

Abstract
Background
Based on molecular characteristics, deficient DNA mismatch repair (dMMR) solid tumors are largely divided into three categories: somatically MLH1-hypermethylated tumors, Lynch syndrome (LS)-associated tumors, and Lynch-like syndrome (LLS)-associated tumors. The incidence of each of these conditions and the corresponding pathogenic genes related to LLS remain elusive.
Methods
We identified dMMR tumors in 3609 tumors from 9 different solid organs, including colorectal cancer, gastric cancer, small-bowel cancer, endometrial cancer, ovarian cancer, upper urinary tract cancer, urinary bladder cancer, prostate cancer, and sebaceous tumor, and comprehensively summarized the characterization of dMMR tumors. Characterization of dMMR tumors were performed as loss of at least one of MMR proteins (MLH1, MSH2, MSH6, and PMS2), by immunohistochemistry, followed by MLH1 promotor methylation analysis and genetic testing for MMR genes where appropriate. Somatic variant analysis of MMR genes and whole exome sequencing (WES) were performed in patients with LLS.
Results
In total, the incidence of dMMR tumors was 5.9% (24/3609). The incidence of dMMR tumors and the proportion of the three categorized dMMR tumors varied considerably with different tumor types. One to three likely pathogenic/pathogenic somatic MMR gene variants were detected in 15 out of the 16 available LLS tumors. One patient each from 12 patients who gave consent to WES demonstrated non-MMR germline variants affect function (POLQ or BRCA1).
Conclusions
Our data regarding the LS to LLS ratio would be useful for genetic counseling in patients who are suspected to have LS, though the genetic backgrounds for the pathogenesis of LLS need further investigation.
Supplementary Information
The online version contains supplementary material available at 10.1007/s10147-024-02518-y.
Keywords: Deficient DNA mismatch repair (dMMR), dMMR tumors, Lynch-like syndrome
Introduction
Recently, utilization of immune checkpoint inhibitors in the treatment of DNA mismatch repair deficient (dMMR) solid tumors has gained significant attention [1, 2]. Currently, two major methods are used to identify dMMR in the tumors, molecular testing of microsatellite instability (MSI) and immunohistochemistry (IHC) of MMR proteins (MLH1, MSH2, MSH6, and PMS2). dMMR is usually defined as high-frequency MSI (MSI-H) or loss of expression (MMR-D) of at least one MMR protein. MSI-H and MMR-D yield high concordant results (95–99%) in colorectal cancer (CRC) and endometrial cancer (EC) [3, 4].
Based on molecular characteristics, MMR-D and/or MSI-H solid tumors are divided into three categories: 1. somatically MLH1-hypermethylated tumors, Lynch syndrome (LS)-associated tumors, and Lynch-like syndrome (LLS)-associated tumors. MLH1-hypermethylated tumor is the most common type, it is non-heritable and caused by aberrant hypermethylation of the MLH1 promoter region, leading transcriptional inactivation [5, 6]. LS is an autosomal dominant inherited disorder, mainly caused by a germline pathogenic variant of MMR genes, though 3′ deletion of EPCAM located upstream of MSH2 is also known to be another cause of LS [7]. Biallelic inactivation of MMR genes by germline and somatic variants leads to a high frequency of replication errors in microsatellite regions and repeated sequences in the coding regions of various, growth-related target genes [8], resulting in the development of dMMR tumors in various organs. LLS has recently been proposed as a third dMMR tumor type that do not harbor germline MMR gene variants or MLH1 hypermethylation [9, 10]. Somatic MMR gene variants frequently occur in LLS [11, 12]. However, there have been a limited number of reports [13] regarding MMR unrelated germline pathogenic variants in LLS tumorigenesis.
Recently, several large-scale investigations have reported the incidence of MSI-H among various solid tumors [14, 15]. However, the proportion of sporadic dMMR, LS-associated tumors (LS-AT), and LLS-associated tumors (LLS-AT), along with the proportion of dMMR-associated MMR genes, remains unknown. In addition, germline pathogenic variants unrelated to MMR have rarely been investigated. Using MMR-IHC, we previously performed universal tumor screening for identification of LS, and reported the proportion of at least three categorized dMMR tumors in nine different tumors, including CRC, gastric cancer (GC), small-bowel cancer (SBC), EC, ovarian cancer (OC), upper urinary tract cancer (UUTC), urinary bladder cancer (UBC), prostate cancer (PC), and sebaceous tumor (ST) [12, 16–23]. In the present study, we comprehensively analyzed data from previous publications. Furthermore, using whole exome sequencing, we investigated the presence of non-MMR germline pathogenic variants in LLS tumorigenesis.
Patients and methods
Ethical consideration
The current study and the associated studies (previously published) [12, 16–23] have been approved by the local ethics committee of the Saitama Medical Center (No. 924, No. 925, and No. 926) and the Saitama Medical University (No. 592 and No. 747). Informed consent was obtained from all patients before MMR genetic testing and whole exome sequencing. Consent was obtained from family members of deceased patients.
Patients
In this study, we analyzed a total of 3609 tumor specimens, resected from nine different solid organs: CRC (n = 1234), GC (n = 513), SBC (n = 30), EC (n = 395), OC (n = 305), UUTC (n = 164), UBC (n = 618), PC (n = 337), and ST (n = 13) (supplementary Table 1). All tumor specimens were resected and stored at Saitama Medical Center of Saitama Medical University.
IHC for MMR proteins
IHC was performed to examine the expression of four MMR proteins (MLH1, MSH2, MSH6, and PMS2). Tumor sections (4 μm) were fixed with formalin and embedded in paraffin (FFPE). Then, the sections were stained using a Staining Automat (BOND III, Leica Biosystems Melbourne Pvt. Ltd, Melbourne, Australia), according to the manufacturer’s protocol. The antibodies used for detecting MMR proteins were described previously [12, 16–23].
The normal expression pattern for MMR proteins is nuclear. Complete loss of nuclear expression in tumor cells with the presence of nuclear expression in non-neoplastic cells, such as normal epithelial cells, lymphocytes, or stromal cells was considered to represent an abnormal pattern (MMR-D).
Classification of dMMR tumors
MLH1-hypermethylated sporadic tumors
Methylation analysis of the MLH1 promoter region was performed in the FFPE tumor specimens using real-time PCR-based, MethyLight, or bisulfite-Sanger sequencing; as previously described [12, 16–23]. Based on the Sanger sequencing results, samples were classified as hypermethylated, heterozygously methylated, or unmethylated [12]. In the present study, “heterogeneously methylated” tumors were classified as “unmethylated”. MethyLight analysis was used to determine the percentage of methylated reference (PMR). Based on validated data, a positive PMR cut-off of 10% was used. Therefore, samples were considered positive if the PMR was > 10%.
