

Abstract
Nanocarriers with intrinsic immune adjuvant properties can activate the innate immune system while delivering tumor antigen, thus efficiently facilitating antitumor adaptive immunity. Bacteria-derived outer membrane vesicles (OMVs) are an excellent candidate due to their abundance of pathogen associated molecular patterns. However, during the uptake of OMVs by dendritic cells (DCs), the interaction between lipopolysaccharide and toll-like receptor 4 induces rapid DC maturation and uptake blockage, a phenomenon we refer to as “maturation-induced uptake obstruction” (MUO). Herein we decorated OMV with the DC-targeting αDEC205 antibody (OMV-DEC), which endowed the nanovaccine with an uptake mechanism termed as “not restricted to maturation via antibody modifying” (Normandy), thereby overcoming the MUO phenomenon. We also proved the applicability of this nanovaccine in identifying the human tumor neoantigens through rapid antigen display. In summary, this engineered OMV represents a powerful nanocarrier for personalized cancer vaccines, and this antibody modification strategy provides a reference to remodel the DC uptake pattern in nanocarrier design.
Keywords: Tumor vaccine, Outer membrane vesicles, Antibody modification, Antigen display, Dendritic cell uptake, Myeloid derived suppressor cells
Graphical abstract
1. Introduction
The promising approach of personalized tumor immunotherapy entails a tumor vaccine that activates tumor-specific adaptive immunity, in which a tumor antigen is the key messenger for transmitting the tumor cell identification to the immune system [1], [2], [3]. However, a regimen of tumor antigen alone often elicits disappointing antitumor immunity, so popular approaches have adopted the use of immune adjuvants and/or vaccine nanocarriers to enhance the immunogenicity of tumor antigens [4,5]. Indeed, combinational application with an immune adjuvant can activate the innate immune system to provide the necessary co-stimulatory signals for antigen presentation [6], while the delivery of a vaccine nanocarrier improves the uptake and processing efficiency of the tumor antigen by the immune system [7]. Currently, the design of tumor nanovaccines tends to integrate the nanocarrier with the immune adjuvant to ensure that the immune adjuvant and antigen enter the same antigen presenting cells (APCs) [8,9]. In addition to using a nanocarrier to co-deliver the immune adjuvant and antigen, this synergistic strategy can be achieved through the development of a nanocarrier with intrinsic immune adjuvant properties, such as polymeric and lipid nanoparticles with stimulator of interferon genes (STING) pathway activation ability [10,11]. Inspired by the body's natural immune defenses against bacterial invasion, we have explored the utilization of bacteria-derived outer membrane vesicles (OMVs) as the nanocarrier to transport tumor antigen in a previous study [12]. OMVs possess large amounts of pathogen-associated molecular patterns (PAMPs) that can act as immune adjuvants to activate innate immunity, thus stimulating strong antitumor adaptive immunity [13,14].
Despite these advancements, nanocarriers with intrinsic immune adjuvant function are still imperfect for vaccine delivery. During the nanocarrier uptake process by dendritic cells (DCs), the maturation of cells is rapidly induced by the immune adjuvant [13,15], and their uptake capacity declines sharply, a phenomenon we termed “maturation-induced uptake obstruction” (MUO), which ultimately inhibits the persistent antigen presentation and results in a limited antigen-specific immune response. As for OMV-based tumor nanovaccines, the MUO phenomenon is even more critical because of the presence of toll-like receptor 4 (TLR4) on the surface of DCs; TLR4 is activated by the lipopolysaccharide (LPS) on the OMV surface at the initiation of DC recognition and uptake [14,16,17]. DEC205, classified as a type I C-type lectin receptor, is highly expressed on the DC surface; αDEC205 antibody is often integrated into vaccine nanocarriers via surface decoration technology to favor DC uptake and potentiate immune activation [18,19]. In addition, DEC205 expression is upregulated downstream from TLR pathway activation in mature DCs [20,21]. Therefore, modification with αDEC205 antibody may endow OMV with an ectopic uptake pathway, enabling persistent DCs uptake, especially when the natural uptake pathway is impeded in mature DCs, thereby overcoming the MUO phenomenon. Few studies have focused on the modification of αDEC205 antibody to remodel DC uptake patterns, especially in nanocarriers with intrinsic immune adjuvant function.
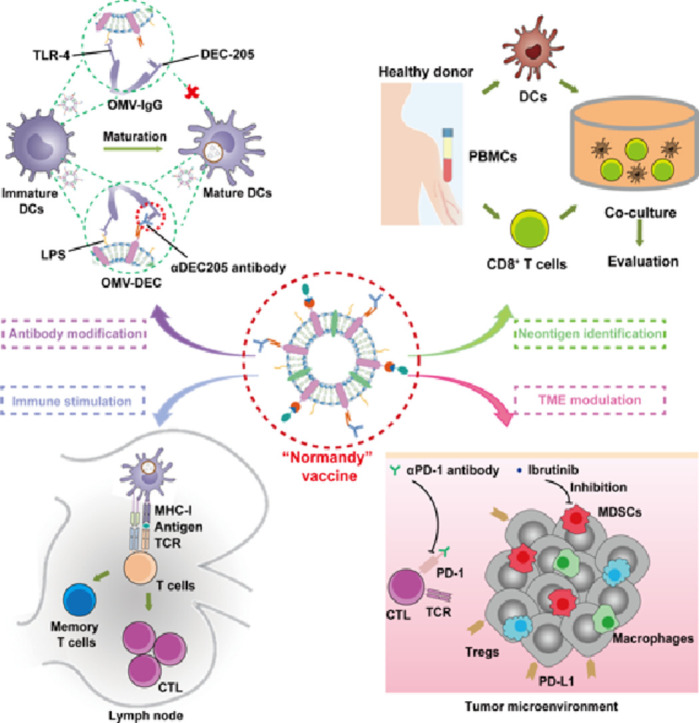
Herein we report a judiciously designed OMV-based tumor vaccine delivery platform, in which the DC uptake pattern is remodeled. We genetically engineered bacteria to express the surface protein Cytolysin A (ClyA) fused with domain B of Staphylococcal protein A (SPAb) (ClyA-SPAb) [22], [23], [24] on OMVs. The SPAb-modified OMVs (OMV-SPAb) were then able to efficiently bind the Fc fragment of the αDEC205 antibody (OMV-DEC), and the outward-facing Fab fragment aided in the recognition and uptake by DCs [25]. In this situation, the DC uptake of OMV-DEC was not restricted to the interaction between LPS and TLR4 (the natural pathway) but was also mediated by targeting antibody recognition (the “artificial” pathway). Therefore, OMV-DEC can continue to be taken up even after DC maturation, and the DC uptake pattern of OMV-DEC is referred to a mechanism termed as “not restricted to maturation via antibody modifying” (Normandy). As a result, the OMV-DEC exhibited greater uptake by DCs than OMV, leading to increased antigen presentation and subsequent cytotoxic T lymphocyte (CTL) and memory T cell activation, ultimately efficiently impeding metastasis in a pulmonary melanoma model. Additionally, we employed molecular glue technology to construct a "Plug-and-Display" function in the OMV-based platform [12], which achieved rapid display of tumor antigens and was further used to perform timely and efficient human tumor neoantigen identification. We assessed the therapeutic potential of the OMV-DEC-based tumor nanovaccine in a subcutaneous melanoma model, in which we panoramically analyzed the immunosuppressive tumor microenvironment and then formulated a synergistic strategy of OMV-DEC nanovaccine combined with an immune checkpoint inhibitor and Ibrutinib to deplete myeloid-derived suppressor cells (MDSCs), dramatically inhibiting subcutaneous melanoma growth.
