

Abstract
Microbial transformation of bile acids affects intestinal immune homeostasis but its impact on inflammatory pathologies remains largely unknown. Using a mouse model of graft-versus-host disease (GVHD), we found that T cell-driven inflammation decreased the abundance of microbiome-encoded bile salt hydrolase (BSH) genes and reduced the levels of unconjugated and microbe-derived bile acids. Several microbe-derived bile acids attenuated farnesoid X receptor (FXR) activation, suggesting that loss of these metabolites during inflammation may increase FXR activity and exacerbate the course of disease. Indeed, mortality increased with pharmacological activation of FXR and decreased with its genetic ablation in donor T cells during mouse GVHD. Furthermore, patients with GVHD after allogeneic hematopoietic cell transplantation showed similar loss of BSH and the associated reduction in unconjugated and microbe-derived bile acids. Additionally, the FXR antagonist ursodeoxycholic acid reduced the proliferation of human T cells and was associated with a lower risk of GVHD-related mortality in patients. We propose that dysbiosis and loss of microbe-derived bile acids during inflammation may be an important mechanism to amplify T cell-mediated diseases.
Introduction
Bile acids (BAs) are cholesterol-derived molecules with important physiological functions1–3. Host-derived BAs are synthesized in the liver and secreted into the small intestine (SI) conjugated to either taurine or glycine (Fig. 1a)4,5. BAs are reabsorbed via the apical sodium BA transporter (ASBT) and transported into the portal circulation by the organic solute transporter (OST) α/β. Approximately 5% of BAs escapes the enterohepatic circulation and undergoes microbial modifications. Key reactions include glycine or taurine deconjugation via the amidate hydrolase activity of bile-salt hydrolase (BSH) enzymes6 and the removal of the 7α-OH group by commensals harboring the bile acid inducible (bai) operon1,7. Additional alterations involve epimerization of the 3-OH or 6-OH groups by hydroxysteroid dehydrogenases (HSDHs),8 and re-conjugation to tyrosine, phenylalanine, leucine or glycine mediated by the aminoacyltransferase activity of BSH9–11. Host- and microbe-derived BAs differentially activate membrane-bound (e.g., G protein-coupled bile acid receptor 1) and nuclear (e.g., the farnesoid X receptor, FXR, and the retinoic acid receptor related orphan receptor, RORγt) receptors controlling central aspects of gut physiology, including motility and barrier function12. BAs were recently implicated in regulating the differentiation of intestinal CD4+ T helper cells. The microbe-derived BAs 3-oxolithocholic acid (3-oxoLCA) and isoLCA limited pro-inflammatory T helper 17 cell polarization by modulating RORγt13,14. Moreover, isoalloLCA, ω-muricholic acid (ω-MCA), and isodeoxycholic acid (isoDCA) enhanced regulatory T cell generation, with the later antagonizing FXR activity in antigen presenting cells13,15. These findings suggest that BA signaling might contribute to T cell-driven pathologies, and that changes in the BA pool during disease may be physiologically relevant.
Fig. 1. T cell-driven inflammation alters the intestinal BA pool.
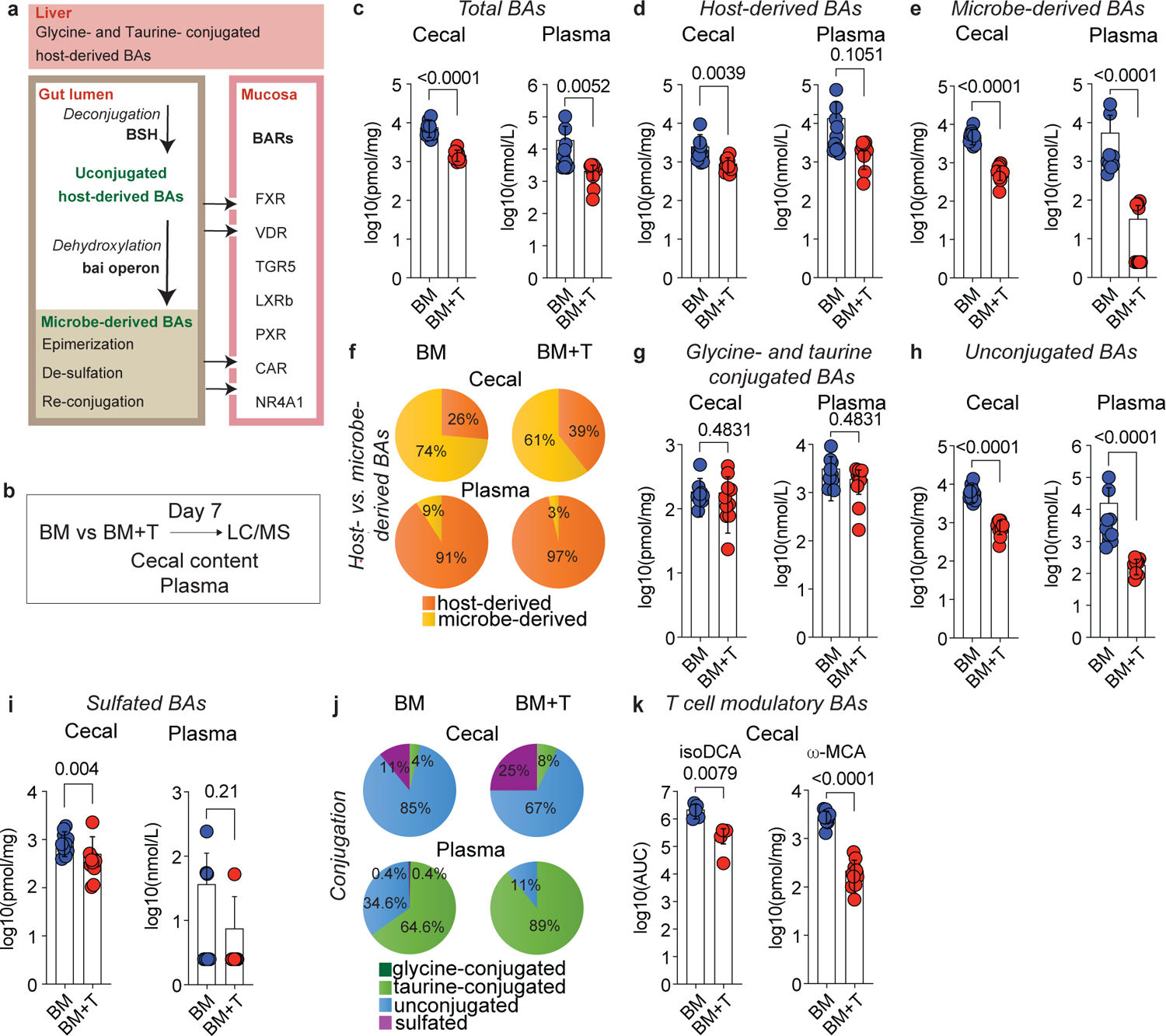
Lethally irradiated 6–8 week old female BALBc mice were transplanted with 10 × 106 BM cells alone (BM) or together with 1 × 106 T cells (BM+T). (a) Simplified schematic overview of BA metabolism. In humans, the host-derived BA pool is primarily composed of CA and CDCA, while in mice α- and β-MCA are dominant. Approximately 95% of the BAs are absorbed in the terminal ileum and enter the enterohepatic circulation, while the remaining 5% are transformed by gut bacteria into various microbe-derived BAs. BAs can bind to several BA receptors (BARs) including the Farnesoid X Receptor (FXR), the Vitamin D Receptor (VDR), the G protein-coupled BA Receptor 1 (TGR5), the Liver X Receptor β (LXRβ), the Pregnane X Receptor (PXR), the Constitutive Androstane Receptor (CAR), and the Nuclear Receptor 4A1 (NR4A1). (b) Experimental workflow for BA analyses during preclinical GVHD. (c-k) Analysis of cecal contents and plasma on day 7 by LC-MS. Concentration of total (c), host-derived (d), and microbe-derived BAs (e). Pie chart showing the relative abundances of host- and microbe-derived BAs (f). Concentration of glycine- and taurine-conjugated (g), unconjugated (h), and sulfated BAs (i). Pie chart showing the relative abundances of glycine- and taurine-conjugated, unconjugated, and sulfated BAs (j). Cecal levels of the T cell immunomodulatory BAs isoDCA and ω-MCA (k). (c-k) Data combined from two independent experiments (n=10) and shown as mean ± S.D or averaged percentages. IsoDCA levels in (k) are reported as AUCs and representative of two independent experiments (n=5). Statistical significance was determined by a two-tailed Mann Whitney test.
Allogeneic hematopoietic cell transplantation (allo-HCT) is a curative treatment for various malignant and hematological diseases16–18. This process significantly disrupts the fecal microbiome, with the ensuing loss of diversity being linked to a higher incidence of graft-versus-host disease (GVHD)19–25. This potentially life-threatening complication occurs when donor T cells initiate an immune response targeting various organs, particularly the skin, gastrointestinal tract, and liver. GVHD affects approximately 50% of allo-HCT patients, and steroid-refractory disease is associated with high mortality26,27. While the association between peri-transplant dysbiosis and GVHD is broadly recognized, the mechanisms by which microbiome changes contribute to disease onset are not fully understood. This study aims to investigate the relevance of the microbial BA metabolism and FXR signaling in T cell responses during GVHD.
Results
T cell-driven inflammation alters the BA pool
We hypothesized that T cell-driven inflammation would alter the gut microbiome and the BA pool. To explore this, we employed a preclinical GVHD mouse model in which C57BL/6 (B6, H2b) bone marrow (BM) is transferred into BALB/cJ (BALBc, H2d) recipients together with T cells (BM+T). This major histocompatibility complex (MHC)-disparate GVHD model typically induces disease around day 7 post-transplant with high mortality by day 9028. We quantified the major species of host- and microbe-derived BAs (Extended Data Table 1) in cecal contents and plasma on day 7 post-transplant using liquid chromatography-mass spectrometry (LC-MS, Fig. 1b). Compared to the control group receiving only BM grafts, mice receiving donor T cells developed GVHD and exhibited lower total BA concentrations in their cecum and plasma (Fig. 1c). Host-derived BAs were significantly lower in the cecum of BM+T mice (Fig. 1d), while microbe-derived BAs were lower in both cecum and plasma (Fig. 1e). The ratio of microbe- to host-derived BAs was markedly lower in both compartments (Fig. 1f, Extended Data Fig. 1a), indicating a systemic remodeling of the BA pool. Since deamidation is the first step in the formation of microbe-derived BAs, we analyzed the abundance of unconjugated BA species. BM+T mice showed lower unconjugated BAs levels compared to BM only recipients, but no difference in glycine- and taurine-conjugated specimens (Fig. 1g, h, Extended Data Fig. 1b). Sulfated BAs were lower in the cecum of BM+T recipients (Fig.1i, j), suggesting reduced intestinal BA output since sulfation facilitates metabolite excretion29. Transplantation of donor T cells pervasively affected several intestinal and circulating BA species (Supplementary Fig. 1). Importantly, microbe-derived BAs with T cell modulating functions including isoDCA15 and ω-MCA15 were reduced in the ceca of BM+T mice compared to controls (Fig. 1k). Although the levels of 3-oxoLCA and isoLCA were below our limit of detection, these immunoregulatory BAs were also lower in the GVHD group (Extended Data Fig. 1c). Overall, these results show that GVHD drastically affects the composition of the BA pool and leads to the loss of microbiota-dependent BAs.
BA pool shifts are linked to microbiome and liver metabolism
To determine if these changes were linked to alterations in microbial BA metabolism, we examined BSH and bai operon gene abundances in the cecal contents of GVHD mice. Consistent with lower unconjugated BA levels, BSH genes were less abundant in BM+T mice compared to controls (Fig. 2a), while bai operon genes were unchanged (Fig. 2b, c). These results suggest that lower BSH likely drives the loss of microbe-derived BAs in GVHD.
Fig. 2. Alloreactive T cells alter host and microbial BA metabolism.
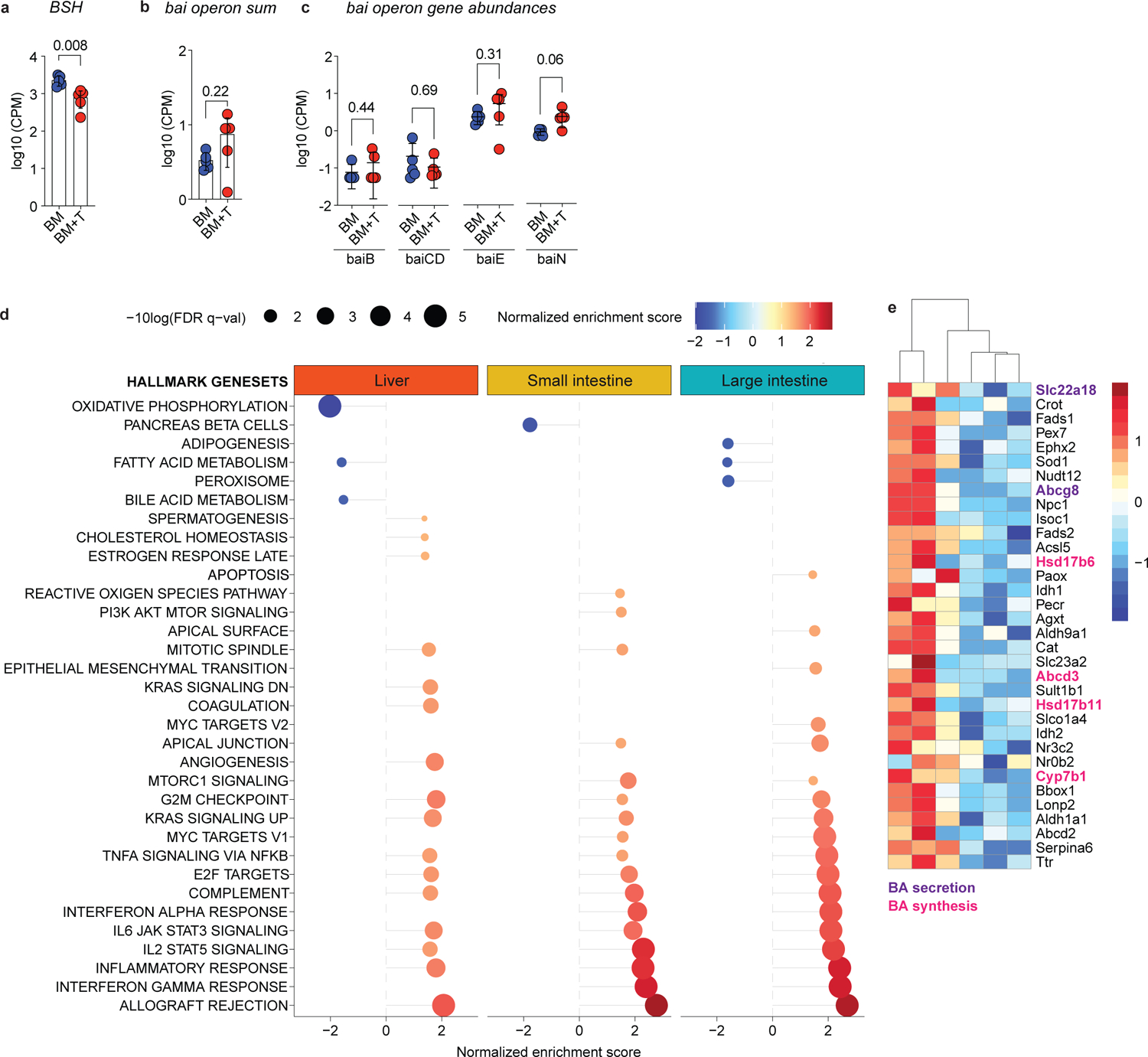
Lethally irradiated 6–8 week old female BALBc mice were transplanted with 10 × 106 BM cells alone (BM) or together with 1 × 106 T cells (BM+T). Mice were analyzed on day 7 post-transplant. (a-c) Metagenomic analyses of intestinal microbiota from cecal contents. Abundance of BSH (a), sum of the gene counts mapping to the bai operon (b) and counts of individual bai operon genes (c). (d, e) Bulk RNA-sequencing analyses of liver tissue and epithelial fractions of the small and large intestines. (d) Gene Set Enrichment Analysis of pathways differentially regulated between BM and BM+T mice. Displaying significant pathways. (e) Heatmap showing hepatic BA metabolism genes enriched among differentially abundant transcripts. Data are from one experiment (a-c, n=5), (d, e, n=3). Data in (a-c) shown as mean ± S.D and statistical significance was determined by a two-tailed Mann Whitney test.
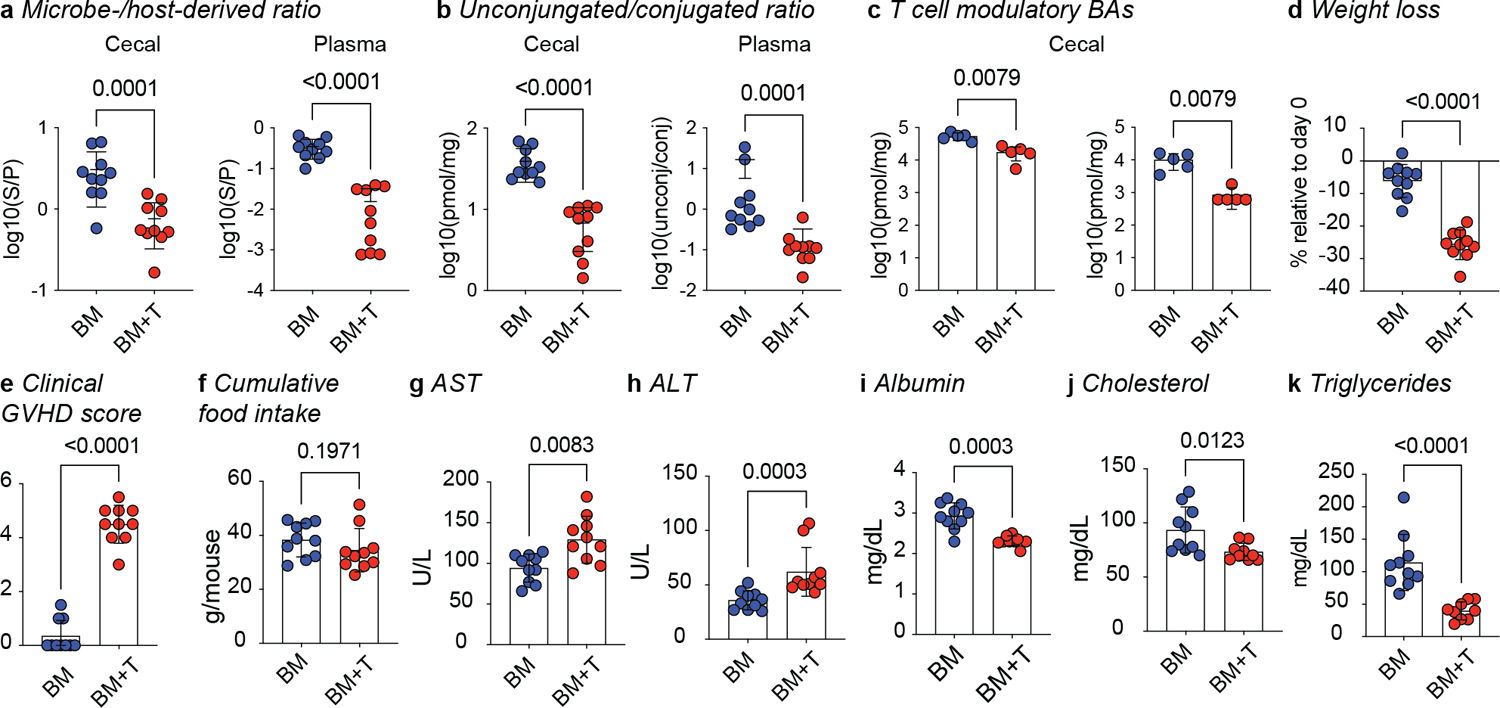
Next, we investigated how changes in host physiology affected the BA pool during T cell-driven inflammation. While BM+T recipients lost more weight and had higher clinical GVHD scores on day 7, cumulative food intake on that day 7 was similar to BM-only mice (Extended Data Fig. 1d–f). GVHD targets the liver and the intestines, which are central metabolic organs involved in BA homeostasis. Indeed, the plasma liver injury markers aspartate transaminase and alanine transaminase were elevated in BM+T recipients on day 7 (Extended Data Fig. 1g, h). Further, albumin levels were lower in BM+T recipients, suggesting reduced hepatic function and/or increased intestinal loss (Extended Data Fig. 1i). Total cholesterol and triglycerides levels were also reduced in BM+T mice (Extended Data Fig. 1j, k), indicating disrupted lipid homeostasis. To comprehensively investigate the effects of GVHD on host metabolism, we carried out RNA-sequencing (RNA-seq) analysis of bulk liver and epithelial cell fractions of the SI and large intestine (LI). A comparison between BM+T and BM mice found 2,247 differentially expressed genes in the liver, 4,270 in the SI and 6,235 in the LI (Supplementary Tables). Gene Set Enrichment Analysis (GSEA)30,31 revealed enrichment in pathways related to allograft rejection and responses to IL-2, IL-6, and TNFα in all three organs from BM+T mice, indicating widespread immune activation (Fig. 2d). SI and LI also showed higher levels of transcripts associated with IFNγ response. Liver transcripts mapping to peroxisome-related processes and BA metabolism were less abundant in GVHD mice, agreeing with altered cholesterol, triglycerides, and BA levels (Fig. 2d). In line with reduced host-derived BAs, the expression of genes related to the GSEA hallmark gene set BA metabolism was reduced in the livers of BM+T recipients compared to the BM group, including transcripts for genes involved in BA synthesis (Abcd3, Hsd17b11, Cyp7b1, Hsd17b6) and transport (Abcg8, Slc22a18) (Fig. 2e). Altogether, these findings indicate that T cell-driven inflammation impacts both host and microbial BA metabolism.
FXR signaling in donor T cells modulates T cell responses
Beyond its role in BA homeostasis, the nuclear receptor FXR also affects host immunity32 and is upregulated in T cells upon activation33. The host-derived BA chenodeoxycholic acid (CDCA) is a potent natural FXR agonist, while a few other BAs were reported to antagonize FXR transcriptional activity15,34. We hypothesized that changes in BA metabolism during T cell-driven inflammation may affect the balance of FXR activating and antagonizing BAs. Therefore, we utilized a cell line stably transfected to express luciferase under the control of an FXR-responsive element to assess the impact of individual BAs on FXR transcriptional activity (Extended Data Fig. 2a). As expected, CDCA induced a strong, dose-dependent increase in luciferase units (Extended Data Fig. 2b), while other BAs weakly induced FXR activation (Fig. 3a). In contrast, several microbe-derived BAs lowered CDCA-induced FXR activation (Fig. 3b), including the previously reported FXR antagonist isoDCA15 and other BAs depleted during GVHD (e.g.,DCA, 3-oxoDCA, LCA, 7-oxoLCA, and 12-oxoLCA). These results suggest that bacterial metabolism of BAs can alter the FXR-activating properties of the BA pool.
