

Abstract
To elucidate the evolutionary mechanisms of the human immunodeficiency virus type 1 gp120 envelope glycoprotein at the single-site level, the degree of amino acid variation and the numbers of synonymous and nonsynonymous substitutions were examined in 186 nucleotide sequences for gp120 (subtype B). Analyses of amino acid variabilities showed that the level of variability was very different from site to site in both conserved (C1 to C5) and variable (V1 to V5) regions previously assigned. To examine the relative importance of positive and negative selection for each amino acid position, the numbers of synonymous and nonsynonymous substitutions that occurred at each codon position were estimated by taking phylogenetic relationships into account. Among the 414 codon positions examined, we identified 33 positions where nonsynonymous substitutions were significantly predominant. These positions where positive selection may be operating, which we call putative positive selection (PS) sites, were found not only in the variable loops but also in the conserved regions (C1 to C4). In particular, we found seven PS sites at the surface positions of the α-helix (positions 335 to 347 in the C3 region) in the opposite face for CD4 binding. Furthermore, two PS sites in the C2 region and four PS sites in the C4 region were detected in the same face of the protein. The PS sites found in the C2, C3, and C4 regions were separated in the amino acid sequence but close together in the three-dimensional structure. This observation suggests the existence of discontinuous epitopes in the protein's surface including this α-helix, although the antigenicity of this area has not been reported yet.
The envelope glycoprotein of human immunodeficiency virus type 1 (HIV-1) interacts with receptors on the target cell and mediates virus entry by fusing the viral and cell membranes. To maintain viral infectivity, amino acids that interact with receptors are expected to be more conserved than other sites on the protein surface. Amino acid changes that reduce the affinity for the receptor will decrease infectivity or survivability, implying that negative selection is operating against amino acid changes on sites for receptor binding. The primary receptor for HIV is CD4 (9), and the secondary receptors are chemokine receptors. The main second receptor for the macrophage-tropic strains is CCR5 (11, 13) and that for T-cell-tropic strains is CXCR4 (15). In contrast to the functional constraint of amino acids for receptor binding, some amino acid changes in this protein may produce antigenic variations that enable the virus to escape from recognition by the host immune system. Variants with such mutations at antigenic sites will have a higher fitness than others, implying that positive selection is operating against amino acid changes at the antigenic sites. Therefore, both positive and negative selections against amino acid changes are taking place during the evolution of the surface proteins of parasites (48, 66).
The relative importance of positive and negative selection at each position in the gp120 presumably affects the degree of amino acid variation. We can imagine that the amino acid sites for receptor binding are relatively conserved because of the functional importance and that antigenic sites are relatively variable. Analysis of amino acid variation at each position would be helpful in predicting antigenic sites, as the analysis of the amino acid variability of the immunoglobulin molecule predicted the complementarity-determining regions (64). The conserved and variable regions of gp120 were originally assigned by considering the proportion of conserved amino acid sites and the frequencies of insertions and deletions in the amino acid sequences of seven isolates from five patients (40, 56). Lauder et al. (34) also evaluated the amino acid variability of this protein by analyzing 63 sequences of various subtypes. They found that the assignment of conserved and variable regions by Modrow et al. (40) was still valid, although they pointed out that the region between the V3 and V4 regions (called the C3 region in this paper) was less conserved. However, the level of amino acid variability or selection mechanism can be quite different among amino acid sites in a short region, and it is possible that hypervariable sites with adaptive significance exist even in the conserved regions.
Comparison of the relative variability among amino acid sites is not sufficient to clarify the relative importance of positive and negative selection for amino acid changes. When we observe a higher degree of amino acid variation at some sites than at others, positive selection is one of the possible explanations. However, from the standpoint of the neutral theory of molecular evolution (27, 28), most such cases can be explained by different levels of functional constraint of amino acids. In general, the surface-exposed amino acid residues of the protein are more variable and hydrophilic than the interior ones (18). Comparison of the rates of silent (synonymous) and amino-acid-altering (nonsynonymous) substitutions (25, 37, 39, 42) enables us to test whether nucleotide variation in the protein-coding region is compatible with the neutral theory (27, 28). This test is based on the prediction by the neutral theory of molecular evolution that the rate of nonsynonymous substitution is not higher than that of synonymous substitution. In general, the preponderance of synonymous substitutions have been reported in the majority of the genes of various organisms, including viruses (14, 19, 20, 37), but there is an increasing number of the reports suggesting the existence of positive selection or overdominant selection at the molecular level (14, 23, 24). Representative examples that showed an excess of nonsynonymous substitutions were molecules involved in the immune system and surface proteins of parasites (14, 24).
There is a rapidly increasing number of reports of comparisons between the rates of synonymous and nonsynonymous substitutions in the env gene of HIV (35, 38, 55). Over the entire region of gp120, the number of synonymous substitutions per site was larger than that of nonsynonymous substitutions (35, 38), implying that negative selection is predominant in the whole protein. However, several studies reported an excess of nonsynonymous substitutions with statistical significance in major epitopes such as the V2 (51) and V3 (3, 4, 49, 51, 52, 68) regions in gp120. These observations imply that positive selection may be dominantly operating on these two regions. On the other hand, an excess of synonymous substitutions has been reported for regions other than the variable regions (51). This observation implies that negative selection is predominant in the regions other than the V2 and V3 regions. However, we cannot definitely say that positive selection is not operating in the conserved regions because positive selection acting at a single site is difficult to determine by comparing the numbers of synonymous and nonsynonymous substitutions at more than one amino acid site.
The relative importance of positive and negative selection or the level of functional constraint of amino acids may be different among amino acid positions within a short region. In fact, the physical locations or the directions of the side chains of the residues can be quite different among the residues within a short region in sequence. Recent studies revealed that there was considerable heterogeneity in substitution rate among amino acid positions (32, 69). These facts suggest that an analysis of variation at the single-amino-acid-site level would be very important to understanding the nature of variations and elucidating the evolutionary mechanism (5, 16, 21, 44). Recently, methods to detect selection at the single-site level by comparison of synonymous and nonsynonymous substitutions have been developed (44, 57), and these methods were applied to the sequences of HIV-1, including the V3 region, which were periodically sampled from individual patients (22).
In this study, to elucidate the evolutionary mechanisms of the whole HIV-1 gp120 envelope glycoprotein at the single-site level, we collected and analyzed all available sequence data for the protein. By analyzing 186 sequences of HIV-1 gp120 (subtype B), we reevaluated amino acid variability at the single-site level and estimated the numbers of synonymous and nonsynonymous substitutions at each codon position to detect positive and negative selection. Thirty-three amino acid positions which may be under positive selection were identified, and we call these sites putative positive-selection (PS) sites. These PS sites were found not only in the variable loops but also in the conserved regions. Confirmation of the physical locations of the PS sites in the three-dimensional structure revealed that several PS sites in the conserved regions, which were far apart in the amino acid sequence, were close together in the three-dimensional structure. Here we discuss the adaptive significance of these PS sites and the possibility of the existence of unidentified epitopes.
MATERIALS AND METHODS
Sequence data and multiple alignment.
Nucleotide sequences coding the entire HIV-1 gp120 of subtype B were collected from the HIV sequence database (30). Nucleotide sequences that contain frameshift mutations or in-frame stop codons were excluded, and 186 sequences were selected for the present analysis. The amino acid sequences deduced from the nucleotide sequences were aligned with each other by using CLUSTAL W, version 1.6 (60), with subsequent inspection and manual modifications. The region for the signal peptide (positions 1 to 28 in strain HXBc2) was excluded from analyses. Alignment was successful for 425 amino acid sites among 483 sites in gp120; however, alignments were not possible for four regions where insertions, deletions, and partial duplications might be frequent (positions 133 to 153, 185 to 190, 392 to 415, and 459 to 465 in HXBc2). Positions 310 Gln, 311 Arg, and 355 Asn were also excluded from analyses because these positions are gap positions in more than half of the sequences. In total, variations at 422 amino acid sites were examined. Nucleotide sequences were aligned by converting amino acids in the alignment into corresponding nucleotides. The gp120 amino acids are numbered according to the sequence of the HXBc2 gp120 glycoprotein, where residue 1 is the methionine at the amino terminus of the signal peptide.
Inferring phylogenetic relationships.
To estimate the number of amino acid or nucleotide substitutions that occurred at each amino acid or nucleotide position, we inferred phylogenetic relationships and estimated the ancestral state of amino acids or nucleotides at internal nodes of the phylogeny. The nucleotide sequence HIVELICG of subtype D was added to the phylogenetic analysis as an outgroup because subtype D was known to be closely related to subtype B. The 1,188 nucleotide sites where an alignment was possible with the outgroup were used for the phylogenetic analysis. Before the phylogenetic analysis, we examined whether there are any effects of recombination in estimating the phylogenetic relationship among the sequences. We examined the distribution of phylogenetically informative sites among all possible combinations of four sequences along sliding windows of 400 bp. We checked whether the distribution of informative sites is significantly different between the 5′-half window and the 3′-half window and did not observe any apparent cases of recombination. A phylogenetic tree was first estimated from nucleotide sequences by the neighbor-joining method (47). Pairwise distances were estimated by the maximum-likelihood methods, taking the transition/transversion ratio into consideration. In order to look for the maximum-likelihood tree topology, a local rearrangement search with the maximum-likelihood approach (1) was conducted by starting from the topology of the neighbor-joining tree. This phylogenetic analysis was conduced by using MOLPHY (1).
