

Abstract
Polygenic risk scores (PRS) estimate genetic susceptibility of an individual to disease and are increasingly proving clinical utility. However, their performance, computation, and reporting in diverse populations remain challenging. Here, we present a pragmatic approach to optimize a PRS for a population of interest that leverages publicly available data and methods and consists of seven steps that are easily implemented without requirement of expertise in complex genetics.
Step 1: Selecting source GWAS and imputation
Step 2: Selecting methods to compute polygenic score
Step 3: Adjusting scores using principal components of ancestry
Step 4: Selecting the best performing score
Step 5: Defining percentiles of a population distribution
Step 6: Validating performance of the optimized polygenic score
Step 7: Implementing the optimized polygenic score in clinical practice
Keywords: Polygenic risk scores, genomic risk prediction, genome wide association studies, ancestry, diversity
INTRODUCTION:
Polygenic risk scores (PRS) – which estimate the genetic susceptibility of an individual to a disease or trait by combining the effects of common variation across the genome – are now commonly used in translational research and starting to penetrate clinical practice.(Patel and Khera, 2022; Klarin and Natarajan, 2022; Kumuthini et al., 2022; Adeyemo et al., 2021; O’Sullivan et al., 2022) However, a major barrier to the widespread adoption of PRS is their reduced performance when computed in populations that were not represented in the discovery genome-wide association studies (GWAS).(Martin et al., 2019; Adeyemo et al., 2021; Novembre et al., 2022) Because more than 80% of the participants of GWAS in the world are of European ancestry, commonly available polygenic risk scores have the strongest performance in individuals of European ancestry and reduced performance in ancestries outside of Europe.(Martin et al., 2019; Fahed Akl C. et al.; Dikilitas et al., 2020) This problem is now considered a top priority area in genomics, with ongoing efforts to increase representation in genomic studies and improve methods of computing polygenic scores.(Adeyemo et al., 2021; Wand et al., 2021; Patel et al., 2023; Ruan et al., 2022) Those efforts are already showing promise for some traits/scores such as increased representation of East Asians,(Ishigaki et al., 2020; Chen et al., 2011) and improved multi-ancestry scores for lipids and coronary artery disease.(Patel et al., 2023) Beyond continental ancestries however, subcontinental population groups, admixed populations, and populations with distinct gene-environment interactions continue to struggle to use an “out-of-the-box” PRS for a trait of interest in that population. A more pragmatic approach that leverages the use of existing datasets to optimize a PRS for a specific population is necessary to help advance equitable implementation of PRS around the world.
There are at least four reasons why PRS developed in one population have reduced performance in another population.(Martin et al., 2019; Salehi Nowbandegani et al., 2023) First, the effect size of a genetic variant in one population might be different from another population. Effect size estimates for genetic variants are obtained from GWAS studies, and those are used to derive PRS. GWAS are often very large and not representative of global populations with strong Eurocentric bias, and as such, the effect size estimated from those might not represent the true effect size in the target population. Second, even for known variants associated with a trait, allele frequencies might differ among populations. In this case, a variant more strongly associated with the trait in the discovery GWAS, might be present but at much lower frequency in a target population. Third, the correlation structure of the genome known as linkage disequilibrium (LD) also varies across populations, such that there are different tagging vs. causal variants. These LD differences result in differences in effect size estimates from GWAS to target population. Fourth, gene-environment interactions are different across populations and also affect the predictive accuracy for PRS. Those include evolutionary gene-environment correlations, differences in prevalence or distribution of a trait in population, or simply comparisons across environmentally stratified cohorts such as a national biobank where there is healthy selection bias vs. a hospital biobank where there is enrichment for disease. Any approach to optimize a PRS for a population of interest should take into consideration those limitations and consider methods and datasets that systematically enable a correction of those factors that typically reduce performance.
MULTI STEP APPROACH TO OPTIMIZE PRS FOR POPULATION OF INTEREST
Here, we present a pragmatic approach to optimize a PRS for a population of interest that leverages publicly available data and methods and consists of seven steps that are easily implemented without requirement of expertise in complex genetics (Figure 1). First, one or more source GWAS for the trait of interest are selected. Second, multiple methods for computing PRS are explored and many candidate PRS are computed. Third, the computed raw scores are adjusted using principal components of ancestry to generate ancestry-specific distributions of PRS. Fourth, in a training dataset, a best-performing score is selected from a list of multiple candidate scores and reported as the final optimized PRS in a validation dataset. Fifth, a population distribution is selected to define percentiles of the PRS and identify risk estimates associated with different population cut-offs (e.g., top 20% of the population with three-fold increased risk). Sixth, external validation of PRS performance provides additional confirmation that the optimized PRS performance is robust for the population of interest and could be moved into clinical implementation. Seventh, efforts to establish analytic validity of testing as well as reporting standards should be established and might be context-specific for a population.
Figure 1. Pragmatic Multi-Step Approach to Optimize Polygenic Risk Scores to Populations of Interest.

A pragmatic approach to optimize a polygenic risk score for a population of interest leverages publicly available data and methods is described in seven steps that are easily implemented without requirement of expertise in complex genetics. GWAS: Genome Wide Association Studies, PRS: Polygenic Risk Score
While several tutorials and methods have been published to inform PRS development and optimization, they often require strict dataset requirements which are rarely available for most populations.(Choi et al., 2020; Patel and Khera, 2022; Hao et al., 2022; Page et al., 2022) In this pragmatic multi step approach, we will present a framework that is flexible in accommodating variable data availability across populations. This is important because different populations might have drastically different datasets available at their disposal. At every step, we will highlight (i) the best-case scenario that could be performed in the presence of comprehensive data, (ii) what could be done with limited data, and (iii) what cannot be done due to risk of introducing bias and inaccuracy. Readers can then decide where they fall on the spectrum for each step based on unique aspects of their population and datasets available at their disposal. For example, in the case of an indigenous Arabs population, where datasets are limited but environmental risk factors are unique, it might be necessary to borrow effect estimates from global GWAS studies.(Saad et al.) In South Asian or East Asian populations where there are emerging larger datasets, using population-specific GWAS to update effect size estimates might be feasible.(Weissbrod et al., 2022; Koyama et al., 2020)
STEP 1: SELECTING SOURCE GWAS AND IMPUTATION
Genome-wide association studies (GWAS) form the foundation of input data for constructing polygenic scores. In a GWAS, a logistic or linear regression is performed between a genotype with an outcome or continuous variable, respectively. The strength or effect size and statistical significance of these associations are determined for a selection of over millions of individual variants found at common frequencies across different populations. These summary statistics inform the input weights for polygenic score calculation. The accuracy of these summary statistics reflects the integrity of phenotyping as well as the sample size, with larger datasets being able to achieve greater resolution in common variants with smaller effect sizes or rare variants with moderate effect sizes.
