).
The results of this study have been presented only as an abstract.
The authors state that ganaxolone did not significantly decrease spasm cluster frequency, nor did it increase responder rate, compared with placebo. Somnolence, lethargy, and irritability were reported slightly more frequently in ganaxolone‐treated participants.
Vigabatrin with Flunarizine versus vigabatrin alone
One study was included (Bitton 2012).
Effects on cessation of spasms: The Bitton study (Bitton 2012) found that in 17 (50%) of 34 patients treated with vigabatrin and flunarizine, spasms stopped within a two‐week period compared with 21 (62%) of 34 patients treated with vigabatrin alone.
Effects on time taken to achieve cessation of spasms was not reported as an outcome in this study.
Effects on reduction in the number of spasms were not reported as an outcome in this study.
Effects on relapse rates were not reported as an outcome in this study.
Effects on resolution of EEG were not reported as an outcome in this study.
Effects on psychomotor development: Bayley and Vineland results were available at baseline and at 24 months in 45 children. No significant difference in the BSID developmental quotient were noted between flunarizine and placebo‐treated children at baseline (44.3 ± 35.5 vs 30.9 ± 29.8; P = 0.18) or 24 months later (56.9 ± 33.3 vs 46 ± 34.2; P = 0.29). However, the 10 flunarizine‐treated children with no identified etiology had better outcomes than eight controls at 24 months on both the Vineland Scale (84.1 ± 11.3 vs 72.3 ± 9.8; P = 0.03) and the Bayley Scale (87.6 ± 14.7 vs 69.9 ± 25.3; P = 0.07).
Effects on subsequent seizure rates were not reported as an outcome in this study.
Side effects were not reported in this study.
Six deaths were reported in this study—three in each group—but the causes of death are not given.
ACTH with magnesium sulphate versus ACTH alone
One study was included (Zou 2010).
Effects on cessation of spasms: a total of 38 participants had complete cessation of spasms at 4 weeks among 8 of 19 (42%) treated with ACTH alone compared with 12 of 19 (63%) treated with ACTH and magnesium sulphate (Peto OR, 95% CI). Effects on time taken to achieve cessation of spasms were not reported as an outcome in this study. Effects on reduction in the number of spasms: At 4 weeks, seven participants treated with ACTH alone had greater than 50% reduction in spasms among ten still with spasms, and three treated with ACTH and magnesium sulphate had greater than 50% reduction in spasms among the six still with spasms.
Effects on relapse rates: None of eight responders receiving ACTH alone but two of twelve responders given ACTH and magnesium sulphate relapsed.
Effects on time taken to relapse were not reported as an outcome in this study. Effects on resolution of EEG: Five of the 19 patients treated with ACTH alone had "normalisation" of the EEG, compared with 9 of 19 receiving ACTH and magnesium sulphate.
Effects on psychomotor development were reported as an outcome in this study, but were given as within‐group comparisons before and after treatment. Effects on subsequent seizure rates were not clearly reported as an outcome in this study. No side effects severe enough to warrant stopping treatment were reported in this study. No deaths were reported.
Discussion
Infantile spasms were first described in 1841 in a letter that Dr West wrote to The Lancet. Since that time, at least 30 drugs have been tried and reported in the literature, but treatment remains problematic because of the variable availability of treatments, adverse reactions, incomplete response rates, and the long‐term prognosis, which remains poor for both psychomotor development and subsequent development of other seizure types.
Despite the wealth of literature on this subject, most studies have been open prospective or retrospective trials. Most of the RCTs that have been undertaken include only small numbers of participants, with the result that individually, they have little power. In addition, studies have used widely varying treatments, dosage regimens, and outcome measures, making meta‐analysis difficult to perform. We found no RCTs comparing therapies such as pyridoxine that are first‐line therapies in some countries, including Japan. Some therapies (e.g. gamma‐globulins, pyridoxine, lamotrigine, topiramate) have been described in case reports or in uncontrolled trials. In addition, many different preparations have been used. This is due in part to the availability of different compounds in different countries. In Europe and Japan, synthetic ACTH may be available, whilst in the United States, natural ACTH derivatives have been used until recently, when costs made them less attractive. Vigabatrin was not licensed for use in the United States or in Japan until recently. ACTH can be given only as an injection (intramuscularly, intravenously, or into the peritoneal cavity). Infants find injections distressing, and many centres use oral preparations (usually prednisolone) in preference: The use of prednisone probably should not be recommended. This makes analysis of the efficacy of individual 'steroid' preparations difficult.
Despite an extensive literature search, we found only 18 RCTs, and the overall reported methodology was poor. Only two studies had considered the number of participants required to determine whether one treatment was more efficacious than another. One study based its calculations on the assumption that the trial drug would be 50% more efficacious than the comparator drug in stopping the spasms (this trial did not find such a difference). The other study based its calculations on the assumption that 250 infants would be required to give 90% power for finding a 20% difference between the two treatment groups. Investigators were able to recruit 107 infants, giving nearly 80% power to see the reported difference of 23%.
Although all studies claimed to be randomised, less than half stated how randomisation had been performed, and only four studies reported adequate concealment of randomisation. Ideally, to minimise performance bias, trials should be double‐blinded, although perhaps it is not surprising that only four such trials have been performed. Blinding recipients (or parents) and staff administering the therapies remains problematic. ACTH can be given only by intramuscular injection, but the other drugs used in the included RCTs are administered orally; nowadays, it is generally considered to be unethical to administer placebo intramuscular injections to young infants. In addition, many of the side effects of steroids, for example, weight gain and hypertension, are common and are easily recognised, making blinding difficult.
Some would argue that it is now proved that spasms are causally linked to the development of severe learning difficulties. Thus delaying treatment for long or administering a placebo would now be considered unethical. Finally, six studies stated that they had lost participants to follow‐up. Not all remaining studies clearly stated that all participants entered into the trials had been successfully followed up for the duration of the study. We had planned to look for possible sources of clinical heterogeneity between studies, for example, sex, age at spasm onset, age at trial entry, and delay to treatment, and whether or not participants had received previous treatment. Unfortunately, this information often was not available, and complex analysis would have been required for any conclusions to be drawn. However, it is possible that some of these sources of heterogeneity are important in determining the outcome. For example, researchers in the Baram study (Baram 1996) found ACTH to be more efficacious than prednisone but had three times as many girls as boys in the ACTH group and roughly equal numbers in the prednisone group. It is possible, therefore, that if girls have a better outcome than boys, this may have contributed to the differences in response noted between the two groups.
When developing our protocol, we tried to provide definitions for terms such as infantile spasms, cessation of spasms, relapse, and so forth. We had planned to establish whether different definitions used by authors for infantile spasms might affect which participants would be included in a trial, thereby affecting outcomes. It became clear from our review of the literature that few authors had clearly defined the term infantile spasms. It was clear that some trials using entry criteria that defined the need for hypsarrhythmia in fact entered infants who did not meet this criterion. Some of these issues have now been considered by an expert group using Delphi methodology to avoid bias (Lux 2004b). No consensus was obtained on a definition of hypsarrhythmia, but other recommendations will be incorporated in our next update. This will require a revised protocol. Inclusion and exclusion criteria varied, and some studies excluded infants with other seizure types. This is likely to have an impact on the outcome. For the purpose of this review, we accepted that any RCT stating that its participants had infantile spasms would be included, whether or not other seizure types were included.
The exact relationship between clinical spasms and typical or even modified (or atypical) hypsarrhythmia on the EEG is not clear. Not all infants with clinical evidence of infantile spasms are reported to have hypsarrhythmia. For those in whom it is present, it is not always present all of the time. It is not known whether the presence or absence of hypsarrhythmia affects long‐term outcomes. Again, we had hoped that collecting and combining data from studies might help clarify these areas of uncertainty. Unfortunately, few studies provided sufficient data to allow any conclusions to be drawn. Neither will meta‐analysis be reliable until hypsarrhythmia is reliably defined.
Infantile spasms are associated with a poor long‐term prognosis. Most affected infants will also have severe learning difficulties, with high cost implications not only for patients and their families and their quality of life, but also for health services. Therefore, if any single treatment was shown to improve long‐term psychomotor development and to reduce the risk of development of further seizure types, it would probably be considered to be more efficacious than other treatments, regardless of its effect on the spasms themselves or on EEG features. Although with time the spasms will resolve, many patients continue to have other forms of epilepsy into adult life, and about one‐fifth will progress to Lennox Gastaut syndrome (Beaumanoir 1992). Therefore, these patients will require anticonvulsants and will continue to suffer the effects of epilepsy, often for life. Very little information on developmental outcome is available; the most important information has come from the Lux studies (Lux 2004, 2005). This suggests that early control of spasms might improve development in those with no proven underlying aetiology, but the finding is not robust and longer‐term follow‐up is awaited. The effect of time from onset of spasms to the start of treatment has not been reported, except in Lux 2005, in which no difference between less than one month and longer than one month was reported. Little information is available on subsequent seizure rates, but the literature suggests that this may not be influenced by treatment.
The spasms themselves are certainly distressing for parents and for caregivers. It is possible that they are also distressing for the infant, as many cry immediately following a spasm or appear to be in pain and distressed. The long‐term benefits of different therapies remain uncertain, and more research on this topic is needed. Two studies have shown that placebo is not as good as active treatment in resolving the spasms. The strongest evidence we found suggests that hormonal treatment leads to resolution of spasms faster and in more infants than treatment with vigabatrin. Response without subsequent relapse may be no different, but if child development might be better after hormonal treatment, this might make hormonal treatments more attractive. More information and further research are required. We found no other RCTs comparing other forms of therapies, such as sodium valproate, benzodiazepines, pyridoxine, or phenobarbitone, in terms of cessation of spasms. Although four studies did consider reduction in the number of spasms, it is no longer clear to us that a reduction in spasms is of any real benefit to a patient. Complete control of spasms has to be the main objective.