Estimation of ancestral amino acids and nucleotides at ancestral nodes.
The ancestral state of an amino acid or a nucleotide at the internal nodes of the phylogeny was estimated by the maximum-parsimony principle. If the ancestral amino acids or nucleotides were not unique, equal probabilities were given to all equally parsimonious states.
Analysis of amino acid variability at the single-site level.
We evaluated the amino acid variability of HIV-1 gp120 by the three simple measures acceptability, changeability, and diversity.
(i) Number of different amino acids (acceptability).
To estimate the range of acceptable amino acids at each position, the number of different amino acids was counted. Note that this value depends on the number of sequences.
(ii) Number of amino acid substitutions (changeability).
To see how changeable each position is, we estimated the number of amino acid substitutions that occurred by estimating the number of amino acid substitutions that have occurred throughout the phylogeny. This changeability of amino acid site depends on mutability and natural selection, which is operating against amino acid changes. To evaluate the nonrandomness of the distribution of substitutions among sites, we compared the distribution of amino acid substitutions among sites with the expectation of the Poisson process (29). Hypervariable sites were identified (68) by using the expectation from the Poisson process. We calculated the upper limit of confidence (99%) of the substitution number from the Poisson distributions with the average number of substitutions. Hypervariable sites were defined as sites where the number of amino acid substitutions was larger than the upper limit.
(iii) Diversity of amino acids.
To evaluate the level of amino acid variation in a population, we defined the diversity of amino acids at a given position as the proportion of pairs having different amino acids in two sequences randomly chosen from the sample. This value is estimated from the frequencies of different amino acids or gaps at each position. Basically, this quantity is analogous to nucleotide diversity and heterozygosity or gene diversity in population genetics (43). The amount of diversity estimated by these methods is independent of the number of sequences if the sequences are randomly sampled. Though some sites in alignments may include gaps, the diversity of gaps and amino acids can be estimated separately by this method. Diversity of gaps (Dgap) can be calculated as
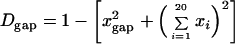 |
where xi represents the proportion of the ith amino acid at a given position and xgap represents the proportion of the gap at a given position. Diversity of amino acids (Daa) can also be estimated separately:
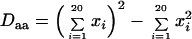 |
We defined monomorphic sites as sites where the diversity of amino acids is less than 0.05. This is analogous to a monomorphic locus within a population (43).
Estimation of the numbers of synonymous and nonsynonymous substitutions at each codon position.
The actual numbers of synonymous and nonsynonymous substitutions throughout the phylogeny were estimated by using the ancestral states in the interior nodes of the phylogeny. If the codons were not unique at ancestral or descendant nodes, equal probabilities were given to all possible substitutions. The numbers of potential synonymous and nonsynonymous sites at each ancestral node were also estimated. If the estimated codon was not unique, the average numbers of synonymous and nonsynonymous sites among equally parsimonious codons were calculated. We took the transition/transversion ratio into consideration to estimate the numbers of synonymous and nonsynonymous sites. This is because analysis of the substitution pattern of HIV (41) showed that transition occurs much more frequently than transversion and the transition/transversion ratio affects the estimation of the relative numbers of synonymous and nonsynonymous sites (25). To obtain a reliable ratio of transitional mutations to transversional mutations that is not affected by functional constraint of amino acids, we analyzed the substitution pattern at fourfold-degenerate sites. The transition/transversion ratio was estimated to be 6.1, and this ratio was used in estimating the numbers of synonymous and nonsynonymous sites.
Comparison of the numbers of synonymous and nonsynonymous substitutions.
To examine whether the rates of synonymous and nonsynonymous substitution were significantly different, statistical tests of independence were conducted. If the rates of synonymous and nonsynonymous substitutions are equal, the following relationships are expected: Sc:St = Nc:Nt or Sc:Nc = St:Nt, where Sc is the total number of synonymous changes throughout the phylogeny, Nc is the total number of nonsynonymous changes throughout the phylogeny, St is the total number of synonymous sites at all internal nodes of the phylogeny, and Nt is the total number of nonsynonymous sites at all internal nodes of the phylogeny. These values were rounded off to the integer, and Fisher's exact test of independence was applied to the data for each codon position. We identified the codon positions where equal rates of synonymous and nonsynonymous substitutions were rejected at the 1% level. In such cases, if nonsynonymous substitutions were predominant, we identified these sites as putative PS sites. On the other hand, if synonymous substitutions were predominant, these sites were identified as putative negative selection (NS) sites.
The codons for methionine and tryptophan have no synonymous codons. The codon sites where methionine or tryptophan is very frequent have few synonymous sites. Therefore, we did not test eight codon positions (35 Trp, 69 Trp, 95 Met, 96 Trp, 100 Met, 427 Trp, 434 Met, and 475 Met) for which the average number of synonymous sites was less than 0.1, although there were nucleotide variations at these codon positions.
Computer programs and statistics.
The computer programs and statistics used in this study are available from the author on request.
RESULTS
Amino acid variabilities at the single-site level.
The levels of amino acid variabilities at each position of HIV-1 gp120 were evaluated by three measures (acceptability, changeability, and diversity) (Fig. 1). We analyzed 422 of 483 amino acid sites of HIV-1 gp120, excluding the four regions where the occurrence of insertions, deletions, and partial duplications might be frequent. The estimated values of amino acid variabilities by the three measures had highly significant correlations with each other. All three measures showed that the level of amino acid variability was very different from site to site in the conserved and variable regions (Fig. 1, Table 1). Note that the standard deviations (SDs) of substitution numbers among sites in the four conserved regions (C1 to C4) were not much smaller than those of the all sites analyzed (Table 1).
FIG. 1.




Amino acid variabilities of HIV-1 gp120 at the single-amino-acid-site level by three different measures. At the top of each section is shown the amino acid sequence of HXB2. The locations of the conserved (C1 to C5) and variable (V1 to V5) regions are shown. Lowercase letters represent amino acid sites that were not analyzed in this study. Below the sequence are shown amino acid variabilities for sites within that sequence as estimated by three measures, acceptability (number of different amino acids), changeability (number of substitutions), and diversity.
TABLE 1.
Amino acid variabilities of HIV-1 gp120 in the conserved (C1 to C5) and variable (V1 to V5) regions
| Region | No. of sites analyzed/no. in region | Kindsa | Substitutionsb | Diversityc | No. of sites/no. analyzed
|
|
|---|---|---|---|---|---|---|
| Monomorphic | Hypervariable | |||||
| C1 | 97/97 | 2.80 ± 2.08 | 5.05 ± 8.99 | 0.085 ± 0.158 | 65/97 | 9/97 |
| V1 | 11/32 | —d | — | — | 6/11 | 3/11 |
| V2 | 33/39 | 5.48 ± 2.81 | 13.34 ± 9.80 | 0.261 ± 0.193 | 6/33 | 13/33 |
| C2 | 99/99 | 2.96 ± 1.81 | 6.81 ± 8.77 | 0.125 ± 0.179 | 56/99 | 20/99 |
| V3 | 33/35 | 5.39 ± 2.32 | 14.92 ± 13.90 | 0.251 ± 0.207 | 6/33 | 12/33 |
| C3e | 53/54 | 4.11 ± 2.72 | 12.33 ± 14.73 | 0.190 ± 0.220 | 19/53 | 17/53 |
| V4 | 9/33 | — | — | — | 3/9 | 2/9 |
| C4 | 41/43 | 2.83 ± 2.20 | 5.58 ± 9.93 | 0.111 ± 0.189 | 27/41 | 6/41 |
| V5 | 6/11 | — | — | — | 4/6 | 1/6 |
| C5 | 40/40 | 2.25 ± 1.06 | 3.38 ± 4.91 | 0.050 ± 0.089 | 30/40 | 2/40 |
| All | 422/483 | 3.39 ± 2.33 | 7.88 ± 10.76f | 0.137 ± 0.187 | 222/422 | 85/422 |
Average number of different amino acids ± SD among sites.
Average number of amino acid substitutions ± SD among sites.
Average diversity of amino acids ± SD among sites.
—, average values for the V1, V4, and V5 regions are not shown because too few sites were analyzed.
In the original paper (40), this region was not classified as either conserved or variable. This region is now often called the C3 region.
If we assume a Poisson distribution with this average number of amino acid substitutions, the probability that the number of amino acid substitutions at a given site is equal to or larger than 15 is less than 0.01. We call these sites hypervariable sites.