Fig. 3. FXR activity contributes to GVHD.
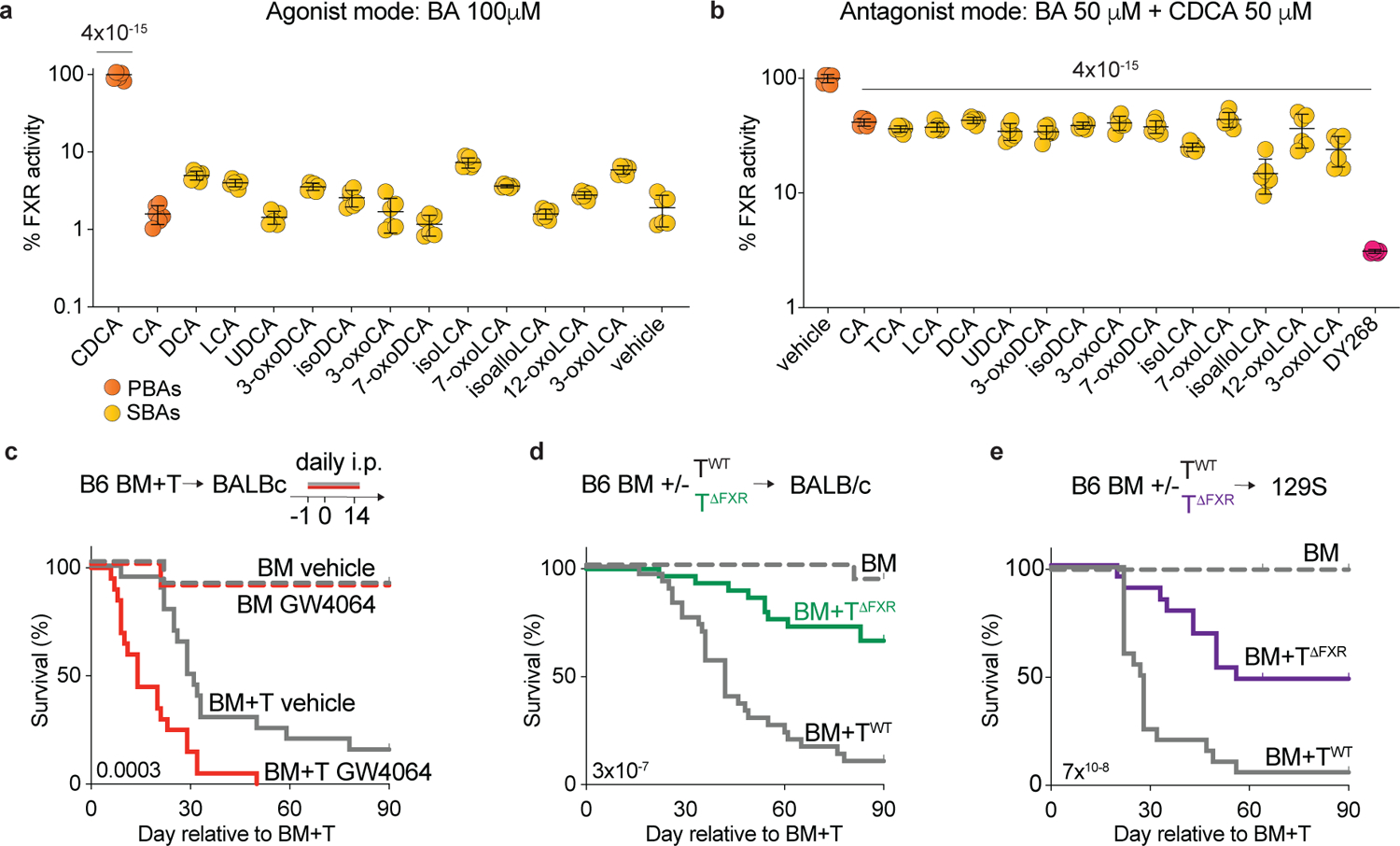
(a, b) Stably transfected HepG2 cells expressing luciferase under the control of an FXR-responsive element were treated with the indicated BAs for 16h. Endpoint luciferase activity was normalized by live cell numbers determined by Hoechst 33342 staining. (a) Assay in agonist mode. FXR activation induced by the indicated BAs at 100μM. Showing % of FXR activity relative to the positive control (CDCA). (b) Assay in antagonist mode. FXR activation induced by a suboptimal concentration of CDCA (50μM) was counteracted by equimolar amounts of the indicated BA or the synthetic FXR antagonist DY268 (20nM). Showing % of FXR activity relative to the positive control (CDCA). (c-e) Lethally irradiated BALBc mice received B6 BM alone (BM) or in combination with T cells (BM+T). (c) Survival of BALBc mice that were treated with the FXR agonist GW4064 30mg/kg intraperitoneally once daily or vehicle for 15 days starting day −1 relative to transplant. (d) Survival of BALBc mice transplanted with donor T cells from either Nr1h4fl/fl (BM+TWT) or Cd4-Cre Nr1h4fl/fl (BM+TΔFXR) mice on a B6 background. (e) Survival of 129S mice transplanted with B6 BM+TWT or TΔFXR. (a, b) Data shown as mean ± S.D, pooled from two independent experiments carried out in technical triplicates. Statistical significance in (a, b) was determined by a one-way ANOVA followed by the Dunnett’s multiple comparisons test. Data combined from two (c, e, n=20) or three (d, n=30) independent experiments. Statistical significance in (c-e) was determined using the log-rank test.
We thus hypothesized that increased FXR activity might exacerbate GVHD. To evaluate this, we administered the selective FXR agonist GW4064 to BM and BM+T recipients from day −1 to day 14 post-transplant. As anticipated, GW4064-treated BM+T recipients exhibited higher mortality (Fig. 3c). We then tested whether FXR activity in donor T cells contributed to increased GVHD lethality by transferring BM recipients with T cells isolated from Cd4Cre Nr1h4fl/fl mice (TΔFXR) or their wild type (WT) control littermates (Nr1h4fl/fl, TWT). In TΔFXRmice, FXR is conditionally ablated in all T cells via Cre recombinase expression driven by the Cd4 promoter15. FXR deletion in donor T cells significantly improved GVHD survival (Fig. 3d). Accordingly, recipients of BM+TΔFXR exhibited less weight loss and lower clinical GVHD scores relative to BM+TWT (Extended Data Fig. 2c, d). Similar results were observed when transferring BM+TΔFXR into 129S1/SvImJ (129S1) recipients, a MHC-matched, minor histocompatibility antigen-disparate GVHD model35 (Fig. 3e). Notably, total body FXR knockout B6 mice (Nr1h4−/−, ΔFXR) transplanted with BALBc BM+T cells showed no differences in survival when compared to WT B6 recipients (Extended Data Fig. 2e), suggesting that FXR signaling in host cells is not relevant for GVHD mortality. Overall, these findings suggest that FXR signaling in donor T cells contributes to GVHD.
Next, we dissected the effects of T cell-intrinsic FXR deficiency on the pathophysiology of GVHD. BM+TΔFXR mice had reduced GVHD-related histopathology compared to BM+TWT on day 28 post allo-HCT (Extended Data Fig. 2f, g). Attenuated LI and liver disease contributed to the overall lower GVHD score of the BM+TΔFXR group, while other organs were not significantly different (Extended Data Fig. 2f). Consistently, T cell infiltration as measured by CD3 staining was reduced in the colons of BM+TΔFXR recipients (Extended Data Fig. 2g). We further profiled T cell effector function by flow cytometry on day 14 post-transplant. In line with our histopathological findings, we found that FXR ablation in CD4 and CD8 donor T cells led to lower IFNγ production in the LI but not in the SI lamina propria (Extended Data Fig. 2h). Altogether, these results suggest that cell-intrinsic FXR signaling affects T cell responses in tissues exposed to high levels of microbe-derived BAs in an allogeneic setting.
GVHD patients have an altered BA pool
To validate our pre-clinical findings in allo-HCT patients, we employed a nested case-control study with prospectively collected fecal samples (Extended Data Fig. 3). We identified 63 acute GVHD patients sampled within 10 days of disease onset (median GVHD onset day 29 post allo-HCT, range 14–90), and 94 GVHD patients sampled around neutrophil engraftment (median GVHD onset day 32 post allo-HCT, range 13–90). These patients were respectively matched with 61 and 98 control patients who never developed acute GVHD. Controls were sampled either during a pseudo-peri-GVHD onset period (assigned based on their paired GVHD match) or in the peri-engraftment period. Additionally, 81 patients were sampled at both timepoints, and 36 met criteria for both peri-engraftment and peri-GVHD onset timepoints. Patient characteristics are summarized in Extended Data Table 2. The CONSORT diagram is shown in Extended Data Fig. 3.
Most patients in this cohort (n=217/235, 92%) received prophylaxis for hepatic sinusoidal obstructive syndrome with ursodeoxycholic acid (UDCA), a microbe-derived BA in humans36,37. Thus, our initial investigation addressed the impact of exogenous UDCA administration on the BA pool. As expected, UDCA exposure correlated with higher fecal UDCA (Extended Data Fig. 4a) and an expansion of the total microbe-derived BA pool (Extended Data Fig. 4b). Similarly, fecal UDCA levels and the total microbe-derived BA pool were increased in samples from patients exposed to UDCA up to a month before stool collection (Extended Data Fig. 4a, b). We also observed a positive correlation between the concentrations of glycine- and taurine-conjugated UDCA and unconjugated UDCA (R=0.5, Extended Data Fig. 4c)38. We then explored the associations between UDCA levels and BA pools. UDCA concentration correlated with total BAs (R=0.6), microbe-derived BAs (R=0.83) and host-derived BAs (R=0.23, Extended Data Fig. 4d–f). These correlations remained significant even after removing UDCA from the total BA (R=0.29) and microbe-derived BA pools (R=0.18, Extended Data Fig. 4g, h). After subtracting UDCA, the ratio of host-derived BAs to the remaining microbe-derived BAs showed no correlation with UDCA concentration (Extended Data Fig. 4i), suggesting independence from UDCA exposure. While investigating the relationship between UDCA and individual BA species, we found moderate to strong correlations between UDCA and several BAs, including the host-derived cholic acid (CA) and CDCA (R=0.43 and R=0.57 respectively, Extended Data Fig. 4j, k). While CDCA is a stereoisomer of UDCA, CA has an additional −OH and comes from a separate hepatic pathway. Overall, these findings suggest that UDCA treatment increases total BA levels by directly contributing to the pool and by regulating host-derived BAs.
Since most patients received UDCA prophylaxis, our analysis was confined to UDCA-exposed samples, including those collected within one month after UDCA discontinuation. No differences in clinical characteristics were observed between GVHD cases and controls in this subcohort (Extended Data Table 3). GVHD patients displayed lower total BA levels compared to controls at disease onset (Fig. 4a), consistent with our preclinical results. While the host-derived BA pool was not significantly changed, both microbe- (including UDCA) and non-UDCA microbe-derived BAs were decreased in GVHD patients relative to controls (Fig. 4b–d, Extended Data Fig. 5a–c). This suggests that lower total BA concentrations in GVHD patients were driven by a reduction in the microbe-derived BA compartment. Given that UDCA constitutes a significant portion of the total BA and microbe-derived BA pools in UDCA-exposed patients, we excluded this BA from further downstream analyses, denoted henceforth by an asterisk (e.g., total BA* = total BA minus UDCA). Next, we assessed BA conjugation status. Consistent with our preclinical observations, unconjugated BAs were lower in GVHD patients compared to controls, while glycine- and taurine-conjugated BAs showed no significant differences (Fig. 4e, f, Extended Data Fig.5d). Sulfated BAs were also reduced in GVHD patients (Fig. 4g, h), again matching our preclinical findings. At peri-GVHD onset, GVHD patients had significantly lower levels of microbe-derived T cell immunomodulatory BAs, including 3-oxoLCA13, isoLCA14, isoDCA15 and ω-MCA15 compared to controls (Fig. 4i). After adjustment for multiple testing using the Bonferroni-Hoffmann (BH) method, several BAs remained significantly lower in GVHD patients compared to controls, including DCA, LCA, 3-oxoLCA, isoLCA and ω-MCA (Extended Data Table 4). Accounting for variables affecting GVHD incidence such as conditioning intensity and graft characteristics27, we found that controls had a higher abundance of microbe-derived BAs like DCA, LCA, ω-MCA and isoLCA (Extended Data Fig. 5e, f).
Fig. 4. Comparison of BA pools in patients with GVHD vs controls.
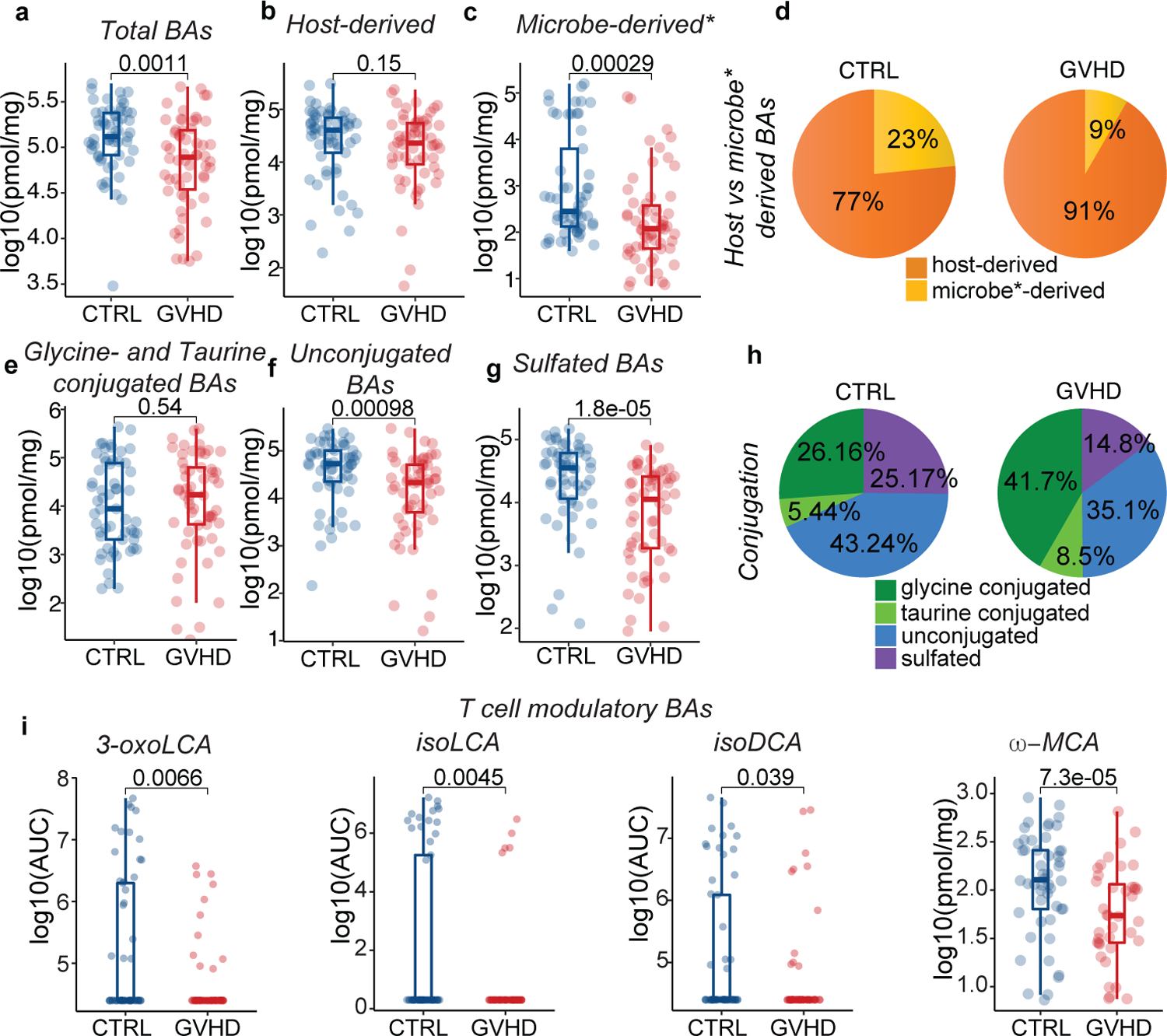
Concentration of total (a), host-derived (b), and microbe-derived* (non-UDCA) (c) BAs. (d) Pie chart showing the averaged percentages of host- and microbe-derived* BA pools in patients with GVHD vs. controls in peri-GVHD onset samples. (e) Glycine- and taurine-conjugated, (f) unconjugated, and (g) sulfated BA levels. (h) Pie chart showing the averaged percentages of glycine- and taurine-conjugated, unconjugated and sulfated BAs in patients with GVHD vs controls in peri-GVHD onset samples. (i) Levels of T cell immunomodulatory BAs in peri-GVHD onset samples of patients with GVHD vs controls. The boxplot center line corresponds to the median, box limits correspond to the 25th and 75th percentile, and whiskers correspond to 1.5x interquartile range. Data representative of 57 control and 58 GVHD patients. Statistical significance was determined with the 2-sided Wilcoxon Rank-sum test.
We then asked whether differences in the BA pool emerged earlier during disease progression. GVHD and control patients had similar BA pools during the peri-engraftment period (7–21 days post-transplant, median of 14 days), but unconjugated BA levels were significantly decreased in the former group (Extended Data Fig. 6i). Nonetheless, no individual BA was differentially abundant in GVHD patients versus controls (Extended Data Table 5), and we obtained similar results when adjusting for clinical parameters using a linear model (Extended Data Fig. 6 l, m). These findings indicate that the loss of unconjugated BAs precedes the BA pool rmodeling during GVHD, manifesting before clinical signs of disease.
Microbial BA metabolism genes are reduced in GVHD patients
We next assessed the abundance of microbial genes involved in BA metabolism using shotgun metagenomic sequencing. Consistent with changes in unconjugated BAs levels, we found that BSH abundance was lower in peri-GVHD onset stool samples of GVHD patients versus controls, (Fig. 5a), but it was not yet statistically different in peri-engraftment samples (p=0.1, Fig. 5b). We focused our analyses on the peri-GVHD timepoint since the loss of microbe-derived BAs became prominent then. We found a significant correlation between bai operon gene abundance and the concentration of microbe-derived BAs* (R=0.72, Fig. 5c), aligning with the current understanding that levels of these BA species are directly linked to microbial 7-α dehydroxylation activity. GVHD patients showed a significant decrease in bai operon gene abundance compared to controls (Fig. 5d–e). HSDH activity required for the generation of isoBAs sparsely encoded in the microbiome and difficult to detect with standard homology-based identification tools due to their low degree of conservation8. So far, HSDHs were reported in two bacteria, Eggerthella lenta and Ruminococcus gnavus8. While E. lenta levels were comparable, the abundance of R. gnavus was significantly lower in patients with GVHD (Extended Data Fig. 7a, b), suggesting reduced potential to produce T cell immunoregulatory BAs (e.g., 3-oxoLCA, isoLCA and isoDCA). We then evaluated the association between metabolomics, metagenomics, microbiome and GVHD incidence. Higher bai operon gene abundance segregated alongside higher microbe-derived BAs, higher microbiota α-diversity and lower GVHD incidence (Fig. 5f–j, Extended Data Fig. 7c). Similarly, mono-dominated samples (defined by a relative abundance of Enterococcus, Streptococcus or any Proteobacteria genus ≥30%24) were associated with lower α-diversity, lack of bai genes and lower microbe-derived BAs* (Extended Data Fig. 7d–f). Overall, our findings suggest that unconjugated BAs and BSH abundance are reduced earlier in the disease course, while overt gastrointestinal inflammation at the time of GVHD onset is further linked to lower bai operon representation and decreased microbe-derived BAs.
Fig. 5. BA biotransformation potential in transplant patients with GVHD vs controls.
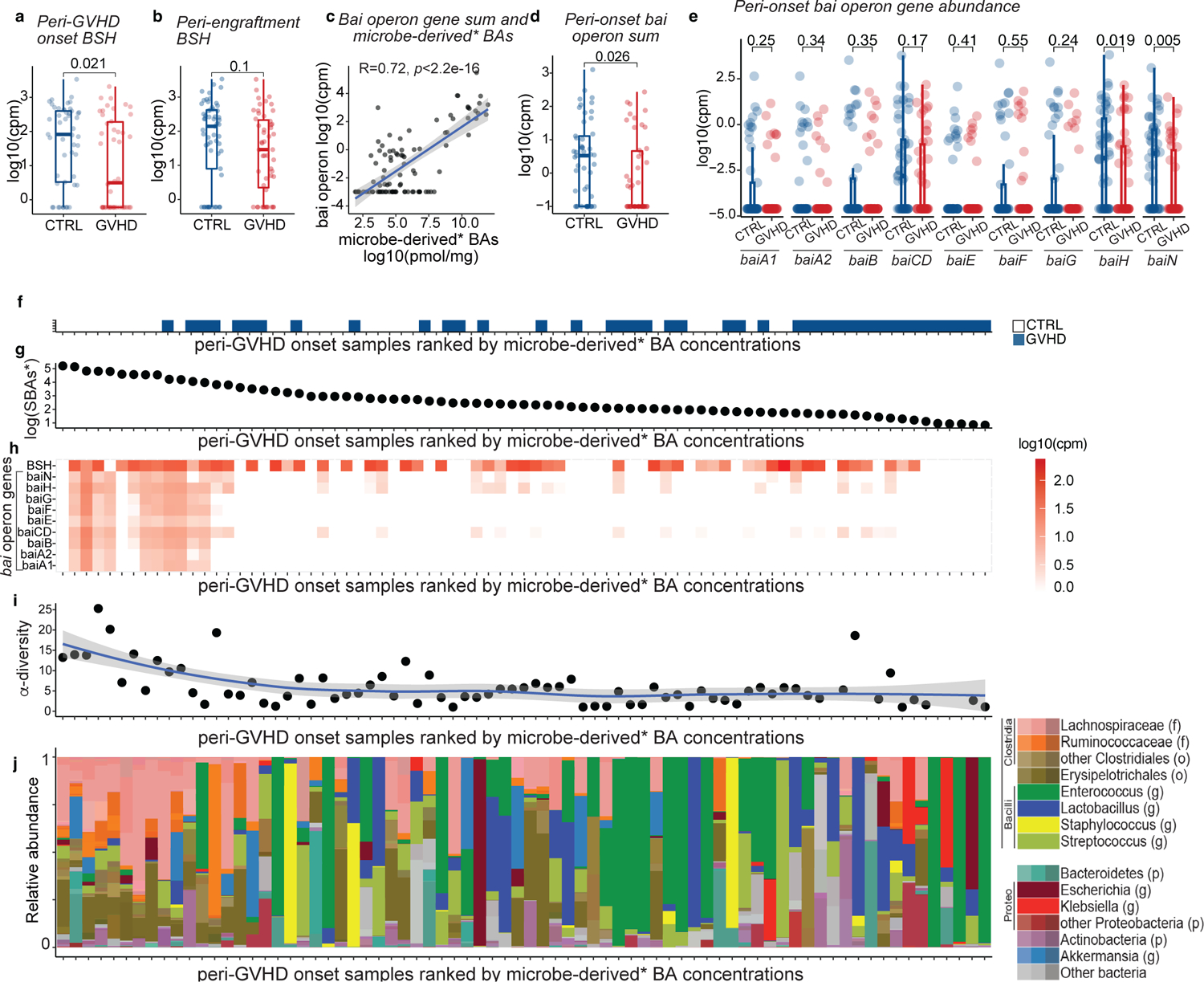
BSH gene abundance in (a) peri-onset (n=46 with GVHD and n=49 controls) and (b) peri-engraftment (n=51 with GVHD and n=57 controls) samples of patients with GVHD vs controls. (c) Correlation between the sum of the gene counts mapping to the bai operon and microbe-derived* BAs. The solid line represents a linear regression model fitted to the data. The shaded region surrounding the line indicates a 95% confidence interval for the regression line. bai operon gene sum (d) and abundance of bai operon genes (e) in patients with GVHD vs controls in peri-GVHD onset samples (n=46 with GVHD and n=49 controls). The boxplot center line corresponds to the median, box limits correspond to the 25th and 75th percentile, and whiskers correspond to 1.5x interquartile range. Statistical significance determined with the 2-sided Wilcoxon Rank-sum test. R was calculated with Pearson correlation. (f-j) Landscape of sample-specific characteristics in peri-GVHD onset samples, ranked from higher to lower microbe-derived* BAs. Showing GVHD incidence (f), concentration of microbe-derived*(g), and BSH and bai operon gene abundance (h). α-diversity calculated with the Simpson reciprocal index (i) and microbiome composition (j). Microbe-derived* BAs: microbe-derived BAs excluding UDCA. The solid line in (i) represents a smoothed trend line generated using the loess method. The shaded region surrounding the line indicatesa 95% confidence interval.