2. Material and methods
2.1. Materials
OVA257-264 (SIINFEKL), SpyTag-OVA257-264 (SIINFEKLGGVPTIVMVDAYKRYK), SpyTag-EGFR-T790M (MQLMPFGSLLGGVPTIVMVDAYKRYK), SpyTag-TP53-R175H (HMTEVVRHCGGVPTIVMVDAYKRYK), SpyTag-CDC73-Q254E (NIFAILESVGGVPTIVMVDAYKRYK), SpyTag-TMEM48-F169L (CLNEYHLFLGGVPTIVMVDAYKRYK), SpyTag-SEC24A-P469L (FLYNLLTRVGGVPTIVMVDAYKRYK), SpyTag-EXOC8-Q656P (IILVAVPHVGGVPTIVMVDAYKRYK) and SpyTag-gp100-A288V (YLEPGPVTVGGVPTIVMVDAYKRYK) peptides were synthesized from Shanghai Top-peptide Biotechnology Co., Ltd. αDEC205 antibody (catalog No. CUS-HB-290) and IgG isotype control (catalog No. BE0086) were purchased from BioXcell (Lebanon, NH, USA). Anti-HA-Tag (catalog No. ab236632), anti-Cmyc-Tag (catalog No. ab9106), anti-Flag-Tag (catalog No. ab18230), HRP conjugated goat anti-mouse IgG (catalog No. ab205719) and HRP conjugated goat anti-rabbit IgG (catalog No. ab6721) antibodies were obtained from Abcam (Cambridge, UK). HRP conjugated Rat IgG (catalog No. 20005-13-HP) was purchased from Alpha Diagnostic International (San Antonio, TX, USA). Murine IL-4 (catalog No. 214-14-20) and Murine GM-CSF (catalog No. AF-315-03-20) were purchased from Peprotech (Rocky Hill, NJ). DAPI and Dil were purchased from Solarbio (Beijing, P. R. China). FITC-anti-mouse TLR4 (catalog No. sc-13591) antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Purified anti-human CD205 (catalog No. 342202), PE anti-mouse CD205 (catalog No. 138214), APC-anti-mouse CD3 (catalog No. 100204), APC-anti-mouse CD3 (catalog No. 100235), FITC-anti-mouse CD8a (catalog No. 100706), PE/Cy7-anti-mouse CD8a (catalog No. 100722), FITC-anti-mouse CD11c (catalog No. 117306), APC-anti-mouse CD11c (catalog No. 117310), FITC-anti-mouse CD80 (catalog No. 104706), PE OVA257-264-specific MHCI complex (MHCI-OVA) (catalog No. 141604), PE/Cy7-anti-mouse CD86 (catalog No. 105014), PE-anti-mouse CD69 (catalog No. 104508), PE anti-human CD137 (4-1BB) (catalog No. 309804), APC anti-mouse CD45 (catalog No. 103112), FITC anti-mouse/human CD11b (catalog No. 101206), PE/Cyanine7 anti-mouse Gr-1 (catalog No. 108416), PE/Cyanine7 anti-mouse F4/80 (catalog No. 123114), PE anti-mouse CD206 (catalog No. 141706), FITC anti-mouse CD4 (catalog No. 100406), APC anti-mouse CD4 (catalog No. 100516), PE/Cyanine7 anti-mouse IFN-γ (catalog No. 505826), PE anti-mouse FoxP3 (catalog No. 126404), APC anti-mouse CD62L (catalog No. 104412) and PE/Cy7 anti-mouse CD44 (catalog No. 103030) antibodies were purchased from BioLegend (USA). T-Select PE OVA-MHCI complex tetramer (OVA-tetramer; catalog No. TS-5001-1C) was purchased from MBL Beijing Biotech Co., Ltd (China). Cell TraceTM CFSE Cell Proliferation Kit (catalog No. C34570) was purchased from Invitrogen (USA). The mouse IFN-γ precoated ELISPOT kit (catalog No.2210005) and human IFN-γ precoated ELISPOT Kit (catalog No.2110002) were purchased from Dakewe Biotech Co., Ltd (Shenzhen, China). Ficoll-Paque PLUS was purchased from Solarbio (China). Human CD8 MicroBeads kit (catalog No. 130-045-201) was purchased from Bergisch-Gladbach, Germany.
2.2. Animals and cells
Female C57BL/6 mice aged 6-8 weeks were obtained from SPF (Beijing) Biotechnology Co., Ltd. All animal protocols were approved by the Institutional Animal Care and Use Committee of the National Center for Nanoscience and Technology. BMDCs were generated from the femurs and tibias of C57BL/6 mice (6-8 weeks) and cultured in RPMI medium 1640 supplemented with GM-CSF (20 ng/mL) and IL-4 (10 ng/mL). The B16-F10 cell line was purchased from the ATCC (Manassas, USA). B16-OVA cells were a gift from Wang Hao at the National Center for Nanoscience and Technology. Cells were cultured in RPMI medium 1640 supplemented with 10% fetal bovine serum, 100 U mL–1 penicillin G sodium and 100 µg mL–1 streptomycin. All cells were incubated at 37°C with 5% CO2 and were identified frequently with their different morphological features.
2.3. Preparation and characterization of the engineered OMV platform
ClyA-SpyC-3Cmyc/ClyA-SPAb-3Flag were cloned into pETDuet-1 expression plasmids (Genewiz, Suzhou, China). The E. coli strain, Rosetta (DE3; Tiangen Biotech Co. Ltd., Beijing, China), was transformed with the expression plasmids, and then grown on solid LB medium at 37°C. The bacteria were then cultured in a shaker (180 rpm) at 37°C for 6 h in liquid LB medium, which was refreshed with LB medium at a 1:100 dilution. When the OD600 value was approximately 0.6, 0.1 mM of isopropyl β-D-1-thiogalactopyranoside (IPTG) was added and the cells were incubated at 16°C for 16 h with shaking at 160 rpm. The flasks contained 75 µg/mL of ampicillin.
OMVs were prepared and purified as follows: briefly, E. coli culture medium was centrifuged at 8000 × g for 5 min at 4°C to remove the bacteria and then filtered through a 0.45 µm sterile filter (EPS, Millipore). The filtrate was further concentrated with an ultrafiltration tube with a molecular weight cutoff (MWCO) of 100 kDa and filtered through a 0.22-µm EPS membrane. OMVs were obtained from the medium by centrifuging at 150,000 × g for 3 h at 4°C. The vesicles were resuspended in PBS and stored at -20°C until use. The protein concentration of the collected OMVs was determined using a Micro BCA Protein Assay Kit (Thermo Fisher Scientific). Dynamic light-scattering (DLS; Zetasizer Nano ZS90, Malvern, UK) and transmission electron microscopy (TEM; Tecnai G2 F20 U-TWIN, FEI, USA) were used to characterize the size and morphology of OMVs, respectively. For western blot analysis, OMV samples were resolved by SDS-PAGE (10% acrylamide) and then transferred onto a polyvinylidene fluoride (PVDF) membrane. The membranes were blocked in 3% BSA/PBST, and anti-Cmyc-Tag (1:2000) or anti-Flag-Tag (1:2000) antibodies were added for 2 h at room temperature. The corresponding HRP conjugated goat anti-rabbit IgG (1:3000) or HRP conjugated goat anti-mouse IgG (1:3000) were then added for 1 h at room temperature. Immunoreactive bands of proteins were visualized via Super Signal West Pico Chemiluminescent Substrate (Thermo Scientific, Rockford, USA). To further demonstrate the successful binding of αDEC205 antibody on OMV-SpyC/SPAb, αDEC205 antibody was modified with the free sulfhydryl group by Traut's reagent (catalog No.4781-83-3), and then labeled with 10 nm gold nanoparticles.
2.4. Preparation and identification of the OMV-based nanovaccine
To construct the OMV-based nanovaccine, the above OMV platform was further incubated with heterogenous antigens at a weight ratio of OMV protein and peptide at 1: 1 and αDEC205 antibody/IgG at a weight ratio of OMV protein and antibody at 1: 1, respectively. To verify the binding of the antibody to SPAb, a one-step western blot assay was used to detect the SPAb protein. Similarly, HRP-conjugated Rat IgG antibodies (1:1000) were used to decorate the membrane for 2 h at room temperature. To evaluate SpyCatcher/SpyTag reactivity, we employed a western blot assay as described above, but using an anti-HA-Tag antibody (1:1000) as the primary antibody. To quantify the SpyTag-OVA peptide bonded on OMV, Cy5.5-NHS was used to label SpyTag-OVA peptide. After the display on OMV-SpyC/SPAb. the unbonded SpyTag-OVA peptide was removed by ultrafiltration (100 kDa MWCO) or dialysis (10 kDa MWCO) and quantitatively measured by High Performance Liquid Chromatography (HPLC, Shimadzu, Kyoto, Japan). The OVA peptide was separated using a symmetry C18 reverse phase column (5 μm; 10 × 250 mm Thermo Scientific) and eluted using an acetonitrile 70%, water 30% and trifluoroaceticacid (0.1% v/v) eluent at the flow rate of 0.5 mL/min, and the eluent was monitored at 280 nm. The display efficiency (DE) was calculated according to the following formula: DE (%) = (mass of OVA peptide added - mass of unbonded OVA peptide on OMV / mass of OVA peptide added) × 100%.