One small study that looked at participants with infantile spasms and an underlying diagnosis of tuberous sclerosis found vigabatrin to be more efficacious than hydrocortisone in stopping the spasms in this group of participants—a finding also reported in some open studies (e.g. Hancock 1999). One study (Elterman 2010) suggested that vigabatrin was more effective in stopping the spasms in participants with tuberous sclerosis as compared with participants without a diagnosis of tuberous sclerosis. However, in participants without a diagnosis of tuberous sclerosis, the best available therapy remains unclear. Another study excluded participants with a known diagnosis of tuberous sclerosis or at high risk for tuberous sclerosis, and researchers suggested that hormonal treatment (tetracosactide or prednisolone) might be more efficacious in people who do not have an underlying diagnosis of tuberous sclerosis. No study has looked specifically at any other subgroup(s) of participants.
Some open, prospective studies have suggested that vigabatrin might control spasms more quickly than steroids. Five RCTs considered the time taken to cessation of spasms. Two small studies showed no difference between vigabatrin and a hormonal treatment, but one study found hormonal treatments to be quicker at resolving spasms than vigabatrin. This finding would be supported by the speed of response to hormonal treatment described by Baram 1996.
When we developed our protocol for this review, we decided to look at relapse rates as it was felt that this would be a useful outcome for clinicians. One treatment may be more efficacious in stopping the spasms in the short term, but if most of those patients subsequently relapse, another treatment with a lower but more sustained success rate might be considered more beneficial. When developing our analysis plan, we decided that we would analyse this information as dichotomous data. However, when we came to extract and analyse these data from the trials, it became apparent that they are censored data, that is, not all participants entered into a trial would be eligible for this outcome; only those in whom the spasms stopped could potentially relapse. We have, therefore, attempted to clarify this by also reporting how many participants remained spasm free, that is, never relapsed (an outcome for which all participants entered into any one trial were eligible). However, we believe that a more appropriate method of analysis would be to look at survival curves, and we will consider this as a more appropriate method of analysis in future updates of this review if individual patient data become available.
We had also planned to look at subgroup analysis by aetiological subgroup and by clinical features, but unfortunately, because of the small numbers of trials and patients and the lack of available detail, this was not possible. It was possible to consider participants with an underlying diagnosis of tuberous sclerosis as a subgroup in only two studies. One underpowered study (Chiron 1997) showed vigabatrin to be more efficacious than hydrocortisone in stopping the spasms in this group of participants. All participants in this study who were treated with vigabatrin had complete cessation of spasms, and only one participant relapsed within the study period. However, the numbers were small, with only eleven participants in each group, and these results should be treated with caution, even though this result mimics the findings of open studies. Further trials with larger numbers of patients are required to confirm (or refute) the findings of this study. The second study (Elterman 2010) found that 52% of 25 participants with tuberous sclerosis responded to vigabatrin within two weeks of treatment compared with 16% of 117 participants without tuberous sclerosis. Researchers stated that within three months, all participants with tuberous sclerosis who continued with vigabatrin were free of infantile spasms; however, two participants had been withdrawn from vigabatrin at this point, at least one because of lack of efficacy. These participants might also have received other treatments by three months. It is not clear, therefore, whether vigabatrin was responsible for the cessation of their spasms.
Side effects (adverse reactions) must always be taken into account. Any drug with unacceptable side effects will not be used in clinical practice. Likewise, if two drugs show similar beneficial effects, the one with the safer drug profile should be used in preference. Within these studies, few side effects or deaths were reported. However, both steroids and vigabatrin have potentially serious side effects. Steroids are associated with potentially life‐threatening problems, including depression of the immune system and modified response to infection leading to overwhelming sepsis. Less serious side effects, for example, hypertension, are often transient but nevertheless have the potential to cause morbidity. Minor side effects, estimated to occur in two‐thirds of patients, include behavioural changes, especially irritability, changes in appetite, weight gain, and alteration in sleep patterns. In addition, some forms of steroids (e.g. ACTH) involve daily or alternate‐day intramuscular injections. However, reports have described both asymptomatic and symptomatic visual field defects with loss of peripheral vision to varying degrees in adults and children treated with vigabatrin. This appears to occur most commonly in patients who have been treated with vigabatrin for longer than six months; it does not appear to be reversible on withdrawal of the vigabatrin. The main difficulty in infants is that they cannot be tested or monitored for visual field defects (a child must be at least 11 years old to be able to co‐operate and complete a reliable test). However, on balance, the devastating effects of infantile spasms and the poor response rate on placebo suggest that the risks of developing the severe side effects of these therapies are less important than the risk of not using the therapies.
The overwhelming conclusion is that more studies are required on this disorder, in particular studies that report long‐term developmental and epilepsy outcomes.
Authors' conclusions
Implications for practice.
Hormonal treatment (prednisolone (not prednisone), tetracosactide depot, and ACTH) resolves spasms faster than vigabatrin and in more infants, but it is not yet clear whether this will result in better long‐term outcomes. Hormonal treatment might improve development in those with no proven underlying neurological disease, but the evidence is not robust. It is important to note that if prednisolone or vigabatrin is used, high dosage is recommended. The optimum dose of ACTH (or tetracosactide) is not yet known. Vigabatrin may be the treatment of choice in tuberous sclerosis. Other treatments cannot be recommended on the basis of the evidence reviewed. No evidence from the RCTs indicates that it is necessary to treat EEG features rather than spasms. A clear statement on the optimum treatment for infantile spasms requires further research. Until additional study results become available, other factors (in particular, underlying aetiology and adverse reactions) will influence parents and clinicians about individual treatment choices.
Implications for research.
Further trials are needed with good methodology, standardised and valid outcome measures, larger numbers of participants, and detailed reporting, including other underlying neurological diseases, gender, time to treatment, time to response, EEG outcome, deaths, epilepsy outcome, and development. Adverse reactions need to be fully reported. Long‐term follow‐up of development is essential. As has been stated, infantile spasms are a relatively rare disorder, so if investigators are to recruit large numbers of patients into randomised controlled trials, continuing multi‐centre collaboration is required.
What's new
| Date | Event | Description |
|---|---|---|
| 15 January 2014 | Amended | Errors in 'Data and Analyses' section checked and corrected. Conclusions remain the same. |
Acknowledgements
We would like to acknowledge Philip Milner who was an author on a previous version of this review (see Contributions of authors).
Bath Unit for Research in Paediatrics.
Cow and Gate.
The Tuberous Sclerosis Association.
The Castang Foundation.
The NIHR through local CLRN funding.
Appendices
Appendix 1. CENTRAL search strategy
#1 MeSH descriptor Spasms, Infantile explode all trees #2 (infantile spasm*) #3 "West syndrome" or "West's syndrome" #4 (salaam spasm*) #5 (hypsarrhythmia) #6 (#1 OR #2 OR #3 OR #4 OR #5)
Appendix 2. MEDLINE search strategy
The following search is based on the Cochrane highly sensitive search strategy for identifying randomised trials in MEDLINE as set out in Appendix 5b of the Cochrane Handbook for Systematic Reviews of Interventions (version 4.2.4, updated March 2005) (Higgins 2005).
1. Spasms, Infantile/
2. West$ syndrome.tw.
3. salaam spasm$.tw.
4. hypsarrhythmia.tw.
5. infantile spasm$.tw.
6. 1 or 2 or 3 or 4 or 5
7. randomized controlled trial.pt.
8. controlled clinical trial.pt.
9. exp Randomized Controlled Trials/
10. exp Random Allocation/
11. exp Double‐Blind Method/
12. exp Single‐Blind Method/
13. 7 or 8 or 9 or 10 or 11 or 12
14. (animals not human).sh.
15. 13 not 14
16. clinical trial.pt.
17. Clinical Trial/
18. (clin$ adj trial$).ab,ti.
19. ((singl$ or doubl$ or trebl$ or tripl$) adj (blind$ or mask$)).ab,ti.
20. exp PLACEBOS/
21. placebo$.ab,ti.
22. random$.ab,ti.
23. exp Research Design/
24. 16 or 17 or 18 or 19 or 20 or 21 or 22 or 23
25. (animals not human).sh.
26. 24 not 25
27. 15 or 26
28. 27 and 6
Data and analyses
Comparison 1. Vigabatrin versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Cessation of spasms | 1 | 40 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 4.05 [0.93, 17.52] |
| 2 Resolution of hypsarrhythmia | 1 | 9 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 2.36 [0.10, 54.60] |
| 3 Relapse rates ‐ number of patients who remain spasm free | 1 | 40 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 8.23 [0.81, 84.07] |
1.1. Analysis.
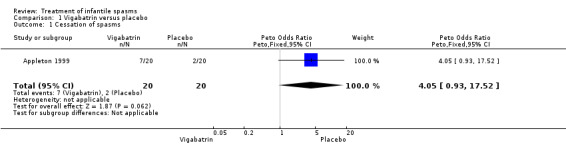
Comparison 1 Vigabatrin versus placebo, Outcome 1 Cessation of spasms.
1.2. Analysis.
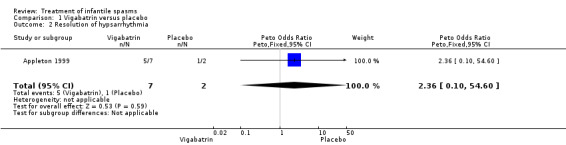
Comparison 1 Vigabatrin versus placebo, Outcome 2 Resolution of hypsarrhythmia.