During CRC analysis [12], MLH1-methylation analysis was omitted in the tumor samples exhibiting BRAFV600E. However, subsequent MethyLight analysis confirmed that these tumors were methylation positive.
As a major principle, we did not perform LS genetic testing for patients with MLH1-methylated tumors. However, two patients with MLH1-methylated tumors underwent genetic testing: Patient 1, a 50-year-old man with CRC exhibiting loss of function of the MLH1/PMS2 protein dimer, with heterogeneously methylated molecular characteristics. A germline MLH1 pathogenic variant was confirmed, leading to an LS diagnosis. Patient 2, a 70-year-old man with SC exhibiting MLH1/PMS2 and MLH1-methylated results. Due to the lack of sufficient dMMR data and aberrant MLH1 methylation in the field of ST, genetic testing was performed to confirm the patient’s LS status [23].
Lynch syndrome-associated tumors
Following genetic counseling and informed consent, we conducted genetic testing: Sanger sequencing and multi-gene panel sequencing with/without RNA-sequencing using DNA from blood leukocytes where appropriate. If only FFPE tissue specimens were available, Sanger sequencing was performed to determine MMR genes, according to the IHC patterns of the MMR proteins. A multiplex ligation-dependent probe amplification (MLPA) method was used to analyze the copy number variation of the MLH1, MSH2, and EPCAM exons, using Salsa MLPA P003 MLH1/MSH2 probemix (MRC-Holland, Amsterdam, Netherlands) andP008 PMS2 for PMS2 (MRC-Holland). Patients with LS-associated class 4 (likely pathogenic) or class 5 (pathogenic) germline variants, based on the InSiGHT classification criteria, were diagnosed with LS.
LS diagnoses were not possible for deceased patients with isolated loss of MSH6 in the UUTC at the time of publication due to low quality urothelial tissue (normal) [20]. Thereafter, the genetic analysis of this proband, demonstrated the insertion of retrotransposon in the exon 5 of MSH6, which could be explained as a cause of LS [24]. Therefore, the patient was regarded as LS in the present study. Germline variants were categorized into class 3, according to the InSiGHT classification criteria, were regarded as variants of uncertain significance (VUS). Patients with VUS were not categorized into either LS or LLS.
LLS-associated tumors
Tumors without germline MMR pathogenic variants or somatic MLH1 hypermethylation were classified as LLS-associated tumors. Somatic variants and copy number variations were investigated using Sanger sequencing (performed using DNA extracted from the micro dissected tissue specimens) and the MLPA method as described previously [12, 16–23].
Whole exome sequencing for LLS cases
WES analysis was conducted at a private laboratory, Novogene Co., Ltd. via Chemical Dojin Co., Ltd., as follows: The Agilent SureSelect Human All Exon V6 (58 M) kit (Agilent Technologies Inc., Santa Clara, CA, USA) was used for DNA target enrichment, followed by sequencing with an Illumina HiSeq4000 sequencer (Illumina, Inc.) as described previously [25].
Results
Incidence of MMR-D tumors
The total MMR-D incidence among the study population was 5.9% (24/3609). The MMR-D incidence in descending order were as follows: ST (38.5%, 5/13), EC (17.2%, 68/395), GC (11.3, 58/513), SBC (6.7%, 2/30), CRC (4.9%, 61/1234), UUTC (2.4%, 4/164), UBC (1.5%, 9/618), PC (1.2%, 4/337), and OC (1.0%, 3/305) (Fig. 1).
Fig. 1.

Percentage of patients with MMR-D tumor by different organs. ST sebaceous tumor, OC ovarian cancer, CRC colorectal cancer, EC endometrial cancer, SBC small-bowel cancer, GC gastric cancer, PC prostatic cancer, UUTC upper urinary tract cancer, UBC urinary bladder cancer, MMR mismatch repair, MMR-D loss of expression of at least one MMR protein, MMR-P expression of all MMR proteins
Pattern of MMR-D by MMR-IHC
Figure 2 demonstrated the pattern of MMR-D, identified using MMR-IHC. Upon analyzing tumors collectively, the identified MMR-D pattern was predominantly loss of expression of the MSH2/MSH6 dimer (90.0%, 9/10) followed by isolated loss of MSH6 (10.0%, 1/10) in UUTC, PC, and SBC. In contrast, in GC, CRC, and EC, the MMR-D pattern was predominantly MLH1/PMS2 (86.6%, 162/187). The MMR-D pattern in UBC varied, demonstrating loss of MLH1/PMS2 (n = 3), MSH2/MSH6 (n = 1), isolated loss of MSH6 (n = 3), and isolated loss of PMS2 (n = 2).
Fig. 2.

Number and MMR-D pattern by cancer type. ST sebaceous tumor, OC ovarian cancer, CRC colorectal cancer, EC endometrial cancer, SBC small-bowel cancer, GC gastric cancer, PC prostatic cancer, UUTC upper urothelial cancer, UBC urinary bladder cancer, MMR-D loss of expression of at least one MMR protein
MLH1-methylated tumors
It can be seen in Fig. 3 that MLH1-hypermethylated tumors among MMR-D tumors were common in GC (94.8%, 55/58), CRC (82.0%, 50/61), and EC (75.0%,51/68). In contrast, MLH1-unmethylated tumors were common in UUTC (100%, 4/4), PC (100%, 4/4), SBC (100%, 2/2), and UBC (88.9%, 8/9).
Fig. 3.