Modulation of FXR activity affects human T cell responses
We next assessed the effects FXR signaling on human T cells. Total T cells from healthy donors were activated in vitro with anti-CD3/CD28 antibodies and IL-2 for 2 days, followed by exposure to BAs or FXR ligands (Extended Data Fig. 8a). Based on our findings, we selected two BAs as natural FXR ligands, CDCA as an agonist and UDCA as an antagonist (Fig. 3a,b). Since BAs also bind to other receptors, we employed the selective FXR agonist GW4064 and the antagonist DY268 to directly test whether FXR activity modulated T cell responses.
FXR agonism by CDCA or GW4064 led to higher T cell numbers relative to vehicle, while FXR inhibition with UDCA or DY268 reduced T cell expansion (Fig. 6a and b, Extended Data Fig. 8b and c). None of the treatments affected cell viability (Extended Data Fig. 8d–f). We then asked whether the differences in proliferation were related to changes in T cell activation. CDCA and GW4064 increased CD25 levels on a per cell basis, while UDCA and DY268 decreased it (Fig. 6c–f). The frequencies of CD25+ cells remained unchanged (Extended Data Fig. 8m,n). Using FXR-deficient T cells from TΔFXR mice, we confirmed that CDCA and UDCA exposure altered CD25 expression in an FXR-dependent manner (Extended Data Fig. 8o). These results suggest that FXR controls T cell activation and expansion in response to BAs.
Fig. 6. UDCA limits human effector T cell responses and is associated with improved GVHD-related mortality.
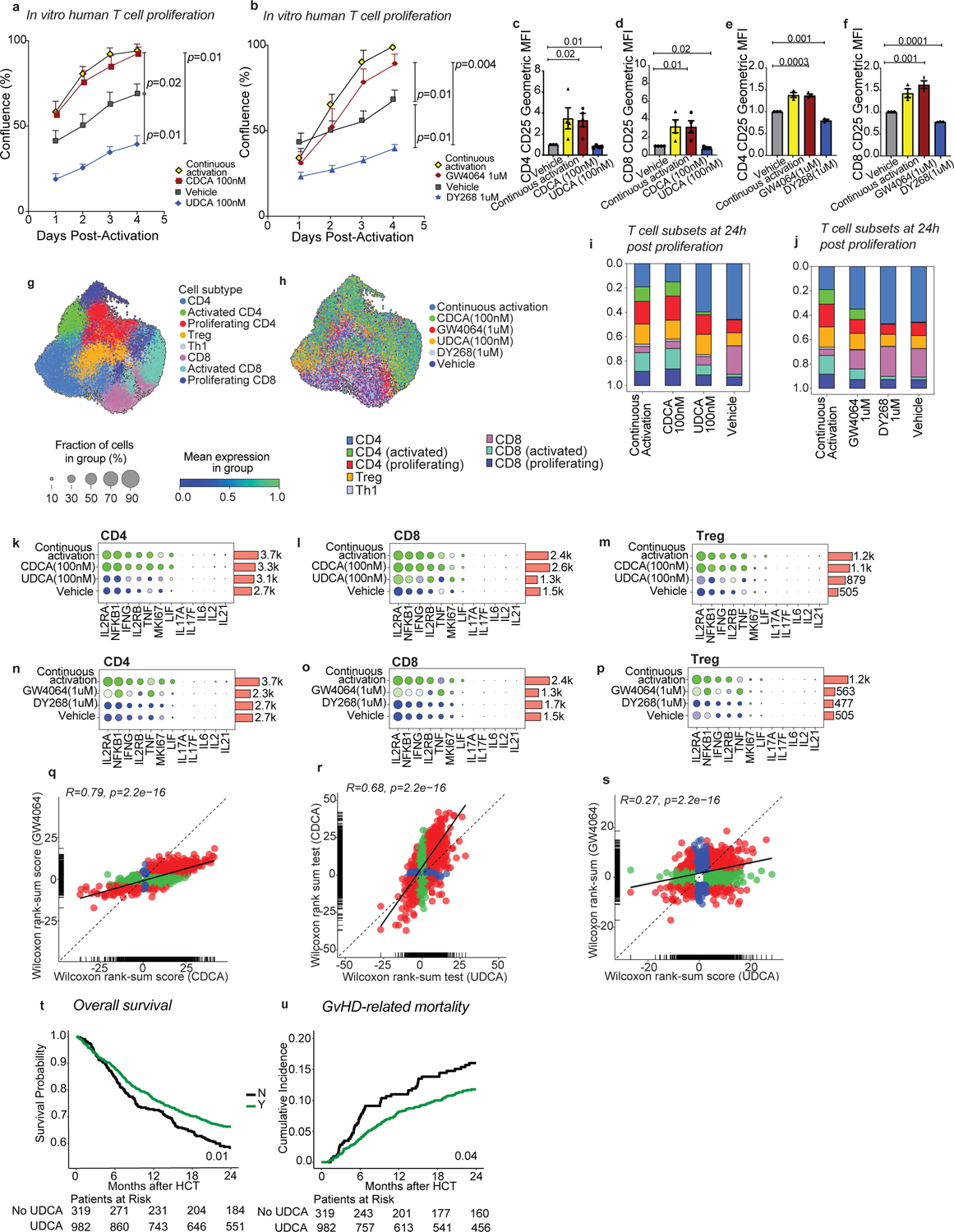
(a-s) Human T cells were activated and further cultured either in the presence of anti-CD3/anti-CD28 (continuous activation control) or in their absence (vehicle control) with or without the indicated compounds for 96 hours. T cell confluence in response to CDCA and UDCA (a) or GW4064 and DY268 after 96 hours (b). CD25 expression of CD4+ and CD8+ T cells after 96 hours of treatment with CDCA and UDCA (c-d), or GW4064 and DY268 (e-f). CD25 expression was measured as geometric mean fluorescence intensity (MFI) of CD25 in CD25+ T cells normalized to the MFI of the vehicle-treated group. (g-s) Single cell RNA-sequencing profiling of activated T cells after 24 hours of treatment with CDCA, UDCA, GW4064 or DY268. Uniform Manifold Approximation and Projection (UMAPs) showing the distribution of the major T cell populations (g) according to treatment group (h). Relative abundances of various T cell clusters in CDCA and UDCA (i), or GW4064 and DY268 (j) treated samples compared to the continuous activation and vehicle. Relative gene expression of selected immune-related genes in CD4 (k,n), CD8 (l,o) and regulatory (m,p) T cells in response to BAs (k-m) and selective FXR ligands. (n-p) Fold-change (FC) vs FC plots comparing the gene expression changes elicited by (q) CDCA and GW4064, (r) CDCA and UDCA, and (s) GW4064 and UDCA relative to vehicle in CD4 T cells. Overall survival (t) and cumulative incidence of GVHD-related mortality (u) in patients who did (n=982) or did not (n=319) receive UDCA. Statistical analysis was performed by two-way (a,b) or one-way ANOVA followed by multiple t-test with Bonferoni correction (c-f). Each data point in (a-f) shows the average of technical duplicates for a single donor. Bars denote the standard error of the mean. Data representative of 4 independent experiments with a total of 4 PBMC donors (a,c,d) or 3 independent experiments with a total of 3 PBMC donors (b,e,f). (g-s) Data from 1 experiment including 2 PBMC donors. (q-s) Statistical analysis was conducted using a 2-sided Wilcoxon rank sum test, with correction for multiple hypotheses using the False Discovery Rate (FDR) method. Outcome analysis was conducted using a log-rank test (t) and the Gray’s test (u).
To gain deeper insights into the immunoregulatory effects of FXR ligands, we next carried out single cell RNA-seq (scRNA-seq) profiling of in vitro activated human T cells treated with CDCA, UDCA, GW4064, and DY268 for 24h. After quality control filtering (Extended Data Fig. 9), we identified major T cell populations and subsets based on marker genes (Fig. 6g–j and Extended Data Fig. 10a–c). CDCA treatment increased the representation of activated CD4 and CD8 T cells relative to vehicle and UDCA (Fig. 6i). Similarly, GW4064 increased the fraction of activated CD4 and CD8 T cell subsets compared to vehicle or DY268 (Fig. 6j). We then assessed the expression of inflammatory markers focusing on genes related to T cell expansion (IL2RA, IL2RB, MKi67, and IL-2) and effector function implicated in GVHD pathogenesis (NFKB, IFNG, TNF, LIF, IL-6, IL-21, IL-17A, and IL-17F). FXR activation with CDCA or GW4064 increased the expression of inflammatory markers in both CD4 and CD8 T cells compared to vehicle or the FXR antagonists UDCA or DY268 (Fig. 6k–p). Next, we compared CDCA and GW4064-induced transcriptional changes to assess the extent by which direct FXR engagement contributes to the responses induced by this BA. Many of the genes affected by CDCA treatment also showed differential expression between GW4064 and vehicle (R=0.79, p<0.001), supporting a role for FXR in CDCA-induced transcriptional changes (Fig. 6q). A number of genes (n=7,524) were exclusively modulated by CDCA, likely reflecting regulation via additional BA receptors. UDCA is considered a weak FXR agonist and has been proposed to reduce FXR activation by diluting the BA pool39. Indeed, while the transcriptional profiles induced by UDCA and CDCA were moderately correlated (R=0.68, p<0.001, Fig. 6r), the shared genes induced by both BAs were overall more highly expressed in CDCA-treated cells. Further, the gene expression changes induced by UDCA and GW4064 were lowly correlated (R=0.27, p<0.001, Fig. 6s), corroborating the notion that UDCA likely dampens FXR activation by preventing its interaction with full agonists. Very few genes were significantly changed in DY268-treated cells compared to the vehicle (n=27), making it difficult to compare the transcriptional signatures induced by UDCA and DY268. Nonetheless, genes downregulated by DY268 were associated with inflammation (PPIA, PHB, CORON1), while upregulated genes were linked to immunosuppression (MALAT1) or cell growth inhibition (IFI44L, Extended Data Table 6). Finally, we performed GSEA to uncover the biological processes linked to transcriptional changes induced by FXR modulation. This analysis includes all normalized gene counts and reports differentially enriched processes contributed by significantly altered transcripts and also by modest changes in gene expression. We sought to identify pathways regulated by the FXR agonists CDCA and GW4064 displaying opposite patterns in UDCA and DY268-treated samples. FXR activation was associated with positive enrichment for mTORC1 signaling, DNA repair, and oxidative phosphorylation pathways in conventional CD4 T cells (Extended Data Fig. 10d), while Treg cells also showed a positive enrichment for DNA repair and negative enrichment for the interferon alpha response pathway upon FXR agonist treatment. No pathway followed this pattern in CD8 T cells. Our findings suggest that targeting FXR activity with natural or pharmacological ligands can alter human T cell activation and proliferation, potentially impacting T cell-driven pathologies.
UDCA is associated with better clinical outcomes post-HCT
In light of the findings above, we hypothesized that UDCA-exposed patients would show improved GVHD-related mortality (GRM). We analyzed an observational cohort of 1,301 allo-HCT recipients, including 319 patients who did not receive UDCA. UDCA-exposed patients (n=982) exhibited enhanced overall survival (OS, p=0.02) and reduced GRM (p=0.04, Fig. 6t, u). To account for clinical factors influencing transplantation outcomes, we conducted multivariable analyses using the Cox model for OS40 and the Fine-Gray model for GRM41. We found that UDCA treatment was significantly associated with improved OS (HR 0.68; 95% CI 0.55–0.85, p<0.001) and reduced GRM (HR 0.66; 95% CI 0.46–0.94 p=0.02, Extended Data Table 7, 8). These findings align with a randomized prospective study demonstrating superior OS and a lower incidence of severe GVHD in UDCA-treated patients36,37 and preclinical findings showing that UDCA treatment reduced histopathological GVHD scores42. These results further confirm our preclinical data that FXR inhibition is correlated with amelioration of GVHD lethality, providing mechanistic support for the continued use of UDCA peri-transplant.
Discussion
Microbiome features and particularly lower α-diversity have been linked to greater incidence and severity of T cell-driven pathologies43,44. Although the association between highly diverse microbiome states and greater representation of bacterial metabolites is well known45, the mechanisms by which the microbiome affects the outcomes of T cell-driven pathologies are not fully understood. Our preclinical GVHD model allowed us to determine the impact of T cell driven-inflammation on the BA pool in the absence of other confounding variables. Remarkably, we also found that GVHD patients showed a similar reduction in unconjugated and microbe-derived BAs upon disease onset, accompanied by the expected changes in microbiome enzymatic potential.
Modeling T cell-driven inflammation suggested that lower BSH abundance is sufficient to affect the levels of microbe-derived BAs. In GVHD patients, loss of BSH and unconjugated BAs was observed before widespread remodeling of the microbiome and the BA pool. While it is possible that bacteria with BSH activity are more sensitive to inflammation relative to commensals carrying the bai operon, an alternative explanation would be that a chronic reduction of BSH-dependent metabolites negatively affects 7-α dehydroxylating bacteria. Interestingly, genetically driven loss of BSH from gut bacteria was shown to lower circulating triglycerides and cholesterol levels46. These findings suggest that reduced BSH activity may also contribute to the systemic metabolic changes observed in our preclinical GVHD model.
The marked differences in BA pool composition during inflammation suggested that signaling via BA-sensing receptors would be altered. Indeed, we found that several microbe-derived BA species reduced during GVHD antagonized FXR transcriptional activity. Notably, in our FXR reporter assay the microbe-derived BA UDCA, which is clinically used to prevent hepatic complications of transplantation, also reduced CDCA-mediated FXR activation. We further showed that prophylactic UDCA treatment was associated with an expansion of the total BA pool in GVHD patients, including host-derived BAs. Recently published work further supports the notion that UDCA increases BA synthesis and accelerates enterohepatic circulation by inhibiting FXR signaling47,48. Since BA synthesis genes were suppressed in the liver of GVHD mice, augmented BA production may also add to the clinical benefits UDCA.
Using mouse genetic models, we demonstrated that FXR signaling in donor T cells, but not in host tissues, affects GVHD lethality, suggesting a role for FXR in T cell-intrinsic alloreactivity. Modulation of FXR activity with BAs or pharmacological ligands also directly affected human T cells, with CDCA stimulating and UDCA potently decreasing T cell activation and proliferation. Interestingly, reduced FXR signaling in both murine and human T cells limited IFNγ production, supporting an immunoregulatory role for this BA-sensing pathway. Lastly, we showed that patients exposed to UDCA during allo-HCT had lower rates of death from GVHD and prolonged OS36,37. The beneficial effect of UDCA on GVHD incidence and OS is further supported by a prospective randomized clinical trial36,37. Additionally, in preclinical models, the administration of tauro-UDCA and UDCA reduced GVHD by decreasing antigen presentation and protecting the intestinal epithelium42. This data supports the notion that FXR inhibition may be a viable strategy to ameliorate GVHD.
The relationship between BAs and the host is multifaceted, and the activation of other BA receptors in immune cells and in the colonic mucosa is anticipated to have diverse effects on host physiology13,49,50. Moreover, microbially amidated BAs conjugated to leucine, tyrosine or phenylalanine were recently discovered to activate FXR9,11. Since FXR activation worsened GVHD in our pre-clinical model, we posit that FXR-dependent immunopathology is likely fueled by the loss of BSH-dependent immunoregulatory metabolites (e.g.., 3-oxoLCA and isoLCA) rather than a paucity of microbially amidated BAs. Overall, our findings add to the complexity of host-microbiota interactions and open avenues of exploration and intervention.
Allo-HCT is a potentially curative treatment for hematologic malignancies. However, conventional GVHD management often fails to control allogenic T cell-driven inflammation and other adverse events including relapse and infections26. Microbiome therpay has the potential to prevent and treat diseases but has been hampered by a lack of mechanistic insights. Our study mapped the functional consequences of microbial BA metabolism changes and identified a mechanism by which this abundant group of metabolites affects GVHD. We found that diversification of the BA pool by the microbiome limits FXR activation. Further, we discovered that the clinically used drug UDCA has additional immunoregulatory benefits, supporting its continued use peri-transplant and paving the way for other therapies targeting the BA-FXR signaling axis in T cell-driven inflammation beyond allo-HCT51–53. Altogether, our work highlights that microbiota-based strategies and inhibition of FXR activation may have a significant role in the prevention and management of T cell-driven pathologies.
Methods
All experiments involving mice were performed in accordance with relevant guidelines and regulations. All studies performed at MSKCC were carried out under protocol 99-07-025 and approved by the Sloan Kettering Institute Institutional Animal Care and Use Committee. Studies performed at the University of Michigan were reviewed and approved by the University Committee on Use and Care of Animals of the University of Michigan. Research involving human research participants has been performed in accordance with the Declaration of Helsinki. The study was approved by the Memorial Sloan Kettering Institutional Review Board.
Mice
B6, BALBc, and 129S1 were obtained from the Jackson Laboratory. B6N were purchased from Charles River. Mice were housed at the Research Animal Resource Center for Memorial Sloan Kettering Cancer (MSKCC) with 12-h light/dark cycles under ambient conditions (temperatures of 18–23°C with 40–60% humidity) and ad libitum access to food and water. Female mice were used for these experiments at an age of 6–8 weeks. Mice were randomly assigned to experimental groups at the beginning of treatments. Investigators were not blinded during group allocation and data analysis due to experimental design complexity. Experimental mice were maintained on a standard rodent diet (5053, LabDiet). CD4cre Nr1h4fl/fl experimental animals were generated by mating mice homozygous for the Nr1h4fl/fl allele, with the male breeder carrying one copy of the Cre-driver gene. Nr1h4fl/fl mice were used as littermate WT controls. STOCK Nr1h4tm1Gonz/J (Strain #:004144) were purchased from the Jackson Laboratory and bred at the University of Michigan. All animals used had no previous history of experimentation.
Mouse BM transplant experiments
Mouse BM transplant experiments for BA profiling and FXR KO in T cells were performed at MSKCC54. Conditioning regimens included split-dosed lethal irradiation with 900 cGy for BALBc recipients and 1000 cGy for 129S1 recipients. BM was T cell-depleted with anti-Thy-1.2 and Low-Tox-M rabbit complement (CEDARLANE Laboratories). Donor T cells were prepared from total splenocytes using the Pan T Cell Isolation Kit II (Miltenyi). T cell purity averaged >90%. BALBc hosts received 10 × 106 BM cells with or without 1 × 106 T cells. 129S1 hosts received 5 × 106 BM cells with or without 4 × 106 T cells. BM or BM+T were infused via tail vein injection. Mice were monitored daily for survival and weekly for determination of GVHD clinical scores54. For FXR agonist experiments, animals received either vehicle (5% DMSO + sterile PBS) or GW4064 (Sigma-Aldrich, 30 mg/kg dissolved in 5% DMSO + sterile PBS) intraperitoneally once a day from day −1 to day 14 relative to BM+T.
BM+T experiments with ΔFXR recipients were performed at University of Michigan Comprehensive Cancer Center55. WT or ΔFXR recipient mice were co-housed for 3 weeks prior to BM+T and were irradiated on day −1 with 1000 cGy single dose followed by 5 × 106 BALBc BM cells and 2 × 106 BALBc T cells via tail vein injection on day 0.
Patient selection
Recipients of allo-HCT at MSKCC between 2009 and 2019 with written consent to a biospecimen-collection protocol approved by the institutional review board were screened for inclusion. Participation in this study was voluntary and carried no compensation. Exclusion criteria comprised of early death (within 21 days after allo-HCT), CD34-selected grafts, non-available or non-evaluable stool samples. From the included cohort, patients with available stool samples between engraftment day and day 100 were classified as cases if they had lower gastrointestinal GVHD of any grade or controls if they did not develop any organ GVHD. We excluded controls that died within 48 days after allo-HCT (median of time-to-GVHD onset in this cohort plus one standard deviation). Nested case design was performed based on age, graft, and donor match. Peri-engraftment samples were selected between days 7–21 after allo-HCT. For the peri-GVHD onset analyses, we selected a sample collected +/− 10 days from the time of disease onset. For the control group, a pseudo-date was assigned based on their paired GVHD case match, and then a sample collected +/− 10 days from the pseudo-date was selected for analysis. Consort diagram is shown in Extended Data Fig 3 and patient characteristics are described in Extended Data Table 2.
Bile acid analysis by LC-MS
Materials:
All reagents and solvents were MS grade. Labeled and unlabeled BA standards were purchased from Cambridge Isotope Laboratories, Steraloids, Toronto Research Chemicals, Isosciences, Cayman Chemical and Avanti Polar Lipids.