1.3. Analysis.
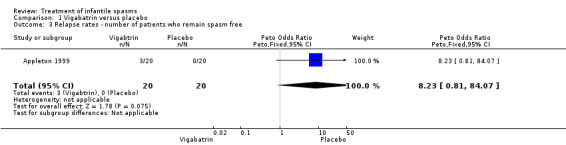
Comparison 1 Vigabatrin versus placebo, Outcome 3 Relapse rates ‐ number of patients who remain spasm free.
Comparison 2. Vigabatrin versus hydrocortisone.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Cessation of spasms | 1 | 22 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 13.80 [2.21, 86.35] |
| 2 Time taken to cessation of spasms | 1 | 16 | Mean Difference (IV, Fixed, 95% CI) | ‐8.8 [‐19.22, 1.62] |
2.1. Analysis.
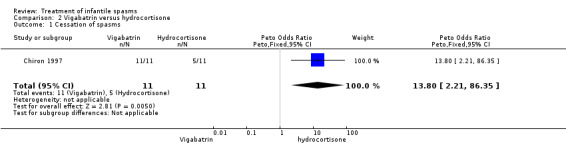
Comparison 2 Vigabatrin versus hydrocortisone, Outcome 1 Cessation of spasms.
2.2. Analysis.
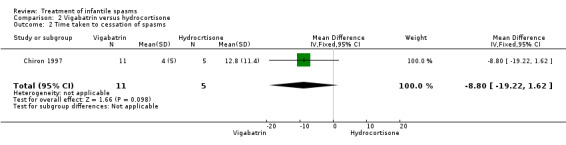
Comparison 2 Vigabatrin versus hydrocortisone, Outcome 2 Time taken to cessation of spasms.
Comparison 3. ACTH versus low‐dose prednisone.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Cessation of spasms | 2 | 53 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 4.19 [1.42, 12.34] |
| 2 Time taken to cessation of spasms | 1 | 17 | Mean Difference (IV, Fixed, 95% CI) | ‐0.80 [‐3.29, 1.69] |
| 3 Relapse rates ‐ number of patients who remain spasm free | 2 | 53 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 2.57 [0.81, 8.11] |
| 4 Resolution of hypsarrhythmia | 1 | 29 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 10.10 [2.36, 43.19] |
3.1. Analysis.
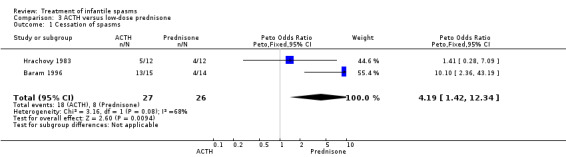
Comparison 3 ACTH versus low‐dose prednisone, Outcome 1 Cessation of spasms.
3.2. Analysis.
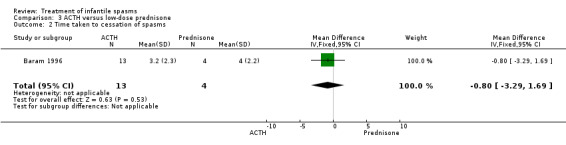
Comparison 3 ACTH versus low‐dose prednisone, Outcome 2 Time taken to cessation of spasms.
3.3. Analysis.
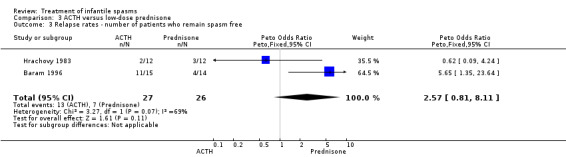
Comparison 3 ACTH versus low‐dose prednisone, Outcome 3 Relapse rates ‐ number of patients who remain spasm free.
3.4. Analysis.
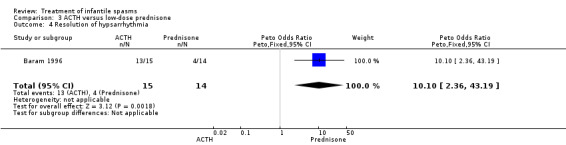
Comparison 3 ACTH versus low‐dose prednisone, Outcome 4 Resolution of hypsarrhythmia.
Comparison 4. High‐dose ACTH versus low‐dose ACTH.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Cessation of spasms | 2 | 85 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.06 [0.44, 2.56] |
| 2 Relapse rates ‐ number of patients who remain spasm free | 2 | 85 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.79 [0.33, 1.88] |
| 3 Resolution of hypsarrhythmia | 2 | 52 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.94 [0.60, 6.20] |
4.1. Analysis.
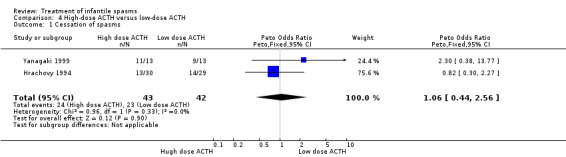
Comparison 4 High‐dose ACTH versus low‐dose ACTH, Outcome 1 Cessation of spasms.
4.2. Analysis.
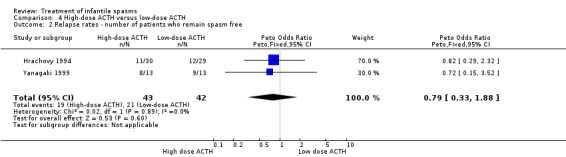
Comparison 4 High‐dose ACTH versus low‐dose ACTH, Outcome 2 Relapse rates ‐ number of patients who remain spasm free.
4.3. Analysis.
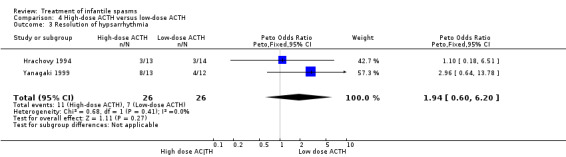
Comparison 4 High‐dose ACTH versus low‐dose ACTH, Outcome 3 Resolution of hypsarrhythmia.
Comparison 5. Nitrazepam versus ACTH.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Reduction in spasms | 1 | 51 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.96 [0.65, 5.93] |
5.1. Analysis.
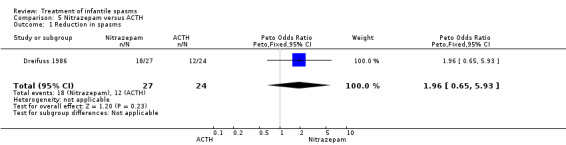
Comparison 5 Nitrazepam versus ACTH, Outcome 1 Reduction in spasms.
Comparison 6. Methysergide versus methylparatyrosine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Cessation of spasms | 1 | 24 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.48 [0.05, 5.15] |
| 2 Reduction in spasms | 1 | 24 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.62 [0.24, 11.17] |
| 3 Relapse rates ‐ number of patients who remain spasm free | 1 | 24 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.14 [0.00, 6.82] |
6.1. Analysis.
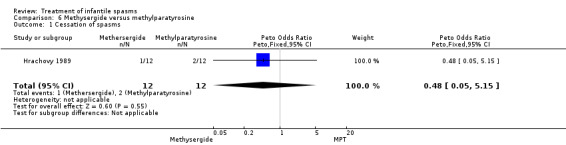
Comparison 6 Methysergide versus methylparatyrosine, Outcome 1 Cessation of spasms.
6.2. Analysis.
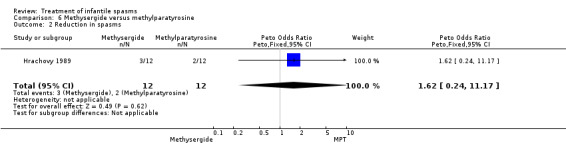
Comparison 6 Methysergide versus methylparatyrosine, Outcome 2 Reduction in spasms.
6.3. Analysis.
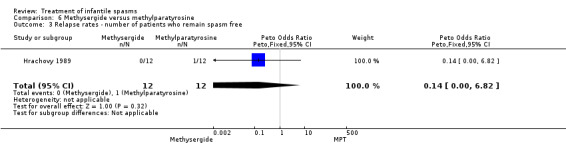
Comparison 6 Methysergide versus methylparatyrosine, Outcome 3 Relapse rates ‐ number of patients who remain spasm free.
Comparison 7. Low‐dose vigabatrin versus high‐dose vigabatrin.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Cessation of spasms | 1 | 221 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.41 [0.18, 0.95] |
| 2 Resolution of EEG | 1 | 142 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.24 [0.11, 0.52] |
| 3 Relapse rates | 1 | 25 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 2.57 [0.27, 24.32] |
7.1. Analysis.
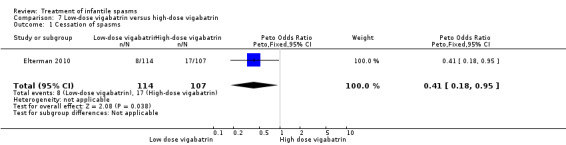
Comparison 7 Low‐dose vigabatrin versus high‐dose vigabatrin, Outcome 1 Cessation of spasms.
7.2. Analysis.
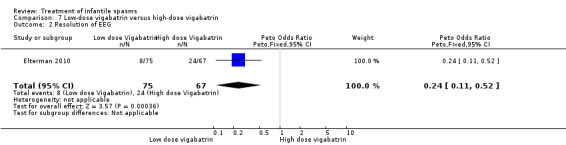
Comparison 7 Low‐dose vigabatrin versus high‐dose vigabatrin, Outcome 2 Resolution of EEG.
7.3. Analysis.
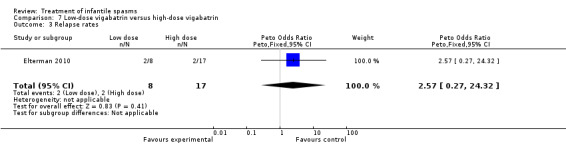
Comparison 7 Low‐dose vigabatrin versus high‐dose vigabatrin, Outcome 3 Relapse rates.