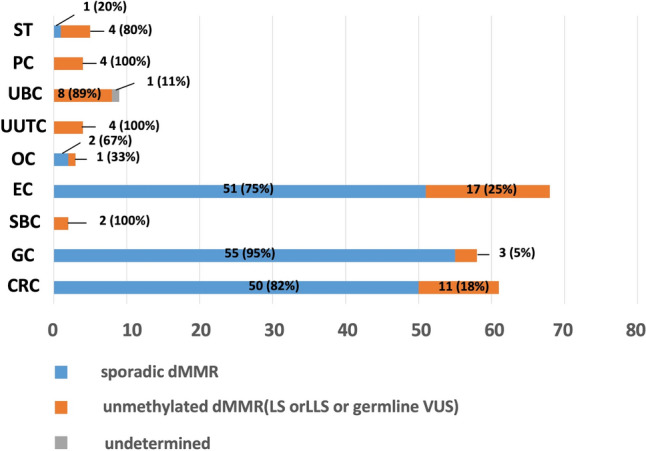
Number of cases by subtypes of MMR-D LS-related tumors. ST sebaceous tumor, OC ovarian cancer, CRC colorectal cancer, EC endometrial cancer, SBC small-bowel cancer, GC gastric cancer, PC prostatic cancer, UUTC upper urothelial cancer, UBC Urinary bladder cancer, dMMR deficient DNA mismatch repair, LS Lynch syndrome, LLS Lynch-like syndrome, VUS variants of uncertain significance
MLH1-unmethylated MMR-D tumors according to genetic testing
Genetic testing was conducted for 47 out of the 58 patients with MLH1-unmethylated dMMR tumors. The patients were divided into three categories: LS (n = 26, 55%), LLS (n = 18, 38%), and germline VUS (n = 3, 7%) (Fig. 4). Germline VUS was exclusively identified in EC (n = 2) and UBC (n = 1). The incidence of LS among the MLH1-unmethylated patients was highest in the UUTC (100%, 4/4), followed by CRC (82%, 9/11), and UBC (75%, 3/4). Meanwhile, the incidence of LLS was highest in PC (100%, 4/4) and OC (100%, 1/1), followed by ST (75%, 3/4).
Fig. 4.

Number of cases by classification of LS, LLS and germline VUS after genetic testing for Lynch syndrome. ST sebaceous tumor, OC ovarian cancer, CRC colorectal cancer, EC endometrial cancer, SBC small-bowel cancer, GC gastric cancer, PC prostatic cancer, UUTC upper urothelial cancer, UBC urinary bladder cancer, LS Lynch syndrome, LLS Lynch-like syndrome, VUS variants of uncertain significance
Summary of MMR-D tumors based on molecular characteristics
Table 1 summarizes the molecular characteristics of the MMR-D tumors. That is, proportion of MMR-D, pattern of loss of MMR protein expression, proportion of MLH1-methylated tumors among all MMR-D tumors, proportion of MLH1-unmethylated tumors among all MMR-D tumors, proportion of LS and LLS among all MLH1-unmthylated tumors. The proportion of germline VUS is shown for EC and UBC.
Table 1.
Summary of molecular characteristics in MMR-D tumors
| Type of tumor | Pattern of MMR-D | MLH1-methylated MMR-D among all MMR-D cases | MLH1-unmethylated MMR-D among all MMR-D cases | Type of MLH1-unmethylated MMR-D | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| MMR-D (%) | Loss of MLH1/PMS2 among all MMR-D cases | Loss of MSH2/MSH6 among all MMR-D cases | Loss of MSH6 among all MMR-D cases | Loss of PMS2 among all MMR-D cases | LS (A%, B%, C%) | LLS (A%, B%, C%) | Germline VUS (A%, B%, C%) | N.A (genetic testing not performed) (A%, B%, C%) | |||
| CRC | 4.9 (61/1234) | 52 (85.2) | 6 (9.8) | 3 (4.9) | 0 (0) | 50 (82.0) | 11 (18.0) | 9 (81.8%, 14.8%, 0.7%) | 2 (18.2%, 3.3%, 0.2%) | 0 | 0 |
| GC | 11.3 (58/513) | 57 (98.3)a | 0 (0) | 0 (0) | 1 (1.7) | 55 (94.8) | 3 (5.2) | 1 (33.3%, 1.7%, 0.2%) | 2 (66.7%, 3.4%, 0.4%) | 0 | 0 |
| SBC | 6.7 (2/30) | 0 (0) | 2 (100) | 0 (0) | 0 (0) | 0 (0) | 2 (100) | 1 (50.0%, 50.0%, 2.9%) | 1 (50.0%, 50.0%, 2.9%) | 0 | 0 |
| EC | 17.2 (68/395) | 53 (77.9) | 11 (16.2)b | 3 (4.4) | 1(1.5) | 51 (75.0) | 17 (25.0) | 5 (29.4%, 7.4%, 1.3%) | 7 (41.2%, 10.3%, 1.8%) | 2 (11.8%, 2.9%, 0.5%) | 3 (17.6%, 4.4%, 0.8%) |