Mouse cecal and human fecal sample extraction:
Approximately 150 mg (wet weight) of mouse cecal and human fecal samples were weighed into 2 mL bead tubes containing 1.4 mm ceramic beads (Omni International) and extracted using 80:20 methanol:water containing internal standards (ISTDs) to a final concentration of 100 mg/mL. Samples were homogenized using a Bead Ruptor 24 (Omni International) and slurries were stored at −80°C overnight. Extracts were vortexed and after centrifugation (20,000 × g), 100 μL of supernatant were transferred into a 96-well collection plate (Waters). Samples were dried using a Genevac (EZ-2 Elite HCI Compatible Personal Evaporator) and reconstituted with 200 μL of 50:50 methanol:water. Samples were mixed (400rpm) at 4°C for 20 min, centrifuged (1349 × g) at 8°C for 30 min and 180 μL of sample was transferred to an Agilent injection plates (Agilent). Pooled quality controls (PQC) were assembled by collecting 5 μL of each sample.
Mouse plasma sample extraction:
Plasma samples are thawed for 1 hour in wet ice and vortexed for 5 seconds. 100 μL of sample were transferred into pre-labeled 1.5 mL Eppendorf tubes containing 1mL of extraction solvent (7:2:1, acetonitrile:1% formic acid:methanol with ISTDs). Samples are vortexed for 5 seconds and incubated in the −80°C freezer overnight. Plasma extracts were incubated on wet ice for 1 hour, vortexed, and centrifuged at 4°C for 20 min at 20,000 × g. One mL of the extract was transferred into a 96-well collection plate and dried using a Genevac. Samples were reconstituted with 100 μL of 50:50 methanol:water, mixed at 4°C for 20 min at a mixing frequency of 400 rpm and centrifuged (1349 × g) at 8°C for 30 minutes, and 80 μL was transferred to an Agilent injection plate. PQCs were assembled by collecting 5 μl of each sample.
LC/MS:
Samples were analyzed by LC-MS analysis. We have developed two orthogonal chromatographic separations with retention times confirmed using authentic standards on both methods, in addition to accurate mass measurements made at the MS level only. For each BA species, we report data from the method where we observe the best chromatographic performance, usually defined as the method that achieves the best baseline separation from isobaric species. Most of the abundant human and mouse BAs are well-resolved on both methods and we observed a high degree of correlation between peak areas from the two separations. We used two chromatographic separations extracted for in an Agilent 1290 Infinity II LC system coupled to an Agilent 6546 Q-TOF. Method 1 uses a 1.6 μm, 2.1 × 50 mm CORTECS T3 column (Waters) and method 2 uses a 1.7 μm, 2.1 × 150 mm ACQUITY UPLC BEH Shield RP18 column (Waters). Active reference mass correction was used according to the manufacturer’s instructions. Mobile phase A was 0.1% formic acid in water, mobile phase B was 0.1% formic acid in acetone. LC gradient for Method 1: 30% B to 65% B over 15 min (flow rate: 0.3 ml/min) and for method 2: 30% B to 57% B over 15 min (flow rate: 0.3 ml/min). Both methods ended with 100% B for 1.5 min at double-flow and re-equilibration for 4.5 min. The acquisition was from m/z 50–1,700 at 10 GHz. The sample injection volume was 5 μL, and data was collected in negative ionization mode. The PQC sample was injected regularly throughout the batch to monitor instrument performance. Data files were converted to Agilent SureMass format and analyzed in Agilent MassHunter Quantitative Analysis software (version 10.1, Agilent Technologies). We restricted our quantitative analysis to the 30 BAs for which we have isotopically labeled ISTDs which were included in the extraction solvent at known and physiologically relevant concentrations. For each of these BAs, quantitative data is generated by calculating a ratio of the analyte peak area to the isotopically labeled internal standard peak area. These were the only BAs that were used when calculating the BA pools. BAs for which specific standards were not included as ISTDs are reported as AUC.
Metabolomics data analysis:
We focused on BA reported to have immunomodulatory properties13,15,49 that could be confidently identified with our LC-MS method. Whenever a BA showed absolute or AUC values below the assay lower limit of detection (LOD), its concentration was reported as a 0. Log transformation was applied to all concentrations and AUCs for downstream analyses. Pearson correlation was used to assess pairwise associations. Wilcoxon rank-sum test was performed to test differential abundance of BAs between GVHD and control groups for peri-engraftment and peri-GVHD onset cohorts. BA differential abundance was assessed between the two groups using linear models (limma) and adjusted for patient characteristics and clinical variables, including age, gender, conditioning intensity, donor match, and graft type.56 The model was run separately for the peri-engraftment and peri-GVHD onset cohorts. Benjamini and Hochberg’s correction57 was conducted for p-value adjustments for both Wilcoxon and limma-based tests.
Mouse GVHD histopathology
Approximately 2 cm of the distal small intestine, 2 cm of the proximal colon, a piece of liver and a piece of ear were harvested day 28 after allo-HCT. After fixation in 10% formalin followed by embedding in paraffin and sectioning at 5 μm, hematoxylin and eosin (H&E) staining, Ki67 (Cell Signaling Cat# 12202; RRID:AB_2620142; 1:500) and CD3 (Abcam Cat# ab135372; RRIDL AB_2884903; 1:200) immunohistochemical (IHC) staining and terminal deoxyribonucleotidyltransferase mediated dUTP-biotin nick end labeling (TUNEL) were performed by the Laboratory of Comparative Pathology facility at MSKCC54. Slides were examined by board-certified veterinary pathologist (S.M.) who was blinded to group treatments during evaluation.
Cell isolation from mouse tissues and flow cytometry
Spleen and mesenteric lymph nodes (MLN):
Spleen and MLN were mashed and passed through a 70-μm strainer. Tissue was incubated for 2 min with ACK lysing buffer (ThermoFisher) and rinsed with harvest media [1× RPMI with 10% FBS (ThermoFisher), 25 mM HEPES buffer (ThermoFisher), 1% penicillin/streptomycin (ThermoFisher), 1% L-glutamine (ThermoFisher)].
Small intestine and large intestine:
Approximately 12 cm of the distal small intestine and the combined cecum and colon were processed as the small intestine and large intestine, respectively. After removal of adherent adipose tissue and resection of Peyer’s patches, intestines were opened longitudinally and shaken vigorously in PBS to release contents. Tissues were incubated in 25 ml intestinal intraepithelial lymphocyte (IEL) solution [1× PBS with 2% FBS (ThermoFisher), 10 mM HEPES buffer (ThermoFisher), 1% penicillin/streptomycin (ThermoFisher), 1% L-glutamine (ThermoFisher), plus 1 mM EDTA (Sigma) and 1 mM dithiothreitol (DTT; Sigma) added immediately before use] for 15 min at 37 °C with vigorous shaking (250 rpm). Intestines were removed from IEL suspension, rinsed in PBS and transferred into 50 ml tubes containing 3× one-quarter-inch ceramic beads (MP Biomedicals) and 25 ml collagenase solution [1× RPMI 1640 with 2% FBS (ThermoFisher), 10 mM HEPES buffer (ThermoFisher), 1% penicillin/streptomycin (ThermoFisher), 1% L-glutamine (ThermoFisher), 1 mg ml−1 collagenase A (Sigma) and 1 U ml−1 DNase I (Sigma)]. Following incubation for 30 min at 37 °C with vigorous shaking (250 rpm), digested lamina propria samples were passed through a 100-μm strainer and centrifuged to remove collagenase solution. The lamina propria fractions were washed by centrifugation (5 min at 450g) in 44% Percoll (ThermoFisher) in PBS to remove debris and excess epithelial cell contamination before ex vivo restimulation for analysis of cytokine production.
Flow cytometry:
For cytokine production analyses, cells were incubated for 3 h at 37 °C/5% CO2 in restimulation media [complete RPMI 1640 with 5% FBS, 50 ng ml−1 PMA (Sigma), 500 ng ml−1 ionomycin (Sigma), 1 μg ml−1 brefeldin A (Sigma) and 2 μM monensin (Sigma)]. Cells were transferred into V-bottom plates (Fisher), pelleted by centrifugation, and incubated with extracellular antibody staining mix containing Ghost Dye™ Violet 510 (TonboBio). For cytokine production cells were fixed and permeabilized with BD Cytofix/Cytoperm for 20 min at 4 °C. Antibodies against intracellular antigens were diluted in 1× BD Perm/Wash buffer and cells were stained for 30 min at 4 °C. Antibodies used included anti-mouse CD4 (BD Biosciences Cat# 564667; RRID AB_2722549; 1:500); anti-mouse CD8a (Cat# 564297; RRID: AB_2722580; 1:800); anit-mouse IFN gamma (Tonbo Biosciences; Cat# 75–7311; RRID: AB_2621970; 1:400); anti-mouse CD45 (BioLegend Cat# 103136; RRID: AB_2562612; 1:600); anti-mouse MHC Class I H2kb (ThermoFisher Cat# 46-5958-82; RRID: AB_2016714; 1:200); anti-mouse/human TCR beta (ThermoFisher Cat# 61-5961-82; RRID: AB_2574644; 1:500); anti-mouse CD90.2 (ThermoFisher Cat# 47-0902-82; RRID: AB_1272187; 1:400); anti-mouse CD3 (Biolegend Cat# 100222; RRID: AB_2242784; 1:100) and anti-mouse CD25 (Cat# 102049; RRID AB_2564130; 1:200).
Bulk RNA sequencing
After perfusion with PBS, a piece of liver weighing approximately 50mg was collected into Trizol (Invitrogen). IEL isolation was carried out as described above. Approximately 1/10 of the IEL suspension was added to Trizol LS (Invitrogen) according to manufacturer’s instructions. Liver and IEL samples were submitted for RNA extraction and sequencing at the IGO at MSKCC.
RNA extraction:
Tissue suspended in TRIzol was homogenized with metal beads. Phase separation in homogenized tissue or in cells lysed in TRIzol was induced with 200 μL chloroform and RNA was extracted from the aqueous phase using the MagMAX mirVana Total RNA Isolation Kit (ThermoFisher) on the KingFisher Flex Magnetic Particle Processor (Thermo Scientific catalog) according to the manufacturer’s protocol. Samples were eluted in 34 μL elution buffer.
Transcriptome sequencing:
After RiboGreen quantification and quality control by Agilent BioAnalyzer, 500 ng of total RNA with RIN values of 7.2–10 underwent polyA selection and TruSeq library preparation according to instructions provided by Illumina (TruSeq Stranded mRNA LT Kit, catalog # RS-122–2102), with 8 cycles of PCR. Samples were barcoded and run on a NovaSeq 6000 in a PE100 run, using the NovaSeq 6000 S2 Reagent Kit (200 Cycles) (Illumina). An average of 30 million paired reads was generated per sample. Ribosomal reads represented 0.2–0.9% of the total reads generated and the percent of mRNA bases averaged 93%.
Data analysis:
Quality control was performed with FastQC v0.11.958. Raw reads were aligned with STARaligner (v2.7.0e)59 against the mouse genome (GRCm38.p5) and aligned reads were assigned to genes using featureCounts v1.6.359. The resulting gene count tables were imported to R for further processing, analysis, and visualization. Lowly expressed genes were filtered out using the edgeR’s filterByExpr() function and normalization factors for library size scaling were calculated using the edgeR’s calcNormFactors() function. Differential expression analysis was performed using the limma’s voom() function and a threshold of FDR ≤0.05 was set to define significantly changed genes between conditions. The table with all differentially expressed genes and relative fold changes was used as a pre-ranked list for GSEA v4.3.230 mouse gene set hallmark resource (mh.all.v2022.1Mm.symbols.gmt) to predict signaling pathways enriched in any of the pairwise comparisons. Pathways were defined as enriched if FDR ≤0.05.
FXR luciferase assays
3 × 104 cells expressing a human FXR luciferase reporter (Signosis Catalog Number: SL-0055) were seeded into white 96-well plates (Thermo Scientific) in 50uL of DMEM with 10% FBS (Gibco). After an overnight incubation, cells were exposed to BA or DY268 (Tocris, 20nM) in 50μL for another 22–24h. Viability was determined using Hoechst 33342 (Invitrogen). Cells were processed according to the manufacturer’s instructions using the cell lysis buffer (Signosis) and Luciferase substrate (Signosis). Fluorescence (excitation 350 nm, bandwidth 20.0 nm; emission 470 nm, bandwidth 20.0 nm; number of flashes: 30; integration time: 40μs; settle time: 0 ms; Mode: top. Gain: optimal. Z-position: 20000um: Z position mode: manual) and luminescence were read in a Spark multimode microplate reader (Tecan).
Microbiome sequencing and analyses
Samples were processed for DNA extraction and sequenced in a central laboratory20,60. The 16S ribosomal-RNA gene region V4–V5 was amplified and sequenced on the iIlumina MiSeq platform. Amplicon sequencing variants (ASV) were called using the DADA2 pipeline and further mapped using the National Center for Biotechnology information 16s rRNA reference genome and BLAST61–63. α-diversity was calculated with the Simpson reciprocal index. Metagenomic sequencing was performed64. Wilcoxon rank-sum test was used to assess differential expression of genes.
The species that are significantly different between the BM and BM+T mice group on day 7 were identified with three tools metagenomeSeq65 (V 1.38.0), MaAsLin266 (1.10.0) and Corncob67 (V 0.2.0). Species relative abundance and counts data were obtained using MetaPhlAn68 (V 3.0). Estimated number of reads from the clade were used in the Corncob and metagenomeSeq tools, whereas relative abundance was used in the MaAsLin2. The features with adjusted p values less than 0.25 were considered significant. A Venn diagram was made to compare the similarity of the significant features found by the three tools using ggvenn package (V 0.1.9). To compare the significance level of same feature among different tools, the coefficients for each feature were scaled to relative portion to the absolute maximum value of the coefficient identified within the same tool. Then a dot chart was made to visualize the results using the ggpubr package (V 0.4.0), illustrating the relative coefficient of each significant feature per tool, and the associated corrected p values.
Shotgun metagenomic sequencing was performed using the Illimina TruSeq DNA library preparation kit and the Illumina platform, targeting approximately 10–20 million reads per sample.64 Functional annotation of the samples was retrieved using HUMAnN68 (V 3.0) with the UniRef90 database. The gene family results were converted to KEGG Ortholog (KO) results using the humann_regroup_table function. And then the converted KO results were normalized using the human_renorm_table function in this tool to counts per million (CPM) units. The feature with KO ID as K01442 was identified as the BSH gene. The normalized KO abundance results were also stratified to show the species contribution with humann_split_stratified_table function.
Since bai operon genes are not diverse, we used Diamond69,70 to identify similar alignments based on a single homolog per gene. We used the reference sequences for eight experimentally verified representative genes. After alignment with Diamond, we filtered for alignments with at least 70 percent identity matches and alignment length of 30 (amino acids). Additionally, we ensured that the alignment for any sequence in the first read (R1) matches the alignment in the corresponding sequence in the second read (R2) before quantification and summation. Kaiju was used for metagenomic taxonomy after applying Kneaddata (to remove known contaminants).71
In vitro human T cell assays
Human T cells from healthy donors PBMCs (New York Blood Center) were freshly isolated for each assay using the Pan T Cell Isolation Kit (Miltenyi) according to manufacturer’s instructions. Typical purity was confirmed to be at least 90%. Plates (24-well, Falcon) were coated with 2μg/mL of anti-CD3 human antibody (Biolegend) for 2 hours at 37°C. T cells (1 × 106) were then seeded in 1mL of TexMACS medium (Miltenyi) supplemented with 1μg/mL of anti-CD28 human antibody (Biolegend), 100 IU of recombinant human IL-2 (Miltenyi Biotec). Cells were treated with CDCA (Cambridge Isotope Laboratories), UDCA (Cambridge Isotope Laboratories), GW4064 (Sigma-Aldrich), DY268 (Tocris), or vehicle (DMSO) and incubated for 48 hours at 37°C/5% CO2 during the activation phase. Activated T cells were then transferred into fresh uncoated 24 wells plates and allowed to proliferate for another 96 hours in the presence of drugs, BAs or DMSO. T cell proliferation was measured by quantifying the cell confluence using the Incucyte S3 Live-Cell Analysis Instrument (Sartorius). Cell viability was assessed with the Incucyte Cytotox Green Dye (Sartorius). Cells activated for only 48 hours in the presence of DMSO were used as vehicle controls. Cells subjected to continuous activation by adding anti-CD3/CD28 and IL-2 during the proliferative phase were used as proliferation controls. For flow cytometric analysis, cells were stained with anti-CD3 (BD Biosciences Cat# 612893; RRID: AB_2870181; 1:200), Zombie UV viability dye (Biolegend) and anti-CD25 (BD Biosciences Cat# 612918; RRID: AB_2870203; 1:100) and acquired on a Cytek Aurora (Cytek Biosciences). The flow cytometry data were analyzed using FlowJo™ v10.8 Software (BD Life Sciences).
Single cell RNA sequnencing
Human T cells were activated for 48 hours and transferred to fresh uncoated plates as above, and then allowed to proliferate in the presence of drugs, BAs, or DMSO for 24 hours. During the proliferation phase, cells were treated with vehicle (DMSO), CDCA (100nM), UDCA (100nM), GW4064 (1uM), DY268 (1uM) or activating signals (2ug/mL of anti-CD3 human antibody, 1ug/mL of anti-CD28, 100 IU of recombinant human IL-2; continuous activation). After 24 hours of proliferation, cells were collected for single cell analysis.
Single cell RNA sequencing (scRNA-seq) of T cell suspensions was performed on Chromium instrument (10X genomics) per user guide manual for 3′ v3.1. In brief, human T cells were washed once with PBS containing 1% bovine serum albumin (BSA) and resuspended in PBS containing 1% BSA to a final concentration of 1,000 cells per ul. Live cells were enriched applying the LeviCell v1 system following the user guide manual (20’ incubation time in Levitation Buffer with concentration of 150mM Levitation Agent). The viability of cells after enrichment was above 80%, as confirmed with 0.2% (w/v) Trypan Blue staining (Countess II). Cells were captured in droplets. Following reverse transcription and cell barcoding in droplets, emulsions were broken and cDNA purified using Dynabeads MyOne SILANE followed by PCR amplification per manual instruction. A total of 30,000 cells were targeted for each sample.
For scRNA-seq seq, individual cell suspension were incubated for 30 min with hashtag oligonucleotide-conjugated antibodies (Extended Data Table 9) in addition to FACS antibodies. Samples were multiplexed together on one lane of 10X Chromium (using Hash Tag Oligonucleotides - HTO)72. Final libraries were sequenced on Illumina NovaSeq S4 platform (R1 – 28 cycles, i7 – 8 cycles, R2 – 90 cycles). Viable cells were identified based on library size and complexity, whereas cells with >20% of transcripts derived from mitochondria were excluded from further analysis.
Hastag antibodies used:
TotalSeqTM-B0301 anti-mouse Hashtag 1 (Cat#155831; 1:50); TotalSeqTM-B0301 anti-mouse Hashtag 2 (Cat#155833; 1:50); TotalSeqTM-B0301 anti-mouse Hashtag 3 (Cat#155835; 1:50); TotalSeqTM-B0301 anti-mouse Hashtag 4 (Cat#155837; 1:50); TotalSeqTM-B0301 anti-mouse Hashtag 5 (Cat#155839; 1:50); TotalSeqTM-B0301 anti-mouse Hashtag 6 (Cat#155841; 1:50) and TotalSeqTM-B0301 anti-mouse Hashtag 7 (Cat#155843; 1:50).
Generation of single-cell data:
Raw sequencing data from cDNA and hash libraries was demultiplexed and aligned with the 10X Genomics Cell Ranger pipeline (v.7.1.0) to a GRCh38–2020-A reference genome. Single-cell cDNA library preparation and sequencing have been, respectively, performed at the Single-Cell Research Initiative (SCRI) and Integrated Genomics Operation (IGO) core facilities of Memorial Sloan Kettering Cancer Center.
Processing and quality control of single-cell data:
Filtered barcode-cell matrices returned by the CellRanger pipeline were used as starting point for analysis. To verify that no cell populations were systematically lost during the CellRanger filtering step, cell calling using the emptyDrops package73 was performed on a subset of the data. Low quality cells were identified and removed based on the following criteria: (1) less than 500 UMIs, (2) more than 20% of reads originating from mitochondrial RNA, and (3) lower than normal molecular complexity, as defined by a loess fit of the relationship between number of unique genes and number of unique molecules.
Single cell RNA-seq preprocessing and downstream data analysis:
The Cell Ranger generated filtered_feature_bc_matrix.h5 files were processed per instructions on the shunPykeR GitHub repository74, an assembled pipeline of publicly available single cell analysis packages, allowing reproducible data analysis using Python and R code. Genes not expressed in any cell, or genes related to ribosome or hemoglobin were removed from downstream analysis. Each cell was normalized to a total library size of 10,000 reads and then gene counts were log-transformed using the log(X+1) formula, in which log refers to the natural logarithm. To reduce noise prior to data clustering, principal component analysis was applied. The knee point (eigenvalues smaller radius of curvature) was used, to select the optimal number of principal components to retain for each datase. We then identified clusters within the PCA-reduced data applying the Leiden clustering75. The quality of the single cells was computationally assessed based on total counts, number of genes, mitochondrial and ribosomal fractions per cell. Negative indicators of cell quality was the low total counts, low number of genes (≤1000) and high mitochondrial content (≥0.2). Cells characterized by more than one negative indicator were considered as “bad” quality cells. We assessed bad quality cells and contaminants in a cluster basis (instead of individually) to avoid bias. Clusters having a bad quality profile and/or a high number of contaminating cells were eliminated. Cells marked as doublets based on their hash id or scrublet,76 as well as cells expressing markers for Natural Killer (NK) and B cells were removed from downstream analysis (Extended Data Fig. 9). After eliminating these cells, PCA and unsupervised clustering was reapplied to the filtered data as described above, followed by batch effect correction across all samples using harmony77 to allow annotation of cell type subsets within the dataset.
Differential expression analysis:
Differential expression analysis was performed using Scanpy’s rank_gene_groups() function with default parameters and using the 2-sided Wilcoxon rank-sum method78. Significance was defined by FDR-adjusted p-value less than 0.05.
Pathway enrichment analysis:
Pathway enrichment analysis was performed with GSEA (v4.3.2)30 as per gene list and rank metric provided. The GSEA Preranked module was used to predict pathway enrichment in threshold free comparisons: (a) CDCA vs vehicle, (b) UDCA vs vehicle, (c) GW4064 vs vehicle and (d) DY268 vs vecicle. Rankings were created for differentially expressed genes using the Wilcoxon Z-score in descending order. Predicted pathways with an FDR<=0.05 were considered as significantly enriched.
In vitro mouse T cell assay
T cells from TΔFXR mice or their WT control littermates were prepared from total splenocytes using the Pan T Cell Isolation Kit II (Miltenyi). T cells were activated with plate-bound antibodies (2.5ug/mL of anti-CD3, Biologend; 1.5ug/mL of anti-CD28, Biolegend) and 100 IU of IL-2 (Miltenyi Biotec), in a 48 wells plate (0.5×106 per well). After 48 hours, T cells were transferred to a new plate without plate-bound antibodies (except positive control) to allow cells to proliferate. During the activation and proliferation phases, cells were treated with CDCA (100nM), UDCA (100nM) or vehicle (DMSO). For flow cytometric analysis, cells were collected after 48 hours of proliferation, stained with anti-CD3 (BioLegend Cat# 317301; RRID: AB_571926; 2.5ug/ml), Zombie UV viability dye and anti-CD28 (BioLegend Cat# 302943; RRID: AB_2616667; 1.5ug/ml) and acquired on a Cytek Aurora (Cytek Biosciences). The data were analyzed using FlowJo™ v10.8 Software (BD Life Sciences).