Comparison 8. Sulthiame versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Cessation of spasms | 1 | 51 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 5.13 [1.22, 21.47] |
| 2 Resolution of EEG | 1 | 51 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 5.13 [1.22, 21.47] |
8.1. Analysis.
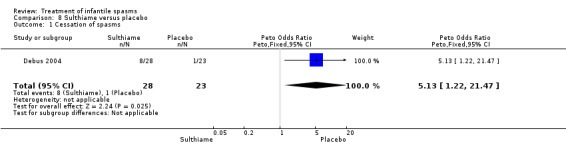
Comparison 8 Sulthiame versus placebo, Outcome 1 Cessation of spasms.
8.2. Analysis.
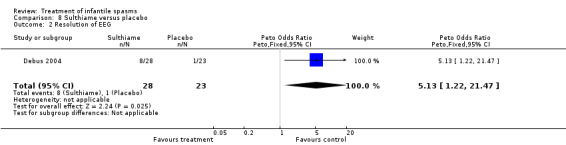
Comparison 8 Sulthiame versus placebo, Outcome 2 Resolution of EEG.
Comparison 9. Vigabatrin versus hormonal treatment.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Cessation of spasms | 3 | 158 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.42 [0.21, 0.80] |
| 2 Resolution of EEG | 3 | 94 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.38 [0.15, 0.99] |
| 3 Seizures at follow‐up | 3 | 158 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.69 [0.36, 1.32] |
9.1. Analysis.
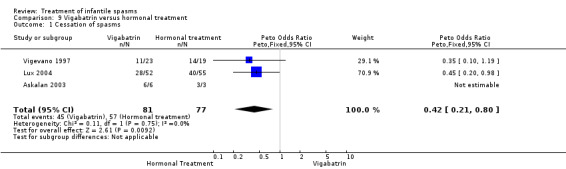
Comparison 9 Vigabatrin versus hormonal treatment, Outcome 1 Cessation of spasms.
9.2. Analysis.
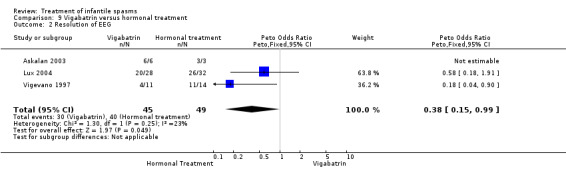
Comparison 9 Vigabatrin versus hormonal treatment, Outcome 2 Resolution of EEG.
9.3. Analysis.
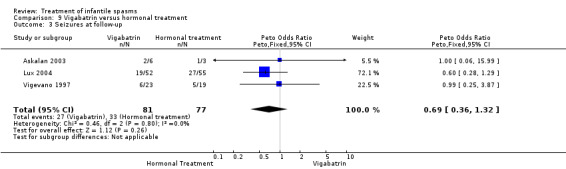
Comparison 9 Vigabatrin versus hormonal treatment, Outcome 3 Seizures at follow‐up.
Comparison 10. ACTH versus high‐dose prednisolone.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Cessation of infantile spasms | 1 | 55 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.35 [0.41, 4.38] |
| 2 Resolution of EEG | 1 | 32 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 3.04 [0.52, 17.66] |
10.1. Analysis.
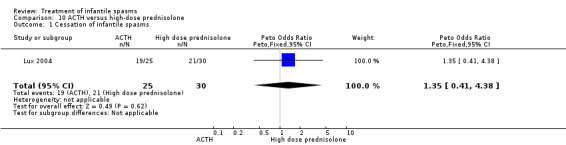
Comparison 10 ACTH versus high‐dose prednisolone, Outcome 1 Cessation of infantile spasms.
10.2. Analysis.
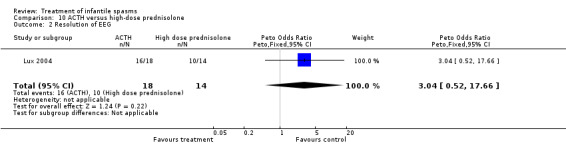
Comparison 10 ACTH versus high‐dose prednisolone, Outcome 2 Resolution of EEG.
Comparison 11. ACTH with magnesium sulphate versus ACTH alone.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Cessation of spasms | 1 | 38 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.34 [0.09, 1.24] |
| 2 Resolution of EEG | 1 | 38 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.41 [0.11, 1.52] |
11.1. Analysis.
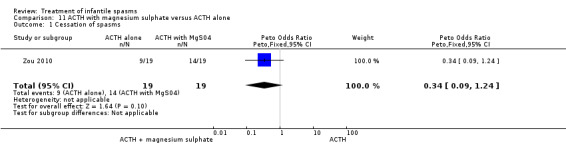
Comparison 11 ACTH with magnesium sulphate versus ACTH alone, Outcome 1 Cessation of spasms.
11.2. Analysis.
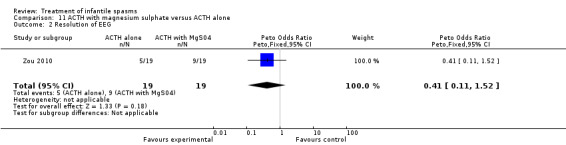
Comparison 11 ACTH with magnesium sulphate versus ACTH alone, Outcome 2 Resolution of EEG.
Comparison 12. Vigabatrin +Flunarizine vs Vigabatrin + Placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Cessation of spasms | 1 | 68 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.62 [0.24, 1.62] |
12.1. Analysis.
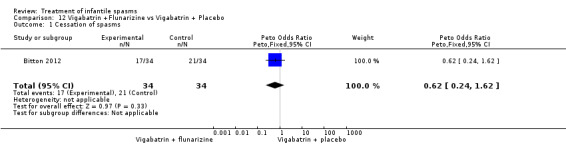
Comparison 12 Vigabatrin +Flunarizine vs Vigabatrin + Placebo, Outcome 1 Cessation of spasms.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Appleton 1999.
| Methods | RCT | |
| Participants | 40 participants with a clinical definition of infantile spasms and classical or modified hypsarrhythmia on EEG | |
| Interventions | Vigabatrin Placebo | |
| Outcomes | (1) Cessation of spasms. (2) Reduction in spasms. (3) Relapse rate. (4) Resolution of EEG. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Low risk | A ‐ Adequate |
Askalan 2003.
| Methods | RCT | |
| Participants | 9 participants with a clinical definition of infantile spasms and video‐EEG with hypsarrhythmia | |
| Interventions | ACTH Vigabatrin | |
| Outcomes | (1) Cessation of spasms. (2) Resolution of EEG. (3) Cognitive outcomes ‐ short term. (4) Language scores. (5) Evolution of epilepsy and autism. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Baram 1996.
| Methods | RCT | |
| Participants | 29 participants with a diagnosis of infantile spasms confirmed by hypsarrhythmia on video‐EEG | |
| Interventions | ACTH Prednisone | |
| Outcomes | (1) Cessation of spasms. (2) Relapse rates. (3) Resolution of EEG. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Low risk | A ‐ Adequate |
Bitton 2012.
| Methods | RCT | |
| Participants | 68 patients with infantile spasms | |
| Interventions | Flunarizine Placebo |
|
| Outcomes | Developmental outcome at 24 months after initiation of treatment | |
| Notes | ||
Chiron 1997.
| Methods | RCT | |
| Participants | 22 participants with a clinical diagnosis of infantile spasms or epileptic spasms or diffuse interictal activity on EEG | |
| Interventions | Vigabatrin Hydrocortisone | |
| Outcomes | (1) Cessation of spasms. (2) Relapse rates. (3) Resolution of EEG. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Debus 2004.
| Methods | RCT | |
| Participants | 51 participants with a clinical diagnosis of infantile spasms and hypsarrhythmia on EEG | |
| Interventions | Sulthiame Placebo | |
| Outcomes | (1) Cessation of spasms. (2) Resolution of EEG. (3) Relapse rates. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Low risk | A ‐ Adequate |
Dreifuss 1986.
| Methods | RCT | |
| Participants | 52 participants with a diagnosis of hypsarrhythmia or modified hypsarrhythmia on EEG | |
| Interventions | ACTH Nitrazepam | |
| Outcomes | Reduction in seizures | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Dyken 1985.
| Methods | RCT | |
| Participants | 17 participants with a clinical definition of infantile spasms and hypsarrhythmia on EEG | |
| Interventions | Valproate Placebo | |
| Outcomes | Reduction in spasms | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Elterman 2010.
| Methods | RCT | |
| Participants | 221 participants with a clinical diagnoses of infantile spasms and hypsarrhythmia, modified hypsarrhythmia, or multifocal spike wave discharges on EEG | |
| Interventions | Low‐dose vigabatrin High‐dose Vigabatrin | |
| Outcomes | (1) Cessation of spasms. (2) Time to cessation of spasms. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Hrachovy 1983.
| Methods | RCT | |
| Participants | 24 participants with a clinical definition of infantile spasms and hypsarrhythmia on EEG | |
| Interventions | ACTH Prednisone | |
| Outcomes | Cessation of spasms | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Hrachovy 1989.
| Methods | RCT | |
| Participants | 24 participants with a clinical definition of infantile spasms and hypsarrhythmia on EEG | |
| Interventions | Methysergide Alpha‐methylparatyrosine | |
| Outcomes | (1) Cessation of spasms. (2) Reduction in spasms. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Hrachovy 1994.
| Methods | RCT | |
| Participants | 59 participants with a clinical definition of infantile spasms and hypsarrhythmia on EEG | |
| Interventions | ACTH high dose ACTH low dose | |
| Outcomes | (1) Cessation of spasms. (2) Relapse rates. (3) Resolution of EEG. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Lux 2004.
| Methods | RCT | |
| Participants | 107 participants with a clinical definition of infantile spasms and hypsarrhythmia on EEG | |
| Interventions | Vigabatrin Hormonal treatment | |
| Outcomes | (1) Cessation of spasms. (2) Resolution of EEG. (3) Relapse rates. (4) Development 14 months. (5) Seizure rates. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Low risk | A ‐ Adequate |
Shu 2009.