We evaluated the nonrandomness of the distribution of amino acid substitutions among sites by comparing the expectation from the Poisson distribution with our findings. The average number of amino acid substitutions over sites was 7.88. The ratio of the variance to the average number of substitutions was 14.70, implying that the distribution of the substitution number was highly heterogeneous. According to the Poisson process, the probability that the number of amino acid substitutions is equal to or more than 15 is less than 0.01 with this average number. By using this upper limit of confidence, we identified 85 hypervariable sites as sites where the number of amino acid substitutions was equal to or greater than 15, the upper limit of confidence (Fig. 1). Among 422 positions examined, 222 positions were detected as monomorphic sites, where diversity is less than 0.05, implying that about half of the amino acid sites in this protein show little variation. These observations show that amino acid substitutions were concentrated on the particular variable sites in this protein.
The level of amino acid variation in the C3 region was closer to that in the V2 or V3 region than that in the other conserved regions. In particular, the number of amino acid substitutions in the C3 region was closer to that in the V2 region. In fact, amino acid substitutions in the C3 region were concentrated on the residues located on the surface of the α-helix (positions 335 to 347), although this α-helix region includes three monomorphic sites in its interior positions. This α-helix is located along the surface of the protein, with one side of the helix facing the outside and the other side facing the hydrophobic interior of the protein (33). Therefore, the pattern of the presence of variable and monomorphic sites seems to be consistent with the pitch of an α-helix, namely, 3.6 residues per turn.
The level of amino acid variation at the residues involved in receptor binding (CD4 and CCR5) (33, 46) was examined (Table 2). To see the relative variability of the receptor binding sites, the average variabilities at the receptor binding sites were compared with those of the whole gp120. On the average, the residues that made direct contact with the CD4 molecule were more conserved than the other sites in this protein. In fact, six positions (366 Gly, 367 Gly, 428 Gln, 430 Val, 456 Arg, and 458 Gly) had no amino acid variations. However, these CD4 binding sites include several hypervariable sites (279 Asp, 281 Ala, 283 Thr, 365 Ser, 426 Asn, and 429 Lys). To see the relative variabilities for the critical sites for second-receptor binding, variabilities where the substitutions were critical for CCR5 binding were examined (Table 2). Lower variabilities were observed at these sites with the exception of two positions, 317 Phe and 440 Ser. In particular, five positions (257 Thr, 381 Glu, 420 Ile, 438 Pro, and 441 Gly) had no amino acid variations. Position 317 Phe in the V3 loop and position 440 Ser in the C4 region had very high substitution numbers.
TABLE 2.
Amino acid variabilities of CD4 and CCR5 binding sitesa
| Protein | Siteb | Kinds | Substitutions | Diversity |
|---|---|---|---|---|
| CD4 | 125/L | 2 | 2.00 | 0.032 |
| 279/D | 4 | 26.00 | 0.603 | |
| 280/N | 2 | 2.00 | 0.021 | |
| 281/A | 7 | 21.00 | 0.483 | |
| 283/T | 5 | 25.00 | 0.525 | |
| 365/S | 5 | 6.00 | 0.084 | |
| 366/G | 1 | 0.00 | 0.000 | |
| 367/G | 1 | 0.00 | 0.000 | |
| 368/D | 2 | 1.00 | 0.010 | |
| 370/E | 2 | 1.00 | 0.021 | |
| 371/I | 3 | 6.00 | 0.063 | |
| 425/N | 2 | 1.00 | 0.011 | |
| 426/N | 5 | 15.00 | 0.487 | |
| 427/W | 3 | 5.00 | 0.053 | |
| 428/Q | 1 | 0.00 | 0.000 | |
| 429/K | 5 | 23.00 | 0.501 | |
| 430/V | 1 | 0.00 | 0.000 | |
| 455/T | 4 | 3.00 | 0.032 | |
| 456/R | 1 | 0.00 | 0.000 | |
| 457/D | 2 | 1.00 | 0.011 | |
| 458/G | 1 | 0.00 | 0.000 | |
| 459/G | NAc | NA | NA | |
| 469/R | 2 | 2.00 | 0.021 | |
| 472/G | 2 | 1.00 | 0.011 | |
| 473/G | 3 | 2.00 | 0.021 | |
| 474/D | 2 | 14.00 | 0.175 | |
| Avg, CD4 bindingd | 2.62 | 6.28 | 0.127 | |
| Avg, gp120e | 3.39 | 7.88 | 0.137 | |
| CCR5f | 121/K | 3 | 4.00 | 0.042 |
| 123/T | 3 | 2.00 | 0.021 | |
| 207/K | 2 | 2.00 | 0.021 | |
| 257/T | 1 | 0.00 | 0.000 | |
| 317/F | 8 | 30.00 | 0.381 | |
| 381/E | 1 | 0.00 | 0.000 | |
| 383/F | 2 | 2.00 | 0.021 | |
| 420/I | 1 | 0.00 | 0.000 | |
| 421/K | 3 | 7.00 | 0.121 | |
| 422/Q | 3 | 2.00 | 0.021 | |
| 438/P | 1 | 0.00 | 0.000 | |
| 440/S | 10 | 52.83 | 0.781 | |
| 441/G | 1 | 0.00 | 0.000 | |
| Avg, CCR5 bindingg | 3.00 | 7.83 | 0.108 | |
| Avg, CCR5 bindingh | 1.91 | 1.73 | 0.022 | |
| Avg, gp120 | 3.39 | 7.88 | 0.137 |
Amino acid sites that contact the CD4 molecule (33) or that may be critical for CCR5 binding are listed. Sites at which substitutions resulted in a greater than 90% decrease in CCR5 binding are listed. (46).
Position/amino acid in strain HXB2. Also see Table 1, footnotes a, b, and c.
NA, not analyzed.
Average values for CD4 binding sites.
Average values for the entire gp120.
Position 317 Phe in the V3 loop and position 440 Ser in the C4 region may be involved in usage of the second receptor.
Average values of the 13 critical sites for CCR5 binding.
Average values of the 11 critical sites for CCR5 binding with data for positions 317 Phe and 440 Ser excluded.
Distribution of synonymous and nonsynonymous substitutions among codon positions.
We estimated the numbers of synonymous and nonsynonymous substitutions per site at each codon position in the gp120 gene and examined the distribution of synonymous and nonsynonymous substitutions among codon positions. The average numbers of synonymous and nonsynonymous substitutions per site in this gene were 4.46 and 4.02, respectively (Table 3). The numbers of synonymous and nonsynonymous substitutions were significantly correlated with each other (r = 0.496, P < 0.01). To see the nonrandomness of the substitutions among codon sites, the distributions of synonymous and nonsynonymous substitutions were compared with the expectation from the Poisson process. The variance/mean ratios for synonymous and nonsynonymous substitutions were 4.33 and 8.00, respectively (Table 3). This higher variance/mean ratio for nonsynonymous substitutions indicates that the distribution of nonsynonymous substitutions was highly heterogeneous among sites because the level of functional constraint or selection mechanism for amino acid changes may be very different among sites. The variance/mean ratio for synonymous substitutions was much smaller than that of nonsynonymous substitutions, however; this ratio of 4.33 was larger than 1. The distribution of synonymous substitutions also seems to be heterogeneous among codon sites. However, there is a possibility that nonsynonymous substitutions affected the estimation of the number of synonymous substitutions which occurred at the same codon, because assignment of synonymous or nonsynonymous substitution is not clear when two codons differ at more than one nucleotide site. To see whether the distribution of synonymous substitutions is really heterogeneous among sites, we also examined the codon sites where the third position is fourfold degenerate and there is no amino acid variation, because the estimated numbers of synonymous substitutions at these sites are not affected by nonsynonymous substitutions. The variance/mean ratio of synonymous substitutions at the fourfold-degenerate sites was estimated to be 2.05. This ratio was still larger than 1. This observation indicates that the distribution of synonymous substitutions was not uniform unless the level of heterogeneity was much lower than that of nonsynonymous substitutions.
TABLE 3.
Distribution of synonymous and nonsynonymous substitutions among codon sites in gp120a
| Type of substitution | No. of codons | Avg | Variance | Ratio (variance/ average)b |
|---|---|---|---|---|
| Synonymous (all)c | 414 | 4.46 | 19.30 | 4.33 |
| Synonymous (fourfold)d | 34 | 3.21 | 6.59 | 2.05 |
| Nonsynonymous | 422 | 4.02 | 32.15 | 8.00 |
The ratio of the number of nonsynonymous substitutions to that of synonymous substitutions (KA/KS ratio) in the entire region analyzed was estimated to be 0.90. The correlation coefficient between the numbers of synonymous and nonsynonymous substitutions was 0.496.
If the distribution is compatible with the Poisson process, this ratio is theoretically expected to be 1.
All codon sites analyzed.
Fourfold-degenerate codon sites where no amino acid variations were observed.
Codon sites where nonsynonymous substitutions were predominant.