Ancestry can have a significant impact on the genetic architecture of traits and diseases. The allele frequencies of variants and haplotype blocks, or blocks of genetic material which are inherited together across generations of meiosis, differ across ancestries. The closer the ancestry of the source GWAS is to the target ancestry, the better the predictive power the final polygenic score will be achieved. To date, the bulk of GWAS participants are of European ancestry. As a result, polygenic scores using these largely European datasets have been shown to underperform in non-European ancestry populations.(Martin et al., 2019)
Ideally, when selecting a source GWAS, [*Copy Editor: Please query the authors if the course GWAS are from the GWAS Catalogue at EBI and if that could be included here.] the GWAS population will be large and closely match the target population. More complex and polygenic traits require a larger source GWAS to get more accurate estimates of weak effect sizes to develop a meaningfully predictive polygenic score when compared with traits whose variance is more fully explained by fewer variants of large effect.(Dudbridge, 2013; Zhang et al., 2023) However, in the absence of this, using summary statistics from a large consortium-based GWAS, which often incorporates meta-analysis of data across different ancestries is another feasible option. Furthermore, recent studies have demonstrated the potential in integrating summary statistics from multiple ancestries to help boost production in target ancestry using methods taking advantage of meta-analyzing summary statistics, leveraging LD patterns, and mixing polygenic scores.(Patel et al., 2023; Ruan et al., 2022; Weissbrod et al., 2022) The method of mixing multi ancestry polygenic scores is of particular relevance when designing scores for mixed populations. For example, a polygenic score informed by GWAS from multiple ancestries and multiple traits had significantly improved performance in predicting CAD in individuals of Hispanic ancestry when compared to previously published scores informed by only European ancestry data.(Patel et al., 2023) PRS computation methods from source GWAS will be described in the following section in STEP 2. Since many traits are genetically related, using multiple source GWASs of related traits to a trait of interest is another approach to further improve the performance of PRS. For example, we recently used this multi-trait approach to improve the performance of a coronary artery disease (CAD) PRS by borrowing genetic information from known CAD risk factors such as diabetes mellitus, body mass index, blood pressure and cholesterol, as well as other atherosclerotic diseases in different vascular beds such as peripheral artery disease and ischemic stroke.(Patel et al., 2023)
Available genotyping arrays only directly genotype hundreds of thousands to a few million common variants in each individual, however the alleles of millions of variants can be inferred through imputation. Genetic imputation algorithms leverage patterns of linkage disequilibrium (LD) observed in the reference panel. LD refers to the non-random association of alleles at different loci due to their physical proximity on the same chromosome. By comparing the genotypes of directly genotyped markers in the study dataset with a reference panel of whole genome sequences in a similar ancestry population, the algorithm can infer the genotypes of untyped markers that are in LD with the known ones. Prior to performing imputation, it is important to ensure high quality input data both from the source GWAS as well as the training dataset, including removal of individuals with discordant reported versus genotypic sex, low genotyping rate (<95% or higher cutoff), putative sex chromosome aneuploidy, heterozygosity outliers, excess relatedness (second degree or closer) and removing variants with low minor allele frequency (<1%) and low call rates (<95% or higher cutoff). It is also important to ensure genome build is consistent across discovery, training, and target populations, otherwise the LiftOver tool can be used for conversion between builds.(Hinrichs et al., 2006)
Over the years, several genome reference panels have been created to facilitate genetic imputation, with recent datasets incorporating genomes from more diverse populations (Table 1). The 1000 Genomes Project is one of the oldest reference panels, providing detailed sequencing data for 2,504 individuals from 26 diverse populations.(Auton et al., 2015) The UK Biobank, which is a large-scale genetic and health data resource of largely European-ancestry individuals, has also released whole-genome sequencing data for almost 500,000 participants which can be used for imputation.(Bycroft et al., 2018) More recently, efforts have centered around collecting whole-genome sequencing data for a diverse set of populations. The Trans-Omics for Precision Medicine (TOPMed) program is an initiative by the National Heart, Lung, and Blood Institute (NHLBI) that provides whole-genome sequencing data for 53,831 individuals of diverse ancestry.(Taliun et al., 2021) Similarly, the GenomeAsia 100K Project has assembled genome sequencing reference data from 1,739 individuals of 219 population groups across Asia.(Wall et al., 2019) The African Genome Variation Project (AGVP) is an ongoing effort to provide whole-genome sequencing data for diverse African populations to help boost imputation accuracy in this group.(Gurdasani et al., 2015) After imputation is performed, imputation quality of each variant is assessed with “info score”, of which >0.8 is recommended for inclusion in subsequent PRS analyses.
Table 1.
Select Imputation Reference Panels
| Reference Panel | Number of genome sequences | Ancestral distribution |
|---|---|---|
| 1000 Genomes (Auton et al., 2015) | 2,504 | 26 world populations |
| Trans-Omics for Precision Medicine (Taliun et al., 2021) | 97,256 | 4 continental populations |
| GenomeAsiaV2 (Wall et al., 2019) | 6,461 | 219 Asian populations |
| NyuWa (Zhang et al., 2021) | 2,999 | Chinese population |
| Haplotype Reference Consortium (McCarthy et al., 2016) | 32,488 | European population |
| CAAPA (Mathias et al., 2016) | 883 | African American population |
With advances in technology and falling costs of sequencing, whole genome sequencing (WGS) has become more readily available. As WGS datasets provide the sequence of each variant in the genome for each individual, population-specific effects from LD patterns will play less of a role in polygenic score analyses. In addition to the common variants, rare variants will also be able to be included in the genome-wide association studies and resulting polygenic scores to ultimately result in more accurate genetic prediction. Another available technology is low coverage whole genome sequencing (lcWGS) which also enables polygenic score analyses without the need for imputation and at a markedly lower cost compared to WGS.(Homburger et al., 2019)
STEP 2: SELECTING METHODS TO COMPUTE POLYGENIC SCORE
Numerous methods to develop polygenic scores have been developed over the years (Table 2). These methods can be split up into three main frameworks based on their statistical approach: frequentist, Bayesian, and hybrid.
Table 2.