| OC | 1.0 (3/305) | 2 (66.7) | 1 (33.3) | 0 (0) | 0 (0) | 2 (66.7) | 1 (33.3) | 0 | 1 (100%, 33.3%, 0.3%) | 0 | 0 |
| UUTC | 2.4 (4/164) | 0 (0) | 3 (75.0) | 1 (25.0) | 0 (0) | 0 (0) | 4 (100) | 4 (100%, 100%, 2.4%) | 0 | 0 | 0 |
| UBC | 1.5 (9/618) | 3 (33.3) | 1 (11.1) | 2 (22.2) | 3 (33.3) | 0 (0) | 8 (88.9) | 3 (37.5%, 33.3%, 0.5%) | 0 | 1 (12.5%, 11.1%, 0.2%) | 4 (50.0%, 44.4%, 0.6%) |
| PC | 1.2 (4/337) | 0 (0) | 4 (100) | 0 (0) | 0 (0) | 0 (0) | 4 (100) | 0 | 2 (50.0%, 50.0%, 0.6%) | 0 | 2 (50.0%, 50.0%, 0.6%) |
| ST | 38.5 (5/13) | 1 (20.0) | 4 (80.0) | 0 (0) | 0 (0) | 1 (20.0) | 4 (80.0) | 1 (25.0%, 20.0,% 7.7%) | 3 (75.0%, 60.0%, 23.1%) | 0 | 0 |
| Total | 5.9 (214/3395) | ||||||||||
A%: Proportion among MLH1-unmethylated cases
B%:Proportion among MMR-D cases
C%: Proportion among all cases
ST sebaceous tumor, OC ovarian cancer, CRC colorectal cancer, EC endometrial cancer, SBC small-bowel cancer, GC gastric cancer, PC prostatic cancer, UUTC upper urinary tract cancer, UBC urinary bladder cancer, MMR-D loss of expression of at least one MMR protein
aLoss of MLH1/PMS2/MSH6 was categorized as loss of MLH1/PMS2
bLoss of isolated MSH2 were categorized as loss of MSH2/MSH6
Characteristics of LLS and WES
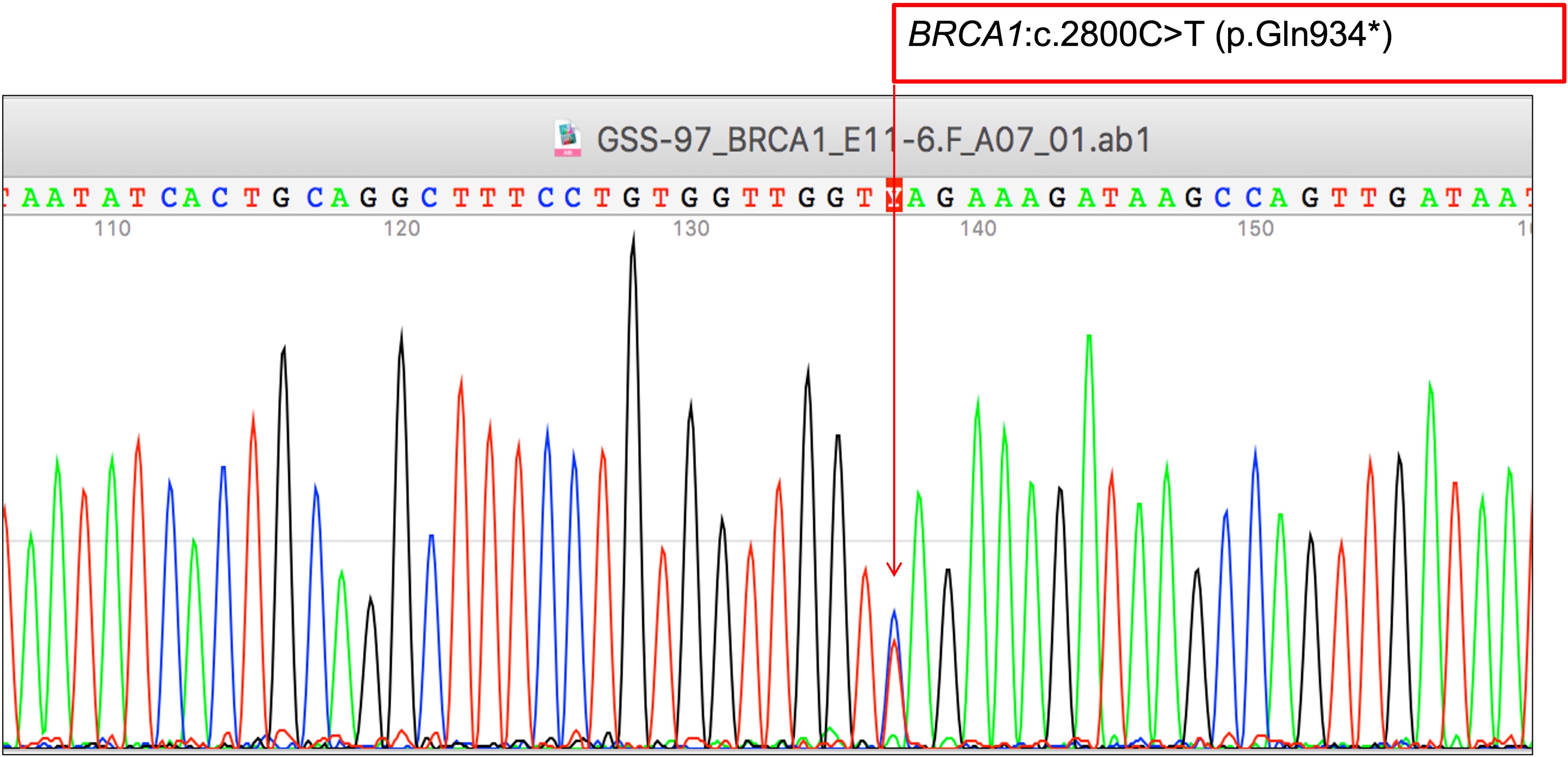
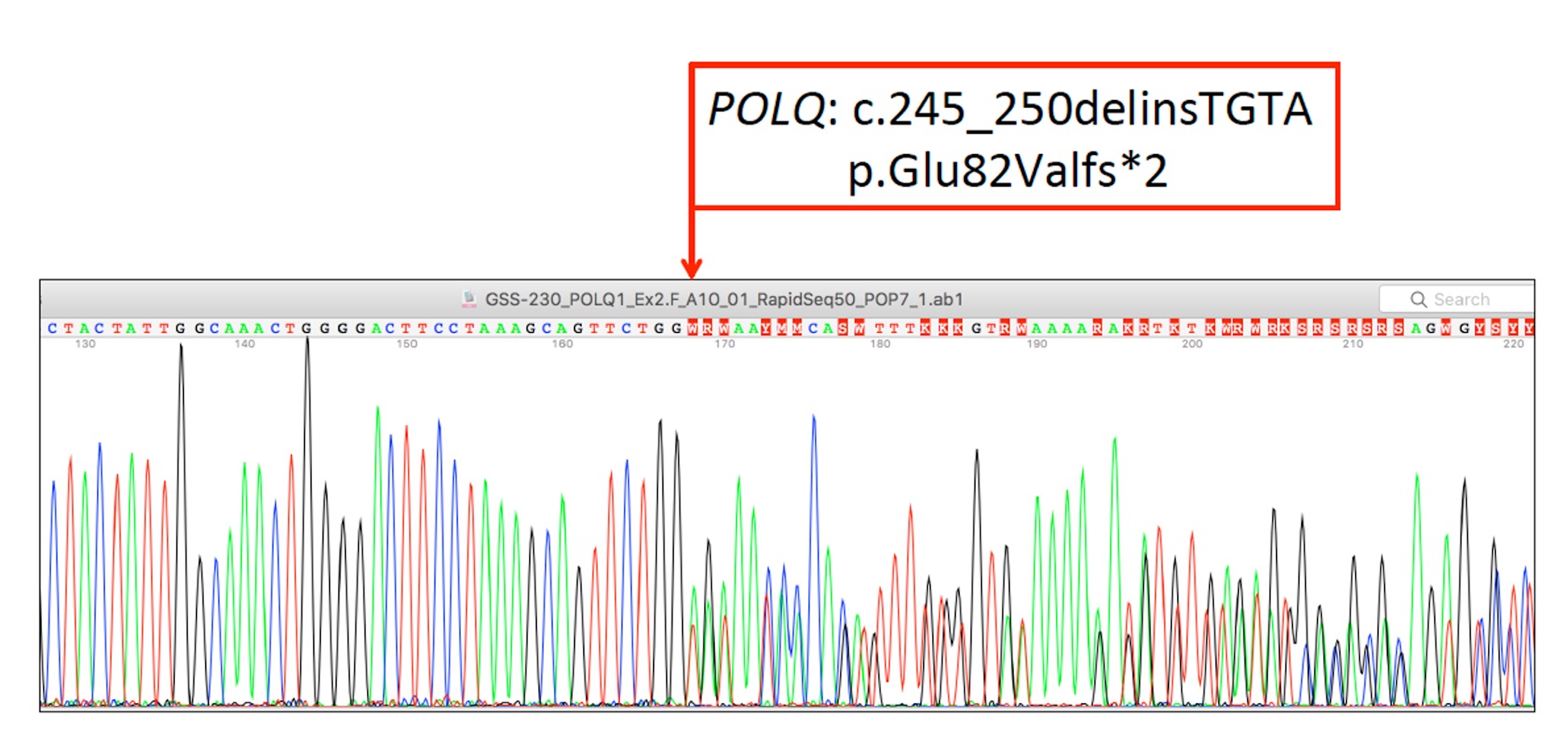
The characteristics of 18 patients with LLS are demonstrated in Table 2. Age at diagnosis of the index tumor ranged from 42 to 82 years (median, 66 years) and the male to female ratio was 8:10. CRC was confirmed, from medical history, in two patients with LS-associated tumors. No patients fulfilled the revised Amsterdam criteria [26]. Somatically inactivated MMR variants were successfully conducted in 16 patient samples. In 15 samples, one to two Class 4/Class 5 MMR gene variants were detected. In the remaining sample, two Class 3 variant was detected. Whole exome sequence was performed for 12 patient samples. Two patients demonstrated non-MMR germline pathogenic variants. A 63-year-old man (Case 3, III-4) (Fig. 5) with sebaceous cancer (MSH2/MSH6) was found to carry a BRCA1(NM_007294.4:c.2800C>T) pathogenic nonsense variant (supplementary Fig. 1), and a 57-year-old man (Case 14, III-6) (Fig. 6) with small-bowel cancer (MSH2/MSH6 loss) was found to carry a POLQ variant (NM_199420: c.245_250delinsTGTA) which probably affects function (supplementary Fig. 2). Multiple sebaceous tumors (MSH2/MSH6 loss) were identified in Case 3 and genetic analysis was performed, as previously reported [23], and an additional sebaceous tumor developed at the age of 57. Case 14: The older male sibling (III-5, Fig. 6) underwent respective surgery for UUTC at the age of 45 and died of the disease at the age of 50. The father of the siblings (II-3, Fig. 6) suffered a CRC-related mortality at the age of 42. The UUTC of the III-5 demonstrated loss of MSH2/MSH6 (supplementary Fig. 3). In addition, Sanger sequencing detected the POLQ variant, as identified in Case 14 (supplementary Fig. 4).
Table 2.
Summary of patients with LLS