Time-to-event data analysis
Kaplan-Meier estimator and the log-rank test were used to compare overall survival (OS) after transplantation between the UDCA and the non-UDCA groups. A multivariable Cox regression model was fitted to investigate the association between ursodiol and OS after adjusting for clinical variables, including age, gender, donor match, graft, and intensity. Four competing risk outcomes were defined, namely censored, relapse or progression of disease (relapse/POD), GVHD-related mortality (GRM) and others. A patient was censored if alive at the end of follow-up without any relapse/POD. The relapse/POD failure type was observed when a patient had relapse/POD during follow-up or died from POD. GRM was defined as death in patients with GVHD. Patients not having the above three failure types were categorized into others. The Gray’s test was performed to test the difference of cumulative incidence rates of GRM between the UDCA and non-UDCA groups. A multivariable Fine-Gray proportional sub-distribution hazards model was fitted to investigate the association between UDCA use and GRM, adjusting for age, gender, donor match, graft, and intensity.
Statistics & Reproducibility
Analysis of processed data were conducted using R v4.3.0, Graphpad Prism v7.5.0. The flow cytometry data were analyzed using FlowJo™ v10.8 Software (BD Life Sciences). Group size for murine BM+T experiment is based upon the statistical premise that 20 animals are required to detect a 50% difference with statistical significance. In the clinical portion of the study, a case-control design was employed, to select appropriate controls for patients with GI GVHD. We excluded from the analysis patients who died early (<48 days which corresponds to the median of the time to GVHD onset plus a standard deviation), recipients of T cell depleted grafts due to their lower GVHD incidence; individuals lacking available or evaluable samples, and those who received combined grafts (bone marrow graft and peripheral blood stem cell graft). No data was excluded from the rest of the analysis. The Investigators were not blinded to allocation during experiments and outcome assessment.
Statistical significance was established at a p value < 0.05. For comparisons between mice receiving BM vs BM+T, a two-tailed Mann-Whitney test was utilized. Continuous variables between patient groups were compared using a Wilcoxon rank-sum test. In vitro T cell assay confluence was assessed and compared between different treatment groups using two-way or one-way ANOVA, followed by multiple t-tests with Bonferroni correction. Gene expression analysis utilized a Wilcoxon rank-sum test, with significance set at FDR ≤ 0.05. Pearson correlation was employed for calculating R values. OS was compared using Kaplan-Meier estimator and log-rank test for univariable analysis, and Cox regression model for multivariable analysis. Cumulative incidence of GRM was assessed using Gray’s test, and multivariable analysis was performed with the Fine-Gray proportional sub-distribution hazards model.
Extended Data
Extended Data Fig. 1. Alloreactive T cells alter intestinal and circulating BAs and host metabolism.
Lethally irradiated 6–8 week old female BALBc mice were transplanted with 10 × 106 BM cells alone (BM) or together with 1 × 106 T cells (BM+T). (a-c) BAs were quantified on day 7 post-transplant by liquid chromatography-mass spectrometry (LC-MS) in the cecal contents and plasma. (a) Ratio of microbe- to host-derived BAs (b) Ratio of unconjugated to glycine- and taurine-conjugated BA. (c). Estimated cecal levels of the T cell modulatory BAs 3-oxoLCA and isoLCA (below limit of detection). Weight loss (a), clinical GVHD scores (b) and cumulative food intake per mouse at day 7 post-transplant (c). Plasma levels of AST (d), ALT (e), albumin (f), cholesterol (h), and triglycerides (g). Data combined from two independent experiments (n=10). Data shown as mean ± S.D and statistical significance determined by two-tailed Mann-Whitney test.
Extended Data Fig. 2.
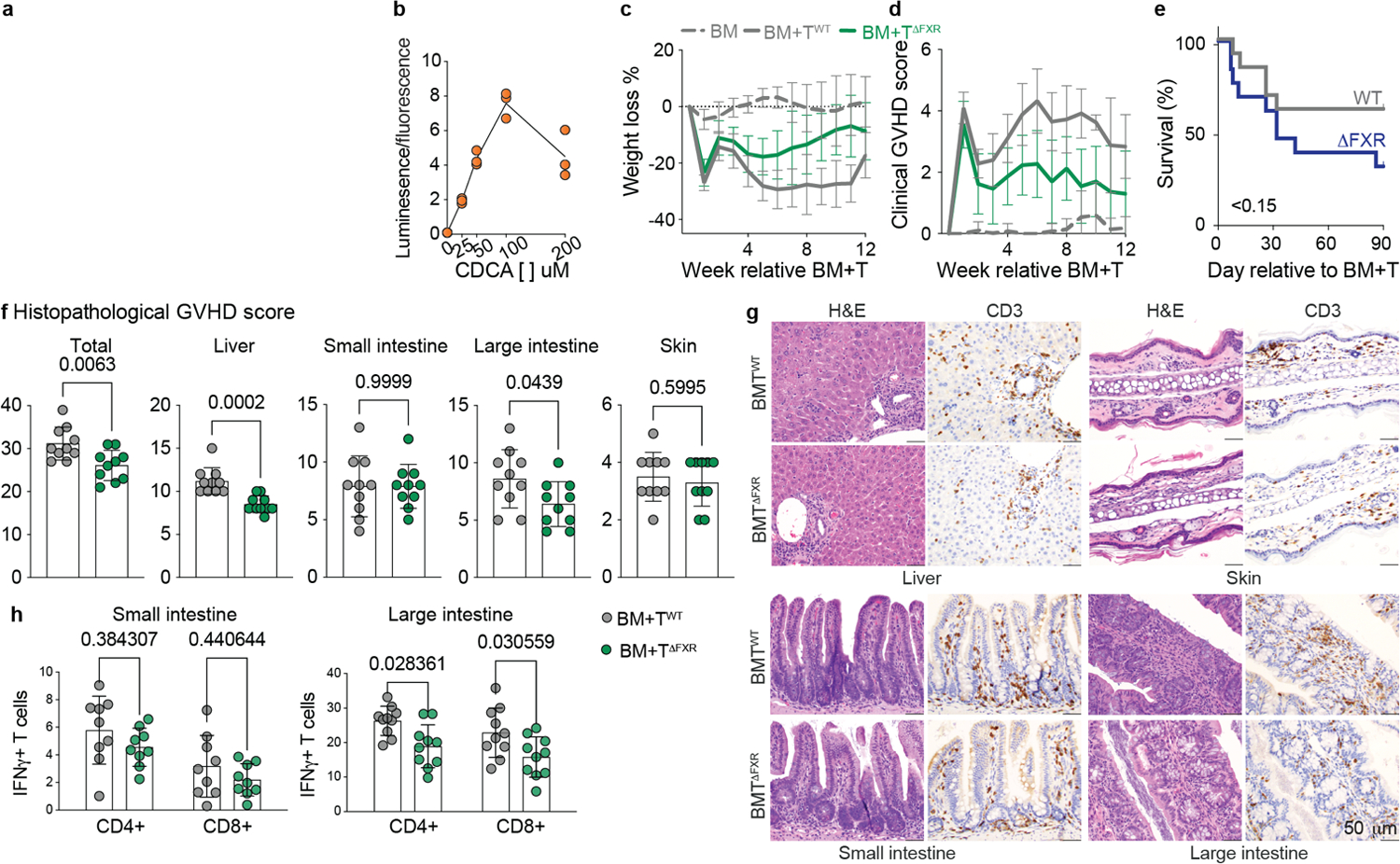
(a) Principle of the FXR luciferase reporter assay used in Fig. 3a and b and Extended Data Fig. 2b. (b) Stably transfected HepG2 cells expressing luciferase under the control of an FXR-responsive element were treated with the indicated doses of CDCA. Luciferase units (luminescence) were normalized to cell viability assessed by Hoechst 33342 staining (fluorescence). Data representative of two independent experiments and presented as technical triplicates with medians connected. Weight loss (c) and clinical GVHD score (d) of survival experiment shown in Fig. 3. Mice transplanted transplanted with 10 × 106 B6 BM cells alone or together with 1 × 106 T cells from either Nr1h4fl/fl (BM+TWT) or Cd4Cre Nr1h4fl/fl (BM+TΔFXR) mice on a C57Bl/6N background. Data combined from three independent experiments (BM group n=20, BM+T groups n=30 per group) and connected as means ± S.D. (e) Survival of cohoused WT or nr1h4−/−l (Δ FXR) B6 mice receiving BALBc BM+T. Data combined from three independent experiments (n=13 per group). Statistical significance was determined using log-rank test. (f-h) BALBc recipient mice transplanted with 10 × 106 B6 BM cells alone or together with 1 × 106 T cells from either Nr1h4fl/fl (BM+TWT) or Cd4Cre Nr1h4fl/fl (BM+TΔFXR) mice on a C57Bl/6N background. (f) Organ-specific and compound histopathological scores at day 28 post-transplant of transplanted mice with representative histology images (g). Data from one experiment (n=10 per group) and statistical significance was determined by by two-tailed Mann-Whitney test. (h) Production of IFNγ by CD4+ and CD8+ T cells in the smallI and large intestine lamina propria 14 days after transplant. Data combined from two (n=10 per group) and presented as mean ± S.D. Statistical significance was determined by by two-tailed Mann-Whitney test.
Extended Data Fig. 3.
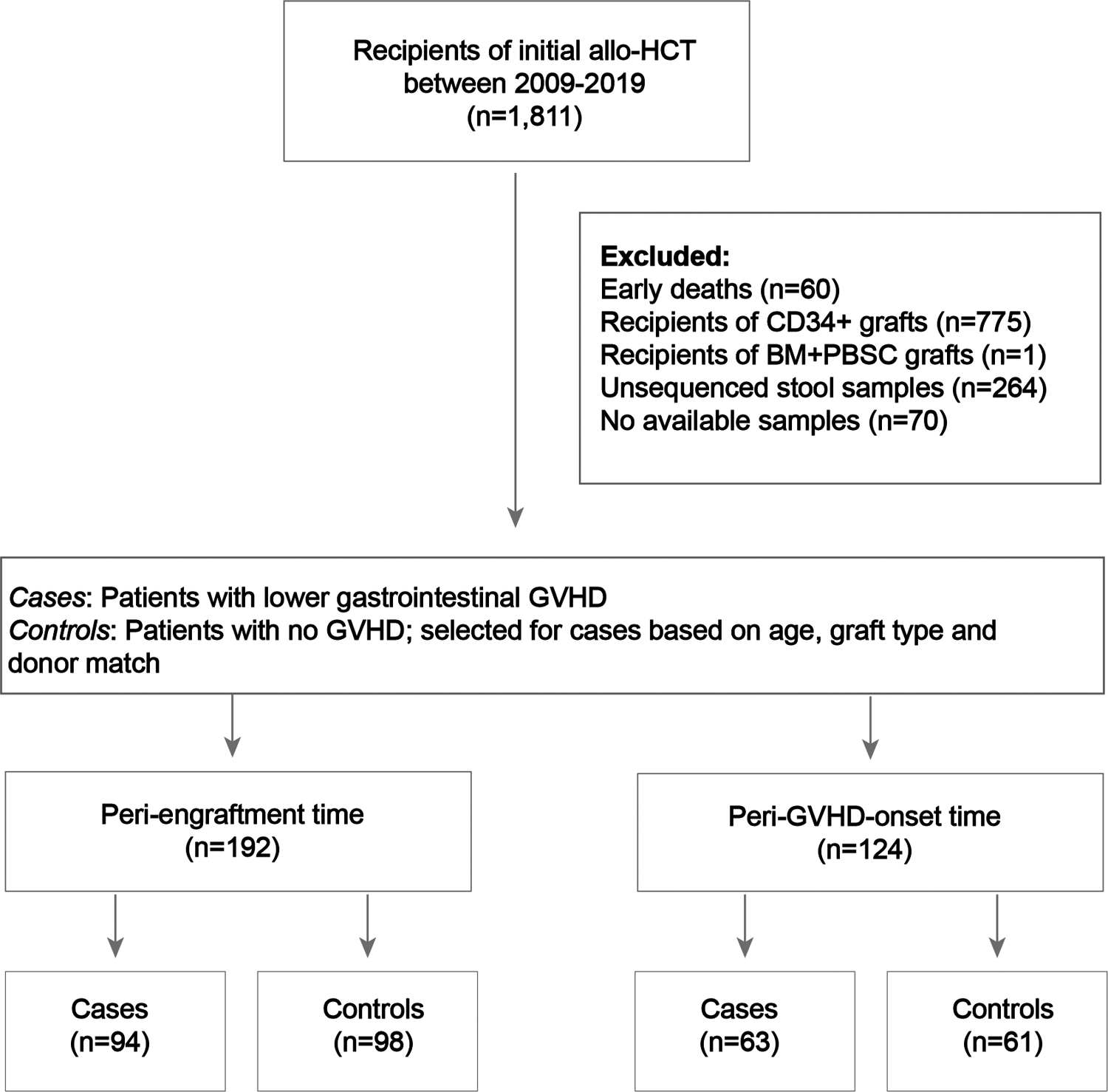
Consort diagram for the data shown in Fig. 4 and 5 (and Extended Data Fig. 5–8). PBSC: peripheral blood stem cell graft.
Extended Data Fig. 4.
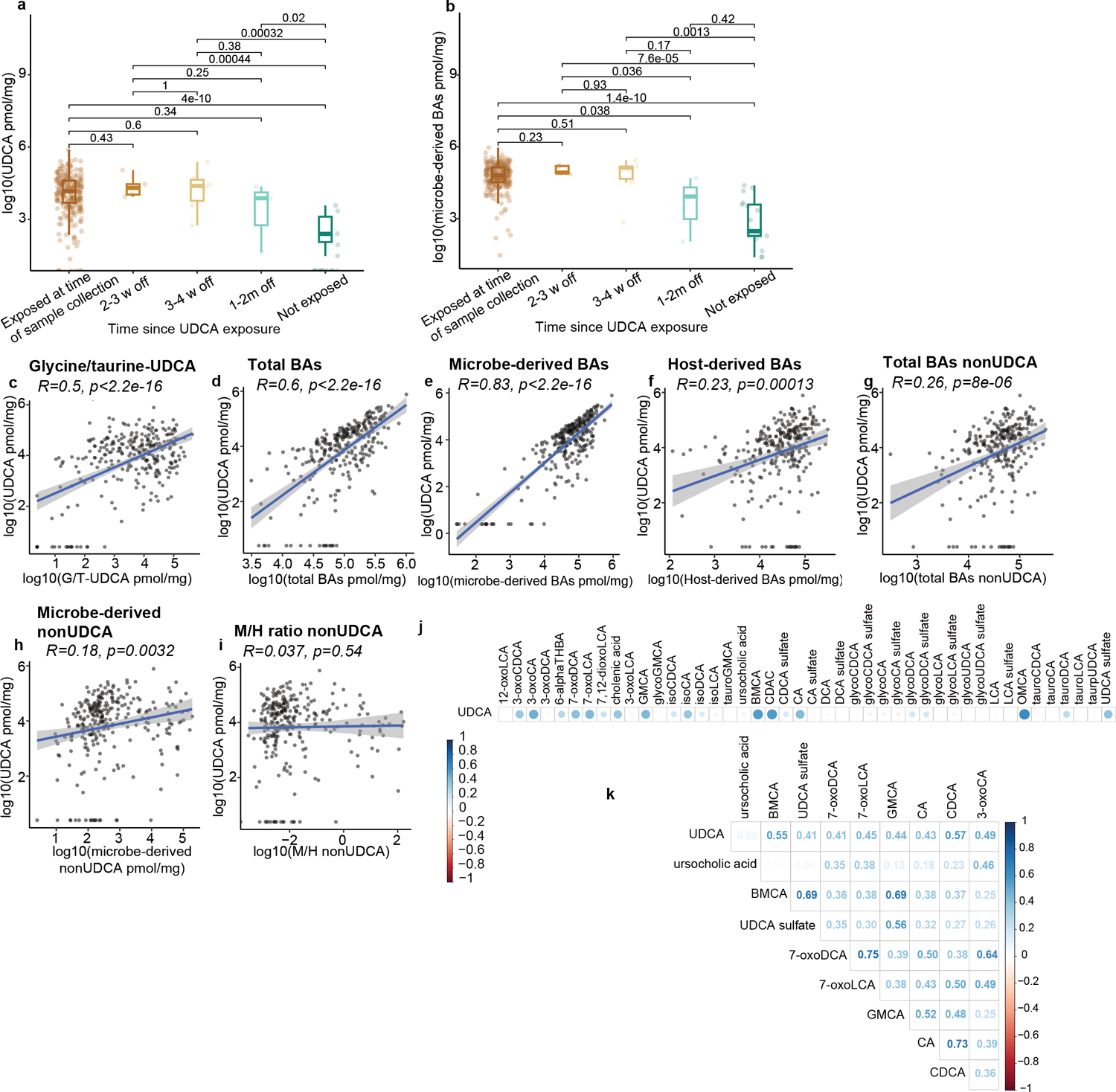
Effects of UDCA exposure on the intestinal BA pool in n=280 samples from either peri-engraftment or peri-GVHD onset timepoints. Fecal concentrations of UDCA (a) and microbe-derived BAs (b). UDCA exposure status is shown in the x-axis (w = weeks; m = months since last exposure). Statistical significance determined by the 2-sided Wilcoxon Rank-sum test. The boxplot center line corresponds to the median, box limits correspond to the 25th and 75th percentile, and whiskers correspond to 1.5x interquartile range. Correlation of fecal UDCA concentrations with the levels of conjugated UDCA (conj-UDCA, c), total BAs (d), microbe-derived BAs (e), host-derived BAs (f), nonUDCA total BAs (g) nonUDCA microbe-derived BAs (h), and microbe- to host- derived (M/H) ratio excluding UDCA (nonUDCA, i). The solid line represents a linear regression model fitted to the data. The shaded region surrounding the line indicates a 95% confidence interval for the regression line. Total BAs nonUDCA (g) are measured in pmol/mg. (j,k) Correlation matrix of the BA species covarying with UDCA. (k) Showing BA species with a Pearson correlation coefficient (R>0.4).
Extended Data Fig. 5.
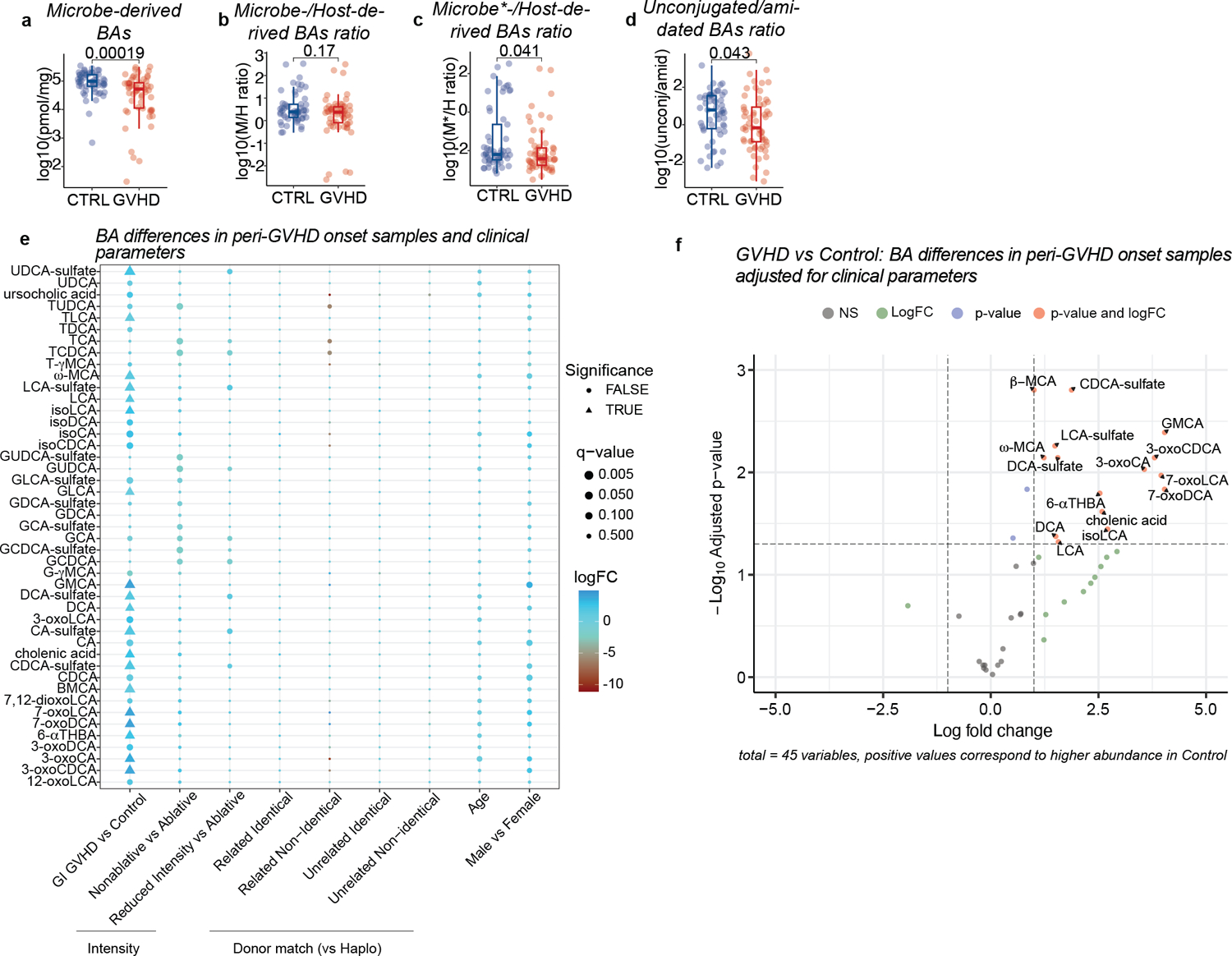
Fecal BA profiles at the peri-GVHD onset time point. Showing the levels of total microbe-derived BAs (a), microbe- to host-derived (M/H) BA ratio (b), M*/H (i.e., nonUDCA M/H) BA ratio (c), and the ratio of unconjugated to amidated BAs (d). Data representative of 57 control and 58 GVHD patients. Statistical significance determined with the 2-sided Wilcoxon Rank-sum test. The boxplot center line corresponds to the median, box limits correspond to the 25th and 75th percentile, and whiskers correspond to 1.5x interquartile range. (e-f) Differential abundance of BAs between GVHD and controls in peri-GVHD onset samples after multivariate adjustment. (e) Grid plot showing significance status, q-values, and log fold changes of BAs relative to indicated clinical variables. (f) Volcano plot showing log-transformed adjusted p-values vs log fold changes of BAs between GVHD and control patients. Statistical comparison was made using the two-sided empirical Bayes moderated t-test and p-values were adjusted using the Benjamini-Hochberg method.
