

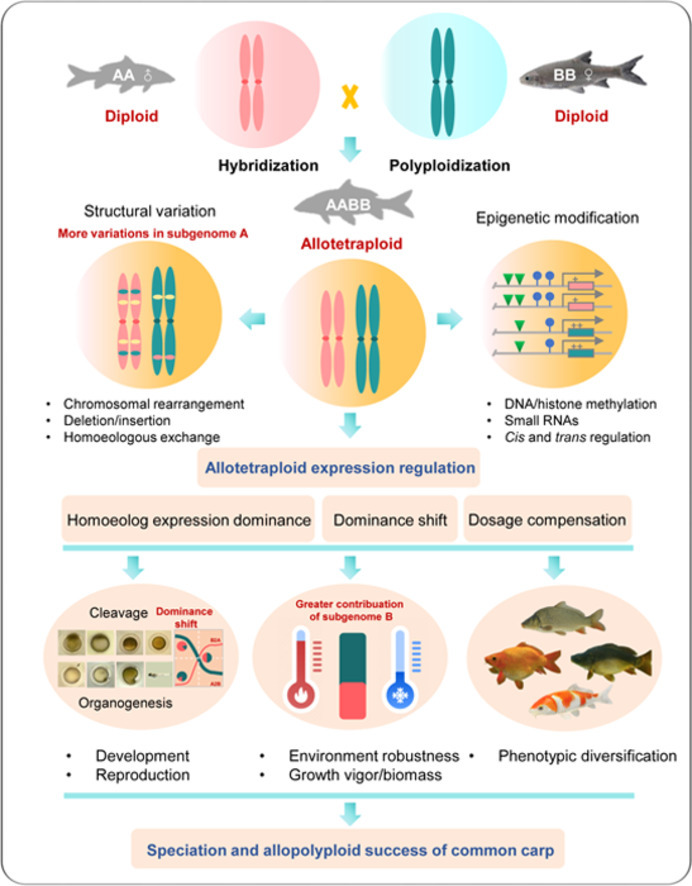
Abstract
Hybridization and polyploidization have made great contributions to speciation, heterosis, and agricultural production within plants, but there is still limited understanding and utilization in animals. Subgenome structure and expression reorganization and cooperation post hybridization and polyploidization are essential for speciation and allopolyploid success. However, the mechanisms have not yet been comprehensively assessed in animals. Here, we produced a high-fidelity reference genome sequence for common carp, a typical allotetraploid fish species cultured worldwide. This genome enabled in-depth analysis of the evolution of subgenome architecture and expression responses. Most genes were expressed with subgenome biases, with a trend of transition from the expression of subgenome A during the early stages to that of subgenome B during the late stages of embryonic development. While subgenome A evolved more rapidly, subgenome B contributed to a greater level of expression during development and under stressful conditions. Stable dominant patterns for homoeologous gene pairs both during development and under thermal stress suggest a potential fixed heterosis in the allotetraploid genome. Preferentially expressing either copy of a homoeologous gene at higher levels to confer development and response to stress indicates the dominant effect of heterosis. The plasticity of subgenomes and their shifting of dominant expression during early development, and in response to stressful conditions, provide novel insights into the molecular basis of the successful speciation, evolution, and heterosis of the allotetraploid common carp.
Keywords: Allotetraploid, Subgenome structural evolution, Homoeologous expression, Expression dominance shift, Environmental adaptation
Graphical abstract
1. Introduction
Aquaculture has made and will continue to make major contributions to global nutrition supply and agricultural sustainability for the growing human population [1,2]. Freshwater aquaculture has contributed more than mariculture [3] but it is facing serious challenges imposed by climate change [4]. Cyprinidae accounts for the majority of freshwater fish species, and it is believed that their genome duplication is responsible for species divergence and biodiversity [5]. Common carp (Cyprinus carpio) is one of the most successful species in the Cyprinidae family and is an important food and ornamental fish cultured worldwide. It is an allotetraploid species, containing 50 homoeologous groups arranged into two subgenomes, A and B (2n = 4X = 100) [6]. This species is derived from the hybridization of a Barbinae-like species (BB) and an undetermined donor AA species (unknown or extinct) followed by whole genome duplication (WGD) ∼12.4 million years ago [7]. Allotetraploid common carp can survive in extremely harsh environments with thermal tolerance ranging from 3 °C to 35 °C [8,9], allowing them to successfully radiate worldwide, except for Antarctica [10]. While diploid Cyprinidae fishes are more often narrow-range endemic fishes, especially for the progenitor-like Barbinae-like species that are naturally distributed in stream systems, they may not be capable of surviving under climate change because of their poor resilience and reliance on the hydrological connectivity for movement [11]. Genetic improvement of these environmentally sensitive diploid fish would be an effective approach for addressing climate change.
Many allopolyploidy events occurred during major periods of global geology-climate change, reinforcing the importance of stress response to the establishment and the success of polyploids [12], [13], [14], [15], [16]. Evidence further supporting this notion came from comparative studies between polyploids and their diploid relatives in response to biotic/abiotic stress [17,18]; multiple subgenomes in polyploids could increase genetic variation available for selection and buffer the effect of the duplicated genes against deleterious mutations, leading to an increased resilience of polyploid organisms to extreme environments, rapid species diversification and desirable production traits [19,20]. Allopolyploids emerging from hybridization and polyploidization are “doubled interspecific hybrids”, leading to a permanent fixation of heterosis [21], [22], [23]. Ecological niche differentiation and karyotypic evolution of newly produced polyploids provide strong reproductive barriers between the polyploids and their ancestors, which is important for speciation and polyploid success, but not for homoploid hybrids [24], [25], [26]. In addition to providing an evolutionary advantage, polyploidy has been widely exploited in agriculture to improve growth vigor and adaptation [27].
Whole-genome duplication and hybridization both contribute to heterosis and genome evolution, but through different mechanisms; genome dosage alone can trigger heterosis effects [28]. Recent studies with plants comparing F1 hybrids, resynthesized polyploids, natural allopolyploids, and their progenitors have suggested that the most predominant advantage of allopolyploid species is through biased subgenome evolution post-WGD, including homoeologous gene expression, DNA methylation patterns, transposable element (TE) distribution, homoeologous. exchanges (HEs), and small RNA targeting [29], [30], [31], [32], [33], [34], [35]. Subgenome dominance has major implications in diverse fields, from ecology to crop improvement efforts [36]. Both expression divergence inherited from parental species and those independently induced by allopolyploidy can result in subgenome expression dominance [32,37]. In allopolyploid crops, the dominant subgenome, also known as the evolutionarily stable subgenome, may be responsible for the most important agronomic traits (morphology and growth), whereas the minor subgenome with more variety may contribute more to environmental changes (disease resistance and ecological adaptability) [[37], [38]]. Hybridization can significantly reduce the expression divergence of parental origin genes, while polyploidization can recover the divergence to a parental-like state, showing that polyploidization may better reflect the direction of parental origin [39]. However, the mechanisms have not been fully dissected in polyploid animal systems with only a few cases having been reported [7,[40], [41], [42], [43]]. Distant hybridization, which refers to crosses between two different species, genera, or higher-ranking taxa, is the most practical and effective way to generate allopolyploid fish [44]. Such processes have been successful in cyprinid fish, providing multiple new germplasm resources and contributing greatly to biological evolution and genetic breeding [44,45]. Hybrid offspring of goldfish and common carp show paternally biased expression, while maternally biased expression has been observed after polyploidization of the hybrid [46]. Allotetraploidization has been accompanied by gradual decreases in cis-regulation and increases in trans-regulation in allopolyploids derived from a hybrid of goldfish and common carp following polyploidization [43]. These results suggest a more complex and divergent regulatory mechanism of polyploidization compared with hybridization, and the mechanisms have not yet been comprehensively assessed.
Common carp, as an allotetraploid and the most cultured fish, is an ideal model for studying structural and functional changes in polyploid vertebrates, and contributes to the understanding of successful speciation and evolution of polyploids in animals. Improved whole genome sequences of common carp were generated [6,7,47]. The scope of functional and evolutionary analyses, particularly those related to genome organization, subgenome evolution and homoeologous expression, is limited by the lack of a high-integrity, high accuracy and subgenome-phased reference genome sequence. Accordingly, in this study, we developed a high-fidelity (HiFi) genome assembly of common carp with base-level accuracy and excellent contiguity. This new reference genome allowed us to untangle subgenome organization and homoeologous gene expression in the allotetraploid common carp. Here, we report asymmetrical structural rearrangements of subgenomes and homoeologous exchanges, homoeologous expression plasticity, and regulated expression of subgenomes during development and in response to thermal stress. These results provide novel insights into the molecular mechanisms underpinning the successful speciation and adaptation of allotetraploid carp, deepen our understanding of genome evolution and heterosis in allopolyploid vertebrates, and ultimately should aid in the design of advanced breeding strategies in Cyprinidae to meet the global challenges of food security in the face of climate change.
2. Materials and methods
2.1. Animals and tissues
This study was carried out in accordance with the recommendations of the care and use of animals for scientific purposes provided by the Animal Care and Use Committee of Xiamen University. The fish were euthanized in MS222 solution before samples were collected. Individuals of adult Yellow River carp were collected from the Hatchery Station of Henan Academy of Fishery Sciences at Zhengzhou, China.
2.2. Genome sequencing and assembly
High molecular weight DNA was isolated from the muscle tissue of an adult female fish using QIAGEN DNeasy Blood & Tissue Kits (Qiagen). HiFi sequencing libraries were generated with an average insert size of ∼15 kbp, and sequenced on two SMRT Cells (PacBio) using the CCS (circular consensus sequencing) algorithm on the PacBio Sequel II Sequencing System in Annoroad (Wuhan, China). A total of 39 Gbp of CCS reads were generated with 537 Gb of raw data, and processed using CCS 5.00 software (https://github.com/PacificBiosciences/ccs). Sequence data were deposited in GenBank via the National Center for Biotechnology Information (NCBI) SRA database with project number of PRJNA823855. HiFi reads were assembled using hifiasm v0.12 [48] with the “-l 0” option to disable the purge duplication step of the software default settings, and then one round of duplicate purging using purge_dups v1.25. In total, 1,578.89 Mb of high-fidelity contigs were produced for subsequent assembly, with an N50 of 14.38 Mb. The contigs were anchored and oriented using 3D-DNA and Juicebox. The final assembly was assessed using Benchmarking Universal Single-Copy Orthologs (BUSCO) [49] with the lineage database Actinopterygii_odb10.