| Methods | RCT | |
| Participants | 30 patients with infantile spasms | |
| Interventions | Conventional (high)‐dose ACTH Low‐dose ACTH |
|
| Outcomes | (1) Initial response. (2) EEG findings. (3) Relapse rates. (4) Time to relapse. (5) Long‐term outcomes. (6) Adverse effects. |
|
| Notes | ||
Tsai 2009.
| Methods | ||
| Participants | 56 infants with infantile spasms | |
| Interventions | Ganaxolone Placebo |
|
| Outcomes | (1) Spasm cluster frequency. (2) Adverse effects. |
|
| Notes | ||
Vigevano 1997.
| Methods | RCT | |
| Participants | 42 participants with a diagnosis according to the ILAE classification | |
| Interventions | Vigabatrin ACTH | |
| Outcomes | (1) Cessation of spasms. (2) Relapse rates. (3) Subsequent epilepsy rates. (4) Resolution of hypsarrhythmia. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Yanagaki 1999.
| Methods | RCT | |
| Participants | 26 participants with a diagnosis confirmed by hypsarrhythmia on video‐EEG | |
| Interventions | ACTH high dose ACTH low dose | |
| Outcomes | (1) Cessation of spasms. (2) Relapse rates. (3) Resolution of hypsarrhythmia. (4) Developmental outcome. | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | B ‐ Unclear |
Zou 2010.
| Methods | RCT | |
| Participants | 38 infants with infantile spasms and hypsarrhythmia | |
| Interventions | ACTH alone compared with ACTH with magnesium sulphate | |
| Outcomes | Cessation of spasms, normalisation of EEG and development | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Allocation concealment (selection bias) | Unclear risk | "Treatment was assigned using a table of numbers following a central 1:1 randomisation." |
EEG: electroencephalogram RCT: randomised controlled trial
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Al Ajlouni 2005 | Not an RCT |
| Albsoul‐Younes 2004 | Not an RCT |
| Appleton 1993 | Not an RCT |
| Arvio 2005 | Not an RCT |
| Azam 2005 | Not an RCT |
| Bachman 1982 | Not an RCT |
| Bensch 1977 | Not infantile spasms |
| Besag 2004 | Not an RCT |
| Bingel 2003 | Not an RCT |
| Blennow 1986 | Not an RCT |
| Bobele 1994 | Not an RCT |
| Boel 2004 | Not an RCT |
| Brodie 1996 | Not an RCT |
| Browne 1978 | Not an RCT |
| Buoni 2004 | Not an RCT |
| Chamberlain 1996 | Not an RCT |
| Chevrie 1984 | Not an RCT |
| Connelly 1993 | Not an RCT |
| Debus 2002 | Not an RCT |
| Duncan 2002 | Not an RCT |
| Duse 1996 | Not an RCT |
| Francois 2003 | Not an RCT |
| Gibbs 1965 | Not an RCT |
| Glauser 2002 | Not an RCT |
| Glaze 1985 | Not an RCT |
| Glaze 1986 | Considered only changes on CT as an outcome measure |
| Grosso 2005 | Not an RCT |
| Haines 1994 | Not an RCT |
| Hosain 2006 | Not an RCT |
| Hrachovy 1979 | Not an RCT |
| Inanaga 1989 | Not infantile spasms |
| Ishii 2002 | Not an RCT |
| Klein 1970 | Not an RCT |
| Li 2004 | Not an RCT |
| Lombroso 1983 | Not an RCT |
| Lortie 1997 | Not an RCT |
| Lotze 2004 | Not an RCT |
| Luna 1989 | Not infantile spasms |
| Mackay 2004 | Not an RCT |
| Matsumoto 1987 | Not an RCT |
| Mikaeloff 2003 | Not an RCT |
| Mikati 2002 | Not an RCT |
| Mimaki 1998 | Not an RCT |
| Mumford 1989 | Not an RCT |
| Mumford 1994 | Not an RCT |
| Nabbout 2003 | Not an RCT |
| Nolte 1988 | Not an RCT |
| O'Donohoe 1977 | Not an RCT |
| Oguni 2006 | Not an RCT |
| Ohtahara 2006 | Not an RCT |
| Oka 2004 | Not an RCT |
| Pietz 1993 | Not an RCT |
| Ramsay 1984 | Not an RCT |
| Scher 2003 | Not an RCT |
| Schlanger 2002 | Not an RCT |
| Schlumberger 1994 | Not an RCT |
| Suzuki 1996 | Not an RCT |
| Suzuki 2002 | Not an RCT |
| Tada 1991 | Not an RCT |
| Trevanthan 2003 | Not an RCT |
| Trompetter 1977 | Not infantile spasms |
| Vassella 1973 | Not an RCT |
| Veggiotti 1994 | Not an RCT |
| Wallace 1995 | Not an RCT |
| Weinmann 1967 | Not an RCT |
| Willig 1980 | Not an RCT |
| Wohlrab 1998 | Not an RCT |
| Yagi 2004 | Not an RCT |
| Yamamoto 1998 | Not an RCT |
| Yamauchi 2004 | Not an RCT |
| Zafeiriou 1996 | Not an RCT |
RCT: randomised controlled trial
Characteristics of ongoing studies [ordered by study ID]
ICISS.
| Trial name or title | International Collaboration Infantile Spasms Study |
| Methods | |
| Participants | Infants with a diagnosis of infantile spasms |
| Interventions | Vigabatrin High‐dose prednisolone Tetracosactide |
| Outcomes | Cessation of spasms Long‐term development Long‐term seizures |
| Starting date | May 2007 |
| Contact information | Dr Finbar O'Callaghan, Chief Investigator, ICISS Children's Centre, Royal United Hospital Bath BA1 3NG UK |
| Notes |
ISRCTN 36757519.
| Trial name or title | |
| Methods | |
| Participants | |
| Interventions | |
| Outcomes | |
| Starting date | |
| Contact information | |
| Notes |
ISRCTN Surgery/AEDs.
| Trial name or title | |
| Methods | |
| Participants | |
| Interventions | |
| Outcomes | |
| Starting date | |
| Contact information | |
| Notes |
EEG: electroencephalogram
Differences between protocol and review
We have included information on studies comparing combination treatment with a single treatment as this is now an obvious way forward for improving the treatment of infantile spasms.
Contributions of authors
Eleanor Hancock was primarily responsible for all aspects of this review including protocol design, undertaking the searches, the database of studies, data collection and extraction, data analysis, and except for this review, presentation of the results.
John Osborne agreed and approved the review at all stages, independently evaluated which studies should be included and excluded from the review, and also independently extracted the data from the included studies. He edited the presentation of the results for this review.
Philip Milner agreed on and approved the review, independently evaluated which studies should be included in and excluded from the review, and also independently extracted the data from the included studies up until February 2002 but including some in this review.
Stuart Edwards replaced Philip Milner and has agreed and approved the review, independently evaluated which studies should be included and excluded from the review, and also independently extracted data from the included studies from February 2002 until February 2010.
Sources of support
Internal sources
Royal United Hospital, Combe Park, Bath, UK.
Chelsea and Westminster Hospital, Fulham Road, London, UK.
University of Bath, School of Health, UK.
Great Ormond Street Hospital, Great Ormond Street, London, UK.
Surrey Heath and Woking Primary Care Trust, UK.
External sources
-
Bath Unit for Research into Paediatrics, UK.
Salary funding for all reviews
-
The Tuberous Sclerosis Association, UK.
Salary funding for first review
-
Cow and Gate, UK.
Salary funding for first review
-
NHS Research and Development Funding, UK.
Salary funding for latest review
-
Castang Foundation, UK.
Funding for salary
Declarations of interest
Dr Hancock, Professor Osborne, and Dr Edwards are members of the Trial Steering Committee for two multi‐centre, randomised, controlled trials in infantile spasms (United Kingdom Infantile Spasms Study (UKISS) and the International Collaborative Infantile Spasms Study).
Professor Milner was the chairman of the Data Monitoring and Ethics Committee for UKISS.
Professor Osborne is also a co‐author for the Consensus Statement of the West Delphi Group.