Extended Data Fig. 6.
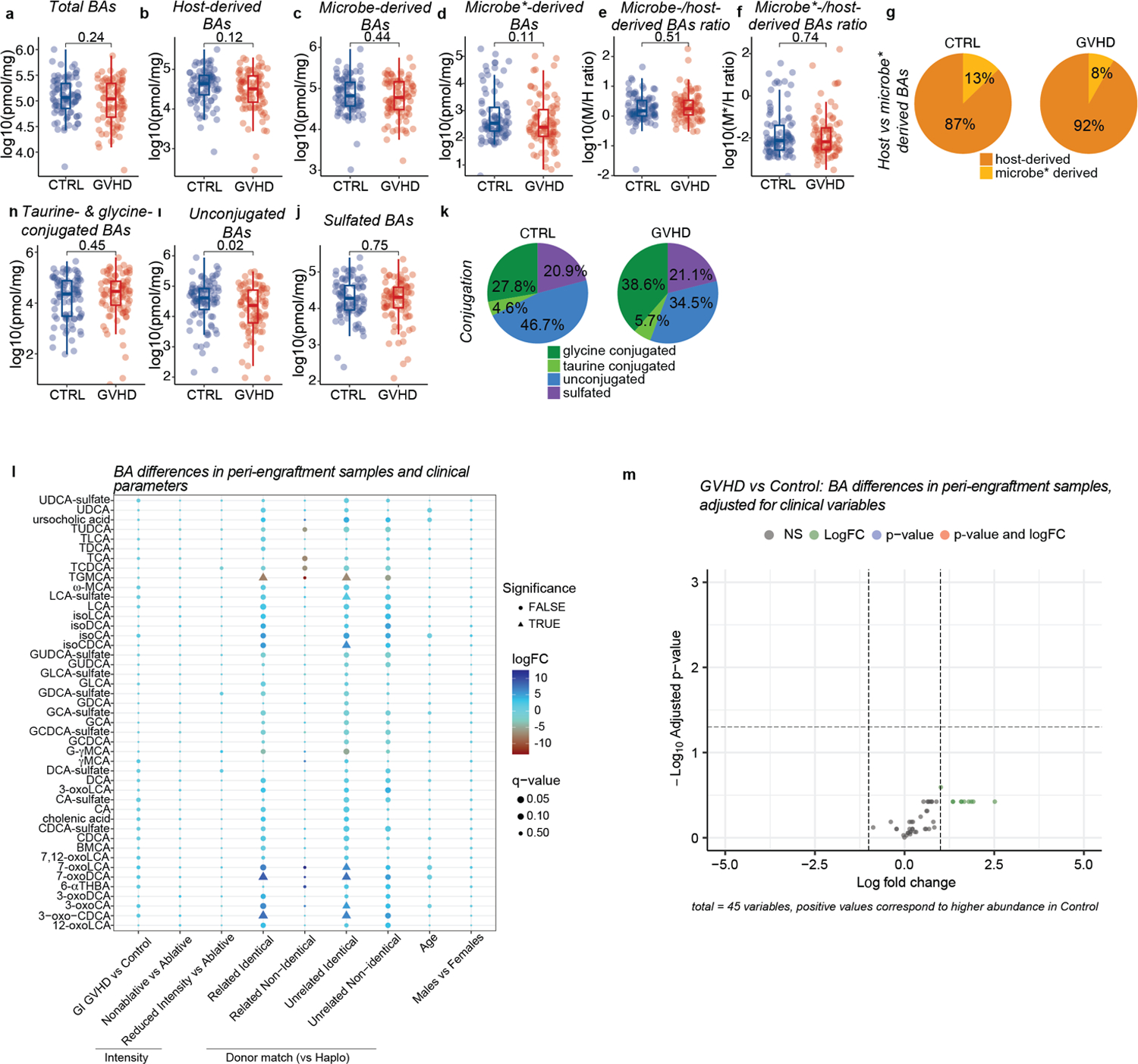
Fecal BA profiles at the peri-engraftment time point. Total BAs (a), host-derived (b), microbe-derived (c), microbe*-derived (d), microbe-derived to host-derived (M/H) BA ratio (e), M*/H (nonUDCA M/H) BA ratio in patients that develop GVHD vs controls (f). (g) Pie chart showing the averaged relative contributions of host-derived and microbe*-derived to the calculated total BA pool. Glycine- and taurine-conjugated (h), unconjugated (i), and sulfated (g) BAs. Pie chart showing the averaged percentages of glycine- and taurine-conjugated, unconjugated and sulfated BAs in patients with GVHD vs controls in peri-GVHD onset samples (k). Microbe*-derived BAs: Microbe-derived BAs excluding UDCA. Data representative of 90 control and 86 GVHD patients. The boxplot center line corresponds to the median, box limits correspond to the 25th and 75th percentile, and whiskers correspond to 1.5x interquartile range. Statistical significance determined with the 2-sided Wilcoxon Rank-sum test. (l,m) Differential abundance of BAs between GVHD and control patients in peri-engraftment samples after multivariate adjustment (l) Grid plot showing significance status, q-values, and log fold changes of BAs relative to indicated clinical variables. (m) Volcano plot showing log-transformed adjusted p-values vs log fold changes of BAs between GVHD and control patients. Statistical comparison was made using the two-sided empirical Bayes moderated t-test and p-values were adjusted using the Benjamini-Hochberg method.
Extended Data Fig. 7.
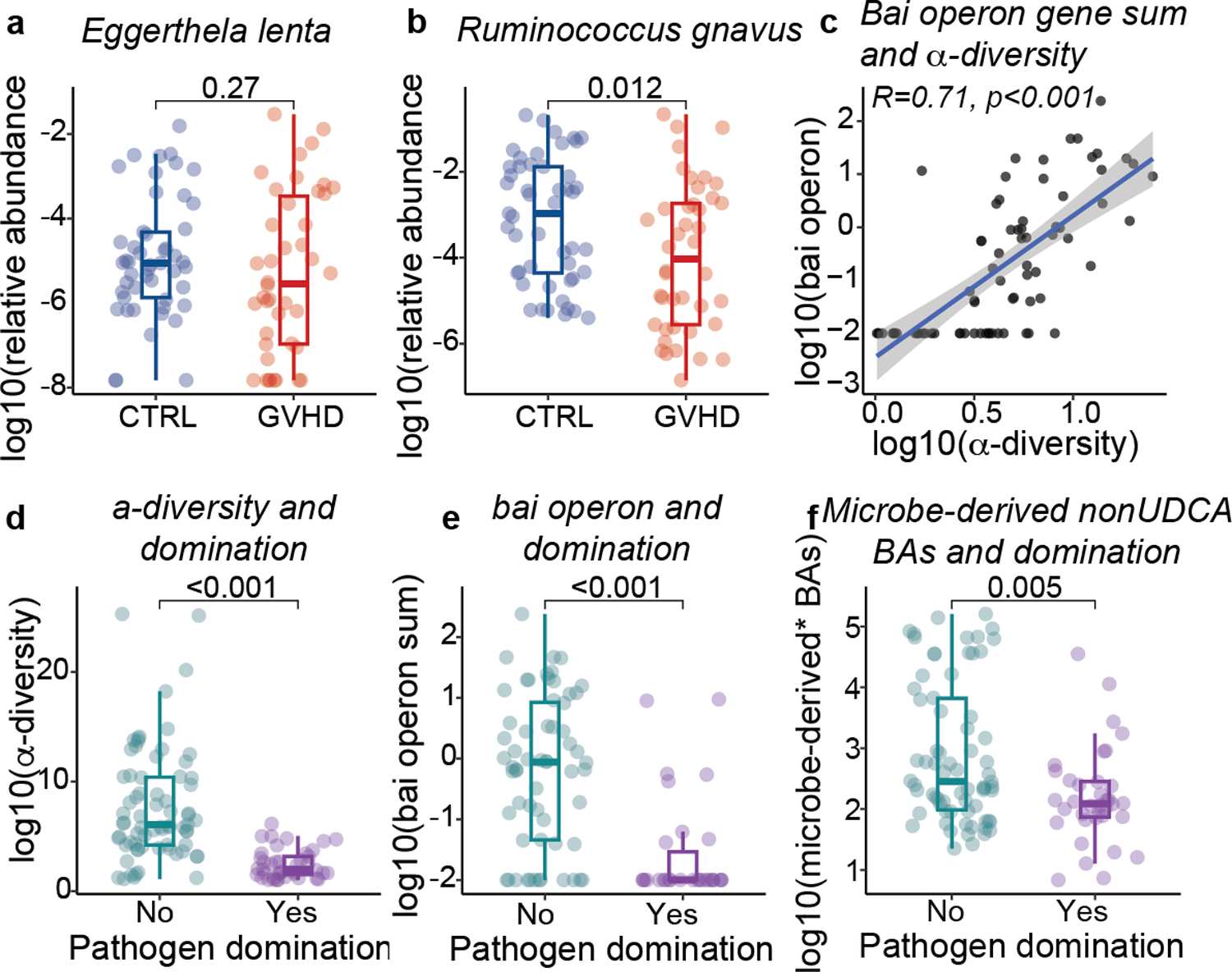
Microbiome features in peri-GVHD onset samples. Relative abundance of (a) Eggerthella lenta and (b) Ruminococcus gnavus. Data representative of 49 control and 42 GVHD patients (c) Correlation of the sum of bai operon gene and α-diveristy as measured by the Simpson reciprocal index. The solid line represents a linear regression model fitted to the data. The shaded region surrounding the line indicates a 95% confidence interval for the regression line. Data representative of 82 patients with peri-GVHD onset samples. (d) α-diversity, (e) sum of bai operon genes identified by shotgun metagenomic analysis (measured in counts per million), and (f) levels of microbe-derived BAs* (pmol/mg) in patients with or without intestinal pathogen domination. Data representative of 41 patients with and 74 patients without pathogen domination. Microbe-derived BAs*= microbe-derived BAs excluding UDCA. Statistical significance determined with the univariate 2-sided Wilcoxon Rank-sum test. The boxplot center line corresponds to the median, box limits correspond to the 25th and 75th percentile, and whiskers correspond to 1.5x interquartile range. R correspond to Pearson’s correlation coefficient.
Extended Data Fig. 8.
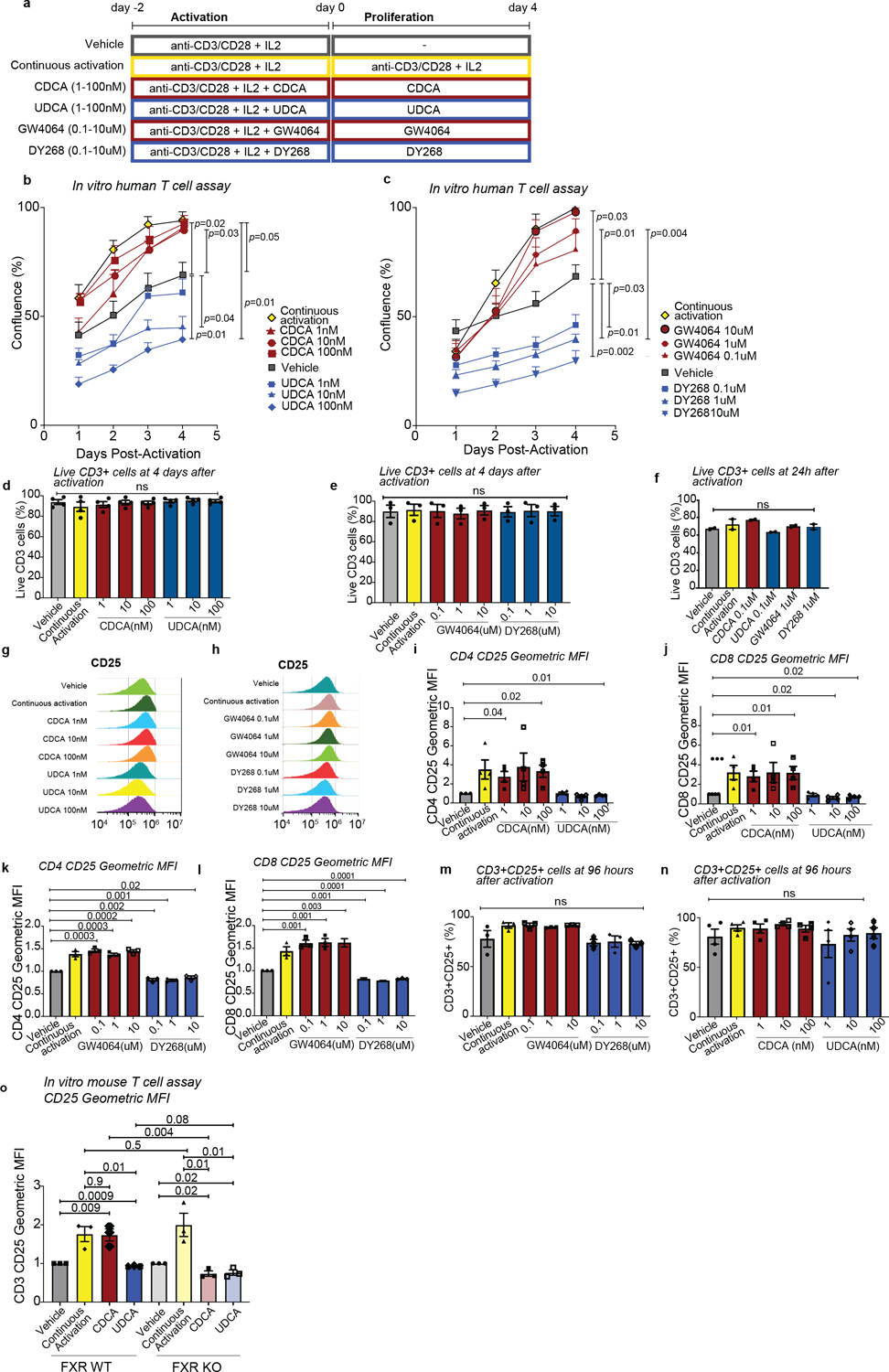
In vitro human T cell proliferation in response to FXR activation or inhibition with drugs or BAs. (a) Experimental design. Purified human T cells were activated with anti-CD3 and anti-CD28 antibodies in the presence of recombinant IL-2 for 2 days and further cultured either in the presence of anti-CD3/anti-CD28 antibodies (continuous activation control) or in their absence (vehicle control) with or without the indicated compounds for 96 hours. Showing T cell confluence in response to CDCA and UDCA (b) or GW4064 and DY268 (c) at the indicated concentrations. Cell viability (d, e, f) and representative histograms (g, h) showing CD25 levels determined by flow cytometric analysis. (i-l) CD25 expression in CD4+ and CD8+ T cells after 96 hours of treatment with CDCA and UDCA (i-j), or GW4064 and DY268 (k-l). Showing the geometric mean fluorescence intensity (MFI) of CD25 in CD25+ T cells. Values were normalized to the MFI of the vehicle-treated group. (m, n) Frequency of CD25 positive celles on day 4 post-activation. (o) CD25 expression from T cells of FXRWT or FXRWT mice treated with anti-CD3 and anti-CD28 antibodies in the presence of IL-2 for 2 days before incubation with CDCA (100nM), or UDCA (100nM), anti-CD3, anti-CD28 and IL-2 (continuous activation), or vehicle for 2 more days. CD25 expression was measured as geometric mean fluorescence intensity (MFI) of CD25 in CD25+ T cells normalized to the MFI of the vehicle-treated group. Statistical analysis was performed by two-way (b,c) or one-way ANOVA followed by multiple t-test with Bonferoni correction (d-f, i-o). Each data point in (i-n) shows the average of technical duplicates for a single donor. Bars denote the standard error of the mean. Data representative of 4 independent experiments with a total of 4 PBMC donors. Each data point in (g) shows the average of technical triplicates from two mice. Bars denote the standard error of the mean. Data representative of 3 independent experiments with a total of 6 mice.
Extended Data Fig. 9.
Quality control of single cell RNA-sequencing profiling of in vitro activated T cells treated with FXR ligands, DMSO or activating signals for 24h. Visualization of 60,767 cells using a uniform manifold approximation and projection (UMAP) of (a) cells from the two donors and (b) per hashtag before eliminating any cells. (c) Total counts (log10 scale) (d) total genes, (e) ribosomal fraction, (f) mitochondrial fraction per cell, (g) predicted doublet and (h) doublet score. (i-k) Cells expressing markers for B and Natural Killer cells were defined as contaminants. (l-m) UMAP and stacked plot showing the fraction of retained (33,634) and removed cells (27,133).
Extended Data Fig. 10.
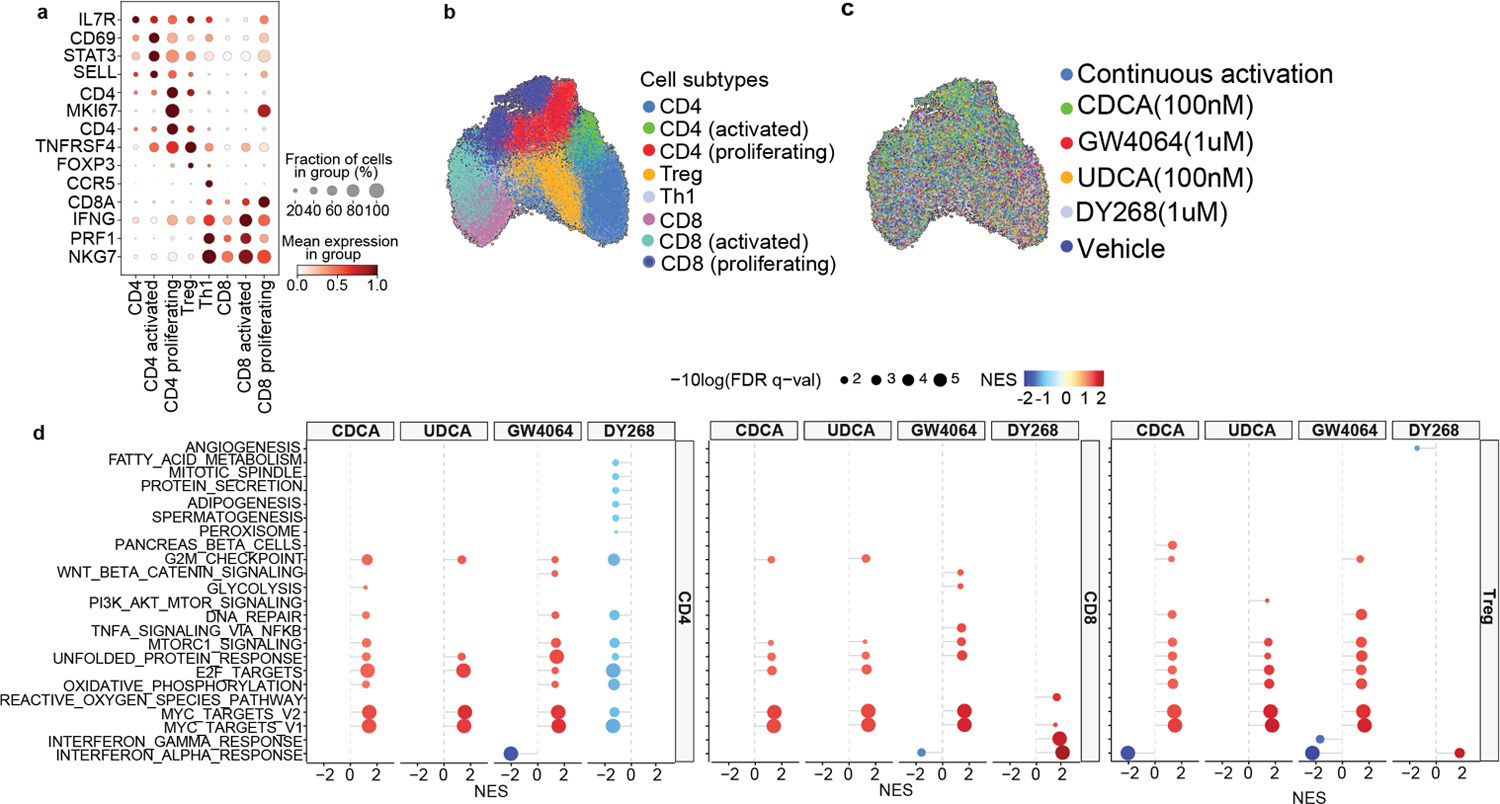
Single cell RNA-sequencing profiling of in vitro activated T cells treated with CDCA (100nM), UDCA (100nM), GW4064 (1uM) and DY268 (1uM) for 24h. (a) Gene markers used to identify cell populations. Visualization of annotated cells using a uniform manifold approximation and projection (UMAP) of (b) subtypes after batch correction and (c) per treatment arm. (d) Gene Set Enrichment Analysis of pathways differentially regulated in the different conditions (CDCA 100nM, UDCA 100nM, GW4064 1uM, DY268 1uM) relative to the vehicle control in CD4+, CD8+ and regulatory T cell populations. Displaying significant pathways.
Extended Data Table 1.