2.3. Functional annotation
Repeat elements of the common carp genome were first identified using RepeatModeler to construct a de novo repeat library, and the unknown consensus sequences were further classified using TEClass software [50]. The results, along with a custom library from RepeatMasker [51], were eventually merged together. We used the comprehensive species-specific repeat element library to mask the repeats from known families (replaced with N), and their location information was labeled intergenic. All repetitive regions were soft-masked before annotation of protein-coding genes. Structural annotation was conducted by three strategies: ab initio, homology, and RNA-seq-based prediction. To conduct ab initio and homology-based gene prediction, the protein sequences of Danio rerio, Carassius auratus, Onychostoma macrolepis, Labeo rohita, and Poropuntius huangchuchieni and RNA-seq datasets were input to Braker2 pipeline v2.1.5 containing Augustus [52]. RNA-seq libraries were also aligned to the soft-masked genome with HISAT2 and de novo assembled with StringTie v2.1.4 [53]. A comprehensive transcriptome database was processed with PASA v2.4.1 and sent to TransDecoder to predict open reading frames (ORFs). Finally, an integrated gene set was produced using EVidenceModeler v1.1.1 [54]. Functional annotation was performed using Diamond v2.0.6 and InterProScan by alignment against databases including NR (Non-Redundant Protein Sequence Database), TrEMbl, SwissProt and GO. RNAmmer and tRNAScan were executed for rRNA and tRNA prediction, respectively. Other noncoding RNAs were detected by alignment to the Rfam database (http://eggnogdb.embl.de/). To identify transcription factors (TFs) in common carp, we applied the deep learning-based tool DeepTFactor for prediction of transcription factors [55].
2.4. Whole-genome collinearity analysis among allotetraploid cyprininae fishes and their diploid progenitors
Genome data of C. carpio (NGS version), O. macrolepis, and P. huangchuchieni were obtained from BIGD Genome Warehouse under accession numbers GWHAATB00000000, GWHALOK00000000 and GWHAOPL00000000. The genome of C. auratus was obtained from NCBI with SAMN12618612. Homoeologous. gene pairs were constructed through a reciprocal best hit (RBH) approach using all-against-all BLASTP (v2.10.1+). For homoploid species, the best mutual was accepted; for each diploid species, the best two mutual homologs in tetraploid species were accepted, while for each tetraploid species, only the best mutual homolog in diploid species was accepted; for both subgenomes in tetraploid species, the best mutual homolog was accepted. Pairwise collinear analysis was performed by MCScanX [56] (http://chibba.pgml.uga.edu/mcscan2/) with the parameters ``-s 4 -m 20 -e 1e-10 -b 2”; (at least 4 consecutive homoeologous. gene pairs were required for a collinear block with a maximum of 20 gaps allowed) and visualized by jcvi (https://github.com/tanghaibao/jcvi/wiki/MCscan-(Python-version).
2.5. Analysis of chromosomal rearrangements
2.5.1. Detection of chromosomal rearrangements
Chromosomal rearrangements were identified in each collinear block resolution (with a maximum of 5 homoeologous. gene pairs overlapping) following a simplified strategy described by Goel et al. [57]. First, we identified two chromosomes of pairs of species with the largest number of homoeologous. gene pairs as a pair of homoeologous. chromosomes; collinear blocks between two nonhomoeologous. chromosomes were defined as interchromosomal translocations. Second, between a pair of homoeologous. chromosomes, the longest forward collinear block was defined as a syntenic region and selected as the basal block for further detection of rearranged regions; the rest of the collinear blocks were defined as either syntenic regions or intrachromosomal translocations according to whether they consisted of or contradicted with the basal block regardless of their orientations. Finally, we defined the reverse “syntenic” regions from the second step as inversions.
2.5.2. Timing of chromosomal rearrangements
Based on the phylogenetic relationships among diploid species and both subgenomes of tetraploid species, pairwise chromosomal rearrangements were classified into homoploid-specific, diploid-subgenome B and subgenome A-ancestor B according to the following instructions, with each type of rearrangement marked on the branches representing corresponding evolutionary periods:
Homoploid- (diploid-, subgenome B- or subgenome A-) specific: rearrangements that occurred in only one species, while the corresponding regions in other species retained in synteny;
Diploid-subgenome B: rearrangements that occurred between two diploid species and subgenome B in two tetraploid species, where two diploids and subgenome B in two tetraploids retained synteny;
Subgenome A-ancestor B: rearrangements that occurred only in subgenome A in two tetraploid species, where subgenome A in two tetraploids and 4 lineage B species (or subgenomes) retained synteny.
2.5.3. Analysis of structural features of rearranged boundaries between the two subgenomes of C. carpio
We chose regions spanning 50 kb up- and downstream of the breakpoints of collinear blocks as boundary regions, while the regions within collinear blocks (both syntenic and rearranged blocks) excluding up- and downstream boundaries were considered inner regions. We calculated TE densities and non-synonymous/synonymous (Ka/Ks) ratios of homoeologous gene pairs in each type of the above genomic regions.
2.6. Identification of homoeologous exchanges
Homoeologous exchanges occur via mispairing between ancestrally related chromosomes (also called homoeologous chromosomes) descended from two distinct diploid progenitors, resulting in either balanced or reciprocal translocations where the two homoeologous intervals swap their locations or unbalanced or nonreciprocal translocations where a chromosome region is replaced with a duplicated copy from the corresponding homoeologous region [58]. With the absence of the subgenome A diploid progenitor, we uniquely mapped 150-bp paired-end reads from the common carp subgenome B progenitor-like species O. macrolepis to both subgenomes of C. carpio. Upper and lower coverage (X) thresholds were chosen based on the sequencing depth distribution (Fig. S8). Whole-genome homoeologous regions were constructed by LASTZ (v1.04.03) [59] with the parameters “–strand = both –ambiguous = n – step = 10 – maxwordcount = 500 –m asking = 10 – gfextend – chain – gapped – identity = 70..100”;. For a given homoeologous interval, higher coverage (> 17X) on subgenome A indicates a candidate subgenome B to A-HE region; similarly, relatively lower coverage (< 10X) on subgenome B indicates a candidate subgenome A to B-HE region. Nonreciprocal HEs were defined when both homoeologous. intervals displayed higher (B-to-A) or lower (A-to-B) coverage, while reciprocal HEs were defined when the two homoeologous. intervals showed opposite coverage depths. The average depth was calculated in 10-kb windows, adjacent candidate windows were linked together with at most 5 intervening windows, and only regions containing at least 4 candidate windows were retained. We validated these HE regions in both subgenomes of C. carpio by assessing the sequencing read coverage of two related diploids, O. macrolepis and P. huangchuchieni. Both diploids exhibited significantly higher coverage in B-to-A-HE regions than in non-HE regions in subgenome A, while the coverage in A-to-B-HE regions was significantly lower than that in non-HE regions in subgenome B (Wilcoxon rank-sum test, p value < 0.01).
2.7. RNA sequencing and expression analysis
Total RNA was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) from the common carp morula (S1), blastula (S2), gastrula (S3), neurula (S4), optic vesicle (S5), tail bud (S6), muscle contraction (S7) and hatched larvae (S8) stages at seven temperature conditions (12 ℃, 16 ℃, 20 ℃, 24 ℃, 27 ℃, 30 ℃, and 33 ℃) with three replicates for each stage. To attain a sufficient amount of RNA, approximately 15 common carp embryos were used for RNA isolation for each sample. RNA-seq libraries were constructed using the Illumina TruSeq library construction kit according to the manufacturer's instructions. Paired-end sequencing was conducted on an Illumina NovoSeq 6000 with 150-bp paired-end reads. Clean RNA-Seq reads from each library were aligned to the common carp reference genome sequence using HISAT2 [60]. TPM (transcripts per million) values were calculated by StringTie [53] software for each sample and transformed to raw counts using a Python script provided by StringTie. Normalized raw counts for each sample were processed for differential expression analysis using DESeq2 after filtering out nonexpressed genes and very lowly expressed genes (RNAs with raw counts <= 1/gene) [61]. DEGs (differentially expressed genes) were identified with thresholds of FDR (false discovery rate)-adjusted p value (q-value) < 0.05 and log2 [fold change] > 1 as previously reported [62]. A gene was classified as ‘expressed’ if the TPM value of at least one stage was above 1.0. Sample structure was visualized using hclust and PCA in the R environment. Heatmaps were plotted using the R function pheatmap with scale = “row”. Clustering for DEGs was conducted using Mfuzz [63]. Stage-specifically expressed genes (SEGs) were identified using PaGeFinder [64] with a dpm value greater than 0.9.
To obtain a full set of homoeologous genes in common carp, we used both the gene sets from the progenitor-like reference genome of O. macrolepis and P. huangchuchieni and identified orthologous gene triplets in syntenic regions of each genome (subgenome). Finally, 14,036 homoeologous gene pairs in both subgenomes of common carp were used in the homoeologous expression analysis. Homoeologous genes with expression level changes greater than two-fold compared with their ohnologs in the other subgenome were defined as dominantly expressed in accordance with previous studies [7,31].
2.8. Identification of homoeologs under selection
The homoeolog pairs of common carp were used for estimating Ka/Ks values. Each pair of the sequences was aligned using the MUSCLE alignment software and then transferred to the KaKs calculator using the ParaAT (Parallel Alignment and back-Translation) pipeline [65].
2.9. Functional enrichment of candidate genes
GO and KEGG enrichment analyses were performed using ClusterProfiler 4.0 [66] and the OmicShare tool (https://www.omicshare.com/tools). GO terms with P < 0.05 (FDR < 0.05) were considered significant.
2.10. Data availability
All genomic datasets of C. carpio will be available under NCBI accession number PRJNA823855 and in the BIGD Genome Warehouse under accession number GWHBHRW00000000 upon publication. Transcriptomes were deposited in the NCBI database under accession number PRJNA824204.
3. Results and discussion
3.1. HiFi assembly of allotetraploid common carp (CC 3.0)
To generate a high-fidelity and high-accuracy genome assembly of common carp, 7.44 million PacBio HiFi reads were generated (537 Gb in total), equivalent to approximately 26-fold CCS coverage of the common carp genome (Table S1). The HiFi assembly of the common carp genome (CC 3.0) (Fig. 1a) covered a genome size of 1,579 Mb, including 1,477 scaffolds with an N50 size of 28.32 Mb (Table 1). This represents an increase of 196 Mb additional sequence (13%), as compared to CC 2.0 (NGS version) [7] (Fig. 1; Table S2). A large majority of the newly assembled additional sequences were repetitive elements, as expected (Fig. 1c, 1d; Table S3). In CC 3.0, approximately 98.4% (3,581 of 3,640) of Actinopterygii BUSCO genes [67] were detected, suggesting the high quality of the assembly. The CC 3.0 genome sequences showed overall consistency with the high-density genetic linkage map and the CC 2.0 genome [68] (Figs. 1e and S1a). Further mapping to genome data from the diploid relatives revealed higher coverage of CC 3.0 than the CC 2.0 (Fig. S1b) [7].
Fig. 1.

Characterization of the common carp HiFi assembly. (a) Features of homoeologous chromosomes in the HiFi genome, CC 3.0. From outer to inner: I, gene density; II, repeat density; III, SNP density; IV, indel density; V, GC content; VI, gene expression based on adult muscle tissue; VII, best-hit gene pairs between two subgenomes. Gene density and repeat distribution were calculated within each 100-kb sliding window. SNP and indel density were based on previously published resequencing data for Yellow River carp [7]. (b) Chromosome length comparison between the HiFi and NGS versions of common carp genomes. (c) Repeat coverage of syntenic and nonaligned regions between the HiFi and NGS version genomes. (d) Alignment of chromosome B22 between the HiFi and NGS genomes. The center shows syntenic blocks, with orange lines for gene density, and purple for repeat density. (e) Example showing the relationship between the HiFi genome and linkage map [68]. The linkage map is on the left.