ST sebaceous tumor, OV ovarian cancer, CC colon cancer, EC endometrial cancer, SBS small-bowel cancer, GC gastric cancer, PC prostatic cancer, LLS Lynch-like syndrome
Fig. 5.

The pedigree of case 3. PV pathogenic variant
Fig. 6.

The pedigree of case 14. PV pathogenic variant
Discussion
We provided data on the incidence of dMMR tumors, assessed by MMR-IHC, among resected specimens of nine different tumor types. CRC, GC, SBC, EC, OC, UUTC, and ST are known to be LS-associated tumors, as listed in the revised Bethesda guidelines [27]. It remains controversial whether PC and UBC are associated with LS; however, and increased risk of PC and UBC in individuals with MSH2 germline pathogenic variants has been reported [28, 29]. MMR-IHC allows for the identification of genes related to the alteration in dMMR function. Therefore, using MMR-IHC and MLH1-methylation analysis, we efficiently selected patients with dMMR tumors who were candidates for LS genetic testing. It is useful to determine the ratio of LS to LLS among LS suspected patients before genetic testing is performed.
To the best of our knowledge, this is the first comprehensive study that demonstrated the proportion of molecular-genetics-based categorized dMMR tumors across various tumors. The ratio of LS to LLS was 4:3. This suggests that approximately 50% of patients with a MLH1-unmethylated dMMR tumor would not have LS. Studies, with a specific focus on CRC and /or EC, demonstrated an LS to LLS ratio at 9:30 [9, 30]. Rodrigez et al. [9] analyzed 1705 CRC cases and reported an LS to LLs of 16:43. This ratio is lower than that (9:2) reported by Chika et al. [12]. It should be noted that the ratio can be influenced by the method and quality of LS genetic testing.
The term “LLS” is used for patients with dMMR tumors (specifically CRC and EC) with no MLH1, MSH2, MSH6, PMS2, or EPCAM deletion variants or MLH1 somatic methylation. At least three molecular genetic possibilities have been considered to explain LLS-AT: cryptic/undetected germline pathogenic variants such as non-cording regions, inversions, or translocations of MMR genes that are not routinely detectable by current genetic testing, (occult/undetected LS) [31]; biallelic somatic variants in MMR genes of unknown etiology [32]; and germline pathogenic variants in non-MMR genes (heritable predisposition) [33]. The latter two may overlap in some patients.
The underlying germline variants of genes involved in LLS have been poorly explored. Some LLS-based studies [34, 35] reported the presence of biallelic germline variants in MUTYH. The MUTYH-associated polyposis can overlap with the LS phenotype, by somatic inactivation of MMR genes [34]. LLS patients carrying POLE and POLD1 germline variants have also been identified [36, 37]. Whole exome sequencing identified germline pathogenic/likely pathogenic variants in DNA repair genes, such as MCM8, MCM9, WRN, MCPH1, BARD1, REV3L, EXO1, POLD1, RFC1, RPA1, ATM, and MLH3. In addition, other cancer-related genes, such as PPARG, CTC1, DCC and ALPK, were identified as candidate genes for LLS [13, 37–39].
