

Key Clinical Message
This case report presents a progressively declining 17‐year‐old patient with membrane protein‐associated neurodegeneration who demonstrated symptomatic improvements in her dysarthria, dysphagia, and gait, and objective improvements in her 6‐minute walk test and 5 times sit‐to‐stand test during elamipretide treatment.
Keywords: case report, elamipretide, MPAN, NBIA
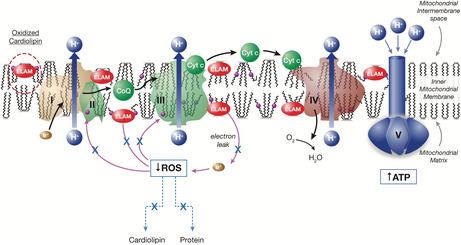
Elamipretide mechanism of action: Restoration of physiologically normal energetics.
1. INTRODUCTION
Neurodegeneration with brain iron accumulation (NBIA) is a group of rare, inherited disorders in which iron accumulates in the basal ganglia. 1 Membrane protein‐associated neurodegeneration (MPAN) is a type of NBIA (NBIA‐4) caused by pathogenic variants in the C19orf12 gene. 2 The age of onset in MPAN patients may vary from childhood to adulthood, and the disorder is most commonly characterized by progressive spastic paresis, gait changes, neuropsychiatric abnormalities, progressive dystonia, and cognitive decline. 1 Other symptoms include optic atrophy, dysphagia, dysarthria, bladder incontinence, axonal neuropathy, and parkinsonism. 1
Currently, there are no curative therapies for the treatment of patients with MPAN and management consists of supportive care with limited clinical impact. The US Food and Drug Administration (FDA) has provided guidance on expanded access for the use of investigational drugs for patients with serious diseases where a therapeutic alternative is lacking and there is a biologic plausibility to predict efficacy. 3 Elamipretide is an investigational agent in development for the treatment of rare mitochondrial and neuromuscular diseases, such as primary mitochondrial myopathy (PMM) 4 and Duchenne muscular dystrophy. 5 This mitochondria‐targeting peptide binds to cardiolipin, resulting in improved membrane stability, enhanced adenosine triphosphate (ATP) production, and reduced production of pathogenic reactive oxygen species (ROS) (Figure 1). 5 , 6 , 7 In this case report, we present the use of elamipretide under an expanded access program (EAP) in a patient with MPAN, with the goal of providing support for this agent's therapeutic role in patients with rare neuromuscular disorders, such as MPAN.
FIGURE 1.
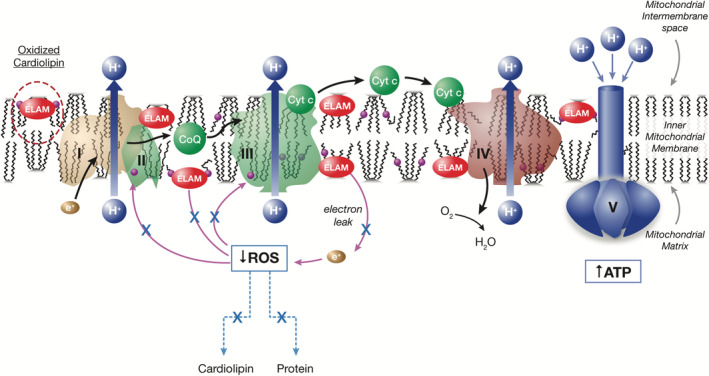
Elamipretide mechanism of action: Restoration of physiologically normal energetics. ATP, adenosine triphosphate; CoQ, coenzyme Q10; Cyt c, cytochrome c; ELAM, elamipretide; ROS, reactive oxygen species.
2. CASE HISTORY/EXAMINATION
A female patient with a past medical history of optic nerve atrophy first presented to our neurology department at 17 years of age for evaluation due to a progressive 4‐year history of difficulty rising from the floor and multiple falls. According to her parents, she appropriately met her early developmental milestones, including walking by 13 months of age, but was described as clumsy throughout her childhood. At 12 years of age, she developed gait imbalance resulting in multiple falls. At approximately 13 years of age, the patient complained of blurry vision, and there was concern for optic atrophy. However, magnetic resonance imaging demonstrated normal optic nerves and brain architecture. At this point, her neurologic exam was positive for pale optic disks on fundoscopy, mild dysarthria, slowed cognition, dystonic posturing of her ankles, strength 4/5 in all extremities proximally and distally, hyperreflexia, positive Hoffman and Babinski signs bilaterally, and unsteady gait with a positive Trendelenburg sign. Labs were remarkable for mildly elevated creatine kinase (299 U/L, reference range 12–191 U/L), while lactic acid (1.5 mmol/L, reference range 0.5–2.2 mmol/L) and aldolase (6.4 U/L, reference range 1.2–7.6 U/L) were within normal limits. Her electromyography (EMG)/nerve conduction study showed fibrillations and positive sharp waves, suggesting an axonal motor neuropathy.