To detect amino acid sites where positive selection may be operating (PS sites), we identified codon positions where the rate of nonsynonymous substitution was significantly higher than that of synonymous substitution. Thirty-three amino acid positions were detected as PS sites (Fig. 2, Table 4) among 414 codon positions tested. The probability that getting 33 sites out of 414 is significant at the 1% level is very low (P < 10−14), suggesting that detecting 33 PS sites out of 414 is not merely a stochastic effect and that most of the PS sites have adaptive significance. Eight of the PS sites were found in the variable loops (position 132 Thr in the V1 loop; positions 172 Glu and 178 Lys in the V2 loop; and positions 303 Thr, 306 Arg, 308 Arg, 319 Thr, and 322 Lys in the V3 loop). However, most of the PS sites (22 of 33) were found outside the variable loops. We confirmed the physical locations of the PS sites in the three-dimensional structure (33) by using the computer package Rasmol (50) and found that PS sites were located on the surface of this protein (Fig. 3). Furthermore, in most cases, the side chains of these PS sites were exposed to the solvent. The distribution of the PS sites was not uniform on the protein surface, and many of the PS sites were in the opposite face for CD4 binding in the outer domain (Fig. 3C). In particular, eight PS sites in the C3 region (333 Ile, 335 Arg, 336 Ala, 337 Lys, 340 Asn, 343 Lys, 346 Ala, and 347 Ser) were found within a short region. Seven of them were on the surface-exposed face of the α-helix. In addition, position 333 Ile was in the β-strand that is just N-terminal of this α-helix. Two PS sites (291 Ser and 293 Glu) in the C2 region were close to the eight PS sites in the C3 region, and position 291 Ser was in the β-strand that was parallel to the β-strand including residue 333 Ile. Furthermore, positions 446 Ser and 444 Arg were detected as PS sites in the β-strand that is parallel to the β-strand including residue 291 Ser (Fig. 3C). In the region just upstream of these two PS sites, 442 Gln and 440 Ser were also detected, although the substitution at position 440 Ser was known to be responsible for CCR5 binding. On the face where CD4 binding occurs, four PS sites (281 Ala, 283 Thr, 429 Lys, and 387 Ser) were detected, although three of them were shown to interact with the CD4 molecule (33). Three PS sites (130 Lys, 195 Ser, and 200 Val) were found in the stem of the V1/V2 loop. Only one PS site (240 Thr) was detected in the inner domain that is inside the trimer complex. In the N-terminal region, where the structure was not solved, three PS sites (31 Thr, 85 Val, and 87 Val) were also detected.
FIG. 2.

PS sites and NS sites in HIV-1 gp120. PS sites (red +) and NS sites (blue −) are shown in the amino acid sequence of strain HXBc2. Amino acid positions for CD4 binding (33) are indicated by orange asterisks. Amino acid positions where substitutions are critical for CCR5 binding (46) are indicated by green asterisks. Amino acid positions for disulfide bonding (36) are indicated by purple ^. Lowercase letters represent amino acid sites that were not tested. Approximate locations for the outer domain, the inner domain, and the bridging sheet (33) are shown by pink, light blue, and yellow-green bars, respectively.
TABLE 4.
Amino acid positions with a significant excess of nonsynonymous substitutions (PS sites)
| Positiona | Region | Synb | Nonsync | Pd | Structuree | Antibodyf | CTLf | Function |
|---|---|---|---|---|---|---|---|---|
| 31/T | C1 | 2.04 | 20.95 | 0.000011 | + | + | ||
| 85/V | C1 | 2.99 | 25.39 | 0.000001 | + | + | ||
| 87/V | C1 | 2.34 | 20.52 | 0.000061 | + | − | ||
| 130/K | V1 | 1.33 | 10.24 | 0.009989 | − | − | ||
| 132/T | V1 | 2.01 | 10.46 | 0.008358 | V1 loop | − | − | |
| 172/E | V2 | 1.25 | 12.27 | 0.002402 | V2 loop | + | − | |
| 178/K | V2 | 0.00 | 12.08 | 0.000450 | V2 loop | + | − | |
| 195/S | V2 | 1.54 | 12.83 | 0.002983 | V1/V2 stem | − | + | |
| 200/V | C2 | 1.50 | 9.74 | 0.003907 | V1/V2 stem | − | + | |
| 240/T | C2 | 2.74 | 17.49 | 0.000241 | − | + | ||
| 281/A | C2 | 1.03 | 11.80 | 0.000979 | + | − | CD4 binding (33) | |
| 283/T | C2 | 3.12 | 12.75 | 0.008117 | − | − | CD4 binding (33) | |
| 291/S | C2 | 3.00 | 14.00 | 0.003021 | β-Strand | − | + | |
| 293/E | C2 | 2.55 | 14.90 | 0.002740 | β-Strand | − | + | |
| 303/T | V3 | 0.00 | 6.93 | 0.004304 | V3 loop | − | + | |
| 306/R | V3 | 2.55 | 19.39 | 0.000280 | V3 loop | + | + | Cell tropism (10, 17) |
| 308/R | V3 | 8.05 | 31.92 | 0.000091 | V3 loop | + | + | Antigenicity (63) |
| 319/T | V3 | 2.78 | 13.98 | 0.001193 | V3 loop | + | + | |
| 322/K | V3 | 5.23 | 29.41 | 0.000049 | V3 loop | + | + | Antigenicity, cell tropism (53) |
| 333/I | C3 | 1.11 | 11.93 | 0.001333 | β-Strand | − | − | |
| 335/R | C3 | 1.14 | 17.45 | 0.000051 | α-Helix | − | − | |
| 336/A | C3 | 2.50 | 19.74 | 0.000025 | α-Helix | − | − | |
| 337/K | C3 | 3.96 | 17.53 | 0.003839 | α-Helix | − | − | |
| 340/N | C3 | 3.91 | 16.56 | 0.005047 | α-Helix | − | + | |
| 343/K | C3 | 2.63 | 21.88 | 0.000100 | α-Helix | − | + | |
| 346/A | C3 | 5.51 | 16.23 | 0.007525 | α-Helix | − | + | |
| 347/S | C3 | 2.88 | 32.97 | 0.000001 | α-Helix | − | + | |
| 387/S | V4 | 2.00 | 11.50 | 0.004150 | + | + | ||
| 429/K | C4 | 1.98 | 10.48 | 0.009522 | + | + | CD4 binding (33) | |
| 440/S | C4 | 9.46 | 30.91 | 0.000251 | Loop | + | + | CCR5 binding (46) |
| 442/Q | C4 | 1.91 | 10.15 | 0.009631 | + | + | ||
| 444/R | C4 | 2.35 | 11.64 | 0.009553 | β-Strand | − | + | |
| 446/S | C4 | 1.00 | 9.00 | 0.005346 | β-Strand | − | + |
Position/amino acid in strain HXB2.
Number of synonymous changes throughout the phylogeny/average number of synonymous sites.
Number of nonsynonymous changes throughout the phylogeny/average number of nonsynonymous sites.
P values in the exact test.
Information about the protein structure, such as notations of variable loops or secondary structures in the gp120 core, were described.
+, site reported as an epitope for antibodies or CTL, including sites in the HIV immunology database (31). Note that the locations of the epitopes in the database were approximate and that not all of the epitopes have been reported to have neutralizing activity.
FIG. 3.


Locations of PS sites and NS sites of HIV-1 gp120 in the three-dimensional structure. Molecular graphics were produced using Rasmol (50). The locations of PS sites (red) and NS sites (blue) are shown. Green, amino acid sites where no significant difference was detected. Gray, not tested (A) All amino acid sites of gp120 are shown. Circled single-letter amino acid codes represent positions where structures were not solved (33). (B through D) Locations of PS sites and NS sites in the gp120 core shown in four different views. Four views of the molecule are given. The PS sites are labeled. (B) Face for CD4 binding. (C) Face opposite that of CD4 binding. (D) Outer domain. (E) Inner domain.
Codon sites where synonymous substitutions were predominant.
We identified 63 codon sites where the rate of synonymous substitutions was significantly higher (NS sites) (Fig. 2 and 3). These NS sites were found in the CD4 binding sites, critical sites for CCR5 binding in and around the bridging sheet, interior positions in this protein, and cysteine residues that form disulfide bridges. Moreover, NS sites were found on the surface in the monomer but presumably in interior locations in the trimer complex. In particular, the distribution of NS sites in the α-helix region (positions 103, 106, and 109) in the inner domain was a striking contrast to the distribution of the PS sites of another α-helix region (positions 335 to 347) in the outer domain. The two NS sites in the α-helix region (positions 103 and 106) were exposed in a monomer unit; however, these residues are considered to form an interface with the trimer complex.
DISCUSSION
We have conducted the most extensive analysis yet of amino acid variation of HIV-1 gp120 at the single-site level from 186 sequences of subtype B by three different measures. Our analysis of amino acid variation revealed that the level of variability was very different from site to site in both conserved and variable regions that were assigned previously (40, 56). We evaluated the level of nonrandomness of the distribution of amino acid substitutions among sites of the protein. This heterogeneity in the substitution rate among sites indicates that the selection mechanism or the level of functional constraint is different among amino acid sites in a short region. The approach to evaluating amino acid variability in this study is different from that in previous work by Lauder et al. (34) in two ways. First, to evaluate amino acid variation at a given site, they took the neighboring amino acid sites into consideration. However, we evaluated amino acid variability regardless of the neighboring sites because we are interested in identifying variable and conserved sites rather than variable and conserved regions. This point is important when we examine the relationship between sequence variability at each position of the protein and the physical location of the position in the three-dimensional structure. Second, we took into account phylogenetic relationships among sequences to evaluate changeability of amino acids. Inferring phylogenetic relationships and estimating ancestral states at each node of the phylogeny allow us to estimate the actual number of amino acid changes.