In the present study, Case 3 carried a BRCA1 (c.2800C>T; p.Q934X) pathogenic variant, which was reported as a founder mutation in the Japanese population [40]. It is well-known that BRCA1/2 is involved in homologous recombination (HR), which is an error-free repair mechanism for DNA double-strand breaks (DSBs) [31]. Colorectal cancer (CRC) with HR deficiency (HRD), developed via biallelic somatic variants of HR-related genes, including BRCA1/2, were more frequent in MSI-H/dMMR than in MMS/pMMR, suggesting a significant association between alterations in the HRD pathway and dMMR [41, 42]. Although BRCA1/2 variants might affect MSI status, there is no evidence to the support that BRCA1/2 germline variants induce MMR somatic variants. It is possible that HRD cells rely on more error-prone alternative DNA damage response pathways, such as nonhomologous end joining, to repair DNA breaks and avoid mitotic catastrophe and cell death. Thus, increasing mutagenesis and genomic instability, inactivating BRCA and other factors to induce MMR variants. A patient suffered from a ST with loss of MSH2/MSH6 proteins, caused by two somatic pathogenic mutations, that is, a nonsense variant (c.2038C>T/p.Arg680*) and a deletion of exons 14–15 in MSH2 [23]. Subsequently, four primary STs were resected, but none with confirmed MSH2 [23]. A previous report [43] suggested that somatic inactivation of the fragile histidine triad (FHIT) gene associated with MSS or inactivation of the MMR system resulting in MSI contributes to the development of periocular sebaceous gland carcinomas in presumptive Muir-Torre syndrome. Moreover, Becker et al. [44] reported that somatic BRCA1 deleterious mutations were associated with candidate tumor suppressor FHIT inactivation in sebaceous gland carcinomas with MSS. Therefore, there may be two possible mechanisms for ST pathogenesis. ST develops either on the background of a dMMR system with MSI via inactivating variants preferentially in MSH2 or on the basis of HRD via deletions of BRCA1 associated with MSS. Based on these pathways, the first tumor may have developed by the former mechanism and the remaining three tumors by the latter mechanism, though the FHIT inactivation was not examined.
We identified the heterozygous POLQ germline frameshift variant in a LLS patient (Case 14). POLQ is involved in the repair of DSBs. Most DSBs are repaired by two pathways: one is the canonical nonhomologous end joining (c-NHEJ), which directly relegates DNA ends without extensive processing; the other is the HR pathway, which is the only precise DSB repair pathway. POLQ was found to be s involved in a third pathway termed polymerase theta-mediated end joining (TMEJ) [45]. Whether POLQ suppresses or promotes genomic instability still remains unknown. TMEJ repair is error prone and is known to generate genomic translocations. Raskin et al. [46] reported that POLQ germline pathogenic variants were detected in each LLS patient and sporadic colorectal cancer patient with MSS. Belhadj et al. [47] identified seven POLQ germline pathogenic variants in patients with suspected LS along with diagnosis based on AC I/II (n = 58) and BG (n = 385) criteria. Since the POLQ variants were not found to be significantly enriched in the study population, lack of association with CRC predisposition was suggested. POLQ has multiple functions, which varies based on the coordinating proteins. This suggests that POLQ may be one of the causative factors for LLS; however, it may not be directly related to the development of LLS.
The limited data on germline alterations in patients with LLS suggest that hereditary factors should not be excluded; however, further investigations are required. In addition, patients suspected to have LLS should be offered genetic counseling that discusses updated germline variant data, which includes non-MMR genes.
The present study has several limitations. First, the data was obtained from a single institution, analyzed retrospectively with a small sample size for each tumor type. Second, data of clinicopathological and personal/family histories regarding the development of malignant neoplasms could not be compared between patients with LS and LLS, especially due to the lack of data in patients with LLS. Furthermore, LS-associated tumors, such as brain, bile duct, and pancreatic tumors could not be analyzed.
Nevertheless, this is the first report that comprehensively documents the characteristics of various dMMR tumors. Notably, our data regarding the LS to LLS ratio would be useful for genetic counseling in patients who are suspected to have LS. Genetic mechanisms for the pathogenesis of LLS needs further investigation.
Supplementary Information
Below is the link to the electronic supplementary material.
{kind=link}
Supplementary file2 Supplementary Figure 1 Confirmation of BRCA1:c.2800C>T (p.Gln934*) in case 3 (JPG 1686 KB)
{kind=link}
Supplementary file3 Supplementary Figure 2 Confirmation of POLQ: c.245_250delinsTGTA (p.Glu82Valfs*2) in case 14 (JPG 1699 KB)
{kind=link}
Supplementary file4 Supplementary Figure 3 Immunohistochemistry for MMR protein in UUTC of the III-5. Loss of expression of MSH2 and MSH6 proteins was observed (JPG 5336 KB)
{kind=link}
Supplementary file5 Supplementary Figure 4 Confirmation of POLQ: c.245_250delinsTGTA (p.Glu82Valfs*2) in the III-5 (JPG 1119 KB)
Acknowledgements
We thank Ms. Aya Saitoh, Fumiyo Fukui, and Akemi Takahashi for their skillful technical assistance.
Funding
Open Access funding provided by Saitama Medical University. This work was supported in part by a grant-in-aid for the Support Project of the Strategic Research Center in Private Universities from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan awarded to the Saitama Medical University Research Center for Genomic Medicine [S1311002] and AMED [grant number JP18kk0205004].
Data availability
The datasets during and/or analyzed during the current study available from the corresponding author on reasonable request.
Declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–413. doi: 10.1126/science.aan6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maio M, Ascierto PA, Manzyuk L, et al. Pembrolizumab in microsatellite instability high or mismatch repair deficient cancers: updated analysis from the phase II KEYNOTE-158 study. Ann Oncol. 2022;33:929–938. doi: 10.1016/j.annonc.2022.05.519. [DOI] [PubMed] [Google Scholar]
- 3.Bartley AN, Luthra R, Saraiya DS, et al. Identification of cancer patients with Lynch syndrome: clinically significant discordances and problems in tissue-based mismatch repair testing. Cancer Prev Res. 2012;5:320–327. doi: 10.1158/1940-6207.CAPR-11-0288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bruegl AS, Ring KL, Daniels M, et al. Clinical challenges associated with universal screening for Lynch syndrome-associated endometrial cancer. Cancer Prev Res (Phila) 2017;10:108–115. doi: 10.1158/1940-6207.CAPR-16-0219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Herman JG, Umar A, Polyak K, et al. Incidence and functional consequences of hMLH1 promoter hypermethylation in colorectal carcinoma. Proc Natl Acad Sci USA. 1998;95:6870–6875. doi: 10.1073/pnas.95.12.6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Esteller M, Levine R, Baylin SB, et al. MLH1 promoter hypermethylation is associated with the microsatellite instability phenotype in sporadic endometrial carcinomas. Oncogene. 1998;17:2413–2417. doi: 10.1038/sj.onc.1202178. [DOI] [PubMed] [Google Scholar]
- 7.Eguchi H, Kumamoto K, Suzuki O, et al. Identification of a Japanese lynch syndrome patient with large deletion in the 3 region of the EPCAM gene. Jpn J Clin Oncol. 2016;46:178–184. doi: 10.1093/jjco/hyv172. [DOI] [PubMed] [Google Scholar]
- 8.Yamaguchi T, Iijima T, Mori T, et al. Accumulation profile of frameshift mutations during development and progression of colorectal cancer from patients with hereditary nonpolyposis colorectal cancer. Dis Colon Rectum. 2006;49:399–406. doi: 10.1007/s10350-005-0293-4. [DOI] [PubMed] [Google Scholar]
- 9.Rodríguez-Soler M, Pérez-Carbonell L, Guarinos C, et al. Risk of cancer in cases of suspected lynch syndrome without germline mutation. Gastroenterology. 2013;144:926–932. doi: 10.1053/j.gastro.2013.01.044. [DOI] [PubMed] [Google Scholar]
- 10.Kang SY, Park CK, Chang DK, et al. Lynch-like syndrome: Characterization and comparison with EPCAM deletion carriers. Int J Cancer. 2015;136:1568–1578. doi: 10.1002/ijc.29133. [DOI] [PubMed] [Google Scholar]
- 11.Mensenkamp AR, Vogelaar IP, van Zelst-Stams WA, et al. Somatic mutations in MLH1 and MSH2 are a frequent cause of mismatch-repair deficiency in Lynch syndrome-like tumors. Gastroenterology. 2014;146:643–646. doi: 10.1053/j.gastro.2013.12.002. [DOI] [PubMed] [Google Scholar]
- 12.Chika N, Eguchi H, Kumamoto K, et al. Prevalence of Lynch syndrome and Lynch-like syndrome among patients with colorectal cancer in a Japanese hospital-based population. Jpn J Clin Oncol. 2017;47:108–117. doi: 10.1093/jjco/hyw178. [DOI] [PubMed] [Google Scholar]
- 13.Golubicki M, Bonjoch L, Acuña-Ochoa JG, et al. Germline biallelic Mcm8 variants are associated with early-onset Lynch-like syndrome. JCI Insight. 2020;5:e140698. doi: 10.1172/jci.insight.140698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vanderwalde A, Spetzler D, Xiao N, et al. Microsatellite instability status determined by next-generation sequencing and compared with PD-L1 and tumor mutational burden in 11,348 patients. Cancer Med. 2018;7:746–756. doi: 10.1002/cam4.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hause RJ, Pritchard CC, Shendure J, et al. Classification and characterization of microsatellite instability across 18 cancer types. Nat Med. 2016;22:1342–1350. doi: 10.1038/nm.4191. [DOI] [PubMed] [Google Scholar]
- 16.Ito T, Suzuki O, Kamae N, et al. Comprehensive analysis of DNA mismatch repair-deficient gastric cancer in a Japanese hospital-based population. Jpn J Clin Oncol. 2021;51:886–894. doi: 10.1093/jjco/hyab026. [DOI] [PubMed] [Google Scholar]
- 17.Ito T, Ishida H, Suzuki O, et al. Prevalence and molecular characterization of defective DNA mismatch repair in small-bowel carcinoma in a japanese hospital-based population. J Anus Rectum Colon. 2020;4:165–173. doi: 10.23922/jarc.2020-026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamamoto A, Yamaguchi T, Suzuki O, et al. Prevalence and molecular characteristics of DNA mismatch repair deficient endometrial cancer in a Japanese hospital-based population. Jpn J Clin Oncol. 2021;51:60–69. doi: 10.1093/jjco/hyaa142. [DOI] [PubMed] [Google Scholar]
- 19.Tajima Y, Eguchi H, Chika N, et al. Prevalence and molecular characteristics of defective mismatch repair epithelial ovarian cancer in a Japanese hospital-based population. Jpn J Clin Oncol. 2018;48:728–735. doi: 10.1093/jjco/hyy081. [DOI] [PubMed] [Google Scholar]
- 20.Ito T, Kono K, Eguchi H, et al. Prevalence of Lynch syndrome among patients with upper urinary tract carcinoma in a Japanese hospital-based population. Jpn J Clin Oncol. 2020;50:80–88. doi: 10.1093/jjco/hyz140. [DOI] [PubMed] [Google Scholar]
- 21.Kagawa M, Kawakami S, Yamamoto A, et al. Identification of Lynch syndrome-associated DNA mismatch repair-deficient bladder cancer in a Japanese hospital-based population. Int J Clin Oncol. 2021;26:1524–1532. doi: 10.1007/s10147-021-01922-y. [DOI] [PubMed] [Google Scholar]
- 22.Kagawa M, Kawakami S, Yamamoto A, et al. Prevalence and clinicopathological/molecular characteristics of mismatch repair protein-deficient tumours among surgically treated patients with prostate cancer in a Japanese hospital-based population. Jpn J Clin Oncol. 2021;51:639–645. doi: 10.1093/jjco/hyaa207. [DOI] [PubMed] [Google Scholar]
- 23.Kuwabara K, Suzuki O, Chika N, et al. Prevalence and molecular characteristics of DNA mismatch repair protein-deficient sebaceous neoplasms and keratoacanthomas in a Japanese hospital-based population. Jpn J Clin Oncol. 2018;48:514–521. doi: 10.1093/jjco/hyy055. [DOI] [PubMed] [Google Scholar]