2.5. Immune analysis of OMV-based nanovaccine
For in vitro cellular uptake experiments, we used a PE-labeled anti-DEC205/IgG antibody to prepare the OMV-based nanovaccines for flow cytometry analysis (Attune NxT flow cytometer; Thermo Fisher, Waltham, USA). BMDCs were incubated with OMV-IgG, OMV-DEC, OMV-OVA-IgG or OMV-OVA-DEC nanovaccines at 37°C for 36h (5 µg/mL equivalent OVA or OMV). Cells were collected and washed with PBS before flow cytometry analysis. For further maturation experiments, APC-anti-mouse CD11c, FITC-anti-mouse CD80 or PE/Cy7-anti-mouse CD86 antibodies were used to stain the BMDCs to evaluate cell maturation.
To examine the co-localization of OMV-based nanovaccines with the TLR4 or DEC205 in DCs, the OMV-OVA-IgG and OMV-OVA-DEC nanovaccines were labeled with Dil (red). After a 1 h incubation with DC2.4 cells, the cell nuclei were stained with DAPI (blue), and the TLR4 and DEC205 were detected using a FITC-labeled antibody (cyan) and a PE-labeled antibody (green), respectively. The cells were then examined with a laser scanning confocal microscope (Nikon, Japan).
For uptake pathway analysis, BMDCs were pre-incubated with 1 µM TLR4 inhibitor (TAK-242) or 5 µg/mL αDEC205 antibody for 1 h. After blocking TLR4 or DEC205, OMV-IgG and OMV-DEC were added for a 12 h incubation to evaluate the cellular uptake efficiency (5 µg/mL equivalent OMV). For detection of the DEC205 molecule in BMDCs, PE-anti-mouse DEC205 antibodies were applied to the BMDCs at 4°C for 30 min before flow cytometry analysis.
For in vivo lymph node draining experiments, Cy5.5-labeled SpyTag-OVA was used to generate OMV-OVA-IgG and OMV-OVA-DEC vaccines, which were then subcutaneously injected into mice at the base of tail. After 6 h, the lymph nodes were isolated for detection of the fluorescent signals. A Maestro imaging system (IVIS; Cambridge Research & Instrumentation, Woburn, MA, USA) was used to measure the fluorescence intensity.
For antigen cross-presentation assays, BMDCs were incubated with the OMV-OVA-IgG or OMV-OVA-DEC vaccines at 37°C for 12 h. Antigen presentation by MHC-I on the cell surface was subsequently evaluated using an OVA257-264-specific MHCI complex (MHCI-OVA) antibody. After incubation with the antibody, flow cytometry data were acquired and analyzed using BD Accuri C6 (BD Biosciences, USA) flow cytometer software.
2.6. In vivo immune response
C57BL/6 mice were injected subcutaneously with different OMV-based nanovaccines (containing 30 µg OVA) on days 0 and 6. On day 12, splenocytes were isolated for T cell activity analysis with 10 µg/mL SIINFEKL re-stimulation. Subsequently, cells were stained with FITC-anti-mouse CD8a, PE-anti-mouse CD69, APC-anti-mouse CD3 or CFSE to assess the activation and proliferative activity of CD8+ T cells. Cells were assessed by flow cytometry analysis. Additionally, these effector cells were mixed with B16-OVA cells (target cells). The CTL activity was evaluated in an LDH cytotoxicity detection assay (Beyotime). Finally, splenocytes from different groups were analyzed for IFN-γ secretion by ELISPOT, according to the manufacturer's instructions. Briefly, splenocytes (1 × 105 cells/well) were seeded in a 96-well plate pre-coated with an IFN-γ antibody. The cells were then incubated with antigen or ionomycin (positive control) for 20 h. The plates were subsequently decorated with a detection antibody followed by alkaline phosphatase conjugated to streptavidin. A purple precipitate appeared as spots with the final addition of a substrate solution. The plates were counted by a Mabtech IRIS FluoroSpot/ELISpot reader (Mabtech, Sweden).
2.7. Lung metastasis inhibition
To construct a lung metastasis model, C57BL/6 mice were injected intravenously with B16-OVA cells (2 × 105) at day 0 and then immunized subcutaneously with different OMV-based nanovaccines (30 µg OVA) on days 6 and 12. On day 18, the lungs were imaged for enumeration of pulmonary tumor nodules, and splenocytes were collected for re-stimulation with 10 µg/mL SIINFEKL, followed by staining with FITC-anti-mouse CD8a, PE-anti-mouse CD69, PE OVA-MHCI complex tetramer or CFSE to assess the activation and proliferative activity of specific CD8+ T cells. Simultaneously, intracellular IFN-γ staining was also performed after cellular surface staining for CD8a and CD4. The cells were resuspended in PBS for flow cytometry analysis (BD Accuri C6, BD Biosciences, USA). IFN-γ ELISPOT analysis was performed as mentioned above.
2.8. Immune memory analysis and tumor re-challenge experiments
Mice were immunized twice with the OMV-based nanovaccines as described above. Splenocytes were collected on day 60 after the final immunization. Next, the cells were stained with FITC-anti-mouse CD8a, PE OVA-MHCI complex tetramer, PE/Cy7 anti-mouse CD44 and APC anti-mouse CD62L at 4°C for 30 min and analyzed by flow cytometry.
To study the antitumor effects of the OMV-OVA-DEC vaccine, C57BL/6 mice were intravenously injected with 2 × 105 B16-OVA cells on day 0 and received OMV-OVA-DEC vaccinations on day 6 and day 12. After the immunotherapy, the mice that survived to day 72 were re-challenged subcutaneously with 5 × 105 B16-OVA or B16F10 cells. Controls were naive mice challenged with 5 × 105 B16-OVA cells. Tumor growth was monitored 16 days after tumor cell inoculation. Tumor volumes were calculated by the formula V = 0.5 × Length × Width2. The mice were euthanized when the tumor volumes reached 2000 mm3.
2.9. Neoantigen-specific priming of CD8+ T cells
To further characterize the specificity of the CD8+ T cell response to the OMV-based neoantigen vaccine, PBMCs of healthy donors who expressed HLA class I allele HLA-A*0201 was isolated from EDTA-K2 blood samples by centrifugation on a Ficoll density gradient and suspended in X-ViVO™15 medium (Lonza). Next, monocyte-derived DCs were generated in theX-VIVO™15 medium by supplementation with 100 ng/ml IL-4 and 100 ng/ml GM-CSF. After incubation with the cells for 2 d and 4 d at 37°C, half of the medium was replaced with fresh DC culture medium. On day 6, the cells were sedimented by centrifugation at 450 × g and resuspended to a titer of 3 × 105 cells per mL, Neoantigen 1 (EGFR-T790M), Neoantigen 2 (TP53-R175H), Neoantigen 3 (CDC73-Q254E), Neoantigen 4 (TMEM48-F169L), Neoantigen 5 (SEC24A-P469L), Neoantigen 6 (EXOC8-Q656P), Neoantigen 7 (gp100-A288V) and the neoantigen-derived OMV-DEC vaccines were separately added to each well and the samples were incubated at 37°C for 16 h.
For CD8+T cell isolation, a human CD8 Microbeads kit (Miltenyi) was used to retain CD8+ T cells, CD8-positive cells were eluted from the LS column by removing the column from the magnetic field. To evaluate the neoantigen-specific priming capacity of CD8 T cells, neoantigen/OMV-neoantigen-DEC vaccine (5 µg/mL equivalent neoantigens) stimulated DCs were resuspended in X-VIVO™15 medium (containing 100 ng/ml IL-4, 100 ng/ml GM-CSF and 50 ng/ml IL-2) and mixed with CD8+ T cells at a 1:5 ratio of DCs: T cells at 37°C for 72 h. IFN-γ release was assessed using a human IFN-γ precoated ELISPOT Kit, and the T cell activation marker 4-1BB was measured by NovoCyte flow cytometry (Agilent Technologies, CA, USA).
2.10. In vivo antitumor activity
Mouse tumor models were established with subcutaneous injections of 5 × 105 B16-OVA cells. For vaccines combined with αPD-1 therapy without or with the addition of ibrutinib experiments, the OMV-OVA-DEC (30 µg OVA) vaccine was administered on the days 6 and 12. αPD-1 antibodies were administered intratumorally beginning on day 6 and continued at two days intervals for four treatments (2.5 mg kg–1 per injection) and Ibrutinib was administered intraperitoneally beginning on day 7 and continued at two days interval for four injections (1.5 mg kg–1 per injection). Tumor size was measured every two days with a caliper and tumor volume was determined by the above formula. The mice were euthanized when the tumor volumes reached to 2000 mm3. Body weight was monitored every two days and survival time was also monitored until the end of the experiment. For tumor microenvironment analysis, tumor tissues were obtained on day 19 or day 20, and then stained to assess the proportion of MDSCs, M2 macrophages and Tregs in the tumor microenvironment using a NovoCyte flow cytometer (Agilent Technologies, CA, USA). In brief, APC anti-mouse CD45, FITC anti-mouse CD11b, PE/Cyanine7 anti-mouse Gr-1, PE/Cyanine7 anti-mouse F4/80 and FITC anti-mouse CD4 were used to stain the cell surface. PE anti-mouse CD206 was used for intracellular staining and PE anti-mouse FoxP3 was used for intranuclear staining. F4/80 and CD206 antibodies were used to detect M2 macrophages, CD4 and FoxP3 antibodies were used to detect Tregs and CD11b and Gr-1 antibodies were used to detect MDSCs.