3. METHODS
Given the presence of weakness, blurry vision, and dystonia, a whole exome sequencing was pursued, demonstrating a de novo pathogenic variant in the C19orf12 gene (c.256C>G, p.Q86X) located in exon 3. This gene has been shown to produce autosomal dominant MPAN. Daily CoQ10 supplementation was initiated, and the patient was enrolled in physical therapy. Into her teenage years, the patient could exercise regularly, alternating between bike riding and weightlifting, but she started to experience worsening gait, stiffness, spasticity, multiple falls, and mild dysphagia.
Due to her continued decline, the patient was enrolled into the intermediate‐size EAP (SPIES‐006: Elamipretide for Subcutaneous [SC] Injection in Patients with Genetically Confirmed Rare Diseases with Known Mitochondrial Dysfunction; Stealth BioTherapeutics), initiating elamipretide therapy at 21 years of age. Prior to beginning treatment with elamipretide, patient consent was obtained and drug administration was approved by the FDA and local institutional review board (IRB). Elamipretide was initiated at a dosage of 40 mg (0.5 mL) SC daily.
Objective changes during elamipretide treatment were measured using the 6‐minute walk test (6MWT) and 5 times sit to stand test (5×SST) because of their accuracy, reproducibility, and ease of administration. The 6MWT is an established functional outcome measure originally developed as an integrated global assessment of cardiac, respiratory, circulatory, and muscular capacity in a wide range of diseases. 8 The 6MWT has been used to evaluate functional capacity in neuromuscular diseases and measures of 6MWT distance have been proposed to be an indicator of quality of life. 9 The 5×SST, on the other hand, is a functional performance measure of leg strength or the force‐generating capacity of muscle which uses the body's weight for resistance during functional activities. 10 A cursory review of the literature indicates that both tests have been used frequently as endpoints to determine functional improvement in patients and thus are appropriate tools to monitor progress during elamipretide treatment in this case. 11 , 12 , 13
4. RESULTS
After 3 months of treatment, improvements were noted in the patient's dysarthria, dysphagia, gait, and ability to complete her activities of daily living. Remarkably, the patient was even able to walk backward without assistance. Her 6MWT distance improved by nearly 10 m and she no longer needed to steady herself using walls during the test. Her 5×SST improved by 0.5 s, but the patient remained unable to complete the test without using her arms to assist herself out of the chair. Her score on the EQ‐5D‐5L Health Questionnaire improved from 72 to 107 in the 3 months on elamipretide.
5. DISCUSSION
This case report describes the expanded‐access use of elamipretide in a patient with MPAN and provides insight into its potential therapeutic benefit in this disorder. MPAN, the fourth most common subtype of NBIA, 1 is caused by pathogenic variants in C19orf12, which encodes for a small transmembrane protein found in high concentrations in mitochondrial membranes and the endoplasmic reticulum. 14 While the exact function of C19orf12 is unknown, variants in this gene are suspected to be associated with impaired mitochondrial respiration; reduced mitochondrial length and cellular ATP; and increased ROS, iron, and lipid peroxidation. 15 Initially, MPAN was thought to be an autosomal recessive condition exclusively 2 ; however, heterozygous pathogenic variants have since been published and a dominant‐negative effect leading to loss of function has been proposed. 16 , 17
Regardless of the inheritance pattern, most patients with MPAN are clinically similar. Our patient demonstrated a clinical presentation consistent with MPAN, as Hartig et al. described the most common signs and symptoms of 67 patients with MPAN to be upper motor neuron signs (spastic paresis and pyramidal signs; 87.9%), cognitive decline (85.9%), dysarthria (82.3%), optic atrophy (75.0%), dystonia (66.7%), psychiatric abnormalities (64.3%), lower motor neuron signs (muscle atrophy, fasciculations, and neurogenic EMG changes; 56.3%), dysphagia (55.6%), and parkinsonism (44.6%). 18 Of note, our patient carries the same pathogenic variant as Subject 227 in Gregory et al. 16 and both patients became symptomatic at the same age and demonstrated optic atrophy as well as progressive gait difficulties.