Our analysis revealed that the level of amino acid variability could be very different among sites within a short region. One of the remarkable examples is the α-helix region (positions 335 to 347) just C-terminal of the V3 loop. In this region, exposed positions of the helix had considerable variations, while the variations in the interior positions were very limited. The arrangement of conserved sites and variable sites seems to be consistent with the fact that the α-helix has 3.6 residues per turn and that its side chains project out of the helix. Insertions or deletions in this region must be very limited because they may affect the arrangement of hydrophobic and hydrophilic amino acid residues that is suitable for the α-helix located on the surface of the protein. Another example of very different variabilities within a short region may be seen in the C4 region (positions 440 to 448), including the β-strand, where variable sites and conserved sites appear alternately (Fig. 1). This observation also seems to be consistent with the finding that the β-strand along the protein surface has residues with side chains protruding out of the protein and residues with side chains buried in an alternate arrangement. About the residue 440 Ser, it is not clear if its side chain projects out of the protein. This residue may interact with the V3 loop and be involved in cell tropisms (6). Although it is well known that variable amino acid sites are located on the surface positions of the protein (18), our results showed that amino acid variability was highly correlated with the solvent accessibility of the side chains of the residues. The arrangement of the variable sites in the sequence seemed to be consistent with the two typical secondary structures, α-helix and β-strand.
The degree of amino acid variabilities at the residues for binding of CD4 and CCR5 was very low except at a few positions. This is considered to be due to the functional constraint for binding. The residues for direct contact with CD4 are located within and near the recessed pocket for CD4 binding (33). The residues in the recessed cavity were highly conserved, and this may be critical for binding. We also observed several hypervariable sites and three PS sites (281 Ala, 283 Thr, and 429 Lys) among the CD4 binding sites (Table 2). This may seem inconsistent with the functional importance for binding, but it may be reasonable, because the existence of epitopes within and near CD4 binding sites has been reported (58). The side chains of residues 281 Ala and 283 Thr project out of the protein, and changes in the side chains at these sites may produce antigenic variations. Therefore, it is possible that the amino acid variations are dominantly affected by antibodies rather than by functional constraint for binding. Moreover, the effect of changing the side chains on binding may be different among these residues for CD4 binding because some of these residues interact with CD4 by their main chains and some of them do so by their side chains. The residue 429 Lys interacts with the CD4 molecule by its main chain, and its side chain is located on the protein surface, pointing to the different direction for CD4 binding. Therefore, changes in the side chain of residue 429 Lys may produce antigenic variations without serious effects on binding. The chemokine receptor binding sites are clustered at the vertex of the trimer predicted to be closest to the target cell. Lower variabilities were also observed in the critical positions for CCR5 binding with the exception of positions 317 Phe and 440 Ser. Among the positions critical for CCR5 binding listed in Table 2, five positions showed no amino acid variation, although the sequences analyzed include the sequences for strains that use CXCR4 rather than CCR5. This suggests that these five invariant sites may be involved in CXCR4 binding, too. Among the residues for CCR5 binding, position 317 Phe in the V3 loop and position 440 Ser in the C4 region were extremely variable, and position 440 Ser was detected as a PS site. This may seem inconsistent with the finding that the substitutions at these positions were shown to lead to a greater than 90% decrease in binding. However, this may be because the nucleotide sequences in this analysis contain the sequences of strains that use CXCR4, the major second receptor for the T-cell-tropic strain, rather than CCR5. The V3 loop is well known as the determinant of cell tropisms that is associated with usage of the second receptor (7). About the residue 440 Ser, there is a report suggesting that this position may also be involved in cell tropism (6). Therefore, the high variabilities at positions 317 Phe and 440 Ser indicate that the amino acids at these positions may be critical for usage of the second receptors.
The high heterogeneity of the nonsynonymous substitution rate among codon positions suggests that the selection mechanisms are very different among sites. Our results also showed that the distribution of synonymous substitutions was not uniform. One of the possible explanations for this heterogeneity in the synonymous substitution rate is that mutation rates among sites might not be uniform. For example, hypermutation (G to A) is known to occur episodically in retroviral replication (62), and the sensitivity of this hypermutation might be different among codon sites. Another explanation may be that synonymous substitutions are not completely neutral. The dinucleotide contents and the usage of synonymous codons of HIV were much biased, implying that some synonymous substitutions may be slightly deleterious (2, 45). The third explanation is that nonsynonymous substitutions affected the estimation of the number of synonymous substitutions at the same codon sites. It is known that there is a positive correlation between the numbers of synonymous and nonsynonymous substitutions (26). In fact, we observed significant positive correlation between the numbers of synonymous and nonsynonymous substitutions (r = 0.496, P < 0.01). If there is variation in the mutation rate among codon sites, the positive correlation between the numbers of synonymous and nonsynonymous substitutions may be explained (26). Another possibility of this positive correlation is that it is an artifact of the estimation of synonymous and nonsynonymous substitutions, which seems to be hard to overcome if it exists. When two codons were different at more than one nucleotide site, we gave an equal probability to all possible multiple paths because we do not know which paths occur more frequently than other paths. Thus, if two codons are different in more than one site, the differences in synonymous and nonsynonymous substitutions are correlated.
The ratio of nonsynonymous substitutions to synonymous substitutions throughout HIV-1 gp120 indicates that the rate of synonymous substitutions is higher than that of nonsynonymous substitutions, but synonymous substitutions are not very predominant. The ratio of 0.90 found in this study was higher than that in the previous report, which showed this ratio to be 0.68 (35) (in the original paper, the KS/KA ratio was presented to be 1.47), although we did not analyze the four variable regions where insertions, deletions, and partial duplications might be very frequent. We think that the KS/KA ratio in this study is more realistic than that given in the previous reports and that the relative number of nonsynonymous substitutions might be underestimated in previous reports for two reasons. First, the numbers of nonsynonymous substitutions by pairwise comparison might be underestimated in the previous report because multiple substitutions in hypervariable sites might be underestimated. In this study, we showed that the nonsynonymous substitutions were concentrated at hypervariable sites and that the distribution of nonsynonymous substitutions was different from that of synonymous substitutions. When the nonsynonymous substitutions were concentrated on particular hypervariable sites, pairwise comparisons underestimated the actual number of multiple substitutions. In such a case, an estimation of the numbers of synonymous and nonsynonymous substitutions that takes phylogenetic relationships into consideration may be better than pairwise comparisons (8). The second reason is that the number of synonymous substitutions might be overestimated in previous reports because the number of synonymous sites might be underestimated by a method that did not consider the higher frequency of transitions than of transversions. We considered the relative frequencies of transition and transversion in estimating the numbers of synonymous and nonsynonymous sites because estimation of the number of synonymous and nonsynonymous sites is much improved by considering the transition/transversion ratio (25).
Our analysis detected 33 amino acid positions that may be under positive selection (PS sites). To see whether these PS sites had been reported to be responsible for antigenic or phenotypic variations, we looked into the literature and the HIV immunology database (31) (Table 4). For the five PS sites in the V3 loop (303 Thr, 306 Arg, 308 Arg, 319 Thr, and 322 Lys), there were many reports of the epitopes for antibodies or cytotoxic T lymphocytes (CTL) (31). Among the five PS sites in the V3 loop, two of them (positions 308 Arg and 319 Thr) had been detected as putative positions under positive selection in a previous study (57). Position 306 Arg was reported to be responsible for cell tropism (10, 17). Position 308 Arg was reported to be responsible for antigenicity (63). Position 322 Lys was reported to be responsible for antigenicity and cell tropism (53). The amino acid variations in the V3 loop were responsible for cell tropism that is associated with second-receptor usage (7). The V3 loop is one of the major epitopes of HIV and includes the epitopes for CTL. The immunological evidence and the effect on cell tropism explain the adaptive significance of amino acid changes in the V3 loop that is exposed to the solvent. The PS sites in the V1/V2 loop (130 Lys, 132 Thr, 172 Glu, and 178 Lys) and the PS sites in the stem part of the loop (195 Ser and 200 Val) may be antigenic because of their surface locations. One of the remarkable findings in this study was that many PS sites were found outside the variable loops. In particular, 15 PS sites were clustered in the outer domain of the gp120 core, opposite the CD4 binding sites. In the α-helix region (positions 335 to 347), seven PS sites (335 Arg, 336 Ala, 337 Lys, 340 Asn, 343 Lys, 346 Ala, and 347 Ser) were exposed, although this region contains three highly conserved sites (338 Trp, 341 Thr, and 342 Leu) in the interior locations. However, it seems that there is no clear evidence of antigenic or phenotypic variation for this α-helix region (positions 335 to 347), although this region is one of the most variable regions in gp120. Furthermore, additional PS sites (positions 291 Ser, 293 Glu, 333 Ile, 440 Ser, 442 Gln, 444 Arg, and 446 Ser) were also clustered on the same face of the protein. This observation indicates that there are discontinuous epitopes in this area. Four PS sites (281 Ala, 283 Thr, 387 Ser, and 429 Lys) on the face for CD4 binding may be antigenic sites. Only one PS site (240 Thr) in the inner domain might be in an exterior location in the trimer complex and thus be an antigenic site. In the N-terminal part of the C1 region, three PS sites (31 Thr, 85 Val, and 87 Val) were found. Though the three-dimensional structure for this region was not solved, these sites were reported elsewhere to be included in the epitopes for antibodies (31).