Table 1.
Global statistics for the common carp genome assembly.
| Genomic feature | Value |
|---|---|
| Length of HiFi assembly | 1,578,889,074 |
| Number of contigs | 676 |
| contigs (>= 50,000 bp) | 562 |
| Longest length (bp) | 32,069,724 |
| Length of contig N50 (bp) | 14,383,676 |
| Length of contig N90 (bp) | 1,336,144 |
| L50 | 41 |
| L90 | 181 |
| Length of pseudomolecule assembly | 1,579,378,574 |
| Total length of anchored pseudomolecules | 1,456,720,202 |
| Gap size (bp) | 492,030 |
| Longest length (bp) | 46,234,316 |
| N50 (bp) | 28,322,000 |
| N90 (bp) | 20,266,301 |
| L50 | 24 |
| L90 | 49 |
| Repetitive elements | 43.4% |
| BUSCO | |
| Complete | 98.4% |
| Duplicated | 63.2% |
| Fragmented | 0.7% |
| Missing | 0.9% |
The HiFi sequencing allowed efficient assembly of the allotetraploid genome with a high percentage of repetitive sequences. Approximately 43.4% of the CC 3.0 genome was annotated as repetitive elements, with class Ⅱ DNA transposons accounting for 29.3%, followed by class Ⅰ retrotransposons (7.2%), and Gypsy and Copia retrotransposons (4.5%) (Fig. S2a; Table S4). The content of repetitive sequences in the genome/subgenomes was similar to that in the closely related species and progenitor-like species, such as goldfish, P. huangchuchieni and O. macrolepis (Fig. S3; Tables S5, S6). The LTR assembly index (LAI) is an indicator of repetitive sequence contiguity, as well as the contiguity of the genome assembly [69]. This HiFi genome had LAI values of 10.58 and 10.87 for subgenomes A and B, respectively (Fig. S4), suggesting that CC 3.0 greatly improved the genome quality and continuity and represents an essential reference genome for precise genetic analyses, especially when dealing with the subgenomes. The subgenomic landscape of TE divergence divided the evolutionary timeline into three periods (Fig. S2b). Period Two, with TE divergence rates ranging from 5% to 9%, indicated that the progenitors of both subgenomes diverged as two distinct linages from their common ancestor. This time window is even narrower than the previously reported window of 7%−13% [7], suggesting a more complete TE set in the HiFi genome. In addition, we observed several TE subfamilies showing specific expansion in subgenome A or B (Fig. S5a, S5b), similar to results recently reported [47], and period-specific expansion in subgenome A (Fig. S5a). After polyploidization, subgenome A underwent a dramatic “genomic shock”; thus, the TE subfamily RC/Helitron experienced a recent burst (Fig. S5c, 5d). These results provide additional insights into the progenitor of subgenome A.
In total, 55,981 high-confidence protein-coding genes were predicted in the CC 3.0 genome (Tables S7, S8), and 53,671 of these genes were included in the 50 chromosomes. Transcription factors play central roles in the transcriptional regulation, mastering embryonic development and contributing to eukaryotic evolution and complexity [70,71]. We identified 3,812 TF genes in the common carp genome, approximately 1.5 times than in the zebrafish (Danio rerio) genome, indicating a more complex regulatory network of genes retained after WGD. Of these TFs, 2,192 (57.5%) were homoeologous gene pairs in both subgenomes, and subgenome B possessed slightly more TFs than that in subgenome A (1,935 vs. 1,865).
Recent advances in long-read/high-fidelity sequencing technologies and computational innovations are revolutionizing genomic research on polyploid species with large and high-repeat genomes. The complexity of the genome reorganization, the epigenomic landscape and transcriptional regulation has been increasingly observed, and has been suggested to drive genome evolution and contribute to certain phenotypic variations and increased adaptability of polyploid plants to diverse environments [[72], [73], [74]]. Here, we present a high-quality, high-fidelity reference genome sequence of common carp using CCS, a technology preferred by various sequencing initiatives with base-level accuracy [[75], [76], [77], [78]]. This new reference genome enables precise analyses of subgenome evolution, homoeologous expression divergence and plasticity during embryogenesis and under thermal stress. The genome will support genetic dissection of economic traits and targeted breeding in carps.
3.2. Asymmetrical structural evolution of the allotetraploid subgenomes
With the availability of genomic resources for nonmodel species in the family Cyprinidae, including those of the polyploid common carp [6,7], Sinocyclocheilus cavefish [79], Oxygymnocypris stewartii [80], goldfish [41,42,81], and Schizothorax o'connori [82], and the diploid O. macrolepis [83] and P. huangchuchieni [84], we assessed the origin and structural evolution of polyploidization in Cyprinidae. Previous results indicated that common carp and goldfish shared a common origin, and that matrilineal-like subgenome B in the allotetraploid C. carpio has a potential diploid progenitor assigned to the Barbinae lineage [7,41]. To explore the evolutionary changes in genome structure before and after tetraploidization in common carp, we first performed gene-based whole-genome collinearity analysis among allotetraploids (C. carpio and C. auratus) and their diploid progenitors from Barbinae (O. macrolepis and P. huangchuchieni). Pairwise collinearity revealed that subgenome B shared more collinear gene pairs with Barbinae diploids than subgenome A (Figs. 2a and S6; Table S9). There were more collinear gene pairs shared within subgenomes in allotetraploids when compared with O. macrolepis than compared with P. huangchuchieni (Table S9), suggesting a potentially closer genetic relationship between subgenomes B and O. macrolepis, assuming similar qualities of the genome assemblies for O. macrolepis and P. huangchuchieni. Hereafter, we chose O. macrolepis as the progenitor-like reference genome for further analysis. Using the phylogenetic topology for each of these species (Fig. 2b) as a reference, we defined chromosomal rearrangements into diploid-specific, subgenome B-specific, subgenome A-specific, diploid-subgenome B and subgenome A-ancestor B. The results showed that C. carpio subgenome B preserved a more stable karyotype of the ancestor Barbinae lineage, while C. carpio subgenome A evolved more rapidly. Moreover, both subgenomes of C. auratus exhibited more chromosomal rearrangements than C. carpio, partially reflecting the complex structural variations associated with the longstanding domestication history of goldfish (Figs. 2a and S6a; Table S10).
Fig. 2.

Structural evolution before and after allotetraploidization in Cyprininae and their diploid progenitors. (a) Whole-genome collinear relationships among subgenome B and diploid progenitors. Typical structural rearrangements between diploids and subgenomes B are marked in red. Rearrangements between the two diploids are marked in dark gray. (b) Phylogenetic relationships of the representative species. The numbers on branches indicate the structural rearrangements, which are defined as diploid-specific, subgenome B-specific, subgenome A-specific, diploid-subgenome B and subgenome A-ancestor B. (c) TE distributions in the syntenic, rearranged and boundary regions (pink and cyan boxes represent subgenomes A and B, respectively). (d) Ka/Ks ratios of homoeologous gene pairs in the syntenic, rearranged and boundary regions. (e) Homoeologous exchanges (HEs) between chromosomes A09 and B09 in the HiFi genome. HEs are detected based on the differences in the sequencing depth distribution of the diploid Oma and Phu mapped to the two subgenomes of C. carpio. For a given homoeologous interval (light gray and purple lines represent forward and reverse alignments respectively), relatively higher coverage on A09 (dark red) reveals B-to-A-HE regions, while relatively lower coverage on B09 (dark cyan) reveals A-to-B-HE regions. The significance of c and d was tested with a two-sided Wilcoxon rank-sum test. Abbreviations: Oma, O. macrolepis; Phu, P. huangchuchieni; Cca, C. carpio; Cau, C. auratus. One asterisk indicates a p value < 0.05, two asterisks indicate a p value < 0.01, and three asterisks indicate a p value < 0.001.
Chromosomal rearrangements were identified when the subgenomes of C. carpio were compared, with 19 of those shared by subgenomes A in both C. carpio and C. auratus and 4 shared by subgenome B in both C. carpio and C. auratus (Fig. S6b; Table S11), indicating that these structural differences were likely inherited from the ancestral lineages of the two subgenomes. There are 9 structural rearrangements occurring specifically in C. carpio subgenome A (Table S11). These results imply that most of the differences between C. carpio subgenomes A and B may have existed before tetraploidization when the respective ancestors of the two subgenomes separated into two distinct diploid lineages. The two subgenomes merged with the retention of structural characteristics from each diploid progenitor, after which subgenome A underwent more structural variations, while subgenome B remained well-conserved.
Comparison of TE distributions in different genomic regions showed that TEs were significantly enriched in the vicinities of collinear block boundaries, indicating that TE insertions may be associated with genome structural variants (SVs), consistent with results observed in plants [85]. Moreover, rearranged block regions also had slightly higher TE densities than syntenic block regions (Fig. 2c). Homoeologous gene pairs located near the boundaries of collinear blocks had higher Ka/Ks ratios than those located inside the collinear blocks (Fig. 2d). These results suggest that transposon activation and relaxed selection pressure may serve as potential evolutionary drivers causing structural rearrangements between C. carpio subgenomes. Genomic rearrangements that arise after polyploidization may enable novel cis-regulation and the accumulation of adaptive genes locally [86]. Structural changes with increased fitness for polyploids could be selected and preserved during evolution [87]. Research in sugarcane showed that more than half of the disease resistance genes were located in rearranged regions [88]. Genes in the flanking regions of the chromosomal rearrangements in common carp also exhibited enrichment for genes associated with infectious diseases (Fig. S7), suggesting the sources of higher levels of resistance against disease and abiotic stresses in common carp than in their progenitors.
Homoeologous exchanges occurred along with allopolyploid formation and the following generations [[89], [90], [91]]. Genomic rearrangements generated by HEs are an important source of genetic and phenotypic variation [92] and have been shown to impact important agronomic traits in plants, such as seed quality, flowering time, and disease resistance [92,93]. In the absence of the diploid progenitor of subgenome A, we uniquely mapped paired-end reads from O. macrolepis to both subgenomes of C. carpio and detected candidate HEs as regions with significant coverage alterations (Fig. S8). In CC 3.0, we observed a significant asymmetrical distribution of HEs within subgenomes, with more subgenome A regions replaced by subgenome B regions than vice versa (2.27% vs. 0.68%) (Figs. 2e and S9; Table S12). Similar findings were reported in allopolyploid crops, such as cotton [94], B. napus [95], strawberry [96] and wheat [97]. This result implies that subgenome B in common carp may contribute more to certain phenotypic evolutions, and therefore, the exchanged fragments were retained across generations. Such phenomena have also been reported in allopolyploid crops where retained and fixed HEs were more often the dominantly expressed homoeologs [98]. We further investigated whether these homoeologous exchange regions have potential roles for certain phenotypes or for adaptation to different habitats. We identified 73 A-to-B, 245 B-to-A and 32 reciprocal HE homoeologous genes, respectively (Table S12). These HE genes were highly enriched in functions related to cancer, the cell cycle, the FoxO signaling pathway, the Hippo signaling pathway, and the mTOR signaling pathway (Fig. S10). Considering that more positively selected genes existed in subgenome B than in subgenome A and their potential roles in the immune response and response to stimulus [84], we speculate that subgenome B in common carp might have contributed more than subgenome A to immune-related functions. Such asymmetric structural evolution between subgenomes could be caused by divergent selection pressures and TE frequencies, and might have contributed to phenotypic evolution in carp.