List of polygenic score methods used with a brief description and parameters
Frequentist methods refer to classical statistical approaches that do not involve Bayesian inference. The naive scoring approach calculates polygenic scores by summing the effect sizes of genetic variants weighted by their allele frequencies and genotypes in the target dataset. The PRSice and PLINK software packages allow for these calculations.(Purcell et al., 2007; Choi and O’Reilly, 2019) They also allow for thresholding and clumping to select relevant variants for score calculation based on linkage disequilibrium blocks to improve the selection of relevant variants and improve the accuracy of resulting scores. BOLT-REML (Bayesian and Omnibus Linear regression with Twin and RElated MethoLogies) is a method that uses a linear mixed model to estimate genetic effects while accounting for relatedness between individuals and provides added efficiency when analyzing large-scale datasets.(Loh et al., 2015a)
Bayesian algorithms incorporate prior knowledge about genetic architecture, leveraging population-specific reference data or external databases to assign weights more accurately to variants and enhance prediction accuracy. BayesR models the effects of all genetic variants simultaneously, assuming that only a subset of variants has non-zero effects on the trait, effectively shrinking the coefficients of irrelevant variants to zero. SBayesR (Spike and Slab Bayesian Regression) incorporates a spike-and-slab prior to model the distribution of effect sizes, allowing for sparse polygenic models with a subset of variants having non-zero effects.(Lloyd-Jones et al., 2019) BOLT-LMM (Bayesian and Lasso-Adjusted Linear Mixed Model) combines linear mixed models with a Lasso-penalized regression to estimate the polygenic score while efficiently handling large-scale genetic data and accounting for relatedness between individuals.(Loh et al., 2015b) LDpred (Linkage Disequilibrium score regression) and LDpred2 estimate the effect sizes of genetic variants while accounting for linkage disequilibrium patterns in the genome.(Privé et al., 2020; Vilhjálmsson et al., 2015) PRS-CS (Polygenic Risk Score - Conditional and Sparse) estimates the effect sizes of genetic variants while allowing for sparsity in the polygenic model.(Ge et al., 2019) It identifies a subset of relevant variants with non-zero effects, reducing the number of predictors in the polygenic score. PRS-CSx additionally incorporates genetic effects across populations through a shared continuous shrinkage (CS) prior, allowing for more improved effect size estimation from sharing information between summary statistics and leveraging LD diversity across input GWAS.(Ruan et al., 2022)
Hybrid methods combine elements of frequentist, Bayesian, and additional techniques such as Lasso regularization. For example, SuSiE (Sum of Single Effects) is a hybrid algorithm that uses Bayesian spike-and-slab regression to estimate sparse polygenic models.35 [*Copy Editor: the superscript 35 appears that it might be a citation. Please query the authors.] It selects a subset of genetic variants with non-zero effects, reducing the number of predictors in the polygenic score and improving its interpretability.
For developing polygenic scores for a non-European or admixed dataset, methods that leverage commonalities across ancestries such as PRS-CSx are most likely to yield the strongest performance. The most optimal course of action, however, is to generate scores using multiple methods and pick the modality resulting in the best performance in a target population.
STEP 3: ADJUSTING SCORES USING PRINCIPAL COMPONENTS OF ANCESTRY
The process of polygenic score development, validation, and interpretation occurs in the context of a reference population, and therefore this must be accounted for in analysis to avoid confounding. Genetic architecture varies across different populations due to historical migrations and genetic drift. Important distinctions include differences in allele frequencies and linkage disequilibrium patterns. These differences can result in significant error in assigning polygenic score weights particularly when imputation is used. If not correctly accounted for, different populations appear to have vastly different distributions of genetic risk relative to each other.