2.11. Gating strategy for flow cytometry
The representative gating strategies for flow cytometry were shown in Figs. S12–16 to analyze immune cells including: mature DCs (CD11c+CD80+CD86+) in BMDCs; OVA-presenting DCs (CD11c+MHCI-OVA+) in BMDCs; CD8+ T cells (CD3+CD8+) and activated CD8+ T cells (CD8+CD69+) in splenocytes; IFN-γ+CD8+ and CD4+ T cells, OVA-tetramer+ in CD8+T cells, CFSE-stained CD8+ T cells and CD44+CD62L+ in CD8+ T cells in splenocytes; CD8+ T cells (CD45+CD8+), CD4+ T cells (CD45+CD4+), M2 macrophages (CD45+CD206+F4/80+), Tregs (CD45+CD4+FoxP3+) and MDSCs (Gr-1+CD11b+) in tumor tissues.
3. Results
3.1. Engineering of a multi-faceted OMV chassis for simultaneous antigen display and antibody binding
We first engineered an OMV chassis (OMV-SpyC/SPAb), in which we fused the SpyCatcher (SpyC) and the SPAb proteins to the C-terminus of the OMV surface protein ClyA. The genes encoding ClyA fused with SpyC (ClyA-SpyC) and ClyA fused with SPAb (ClyA-SPAb) were recombined into a co-expression plasmid, pETDuet-1, and the fusion proteins were inductively expressed in the transformed E. coli Rosetta (DE3) strain by adding isopropyl-β-d-thiogalactoside (IPTG) at 16°C for 16 h. OMVs in the culture medium were then separated by ultracentrifugation. By design, the isolated OMV chassis (OMV-SpyC/SPAb) allowed for the SpyTag-labeled heterogenous antigen (SpyTag-antigen) display via the orthogonal reactivity of SpyCatcher/SpyTag and the binding of αDEC205 antibody to SPAb, leading to the final construction of the OMV-based nanovaccine (OMV-antigen-DEC). A schematic illustration of this bioengineered OMV-based nanovaccine was shown in Fig. 1a.
Fig. 1.

Engineering of the multi-faceted OMV chassis for simultaneous antigen display and antibody binding. (a) Schematic of the OMV platform engineering and vaccine preparation, (b) and (c) DLS analysis (b) and TEM examination (c) of OMV-SpyC/SPAb. Scale bar, 50 nm. (d) and (e) Western blot assay to detect ClyA-SpyC-3Cmyc (d) and ClyA-SPAb-3Flag (e) levels in OMV-SpyC/SPAb using anti-Cmyc and anti-Flag antibodies, respectively. (f) One-step western blot assay to analyze the antibody binding function of ClyA-SPAb-3Flag using the HRP-conjugated Rat IgG antibody (Rat IgG-HRP). (g) The observation of αDEC205 antibody binding on OMV-SpyC/SPAb using TEM. αDEC205 antibody was labeled with 10 nm gold nanoparticles. Scale bar, 20 nm. (h) Western blot assay to verify the rapid binding between ClyA-SpyC-3Cmyc and SpyT-HA (ClyA-SpyC-SpyT-HA, ∼60 kDa) using an anti-HA antibody. MW, molecular weight.
As apparent by the dynamic light scattering (DLS) and transmission electron microscopy (TEM) results (Fig. 1b and c), OMV-SpyC/SPAb possessed a bilayer structure and a uniform circular morphology, with a diameter of approximately 27.9 nm. To verify the expression of the fusion proteins, three Cmyc tags and three Flag tags were attached to the C-terminus of ClyA-SpyC (ClyA-SpyC-3Cmyc) and ClyA-SPAb (ClyA-SPAb-3Flag), respectively. As shown in Fig. 1d and e, the ClyA-SpyC-3Cmyc (∼60 kDa) and ClyA-SPAb-3Flag (∼45 kDa) were detected by western blot analysis utilizing anti-Cmyc and anti-Flag antibodies, respectively. In the ClyA-SpyC-3Cmyc detection, a faint band at ∼45 kDa, which may be attributed to the nonspecific antibody binding to ClyA-SPAb-3Flag, was also observed (Fig. 1d).
Next, we employed a one-step western blot assay using a horse radish peroxidase (HRP)-conjugated Rat IgG antibody to verify the antibody binding function of ClyA-SPAb-3Flag, indicated by the band at ∼45 kDa (Fig. 1f). To demonstrate the successful binding of αDEC205 antibody on OMV-SpyC/SPAb, αDEC205 antibody was labeled with 10 nm gold nanoparticles. As shown in TEM image in Fig. 1g, gold-labeling αDEC205 antibody were shown on the surface of OMV-SpyC/SPAb, indicating the successful binding of αDEC205 antibody on OMV-SpyC/SPAb. Then, using an HA tag as a simulated antigen, we synthesized SpyTag-labeled HA tag (SpyT-HA), and observed rapid binding between ClyA-SpyC-3Cmyc and SpyT-HA (ClyA-SpyC-SpyT-HA, ∼60 kDa) by western blot analysis using an anti-HA antibody (Fig. 1h). The above results demonstrate the feasibility of our multi-faceted OMV-based nanovaccine platform which was engineered for the versatile decoration of heterogenous antigens and functional antibodies.
3.2. Mechanism of improved antigen delivery by αDEC205 antibody modification
To analyze the antigen delivery efficacy and the following antigen cross-presentation, we chose an antigenic epitope of ovalbumin (OVA), OVA257-264(SIINFEKL), as a model antigen, which is a major histocompatibility complex I (MHCI)-restricted CD8+ T cell antigen. Thus, we prepared an OVA257-264-displaying OMV-antigen-DEC nanovaccine (OMV-OVA-DEC) and an IgG isotype control (OMV-OVA-IgG). After removing the unbonded antigen peptide by ultrafiltration (100 kDa MWCO) or dialysis (10 kDa MWCO), the display efficiency of OVA antigen peptide was quantitatively determined as about 99.5%. As shown in Fig. 2a, due to the abundant LPS on the OMV surface, OMV-OVA-IgG was rapidly recognized and taken up by DCs, however this process was blocked by DC maturation triggered by TLR4 activation. Compared with OMV-OVA-IgG, the αDEC205 antibody modification endowed OMV-OVA-DEC with an additional DC recognition and uptake pathway through binding with DEC205 on DCs. These features favored a transition from the transient uptake of OMV-OVA-IgG to a persistent uptake of OMV-OVA-DEC by DCs that continues after DC maturation and may lead to enhanced antigen presentation and immune stimulation.
Fig. 2.

Improved antigen delivery of OMV-DEC-based nanocarrier mediated by DEC205/αDEC205 antibody interaction. (a) Schematic illustration depicting the uptake enhancement mediated by αDEC205 antibody modification, due to the transition from transient uptake of OMV-OVA-IgG to persistent uptake of OMV-OVA-DEC by DCs. (b) BMDC uptake of the indicated OMV-based nanovaccines constructed by PE-labeled αDEC205 antibody and IgG isotype, as measured by flow cytometry (n = 3). (c) BMDC uptake of PE-labeled OMV-IgG or OMV-DEC at 12 h with or without pre-blocking with TLR4 inhibitor (1 µM TAK-242) or DEC205 blocking antibody (5 µg/mL αDEC205 antibody) for 1 h (n = 3). (d) Laser scanning confocal microscopy images of DC2.4 cells after 1 h incubation with the indicated OMV-based nanovaccines. OMV-based nanovaccines were labeled with Dil (red). DEC205 was detected with a PE-labeled antibody (green), and TLR4 was detected with a FITC-labeled antibody (cyan). Scale bar, 50 µm. (e)–(g) Lymph node drainage of OMV-OVA-IgG and OMV-OVA-DEC labeled with Cy5.5-labeled OVA at 6 h after subcutaneous administration at the base of tail. Lymph node accumulation was evaluated visually by examining the Cy5.5 fluorescence in vivo (e) and ex vivo (f), and quantitatively analyzed (g). The fluorescence intensity in the OMV-OVA-DEC group was set to 100% (n = 3). (h) and (i) Antigen presentation efficiency in DCs as examined by flow cytometry using an OVA257-264-specific MHCI complex (MHCI-OVA) antibody at 12 h after incubation in BMDCs in vitro (h) or 6 h after subcutaneous vaccination in vivo (i) (n = 3). One-way ANOVA with a Tukey post-hoc test was used for statistical analysis. Data are shown as mean ± SD.*, P < 0.05; ⁎⁎⁎, P < 0.001.