3.3. Expression dynamics across embryonic development
Gene expression dynamics and plasticity of homoeologous genes make major contributions to agronomic trait variations and species adaptive radiation [[99], [100], [101], [102]]. In some circumstances, dominance effects of a single homoeolog can lead to dominant phenotypes [103]. Allotetraploid common carp is a successful example of adaptive radiation in the animal system, and therefore, determination of its genomic and subgenomic expression during embryonic development and under environmental stress is important for both evolutionary understanding and practical applications [104,105]. Firstly, we determined the transcriptomes across eight developmental stages (Fig. 3a, Fig. S11), covering the morula (S1), blastula (S2), gastrula (S3), neurula (S4), optic vesicle (S5), tail bud (S6), muscle contraction (S7), and hatched larvae (S8) stages. We found evidence of expression for 19,024 high-confidence genes, based on expression of > 1 TPM in at least one of the eight developmental stages. Among these expressed genes, 476 to 8,244 were differentially expressed between two adjacent stages according to pairwise comparison (termed stage-related DEGs, SDEGs) (Table S13). SDEGs were observed with clear patterns for a specific stage or transitions among stages (Fig. 3b), including 2,499 stage-specifically expressed genes (SEGs) exclusively expressed in one stage (Fig. S12; Table S14), reflecting their roles in the corresponding developmental stages (Figs. 3c and S13). For example, genes showing the highest expression in the tail bud and muscle contraction stages (cluster 6) were enriched in the PI3K-Akt signaling pathway, signaling pathways regulating pluripotency of stem cells, the Hippo signaling pathway, and the Wnt signaling pathway (Figs. 3c and S13). These pathways play pivotal roles in regulating organ development, tissue homeostasis and regeneration [[106], [107], [108], [109]], suggesting nearly complete cell fate differentiation and organ formation, and readiness to hatch.
Fig. 3.

Expression landscape during embryogenesis in allotetraploid common carp. (a) Sampling stages during embryogenesis. S1, morula; S2, blastula; S3, gastrula; S4, neurula; S5, optic vesicle; S6, tail bud; S7, muscle contraction; S8, hatched larvae. Microscope images were modified according to Jin et al. [110]. (b) Expression patterns of DEGs during embryogenesis. DEGs were identified by pairwise comparison between two adjacent stages at 24 °C and termed stage-related DEGs or SDEGs. The heatmap shows the overall expression of all the SDEGs, indicating the similarity between S6 and S7. (c) Functional enrichment of genes in seven clusters in Fig. 3b. (d) Percentage of single or paired homoeologs in each of the seven clusters. Paired means that the two homoeologous genes in the two subgenomes existed in the same cluster.
Genes with similar expression patterns are commonly found to have analogous functions. We next examined the homoeologous status of each cluster for a better understanding of the functional divergence of duplicated genes in the two subgenomes. Of the 14,036 syntenic homoeologous gene pairs that had a 1:1:1 relationship in subgenomes A and B of common carp and the ancestral species, significantly more single copies (71.7%−90.2%) were found rather than paired copies (9.9%−28.3%) in a cluster (T-test, P = 1.50088 × 10−5) (Fig. 3d; Table S15). This indicated that only a few homoeologous genes were co-expressed and that the majority were divergently expressed during embryonic development. HGs involved in the evolutionarily conserved pathways (Wnt signaling pathway, translation, and ribosome) were more likely to be co-expressed during embryogenesis (Fig. S14). These results provide a broad view of gene expression patterns and a functional atlas during embryonic development.
3.4. Subgenome expression divergence across embryonic development
Subgenome expression dominance has been thoroughly described in allopolyploid plants [111]. To draw a comprehensive picture for subgenomic expression across development, we focused on HGs that had a summed expression of > 1 TPM in at least one stage (9,325 homoeologous gene pairs). The overall expression levels of subgenome B were significantly higher than those of subgenome A throughout the development (Fig. 4a, two-sided Wilcoxon rank-sum test, P < 0.05). In addition to the relative expression levels, most HGs were assigned to the biased categories (homoeologous expression bias, HEB) with expression divergence larger than twofold. The divergent ratios decreased as the development proceeded: the highest ratio was 79.8% in S1 stage (morula), and the lowest ratio was 63.4% in S8 (hatched larvae) (Figs. 4b and S15; Table S16). These results suggest broad asymmetrical expression between subgenomes during embryogenesis and that fetal development reduces the expression differences of parental origin homoeologs in the allotetraploid genome. Interestingly, the number of genes biased toward subgenome B (34.5%−41.6%) was significantly larger than that toward subgenome A (29.0%−38.2%) (Table S16, T test, P = 8.54 × 10−5). Such bias was universal not only across different stages of embryonic development as found here, but also in adult organs [7], suggesting a greater contribution of expression from subgenome B than subgenome A in the allotetraploid genome. These results are consistent with those found in maize embryos, where expression is biased toward the maternally inherited alleles [112].
Fig. 4.

Subgenomic expression divergence during embryogenesis in allotetraploid common carp. (a) Expression levels of the two subgenomes. The boxplot was based on homoeologous gene pairs with a summed expression of > 1 TPM in at least one developmental stage. The expression levels of genes in subgenomes A and B and the sum of both copies are included. (b) Percentage of dominant and balanced expression of genes during embryogenesis. Dominant expression is defined as an expression divergence larger than 2-fold. (c) The expression levels of dominant and balanced expressed homoeologous genes. The expression levels of the balanced genes were higher than those of the suppressed genes, but lower than those of the dominant genes. (d) Expression patterns for HGs switching their dominant patterns during development. In total, 275 HGs showed a dominant switch from A to B, and 202 from B to A. The turning point occurred at the S4 (neurula) stage. One asterisk indicates a p value < 0.05, two asterisks indicate a p value < 0.01, and three asterisks indicate a p value < 0.001. The significance for a and c was tested with a two-sided Wilcoxon rank-sum test.
When genes were divided into three clades based on expression dominance, balanced genes had significantly lower levels of expression than dominant genes (Wilcoxon signed rank sum test, P = 2.22 × 10−16) (Fig. 4c). Intriguingly, the total expression of homoeologous pairs was highest for B-dominant genes, followed by A-dominant genes, with the lowest expression for the balanced homoeologous genes. In hexaploid bread wheat, balanced genes had the highest expression among all gene clades, and the dominant triads are not the result of the increased expression of a single homoeolog, but the relatively lower expression of the other homoeologs [113]. Inconsistency may be the result of karyotype differences or an intrinsic discrepancy between plants and animals. Dominant homoeologous genes with high expression levels may cause a greater advantage in the allotetraploid common carp than in their diploid progenitors. Another possible explanation is that the allotetraploid common carp was from the hybridization of two distant species, where many of the cellular functions require genes from a single parent, and such functional genes are generally expressed at higher levels in one subgenome than in the other one. Additionally, our samples underwent stages of rapid development and differentiation, and the biased subgenome expression may be functionally relevant to development [112]. Previous work with plants suggested that cytoplasmic genomes may cause dominant expression of maternal-origin homoeologous genes [114]. If a similar pattern holds true in common carp, these results would suggest that the dominance status of subgenome B was to some extent inherited from the maternal species, in agreement with the prediction that subgenome B was of maternal origin [41].
We further investigated whether embryonic development led to a change in the dominance pattern of homoeologous genes. All the expressed genes were divided into four groups: 501 (5.4%) HGs with copy A dominantly expressed in all eight stages, 633 (6.8%) HGs with copy B dominantly expressed in all stages, 1,056 (11.3%) homoeologs with balanced expression from both copies in all stages, and the remaining 7,135 (76.5%) HGs with switching expression patterns during embryogenesis (Table S17). Homoeologous genes with consistently biased expression in subgenome A or B shared most of the enriched functions (Fig. S16), implying that hybrids may enable the exploitation of the favorable parental alleles either in subgenome A or B to express at relatively high levels, a phenomenon that can be explained by the dominance hypothesis for heterosis [115]. HGs with switching patterns of homoeologous dominance might be relevant to a specific development or differentiation process, or a functional division in temporal or spatial dimensions or both, and the subfunctionalization of these HGs may be required for development, similar to the behavior of genomically imprinted genes, although there have been no reports of genomic imprinting in teleost fish. Interestingly, two switch patterns were observed in the switching HGs, where 275 showed a dominant switch from A to B, and 202 from B to A, respectively (Fig. 4d). The turning point occurred at the S4 (neurula) stage, consistent with the functional transition from the early cell proliferative stage to differentiation for the development of organs. The significantly higher number of A-to-B HGs (chi-square test, P = 8.305 × 10−4), together with a greater number of B-dominant homoeologs (chi-square test, P = 8.861 × 10−5), suggests that the subgenome B was more important for late embryonic development and differentiation. In the context of genome and subgenome expression in relation to early embryonic development, the findings reported here are novel among animal systems. However, shifts in expression from homoeologs or shifts in homoeolog usage were reported in allopolyploid Glycine dolichocarpa [116] and Arabidopsis [117], and hexaploid wheat [118]. These genes with different switching directions participate in distinctive biological processes (Figs. S17, S18), indicating a development-dependent functional segmentation of WGD-induced duplicated genes. Such altered dominance patterns and the distinguishing functions of HGs are pervasive in polyploids, where subgenome functional partitioning is a major mechanism for HG preservation [119].
3.5. Homoeologous expression plasticity under thermal stress
The temperature tolerance for common carp was reported to be 3 °C to 35 °C [8,9], and the thermal responsiveness of fish embryos was the lowest among all the life stages [120]. To investigate gene expression patterns and how homoeologous genes respond to thermal stress during embryonic development in common carp, bulk transcriptomes were obtained from eggs to larvae under seven temperatures (12 °C, 16 °C, 20 °C, 24 °C, 27 °C, 30 °C and 33 °C), with the 24 °C group as the control (Fig. 5a). It is worth noting that allotetraploid embryos cannot hatch and be stunted at the gastrula stage (S3) in the highest thermal group (33 °C), and at the muscle contraction stage (S7) in the lowest thermal group (12 °C). The diploid embryos of O. macrolepis cannot proceed with development at 28 °C according to our recent experiments, again indicating a higher thermal tolerance for the allotetraploid common carp than its progenitor-like species.
Fig. 5.