Edited (no change to conclusions)
References
References to studies included in this review
Appleton 1999 {published data only}
- Appleton RE, Peters ACB, Mumford JP, Shaw DE. Randomised, placebo‐controlled study of vigabatrin as first‐line treatment of infantile spasms. Epilepsia 1999;40(11):1627‐33. [DOI] [PubMed] [Google Scholar]
Askalan 2003 {published data only}
- Askalan R, Mackay M, Brian J, Otsubo H, McDermott C, Bryson S, et al. Prospective preliminary analysis of the development of autism and epilepsy in children with infantile spasms. Journal of Child Neurology 2003;18(3):165‐70. [DOI] [PubMed] [Google Scholar]
Baram 1996 {published data only}
- Baram TZ, Mitchell WG, Tournay A, Carter‐Snead O, Hanson RA, Horton EJ. High‐dose corticotropin (ACTH) versus prednisone for infantile spasms: a prospective, randomised, blinded study. Pediatrics 1996;97(3):375‐9. [PMC free article] [PubMed] [Google Scholar]
Bitton 2012 {published data only}
- Bitton JY, Sauerwein HC, Weiss SK, Donner EJ, Whiting S, Dooley JM, et al. A randomized controlled trial of flunarizine as add‐on therapy and effect on cognitive outcome in children with infantile spasms.. Epilepsia 2012;53(9):1570‐6. [DOI] [PubMed] [Google Scholar]
- Sauerwein H, Goodyear E, Carmant L, Lassonde M. Study of the neuroprotective effect of the L‐type calcium channel blocker flunarizine on the developmental outcome of infants with West syndrome. Epilepsia 2006;47 Suppl 3:173. [Google Scholar]
- Weiss SK, Whiting S, Wirrell, Donner E, Dooley J, Ronen G, et al. Prospective infantile spasm multicenter trial: what clinical risk factors predict a normal EEG at 6 months?. Epilepsia 2007;48 Suppl 6:64. [Google Scholar]
Chiron 1997 {published data only}
- Chiron C, Dumas C, Jambaque I, Mumford J, Dulac O. Randomized trial comparing vigabatrin and hydrocortisone in infantile spasms due to tuberous sclerosis. Epilepsy Research 1997;26:389‐95. [DOI] [PubMed] [Google Scholar]
Debus 2004 {published data only}
- Debus O, Kurlemann G. Sulthiame in the primary therapy of West syndrome: a randomised double blind placebo‐controlled add‐on trial on baseline pyridoxine medication. Personal Communication. [DOI] [PubMed]
- Debus OM, Kurlemann G. Sulthiame in the primary therapy of West syndrome: a randomised double blind placebo‐controlled add‐on trial on baseline pyridoxine medication. Epilepsia 2004;45(2):103‐8. [DOI] [PubMed] [Google Scholar]
Dreifuss 1986 {published data only}
- Dreifuss F, Farwell J, Holmes G, Joseph C, Lockman L, Madsen J, et al. Infantile spasms. Comparative trial of nitrazepam and corticotropin. Archives of Neurology 1986;43:1107‐10. [DOI] [PubMed] [Google Scholar]
Dyken 1985 {published data only}
- Dyken PR, DuRant RH, Batts Minden D, King DW. Short term effects of valproate on infantile spasms. Pediatric Neurology 1985;1(1):34‐7. [DOI] [PubMed] [Google Scholar]
Elterman 2010 {published data only}
- Elterman RD, Collins SD, Shields D, Mansfield KA, Nakagawa J. Efficacy of vigabatrin in subjects with infantile spasms. Epilepsia 2005;46 Suppl 8:167. [Google Scholar]
- Elterman RD, Shields D, Bittman RM, Torri SA, Sagar SM, Collins SD. Vigabatrin for the treatment of infantile spasms: final report of a randomized trial. Journal of Child Neurology 2010;25(11):1340‐7. [PUBMED: 20404353] [DOI] [PubMed] [Google Scholar]
- Elterman RD, Shields D, Collins S. Vigabatrin effective in multiple etiologies of infantile spasms. Epilepsia 2006;47 Suppl 4:179. [Google Scholar]
- Elterman RD, Shields WD, Mansfield KA, Nakagawa J. Randomized trial of vigabatrin in patients with infantile spasms. Neurology 2001;57(8):1416‐21. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
- Shields D, Collins SD, Elterman RD, Nakagawa J, Mansfield KA. AEs and safety of vigabatrin in subjects with infantile spasms. Epilepsia 2005;46 Suppl 8:161. [Google Scholar]
Hrachovy 1983 {published data only}
- Hrachovy RA, Frost JD, Kellaway P, Zion TE. Double‐blind study of ACTH versus prednisone therapy in infantile spasms. Journal of Pediatrics 1983;103(4):641‐4. [DOI] [PubMed] [Google Scholar]
Hrachovy 1989 {published data only}
- Hrachovy RA, Frost JD, Glaze DG, Rose D. Treatment of infantile spasms with methysergide and alpha‐methylparatyrosine. Epilepsia 1989;30(5):607‐10. [DOI] [PubMed] [Google Scholar]
Hrachovy 1994 {published data only}
- Hrachovy RA, Frost JD, Glaze DG. High‐dose, long‐duration versus low‐dose, short ‐duration corticotropin therapy for infantile spasms. The Journal of Pediatrics 1994;124(5(1)):803‐5. [DOI] [PubMed] [Google Scholar]
Lux 2004 {published and unpublished data}
- Darke K, Edwards E, Hancock, Johnson AL, Kennedy CR, Lux AL, et al. Developmental and epilepsy outcomes at age 4 years in the UKISS trial comparing hormonal treatments to vigabatrin for infantile spasms: a multi‐centre randomised trial. Archives of Disease in Childhood 2010;95(5):382‐6. [PUBMED: 20457702] [DOI] [PubMed] [Google Scholar]
- Lux AL, Edwards SW, Hancock E, Johnson AL, Kennedy CR, Newton RW, et al. The United Kingdom Infantile Spasms Study (UKISS) comparing hormone treatment with vigabatrin on developmental and epilepsy outcomes to age 14 months: a multicentre randomised trial. Lancet Neurology 2005;4(11):712‐7. [DOI] [PubMed] [Google Scholar]
- Lux AL, Edwards SW, Hancock E, Johnson AL, Kennedy CR, Newton RW, et al. The United Kingdom Infantile Spasms Study comparing vigabatrin with prednisolone or tetracosactide at 14 days: a multicentre, randomised controlled trial. Lancet 2004;364(9447):1173‐8. [DOI] [PubMed] [Google Scholar]
Shu 2009 {published data only}
- Shu XM, Li J, Zhang GP, Mao Q. A comparative study of conventional dose and low dose adrenocorticotrophic hormone therapy for West syndrome. Chinese Journal of Contemporary Pediatrics 2009;11(6):445‐8. [PubMed] [Google Scholar]
Tsai 2009 {published data only}
- Tsai J, Pellock JM, Hehn J, Pieribone V, Shaw K, Garofalo E. A double‐blind, placebo‐controlled, dose‐ranging clinical study to evaluate the safety, tolerability, and antiepileptic activity of ganaxolone in treatment of patients with infantile spasms. Neurology 2009;72(11 Suppl 3):A119. [Google Scholar]
Vigevano 1997 {published data only}
- Vigevano F, Cilio MR. Vigabatrin versus ACTH as first line treatment for infantile spasms: a randomised prospective study. Epilepsia 1997;38(12):1270‐4. [DOI] [PubMed] [Google Scholar]
- Vigevano F, Cilio MR, Claps D, Faberi A, Gisondi A. Vigabatrin versus ACTH as first line therapy in West syndrome [Vigabatrin versus ACTH come terapia di prima scelta nella sindrome di West]. Bollettino lega Italiana contro l'epilessia 1994;86/87:115‐6. [Google Scholar]
Yanagaki 1999 {published data only}
- Yanagaki S, Oguni H, Hayashi K, Imai K, Funatuka M, Tanaka T, et al. A comparative study of high‐dose and low‐dose ACTH therapy for West syndrome. Brain and Development 1999;21(7):461‐7. [DOI] [PubMed] [Google Scholar]
Zou 2010 {published data only (unpublished sought but not used)}
- Zou LP, Wang X, Dong CH, Chen CH, Zhao W, Zhao RY. Three‐week combination treatment with ACTH + magnesium sulfate versus ACTH monotherapy for infantile spasms: a 24 week, randomised, open‐label, follow up study in China. Clinical Therapeutics 2010;32(4):692‐700. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Al Ajlouni 2005 {published data only}
- Al Ajlouni S, Shorman A, Daoud AS. The efficacy and side effects of topiramate on refractory epilepsy in infants and young children: a multicenter clinical trial. Seizure 2005;14(7):459‐63. [DOI] [PubMed] [Google Scholar]
Albsoul‐Younes 2004 {published data only}
- Albsoul‐Younes AM, Salem HA, Al‐Safi SA. Topiramate slow dose titration: improved efficacy and tolerability. Pediatric Neurology 2004;31(5):349‐52. [DOI] [PubMed] [Google Scholar]
Appleton 1993 {published data only}
- Appleton RE. The role of vigabatrin in the management of infantile epileptic syndromes. Neurology 1993;43 Suppl 5:21‐3. [PubMed] [Google Scholar]
Arvio 2005 {published data only}
- Arvio M, Sillanpaa M. Topiramate in long‐term treatment of epilepsy in the intellectually disabled. Journal of Intellectual Disability Research 2005;49(3):183‐9. [DOI] [PubMed] [Google Scholar]
Azam 2005 {published data only}
- Azam M, Bhatti N, Krishin J. Use of ACTH and prednisolone in infantile spasms: experience from a developing company. Seizure 2005;14 (8):552‐6. [DOI] [PubMed] [Google Scholar]
Bachman 1982 {published data only}
- Bachman DS. Use of valproic acid in treatment of infantile spasms. Archives of Neurology 1982;39:49‐52. [DOI] [PubMed] [Google Scholar]
Bensch 1977 {published data only}
- Bensch J, Blennow G, Ferngren H, Gamstorp I, Herrlin K‐M, Kubista J, et al. A double‐blind study of clonazepam in the treatment of therapy‐resistant epilepsy in children. Developmental Medicine and Child Neurology 1977;19:335‐42. [DOI] [PubMed] [Google Scholar]