To test the above hypothesis, we used fluorescent dye PE-labeled anti-DEC205 antibody and an IgG isotype to construct OMV-based nanovaccines. We then monitored the uptake of the nanovaccines by bone marrow-derived dendritic cells (BMDCs) using flow cytometry. As shown in Fig. 2b, the BMDC uptake of OMV-OVA-DEC was close to 2.8-fold greater than that of OMV-OVA-IgG, and OMV-DEC was about 7.9-fold greater than that of OMV-IgG, despite the similar level of BMDC maturation in all groups (Fig. S1a). To further verify the effects of αDEC205 antibody modification, we pre-incubated the BMDCs with the TLR4 inhibitor TAK-242 or αDEC205 antibody (Fig. 2c). We found that TLR4 inhibition did not significantly impede the uptake of OMV-DEC. By contrast, BMDC uptake of OMV-DEC was considerably reduced by pre-blockade of DEC205, confirming that the αDEC205 antibody modification played a major role in this process. In addition, we found a higher DEC205 expression in BMDCs treated with the OMV or OMV-OVA-IgG compared with that in PBS-treated BMDCs. However, the DEC205 expression significantly decreased in the OMV-OVA-DEC group, and the change was likely attributed to endocytosis of OMV-OVA together with DEC205 (Fig. S1b).
We observed a co-localization between the OMV-based nanovaccines and DEC205 or TLR4. DC2.4 cells (a murine DC cell line) were incubated with Dil-labeled OMV-based nanovaccines (red) for 1 h, and TLR4 and DEC205 were detected using a FITC-labeled antibody (cyan) and a PE-labeled antibody (green), respectively. The overlay between red fluorescence (OMV) and green fluorescence (DEC205) was significantly stronger in the OMV-OVA-DEC group than that in the OMV-OVA-IgG group (Fig. 2d). However, the overlay between the red (OMV) and cyan fluorescence (TLR4) was similar in different groups. These results indicate that the αDEC205 antibody modification provides an additional recognition pathway of DCs for OMV-DEC nanocarriers through binding with DEC205 on the DCs.
We next evaluated lymph node drainage in vivo using Cy5.5-labeled OVA to prepare OMV-OVA-IgG and OMV-OVA-DEC. As shown in Fig. 2e and f, compared with OMV-OVA-IgG, more OMV-OVA-DEC accumulated in the draining lymph nodes at 6 h after subcutaneous vaccination. The fluorescence intensity of the draining lymph nodes in the OMV-OVA-DEC group was about 1.7-fold greater than that in the OMV-OVA-IgG group (Fig. 2g). These results suggest that the αDEC205 antibody modification can also enhance the lymph node drainage of OMV-DEC nanocarriers in vivo.
We examined the antigen presentation by flow cytometry using the OVA257-264-specific MHCI complex (MHCI-OVA) antibody. Compared with OMV-OVA-IgG, OMV-OVA-DEC vaccination induced an approximately 2.5-fold greater level of MHCI-OVA expression in DCs either in vitro or in vivo (Figs. 2h, i and S1c), demonstrating that the αDEC205 antibody binding on OMV-DEC enabled not only enhanced antigen delivery but also stronger antigen presentation in DCs, laying the foundation for efficient immune stimulation and tumor inhibition.
3.3. Improved immune response and lung metastasis inhibition
We tested whether the OMV-OVA-DEC nanovaccine could elicit a stronger antigen-specific T cell response. The different OMV-based nanovaccines were subcutaneously injected into C57BL/6 mice on days 0 and 6, and the splenocytes were harvested on day 12 and stimulated with OVA257-264. We then performed an analysis of the activation and proliferation of CD8+ T cells (Fig. 3a). As shown in Figs. 3b and S2a, OMV-OVA-DEC (44.7%) or OMV-OVA-IgG (38.1%) nanovaccine stimulated an expansion of CD3+CD8+ T cells compared with PBS (27.1%); the greatest proportion of these cells was achieved in splenocytes from mice immunized with OMV-OVA-DEC. Using CD69 as an activation marker in CD8+ T cells, we found that OMV-OVA-DEC vaccination provoked stronger CD8+ T cell activation (13.1%) than OMV-OVA-IgG (10.1%) or PBS (8.2%) (Figs. 3c and S2b). The cytotoxic effects of splenocytes were also evaluated through co-incubation of splenocytes with the target cells B16-OVA (B16 melanoma cell expressing ovalbumin). As shown in Fig. 3d, the splenocytes from mice administered the OMV-OVA-IgG nanovaccine led to specific lysis in 21.8% of B16-OVA cells, while 60.0% of B16-OVA cells were lysed after incubation with splenocytes from mice treated with the OMV-OVA-DEC nanovaccine. In addition, 5,6-carboxyfluorescein diacetate succinimidyl ester (CFSE) staining was performed to analyze T cell proliferation; an approximate 3.2-fold greater T cell proliferation rate was observed in the OMV-OVA-DEC group (24.1%) compared with that in the OMV-OVA-IgG group (7.5%) (Figs. 3e and S2c). Similarly, although more IFN-γ was produced by splenocytes from mice immunized with OMV-OVA, OMV-OVA-IgG or OMV/OVA/DEC (a control mixture of natural OMV, OVA peptide and αDEC205 antibody) after exposure to OVA257-264 than that in the PBS group, the most IFN-γ spots in the enzyme-linked immunospot (ELISPOT) assay were observed in the OMV-OVA-DEC group (Figs. 3f and S3d). Together, our data suggest that both OMV-OVA-IgG and OMV-OVA-DEC can stimulate a T cell-mediated immune response and marked CTL-mediated specific immune elimination of antigen-positive target cells, with the OMV-OVA-DEC vaccine most potent in this function, likely because of the targeting antibody decoration.
Fig. 3.

Immune response evaluation and lung metastasis inhibition. (a)–(f) Immune response evaluation. C57BL/6 mice were vaccinated with different OMV-based nanovaccines on days 0 and 6, and the immune response analysis was performed on day 12 (a). The splenocytes were harvested and stimulated with OVA257-264 overnight. The percentages of CD8+ in CD3+ T cells (B) and CD69+ in CD8+ T cells (c) were evaluated using flow cytometry (n = 4). The B16-OVA cells (target cells) were incubated with the splenocytes (effector cells) for 12 h at an effector: target cell ratio of 20: 1, and the specific lysis of B16-OVA cells was evaluated using an LDH cytotoxicity detection assay (n = 4) (d). The proliferation in CD8+ T cells was analyzed by flow cytometry using CFSE staining (n = 4) (e). IFN-γ secretion from splenocytes stimulated with OVA257-264 was quantitatively detected by the ELISPOT assay (n = 4) (f). (g)-(n) Antitumor effect of OMV-based nanovaccines in a pulmonary metastasis model. C57BL/6 mice were challenged by tail vein injection of 2 × 105 B16-OVA cells on day 0, and received vaccinations on days 6 and 12 (g). The lungs were collected on day 18 (h). The splenocytes were also harvested and stimulated with OVA257-264 overnight. The percentages of CD69+ in CD8+ T cells (i), IFN-γ+ in CD8+ T cells (j), IFN-γ+ in CD4+ T cells (k) and OVA-tetramer+ CD8+ T cells (l) were measured using flow cytometry (n = 4). The proliferation in CD8+ T cells was analyzed by flow cytometry using CFSE staining (n = 4) (m). IFN-γ secretion from splenocytes stimulated with OVA257-264 was quantitatively detected using the ELISPOT assay (n = 4) (n). One-way ANOVA with a Tukey post hoc test was used for statistical analysis. Data are shown as mean ± SD. N.S., no significance; *, P < 0.05; ⁎⁎, P < 0.01; ⁎⁎⁎, P < 0.001.