The standard way to adjust for population stratification is using principal components of genetic ancestry, which are linear combinations of genetic markers that capture the major sources of genetic variation within a population.(Price et al., 2006) A linear regression model can be used to regress principal components of genetic ancestry with the polygenic score. The model can be used to predict a polygenic score and these values are then subtracted from the raw polygenic score to get an adjusted score. The adjusted score is then scaled so that its mean is 0 and standard deviation is 1 to facilitate analysis. This commonly centers the population-specific polygenic distributions and allows for better comparison of genetic risk across groups (Figure 2).
Figure 2. Raw vs. Ancestry-Adjusted Polygenic Score Population Distributions.
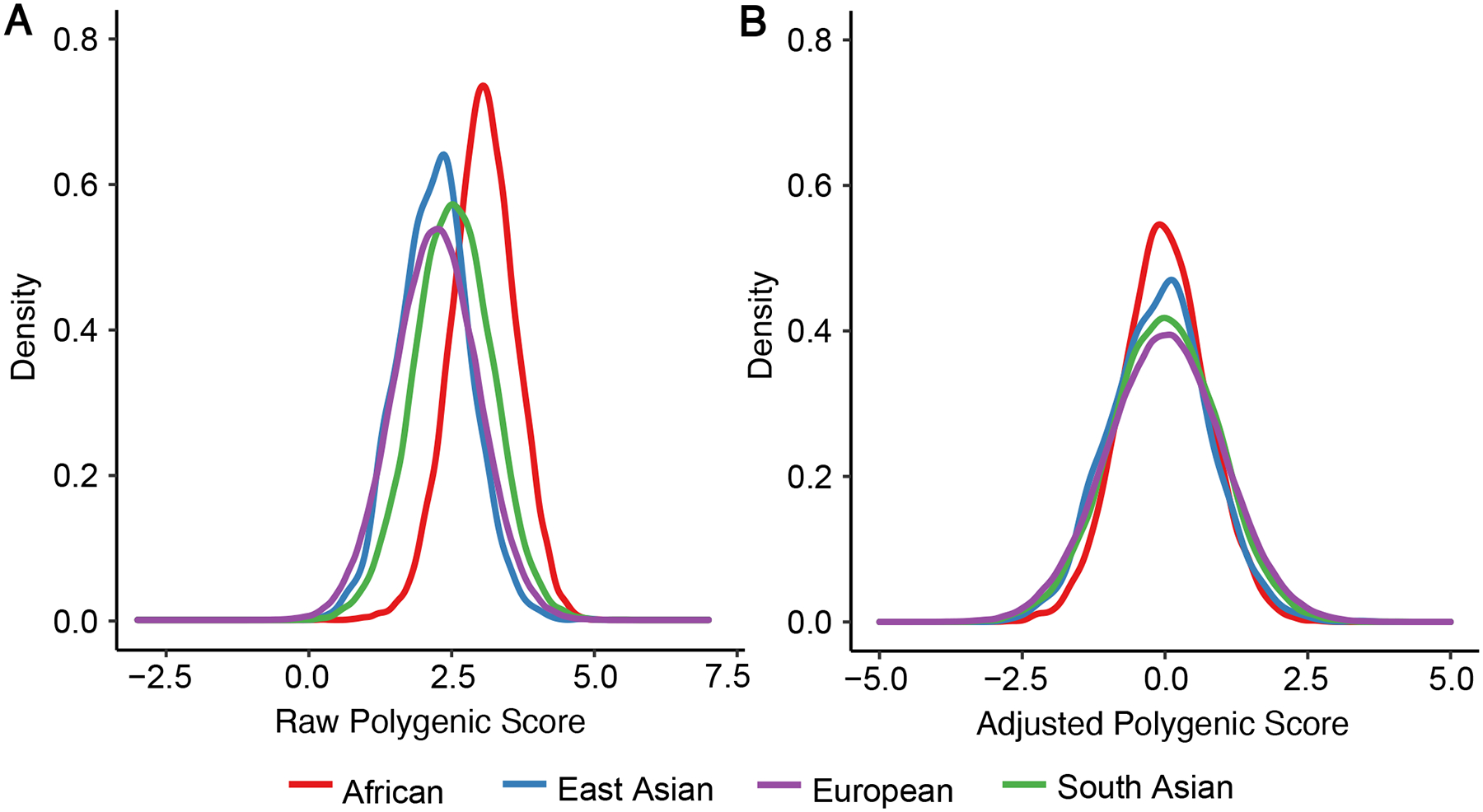
(A) Raw distribution of a recently published polygenic risk score (PRS) for coronary artery disease (GPSMult) in individuals of African (n=7,281), East Asian (n=1,464), European (n=308,264), and South Asian (n=8,982) ancestries shows shifted distributions due population stratification. (B) When the same PRS is adjusted for principal components of ancestry as described in STEP 3 the distributions are overlapping. Briefly, a linear regression model is used to regress principal components of genetic ancestry with the PRS. The model is then used to predict a PRS, and these values are then subtracted from the raw PRS to get an adjusted score. The adjusted PRS is then scaled so that its mean is 0 and standard deviation is 1. This centers the population-specific polygenic distributions and allows for better comparison of genetic risk across groups.
STEP 4: SELECTING THE BEST PERFORMING SCORE
To ensure the accuracy, reliability, and generalizability of the polygenic score, it is important to ensure independence of discovery, training, and validation datasets, as is the case for other predictive models (Figure 3). The discovery datasets refer to the genome-wide association studies that were used to generate the weights for each variant in the score. Only summary level data is required, and these datasets are often publicly available for download, such as in the GWAS catalog.(GWAS Catalog) The training data set refers to the individual-level data used to build the polygenic score model. It is used to identify the relevant genetic variants associated with the trait, estimate their effect sizes, and compute the additional polygenic score algorithm-specific hyperparameters which lead to the best performing score. The validation dataset refers to individual-level data used to evaluate the performance of the polygenic score model. The training and validation datasets could be partitions of an available dataset, or these steps can occur in completely separate studies. For validation of PRS for highly heritable traits, effective sample sizes of at least 100 individuals are needed for meaningful analyses (Choi et al., 2020).
Figure 3. Datasets Used for Development of Polygenic Risk Score.
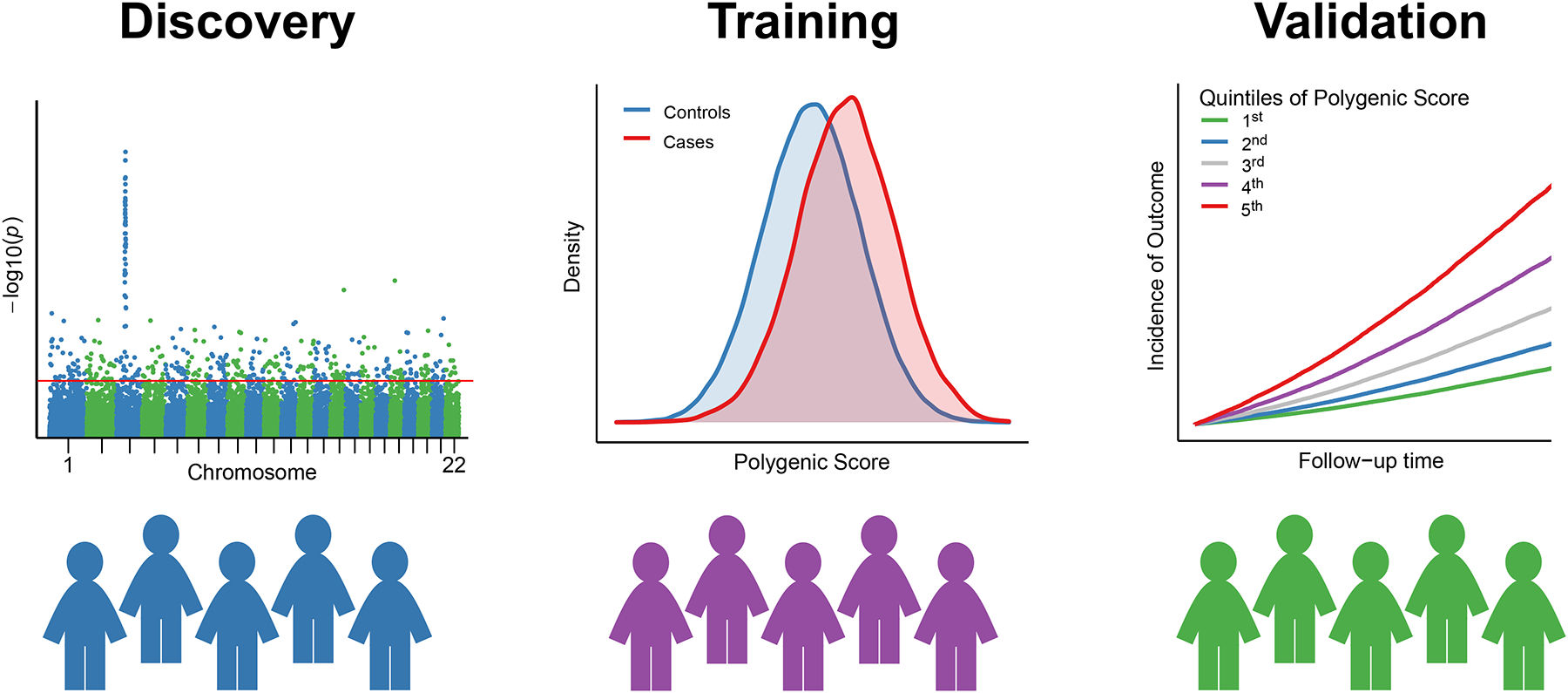
Three datasets are often used to develop a polygenic risk score (PRS). First, a discovery dataset is used to establish the genetic associations of the trait or disease in question, or in other words to perform a genome-wide association study (GWAS). For pragmatic purposes, existing GWAS association statistics that are publicly available are used as described in STEP 1. Second, a training dataset is used to compute multiple PRS and select the best-performing one as described in STEPS 2, 3 and 4. Third, a validation dataset is used to study and report the performance of the optimized PRS. While an external validation dataset is preferable, it is common practice to also split a single large enough dataset into training and validation datasets.