There are no curative or specific treatment options for MPAN, and supportive care is of limited clinical impact. Supportive care for MPAN patients includes pharmacologic treatment of spasticity, dystonia, and parkinsonism (e.g., trihexyphenidyl, baclofen, and intramuscular botulinum toxin injections); psychiatric treatment of neuropsychiatric manifestations; occupational, physical, and speech therapies; nutritional supplements and gastric tube feeding; and management of secretions and aspiration risk (e.g., scopolamine patch, glycopyrrolate, and/or tracheostomy). 1 The therapeutic effect of iron chelation with deferiprone has not been proven in MPAN patients, showing mixed results in the few published case reports. 19 , 20
Elamipretide is a mitochondria‐targeting agent that has offered promise in patients with degenerative neuromuscular disorders, such as PMM. 4 Through its association with cardiolipin, elamipretide improves mitochondrial function through improvement in ATP production and potential reversal of damaging oxidative stress, 6 , 7 issues which may be associated with pathogenic variants in C19orf12. 15 Our case is the first to use elamipretide in the treatment of a patient with MPAN. While our patient has only been on elamipretide for a short period of time, the patient has shown remarkable improvement in several symptoms after years of progressive deterioration. This case also highlights the importance of EAPs, as this patient was not eligible for available clinical trials. The EAP for elamipretide serves an important role in bringing a therapeutic candidate to these patients for whom no effective therapies are available.
6. CONCLUSION
This case report provides support for the therapeutic use of elamipretide in a patient with MPAN, as the agent was well tolerated and may have contributed to improvement in symptoms. This experience may guide the use of elamipretide in future patients enrolled in the EAP and help to inform clinical trial efforts.
AUTHOR CONTRIBUTIONS
Jorge Patino: Investigation; writing – original draft; writing – review and editing. Anna Haertling Clearman: Investigation; writing – review and editing. Lindsey Miller: Investigation; writing – review and editing. Mary Kay Koenig: Investigation; writing – review and editing.
FUNDING INFORMATION
Stealth BioTherapeutics provided support in supplying elamipretide for the patient and publishing this case report. The authors confirm independence from the sponsor. The content of the article has not been influenced by the sponsor.
CONFLICT OF INTEREST STATEMENT
The authors have no conflicts of interest to declare.
ETHICS STATEMENT
FDA and local IRB review and approval were obtained prior to initiating therapy with elamipretide SC daily under this EAP.
CONSENT
Written informed consent was obtained from the patient to publish this report in accordance with the journal's patient consent policy.
ACKNOWLEDGMENTS
The authors wish to thank the patient and her family for participating, as well as Jamie L. Dermatis, DPM, and James A. Shiffer, RPh, Write On Time Medical Communications, LLC, for their writing and editorial assistance during the development of this manuscript.
Patino J, Clearman AH, Miller L, Koenig MK. Expanded‐access use of elamipretide in a patient with membrane protein‐associated neurodegeneration. Clin Case Rep. 2024;12:e9116. doi: 10.1002/ccr3.9116
DATA AVAILABILITY STATEMENT
The data that support the findings of this case report are not publicly available due to the data containing information that could compromise the privacy of the patient, but may be available from the corresponding author upon reasonable request.
REFERENCES
- 1. Gregory A, Klopstock T, Kmiec T, et al. Mitochondrial membrane protein‐associated neurodegeneration. In: Adam MP, Feldman J, Mirzaa GM, eds. GeneReviews®. University of Washington, Seattle; 2014. [PubMed] [Google Scholar]
- 2. Hartig MB, Iuso A, Haack T, et al. Absence of an orphan mitochondrial protein, c19orf12, causes a distinct clinical subtype of neurodegeneration with brain iron accumulation. Am J Hum Genet. 2011;89(4):543‐550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Food and Drug Administration . Expanded Access to Investigational Drugs for Treatment Use, Questions and Answers, Guidance for Industry Draft guidance. 2022. Accessed January 5, 2024 https://www.fda.gov/media/162793/download. [Google Scholar]
- 4. Karaa A, Bertini E, Carelli V, et al. Efficacy and safety of elamipretide in individuals with primary mitochondrial myopathy: the MMPOWER‐3 randomized clinical trial. Neurology. 2023;101(3):e238‐e252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brown DA, Vining A, Mackinnon A, Elustondo P, McKenna M, Nagaraju K. The Mitochondria‐Targeting Peptide Elamipretide Potentiates Dystrophin Expression Induced by an Exon‐Skipping Morpholino in the mdx Mouse Model. Proceedings of Muscular Dystrophy Association (MDA) Clinical & Scientific Conference. 2022. Muscular Dystrophy Association.