Homoeologous expression plasticity to cope with heat stress in allotetraploid common carp. (a) Experimental design of thermal stress during embryogenesis. Seven temperatures along a gradient were set, and the 24 °C group was the control. The maximum lethal temperature for the allotetraploid embryos is 33 °C (development stopped at the gastrula stage), and the minimum lethal temperature is 12 °C (development stopped at the muscle contraction stage). (b) Overlap of homoeologous genes indicating the consistent or changeable dominant pattern under heat stress. Each row corresponds to a possible intersection: the filled-in cells show which set is part of an intersection. Purple numbers represent HGs maintaining their dominant patterns under different temperature conditions. (c) Comparison of functional enrichment for HGs with different dominant patterns under control and heat stress. GO enrichment was performed using ClusterProfiler 4.0. (d) Ka/Ks for HGs with consistent dominant patterns during development. Homoeologous gene pairs were divided into four clades according to whether their dominant patterns had changed during development. One asterisk indicates a p value < 0.05, two asterisks indicate a p value < 0.01, and three asterisks indicate a p value < 0.001. The significance was tested with a two-sided Wilcoxon rank-sum test. (e) HG dominant pattern changes under heat stress at the S3 stage. Purple depicts directional changes in the dominant patterns of HGs under heat stress. Purple in the second quadrant represents a B to A dominant change, and purple in the fourth quadrant represents a A to B dominant change. Green points represent HGs with consistent dominant patterns. Orange points represent HGs changing their patterns from dominant to balanced or from balanced to dominant under stressed conditions. (f) Examples of 16 HGs changing their dominant patterns during development and under heat stress. The 16 HGs included 9 A-to-B and 7 B-to-A HGs showing switching both during embryogenesis and under heat stress conditions. T24 represents 24 °C (control), and T30 represents heat stress.
Under heat stress, a total of 388 (S6) to 22,753 (S1) DEGs were identified in at least two temperature comparison groups in each stage (Table S18), suggesting their potential sensitivity at earlier developmental stages. To (a) simplify the results, (b) facilitate the comparative analysis under control and stress conditions, and (c) avoid disturbances from development, we further focused on the 154 to 21,479 DEGs at each stage that overlapped between the comparisons of 30 °C versus 24 °C (Table S19). The number of DEGs in subgenome B was slightly greater than that in subgenome A (7,904 vs. 7,818). As expected, these DEGs were involved in many pathways related to the heat stress response, including ribosome, oxidative phosphorylation, carbon metabolism, TCA cycle, and thermogenesis (Fig. S19a), as previously reported [121]. Cold stress-induced DEGs were involved in similar functional categories (Fig. S19). Among heat-induced DEGs, 2,095 (1,874 upregulated, 349 downregulated) were transcription factors, accounting for ∼55% of the total TFs (2,095/3,812), indicating the critical role of TFs in the regulation of the stress response in the polyploid genome [122].
In allopolyploid plants, each subgenome may control different sets of traits, and certain phenotypic traits could be largely controlled by a single subgenome [123,124]. Accordingly, we were eager to determine how homoeologous genes in the two subgenomes respond to heat stress. Somewhat to our surprise, the majority of the HGs responded to heat stress with either subgenomic copy (Table S20). In contrast to the significantly greater contributions of homoeologous genes in subgenome B during embryonic development, here in response to stress, the two subgenomes contributed with similar genes, but again with a slight bias for genes of subgenome B (Table S20), suggesting a slight advantage of subgenome B over subgenome A in response to thermal stress. Additionally, for the limited HGs that contributed to the heat response in both cases, most were regulated in the same direction (Table S21). Such “consistent” expression changes in HGs after stress treatment has been previously described in plants [125], and predicted to be regulated by trans-acting factors [126,127]. However, we did find a small set of HGs showing the opposite direction of regulation, with one of the homoeologous gene pairs upregulated while the other was downregulated, suggesting extreme functional divergence (Figs. S20, S21; Table S21). The gene ndufa4 (NADH dehydrogenase [ubiquinone] 1 alpha subcomplex assembly factor 4) provides a good example of this type of opposite direction of regulation (Fig. S20). Taken together, these results imply that the expression or functional divergence of post-WGD preferentially retained most homoeologous genes in stress responses [128,129].
We further examined subgenome dominance under thermal stress. Heat stress increased the number of A- or B-dominant HGs (Figs. S15 and S22). This was also found during cold stress (Fig. S23), indicating that subgenomes in common carp may cope with changing temperatures by driving the divergent expression of HGs in a favorable state. Comparisons of genes with consistent dominant patterns between 24 °C and 30 °C revealed that the majority of HGs maintained their pattern of dominance (Table S22); 436 and 544 HGs biased toward subgenome A and B, respectively, exhibited consistent expression dominance bias shared under the two temperature conditions (Fig. 5b; Table S22). Functional analysis revealed that HGs involved in peptide metabolic processes, translation, and ribosomes were likely not to change their dominant patterns regardless of development or environmental conditions (Fig. 5c). The 980 HGs (436 and 544 HGs with A or B bias) were mostly involved in disease and environmental adaptation pathways, such as thermogenesis, oxidative phosphorylation, bacterial invasion of epithelial cells, and homoeologous. recombination (Fig. S24). Similar to the situation with development, the homoeologous genes showing consistent dominant patterns under temperature conditions suggested that the allotetraploid common carp preferentially uses the favorable copies of the homoeologous genes, and preferentially expresses them at higher levels. This is consistent with the mechanism of heterosis [115] and shows that some allelic expression was already fixed in the allotetraploid genome. Additionally, HGs changing their dominant patterns under heat stress were involved in functional categories related to cellular response toward stimulus and immune response-activating signal transduction. Unique HGs with a bias toward subgenome B (312 HGs) were associated with GO terms such as mitotic nuclear division, transcription from the RNA polymerase II promoter, Hsp90 protein binding, and ATP binding, again suggesting the greater roles of subgenome B in developmental and environmental regulation.
To obtain insight into the mechanisms contributing to the expression divergence of HGs, we performed a selection pressure analysis. Dominant HGs showed significantly higher Ka/Ks values than the balanced HGs (Figs. 5d and S25), suggesting that the homoeologous expression pattern may be induced by a higher rate of sequence divergence caused by asymmetrical evolution. Similar phenomena have been described in tetraploid Salvia splendens and Ethiopian cereal teff, in which homoeologous gene pairs with stronger expression bias were observed with more relaxed selective constraints [85,130]. The significance was expanded by the comparison of B dominant and switching HGs, as well as B-dominant and A-dominant HGs under a high temperature of 30 °C (Fig. 5d). There was a high frequency of B-dominant or switching to B-dominant expression, suggesting that the B subgenome may be more involved in adaptive evolution than the A subgenome in terms of the stress response. In addition, several HGs with Ka/Ks > 1 were found to be involved in response to stimulus and immune system process (Fig. S26), such as hsp70-binding protein 1, diablo homolog: mitochondrial, and T-cell immunomodulatory protein.
For each developmental stage, dominant pattern changes were observed for several HGs under higher temperatures (Figs. 5e and S27; Table S23). Strikingly, there were 9 A-to-B and 7 B-to-A HGs showed switching both during embryogenesis (S3 as a representation of cleavage and S7 for organogenesis) and under heat stress conditions (Figs. 5f and S28). Of these 16 HGs, 10 HGs showed a significant heat response with one or two copies. One example was the ubiquitin-conjugating enzyme E2 B (ube2b) gene, a key gene that plays vital roles in both the development and regulation of immunity and various biotic/abiotic stress responses [131], [132], [133]. Changes in DNA sequence can lead to enduring functional divergence, while expression regulation shaped by transcriptional, post-transcriptional, or translational modifications provides flexibility and plasticity for the species [134], [135], [136]. These results provide insights into how allotetraploids can modulate the expression pattern of HGs to facilitate development and adaptation to changing environmental conditions, a phenomenon that may contribute to heterosis.
Recently, researchers proposed a route to de novo domestication of wild allotetraploid rice [137]. This inspired the application of such a strategy in the Cyprinidae system. Excellent characteristics of carps have been employed to produce various breeding strains, greatly benefiting the breeding industry [138,139]. The utilization of diploid germplasm is very limited. Comprehensive description and elucidation of the molecular rationale for heterosis phenotypes and other advantageous traits of allotetraploid carps are urgent. Causal effector genes associated with agriculturally important traits could benefit targeted breeding and de novo domestication of carps and their diploid relatives for performance improvements. Ideally, such an analysis should be based on a full and thorough comparison between the allotetraploids and their two diploid progenitors. The lack of paternal progenitor species hinders, but does not stop, progress. On the basis of our findings, we can infer that not only genomic/subgenomic plasticity and nucleic acid level diversity, but also the regulation of subgenome expression together promotes organismal development and adaptation to variable environmental conditions, provides genetic robustness, and contribute to the expanded distribution and widespread phenotypic variations of common carp. These results reveal the importance of allopolyploidization in carp species. Considering the dominance of the maternal-origin subgenome B from the Barbinae-like species, and the laggard utilization and improvement of wild-like diploid fish species, such understanding of the subgenome evolution and plasticity of the allotetraploid common carp, as well as further efforts focusing on the a thorough comparison between allotetraploids and their progenitor-like diploids, will: (1) support a greater understanding of speciation and heterosis, (2) facilitate targeted breeding and domestication of carp and diploid relatives, and (3) advance sustainable aquaculture.
4. Conclusion
A comprehensive understanding of the speciation, genome evolution, and subgenome expression plasticity of polyploids is crucial for interpreting the evolutionary consequences and advantages of polyploidization. Such importance has been well demonstrated in polyploid plants, but not in animals. Here, we present a high-quality, high-fidelity genome assembly for common carp, which enabled precise analyses of subgenome evolution. As schematically presented in Fig. 6, we provided evidence for subgenome reorganization, asymmetrical subgenome expression, and shifting expression of subgenomes during development and under thermal stress. Subgenome A was found to have evolved more rapidly than subgenome B, whereas subgenome B contributed greater levels of expression, especially for disease or stress response genes. The majority of genes were expressed with subgenome bias. There was an apparent trend of transition/shift from the expression of subgenome A during early embryonic development with predominant activities of cellular proliferation to that of subgenome B during later embryonic development with predominant activities of tissue/organ differentiation. Such regulation could result from variations in regulatory sequences evolving during evolution and could also involve epigenetic regulation. Future studies on epigenetic modifications and their control over subgenomic expression during development and under stress are warranted. Homoeologous genes with consistent or changing dominant patterns during development and under thermal stress suggest potential fixed heterosis and a dominant effect of heterosis in the allotetraploid genome. Our findings suggest that the reorganized genomic structure and regulatory networks that we report in carp are indicative of processes that drive speciation, adaptation, and phenotype evolution of polyploids compared with their progenitor-like species. Taken together, these analyses provide novel insights into the molecular basis of the successful speciation, distribution, and evolution of the allotetraploid common carp.
Fig. 6.

A schematic depicting the potential mechanisms involved in homoeolog expression that lead to development, growth vigor and enhanced adaptation in allotetraploid common carp. Major findings of this work: revealing variations of the subgenomes, differential expression of subgenomes, and shifting dominant expression of homoeologs during development and under environmental stress, providing insights into the successful speciation and heterosis of allotetraploid common carp. Words in red indicate evidence generated in the present study.
Declaration of competing interest
The authors declare that they have no conflicts of interest in this work.
Acknowledgments
This work was supported by the National Key R&D Program of China (2019YFE0119000), the National Science Fund for Distinguished Young Scholars (32225049), the National Natural Science Foundation of China (31872561) and the Alliance of International Science Organizations (ANSO-CR-PP-2021-03). We thank Dr. Liangsheng Zhang for the valuable discussion on genome assembly.
Biographies

Lin Chen is a PhD graduate of College of Ocean and Earth Sciences at Xiamen University.