When benchmarking polygenic risk scores, it is essential to consider multiple metrics to get a comprehensive understanding of their performance i) in the training dataset to select the best parameters and score version and ii) in the testing dataset to compare with other available scores and clinical risk estimators (Table 3). Simple correlation metrics like Pearson correlation quantify the correlation between the PRS and continuous trait values. The strength of association between polygenic scores and a binary trait can be expressed using measures of risk, such as cox proportional hazards or odds ratios. These metrics are often reported per standard deviation of the polygenic score scaled to a standard deviation of 1 and mean of 0. Individuals in the top percentiles of the score can also be compared with remaining individuals in the population or the middle quintile of individuals when reporting these risk estimates.
Table 3.
Metrics for Studying the Performance of Polygenic Risk Scores
| Metric | Use Case |
|---|---|
| Predictive Ability | |
| Odds Ratio | Logistic regression comparing either percentile groups or reporting per standard deviation of scaled score |
| Hazard ratio | Cox proportional hazards regression comparing either percentile groups or reporting per standard deviation of scaled score |
| Variance explained | |
| Nagelkerke R2 | Phenotypic variance calculated on observed scale, can underestimate if disease prevalence is low |
| Log-liability R2 | Phenotypic variance calculated on the liability scale to allow for more direct comparison with heritability estimates |
| Discrimination | |
| AUC-ROC | Measure of discriminatory capacity of a model in distinguishing between individuals with and without a certain trait or condition |
| Reclassification | |
| Net reclassification index | Classification accuracy achieved by moving individuals into more appropriate risk categories when using polygenic score compared to another model, based on threshold or continuous scale |
R-squared (R2) measures the proportion of variance in the trait or disease explained by the polygenic risk score. A higher R2 indicates that the PRS can better predict the phenotype of interest. Similar to R2, Nagelkerke’s R2 measures the proportion of variance explained by the PRS but is adjusted for the baseline prevalence of the trait. A similar metric, the log liability R2 helps explain the proportion of variance explained to allow for direct comparison on the heritability scale.38 [*Copy Editor: Again, please ask the authors about this superscript.]
Metrics of discrimination are particularly relevant when assessing polygenic scores. The Area Under the Receiver Operating Characteristic Curve (AUC-ROC-AUC) assesses the discriminatory power of the PRS model. It plots the true positive rate against the false positive rate at various prediction thresholds. An AUC value closer to 1 indicates better discrimination. The concordance statistic, or C-statistic, reflects the area under the ROC. A related metric, the Precision-Recall Area Under the Curve (PR-AUC) is useful when dealing with imbalanced datasets.
Finally, measures of reclassification help compare the added value of polygenic scores to available models. The Net Reclassification Improvement (NRI) evaluates whether the addition of PRS improves risk classification compared to a baseline model without the PRS. This can be calculated on the continuous spectrum or based on a threshold. Similarly, the Integrated Discrimination Improvement (IDI) assesses the improvement in risk prediction when including the PRS in the model.
STEP 5: DEFINING PERCENTILES OF A POPULATION DISTRIBUTION
PRS is defined as a percentile of a population distribution which enables quantification of risk in individuals or groups within a population. Given that PRS has a normal distribution in any population, individuals are ranked into 100 groups from lowest to highest PRS value. Individuals with higher percentile numbers have higher risk and vice versa. Commonly, the extremes of the distributions are used to define relative risk, compared to the average population risk or middle of the distribution. For example, the top quintile of the population distribution of a recent CAD PRS was associated with three-fold increased risk of CAD compared to the average (Patel et al., 2023). This means that this PRS can detect 20% of the population at 3-fold increased risk of CAD. Clinical reports of PRS use specific percentile thresholds to define risk depending on the PRS, disease, and reporting mechanisms (Wand et al., 2021; Brockman et al., 2021; Maamari et al., 2022).
The selection of the population distribution used to define percentiles is an important step, especially in the context of ancestrally diverse populations and smaller case-control datasets used for PRS derivation. As a general rule, the dataset used to define the population distribution should be representative of the population in which PRS is being used. In many cases where the training and validation sets are large and not markedly biased for disease, such as in population cohorts or national biobanks, those same datasets can be used to define the percentiles as they are usually representative of the population. For example, polygenic scores derived from the UK Biobank, a prospective nationwide population-based study of half a million middle-aged participants in the UK, typically use the UK Biobank itself to define percentiles (Inouye et al., 2018; Khera et al., 2018).
However, when smaller case-control datasets that are enriched for disease of interest are used to train and validate a PRS, a separate dataset that is representative of the population is needed to define percentiles. Such case-control datasets by definition are heavily skewed to disease and not representative of the population. It is preferable to use a separate dataset not enriched for the disease of interest. In order to have an accurate percentile distribution, it is preferable that this dataset is large enough (usually more than 1000 participants). There are different ways of having a representative population distribution that are context specific. For example, in a recent work optimizing PRS in Saudi Arabia, we used a reference population of 1017 individuals sampled from 28 tribes of Saudi Arabia without consideration of any disorders (Mineta et al., 2021). Similarly in deriving a PRS for South Asians, a separate dataset of 1,733 individuals from a population-based study in India were recruited without consideration of disease status (Wang et al., 2020b).