The 33 PS sites include positions without clear evidence of antigenic or phenotypic variations. The surface area of the α-helix region (positions 335 to 347) might be included in the silent face (67) as a minimally immunogenic area because this area was considered to be heavily glycosylated. In fact, there are several potential glycosylation sites in this area. However, it is not clear whether the silent face is always fully covered by sugar chains and whether there is a space for binding of antibodies. The rate of amino acid substitution in this region may be very fast in intrahost evolution. In fact, putative sites under positive selection were detected by analyzing the nucleotide sequences of HIV samples obtained periodically from a single patient (35). Attempts to examine the antigenicity of these PS sites without clear biological evidence will be encouraged. The neutralizing face (67) includes the CD4 binding sites, CD4-induced epitopes (59), and the binding sites for 2G12 (61), a monoclonal antibody, and interactions between monoclonal antibodies and this neutralizing face have been well characterized. Our results suggest that there are antigenic sites in the silent face and that the real neutralizing face is larger than the previous investigators thought.
The PS sites detected in this study were located on surface positions in this protein, and in most cases, the side chains of the PS sites projected out of the protein. This observation indicates that variations in the side chains of the PS sites dominantly contribute to antigenic variations that enable the virus to escape from recognition by the immune system. Some PS sites may be responsible for other phenotypic variations, such as cell tropism and replication rate. We also identified 63 NS sites where synonymous substitutions were predominant. NS sites were found in the probable contact face of the trimer complex, receptor-binding sites, cysteine residues for disulfide bridges, and interior positions of the protein. The distribution of PS sites and NS sites provides a much better understanding of the evolutionary force of proteins. First, negative selection is operating to maintain the proper folding and structure of the protein. Second, to maintain the ability to bind the receptors, negative selection is operating on amino acid sites for receptor bindings. Third, on the protein surface, positive selection is operating against amino acid changes that are responsible for antigenic or phenotypic variation. Although the conserved amino acid sites for receptor binding are located on the core surface of the protein, there is evidence that the variable loops might cover the conserved faces for receptor binding (65). This relationship between the variable loops and the conserved face for receptor binding is considered very important for the virus to survive. Exposing the conserved face for binding is not a good strategy for the virus, because the immune system may easily recognize the conserved face and neutralize the virus. In virus entry, sequential conformational changes of the envelope glycoprotein may uncover the conserved face for receptor binding only when the receptor binding sites are used. The V1/V2 and V3 variable loops are considered to have a role in protecting the receptor binding sites from antibodies. Furthermore, the sequential conformational changes in the envelope glycoprotein in viral entry may also contribute to antigenic variation.
This study showed that the PS sites in gp120 were dispersed over the protein surface, although the distribution on the surface may not be uniform. Many examples of positive selection at the molecular level showed that positive selection might be operating in only the part of the protein that interacts with other molecules (14, 23, 24). However, in the previous reports, comparisons of synonymous and nonsynonymous substitutions were performed in local regions including many sites. Actually, Seibert et al.'s analysis (51) of HIV gp120 found the V2 and V3 regions where positive selection may be operating, while they concluded that negative selection might be operating largely in the other regions of gp120. Our analysis at the single-site level revealed the distribution of the PS sites in the amino acid sequence and their locations in the three-dimensional structure. Analysis at the single-site level enables us to see the distributions of PS sites and NS sites that have not revealed by the analyses including many sites.
There may be more amino acid sites where positive selection is operating that we could not identify in this study for several reasons. First, if the number of sequences for analysis increases and the observed substitution numbers increase, we may be able to identify additional PS sites for which statistical significance was not detected in this study. Second, though we did not analyze the region where frequencies of insertion, deletion, and partial duplications might be high, changes in the length of the variable loops may be responsible for antigenic variations or cell tropism (54). Third, the comparison of synonymous and nonsynonymous substitutions may not be able to detect all positive selection (8). If only a few amino acids are acceptable at a given site, it may be hard to detect positive selection by comparison of synonymous and nonsynonymous substitutions because many nonsynonymous mutations will be deleterious. Furthermore, we believe that the approach used for detecting selection in this study is aimed at detecting selection which is consistent between hosts and that this approach might overlook selections operating in a host-specific manner.
The approach to detecting selection used in this study is based on the same concept as the method developed by Suzuki and Gojobori (57), in which no assumption was made about the ratio of synonymous to nonsynonymous changes at a given site. There are two significant differences between the methods used in this study and in the previous study. First, we took the transition/transversion ratio into consideration in estimating the numbers of synonymous and nonsynonymous sites to obtain more reliable results. Second, the significance level was set at 1% in this study, while that in Suzuki and Gojobori's study (57) was 5%. We set a higher significance level to avoid the problem of multiple tests, because we tested more than 400 amino acid positions in this study. This approach is applicable for data sets of many sequences, for example, the Env and Nef proteins of HIV, HA1 of influenza virus A, and HLA of humans. The level of divergence is also important for the applicability of this approach. Sequences must be divergent enough for many substitutions to be observed and give for significant results. However, if the sequences are too divergent, the saturation effect of synonymous substitutions may affect the results. One may think that another problem in this approach may arise because the phylogenetic tree topology of many sequences cannot be accurately estimated. When we estimate a phylogenetic tree of many sequences that are closely related, we usually observe many short internal branches where the clusterings are not clear. However, Suzuki and Gojobori (57) conducted computer simulations to compare the efficiencies for detecting selection for the two cases of when we know the phylogeny and when we do not know it. They then found that there was little difference in efficiencies for detecting selection. This suggests that the lack of detailed and accurate information about the tree topology does not affect the results seriously. This robustness of the results may be due to the fact that the relative numbers of substitutions on these short internal branches are usually much smaller than the total number of substitutions throughout the phylogeny.
Evaluation of amino acid variations will also provide useful statistics of an empirical pattern of variations that will enable us to estimate the range of possible variations of proteins in the future. Even if the number of sequences increases to a thousand, the number of amino acids that are acceptable at a given site of the protein would converge to a certain level. In the estimation of number of amino acid changes, the types of amino acid changes are also identified. The empirical pattern of amino acid substitutions will be useful for predicting possible changes. Hundreds of sequences and information about the three-dimensional structure enable us to predict the range of acceptable amino acid variations for each position and predict amino acids that will be deleterious for the virus's survival. If the range of functional constraints of amino acids at each position is properly estimated, we will be able to interpret various ratios of synonymous to nonsynonymous substitutions much better (12) than the studies up to the present have been able to do.
Our analyses of sequence variation at the single-site level revealed that selection pressure against amino acid changes can be very different among positions within a short region, as observed in the α-helix region just C-terminal of the V3 loop. Furthermore, we showed that some PS sites which were far apart in the sequence were close together in the three-dimensional structure, suggesting the existence of unidentified discontinuous epitopes. Examination of the relationships between sequence variation and physical locations in protein structure would be useful not only to elucidate the driving force of protein evolution, but also to predict the biological function of each position.
ACKNOWLEDGMENTS
We thank Takashi Miyata, Kei-ichi Kuma, Tomoyuki Miura, Jun Takehisa, and Masanori Hayami at Kyoto University; Naoko Takezaki and Yoshiyuki Suzuki at the National Institute of Genetics; and Tatsuo Shioda at the University of Tokyo for helpful suggestions and comments on this work. We also thank Silvana Gaudieri at the National Institute of Genetics, Toshinori Endo at Riken, Eiji Ido at Kyoto University, Ziheng Yang at University College London, Keith A. Crandall at Brigham Young University, and Kei Yura at Nagoya University for helpful suggestions on the manuscript. In particular, Y.Y.-K. expresses sincere gratitude to Takashi Miyata and Masanori Hayami for hosting her in their laboratories.
Y.Y.-K. was supported by a Research Fellowship for Young Scientists provided by the Japan Society for the Promotion of Science.