Peng Xu(BRID: 09821.00.15175) is a professor of College of Ocean and Earth Sciences, Xiamen University. His research interest is fish genomics and genetics, genome-assistant breeding and selection, population genetics and evolution genomics, genetic mechanism of adaptive evolution.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.fmre.2023.06.011.
Contributor Information
Zhanjiang Liu, Email: johnliu@syr.edu.
Peng Xu, Email: xupeng77@xmu.edu.cn.
Appendix. Supplementary materials
References
- 1.Edwards P., Zhang W.B., Belton B., et al. Misunderstandings, myths and mantras in aquaculture: Its contribution to world food supplies has been systematically over reported. Mar. Policy. 2019;106:103547. [Google Scholar]
- 2.FAO . FAO; 2020. The State of World Fisheries and Aquaculture 2020 (Sustainability in Action) [Google Scholar]
- 3.Zhang W.B., Belton B., Edwards P., et al. Aquaculture will continue to depend more on land than sea. Nature. 2022;603(7900):E2–E4. doi: 10.1038/s41586-021-04331-3. [DOI] [PubMed] [Google Scholar]
- 4.Barbarossa V., Bosmans J., Wanders N., et al. Threats of global warming to the world’s freshwater fishes. Nat. Commun. 2021;12(1):1701. doi: 10.1038/s41467-021-21655-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ren R., Wang H.F., Guo C.C., et al. Widespread whole genome duplications contribute to genome complexity and species diversity in angiosperms. Mol. Plant. 2018;11(3):414–428. doi: 10.1016/j.molp.2018.01.002. [DOI] [PubMed] [Google Scholar]
- 6.Xu P., Zhang X.F., Wang X.M., et al. Genome sequence and genetic diversity of the common carp, Cyprinus carpio. Nat. Genet. 2014;46(11):1212–1219. doi: 10.1038/ng.3098. [DOI] [PubMed] [Google Scholar]
- 7.Xu P., Xu J., Liu G.J., et al. The allotetraploid origin and asymmetrical genome evolution of the common carp Cyprinus carpio. Nat. Commun. 2019;10(1):4625. doi: 10.1038/s41467-019-12644-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McLarney W.O. Hartley & Marks Publishers; 1998. Freshwater aquaculture: A Handbook for Small Scale Fish Culture in North America. [Google Scholar]
- 9.Froese R. FishBase. world wide web electronic publication. http://www.fishbase.org
- 10.Balon E.K. Origin and domestication of the wild carp, Cyprinus carpio: From Roman gourmets to the swimming flowers. Aquaculture. 1995;129(1):3–48. [Google Scholar]
- 11.Ruiz-Navarro A., Gillingham P.K., Britton J.R. Shifts in the climate space of temperate cyprinid fishes due to climate change are coupled with altered body sizes and growth rates. Glob. Chang. Biol. 2016;22(9):3221–3232. doi: 10.1111/gcb.13230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Novikova P.Y., Hohmann N., Van de Peer Y. Polyploid Arabidopsis species originated around recent glaciation maxima. Curr. Opin. Plant Biol. 2018;42:8–15. doi: 10.1016/j.pbi.2018.01.005. [DOI] [PubMed] [Google Scholar]
- 13.Van de Peer Y., Mizrachi E., Marchal K. The evolutionary significance of polyploidy. Nat. Rev. Genet. 2017;18(7):411–424. doi: 10.1038/nrg.2017.26. [DOI] [PubMed] [Google Scholar]
- 14.Cai L.M., Xi Z.X., Amorim A.M., et al. Widespread ancient whole-genome duplications in Malpighiales coincide with Eocene global climatic upheaval. New Phytol. 2019;221(1):565–576. doi: 10.1111/nph.15357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van De Peer Y., Ashman T.L., Soltis P.S., et al. Polyploidy: An evolutionary and ecological force in stressful times. Plant Cell. 2021;33(1):11–26. doi: 10.1093/plcell/koaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang J., Dong S.Y., Yang L.H., et al. Allopolyploid speciation accompanied by gene flow in a tree fern. Mol. Biol. Evol. 2020;37(9):2487–2502. doi: 10.1093/molbev/msaa097. [DOI] [PubMed] [Google Scholar]
- 17.Hias N., Svara A., Keulemans J.W. Effect of polyploidisation on the response of apple (Malus× domestica Borkh.) to Venturia inaequalis infection. Eur. J. Plant Pathol. 2018;151(2):515–526. [Google Scholar]
- 18.Syngelaki E., Schinkel C.C., Klatt,et S., et al. Effects of temperature treatments on cytosine-methylation profiles of diploid and autotetraploid plants of the alpine species Ranunculus kuepferi (Ranunculaceae) Front. Plant Sci. 2020;11:435. doi: 10.3389/fpls.2020.00435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yves V.d.P., Steven M., Axel M. The evolutionary significance of ancient genome duplications. Nat. Rev. Genet. 2009;10(10):725–732. doi: 10.1038/nrg2600. [DOI] [PubMed] [Google Scholar]
- 20.Zhou L., Gui J.F. Natural and artificial polyploids in aquaculture. Aquacult. Fish. 2017;2(3):103–111. [Google Scholar]
- 21.Chen Z.J. Molecular mechanisms of polyploidy and hybrid vigor. Trends Plant Sci. 2010;15(2):57–71. doi: 10.1016/j.tplants.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Comai L. The advantages and disadvantages of being polyploid. Nature Rev. Genet. 2005;6(11):836–846. doi: 10.1038/nrg1711. [DOI] [PubMed] [Google Scholar]
- 23.Chen Z.J. Genomic and epigenetic insights into the molecular bases of heterosis. Nat. Rev. Genet. 2013;14(7):471–482. doi: 10.1038/nrg3503. [DOI] [PubMed] [Google Scholar]
- 24.Dai X.K., Li X., Huang Y.Q., et al. The speciation and adaptation of the polyploids: A case study of the Chinese Isoetes L. diploid-polyploid complex. BMC Evol. Biol. 2020;20(1):1–13. doi: 10.1186/s12862-020-01687-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rieseberg L.H., Willis J.H. Plant speciation. Science. 2007;317(5840):910–914. doi: 10.1126/science.1137729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baniaga A.E., Marx H.E., Arrig N., et al. Polyploid plants have faster rates of multivariate niche differentiation than their diploid relatives. Ecol. Lett. 2020;23(1):68–78. doi: 10.1111/ele.13402. [DOI] [PubMed] [Google Scholar]
- 27.Julião S.A., Ribeiro C.D.V., Lopes J.M.L., et al. Induction of synthetic polyploids and assessment of genomic stability in Lippia alba. Front Plant Sci. 2020;11:292. doi: 10.3389/fpls.2020.00292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fort A., Ryder P., Mckeown P.C., et al. Disaggregating polyploidy, parental genome dosage and hybridity contributions to heterosis in Arabidopsis thaliana. New Phytol. 2016;209(2):590–599. doi: 10.1111/nph.13650. [DOI] [PubMed] [Google Scholar]
- 29.Osborn T.C., Pires J.C., Birchler J.A., et al. Understanding mechanisms of novel gene expression in polyploids. Trends Genet. 2003;19(3):141–147. doi: 10.1016/s0168-9525(03)00015-5. [DOI] [PubMed] [Google Scholar]
- 30.Doyle J.J., Flagel L.E., Paterson A.H., et al. Evolutionary genetics of genome merger and doubling in plants. Annu. Rev. Genet. 2008;42:443–461. doi: 10.1146/annurev.genet.42.110807.091524. [DOI] [PubMed] [Google Scholar]
- 31.Cheng F., Sun C., Wu J., et al. Epigenetic regulation of subgenome dominance following whole genome triplication in Brassica rapa. New Phytol. 2016;211(1):288–299. doi: 10.1111/nph.13884. [DOI] [PubMed] [Google Scholar]
- 32.Schnable J.C., Springer N.M., Freeling M. Vol. 108. 2011. Differentiation of the maize subgenomes by genome dominance and both ancient and ongoing gene loss; pp. 4069–4074. (Proc. Nat. Acad. Sci.). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Botet R., Keurentjes J.J. The role of transcriptional regulation in hybrid vigor. Front. Plant Sci. 2020;11:410. doi: 10.3389/fpls.2020.00410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ng D.W., Lu J., Chen Z.J. Big roles for small RNAs in polyploidy, hybrid vigor, and hybrid incompatibility. Curr. Opin. Plant Biol. 2012;15(2):154–161. doi: 10.1016/j.pbi.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 35.Kawanabe T., Ishikura S., Miyaji N., et al. Role of DNA methylation in hybrid vigor in Arabidopsis thaliana. Proc. Nat. Acad. Sci. 2016;113(43):E6704–E6711. doi: 10.1073/pnas.1613372113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bird K.A., Vanburen R., Puzey J.R., et al. The causes and consequences of subgenome dominance in hybrids and recent polyploids. New Phytol. 2018;220(1):87–93. doi: 10.1111/nph.15256. [DOI] [PubMed] [Google Scholar]
- 37.Ma X.T., Zhang Z.B., Li G., et al. Spatial and temporal transcriptomic heredity and asymmetry in an artificially constructed allotetraploid wheat (AADD) Front. Plant Sci. 2022;13 doi: 10.3389/fpls.2022.887133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feldman M., Levy A.A. Genome evolution in allopolyploid wheat—a revolutionary reprogramming followed by gradual changes. J. Genet. Genom. 2009;36(9):511–518. doi: 10.1016/S1673-8527(08)60142-3. [DOI] [PubMed] [Google Scholar]
- 39.Xu C.M., Bai Y., Lin X.Y., et al. Genome-wide disruption of gene expression in allopolyploids but not hybrids of rice subspecies. Mol. Biol. Evol. 2014;31(5):1066–1076. doi: 10.1093/molbev/msu085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Session A.M., Uno Y., Kwon T., et al. Genome evolution in the allotetraploid frog Xenopus laevis. Nature. 2016;538(7625):336–343. doi: 10.1038/nature19840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Luo J., Chai J., Wen Y.L., et al. From asymmetrical to balanced genomic diversification during rediploidization: Subgenomic evolution in allotetraploid fish. Sci. Adv. 2020;6(22) doi: 10.1126/sciadv.aaz7677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen Z.L., Omori Y., Koren S., et al. De novo assembly of the goldfish (Carassius auratus) genome and the evolution of genes after whole-genome duplication. Sci. Adv. 2019;5(6):eaav0547. doi: 10.1126/sciadv.aav0547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ren L., Gao X., Cui J.L., et al. Symmetric subgenomes and balanced homoeolog expression stabilize the establishment of allopolyploidy in cyprinid fish. BMC Biol. 2022;20(1):1–17. doi: 10.1186/s12915-022-01401-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen J., Luo M., Li S.G., et al. A comparative study of distant hybridization in plants and animals. Sci. China Life Sci. 2018;61:285–309. doi: 10.1007/s11427-017-9094-2. [DOI] [PubMed] [Google Scholar]
- 45.Liu S.J., Wang S., Liu Q.F., et al. The Research Advances in Animal Distant Hybridization and Polyploid Organisms. Fish Distant Hybridization, Science Press; Beijing: 2022. pp. 1–37. [Google Scholar]
- 46.Liu S.J., Luo J., Chai J., et al. Genomic incompatibilities in the diploid and tetraploid offspring of the goldfish× common carp cross. Proc. Nat. Acad. Sci. 2016;113(5):1327–1332. doi: 10.1073/pnas.1512955113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li J.T., Wang Q., Huangyang M.D., et al. Parallel subgenome structure and divergent expression evolution of allo-tetraploid common carp and goldfish. Nat. Genet. 2021;53(10):1493–1503. doi: 10.1038/s41588-021-00933-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng H.Y., Concepcion G.T., Feng X.W., et al. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods. 2021;18(2):170–175. doi: 10.1038/s41592-020-01056-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Manni M., Berkeley M.R., Seppey M., et al. BUSCO update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 2021;38(10):4647–4654. doi: 10.1093/molbev/msab199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abrusán G., Grundmann N., Demester L., et al. TEclass—a tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics. 2009;25(10):1329–1330. doi: 10.1093/bioinformatics/btp084. [DOI] [PubMed] [Google Scholar]