STEP 6: VALIDATING THE OPTIMIZED POLYGENIC SCORE PERFORMANCE
Validating a polygenic score involves testing it in an independent population to determine its accuracy, reliability, and generalizability. As with training, the polygenic score is modeled along with age, sex, and principal components of genetic ancestry, and performance is benchmarked by a variety of previously described metrics (Table 3). The size of the validation cohort is crucial for obtaining meaningful results. Small sample sizes may lead to unstable estimates and reduced statistical power, making it challenging to draw reliable conclusions about the polygenic score’s performance. Oftentimes, a single cohort is divided into a subset for training and validating. Although convenient, this often is subject to overfitting, and internal validation produces stronger associations which are difficult to reproduce in other datasets. However, in the absence of abundant, independent, ancestry-specific datasets, this is a reasonable option.
For a score to be implemented widely, it is important to benchmark its performance to other scores in external validation datasets, at least in the target ancestry and ideally in multiple ancestries. Comparing the performance of a new score with previously published scores helps identify which predictor would be most useful for future analyses. The Polygenic Score Catalog (http://www.pgscatalog.org/), [*Copy Editor: added the url – please inform the authors.] an open database of published polygenic scores for any trait or disease, is valuable for performing such comparisons (Lambert et al., 2021). It is important to note that even when scores are trained and externally validated in the same ancestral group, the performance of the score can vary based on other factors, such as geographic or environmental differences. For example, a polygenic score for coronary artery disease trained in the UKBB European ancestry individuals had diminished performance when applied in White participants in the Million Veteran Program (Gaziano et al., 2016; Patel et al., 2023).
STEP 7: IMPLEMENTING THE OPTIMIZED POLYGENIC SCORE IN CLINICAL PRACTICE
Moving PRS from a research tool into clinical implementation requires additional considerations that are also relevant in the context of diverse populations (Hao et al., 2022). Those considerations fall under three groups. First, clinical assays need to be constructed which require confirmation of analytical validity.(Hao et al., 2022) The variability in the validity of PRS in diverse ancestries whether non-European or admixed needs to be accounted for, both in the way PRS is calculated for an individual patient and the way it is interpreted. In an ideal world, an ancestry specific PRS would be used and reported based on its published effect size estimates to individuals of each ancestry. However, this is not possible in most cases and multiple alternative approaches are often used: (i) In one approach, a single PRS could be used for all ancestries, preferentially one that performs well (but still variably) across multiple ancestries, and “high-risk” is defined for different percentile cut-offs that result in equivalent risk increase across ancestries. This approach results in a lower proportion of the population where the PRS has a reduced performance being reported as “high-risk” compared to the population where the PRS has a higher performance, but the risk level reported to those individuals is similar. (ii) In another approach, the same percentile cut-off is used but different risk levels are reported for different ancestries. (iii) A modification of this latter approach is having a disclaimer around the lower performance of the score in individuals of non-European ancestries but without specifying the risk level.
Second, clinical reports of PRS need to be created in a way that is understandable by their target audience, patients and/or providers. There are no standards of clinical reports today and practices vary significantly as we have recently reviewed.(Brockman et al., 2021) However, it is critical that reports are designed in a user-centric approach that also takes into consideration the target audience with regards to comprehension and reactions elicited by receiving the result.(Muse et al., 2022; Maamari et al., 2022) The content of the report itself can also vary with regards to the amount of details with regards to recommendations and guidance on next steps vs. limiting to an interpretative service. In the context of diverse populations, reports should be delivered in a user-centric lens with regards to the level of understanding, language barriers, and even cultural considerations on how risk is perceived.
Third, disclosure of PRS results to patients should occur in the context of clinical workflows and resources that support their use for patients and providers. Genetic counseling services are often paired with disclosure of the results. Clinical guidelines will be needed to guide providers on when to order the test and what to do with the results. This still however remains in its infancy as translational research with prospective studies on clinical utility continues to accumulate.
CONCLUSION
Polygenic risk scores are a powerful tool with emerging clinical utility. Leveraging the advantage of DNA information available early in life, they can deliver on the promise of precision medicine across multiple disease areas. Their variable performance across populations, particularly reduced cross-ethnic performance in ancestries outside Europe, is an important limitation to advancing translational research and clinical utility. However, increasing availability of public datasets and tools enables us to optimize a PRS for a population of interest. We presented a pragmatic seven-step approach to optimize a PRS in diverse populations starting from identifying a genome-wide association study for a trait of interest, developing the PRS for that trait that will perform best in the population of interest, and all the way to implementing its disclosure to the next patient in the clinic.
ACKNOWLEDGEMENTS:
Dr. Patel receives funding from the Harvard Catalyst and the National Human Genome Research Institute (grant 1U01HG011719). Dr. Fahed receives funding from the National Heart Lung and Blood Institute (grants K08 HL161448 and R01 HL164629) for work on polygenic risk scores. [*Copy Editor: The authors had included the conflict of interest statement and the acknowledgements in a single CoI section. I created the Acknowledgement section and moved the appropriate information there. Please inform the authors.]
Footnotes
CONFLICT OF INTEREST STATEMENT:
Dr. Fahed reports being co-founder of Goodpath, unrelated to the subject of this manuscript.
Contributor Information
Aniruddh P. Patel, Cardiovascular Research Center, Massachusetts General Hospital, 185 Cambridge Street | CPZN 3.128, Boston, MA 02114.
Akl C. Fahed, Cardiovascular Research Center, Massachusetts General Hospital, 185 Cambridge Street | CPZN 3.128, Boston, MA 02114.