- 24.Yamamoto G, Miyabe I, Tanaka K, et al. SVA retrotransposon insertion in exon of MMR genes results in aberrant RNA splicing and causes Lynch syndrome. Eur J Hum Genet. 2021;29:680–686. doi: 10.1038/s41431-020-00779-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tanakaya K, Kumamoto K, Tada Y, et al. A germline MBD4 mutation was identified in a patient with colorectal oligopolyposis and early-onset cancer: a case report. Oncol Rep. 2019;42:1133–1140. doi: 10.3892/or.2019.7239. [DOI] [PubMed] [Google Scholar]
- 26.Vasen HF. Clinical diagnosis and management of hereditary colorectal cancer syndromes. J Clin Oncol. 2000;18:81s–92s. [PubMed] [Google Scholar]
- 27.Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96:261–268. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Der Post RS, Kiemeney LA, Ligtenberg MJL, et al. Risk of urothelial bladder cancer in Lynch syndrome is increased, in particular among MSH2 mutation carriers. J Med Genet. 2010;47:464–470. doi: 10.1136/jmg.2010.076992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Engel C, Loeffler M, Steinke V, et al. Risks of less common cancers in proven mutation carriers with lynch syndrome. J Clin Oncol. 2017;30:4409–4415. doi: 10.1200/JCO.2012.43.2278. [DOI] [PubMed] [Google Scholar]
- 30.Mills AM, Sloan EA, Thomas M, et al. Clinicopathologic comparison of Lynch syndrome–associated and "Lynch-like" endometrial carcinomas identified on universal screening using mismatch repair protein immunohistochemistry. Am J Surg Pathol. 2016;40:155–165. doi: 10.1097/PAS.0000000000000544. [DOI] [PubMed] [Google Scholar]
- 31.Welcsh PL, Owens KN, King MC. Insights into the functions of BRCA1 and BRCA2. Trends Genet. 2000;16:69–74. doi: 10.1016/s0168-9525(99)01930-7. [DOI] [PubMed] [Google Scholar]
- 32.Rahman N, Stratton MR. The genetics of breast cancer susceptibility. Annu Rev Genet. 1998;32:95–121. doi: 10.1146/annurev.genet.32.1.95. [DOI] [PubMed] [Google Scholar]
- 33.Yurgelun MB, Allen B, Kaldate RR, et al. Identification of a variety of mutations in cancer predisposition genes in patients with suspected Lynch syndrome. Gastroenterology. 2015;149:604–613.e20. doi: 10.1053/j.gastro.2015.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Castillejo A, Vargas G, Castillejo MI, et al. Prevalence of germline MUTYH mutations among Lynch-like syndrome patients. Eur J Cancer. 2014;50:2241–2250. doi: 10.1016/j.ejca.2014.05.022. [DOI] [PubMed] [Google Scholar]
- 35.Morak M, Heidenreich B, Keller G, et al. Biallelic MUTYH mutations can mimic Lynch syndrome. Eur J Hum Genet. 2014;22:1334–1337. doi: 10.1038/ejhg.2014.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jansen AM, van Wezel T, van den Akker BE, et al. Combined mismatch repair and POLE/POLD1 defects explain unresolved suspected Lynch syndrome cancers. Eur J Hum Genet. 2016;24:1089–1092. doi: 10.1038/ejhg.2015.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xavier A, Olsen MF, Lavik LA, et al. Comprehensive mismatch repair gene panel identifies variants in patients with Lynch-like syndrome. Mol Genet Genomic Med. 2019;7:e850. doi: 10.1002/mgg3.850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xicola RM, Clark JR, Carroll T, et al. Implication of DNA repair genes in Lynch-like syndrome. Fam Cancer. 2019;18:331–342. doi: 10.1007/s10689-019-00128-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dos Santos W, de Andrade ES, Garcia FAO, et al. Whole-exome sequencing identifies pathogenic germline variants in patients with Lynch-like syndrome. Cancers (Basel) 2022;14:4233. doi: 10.3390/cancers14174233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sekine M, Nagata H, Tsuji S, et al. Japanese Familial Ovarian Cancer Study Group. Mutational analysis of BRCA1 and BRCA2 and clinicopathologic analysis of ovarian cancer in 82 ovarian cancer families: two common founder mutations of BRCA1 in Japanese population. Clin Cancer Res. 2001;7:3144–3150. [PubMed] [Google Scholar]
- 41.Moretto R, Elliott A, Zhang J, et al. Homologous recombination deficiency alterations in colorectal cancer: clinical, molecular, and prognostic implications. J Natl Cancer Inst. 2022;114:271–279. doi: 10.1093/jnci/djab169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arai H, Elliott A, Xiu J, et al. The landscape of alterations in DNA damage response pathways in colorectal cancer. Clin Cancer Res. 2021;27:3234–3242. doi: 10.1158/1078-0432.CCR-20-3635. [DOI] [PubMed] [Google Scholar]
- 43.Goldberg M, Rummelt C, Foja S, et al. Different genetic pathways in the development of periocular sebaceous gland carcinomas in presumptive Muir-Torre syndrome patients. Hum Mutat. 2006;27:155–162. doi: 10.1002/humu.20281. [DOI] [PubMed] [Google Scholar]
- 44.Becker K, Goldberg M, Helmbold P, et al. Deletions of BRCA1/2 and p53 R248W gain-of-function mutation suggest impaired homologous recombination repair in fragile histidine triad-negative sebaceous gland carcinomas. Br J Dermatol. 2008;159:1282–1289. doi: 10.1111/j.1365-2133.2008.08783.x. [DOI] [PubMed] [Google Scholar]
- 45.Schrempf A, Slyskova J, Loizou JI. Targeting the DNA repair enzyme polymerase θ in cancer therapy. Trends Cancer. 2021;7:98–111. doi: 10.1016/j.trecan.2020.09.007. [DOI] [PubMed] [Google Scholar]
- 46.Raskin L, Guo Y, Du L, et al. Targeted sequencing of established and candidate colorectal cancer genes in the Colon Cancer Family Registry Cohort. Oncotarget. 2017;8:93450–93463. doi: 10.18632/oncotarget.18596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Belhadj S, Terradas M, Munoz-Torres PM, et al. Candidate genes for hereditary colorectal cancer: mutational screening and systematic review. Hum Mutat. 2020;41:1563–1576. doi: 10.1002/humu.24057. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary file2 Supplementary Figure 1 Confirmation of BRCA1:c.2800C>T (p.Gln934*) in case 3 (JPG 1686 KB)
Supplementary file3 Supplementary Figure 2 Confirmation of POLQ: c.245_250delinsTGTA (p.Glu82Valfs*2) in case 14 (JPG 1699 KB)
Supplementary file4 Supplementary Figure 3 Immunohistochemistry for MMR protein in UUTC of the III-5. Loss of expression of MSH2 and MSH6 proteins was observed (JPG 5336 KB)
Supplementary file5 Supplementary Figure 4 Confirmation of POLQ: c.245_250delinsTGTA (p.Glu82Valfs*2) in the III-5 (JPG 1119 KB)
Data Availability Statement
The datasets during and/or analyzed during the current study available from the corresponding author on reasonable request.