- 6. Allen ME, Pennington ER, Perry JB, et al. The cardiolipin‐binding peptide elamipretide mitigates fragmentation of cristae networks following cardiac ischemia reperfusion in rats. Commun Biol. 2020;3(1):389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Szeto HH. First‐in‐class cardiolipin‐protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol. 2014;171(8):2029‐2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McDonald CM, Henricson EK, Abresch RT, et al. The 6‐minute walk test and other clinical endpoints in Duchenne muscular dystrophy: reliability, concurrent validity, and minimal clinically important differences from a multicenter study. Muscle Nerve. 2013;48(3):357‐368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nordastig J, Broeren M, Hensäter M, Perlander A, Österberg K, Jivegård L. Six‐minute walk test closely correlates to “real‐life” outdoor walking capacity and quality of life in patients with intermittent claudication. J Vasc Surg. 2014;60:404‐409. [DOI] [PubMed] [Google Scholar]
- 10. Melo TA, Duarte ACM, Bezerra TS, França F, Soares NS, Brito D. The five times sit‐to‐stand test: safety and reliability with older intensive care unit patients at discharge. Rev Bras Ter Intensiva. 2019;31(1):27‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Newman J, Galna B, Jakovljevic DG, et al. Preliminary evaluation of clinician rated outcome measures in mitochondrial disease. J Neuromuscul Dis. 2015;2(2):151‐155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Montano V, Lopriore P, Gruosso F, et al. Primary mitochondrial myopathy: 12‐month follow‐up results of an Italian cohort. J Neurol. 2022;269(12):6555‐6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Thompson WR, Manuel R, Abbruscato A, et al. Long‐term efficacy and safety of elamipretide in patients with Barth syndrome: 168‐week open‐label extension results of TAZPOWER. Genet Med. 2024;26(7):101138. [DOI] [PubMed] [Google Scholar]
- 14. Venco P, Bonora M, Giorgi C, et al. Mutations of C19orf12, coding for a transmembrane glycine zipper containing mitochondrial protein, cause mis‐localization of the protein, inability to respond to oxidative stress and increased mitochondrial Ca2+ . Front Genet. 2015;19(6):185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Shao C, Zhu J, Ma X, et al. C19orf12 ablation causes ferroptosis in mitochondrial membrane protein‐associated with neurodegeneration. Free Radic Biol Med. 2022;182:23‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gregory A, Lotia M, Jeong SY, et al. Autosomal dominant mitochondrial membrane protein‐associated neurodegeneration (MPAN). Mol Genet Genomic Med. 2019;7(7):e00736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Monfrini E, Melzi V, Buongarzone G, et al. A de novo C19orf12 heterozygous mutation in a patient with MPAN. Parkinsonism Relat Disord. 2018;48:109‐111. [DOI] [PubMed] [Google Scholar]
- 18. Hartig M, Prokisch H, Meitinger T, Klopstock T. Mitochondrial membrane protein‐associated neurodegeneration (MPAN). Int Rev Neurobiol. 2013;110:73‐84. [DOI] [PubMed] [Google Scholar]
- 19. Löbel U, Schweser F, Nickel M, et al. Brain iron quantification by MRI in mitochondrial membrane protein‐associated neurodegeneration under iron‐chelating therapy. Ann Clin Transl Neurol. 2014;1(12):1041‐1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen S, Lai X, Fu J, et al. A novel C19ORF12 mutation in two MPAN sisters treated with deferiprone. BMC Neurol. 2023;23(1):134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the findings of this case report are not publicly available due to the data containing information that could compromise the privacy of the patient, but may be available from the corresponding author upon reasonable request.