DATA AVAILABILITY STATEMENT:
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
LITERATURE CITED
- Adeyemo A, Balaconis MK, Darnes DR, Fatumo S, Granados Moreno P, Hodonsky CJ, Inouye M, Kanai M, Kato K, Knoppers BM, et al. 2021. Responsible use of polygenic risk scores in the clinic: potential benefits, risks and gaps. Nature Medicine 27:1876–1884. [DOI] [PubMed] [Google Scholar]
- Auton A, Abecasis GR, Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, Donnelly P, Eichler EE, et al. 2015. A global reference for human genetic variation. Nature 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockman DG, Petronio L, Dron JS, Kwon BC, Vosburg T, Nip L, Tang A, O’Reilly M, Lennon N, Wong B, et al. 2021. Design and user experience testing of a polygenic score report: a qualitative study of prospective users. BMC Medical Genomics 14:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O’Connell J, et al. 2018. The UK Biobank resource with deep phenotyping and genomic data. Nature 562:203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Chen J, Collins R, Guo Y, Peto R, Wu F, and Li L 2011. China Kadoorie Biobank of 0.5 million people: survey methods, baseline characteristics and long-term follow-up. International Journal of Epidemiology 40:1652–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SW, Mak TS-H, and O’Reilly PF 2020. Tutorial: a guide to performing polygenic risk score analyses. Nature Protocols 15:2759–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SW, and O’Reilly PF 2019. PRSice-2: Polygenic Risk Score software for biobank-scale data. GigaScience 8:giz082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dikilitas O, Schaid DJ, Kosel ML, Carroll RJ, Chute CG, Denny JA, Fedotov A, Feng Q, Hakonarson H, Jarvik GP, et al. 2020. Predictive Utility of Polygenic Risk Scores for Coronary Heart Disease in Three Major Racial and Ethnic Groups. The American Journal of Human Genetics 106:707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudbridge F 2013. Power and Predictive Accuracy of Polygenic Risk Scores. PLoS Genetics 9:e1003348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahed Akl C., Aragam Krishna G., George Hindy, Chen Yii-Der Ida, Kumardeep Chaudhary, Amanda Dobbyn, Krumholz Harlan M., Sheu Wayne H.H., Rich Stephen S., Rotter Jerome I., et al. Transethnic Transferability of a Genome-wide Polygenic Score for Coronary Artery Disease. Circulation: Genomic and Precision Medicine 0. Available at: http://www.ahajournals.org/doi/10.1161/CIRCGEN.120.003092 [Accessed January 5, 2021]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaziano JM, Concato J, Brophy M, Fiore L, Pyarajan S, Breeling J, Whitbourne S, Deen J, Shannon C, Humphries D, et al. 2016. Million Veteran Program: A mega-biobank to study genetic influences on health and disease. Journal of Clinical Epidemiology 70:214–223. [DOI] [PubMed] [Google Scholar]
- Ge T, Chen C-Y, Ni Y, Feng Y-CA, and Smoller JW 2019. Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nature Communications 10:1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurdasani D, Carstensen T, Tekola-Ayele F, Pagani L, Tachmazidou I, Hatzikotoulas K, Karthikeyan S, Iles L, Pollard MO, Choudhury A, et al. 2015. The African Genome Variation Project shapes medical genetics in Africa. Nature 517:327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GWAS Catalog Available at: https://www.ebi.ac.uk/gwas/ [Accessed September 5, 2023].
- Hao L, Kraft P, Berriz GF, Hynes ED, Koch C, Korategere V Kumar P, Parpattedar SS, Steeves M, Yu W, Antwi AA, et al. 2022. Development of a clinical polygenic risk score assay and reporting workflow. Nature Medicine 28:1006–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinrichs AS, Karolchik D, Baertsch R, Barber GP, Bejerano G, Clawson H, Diekhans M, Furey TS, Harte RA, Hsu F, et al. 2006. The UCSC Genome Browser Database: update 2006. Nucleic Acids Research 34:D590–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homburger JR, Neben CL, Mishne G, Zhou AY, Kathiresan S, and Khera AV 2019. Low coverage whole genome sequencing enables accurate assessment of common variants and calculation of genome-wide polygenic scores. Genome Medicine 11:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inouye M, Abraham G, Nelson CP, Wood AM, Sweeting MJ, Dudbridge F, Lai FY, Kaptoge S, Brozynska M, Wang T, et al. 2018. Genomic Risk Prediction of Coronary Artery Disease in 480,000 Adults: Implications for Primary Prevention. Journal of the American College of Cardiology 72:1883–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishigaki K, Akiyama M, Kanai M, Takahashi A, Kawakami E, Sugishita H, Sakaue S, Matoba N, Low S-K, Okada Y, et al. 2020. Large-scale genome-wide association study in a Japanese population identifies novel susceptibility loci across different diseases. Nature Genetics 52:669–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, Natarajan P, Lander ES, Lubitz SA, Ellinor PT, et al. 2018. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nature Genetics 50:1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klarin D, and Natarajan P 2022. Clinical utility of polygenic risk scores for coronary artery disease. Nature Reviews. Cardiology 19:291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama S, Ito K, Terao C, Akiyama M, Horikoshi M, Momozawa Y, Matsunaga H, Ieki H, Ozaki K, Onouchi Y, et al. 2020. Population-specific and trans-ancestry genome-wide analyses identify distinct and shared genetic risk loci for coronary artery disease. Nature Genetics 52:1169–1177. [DOI] [PubMed] [Google Scholar]
- Kumuthini J, Zick B, Balasopoulou A, Chalikiopoulou C, Dandara C, El-Kamah G, Findley L, Katsila T, Li R, Maceda EB, et al. 2022. The clinical utility of polygenic risk scores in genomic medicine practices: a systematic review. Human Genetics 141:1697–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert SA, Gil L, Jupp S, Ritchie SC, Xu Y, Buniello A, McMahon A, Abraham G, Chapman M, Parkinson H, et al. 2021. The Polygenic Score Catalog as an open database for reproducibility and systematic evaluation. Nature Genetics 53:420–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Goddard ME, Wray NR, and Visscher PM 2012. A better coefficient of determination for genetic profile analysis. Genetic Epidemiology 36:214–224. [DOI] [PubMed] [Google Scholar]