Besag 2004 {published data only}
- Besag F. Behvioural effects of the newer antiepileptic drugs: an update. Expert Opinion on Drug Safety 2004;3(1):1‐8. [DOI] [PubMed] [Google Scholar]
Bingel 2003 {published data only}
- Bingel U, Pinter JD, Sotero de Menezes M, Rho JM. Intravenous immunoglobulin as adjunctive therapy for juvenile spasms. Journal of Child Neurology 2003;18(6):379‐82. [DOI] [PubMed] [Google Scholar]
Blennow 1986 {published data only}
- Blennow G, Starck L. High dose B6 treatment in infantile spasms. Neuropediatrics 1986;17:7‐10. [DOI] [PubMed] [Google Scholar]
Bobele 1994 {published data only}
- Bobele GB, Bodensteiner JB. The treatment of infantile spasms by child neurologists. Journal of Child Neurology 1994;9(4):432‐5. [DOI] [PubMed] [Google Scholar]
Boel 2004 {published data only}
- Boel MJ. Behavioural and neuropsychological problems in refractory paediatric epilepsies. European Journal of Paediatric Neurology 2004;8(6):291‐7. [DOI] [PubMed] [Google Scholar]
Brodie 1996 {published data only}
- Brodie MJ. Lamotrigine ‐ an update. The Canadian Journal of Neurological Sciences 1996;23 Suppl 2:6‐9. [DOI] [PubMed] [Google Scholar]
Browne 1978 {published data only}
- Browne TR. Clonazepam. New England Journal of Medicine 1978;299(15):812‐6. [DOI] [PubMed] [Google Scholar]
Buoni 2004 {published data only}
- Buoni S, Zannolli R, Strambi M, Fois A. A combined treatment with vigabatrin and topiramate in West syndrome. Journal of Child Neurology 2004;19(5):385‐6. [DOI] [PubMed] [Google Scholar]
Chamberlain 1996 {published data only}
- Chamberlain MC. Nitrazepam for refractory infantile spasms and the Lennox‐Gastaut Syndrome. Journal of Child Neurology 1996;11(1):31‐4. [DOI] [PubMed] [Google Scholar]
Chevrie 1984 {published data only}
- Chervrie JJ. Therapy of infantile spasms. The Journal of Pediatrics 1984;105(1):168‐9. [DOI] [PubMed] [Google Scholar]
Connelly 1993 {published data only}
- Connelly JF. Vigabatrin. The Annals of Pharmacotherapy 1993;27:197‐204. [DOI] [PubMed] [Google Scholar]
Debus 2002 {published data only}
- Debus OM, Kohring J, Fiedler B, Franssen M. Add‐on treatment with pyridoxine and sulthiame in 12 infants with West syndrome: an open clinical study. Seizure 2002;11(6):381‐3. [DOI] [PubMed] [Google Scholar]
Duncan 2002 {published data only}
- Duncan JS. The promise of new antiepileptic drugs. British Journal of Clinical Pharmacology 2002;53(2):123‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Duse 1996 {published data only}
- Duse M, Notarangelo LD, Tiberti S, Menegati E, Plebani A, Ugazio AG. Intravenous immune globulin in the treatment of intractable childhood epilepsy. Clinical and Experimental Immunology 1996;104 Suppl 1:71‐6. [PubMed] [Google Scholar]
Francois 2003 {published data only}
- Francois II, Manel V, Rouselle C, David M. Ketogenic regime as anti‐epileptic treatment: its use in 29 epileptic children. Achives De Pediatrie 2003;10(4):300‐6. [DOI] [PubMed] [Google Scholar]
Gibbs 1965 {published data only}
- Gibbs FA, Anderson EM. Treatment of hypsarrhythmia and infantile spasms with a librium analogue. Neurology 1965;15(12):1173‐6. [DOI] [PubMed] [Google Scholar]
Glauser 2002 {published data only}
- Glauser TA, Pellock JM. Zonisamide in pediatric epilepsy: review of the Japanese experience. Journal of Child Neurology 2002;17(2):87‐96. [DOI] [PubMed] [Google Scholar]
Glaze 1985 {published data only}
- Glaze D, Zion T. Infantile spasms. In: Lockhart JD editor(s). Current Problems in Pediatrics. 11. Vol. 15, Chicago: Year Book Medical Publisher, 1985:1‐39. [DOI] [PubMed] [Google Scholar]
Glaze 1986 {published data only}
- Glaze DG, Hrachovy RA, Frost JD, Zion TE, Bryan RN. Computed tomography in infantile spasms: effects of hormonal therapy. Pediatric Neurology 1986;2(1):23‐7. [DOI] [PubMed] [Google Scholar]
Grosso 2005 {published data only}
- Grosso S. Efficacy and safety of topiramate in infants according to epilepsy syndromes. Seizure 2005;14(3):183‐9. [DOI] [PubMed] [Google Scholar]
Haines 1994 {published data only}
- Haines ST, Casto DT. Treatment of infantile spasms. The Annals of Pharmacotherapy 1994;28:779‐91. [DOI] [PubMed] [Google Scholar]
Hosain 2006 {published data only}
- Hosain SA, Merchant S, Solomon GE. Topiramate for the treatment of infantile spasms. Journal of Child Neurology 2006;21(1):17‐19. [DOI] [PubMed] [Google Scholar]
Hrachovy 1979 {published data only}
- Hrachovy RA, Frost JD, Kellaway P, Zion T. A controlled study of prednisone therapy in infantile spasms. Epilepsia 1979;20:403‐7. [DOI] [PubMed] [Google Scholar]
Inanaga 1989 {published data only}
- Inanaga K, Kumashiro H, Fukuyama Y, Ohtahara S, Shirouzu M. Clinical study of oral administration of DN‐1417, a TRH analog, in patients with intractable epilepsy. Epilepsia 1989;30(4):438‐45. [DOI] [PubMed] [Google Scholar]
Ishii 2002 {published data only}
- Ishii K, Oguni H, Hayashi K, Shirakawa S, Itoh Y, Osawa M. Clinical study of catastrophic infantile epilepsy with focal seizures. Pediatric Neurology 2002;27(5):369‐77. [DOI] [PubMed] [Google Scholar]
Klein 1970 {published data only}
- Klein R. The effects of ACTH and corticosteroids on eplieptiform disorders. Progress in Brain Research 1970;32:263‐9. [DOI] [PubMed] [Google Scholar]
Li 2004 {published data only}
- Li ST, Chiu NC, Ho CS. Effect of topiramate on intractable seizures in Taianese children. Acta Neurologica Tawanica 2004;13(2):59‐63. [PubMed] [Google Scholar]
Lombroso 1983 {published data only}
- Lombroso CT. A prospective study of infantile spasms: clinical and therapeutic correlations. Epilepsia 1983;24:135‐58. [DOI] [PubMed] [Google Scholar]
Lortie 1997 {published data only}
- Lortie A, Chiron C, Dumas C, Mumford J, Dulac O. Optimizing the indication of vigabatrin in children with refractory epilepsy. Journal of Child Neurology 1997;12(4):253‐9. [DOI] [PubMed] [Google Scholar]
Lotze 2004 {published data only}
- Lotze TE, Wilfong AA. Zonisamide treatment for symptomatic infantile spasms. Neurology 2004;62(2):296‐8. [DOI] [PubMed] [Google Scholar]
Luna 1989 {published data only}
- Luna D, Dulac O, Pajot N, Beaumont D. Vigabatrin in the treatment of childhood epilepsies: a single‐blind placebo‐controlled study. Epilepsia 1989;30(4):430‐7. [DOI] [PubMed] [Google Scholar]
Mackay 2004 {published data only}
- Mackay MT, Weiss SK, Adams‐Webber T, Ashwal S, Stephens D, Ballaban‐Gill K, et al. Practice parameter: medical treatment of infantile spasms: report of the American Academy of Neurology and the Child Neurology Society. Neurology 2004;62(10):1668‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Matsumoto 1987 {published data only}
- Matsumoto A, Kumagai T, Takeuchi T, Miyazaki S, Watanabe K. Clinical effects of thyrotropin‐releasing hormone for severe epilepsy in childhood: a comparative study with ACTH therapy. Epilepsia 1987;28(1):49‐55. [DOI] [PubMed] [Google Scholar]
Mikaeloff 2003 {published data only}
- Mikaeloff Y, Saint‐Martin A, Mancini J, Peudenier S, Pedespan JM, Vallée L, et al. Topiramate: efficacy and tolerability in children according to epilepsy syndromes. Epilepsy Reseach 2003;53(3):225‐32. [DOI] [PubMed] [Google Scholar]
Mikati 2002 {published data only}
- Mikati MA, Fayad M, Koleilat M, Mounla N, Hussein R, Kazma A, et al. Efficacy, tolerability and kinetics of lamotrogine in infants. Journal of Pediatrics 2002;141(1):31‐5. [DOI] [PubMed] [Google Scholar]
Mimaki 1998 {published data only}
- Mimaki T. Clinical pharmacology and therapeutic drug monitoring of zonisamide. Therapeutic Drug Monitoring 1998;20(6):593‐7. [DOI] [PubMed] [Google Scholar]
Mumford 1989 {published data only}
- Mumford JP, Dam M. Meta‐analysis of European placebo controlled studies of vigabatrin in drug resistant epilepsy. British Journal of Clinical Pharmacology 1989;27 Suppl:101‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mumford 1994 {published data only}
- Mumford JP, Cannon DJ. Vigabatrin. Epilepsia 1994;35 Suppl 5:25‐8. [DOI] [PubMed] [Google Scholar]
Nabbout 2003 {published data only}
- Nabbout R, Dulac O. Epileptic encephalopathies: a brief overview. Journal of Clinical Neurophysiology 2003;20(6):393‐7. [DOI] [PubMed] [Google Scholar]
Nolte 1988 {published data only}
- Nolte R, Christen HJ, Doerrer J. Preliminary report of a multi‐center study on the West Syndrome. Brain and Development 1988;10(4):236‐42. [DOI] [PubMed] [Google Scholar]
O'Donohoe 1977 {published data only}
- O'Donohoe NV, Paes BA. A trial of clonazepam in the treatment of severe epilepsy in infancy and childhood. Epilepsy: The Eighth International Symposium. New York: Raven Press, 1977:159‐62.