- 51.Chen N.S. Using Repeat Masker to identify repetitive elements in genomic sequences. Curr. Prot. Bioinformat. 2004;5(1) doi: 10.1002/0471250953.bi0410s05. p. 4.10.1-4.10.14. [DOI] [PubMed] [Google Scholar]
- 52.Stanke M., Keller O., Gunduz I., et al. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006;34(Suppl 2):W435–W439. doi: 10.1093/nar/gkl200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pertea M., Pertea G.M., Antonescu C.M., et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015;33(3):290–295. doi: 10.1038/nbt.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haas B.J., Salzberg S.L., Zhu W., et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biol. 2008;9(1):1–22. doi: 10.1186/gb-2008-9-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim G.B., Gao Y., Palsson B.O., et al. DeepTFactor: A deep learning-based tool for the prediction of transcription factors. Proc. Nat. Acad. Sci. 2021;118(2) doi: 10.1073/pnas.2021171118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Y.P., Tang H.B., Debarry J.D., et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40(7):e49. doi: 10.1093/nar/gkr1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goel M., Sun H., Jiao W.B., et al. SyRI: Finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome Biol. 2019;20(1):1–13. doi: 10.1186/s13059-019-1911-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mason A.S., Wendel J.F. Homoeologous exchanges, segmental allopolyploidy, and polyploid genome evolution. Front. Genet. 2020;11:1014. doi: 10.3389/fgene.2020.01014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Harris R.S. The Pennsylvania State University; 2007. Improved Pairwise Alignment of Genomic DNA. [Google Scholar]
- 60.Kim D., Paggi J.M., Park C., et al. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019;37(8):907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):1–21. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen Z.J., Sreedasyam A., Ando A., et al. Genomic diversifications of five Gossypium allopolyploid species and their impact on cotton improvement. Nat. Genet. 2020;52(5):525–533. doi: 10.1038/s41588-020-0614-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kumar L., Futschik M.E. Mfuzz: A software package for soft clustering of microarray data. Bioinformation. 2007;2(1):5. doi: 10.6026/97320630002005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pan J.B., Hu S.C., Wang H., et al. PaGeFinder: Quantitative identification of spatiotemporal pattern genes. Bioinformatics. 2012;28(11):1544–1545. doi: 10.1093/bioinformatics/bts169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang Z., Xiao J., Wu J., et al. ParaAT: A parallel tool for constructing multiple protein-coding DNA alignments. Biochem. Biophys. Res. Commun. 2012;419(4):779–781. doi: 10.1016/j.bbrc.2012.02.101. [DOI] [PubMed] [Google Scholar]
- 66.Wu T.Z., Hu E.Q., Xu S.B., et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation. 2021;2(3):100141. doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Waterhouse R.M., Seppey M., Simão F.A., et al. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2018;35(3):543–548. doi: 10.1093/molbev/msx319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Peng W.Z., Xu J., Zhang Y., et al. An ultra-high density linkage map and QTL mapping for sex and growth-related traits of common carp (Cyprinus carpio) Sci. Rep. 2016;6(1):1–16. doi: 10.1038/srep26693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ou S.J., Chen J.F., Jiang N. Assessing genome assembly quality using the LTR Assembly Index (LAI) Nucleic Acids Res. 2018;46(21) doi: 10.1093/nar/gky730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.de Mendoza A., Sebé-Pedrós A., Šestak M.S., et al. Transcription factor evolution in eukaryotes and the assembly of the regulatory toolkit in multicellular lineages. Proc. Nat. Acad. Sci. 2013;110(50):E4858–E4866. doi: 10.1073/pnas.1311818110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Meyerowitz E.M. Plants compared to animals: The broadest comparative study of development. Science. 2002;295(5559):1482–1485. doi: 10.1126/science.1066609. [DOI] [PubMed] [Google Scholar]
- 72.Yang J., Moeinzadeh M., Kuhl H., et al. Haplotype-resolved sweet potato genome traces back its hexaploidization history. Nat. Plants. 2017;3(9):696–703. doi: 10.1038/s41477-017-0002-z. [DOI] [PubMed] [Google Scholar]
- 73.Song J.M., Zhang Y.T., Zhou Z.W., et al. Oil plant genomes: Current state of the science. J. Exp. Bot. 2022;73(9):2859–2874. doi: 10.1093/jxb/erab472. [DOI] [PubMed] [Google Scholar]
- 74.Zhang Q., Guan P.P., Zhao L., et al. Asymmetric epigenome maps of subgenomes reveal imbalanced transcription and distinct evolutionary trends in Brassica napus. Mol. Plant. 2021;14(4):604–619. doi: 10.1016/j.molp.2020.12.020. [DOI] [PubMed] [Google Scholar]
- 75.Rhie A., Mccarthy S.A., Fedrigo O., et al. Towards complete and error-free genome assemblies of all vertebrate species. Nature. 2021;592(7856):737–746. doi: 10.1038/s41586-021-03451-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lewin H.A., Richards S., Aiden E.L., et al. The earth BioGenome project 2020: Starting the clock. Proc. Nat. Acad. Sci. 2022;119(4) doi: 10.1073/pnas.2115635118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Exposito-Alonso M., Drost H.G., Burbano H.A., et al. The Earth BioGenome project: Opportunities and challenges for plant genomics and conservation. Plant J. 2020;102(2):222–229. doi: 10.1111/tpj.14631. [DOI] [PubMed] [Google Scholar]
- 78.Wenger A.M., Peluso P., Rowell W.J., et al. Accurate circular consensus long-read sequencing improves variant detection and assembly of a human genome. Nat. Biotechnol. 2019;37(10):1155–1162. doi: 10.1038/s41587-019-0217-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang J., Chen X., Bai J., et al. The Sinocyclocheilus cavefish genome provides insights into cave adaptation. BMC Biol. 2016;14(1):1–13. doi: 10.1186/s12915-015-0223-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu H.P., Xiao S.J., Wu N., et al. The sequence and de novo assembly of Oxygymnocypris stewartii genome. Sci. Data. 2019;6(1):1–11. doi: 10.1038/sdata.2019.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen D., Zhang Q., Tang W.Q., et al. The evolutionary origin and domestication history of goldfish (Carassius auratus) Proc. Nat. Acad. Sci. 2020;117(47):29775–29785. doi: 10.1073/pnas.2005545117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xiao S.J., Mou Z.B., Fan D.D., et al. Genome of Tetraploid Fish Schizothorax o’connori provides insights into early re-diploidization and high-altitude adaptation. Iscience. 2020;23(9):101497. doi: 10.1016/j.isci.2020.101497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Sun L., Gao T., Wang F.L., et al. Chromosome-level genome assembly of a cyprinid fish Onychostoma macrolepis by integration of nanopore sequencing, Bionano and Hi-C technology. Mol. Ecol. Resour. 2020;20(5):1361–1371. doi: 10.1111/1755-0998.13190. [DOI] [PubMed] [Google Scholar]
- 84.Chen L., Li B.J., Chen B.H., et al. Chromosome-level genome of Poropuntius huangchuchieni provides a diploid progenitor-like reference genome for the allotetraploid Cyprinus carpio. Mol. Ecol. Resour. 2021;21(5):1658–1669. doi: 10.1111/1755-0998.13365. [DOI] [PubMed] [Google Scholar]
- 85.Jia K.H., Liu H., Zhang R.G., et al. Chromosome-scale assembly and evolution of the tetraploid Salvia splendens (Lamiaceae) genome. Hortic. Res. 2021;8(1):1–15. doi: 10.1038/s41438-021-00614-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yeaman S. Genomic rearrangements and the evolution of clusters of locally adaptive loci. Proc. Nat. Acad. Sci. 2013;110(19):E1743–E1751. doi: 10.1073/pnas.1219381110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Osborn T.C., Butrulle D.V., Sharpe A.G., et al. Detection and effects of a homeologous reciprocal transposition in Brassica napus. Genetics. 2003;165(3):1569–1577. doi: 10.1093/genetics/165.3.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang J.S., Zhang X.T., Tang H.B., et al. Allele-defined genome of the autopolyploid sugarcane Saccharum spontaneum L. Nat. Genet. 2018;50(11):1565–1573. doi: 10.1038/s41588-018-0237-2. [DOI] [PubMed] [Google Scholar]
- 89.Zhang Z.B., Gou X.W., Xun H.W., et al. Homoeologous exchanges occur through intragenic recombination generating novel transcripts and proteins in wheat and other polyploids. Proc. Nat. Acad. Sci. 2020;117(25):14561–14571. doi: 10.1073/pnas.2003505117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mason A.S., Wendel J.F. Homoeologous exchanges, segmental allopolyploidy, and polyploid genome evolution. Front. Genet. 2020:1014. doi: 10.3389/fgene.2020.01014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wu Y., Lin F., Zhou Y., et al. Genomic mosaicism due to homoeologous exchange generates extensive phenotypic diversity in nascent allopolyploids. Natl. Sci. Rev. 2021;8(5):nwaa277. doi: 10.1093/nsr/nwaa277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Stein A., Coriton O., Rousseau‐Gueutin M., et al. Mapping of homoeologous chromosome exchanges influencing quantitative trait variation in Brassica napus. Plant Biotechnol. J. 2017;15(11):1478–1489. doi: 10.1111/pbi.12732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hurgobin B., Golicz A.A., Bayer P.E., et al. Homoeologous exchange is a major cause of gene presence/absence variation in the amphidiploid Brassica napus. Plant Biotechnol. J. 2018;16(7):1265–1274. doi: 10.1111/pbi.12867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.H. Guo, X.Y. Wang, H. Gundlach, et al., Extensive and biased intergenomic nonreciprocal DNA exchanges shaped a nascent polyploid genome, Gossypium (cotton), Genetics 197 (4) (2014) 1153–1163. [DOI] [PMC free article] [PubMed]
- 95.Chalhoub B., Denoeud F., Liu S., et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science. 2014;345(6199):950–953. doi: 10.1126/science.1253435. [DOI] [PubMed] [Google Scholar]