We next tested the antitumor effect of the OMV-based nanovaccine-triggered immune response in a pulmonary metastasis model of B16-OVA cells. Mice were inoculated by tail vein injection of B16-OVA cells on day 0, and the vaccination and immune response analysis were performed according to the same schedule as the previous experiment (Fig. 3g). As shown in Figs. 3h and S3, the OMV-OVA-DEC vaccination strikingly inhibited the lung metastasis, demonstrating the key contribution of αDEC205 antibody decoration. To clearly characterize the immune mechanism underlying these antitumor effects, we again used the flow cytometry and ELISPOT assay to explore T cell stimulation in splenocytes. As expected, the CD8+CD69+ T cells (Figs. 3i and S4a) and the CD8+IFN-γ+ T cells (Figs. 3j and S4b) were most abundant (13.7% and 9%, respectively) in the OMV-OVA-DEC group, indicating more prominent CD8+ T cell stimulation and activation compared with the other groups. Meanwhile, as shown in Figs. 3k and S4c, the IFN-γ+ in CD4+ T cells (29.3%) in the OMV-OVA-DEC group was also greater than that in the other groups. These CD4+ T cells may assist in the CD8+ T-mediated cellular immunity. Using an OVA-MHCI complex tetramer (OVA-tetramer) to label the OVA-specific T cells, we found that the OMV-OVA-IgG (8.0%) and OMV-OVA-DEC (10.1%) vaccination triggered an obvious increase of OVA-specific CD8+ T cells compared with that in PBS group (3.9%) (Figs. 3l and S5a). This immune stimulation-induced OVA-specific CD8+ T cell increase by OMV-OVA-DEC vaccination (6.2%) was close to 1.5-fold greater than that by OMV-OVA-IgG vaccination (4.1%). In addition, as shown in Figs. 3m, n, S5b, and c, the greater T cell proliferation and IFN-γ production were detected in splenocytes derived from mice immunized with OMV-OVA-DEC than that in OMV-OVA-IgG group. These results demonstrate that the OMV-OVA-DEC nanovaccine the assistance of αDEC205 antibody modification is a promising immunostimulant to drive a robust antitumor effect.
3.4. Evaluation of immune memory
We examined the long-term immune memory after two immunizations with the OMV-based nanovaccines. On day 60 after the second vaccination, splenocytes were collected and stimulated with OVA257-264 to analyze the repertoire of OVA-specific CD8+ T cells and T cell memory. As shown in Fig. 4a and b, the proportion of OVA-tetramer+CD8+ T cells was 11.9% in the OMV-OVA-DEC group. By contrast, only 6.6% OVA-tetramer+CD8+ T cells were detected in the OMV-OVA-IgG group, which was a similar result to that in the previous short-term immune response analysis. This suggests that the OVA-specific T cells stimulated by OMV-OVA-DEC can be activated upon antigen recognition after at least 60 days. We next analyzed the immune memory in the collected splenocytes. Although there was no significant increase in central memory T (TCM) cells (CD44hiCD62Lhi in CD8+ T cells) in the OMV-DEC (18.9%) and OMV-OVA-IgG (19.5%) groups compared with that in the PBS group (17.2%), OMV-OVA-DEC induced additional TCM cells (23.1%; Fig. 4c and d). The percentage of CD44hiCD62Llo in CD8+ T cells (effector memory T cells, TEM) in the OMV-OVA-DEC group (37.2%) was also higher than that in the PBS (30.1%), OMV-DEC (33.0%) and OMV-OVA-IgG (31.7%) groups (Fig. 4e). These results are indicative of an effective immune memory induced by OMV-OVA-DEC vaccination.
Fig. 4.

Long-term immune memory evaluation and resistance to re-challenge. C57BL/6 mice received two vaccinations, and the splenocytes were collected on day 60 after the second vaccination and stimulated with OVA257-264 to analyze the repertoire of OVA-specific CD8+ T cells and T memory cells. (a) and (b) Measurement (a) and quantitative analysis (b) of OVA-tetramer+ CD8+ T cells using flow cytometry in splenocytes stimulated with OVA257-264 (n = 4). (c)-(e) Measurement (c) and quantitative analysis of central memory T cells (CD44hi CD62Lhi) (d) and effector memory T cells (CD44hi CD62Llo) (e) in CD8+ T cells using flow cytometry in splenocytes (n = 4). (f) C57BL/6 mice were challenged by tail vein injection of 1 × 105 B16-OVA cells on day 0, and received OMV-OVA-DEC vaccination on days 6 and 12. OMV-OVA-DEC-cured or healthy control mice were subcutaneously re-challenged with B16-OVA cells or B16F10 cells on day 72 after primary tumor inoculation, and tumor growth was monitored within another 16 days (n = 8). One-way ANOVA with a Tukey post-hoc test was used for statistical analysis. Data are shown as mean ± SD. *, P < 0.05; ⁎⁎, P < 0.01; ⁎⁎⁎, P < 0.001.
We next tested whether the generated immune memory can effectively protect mice from tumor re-challenge. We again administered OMV-OVA-DEC vaccination to mice with lung metastases of B16-OVA cells, and subcutaneously re-challenged the mice with B16-OVA cells or B16F10 cells on day 72 after the primary tumor inoculation. As shown in Fig. 4f, 62.5% of immunized mice rejected the re-inoculated B16-OVA cells, while 100% of B16-OVA tumors rapidly grew in the healthy control mice. Notably, 25% of the immunized mice rejected the re-inoculated B16F10cells without the OVA antigen (Fig. 4f). This finding suggests that antigens released during vaccination-induced B16-OVA cell death can stimulate an antigen-spreading immunity, which is crucial for the inhibition and treatment of tumors with rapid variability and genetic heterogeneity. Collectively, the OMV-based nanovaccine induced a marked immune memory, which is critical for a long-term benefit in the prevention of clinical tumor recurrence.
3.5. Analysis of neoantigen-specific activation in human CD8+ T cells
To further characterize the adaptability of our OMV-based nanoplatform in clinical personalized cancer vaccines, we chose seven human leukocyte antigen (HLA) class I allele HLA-A*0201-restricted neoantigens to prepare OMV-antigen-DEC nanovaccines (OMV-neoantigen-DEC). Those neoantigens were identified to possess high immunogenicity by predicted binding affinity according to in silico algorithms [26], [27], [28], [29], [30]. DCs were collected from a healthy donor with HLA-A*0201, and were incubated with the OMV-neoantigen-DEC nanovaccines. Meanwhile, CD8+ T cells were separated from the same individual using human CD8 microbeads. The nanovaccine-stimulated DCs were subsequently co-cultured with the CD8+ T cells. Finally, the activation and neoantigen-specific immune response were evaluated by flow cytometry and ELISPOT assay to detect 4-1BB expression and IFN-γ secretion in CD8+ T cells, respectively. As shown in Fig. 5a and b, the proportion of 4-1BB+CD8+ T cells was significantly higher in every OMV-neoantigen-DEC group compared with the corresponding neoantigen group, indicating that the OMV-DEC platform plays a deterministic role in the activation of CD8+ T cells. We further assessed the antigen-specific priming by ELISPOT assays (Fig. 5c and d). After re-stimulation with the corresponding neoantigen peptides, IFN-γ secretion was significantly greater in the OMV-neoantigen-DEC group than that in the corresponding neoantigen group. Taken together, these results indicate that the novel OMV-DEC-based vector possesses the ability to deliver human neoantigens and elicit specific immune responses; the rapid antigen display function makes this platform applicable in rapid antigen identification for personalized cancer vaccine development.
Fig. 5.

Neoantigen-specific immune activation of human CD8+T cells. Seven HLA class I allele HLA-A*0201-restrictedneoantigenswere used to prepare OMV-antigen-DEC nanovaccines (OMV-neoantigen-DEC): Neoantigen 1 (EGFR-T790M), Neoantigen 2 (TP53-R175H), Neoantigen 3 (CDC73-Q254E), Neoantigen 4 (TMEM48-F169L), Neoantigen 5 (SEC24A-P469L), Neoantigen 6 (EXOC8-Q656P) and Neoantigen 7 (gp100-A288V). DCs were collected from a healthy donor with HLA-A*0201, and were incubated with these OMV-neoantigen-DEC nanovaccines at 37°C for 16 h. Meanwhile, CD8+ T cells were separated from the same individual using human CD8 microbeads. The nanovaccine-stimulated DCs were subsequently co-cultured with CD8+ T cells at a 1:5 ratio of DCs: T cells at 37°C for 72 h. (a) and (b) Flow cytometry measurements (a) and quantitative analysis (b) of 4-1BB+ cells (n = 3). (c) and (d) Images (c) and quantitative analysis (d) of IFN-γ secretion after re-stimulation with separate neoantigen peptides, as measured by the ELISPOT assay (n = 3). One-way ANOVA with a Tukey post-hoc test was used for statistical analysis. Data are shown as mean ± SD. *, P < 0.05; ⁎⁎, P < 0.01; ⁎⁎⁎, P < 0.001.