REFERENCES
- 1.Adachi J, Hasegawa M. MOLPHY version 2.3: programs for molecular phylogenetics based on maximum likelihood. Computer Science Monographs vol. 28. Tokyo: Institute of Statistical Mathematics; 1996. pp. 1–150. [Google Scholar]
- 2.Akashi H. Inferring weak selection from patterns of polymorphism and divergence at “silent” sites in Drosophila DNA. Genetics. 1995;139:1067–1076. doi: 10.1093/genetics/139.2.1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bagnarelli P, Mazzola F, Menzo S, Montroni M, Butini L, Clementi M. Host-specific modulation of the selective constraints driving human immunodeficiency virus type 1 env gene evolution. J Virol. 1999;73:3764–3777. doi: 10.1128/jvi.73.5.3764-3777.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonhoeffer S, Holmes E C, Nowak M A. Cause of HIV diversity. Nature. 1995;376:125. doi: 10.1038/376125a0. [DOI] [PubMed] [Google Scholar]
- 5.Bush R M, Fitch W M, Bender C A, Cox N J. Positive selection on the H3 hemagglutinin gene of human influenza virus A. Mol Biol Evol. 1999;16:1457–1465. doi: 10.1093/oxfordjournals.molbev.a026057. [DOI] [PubMed] [Google Scholar]
- 6.Carrillo A, Ratner L. Human immunodeficiency virus type 1 tropism for T-lymphoid cell lines: role of the V3 loop and C4 envelope determinants. J Virol. 1996;70:1301–1309. doi: 10.1128/jvi.70.2.1301-1309.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cocchi F, DeVico A L, Garzino-Demo A, Cara A, Gallo R C, Lusso P. The V3 domain of the HIV-1 gp120 envelope glycoprotein is critical for chemokine-mediated blockade of infection. Nat Med. 1996;2:1244–1247. doi: 10.1038/nm1196-1244. [DOI] [PubMed] [Google Scholar]
- 8.Crandall K A, Kelsely C R, Imamichi H, Lane H C, Salzman N P. Parallel evolution of drug resistance in HIV: failure of nonsynonymous/synonymous substitution rates to detect selection. Mol Biol Evol. 1999;16:372–382. doi: 10.1093/oxfordjournals.molbev.a026118. [DOI] [PubMed] [Google Scholar]
- 9.Dalgleish A G, Beverley P C, Clapham P R, Crawford D H, Greaves M F, Weiss R A. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature. 1984;312:763–767. doi: 10.1038/312763a0. [DOI] [PubMed] [Google Scholar]
- 10.De Jong J-J, De Ronde A, Keulen W, Tersmette M, Goudsmit J. Minimal requirement for the human immunodeficiency virus type 1 V3 domain to support the syncytium-inducing phenotype: analysis by single amino acid substitution. J Virol. 1992;66:6777–6780. doi: 10.1128/jvi.66.11.6777-6780.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton R E, Hill C M, Davis C B, Peiper S C, Schall T J, Littman D R, Landau N R. Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996;381:661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- 12.Domingo E, Holland J J. Mutation rates and rapid evolution of RNA virus. In: Morse S S, editor. The evolutionary biology of viruses. New York, N.Y: Raven Press, Ltd.; 1994. pp. 161–184. [Google Scholar]
- 13.Dragic T, Litwin V, Allaway G P, Martin S R, Huang Y, Nagashima K A, Cayanan C, Maddon P J, Koup R A, Moore J P, Paxton W A. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature. 1996;381:667–673. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 14.Endo T, Ikeo K, Gojobori T. Large-scale search for genes on which positive selection may operate. Mol Biol Evol. 1996;13:685–690. doi: 10.1093/oxfordjournals.molbev.a025629. [DOI] [PubMed] [Google Scholar]
- 15.Feng Y, Broder C C, Kennedy P E, Berger E A. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272:872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- 16.Fitch W M, Bush R M, Bender C A, Cox N J. Long term trends in the evolution of H(3) HA1 human influenza type A. Proc Natl Acad Sci USA. 1997;94:7712–7718. doi: 10.1073/pnas.94.15.7712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fouchier R A M, Groenink M, Kootstra N A, Tersmette M, Huisman H G, Miedema F, Schutemaker H. Phenotype-associated sequence variation in the third variable domain of the human immunodeficiency virus type 1 gp120 molecule. J Virol. 1992;66:3183–3187. doi: 10.1128/jvi.66.5.3183-3187.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Go M, Miyazawa S. Relationship between mutability, polarity and exteriority of amino acid residues in protein evolution. Int J Peptide Protein Res. 1980;15:211–224. doi: 10.1111/j.1399-3011.1980.tb02570.x. [DOI] [PubMed] [Google Scholar]
- 19.Gojobori T, Moriyama E N, Kimura M. Molecular clock of viral evolution, and the neutral theory. Proc Natl Acad Sci USA. 1990;87:10015–10018. doi: 10.1073/pnas.87.24.10015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gojobori T, Yamaguchi Y, Ikeo K, Mizokami M. Evolution of pathogenic viruses with special reference to the rates of synonymous and nonsynonymous substitutions. Jpn J Genet. 1994;69:481–488. doi: 10.1266/jjg.69.481. [DOI] [PubMed] [Google Scholar]
- 21.Haydon D, Lea S, Fry L, Knowles N, Samual A R, Stuart D, Woolhouse M E J. Characterizing sequence variation in the VP1 capsid proteins of foot and mouth disease virus (serotype 0) with respect to virion structure. J Mol Evol. 1998;46:465–475. doi: 10.1007/pl00006327. [DOI] [PubMed] [Google Scholar]
- 22.Holmes E C, Zhang L Q, Simmonds P, Ludlam C A, Leigh Brown A J. Convergent and divergent sequence evolution in the surface envelope glycoprotein of human immunodeficiency virus type 1 within a single infected patient. Proc Natl Acad Sci USA. 1992;89:4835–4839. doi: 10.1073/pnas.89.11.4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hughes A L, Nei M. Pattern of nucleotide substitution at major histocompatibility complex class I loci reveals overdominant selection. Nature. 1988;335:167–170. doi: 10.1038/335167a0. [DOI] [PubMed] [Google Scholar]
- 24.Ina Y, Gojobori T. Statistical analysis of nucleotide sequences of the hemagglutinin genes of human influenza A viruses. Proc Natl Acad Sci USA. 1994;91:8388–8392. doi: 10.1073/pnas.91.18.8388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ina Y. New methods for estimating the numbers of synonymous and nonsynonymous substitutions. J Mol Evol. 1995;40:190–226. doi: 10.1007/BF00167113. [DOI] [PubMed] [Google Scholar]
- 26.Ina Y. Correlation between synonymous and nonsynonymous substitutions and variation in synonymous substitutions numbers. In: Nei M, Takahata N, editors. Current topics on molecular evolution. University Park, Pa: Institute of Molecular Evolutionary Genetics, The Pennsylvania State University; 1996. pp. 105–113. [Google Scholar]
- 27.Kimura M. Evolutionary rate at the molecular level. Nature. 1968;217:624–626. doi: 10.1038/217624a0. [DOI] [PubMed] [Google Scholar]
- 28.Kimura M. The neutral theory of molecular evolution. Cambridge, England: Cambridge University Press; 1983. [Google Scholar]
- 29.King J L, Jukes T H. Non-Darwinian evolution. Science. 1969;164:788–798. doi: 10.1126/science.164.3881.788. [DOI] [PubMed] [Google Scholar]
- 30.Korber B, Hahn B, Foley B, Mellors J W, Leitner T, Myers G, McCutchan F, Kuiken C, editors. Human retroviruses and AIDS 1997: a compilation and analysis of nucleic acid and amino acid sequences. Los Alamos, N.M: Theoretical Biology and Biophysics Group, Los Alamos National Laboratory; 1998. [Google Scholar]
- 31.Korber B, Brander C, Haynes B, Koup R, Moore J, Walker B, editors. HIV molecular immunology database 1998. Los Alamos, N.M: Theoretical Biology and Biophysics Group, Los Alamos National Laboratory; 1999. [Google Scholar]
- 32.Koshi J M, Goldstein R A. Models of natural mutation including site heterogeneity. Proteins. 1998;32:289–295. [PubMed] [Google Scholar]
- 33.Kwong P D, Wyatt R, Robinson J, Sweet R W, Sodroski J, Hendrickson W A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393:648–659. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lauder I J, Lin H J, Siwak E B, Hollinger F B. Kernel density analysis of variable and conserved regions of the envelope proteins of human immunodeficiency virus type 1 and associated epitopes. AIDS Res Hum Retroviruses. 1996;12:91–97. doi: 10.1089/aid.1996.12.91. [DOI] [PubMed] [Google Scholar]
- 35.Leigh Brown A, Monaghan P. Evolution of the structural proteins of human immunodeficiency virus: selective constraints on nucleotide substitution. AIDS Res Hum Retroviruses. 1988;4:399–407. doi: 10.1089/aid.1988.4.399. [DOI] [PubMed] [Google Scholar]
- 36.Leonard C K, Spellman M W, Riddle L, Harris R J, Thomas J N, Gregory T J. Assignment of intrachain disulfide bonds and characterization of potential glycosylation sites of the type 1 recombinant human immunodeficiency virus envelope glycoprotein (gp120) expressed in Chinese hamster ovary cells. J Biol Chem. 1990;265:10373–10382. [PubMed] [Google Scholar]