- Lloyd-Jones LR, Zeng J, Sidorenko J, Yengo L, Moser G, Kemper KE, Wang H, Zheng Z, Magi R, Esko T, et al. 2019. Improved polygenic prediction by Bayesian multiple regression on summary statistics. Nature Communications 10:5086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh P-R, Bhatia G, Gusev A, Finucane HK, Bulik-Sullivan BK, Pollack SJ, Schizophrenia Working Group of Psychiatric Genomics Consortium, de Candia TR, Lee SH, Wray NR, et al. 2015a. Contrasting genetic architectures of schizophrenia and other complex diseases using fast variance-components analysis. Nature Genetics 47:1385–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh P-R, Tucker G, Bulik-Sullivan BK, Vilhjálmsson BJ, Finucane HK, Salem RM, Chasman DI, Ridker PM, Neale BM, Berger B, et al. 2015b. Efficient Bayesian mixed-model analysis increases association power in large cohorts. Nature Genetics 47:284–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maamari DJ, Khera AV, and Fahed AC 2022. Clinical Implementation of Combined Monogenic and Polygenic Risk Disclosure for Coronary Artery Disease. JACC: Advances:100068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, and Daly MJ 2019. Clinical use of current polygenic risk scores may exacerbate health disparities. Nature Genetics 51:584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathias RA, Taub MA, Gignoux CR, Fu W, Musharoff S, O’Connor TD, Vergara C, Torgerson DG, Pino-Yanes M, Shringarpure SS, et al. 2016. A continuum of admixture in the Western Hemisphere revealed by the African Diaspora genome. Nature Communications 7:12522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, Kang HM, Fuchsberger C, Danecek P, Sharp K, et al. 2016. A reference panel of 64,976 haplotypes for genotype imputation. Nature Genetics 48:1279–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mineta K, Goto K, Gojobori T, and Alkuraya FS 2021. Population structure of indigenous inhabitants of Arabia. PLoS genetics 17:e1009210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muse ED, Chen S-F, Liu S, Fernandez B, Schrader B, Molparia B, León AN, Lee R, Pubbi N, Mejia N, et al. 2022. Impact of polygenic risk communication: an observational mobile application-based coronary artery disease study. npj Digital Medicine 5:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novembre J, Stein C, Asgari S, Gonzaga-Jauregui C, Landstrom A, Lemke A, Li J, Mighton C, Taylor M, and Tishkoff S 2022. Addressing the challenges of polygenic scores in human genetic research. The American Journal of Human Genetics 109:2095–2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan JW, Raghavan S, Marquez-Luna C, Luzum JA, Damrauer SM, Ashley EA, O’Donnell CJ, Willer CJ, Natarajan P, and American Heart Association Council on Genomic and Precision Medicine; Council on Clinical Cardiology; Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular Radiology and Intervention; Council on Lifestyle and Cardiometabolic Health; and Council on Peripheral Vascular Disease 2022. Polygenic Risk Scores for Cardiovascular Disease: A Scientific Statement From the American Heart Association. Circulation 146:e93–e118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page ML, Vance EL, Cloward ME, Ringger E, Dayton L, Ebbert MTW, Miller JB, and Kauwe JSK 2022. The Polygenic Risk Score Knowledge Base offers a centralized online repository for calculating and contextualizing polygenic risk scores. Communications Biology 5:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AP, and Khera AV 2022. Advances and Applications of Polygenic Scores for Coronary Artery Disease. Annual Review of Medicine. [DOI] [PubMed] [Google Scholar]
- Patel AP, Wang M, Ruan Y, Koyama S, Clarke SL, Yang X, Tcheandjieu C, Agrawal S, Fahed AC, Ellinor PT, et al. 2023. A multi-ancestry polygenic risk score improves risk prediction for coronary artery disease. Nature Medicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, and Reich D 2006. Principal components analysis corrects for stratification in genome-wide association studies. Nature Genetics 38:904–909. [DOI] [PubMed] [Google Scholar]
- Privé F, Arbel J, Aschard H, and Vilhjálmsson BJ 2022. Identifying and correcting for misspecifications in GWAS summary statistics and polygenic scores. Human Genetics and Genomics Advances 3. Available at: https://www.cell.com/hgg-advances/abstract/S2666-2477(22)00052-5 [Accessed January 2, 2023]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Privé F, Arbel J, and Vilhjálmsson BJ 2020. LDpred2: better, faster, stronger. Bioinformatics (Oxford, England) 36:5424–5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, et al. 2007. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. American Journal of Human Genetics 81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan Y, Lin Y-F, Feng Y-CA, Chen C-Y, Lam M, Guo Z, Stanley Global Asia Initiatives, He L, Sawa A, Martin AR, et al. 2022. Improving polygenic prediction in ancestrally diverse populations. Nature Genetics 54:573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saad M, El-Menyar A, Kunji K, Ullah E, Al Suwaidi J, and Kullo IJ Validation of Polygenic Risk Scores for Coronary Heart Disease in a Middle Eastern Cohort Using Whole Genome Sequencing. Circulation: Genomic and Precision Medicine 0:e003712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi Nowbandegani P, Wohns AW, Ballard JL, Lander ES, Bloemendal A, Neale BM, and O’Connor LJ 2023. Extremely sparse models of linkage disequilibrium in ancestrally diverse association studies. Nature Genetics:1–9. [DOI] [PubMed] [Google Scholar]
- Taliun D, Harris DN, Kessler MD, Carlson J, Szpiech ZA, Torres R, Taliun SAG, Corvelo A, Gogarten SM, Kang HM, et al. 2021. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590:290–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilhjálmsson BJ, Yang J, Finucane HK, Gusev A, Lindström S, Ripke S, Genovese G, Loh P-R, Bhatia G, Do R, et al. 2015. Modeling Linkage Disequilibrium Increases Accuracy of Polygenic Risk Scores. The American Journal of Human Genetics 97:576–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall JD, Stawiski EW, Ratan A, Kim HL, Kim C, Gupta R, Suryamohan K, Gusareva ES, Purbojati RW, Bhangale T, et al. 2019. The GenomeAsia 100K Project enables genetic discoveries across Asia. Nature 576:106–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wand H, Lambert SA, Tamburro C, Iacocca MA, O’Sullivan JW, Sillari C, Kullo IJ, Rowley R, Dron JS, Brockman D, et al. 2021. Improving reporting standards for polygenic scores in risk prediction studies. Nature 591:211–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Sarkar A, Carbonetto P, and Stephens M 2020a. A Simple New Approach to Variable Selection in Regression, with Application to Genetic Fine Mapping. Journal of the Royal Statistical Society Series B: Statistical Methodology 82:1273–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Menon R, Mishra S, Patel AP, Chaffin M, Tanneeru D, Deshmukh M, Mathew O, Apte S, Devanboo CS, et al. 2020b. Validation of a Genome-Wide Polygenic Score for Coronary Artery Disease in South Asians. Journal of the American College of Cardiology 76:703–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissbrod O, Kanai M, Shi H, Gazal S, Peyrot WJ, Khera AV, Okada Y, Martin AR, Finucane HK, and Price AL 2022. Leveraging fine-mapping and multipopulation training data to improve cross-population polygenic risk scores. Nature Genetics 54:450–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Zhan J, Jin J, Zhang J, Lu W, Zhao R, Ahearn TU, Yu Z, O’Connell J, Jiang Y, et al. 2023. A new method for multi-ancestry polygenic prediction improves performance across diverse populations. 2022.03.24.485519. Available at: https://www.biorxiv.org/content/10.1101/2022.03.24.485519v6 [Accessed September 6, 2023].
- Zhang P, Luo H, Li Y, Wang Y, Wang J, Zheng Y, Niu Y, Shi Y, Zhou H, Song T, et al. 2021. NyuWa Genome resource: A deep whole-genome sequencing-based variation profile and reference panel for the Chinese population. Cell Reports 37:110017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.