3.6. Development of a combinational immunotherapy strategy for solid tumors
To investigate the antitumor efficiency of OMV-OVA-DEC in solid tumors, we established a subcutaneous B16-OVA-bearing mouse tumor model. Considering the critical role of the immune checkpoint in the immunosuppression of solid tumors, we combined the anti-PD1 antibody (αPD-1) with the OMV-OVA-DEC nanovaccine to induce robust tumor suppression. As shown in Fig. S6a and b, compared with the PBS group, treatment with only OMV-OVA-DEC or αPD-1 moderately delayed tumor growth, while mice in the OMV-OVA-DEC/αPD-1 group exhibited greater tumor inhibition efficiency. The combination of OMV-OVA-DEC and αPD-1 also resulted in prolonged survival compared with the other groups (n=12) (Fig. S6c). In addition, the body weight curves in the different groups indicated that the OMV-OVA-DEC/αPD-1 combination had no obvious toxicity (Fig. S6d). Unfortunately, the extent of the antitumor effect of OMV-OVA-DEC/αPD-1 was not satisfactory, so we next analyzed the immune cell components in the tumor microenvironment panoramically (Figs. S6e–i and S7a–e). The tumor-infiltrating CD8+ T cells (Figs. S6e and S7a) in the OMV-OVA-DEC/αPD-1 group were present at the highest levels, which was an indication that combination with the αPD-1 reversed tumor immunosuppression and improved the antitumor effect of the OMV-OVA-DEC nanovaccine to some extent. Immunosuppressive cells in the tumor microenvironment in the OMV-OVA-DEC/αPD-1 group, such as M2-like macrophages (M2 macrophages; Figs. S6g and S7c) and regulatory T cells (Tregs; Figs. S6h and S7d) were significantly reduced compared with the other groups. Unexpectedly, compared with other groups, the MDSCs (Figs. S6i and S7e) in the OMV-OVA-DEC/αPD-1 group showed no significant change, which may be the main immunosuppressive stress after OMV-OVA-DEC/αPD-1 therapy.
The infiltration of MDSCs can inhibit T cell stimulation and proliferation, subsequently promoting an imbalanced tumor microenvironment. After finding unaltered levels of MDSCs in the panoramic immune microenvironment analysis, we added a MDSC-depleting agent, Ibrutinib, into the combinational strategy (OMV-OVA-DEC/αPD-1/Ibrutinib). Compared with the other formulations, OMV-OVA-DEC/αPD-1/Ibrutinib significantly inhibited the growth of subcutaneous B16-OVA tumors (Fig. 6a and b) and significantly prolonged the survival of mice (Fig. S8). Body weight monitoring did not reveal any apparent anomalous change (Fig. S9). Compared with OMV-OVA-DEC/αPD-1, the assistant effect of Ibrutinib also significantly increased the number of tumor-infiltrating CD8+ T cells (Figs. 6c and S10a) and CD4+ T cells (Figs. 6d and S10b). By contrast, the populations of immunosuppressive cells, M2 macrophages, Tregs and MDSCs also displayed a significant decrease in tumors treated with OMV-OVA-DEC/αPD-1/Ibrutinib compared with that in the other groups (Figs. 6e–g and S11a–c), showing that this combinational therapy was capable of normalizing the immunosuppressive tumor microenvironment. To summarize, when combined with αPD-1 or Ibrutinib, this promising nanovaccine could be a potent immunopotentiator.
Fig. 6.

Therapeutic effects of combinational immunotherapy containing OMV-OVA-DEC nanovaccine combined with αPD-1 and Ibrutinib in a subcutaneous melanoma model. B16-OVA cells were subcutaneously injected into C57BL/6 mice, after which the mice received treatment with the indicated formulations. (a) Tumor growth curves recorded for 20 days (n = 12). (b) Tumors isolated from the mice on day 20 (n = 5). (c)-(g) The percentages of tumor infiltrating CD8+ T cells (c), CD4+ T cells (d), M2 macrophages (e), Tregs (f) and MDSCs (g) in the collected tumor tissues on day 20, as measured by flow cytometry (n = 4). Scale bar, 1 cm. One-way ANOVA with a Tukey post hoc test was used for statistical analysis. Data are shown as mean ± SD. N.S., no significance; *, P < 0.05; ⁎⁎, P < 0.01; ⁎⁎⁎, P < 0.001.
4. Discussion
The development of nanocarriers has revolutionized vaccine delivery technology, and an increasing number of studies have focused on the integration of nanocarriers with immune adjuvants to achieve efficient co-delivery of antigens and immune adjuvants. For example, the adjuvant system (AS) developed by GlaxoSmithKline encapsulates an immune adjuvant into a nanocarrier, such as a liposome [6]. In addition, polymers or phospholipids with STING pathway activation ability have been explored for synthesizing nanocarriers with intrinsic immune adjuvant function [11]. With the advancements in genetic engineering technology, vaccine carriers based on attenuated bacteria or bacteria-derived substances have become a burgeoning field of research, as these carriers possess large amounts of intrinsic PAMPs as immune adjuvants [31]. For instance, the Advaxis company developed Listeria as living carriers to deliver tumor antigens into DCs, exploiting the intracellular infective properties of Listeria [32]. Considering the potential infection risk of viable bacterial carriers, newer studies have begun to use bacteria-derived nanocarriers, such as a 400 nm sized Minicell system derived from bacteria through a complex genetic engineering and isolation process [33], to transport tumor antigens. For rapid and simple vector preparation, we selected OMVs as the vaccine nanocarrier system in our previous work [12]. These nanoplatforms can be produced in large quantities via high-speed centrifugation, and can stimulate rapid antigen display on OMVs through genetic engineering and molecular glue technology. The OMV-based nanocarrier can be efficiently recognized and taken up by DCs via the TLR4/LPS interaction[16, 31]. However, as previously mentioned, the rapid maturation of DCs limits the further antigen uptake of OMVs. Therefore, in this study, we overcome the MUO phenomenon by modifying OMVs with αDEC205 antibody to remodel the OMVs uptake by DCs.
To successfully construct the OMV-DEC nanovaccine vectors, we selected the domain B of SPA to fuse with ClyA on the surface of OMVs. It is well known that domain B of SPA has a strong, non-specific binding affinity for antibody Fc fragments, which is equivalent to the binding affinity between antibody and antigen [34,35]. Thus, the surface ClyA-SPAb efficiently and stably bound the αDEC205 antibody without affecting the function of the outer Fab fragment. In contrast to antibody binding of SPAb, both direct fusion expression and chemical coupling may affect the function of an antibody. Moreover, this flexible antibody modification technology with SPAb as a surface scaffold enables OMV to rapidly bind different antibodies for versatile applications, as we previously demonstrated in a study of rapid antigen display based on a "Plug-and-Display" system; tumor antigen and targeting antibody were quickly displayed on the multi-faceted OMV without cross-correlation interference. Together with the ability of OMVs to be generated on a large scale via bacterial fermentation, this modular design will accelerate the clinical translation of OMV-based tumor nanovaccines.
The modification with αDEC205 antibody remodeled the DC uptake pathway of the OMVs. In immature DCs, OMV or OMV-DEC were mainly taken up through the LPS-TLR4 signaling pathway, due to the large amounts of LPS on the OMV surface. However, as increasing numbers of OMV or OMV-DEC entered DCs, the DCs matured induced by the immune adjuvant effect. Under natural conditions, mature DCs no longer efficiently ingest OMV, however OMV-DEC was still continuously ingested by DCs through an artificial pathway mediated by the αDEC205antibody/DEC205 binding. In addition to the natural pathway mediated by LPS/TLR4 interaction, the modification of αDEC205 antibody provided another artificial pathway (αDEC205 antibody/DEC205 recognition) for the DC uptake of OMV-DEC. Therefore, the DC uptake pattern of OMV-DEC was referred to as “Normandy”. We verified this experimentally by pre-blocking DCs with a TLR4 inhibitor that did not markedly affect DC uptake of OMV-DEC, whereas DEC205 blockage notably decreased the uptake of OMV-DEC. These results suggest that the “Normandy” mechanism in which the recognition by DCs is mediated by multiple pathways is important for an effective vaccine vector and lays the foundation for the general design of vaccine nanocarriers in the future.