- 37.Li W-H, Wu C-I, Luo C-C. A new method for estimating synonymous and nonsynonymous rates of nucleotide substitution considering the relative likelihood of nucleotide and codon changes. Mol Biol Evol. 1985;2:150–174. doi: 10.1093/oxfordjournals.molbev.a040343. [DOI] [PubMed] [Google Scholar]
- 38.Li W-H, Tanimura M, Sharp P M. Rates and dates of divergence between AIDS virus nucleotide sequences. Mol Biol Evol. 1988;5:313–330. doi: 10.1093/oxfordjournals.molbev.a040503. [DOI] [PubMed] [Google Scholar]
- 39.Miyata T, Yasunaga T. Molecular evolution of mRNA: a method for estimating evolutionary rates of synonymous and amino acid substitutions from homologous nucleotide sequences and its application. J Mol Evol. 1980;16:23–36. doi: 10.1007/BF01732067. [DOI] [PubMed] [Google Scholar]
- 40.Modrow S, Hahn B H, Shaw G M, Gallo R C, Wong-Staal F, Wolf H. Computer-assisted analysis of envelope protein sequences of seven human immunodeficiency virus isolates: prediction of antigenic epitopes in conserved and variable regions. J Virol. 1987;61:570–578. doi: 10.1128/jvi.61.2.570-578.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moriyama E N, Ina Y, Ikeo K, Shimizu N, Gojobori T. Mutation pattern of human immunodeficiency virus genes. J Mol Evol. 1991;32:360–363. doi: 10.1007/BF02101275. [DOI] [PubMed] [Google Scholar]
- 42.Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous substitutions. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- 43.Nei M. Molecular evolutionary genetics. New York, N.Y: Columbia University Press; 1987. [Google Scholar]
- 44.Nielsen R, Yang Z. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics. 1998;148:929–936. doi: 10.1093/genetics/148.3.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pedersen A K, Wiuf C, Christiansen F B. A codon-based model designed to describe lentiviral evolution. Mol Biol Evol. 1998;15:1069–1081. doi: 10.1093/oxfordjournals.molbev.a026006. [DOI] [PubMed] [Google Scholar]
- 46.Rizzuto C D, Wyatt R, Hernandez-Ramos N, Sun Y, Kwong P D, Hendrickson W A, Sodroski J. A conserved HIV gp120 glycoprotein structure involved in chemokine receptor binding. Science. 1998;280:1949–1953. doi: 10.1126/science.280.5371.1949. [DOI] [PubMed] [Google Scholar]
- 47.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 48.Sasaki A. Evolution of antigen drift/switching: continuously evading pathogens. J Theor Biol. 1994;168:291–308. doi: 10.1006/jtbi.1994.1110. [DOI] [PubMed] [Google Scholar]
- 49.Sato H, Shiino T, Kodama N, Taniguchi K, Tomita Y, Kato K, Miyakuni T, Takebe Y. Evolution and biological characterization of human immunodeficiency virus type 1 subtype E gp120 V3 sequences following horizontal and vertical virus transmission in a single family. J Virol. 1999;73:3551–3559. doi: 10.1128/jvi.73.5.3551-3559.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sayle R, Milner-White E J. Rasmol: biomolecular graphics for all. Trends Biochem Sci. 1995;20:374. doi: 10.1016/s0968-0004(00)89080-5. [DOI] [PubMed] [Google Scholar]
- 51.Seibert S A, Howell C Y, Hughes M K, Hughes A L. Natural selection on the gag, pol, and env genes of human immunodeficiency virus 1 (HIV-1) Mol Biol Evol. 1995;12:803–813. doi: 10.1093/oxfordjournals.molbev.a040257. [DOI] [PubMed] [Google Scholar]
- 52.Shiino T, Kato K, Kodaka N, Miyakuni T, Takebe Y, Sato H. A group of V3 sequences of human immunodeficiency virus type 1 subtype E non-syncytium-inducing, CCR5-using variants are resistant to positive selection pressure. J Virol. 2000;74:1069–1078. doi: 10.1128/jvi.74.3.1069-1078.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shioda T, Oka S, Ida S, Nokihara K, Toriyoshi H, Mori S, Takebe Y, Kimura S, Shimada K, Nagai Y. A naturally occurring single basic amino acid substitution in the V3 region of the human immunodeficiency virus type 1 Env protein alters the cellular host range and antigenic structure of the virus. J Virol. 1994;68:7689–7696. doi: 10.1128/jvi.68.12.7689-7696.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shioda T, Oka S, Xin X, Liu H, Harukuni R, Kurotani A, Fukushima M, Hasan M K, Shiino T, Takebe Y, Iwamoto A, Nagai Y. In vivo sequence variability of human immunodeficiency virus type 1 envelope gp120: association of V2 extension with slow disease progression. J Virol. 1997;71:4871–4881. doi: 10.1128/jvi.71.7.4871-4881.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Simmonds P, Balfe P, Ludlam C A, Bishop J O, Leigh Brown A J. Analysis of sequence diversity in hypervariable regions of the external glycoprotein of human immunodeficiency virus type 1. J Virol. 1990;64:5840–5850. doi: 10.1128/jvi.64.12.5840-5850.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Starcich B R, Hahn B H, Shaw G M, McNeely P D, Modrow S, Wolf H, Parks E S, Parks W P, Josephs S F, Gallo R C, Wong-Staal F. Identification and characterization of conserved and variable regions in the envelope gene of HTLV-III/LAV, the retrovirus of AIDS. Cell. 1986;45:637–648. doi: 10.1016/0092-8674(86)90778-6. [DOI] [PubMed] [Google Scholar]
- 57.Suzuki Y, Gojobori T. A method for detecting positive selection at single amino acid sites. Mol Biol Evol. 1999;16:1315–1328. doi: 10.1093/oxfordjournals.molbev.a026042. [DOI] [PubMed] [Google Scholar]
- 58.Thali M, Furman C, Ho D D, Robinson J, Tilley S, Pinter A, Sodroski J. Discontinuous, conserved neutralization epitopes overlapping the CD4-binding region of human immunodeficiency virus type 1 gp120 envelope glycoprotein. J Virol. 1992;66:5635–5641. doi: 10.1128/jvi.66.9.5635-5641.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thali M, Moore J P, Furman C, Charles M, Ho D D, Robinson J, Sodroski J. Characterization of conserved human immunodeficiency virus type 1 gp120 neutralization epitopes exposed upon gp120-CD4 binding. J Virol. 1993;67:3978–3988. doi: 10.1128/jvi.67.7.3978-3988.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thompson J D, Higgins D G, Gibson T J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Trkola A, Purtscher M, Muster T, Ballaun C, Buchacher A, Sullivan N, Sriivasan K, Sodroski J, Moore J P, Katinger H. Human monoclonal antibody 2G12 defines a distinctive neutralization epitope on the gp120 glycoprotein of human immunodeficiency virus type 1. J Virol. 1996;70:1100–1108. doi: 10.1128/jvi.70.2.1100-1108.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vartanian J P, Meyerhans A, Asjo B, Wain-Hobson S. Selection, recombination, and G→A hypermutation of human immunodeficiency virus type 1 genomes. J Virol. 1991;65:1779–1788. doi: 10.1128/jvi.65.4.1779-1788.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wolfs T F W, Zwart G, Bakker M, Valk M, Kuiken C L, Goudsmit J. Naturally occurring mutations within HIV-1 V3 genomic RNA lead to antigenic variation dependent on a single amino acid substitution. Virology. 1991;185:195–205. doi: 10.1016/0042-6822(91)90767-6. [DOI] [PubMed] [Google Scholar]
- 64.Wu T T, Kabat E A. An analysis of the sequences of the variable regions of Bence Jones proteins and myeloma light chains and their implications for antibody complementarity. J Exp Med. 1970;132:211–250. doi: 10.1084/jem.132.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wyatt R, Sullivan N, Thali M, Repke H, Ho D, Robinson J, Posner M, Sodroski J. Functional and immunologic characterization of human immunodeficiency virus type 1 envelope glycoproteins containing deletions of the major variable regions. J Virol. 1993;67:4557–4565. doi: 10.1128/jvi.67.8.4557-4565.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wyatt R, Sodroski J. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science. 1998;280:1884–1888. doi: 10.1126/science.280.5371.1884. [DOI] [PubMed] [Google Scholar]
- 67.Wyatt R, Kwong P D, Desjardins E, Sweet R W, Robinson J, Hendrickson W A, Sodroski J G. The antigenic structure of the HIV gp120 envelope glycoprotein. Nature. 1998;393:705–711. doi: 10.1038/31514. [DOI] [PubMed] [Google Scholar]
- 68.Yamaguchi Y, Gojobori T. Evolutionary mechanisms and population dynamics of the third variable envelope region of HIV within single hosts. Proc Natl Acad Sci USA. 1997;94:1264–1269. doi: 10.1073/pnas.94.4.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang Z. Among-site rate variation and its impact on phylogenetic analyses. Tree. 1996;11:367–372. doi: 10.1016/0169-5347(96)10041-0. [DOI] [PubMed] [Google Scholar]