Oguni 2006 {published data only}
- Oguni H, Yanagaki S, Hayashi K, et al. Extremely low‐dose ACTH step up protocol for West syndrome: maximum therapeutic effect with minimal side effects. Brain and Development 2006;28 (1):8‐13. [DOI] [PubMed] [Google Scholar]
Ohtahara 2006 {published data only}
- Ohtahara S. Zonisamide in the management of epilepsy ‐ Japanese experience. Epilepsy Research 2006;68 Suppl:25‐33. [DOI] [PubMed] [Google Scholar]
Oka 2004 {published data only}
- Oka M, Kobayashi K, Akiyama T, Ogino T, Oka E. A study of spike‐density on EEG in West syndrome. Brain and Development 2004;26(2):105‐12. [DOI] [PubMed] [Google Scholar]
Pietz 1993 {published data only}
- Pietz J, Benninger C, Schafer H, Sontheimer D, Mittermaier G, Rating D. Treatment of infantile spasms with high‐dosage vitamin B6. Epilepsia 1993;34(4):757‐63. [DOI] [PubMed] [Google Scholar]
Ramsay 1984 {published data only}
- Ramsey RE. Controlled and comparative trials with valproate: United States. Epilepsia 1984;25 Suppl 1:40‐3. [DOI] [PubMed] [Google Scholar]
Scher 2003 {published data only}
- Scher MS, et al. Uncoupling of EEG‐clinical neonatal seizures after antiepileptic drug use. Pediatric Neurology 2003;28(4):277‐80. [DOI] [PubMed] [Google Scholar]
Schlanger 2002 {published data only}
- Schlanger S, Shinitzky M, Yam D. Diet enriched with omega‐3 fatty acids alleviates convulsion symptoms in epilepsy patients. Epilepsia 2002;43(1):103‐4. [DOI] [PubMed] [Google Scholar]
Schlumberger 1994 {published data only}
- Schlumberger E, Chavez F, Palacios L, Rey E, Pajot N, Dulac O. Lamotrigine in treatment of 120 children with epilepsy. Epilepsia 1994;35(2):359‐67. [DOI] [PubMed] [Google Scholar]
Suzuki 1996 {published data only}
- Suzuki Y, Kita T, Mano T, Arai H, Matsuoka T, Kodaka R, et al. Outcome of initial treatment with high‐dose vitamin B6, valproate sodium or clonazepam in West Syndrome. No To Hattatsu (Brain and Development) 1996;28(5):398‐402. [PubMed] [Google Scholar]
Suzuki 2002 {published data only}
- Suzuki Y, Imai K, Toribe Y, Ueda H, Yanagihara K, Shimono K, et al. Long‐term response to zonisamide in patients with West syndrome. Neurology 2002;58(10):1556‐9. [DOI] [PubMed] [Google Scholar]
Tada 1991 {published data only}
- Tada H, Morooka K, Arimoto K, Matsuo T. Clinical effects of allopurinol on intractable epilepsy. Epilepsia 1991;32(2):279‐83. [DOI] [PubMed] [Google Scholar]
Trevanthan 2003 {published data only}
- Trevathan E. Infantile spasms and Lennox‐Gastaut syndrome. Journal of Child Neurology 2003;17(Suppl 2):2S9‐2S22. [DOI] [PubMed] [Google Scholar]
Trompetter 1977 {published data only}
- Trompetter‐van Woerden ML, Zwan A. Experiences with sodium 2‐propyl valproate in the treatment of epilepsy [Ervaringen met valproinezuur (Depakine) bij de behandeling van epilepsie]. Nederlands Tijdschrift voor Geneeskunde 1977;121(14):578‐90. [PubMed] [Google Scholar]
Vassella 1973 {published data only}
- Vassella F, Pavlincova E, Schneider HJ, Rudin HJ, Karbowski K. Treatment of infantile spasms and Lennox‐Gastaut syndrome with clonazepam (Rivotril). Epilepsia 1973;14:165‐75. [DOI] [PubMed] [Google Scholar]
Veggiotti 1994 {published data only}
- Veggiotti P, Cieuta C, Rey E, Dulac O. Lamotrigine in infantile spasms. Lancet 1994;344:1375‐6. [DOI] [PubMed] [Google Scholar]
Wallace 1995 {published data only}
- Wallace SJ. Management issues in severe childhood epilepsies. Seizure 1995;4:215‐20. [DOI] [PubMed] [Google Scholar]
Weinmann 1967 {published data only}
- Weinmann HM. Mogadon therapy of infantile spasms. Electroencephalography and Clinical Neurophysiology 1967;23(2):183. [PubMed] [Google Scholar]
Willig 1980 {published data only}
- Willig RP, Lagenstein I. Therapeutic trial with a fragment of ACTH (ACTH 4‐10) in early childhood epilepsy [Therapieversuch mit einem ACTH‐Fragment (ACTH 4‐10) bei Fruhkindlichen Anfallen]. Monatsschrift Kinderheilkunde 1980;128(2):100‐3. [PubMed] [Google Scholar]
Wohlrab 1998 {published data only}
- Wohlrab G, Bolthauser E, Schmitt B. Vigabatrin as a first ‐line drug in West Syndrome: clinical and electroencephalographic outcome. Neuropediatrics 1998;29:133‐6. [DOI] [PubMed] [Google Scholar]
Yagi 2004 {published data only}
- Yagi K. Overview of Japanese experience‐controlled and uncontrolled trials. Seizure 2004;13 Suppl 1:11‐5. [DOI] [PubMed] [Google Scholar]
Yamamoto 1998 {published data only}
- Yamamoto H, Asoh M, Murakami H, Kamiyama N, Ohta C. Liposteroid (dexamethasone palmitate) therapy for West Syndrome: a comparative study wWith ACTH therapy. Pediatric Neurology 1998;18(5):415‐9. [DOI] [PubMed] [Google Scholar]
Yamauchi 2004 {published data only}
- Yamauchi T, Aikawa H. Efficacy of zonisamide: our experience. Seizure 2004;13 Suppl 1:41‐8. [DOI] [PubMed] [Google Scholar]
Zafeiriou 1996 {published data only}
- Zafeiriou DI, Kontopoulos EE, Tsikoulas IG. Adrenocorticotropic hormone and vigabatrin treatment of children with infantile spasms underlying cerebral palsy. Brain and Development 1996;18:450‐2. [DOI] [PubMed] [Google Scholar]
References to ongoing studies
ICISS {unpublished data only}
- International Collaboration Infantile Spasms Study. Ongoing study May 2007.
ISRCTN 36757519 {unpublished data only}
- Ongoing study Starting date of trial not provided. Contact author for more information.
ISRCTN Surgery/AEDs {published data only}
- Ongoing study Starting date of trial not provided. Contact author for more information.
Additional references
Aicardi 1994
- Aicardi J. Epilepsy in children: infantile spasms and related syndromes. In: Procopis P, Rapin I editor(s). The International Review of Child Neurology. 2nd Edition. New York: Raven Press, 1994. [Google Scholar]
Beaumanoir 1992
- Beaumanoir A, Dravet C. The Lennox‐Gastaut Syndrome. In: Roger J, Bureau M, Dravet C, Dreifuss F, Pervet H, Wolf P editor(s). Epileptic syndromes in infancy, childhood and adolescence. Second Edition. London: John Libbey, 1992:115‐32. [Google Scholar]
Cowan 1991
- Cowan LD, Hudson LS. The epidemiology and natural history of infantile spasms. Journal of Child Neurology 1991;6:335‐64. [DOI] [PubMed] [Google Scholar]
Go 2012
- Go CY, Mackay MT, Weiss SK, Stephens D, Adams‐Webber T, Ashwal S, et al. Evidence‐based guideline update: medical treatment of infantile spasms. Report of the Guideline Development Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology 2012;78(24):1974‐80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hancock 1998
- Hancock E, Osborne J. Treatment of infantile spasms with high dose oral prednisolone. Developmental Medicine and Child Neurology 1998;40:500. [PubMed] [Google Scholar]
Hancock 1999
- Hancock E, Osborne J. Vigabatrin in treatment of infantile spasms in tuberous sclerosis: literature review. Journal of Child Neurology 1999;14(2):71‐4. [DOI] [PubMed] [Google Scholar]
Higgins 2005
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 4.2.4 [updated March 2005]. The Cochrane Collaboration, 2005. www.cochrane‐handbook.org.
ILAE 1989
- ILAE. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989;30(4):389‐99. [DOI] [PubMed] [Google Scholar]
Lux 2004b
- Lux AL, Osborne JP. A proposal for case definitions and outcome measures in studies of infantile spasms and West syndrome: consensus statement of the West Delphi Group. Epilepsia 2004;45(11):1416‐28. [DOI] [PubMed] [Google Scholar]
Mackay 2004
- Mackay MT, Weiss SK, Adams‐Webber T, Ashwal S, Stephens D, Ballaban‐Gill K, et al. Practice parameter: medical treatment of infantile spasms: report of the American Academy of Neurology and the Child Neurology Society. Neurology 2004;62(10):1668‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
O'Callaghan 2011
- O'Callaghan FJK, Lux AL, Darke K, Edwards SW, Hancock E, Johnson AL, et al. The effect of lead time to treatment and of age of onset on developmental outcome at 4 years in infantile spasms: Evidence from the United Kingdom Infantile Spasms Study. Epilepsia 2011;52(7):1359‐64. [DOI] [PubMed] [Google Scholar]
Riikonen 1996
- Riikonen R. Long‐term outcome of West Syndrome: a study of adults with a history of infantile spasms. Epilepsia 1996;37(4):367‐72. [DOI] [PubMed] [Google Scholar]
Roger 1985
- Roger J, Durvet C, Bureau M, Dreifuss FE, Wolf P. West syndrome: infantile spasms. Epileptic syndromes in infancy, childhood and adolescence. John Libbey Eurotext Ltd. 1985.
References to other published versions of this review
Hancock 2008
- Hancock EC, Osborne JP, Edwards SW. Treatment of infantile spasms. Cochrane Database of Systematic Reviews 2008, Issue 4. [DOI: 10.1002/14651858.CD001770.pub2] [DOI] [PubMed] [Google Scholar]