Bile acid pool
| Bile acid | Abbreviation | Host vs microbe | Conjugation |
|---|---|---|---|
| mouse | |||
| alpha-muricholic acid | AMCA | primary/host | unconjugated |
| beta-muricholic acid | BMCA | primary/host | unconjugated |
| chenodeoxycholic acid | CDCA | primary/host | unconjugated |
| chenodeoxycholic acid-3-sulfate | CDCA-sulfate | primary/host | sulfated |
| cholic acid | CA | primary/host | unconjugated |
| cholic acid-3-sulfate | CA-sulfate | primary/host | sulfated |
| deoxycholic acid | DCA | secondary/microbe | unconjugated |
| deoxycholic acid-3-sulfate | DCA-sulfate | secondary/microbe | sulfated |
| glycochenodeoxycholic acid | GCDCA | primary/host | glycine conjugated |
| glycochenodeoxycholic acid-3-sulfate | GCDCA-sulfate | primary/host | sulfated |
| glycocholic acid | GCA | primary/host | glycine conjugated |
| glycocholic acid-3-sulfate | GCA-sulfate | primary/host | sulfated |
| glycodeoxycholic acid | GDCA | secondary/microbe | glycine conjugated |
| glycodeoxycholic acid-3-sulfate | GDCA-sulfate | secondary/microbe | sulfated |
| glycolithocholic acid | GLCA | secondary/microbe | glycine conjugated |
| glycolithocholic acid-3-sulfate | GLCA-sulfate | secondary/microbe | sulfated |
| glycoursodeoxycholic acid | GUDCA | primary/host | glycine conjugated |
| glycoursodeoxycholic acid-3-sulfate | GUDCA-sulfate | primary/host | sulfated |
| lithocholic acid | LCA | secondary/microbe | unconjugated |
| lithocholic acid-3-sulfate | LCA-sulfate | secondary/microbe | unconjugated |
| omega-muricholic acid | OMCA | secondary/ microbe | unconjugated |
| tauro-alpha-muricholic acid | TAMCA | primary/host | taurine conjugated |
| tauro-beta-muricholic acid | TBAMCA | primary/host | taurine conjugated |
| taurochenodeoxycholic acid | TCDCA | primary/host | taurine conjugated |
| taurocholic acid | TCA | primary/host | taurine conjugated |
| taurodeoxycholic acid | TDCA | secondary/microbe | taurine conjugated |
| taurolithocholic acid | TLCA | secondary/microbe | taurine conjugated |
| tauroursodeoxycholic acid | TUDCA | primary/host | taurine conjugated |
| ursodeoxycholic acid | UDCA | primary/host | taurine conjugated |
| ursodeoxycholic acid-3-sulfate | UDCA-sulfate | primary/host | sulfated |
| human | |||
| beta-muricholic acid | BMCA | primary/host | unconjugated |
| chenodeoxycholic acid | CDCA | primary/host | unconjugated |
| chenodeoxycholic acid-3-sulfate | CDCA-sulfate | primary/host | sulfated |
| cholic acid | CA | primary/host | unconjugated |
| cholic acid-3-sulfate | CA-sulfate | primary/host | sulfated |
| deoxycholic acid | DCA | secondary/microbe | unconjugated |
| deoxycholic acid-3-sulfate | DCA-sulfate | secondary/microbe | sulfated |
| glycochenodeoxycholic acid | GCDCA | primary/host | glycine conjugated |
| glycochenodeoxycholic acid-3-sulfate | GCDCA-sulfate | primary/host | sulfated |
| glycocholic acid | GCA | primary/host | glycine conjugated |
| glycocholic acid-3-sulfate | GCA-sulfate | primary/host | sulfated |
| glycodeoxycholic acid | GDCA | secondary/microbe | glycine conjugated |
| glycodeoxycholic acid-3-sulfate | GDCA-sulfate | secondary/microbe | sulfated |
| glycolithocholic acid | GLCA | secondary/microbe | glycine conjugated |
| glycolithocholic acid-3-sulfate | GLCA-sulfate | secondary/microbe | sulfated |
| glycoursodeoxycholic acid* | GUDCA | secondary/microbe/exogenous | glycine conjugated |
| glycoursodeoxycholic acid-3-sulfate* | GUDCA-sulfate | secondary/microbe/exogenous | sulfated |
| lithocholic acid | LCA | secondary/microbe | unconjugated |
| lithocholic acid-3-sulfate | LCA-sulfate | secondary/microbe | unconjugated |
| omega-muricholic acid | OMCA | secondary/microbe | unconjugated |
| taurochenodeoxycholic acid | TCDCA | primary/host | taurine conjugated |
| taurocholic acid | TCA | primary/host | taurine conjugated |
| taurodeoxycholic acid | TDCA | secondary/microbe | taurine conjugated |
| taurolithocholic acid | TLCA | secondary/microbe | taurine conjugated |
| tauroursodeoxycholic acid* | TUDCA | secondary/microbe/exogenous | taurine conjugated |
| ursodeoxycholic acid* | UDCA | secondary/microbe/exogenous | taurine conjugated |
| ursodeoxycholic acid-3-sulfate* | UDCA-sulfate | secondary/microbe/exogenous | sulfated |
Extended Data Table 2.
Patient characteristics
| Peri-engraftment (n=192) | Peri-GVHD-onset (n=124) | Total | |||||
|---|---|---|---|---|---|---|---|
| GI-GVHD, (n=94) | Control, (n=98) | p-value | GI-GVHD, (n=63) | Control, (n=61) | p-value | (n=235) | |
| Age | 59 (48,66) | 60 (49,67) | 0.50 | 56 (46,66) | 59 (51,66) | 0.40 | 59 (48,66) |
| Sex (F) | 36 (34%) | 33 (38%) | 0.50 | 25 (40%) | 21 (34%) | 0.50 | 89 (38%) |
| Disease | >0.90 | >0.9 | |||||
| Leukemia | 46 (49%) | 44 (45%) | 29 (46%) | 26 (43%) | 110 (47%) | ||
| Lymphoma | 32 (33%) | 32 (34%) | 19 (31%) | 22 (35%) | 76 (32%) | ||
| MDS/MPN | 14 (15%) | 17 (16%) | 9 (14%) | 12 (19.7%) | 38 (16%) | ||
| Multiple myeloma | 1 (1%) | 3 (3%) | 1 (1.6%) | 1 (1.6%) | 4 (1.7%) | ||
| Non-malignant | 1 (1%) | 2 (2%) | 2 (3.2%) | 3 (4.9%) | 7 (2.5%) | ||
| Graft | 0.07 | 0.08 | |||||
| BM | 7 (7.4%) | 15 (15%) | 8 (13%) | 17 (28%) | 38 (16%) | ||
| PBSC | 65 (69%) | 70 (71%) | 41 (65%) | 36 (59%) | 157 (67%) | ||
| UCT | 22 (23%) | 13 (13%) | 14 (22%) | 8 (13%) | 40 (17%) | ||
| Conditioning intensity | 0.40 | 0.20 | |||||
| Ablative | 21 (22%) | 30 (31%) | 18 (28.6%) | 21 (34%) | 64 (27%) | ||
| Non-ablative | 11(12%) | 13 (13%) | 10 (15.9%) | 16 (26%) | 36 (15%) | ||
| Reduced intensity | 62 (66%) | 55 (56%) | 35 (55.6%) | 24 (39%) | 135 (57%) | ||
| Donor match | 0.20 | 0.20 | |||||
| Haploidentical | 9 (9.6%) | 13 (13%) | 9 (15%) | 9 (14%) | 27 (11%) | ||
| Related identical | 14 (15%) | 23 (23%) | 5 (7.9%) | 12 (20%) | 47 (20%) | ||
| Related non-identical | 1 (1%) | 0 (0%) | 1 (1.6%) | 0 (0%) | 1 (0.4%) | ||
| Unrelated identical | 42 (45%) | 44 (45%) | 28 (44%) | 27 (44%) | 103 (44%) | ||
| Unrelated non-identical | 28 (30%) | 18 (18%) | 20 (32%) | 13 (21%) | 57 (24%) | ||
Extended Data Table 3.
Patient characteristics in the sub-cohort of patients that received UDCA.
| Peri-engraftment | Peri-GVHD onset | Total | |||||
|---|---|---|---|---|---|---|---|
| Characteristic | GVHD, n = 861 | Control, n = 901 | p-value2 | GVHD, n = 581 | Control, n = 571 | p-value2 | n=217 |
| Age | 61 (48, 67) | 61 (49, 68) | 0.60 | 57 (41, 66) | 60 (52, 66) | 0.30 | 60 (48,66) |
| Sex (F) | 32 (37%) | 32 (36%) | 0.80 | 22 (38%) | 21 (37%) | >0.9 | 83 (38%) |
| Disease | 0.80 | >0.9 | |||||
| Leukemia | 43 (50%) | 40 (44%) | 27 (47%) | 25 (44%) | 102 (47%) | ||
| Lymphoma | 28 (33%) | 28 (31%) | 19 (33%) | 16 (28%) | 67 (31%) | ||
| MDS/MPN | 13 (15%) | 17 (19%) | 9 (16%) | 12 (21%) | 37 (17%) | ||
| Multiple Myeloma | 1 (1%) | 3 (3.3%) | 1 (1.7%) | 1 (1.8%) | 4 (1.8%) | ||
| Non-Malignant Hematological Disorders | 1 (1%) | 2 (1.2%) | 2 (3.4%) | 3 (5.3%) | 7 (3.2) | ||
| Graft type | 0.04 | 0.07 | |||||
| BM | 6 (7%) | 15 (17%) | 7 (12%) | 16 (28%) | 36 (17%) | ||
| PBSC | 59 (69%) | 63 (70%) | 37 (64%) | 33 (58%) | 143 (66%) | ||
| UCT | 21 (24%) | 12 (13%) | 14 (24%) | 8 (14%) | 38 (18%) | ||
| Conditioning intensity | 0.20 | 0.12 | |||||
| Ablative | 17 (20%) | 27 (30%) | 16 (28%) | 20 (35%) | 57 (26%) | ||
| Non-ablative | 9 (10%) | 11 (12%) | 8 (14%) | 14 (25%) | 31 (14%) | ||
| Reduced Intensity | 60 (70%) | 52 (58%) | 34 (59%) | 23 (40%) | 129 (59%) | ||
| Donor match | 0.8 | 0.30 | |||||
| Related Haploidentical | 9 (10%) | 13 (14%) | 8 (14%) | 9 (16%) | 27 (12%) | ||
| Related Identical | 11 (13%) | 20 (22%) | 4 (6.9%) | 9 (16%) | 40 (18%) | ||
| Related Non-Identical | 1 (1.2%) | 0 (0%) | 1 (1.7%) | 0 (0%) | 1 (0.5%) | ||
| Unrelated Identical | 38 (44%) | 40 (44%) | 25 (43%) | 26 (46%) | 94 (43%) | ||
| Unrelated Non-identical | 27 (31%) | 17 (19%) | 20 (34%) | 13 (23%) | 55 (26%) | ||
n (%); Median (IQR)
Fisher’s exact test; Pearson’s Chi-squared test; Wilcoxon rank sum test
Extended Data Table 4.
Comparisons of bile acid levels in patients with GVHD and controls in peri-GVHD onset samples.
| Bile acid | Patients with GVHD N = 581 | Control N = 571 | p-value2 | q-value3 | Level of confidence6 |
|---|---|---|---|---|---|
| UDCA-sulfate4 | 1,929.0 (287.3, 7,522.3) | 15,357.8 (2,357.7, 34,033.4) | <0.001 | <0.001 | |
| β-MCA4 | 0.3 (0.0, 6.5) | 8.1 (2.0, 21.2) | <0.001 | <0.001 | |
| CA-sulfate4 | 3.9 (0.5, 17.4) | 45.2 (6.4, 243.2) | <0.001 | <0.001 | |
| ω-MCA4 | 28.5 (0.0, 100.8) | 115.3 (31.4, 257.2) | <0.001 | <0.001 | |
| γ-MCA5 | 0.0 (0.0, 136,156.2) | 161,338.0 (0.0, 364,832.0) | <0.001 | 0.001 | |
| 3-oxoCDCA5 | 0.0 (0.0, 0.0) | 69,977.0 (0.0, 712,188.0) | <0.001 | 0.001 | |
| CDCA-sulfate4 | 87.8 (6.6, 742.0) | 756.5 (89.9, 3,279.4) | <0.001 | 0.002 | |
| 3-oxoCA5 | 26,856.5 (0.0, 386,074.8) | 297,807.0 (134,653.0, 1,765,809.0) | 0.001 | 0.004 | |
| 6α-THBA5 | 0.0 (0.0, 0.0) | 0.0 (0.0, 98,567.0) | 0.001 | 0.004 | |
| 7-oxoDCA5 | 0.0 (0.0, 2,538,904.5) | 937,617.0 (0.0, 23,199,642.0) | <0.001 | 0.004 | |
| 7-oxoLCA5 | 0.0 (0.0, 1,248,594.8) | 1,312,422.0 (61,782.0, 9,587,159.0) | <0.001 | 0.004 | |
| DCA4 | 2.5 (0.2, 31.4) | 32.8 (4.0, 360.0) | 0.001 | 0.004 | |
| cholenic acid5 | 0.0 (0.0, 0.0) | 0.0 (0.0, 72,294.0) | 0.002 | 0.007 | |
| LCA-sulfate4 | 14.1 (2.9, 91.8) | 98.6 (9.6, 432.7) | 0.002 | 0.007 | |
| CA4 | 2,443.2 (59.2, 15,819.0) | 14,881.8 (374.4, 33,058.8) | 0.004 | 0.011 | |
| DCA-sulfate4 | 0.6 (0.0, 10.5) | 17.3 (0.5, 138.9) | 0.004 | 0.011 | |
| isoCA5 | 0.0 (0.0, 217,816.0) | 111,039.0 (0.0, 1,769,979.0) | 0.005 | 0.011 | |
| isoLCA5 | 0.0 (0.0, 0.0) | 0.0 (0.0, 179,311.0) | 0.005 | 0.011 | |
| LCA4 | 2.8 (0.2, 11.0) | 6.8 (2.6, 204.3) | 0.004 | 0.011 | |
| 3oxoLCA5 | 0.0 (0.0, 0.0) | 0.0 (0.0, 1,953,269.0) | 0.007 | 0.015 | |
| isoCDCA5 | 0.0 (0.0, 94,188.0) | 0.0 (0.0, 1,153,771.0) | 0.008 | 0.016 | |
| ursocholic acid5 | 0.0 (0.0, 217,720.2) | 234,918.0 (0.0, 1,314,511.0) | 0.011 | 0.023 | |
| CDCA4 | 830.7 (60.4, 3,860.2) | 2,903.2 (364.7, 14,684.4) | 0.012 | 0.023 | |
| 3-oxoDCA5 | 0.0 (0.0, 0.0) | 0.0 (0.0, 169,038.0) | 0.026 | 0.049 | |
| GLCA-sulfate4 | 18.7 (7.6, 70.5) | 47.8 (12.2, 107.0) | 0.038 | 0.067 | |
| isoDCA5 | 0.0 (0.0, 0.0) | 0.0 (0.0, 1,200,227.0) | 0.039 | 0.067 | |
| 12-oxoLCA5 | 0.0 (0.0, 0.0) | 0.0 (0.0, 126,673.0) | 0.064 | 0.11 | |
| 7,12-dioxoLCA5 | 0.0 (0.0, 0.0) | 0.0 (0.0, 55,133.0) | 0.092 | 0.15 | |
| Gγ-MCA5 | 84,721.0 (0.0, 535,719.2) | 0.0 (0.0, 322,213.0) | 0.10 | 0.2 | |
| GCA4 | 3,148.5 (244.6, 17,332.4) | 532.5 (69.2, 6,453.0) | 0.11 | 0.2 | |
| UDCA4 | 10,716.5 (2,622.0, 29,154.2) | 22,632.5 (3,336.6, 46,019.2) | 0.2 | 0.3 | |
| GLCA4 | 0.5 (0.4, 1.1) | 0.6 (0.4, 3.7) | 0.2 | 0.3 | |
| TUDCA4 | 422.6 (37.6, 4,278.1) | 711.4 (207.0, 5,071.7) | 0.2 | 0.3 | |
| TLCA4 | 0.7 (0.4, 1.6) | 1.3 (0.4, 6.1) | 0.3 | 0.4 | |
| Tγ-MCA5 | 0.0 (0.0, 295,257.0) | 0.0 (0.0, 901,258.0) | 0.5 | 0.6 | |
| TCDCA4 | 391.9 (22.4, 2,279.0) | 312.6 (61.4, 2,281.3) | 0.6 | 0.7 | |
| TCA4 | 1,342.8 (180.1, 5,517.0) | 688.7 (84.0, 4,786.7) | 0.6 | 0.7 | |
| GCDCA4 | 982.3 (119.0, 10,338.7) | 502.7 (126.5, 5,975.0) | 0.6 | 0.7 | |
| TDCA4 | 1.0 (0.0, 5.7) | 1.1 (0.0, 28.5) | 0.7 | 0.8 | |
| GUDCA-sulfate4 | 1,535.2 (97.7, 13,577.0) | 1,809.3 (52.7, 13,547.9) | 0.7 | 0.8 | |
| GCDCA-sulfate4 | 177.6 (26.6, 1,201.0) | 222.6 (3.7, 1,484.6) | 0.8 | 0.8 | |
| GCA-sulfate4 | 21.2 (4.4, 64.9) | 30.7 (1.0, 87.1) | 0.8 | 0.9 | |
| GDCA4 | 4.0 (0.0, 22.5) | 2.3 (0.0, 33.0) | >0.9 | >0.9 | |
| GDCA-sulfate4 | 2.8 (0.2, 18.7) | 4.4 (0.0, 21.5) | >0.9 | >0.9 | |
| GUDCA4 | 3,392.3 (324.6, 26,282.2) | 1,973.7 (246.2, 25,565.6) | >0.9 | >0.9 |
Median (IGR);
Wilcoxon rank sum test;
False discovery rate correction for multiple testing;
Measured in pmol/mg;
Measured in AUC,
Schymanski, E. L. et al. Identifying Small Molecules via High-Resolution Mass Spectrometry: Communicating Confidence. Environ. Sci. Technol. 48, 2097–2098 (2014).
Extended Data Table 5.
Comparisons of bile acid levels in patients with GVHD and controls in peri-engraftment samples.
| Bile acid | GVHD, N = 701 | Control, N = 631 | p-value2 | q-value3 | Level of confidence6 |
|---|---|---|---|---|---|
| 3-oxoCA4 | 82,116.0 (0.0, 337,366.0) | 254,728.0 (0.0, 781,042.0) | 0.018 | 0.3 | |
| 7,12-dioxoLCA4 | 0.0 (0.0, 0.0) | 0.0 (0.0, 0.0) | 0.025 | 0.3 | |
| CDCA5 | 1,359.7 (148.7, 5,939.7) | 3,329.9 (678.7, 11,147.0) | 0.028 | 0.3 | |
| CA5 | 4,245.1 (144.6, 18,047.6) | 9,838.1 (1,152.5, 30,946.4) | 0.027 | 0.3 | |
| CA-sulfate5 | 6.1 (2.2, 40.7) | 38.3 (2.9, 170.8) | 0.016 | 0.3 | |
| 3-oxoCDCA4 | 0.0 (0.0, 111,300.0) | 0.0 (0.0, 519,388.5) | 0.073 | 0.4 | |
| 7-oxoDCA4 | 61,062.5 (0.0, 1,950,979.8) | 297,844.0 (0.0, 15,745,451.5) | 0.12 | 0.4 | |
| 7-oxoLCA4 | 0.0 (0.0, 2,087,354.5) | 378,170.0 (0.0, 6,172,302.5) | 0.11 | 0.4 | |
| cholenic acid4 | 0.0 (0.0, 0.0) | 0.0 (0.0, 54,132.5) | 0.10 | 0.4 | |
| isoCA4 | 0.0 (0.0, 39,423.0) | 0.0 (0.0, 1,728,128.0) | 0.089 | 0.4 | |
| LCA5 | 2.1 (0.0, 9.7) | 4.4 (1.0, 16.3) | 0.12 | 0.4 | |
| UDCA5 | 9,761.9 (3,684.7, 39,479.1) | 27,438.3 (6,555.8, 53,308.9) | 0.10 | 0.4 | |
| UDCA-sulfate5 | 5,003.4 (1,404.1, 17,840.6) | 10,458.9 (1,815.4, 31,516.0) | 0.12 | 0.4 | |
| 6α-THBA4 | 0.0 (0.0, 0.0) | 0.0 (0.0, 70,020.0) | 0.2 | 0.5 | |
| DCA-sulfate5 | 7.4 (0.0, 69.3) | 10.0 (1.0, 98.7) | 0.2 | 0.5 | |
| γ-MCA4 | 66,331.0 (0.0, 258,874.2) | 124,410.0 (0.0, 430,093.0) | 0.2 | 0.5 | |
| ω-MCA5 | 47.6 (4.5, 165.0) | 86.7 (13.9, 199.7) | 0.2 | 0.5 | |
| TCA5 | 1,587.0 (259.4, 5,143.3) | 2,612.0 (424.1, 6,142.6) | 0.2 | 0.5 | |
| ursocholic acid4 | 0.0 (0.0, 131,510.0) | 67,075.0 (0.0, 256,186.0) | 0.2 | 0.5 | |
| CDCA-sulfate5 | 476.0 (34.5, 2,055.3) | 1,005.6 (114.1, 3,276.9) | 0.2 | 0.5 | |
| DCA5 | 7.3 (0.8, 85.9) | 19.3 (2.4, 118.2) | 0.2 | 0.5 | |
| GLCA5 | 0.5 (0.4, 1.0) | 0.6 (0.4, 1.2) | 0.2 | 0.5 | |
| GUDCA-sulfate5 | 6,970.3 (1,535.9, 20,655.5) | 5,041.9 (662.0, 16,568.3) | 0.3 | 0.5 | |
| 3oxoLCA4 | 0.0 (0.0, 0.0) | 0.0 (0.0, 0.0) | 0.3 | 0.6 | |
| isoCDCA4 | 0.0 (0.0, 50,789.2) | 0.0 (0.0, 448,105.0) | 0.3 | 0.6 | |
| TCDCA5 | 334.9 (47.0, 1,498.3) | 404.7 (111.0, 2,409.9) | 0.4 | 0.6 | |
| LCA-sulfate5 | 46.4 (5.2, 225.2) | 63.5 (9.0, 212.3) | 0.5 | 0.8 | |
| β-MCA5 | 3.9 (0.0, 10.0) | 3.5 (0.5, 14.7) | 0.6 | 0.9 | |
| isoDCA4 | 0.0 (0.0, 0.0) | 0.0 (0.0, 0.0) | 0.6 | 0.9 | |
| Tγ-MCA4 | 52,790.5 (0.0, 920,961.5) | 0.0 (0.0, 575,946.0) | 0.6 | 0.9 | |
| GCDCA5 | 2,665.9 (176.1, 10,941.5) | 3,094.7 (188.1, 12,619.9) | 0.7 | >0.9 | |
| GCDCA-sulfate5 | 685.3 (101.8, 1,534.2) | 613.3 (70.4, 1,941.1) | 0.7 | >0.9 | |
| GCA-sulfate5 | 59.1 (23.4, 115.4) | 49.7 (19.9, 117.6) | 0.7 | >0.9 | |
| GLCA-sulfate5 | 29.8 (9.3, 99.3) | 56.4 (11.1, 93.4) | 0.7 | >0.9 | |
| isoLCA4 | 0.0 (0.0, 0.0) | 0.0 (0.0, 0.0) | 0.6 | >0.9 | |
| TLCA5 | 0.6 (0.4, 1.5) | 0.7 (0.3, 1.7) | 0.7 | >0.9 | |
| TDCA5 | 1.6 (0.0, 8.6) | 1.4 (0.3, 14.4) | 0.8 | >0.9 | |
| 12-oxoLCA4 | 0.0 (0.0, 0.0) | 0.0 (0.0, 0.0) | >0.9 | >0.9 | |
| 3-oxoDCA4 | 0.0 (0.0, 0.0) | 0.0 (0.0, 0.0) | 0.9 | >0.9 | |
| Gγ-MCA4 | 190,180.0 (0.0, 814,007.5) | 167,944.0 (0.0, 547,274.0) | >0.9 | >0.9 | |
| GCA5 | 3,709.5 (324.9, 19,848.0) | 4,366.8 (427.7, 26,212.4) | 0.8 | >0.9 | |
| GDCA5 | 3.6 (0.0, 33.8) | 6.4 (0.0, 37.2) | >0.9 | >0.9 | |
| GDCA-sulfate5 | 18.6 (1.3, 51.6) | 18.0 (0.9, 49.2) | 0.8 | >0.9 | |
| GUDCA5 | 8,581.4 (3,121.4, 50,297.3) | 14,220.0 (1,355.5, 39,569.6) | >0.9 | >0.9 | |
| TUDCA5 | 1,002.1 (143.0, 6,600.1) | 1,717.8 (196.3, 4,963.6) | >0.9 | >0.9 |
Median (IQR);
Wilcoxon rank sum test;
False discovery rate correction for multiple testing;
Measured in AUC;
Measured in pmol/mg;
Schymanski, E. L. et al. Identifying Small Molecules via High-Resolution Mass Spectrometry: Communicating Confidence. Environ. Sci. Technol. 48, 2097–2098 (2014).