- 96.Tennessen J.A., Govindarajulu R., Ashman T.L., et al. Evolutionary origins and dynamics of octoploid strawberry subgenomes revealed by dense targeted capture linkage maps. Genome Biol. Evol. 2014;6(12):3295–3313. doi: 10.1093/gbe/evu261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gou X.W., Bian Y., Zhang A., et al. Transgenerationally precipitated meiotic chromosome instability fuels rapid karyotypic evolution and phenotypic diversity in an artificially constructed allotetraploid wheat (AADD) Mol. Biol. Evol. 2018;35(5):1078–1091. doi: 10.1093/molbev/msy009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lloyd A., Blary A., Charif D., et al. Homoeologous exchanges cause extensive dosage-dependent gene expression changes in an allopolyploid crop. New Phytol. 2018;217(1):367–377. doi: 10.1111/nph.14836. [DOI] [PubMed] [Google Scholar]
- 99.Borrill P., Adamski N., Uauy C. Genomics as the key to unlocking the polyploid potential of wheat. New Phytol. 2015;208(4):1008–1022. doi: 10.1111/nph.13533. [DOI] [PubMed] [Google Scholar]
- 100.He F., Wang W., Rutter W.B., et al. Genomic variants affecting homoeologous gene expression dosage contribute to agronomic trait variation in allopolyploid wheat. Nat. Commun. 2022;13(1):1–15. doi: 10.1038/s41467-022-28453-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Combes M.C., Dereeper A., Severac D., et al. Contribution of subgenomes to the transcriptome and their intertwined regulation in the allopolyploid Coffea arabica grown at contrasted temperatures. New Phytol. 2013;200(1):251–260. doi: 10.1111/nph.12371. [DOI] [PubMed] [Google Scholar]
- 102.Liu Z.S., Xin M.M., Qin J.X., et al. Temporal transcriptome profiling reveals expression partitioning of homeologous genes contributing to heat and drought acclimation in wheat (Triticum aestivum L.) BMC Plant Biol. 2015;15(1):1–20. doi: 10.1186/s12870-015-0511-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Simons K.J., Fellers J.P., Trick H.N., et al. Molecular characterization of the major wheat domestication gene Q. Genetics. 2006;172(1):547–555. doi: 10.1534/genetics.105.044727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Metzger D.C., Schulte P.M. Maternal stress has divergent effects on gene expression patterns in the brains of male and female threespine stickleback. Proc. R. Soc. B. 2016;283(1839) doi: 10.1098/rspb.2016.1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Schnurr M.E., Yin Y., Scott G.R. Temperature during embryonic development has persistent effects on metabolic enzymes in the muscle of zebrafish. J. Exper. Biol. 2014;217(8):1370–1380. doi: 10.1242/jeb.094037. [DOI] [PubMed] [Google Scholar]
- 106.Heallen T., Zhang M., Wang J., et al. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science. 2011;332(6028):458–461. doi: 10.1126/science.1199010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wang Y., Yu A., Yu F.X. The Hippo pathway in tissue homeostasis and regeneration. Protein Cell. 2017;8(5):349–359. doi: 10.1007/s13238-017-0371-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Naito A.T., Akazawa H., Takano H., et al. Phosphatidylinositol 3-kinase–Akt pathway plays a critical role in early cardiomyogenesis by regulating canonical Wnt signaling. Circ. Res. 2005;97(2):144–151. doi: 10.1161/01.RES.0000175241.92285.f8. [DOI] [PubMed] [Google Scholar]
- 109.Fu V., Plouffe S.W., Guan K.L. The Hippo pathway in organ development, homeostasis, and regeneration. Curr. Opin. Cell Biol. 2017;49:99–107. doi: 10.1016/j.ceb.2017.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jin D.L., Zhang Q.K., Wang Y.F., et al. Observation of embryonic, larva and juvenile development of Opsariichthys bidens. Oceanologia et Limnologia Sinica/Hai Yang Yu Hu Chao. 2017;48(4):838–847. [Google Scholar]
- 111.Alger E.I., Edger P.P. One subgenome to rule them all: Underlying mechanisms of subgenome dominance. Curr. Opin. Plant Biol. 2020;54:108–113. doi: 10.1016/j.pbi.2020.03.004. [DOI] [PubMed] [Google Scholar]
- 112.Springer N.M., Stupar R.M. Allele-specific expression patterns reveal biases and embryo-specific parent-of-origin effects in hybrid maize. Plant Cell. 2007;19(8):2391–2402. doi: 10.1105/tpc.107.052258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ramírez-González R., Borrill P., Lang D., et al. The transcriptional landscape of polyploid wheat. Science. 2018;361(6403):eaar6089. doi: 10.1126/science.aar6089. [DOI] [PubMed] [Google Scholar]
- 114.Sharbrough J., Conover J.L., Tate J.A., et al. Cytonuclear responses to genome doubling. Am. J. Bot. 2017;104(9):1277–1280. doi: 10.3732/ajb.1700293. [DOI] [PubMed] [Google Scholar]
- 115.Shao L., Xing F., Xu C.H., et al. Patterns of genome-wide allele-specific expression in hybrid rice and the implications on the genetic basis of heterosis. Proc. Nat. Acad. Sci. 2019;116(12):5653–5658. doi: 10.1073/pnas.1820513116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Coate J.E., Bar H., Doyle J.J. Extensive translational regulation of gene expression in an allopolyploid (Glycine dolichocarpa) Plant Cell. 2014;26(1):136–150. doi: 10.1105/tpc.113.119966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Coate J.E., Farmer A.D., Schiefelbein J.W., et al. Expression partitioning of duplicate genes at single cell resolution in Arabidopsis roots. Front. Genet. 2020;11:1363. doi: 10.3389/fgene.2020.596150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Caruana L., Orr D.J., Carmo-Silva E. Rubiscosome gene expression is balanced across the hexaploid wheat genome. Photosyn. Res. 2022;152(1):1–11. doi: 10.1007/s11120-022-00897-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Blanc G., Wolfe K.H. Functional divergence of duplicated genes formed by polyploidy during Arabidopsis evolution. Plant Cell. 2004;16(7):1679–1691. doi: 10.1105/tpc.021410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dahlke F.T., Wohlrab S., Butzin M., et al. Thermal bottlenecks in the life cycle define climate vulnerability of fish. Science. 2020;369(6499):65–70. doi: 10.1126/science.aaz3658. [DOI] [PubMed] [Google Scholar]
- 121.Wang L., Ma K.B., Lu Z.G., et al. Differential physiological, transcriptomic and metabolomic responses of Arabidopsis leaves under prolonged warming and heat shock. BMC Plant Biol. 2020;20(1):1–15. doi: 10.1186/s12870-020-2292-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hrmova M., Hussain S.S. Plant transcription factors involved in drought and associated stresses. Int. J. Mol. Sci. 2021;22(11):5662. doi: 10.3390/ijms22115662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Simon R.B., Eli R.M., Jeffrey R.I. Gene fractionation and function in the ancient subgenomes of maize. Mol. Biol. Evol. 2017;34(8):1825–1832. doi: 10.1093/molbev/msx121. [DOI] [PubMed] [Google Scholar]
- 124.Marcussen T., Sandve S., Heier L., et al. Ancient hybridizations among the ancestral genomes of bread wheat. Science. 2014;345(6194) doi: 10.1126/science.1250092. [DOI] [PubMed] [Google Scholar]
- 125.Ye C.Y., Wu D.Y., Mao L.F., et al. The genomes of the allohexaploid Echinochloa crus-galli and its progenitors provide insights into polyploidization-driven adaptation. Mol. Plant. 2020;13(9):1298–1310. doi: 10.1016/j.molp.2020.07.001. [DOI] [PubMed] [Google Scholar]
- 126.Zhao N., Dong Q.L., Nadon B.D., et al. Evolution of homeologous gene expression in polyploid wheat. Genes. 2020;11(12):1401. doi: 10.3390/genes11121401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shi X.L., Ng D.W., Zhang C.Q., et al. Cis-and trans-regulatory divergence between progenitor species determines gene-expression novelty in Arabidopsis allopolyploids. Nat. Commun. 2012;3(1):1–9. doi: 10.1038/ncomms1954. [DOI] [PubMed] [Google Scholar]
- 128.Adams K.L., Wendel J.F. Allele-specific, bidirectional silencing of an alcohol dehydrogenase gene in different organs of interspecific diploid cotton hybrids. Genetics. 2005;171(4):2139–2142. doi: 10.1534/genetics.105.047357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ren G.P., Jiang Y.Y., Li A., et al. The genome sequence provides insights into salt tolerance of Achnatherum splendens (Gramineae), a constructive species of alkaline grassland. Plant Biotechnol. J. 2022;20(1):116–128. doi: 10.1111/pbi.13699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.VanBuren R., Wai C.M., Wang X., et al. Exceptional subgenome stability and functional divergence in the allotetraploid Ethiopian cereal teff. Nat. Commun. 2020;11(1):1–11. doi: 10.1038/s41467-020-14724-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhou G.A., Chang R.Z., Qiu L.J. Overexpression of soybean ubiquitin-conjugating enzyme gene GmUBC2 confers enhanced drought and salt tolerance through modulating abiotic stress-responsive gene expression in Arabidopsis. Plant Mol. Biol. 2010;72(4):357–367. doi: 10.1007/s11103-009-9575-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jue D.W., Sang X.L., Liu L.Q., et al. The ubiquitin-conjugating enzyme gene family in longan (Dimocarpus longan lour.): Genome-wide identification and gene expression during flower induction and abiotic stress responses. Molecules. 2018;23(3):662. doi: 10.3390/molecules23030662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Liu W.G., Tang X., Qi X.H., et al. The ubiquitin conjugating enzyme: An important ubiquitin transfer platform in ubiquitin-proteasome system. Int. J. Mol. Sci. 2020;21(8):2894. doi: 10.3390/ijms21082894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Chiang C., Scott A.J., Davis J.R., et al. The impact of structural variation on human gene expression. Nat. Genet. 2017;49(5):692–699. doi: 10.1038/ng.3834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jeffares D.C., Jolly C., Hoti M., et al. Transient structural variations have strong effects on quantitative traits and reproductive isolation in fission yeast. Nat. Commun. 2017;8(1):14061. doi: 10.1038/ncomms14061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gabur I., Chawla H.S., Lopisso D.T., et al. Gene presence-absence variation associates with quantitative Verticillium longisporum disease resistance in Brassica napus. Sci. Rep. 2020;10(1):1–11. doi: 10.1038/s41598-020-61228-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Yu H., Lin T., Meng X.B., et al. A route to de novo domestication of wild allotetraploid rice. Cell. 2021;184(5):1156–1170. doi: 10.1016/j.cell.2021.01.013. [DOI] [PubMed] [Google Scholar]
- 138.Chen L., Xu J., Sun X.W., et al. Research advances and future perspectives of genomics and genetic improvement in allotetraploid common carp. Rev. Aquacult. 2022;14(2):957–978. [Google Scholar]
- 139.Hu X.S., Ge Y.L., Li C.T., et al. Developments in common carp culture and selective breeding of new varieties. Aquacult. China. 2018:125–148. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All genomic datasets of C. carpio will be available under NCBI accession number PRJNA823855 and in the BIGD Genome Warehouse under accession number GWHBHRW00000000 upon publication. Transcriptomes were deposited in the NCBI database under accession number PRJNA824204.