Extended Data Table 6.
Genes Differentially expressed in DY268 compared to the vehicle, using Wilcoxon rank-sum test.
| Gene | Z-score | Log2 (FC) | Adjusted p-value |
|---|---|---|---|
| FTL | 6.42 | 0.194 | 0.000000855 |
| IFI6 | 5.29 | 0.567 | 0.000566 |
| IFI44L | 4.60 | 0.487 | 0.00858 |
| MALAT1 | 4.12 | 0.132 | 0.0485 |
| ETFB | −4.10 | −0.265 | 0.0485 |
| PKM | −4.11 | −0.250 | 0.0485 |
| MIF | −4.11 | −0.178 | 0.0485 |
| CORO1A | −4.25 | −0.252 | 0.0297 |
| LTB | −4.32 | −0.243 | 0.0232 |
| ACADVL | −4.32 | −0.375 | 0.0232 |
| PFN1 | −4.39 | −0.154 | 0.0178 |
| PPIA | −4.45 | −0.187 | 0.0144 |
| ACTB | −4.55 | −0.146 | 0.00971 |
| GMFG | −4.57 | −0.266 | 0.00935 |
| NAA10 | −4.62 | −0.339 | 0.00826 |
| SUMO2 | −4.67 | −0.218 | 0.00690 |
| LPP | −4.76 | −0.315 | 0.00487 |
Extended Data Table 7.
Cox regression model for overall survival (OS)
| Characteristic | HR | 95%CI | p-value |
|---|---|---|---|
| Age | 1.03 | 1.02–1.04 | <0.001 |
| Sex (males vs females) | 0.95 | 0.79–1.15 | 0.60 |
| Donor match (vs haplo) | |||
| Haplo/MMUD | 1.19 | 0.57–2.47 | 0.60 |
| MMUD | 1.22 | 0.73–2.03 | 0.40 |
| MRD | 0.73 | 0.45–1.18 | 0.20 |
| MUD | 0.81 | 0.51–1.29 | 0.40 |
| Graft (vs BM) | |||
| CD34+ PBSC | 0.90 | 0.60–1.34 | 0.60 |
| PBSC | 1.16 | 0.80–1.67 | 0.40 |
| UCB | 0.75 | 0.44–1.28 | 0.30 |
| Intensity (vs ablative) | |||
| Non-ablative | 0.49 | 0.33–0.73 | <0.001 |
| Reduced Intensity | 1.00 | 0.74–1.35 | >0.9 |
| UDCA (yes vs no) | 0.68 | 0.55–0.85 | <0.001 |
Haplo: haploidentical; MMUD: mismatched unrelated donor; MRD: matched related donor; MUD: matched unrelated donor; PBSC: peripheral blood stem cell; UCB: umbilical cord.
Extended Data Table 8.
Fine-Grey model for transplant related mortality (GRM)
| Characteristic | HR | 95%CI | p-value |
|---|---|---|---|
| Age | 1.02 | 1.01–1.04 | 0.002 |
| Sex (males vs females) | 0.77 | 0.56–1.04 | 0.09 |
| Donor match (vs haplo) | |||
| Haplo/MMUD | 1.86 | 0.60–5.78 | 0.29 |
| MMUD | 1.59 | 0.68–3.69 | 0.28 |
| MRD | 0.60 | 0.27–1.32 | 0.21 |
| MUD | 0.77 | 0.36–1.64 | 0.50 |
| Graft (vs BM) | |||
| CD34 | 1.26 | 0.58–2.72 | 0.55 |
| PBSC | 1.63 | 0.85–3.13 | 0.14 |
| UCB | 0.88 | 0.35–2.16 | 0.77 |
| Intensity (vs ablative) | |||
| Non-ablative | 0.95 | 0.53–1.71 | 0.87 |
| Reduced Intensity | 1.24 | 0.75–2.07 | 0.40 |
| UDCA (yes vs no) | 0.66 | 0.46–0.94 | 0.02 |
Haplo: haploidentical; MMUD: mismatched unrelated donor; MRD: matched related donor; MUD: matched unrelated donor; PBSC: peripheral blood stem cell; UCB: umbilical cord.
Extended Data Table 9.
Hashtag sequences for oligonucleotide-conjugated antibodies.
| Name | Hash Sequence | Manufacturer | Catalog # | Dilution |
|---|---|---|---|---|
| TotalSeq™-B0301 anti-mouse Hashtag 1 | ACCCACCAGTAAGAC | BioLegend | 155831 | 1:50 |
| TotalSeq™-B0302 anti-mouse Hashtag 2 | GGTCGAGAGCATTCA | BioLegend | 155833 | 1:50 |
| TotalSeq™-B0303 anti-mouse Hashtag 3 | CTTGCCGCATGTCAT | BioLegend | 155835 | 1:50 |
| TotalSeq™-B0304 anti-mouse Hashtag 4 | AAAGCATTCTTCACG | BioLegend | 155837 | 1:50 |
| TotalSeq™-B0305 anti-mouse Hashtag 5 | CTTTGTCTTTGTGAG | BioLegend | 155839 | 1:50 |
| TotalSeq™-B0306 anti-mouse Hashtag 6 | TATGCTGCCACGGTA | BioLegend | 155841 | 1:50 |
| TotalSeq™-B0307 anti-mouse Hashtag 7 | GAGTCTGCCAGTATC | BioLegend | 155843 | 1:50 |
Supplementary Material
Acknowledgments
We acknowledge Ronan Chaligne, the single cell analytics innovation laboratory (SAIL) and the use of the Integrated Genomics Operation Core (IGO) as well as the molecular microbiology facility (MMF), which performed RNA sequencing (SAIL, IGO), as well as the 16S and metagenomic shotgun sequencing (MMF, IGO) for mouse and human studies and and is funded by the NCI Cancer Center Support Grant (CCSG, P30 CA08748), Cycle for Survival, and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology. This research was supported by National Cancer Institute award numbers, R01-CA228358, R01-CA228308, P30 CA008748 MSK Cancer Center Support Grant/Core Grant and P01-CA023766; National Heart, Lung, and Blood Institute (NHLBI) award number R01-HL123340 and R01-HL147584; and Tri Institutional Stem Cell Initiative. Additional funding was received from The Lymphoma Foundation, The Susan and Peter Solomon Family Fund, The Solomon Microbiome Nutrition and Cancer Program, Cycle for Survival, Parker Institute for Cancer Immunotherapy, Paula and Rodger Riney Multiple Myeloma Research Initiative, Starr Cancer Consortium, and Seres Therapeutics. S.L. was supported by the Deutsche Forschungsgemeinschaft (DFG, LI 3565/1-1) and DKMS. O.M. was supported by the American Society of Clinical Oncology Young Investigator Award, a Hyundai Hope on Wheels Young Investigator Award, a Cycle for Survival Equinox Innovation Award, a Collaborative Pediatric Cancer Research Program Award, Michael Goldberg Fellowship and Tow Center for Developmental Oncology Career Development Award. K.A.M. was supported by the DKMS and the ASH Scholar Award. J.U.P. reports funding from NHLBI NIH Award K08HL143189.
Competing Interests Statement
M.A.P. reports honoraria from Adicet, Allovir, Caribou Biosciences, Celgene, Bristol-Myers Squibb, Equilium, Exevir, Incyte, Karyopharm, Kite/Gilead, Merck, Miltenyi Biotec, MorphoSys, Nektar Therapeutics, Novartis, Omeros, OrcaBio, Syncopation, VectivBio AG, and Vor Biopharma; he serves on DSMBs for Cidara Therapeutics, Medigene, and Sellas Life Sciences, and the scientific advisory board of NexImmune; he has ownership interests in NexImmune and Omeros; he has received institutional research support for clinical trials from Incyte, Kite/Gilead, Miltenyi Biotec, Nektar Therapeutics, and Novartis. KAM holds equity and is on the advisory board of Postbiotics Plus and has consulted for Incyte. J.U.P. reports research funding, intellectual property fees, and travel reimbursement from Seres Therapeutics, and consulting fees from DaVolterra, CSL Behring, and from MaaT Pharma; he serves on an Advisory board of and holds equity in Postbiotics Plus Research, he has filed intellectual property applications related to the microbiome (reference numbers #62/843,849, #62/977,908, and #15/756,845). M.R.M.B. has received research support and stock options from Seres Therapeutics and stock options from Notch Therapeutics and Pluto Therapeutics; he has received royalties from Wolters Kluwer; has consulted, received honorarium from or participated in advisory boards for Seres Therapeutics, Vor Biopharma, Rheos Medicines, Frazier Healthcare Partners, Nektar Therapeutics, Notch Therapeutics, Ceramedix, Lygenesis, Pluto Therapeutics, GlaskoSmithKline, Da Volterra, Thymofox, Garuda, Novartis (Spouse), Synthekine (Spouse), Beigene (Spouse), Kite (Spouse); he has IP Licensing with Seres Therapeutics and Juno Therapeutics; and holds a fiduciary role on the Foundation Board of DKMS (a nonprofit organization). MSK has institutional financial interests relative to Seres Therapeutics. The remaining authors declare no competing interests.
Footnotes
Code availability
The code and the corresponding figures for Fig. 2d and e, Fig. 4–5, Fig. 6h–v, Extended Data Fig. 4–7, 10 can be accessed at GitHub (https://github.com/orianamiltiadous/BAs_and_GVHD).
Data availability
Metabolomics data including standards (Fig. 1, Fig. 4, Fig. 5, Extended data Fig. 4–7, Extended Table 4 and 5) are available at GNPS (https://gnps.ucsd.edu/) under MassIVE project ID #MSV000092300). The bulk RNAseq data from murine experiments (Fig. 2 and Extended Fig. 2) are available at NCBI GEO under GEO accession GSE218343 and also in Supplementary tables. The 16s and shotgun sequencing data (Fig. 2, Fig.5, Extended Fig. 7) are available at NCBI under accession numbers listed in the Supplementary tables. The processed scRNA-seq files are available under GEO accession GSE253360. For access to raw data, kindly request permission by contacting the contributing author at mvandenbrink@coh.org. Please anticipate a response within 2 weeks. Once legal agreements are approved, raw genomic data can be shared within an additional month. Source data for Figs. 1–3, 6a–c and Extended Data Figs. 1–3, 8 are available in the Supplementary tables. The datasets required to run the code were also made publicly available in github and are also included with Supplementary information. Our institutional data-sharing policies prevent us from posting publicly the patient-level information used to calculate clinical outcomes (Fig 6t,u). However, interested parties may request access by contacting the contributing author at mvandenbrink@coh.org. Please anticipate a response within one month. Data sharing of patient-level information is contingent upon the establishment of a formal data transfer agreement between Memorial Sloan Kettering and the respective parties involved.
References
- 1.Hamilton JP, et al. Human cecal bile acids: concentration and spectrum. Am J Physiol Gastrointest Liver Physiol 293, G256–263 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Chen ML, Takeda K & Sundrud MS Emerging roles of bile acids in mucosal immunity and inflammation. Mucosal Immunol 12, 851–861 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Belkaid Y & Hand TW Role of the microbiota in immunity and inflammation. Cell 157, 121–141 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Russell DW The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem 72, 137–174 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Ridlon JM, Kang DJ & Hylemon PB Bile salt biotransformations by human intestinal bacteria. J Lipid Res 47, 241–259 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Begley M, Hill C & Gahan CG Bile salt hydrolase activity in probiotics. Appl Environ Microbiol 72, 1729–1738 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wells JE & Hylemon PB Identification and characterization of a bile acid 7alpha-dehydroxylation operon in Clostridium sp. strain TO-931, a highly active 7alpha-dehydroxylating strain isolated from human feces. Appl Environ Microbiol 66, 1107–1113 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Devlin AS & Fischbach MA A biosynthetic pathway for a prominent class of microbiota-derived bile acids. Nat Chem Biol 11, 685–690 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Quinn RA, et al. Global chemical effects of the microbiome include new bile-acid conjugations. Nature 579, 123–129 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Foley MH, et al. Bile salt hydrolases shape the bile acid landscape and restrict Clostridioides difficile growth in the murine gut. Nat Microbiol 8, 611–628 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patterson A, et al. Bile Acids Are Substrates for Amine N-Acyl Transferase Activity by Bile Salt Hydrolase. (2022).
- 12.Shin DJ & Wang L Bile Acid-Activated Receptors: A Review on FXR and Other Nuclear Receptors. Handb Exp Pharmacol 256, 51–72 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Hang S, et al. Bile acid metabolites control T(H)17 and T(reg) cell differentiation. Nature 576, 143–148 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paik D, et al. Human gut bacteria produce T(H)17-modulating bile acid metabolites. Nature 603, 907–912 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campbell C, et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 581, 475–479 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gratwohl A, et al. Hematopoietic stem cell transplantation: a global perspective. Jama 303, 1617–1624 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gooley TA, et al. Reduced mortality after allogeneic hematopoietic-cell transplantation. N Engl J Med 363, 2091–2101 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aljurf M, et al. “Worldwide Network for Blood & Marrow Transplantation (WBMT) special article, challenges facing emerging alternate donor registries”. Bone Marrow Transplant 54, 1179–1188 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peled JU, et al. Microbiota as Predictor of Mortality in Allogeneic Hematopoietic-Cell Transplantation. N Engl J Med 382, 822–834 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taur Y, et al. The effects of intestinal tract bacterial diversity on mortality following allogeneic hematopoietic stem cell transplantation. Blood 124, 1174–1182 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jenq RR, et al. Regulation of intestinal inflammation by microbiota following allogeneic bone marrow transplantation. J Exp Med 209, 903–911 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holler E, et al. Metagenomic analysis of the stool microbiome in patients receiving allogeneic stem cell transplantation: loss of diversity is associated with use of systemic antibiotics and more pronounced in gastrointestinal graft-versus-host disease. Biol Blood Marrow Transplant 20, 640–645 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burgos da Silva M, et al. Preservation of fecal microbiome is associated with reduced severity of Graft-versus-Host Disease. Blood (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taur Y, et al. Intestinal domination and the risk of bacteremia in patients undergoing allogeneic hematopoietic stem cell transplantation. Clin Infect Dis 55, 905–914 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ara T & Hashimoto D Novel Insights Into the Mechanism of GVHD-Induced Tissue Damage. Front Immunol 12, 713631 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zeiser R & Blazar BR Acute Graft-versus-Host Disease - Biologic Process, Prevention, and Therapy. N Engl J Med 377, 2167–2179 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Malard F, Holler E, Sandmaier BM, Huang H & Mohty M Acute graft-versus-host disease. Nat Rev Dis Primers 9, 27 (2023). [DOI] [PubMed] [Google Scholar]
- 28.Reddy P, Negrin R & Hill GR Mouse models of bone marrow transplantation. Biol Blood Marrow Transplant 14, 129–135 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palmer RH The formation of bile acid sulfates: a new pathway of bile acid metabolism in humans. Proc Natl Acad Sci U S A 58, 1047–1050 (1967). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mootha VK, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 34, 267–273 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Gadaleta RM, et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 60, 463–472 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Campbell C, et al. FXR mediates T cell-intrinsic responses to reduced feeding during infection. Proc Natl Acad Sci U S A 117, 33446–33454 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sayin SI, et al. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab 17, 225–235 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Shono Y, et al. Increased GVHD-related mortality with broad-spectrum antibiotic use after allogeneic hematopoietic stem cell transplantation in human patients and mice. Sci Transl Med 8, 339ra371 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruutu T, et al. Ursodeoxycholic acid for the prevention of hepatic complications in allogeneic stem cell transplantation. Blood 100, 1977–1983 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Ruutu T, et al. Improved survival with ursodeoxycholic acid prophylaxis in allogeneic stem cell transplantation: long-term follow-up of a randomized study. Biol Blood Marrow Transplant 20, 135–138 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Guzior DV & Quinn RA Review: microbial transformations of human bile acids. Microbiome 9, 140 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Claudel T, Staels B & Kuipers F The Farnesoid X receptor: a molecular link between bile acid and lipid and glucose metabolism. Arterioscler Thromb Vasc Biol 25, 2020–2030 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Cox DR Regression Models and Life-Tables. Journal of the Royal Statistical Society: Series B (Methodological) 34, 187–202 (1972). [Google Scholar]
- 41.Gray JPFRJ A Proportional Hazards Model for the Subdistribution of a Competing Risk. Theory and Method 94, 496–509 (1997). [Google Scholar]
- 42.Haring E, et al. Bile acids regulate intestinal antigen presentation and reduce graft-versus-host disease without impairing the graft-versus-leukemia effect. Haematologica 106, 2131–2146 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gevers D, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 15, 382–392 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peled JU, et al. Intestinal Microbiota and Relapse After Hematopoietic-Cell Transplantation. J Clin Oncol 35, 1650–1659 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCarville JL, Chen GY, Cuevas VD, Troha K & Ayres JS Microbiota Metabolites in Health and Disease. Annu Rev Immunol 38, 147–170 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Yao L, et al. A selective gut bacterial bile salt hydrolase alters host metabolism. Elife 7(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y, et al. Ursodeoxycholic acid accelerates bile acid enterohepatic circulation. Br J Pharmacol 176, 2848–2863 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Winston JA, Rivera A, Cai J, Patterson AD & Theriot CM Secondary bile acid ursodeoxycholic acid alters weight, the gut microbiota, and the bile acid pool in conventional mice. PLoS One 16, e0246161 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Song X, et al. Microbial bile acid metabolites modulate gut RORγ(+) regulatory T cell homeostasis. Nature 577, 410–415 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li W, et al. A bacterial bile acid metabolite modulates T(reg) activity through the nuclear hormone receptor NR4A1. Cell Host Microbe (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee JWJ, et al. Multi-omics reveal microbial determinants impacting responses to biologic therapies in inflammatory bowel disease. Cell Host Microbe 29, 1294–1304.e1294 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gopalakrishnan V, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 359, 97–103 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smith M, et al. Gut microbiome correlates of response and toxicity following anti-CD19 CAR T cell therapy. Nat Med 28, 713–723 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stein-Thoeringer CK, et al. Lactose drives Enterococcus expansion to promote graft-versus-host disease. Science 366, 1143–1149 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mathewson ND, et al. Gut microbiome-derived metabolites modulate intestinal epithelial cell damage and mitigate graft-versus-host disease. Nat Immunol 17, 505–513 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ritchie ME, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Benjamini Y & Hochberg Y Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society: Series B (Methodological) 57, 289–300 (1995). [Google Scholar]
- 58.FastQC. (2015).
- 59.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Peled JU, Gomes ALC & van den Brink MRM Microbiota and Allogeneic Hematopoietic-Cell Transplantation. Reply. N Engl J Med 382, 2378–2379 (2020). [DOI] [PubMed] [Google Scholar]
- 61.Miltiadous O, et al. Early intestinal microbial features are associated with CD4 T-cell recovery after allogeneic hematopoietic transplant. Blood 139, 2758–2769 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Callahan BJ, McMurdie PJ & Holmes SP Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J 11, 2639–2643 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Callahan BJ, et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods 13, 581–583 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dubin KA, et al. Diversification and Evolution of Vancomycin-Resistant Enterococcus faecium during Intestinal Domination. Infect Immun 87(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Paulson JN, Stine OC, Bravo HC & Pop M Differential abundance analysis for microbial marker-gene surveys. Nat Methods 10, 1200–1202 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mallick H, et al. Multivariable association discovery in population-scale meta-omics studies. PLoS Comput Biol 17, e1009442 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Martin BD, Witten D & Willis AD MODELING MICROBIAL ABUNDANCES AND DYSBIOSIS WITH BETA-BINOMIAL REGRESSION. Ann Appl Stat 14, 94–115 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Beghini F, et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. Elife 10(2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Buchfink B, Xie C & Huson DH Fast and sensitive protein alignment using DIAMOND. Nat Methods 12, 59–60 (2015). [DOI] [PubMed] [Google Scholar]
- 70.Buchfink B, Reuter K & Drost HG Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat Methods 18, 366–368 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Menzel P, Ng KL & Krogh A Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat Commun 7, 11257 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stoeckius M, et al. Cell Hashing with barcoded antibodies enables multiplexing and doublet detection for single cell genomics. Genome Biol 19, 224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lun ATL, et al. EmptyDrops: distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data. Genome Biol 20, 63 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kousa AI, & Lemarquis AL. The shunPykeR’s guide to single cell analysis (Version 1.0.0) [Computer software].. (2023).
- 75.Traag VA, Waltman L & van Eck NJ From Louvain to Leiden: guaranteeing well-connected communities. Sci Rep 9, 5233 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wolock SL, Lopez R & Klein AM Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst 8, 281–291 e289 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Korsunsky I, et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ntranos V, Yi L, Melsted P & Pachter L A discriminative learning approach to differential expression analysis for single-cell RNA-seq. Nat Methods 16, 163–166 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Metabolomics data including standards (Fig. 1, Fig. 4, Fig. 5, Extended data Fig. 4–7, Extended Table 4 and 5) are available at GNPS (https://gnps.ucsd.edu/) under MassIVE project ID #MSV000092300). The bulk RNAseq data from murine experiments (Fig. 2 and Extended Fig. 2) are available at NCBI GEO under GEO accession GSE218343 and also in Supplementary tables. The 16s and shotgun sequencing data (Fig. 2, Fig.5, Extended Fig. 7) are available at NCBI under accession numbers listed in the Supplementary tables. The processed scRNA-seq files are available under GEO accession GSE253360. For access to raw data, kindly request permission by contacting the contributing author at mvandenbrink@coh.org. Please anticipate a response within 2 weeks. Once legal agreements are approved, raw genomic data can be shared within an additional month. Source data for Figs. 1–3, 6a–c and Extended Data Figs. 1–3, 8 are available in the Supplementary tables. The datasets required to run the code were also made publicly available in github and are also included with Supplementary information. Our institutional data-sharing policies prevent us from posting publicly the patient-level information used to calculate clinical outcomes (Fig 6t,u). However, interested parties may request access by contacting the contributing author at mvandenbrink@coh.org. Please anticipate a response within one month. Data sharing of patient-level information is contingent upon the establishment of a formal data transfer agreement between Memorial Sloan Kettering and the respective parties involved.