Despite the recent rapid development of bioinformatics technology and MHC-peptide mass spectrometry, the accuracy of tumor neoantigen prediction is still limited [36], [37], [38]. The identification of truly effective tumor neoantigens still relies on the verification of immunogenicity, which requires evaluation of synthetic vaccines in the cultured immune cells [1]. Current tumor nanovaccines mostly encapsulate the neoantigen into the nanocarrier, which is an inefficient method and unable to successfully identify a large number of tumor neoantigens [39], [40], [41]. Our versatile OMV-based nanocarrier enables rapid antigen display and tumor vaccine preparation via the “Plug-and-Display” system. In the present work, we verified the ability of this vector to display real human tumor neoantigens and induce a strong antigen-specific immune response in human immune cells, suggesting the possible application of this platform in clinical neoantigen identifying [30].
Although immune checkpoint inhibitors have achieved remarkable clinical benefits in cancer treatment, they still encounter the challenge of a limited number of infiltrating T cells in the tumor microenvironment [17,42,43]. Like other immune activation strategies, such as chemotherapy- or radiotherapy-induced tumor antigen release and using cytokines to stimulate the immune system, our OMV-based tumor nanovaccine also elicits a robust antitumor immunity, which was buttressed by the therapeutic effects of a checkpoint inhibitor and MDSC depletion agent (Ibrutinib). Indeed, the checkpoint inhibitor and Ibrutinib removed some barriers of immunosuppressor cells, such as MDSCs and Tregs, allowing CTLs to kill tumor cells [36,44,45]. The complementary mechanism resulted in a synergetic antitumor effect, which destroyed subcutaneous melanoma in mice.
5. Conclusion
In conclusion, by fusing the domain B of SPA with the surface ClyA protein, we improved the OMV-based tumor nanovaccine platform. The novel platform not only showed the ability to rapidly identify neoantigen to develop precise immunotherapy, but also became decorated with αDEC205 antibody via binding between SPAb and the Fc fragment (Fig. 7). The αDEC205 antibody modification greatly remodeled the OMVs uptake by DCs to a Normandy pattern, leading to robust antigen presentation and CTL expansion, which ultimately inhibited lung metastasis in a melanoma model and improved the therapeutic effect of the checkpoint inhibitor and MDSC depletion agent in a subcutaneous tumor (Fig. 7). This versatile, OMV-based nanocarrier was built with the intent to achieve individualized cancer vaccines and guide vaccine design for the prophylaxis and therapy of other diseases with highly variable antigens, such as influenza and human immunodeficiency viruses. Our study brings this promising nanovaccine technology a step closer to clinical translation.
Fig. 7.

Schematic diagram of personalized tumor vaccines from the versatile OMV-based nanocarriers with “Normandy” uptake pattern by DCs and their combinational application with other immunotherapeutic agents in solid tumor treatment.
Declaration of Competing Interest
G.N., X.Z., Y.Z. and J.L. are inventors on a filed provisional application China patent no. 202110129743.9 (A general vaccine vector based on bacterial outer membrane vesicles) submitted by the National Center for Nanoscience and Technology that covers the potential diagnostic and therapeutic uses of the vaccine for cancer immunotherapy. The authors declare that they have no other competing interests.
Acknowledgements
This work was supported by grants from the National Key R&D Program of China (Grants No. 2018YFA0208900, 2018YFE0205300, and 2021YFA0909900), the Strategic Priority Research Program of Chinese Academy of Sciences (Grant No. XDB36000000), the CAS Project for Young Scientists in Basic Research (Grant No. YSBR-010), the Beijing Natural Science Foundation of China (Grant No. Z200020), the Beijing Nova Program (Z201100006820031), the National Natural Science Foundation of China (Grants No. 32171384, 31800838, 31820103004, 31730032, and 51861145302), the Key Research Project of Frontier Science of the Chinese Academy of Sciences (Grant No. QYZDJ-SSW-SLH022), and the Innovation Research Group of National Natural Science Foundation (Grant No. 11621505).
Biographies

Jie Liang is a young research fellow at National Center for Nanoscience and Technology, and currently is a Ph.D. student of the Chinese Academy of Sciences. Her research mainly focuses on the Nanomaterial-based delivery vaccine used in tumor immunotherapy.

Guangjun Nie is a professor of the CAS Key Laboratory for Biomedical Effects of Nanomaterials and Nanosafety& CAS Center for Excellence in Nanoscience, National Center for Nanoscience and Technology of China. He has a long standing interest in cancer biology, blood physiology and pathophysiology. His most recent research activities generated a group of interdisciplinary works in nanobiology and nanomedicine fields.
Footnotes
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.fmre.2021.11.032.
Contributor Information
Xiao Zhao, Email: zhaox@nanoctr.cn.
Guangjun Nie, Email: niegj@nanoctr.cn.
Appendix. Supplementary materials
References
- 1.Tanyi J.L., Chiang C.L.L., Chiffelle J., et al. Personalized cancer vaccine strategy elicits polyfunctional T cells and demonstrates clinical benefits in ovarian cancer. NPJ Vaccines. 2021;6(1):36. doi: 10.1038/s41541-021-00297-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waldman A.D., Fritz J.M., Lenardo M.J. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat. Rev. Immunol. 2020;20(11):651–668. doi: 10.1038/s41577-020-0306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang J., Li Y., Nie G. Multifunctional biomolecule nanostructures for cancer therapy. Nat. Rev. Mater. 2021 doi: 10.1038/s41578-021-00315-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu J., Xu B., Tao J., et al. Microneedle-assisted, DC-targeted codelivery of pTRP-2 and adjuvant of paclitaxel for transcutaneous immunotherapy. Small. 2017;13(28) doi: 10.1002/smll.201700666. [DOI] [PubMed] [Google Scholar]
- 5.Martin J.D., Cabral H., Stylianopoulos T., et al. Improving cancer immunotherapy using nanomedicines: progress, opportunities and challenges. Nat. Rev. Clin. Oncol. 2020;17(4):251–266. doi: 10.1038/s41571-019-0308-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garcon N., Chomez P., Van Mechelen M. GlaxoSmithKline adjuvant systems in vaccines: concepts, achievements and perspectives. Expert Rev. Vaccines. 2007;6(5):723–739. doi: 10.1586/14760584.6.5.723. [DOI] [PubMed] [Google Scholar]
- 7.Sun Q., Barz M., De Geest B.G., et al. Nanomedicine and macroscale materials in immuno-oncology. Chem. Soc. Rev. 2019;48(1):351–381. doi: 10.1039/c8cs00473k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu X., Liu C., Zheng Z., et al. Vesicular antibodies: a bioactive multifunctional combination platform for targeted therapeutic delivery and cancer immunotherapy. Adv. Mater. 2019;31(17) doi: 10.1002/adma.201808294. [DOI] [PubMed] [Google Scholar]
- 9.Wculek S.K., Cueto F.J., Mujal A.M., et al. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020;20(1):7–24. doi: 10.1038/s41577-019-0210-z. [DOI] [PubMed] [Google Scholar]
- 10.Miao L., Li L., Huang Y., et al. Delivery of mRNA vaccines with heterocyclic lipids increases anti-tumor efficacy by STING-mediated immune cell activation. Nat. Biotechnol. 2019;37(10):1174–1185. doi: 10.1038/s41587-019-0247-3. [DOI] [PubMed] [Google Scholar]
- 11.Luo M., Wang H., Wang Z., et al. A STING-activating nanovaccine for cancer immunotherapy. Nat. Nanotechnol. 2017;12(7):648–654. doi: 10.1038/nnano.2017.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng K., Zhao R., Li Y., et al. Bioengineered bacteria-derived outer membrane vesicles as a versatile antigen display platform for tumor vaccination via Plug-and-Display technology. Nat. Commun. 2021;12(1):2041. doi: 10.1038/s41467-021-22308-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaparakis-Liaskos M., Ferrero R.L. Immune modulation by bacterial outer membrane vesicles. Nat. Rev. Immunol. 2015;15(6):375–387. doi: 10.1038/nri3837. [DOI] [PubMed] [Google Scholar]
- 14.Kuzmich N.N., Sivak K.V., Chubarev V.N., et al. TLR4 signaling pathway modulators as potential therapeutics in inflammation and sepsis. Vaccines. 2017;5(4):34. doi: 10.3390/vaccines5040034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yoo J.W., Irvine D.J., Discher D.E., et al. Bio-inspired, bioengineered and biomimetic drug delivery carriers. Nat. Rev. Drug Discov. 2011;10(7):521–535. doi: 10.1038/nrd3499. [DOI] [PubMed] [Google Scholar]
- 16.Schwechheimer C., Kuehn M.J. Outer-membrane vesicles from Gram-negative bacteria: biogenesis and functions. Nat. Rev. Microbiol. 2015;13(10):605–619. doi: 10.1038/nrmicro3525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qing S., Lyu C., Zhu L., et al. Biomineralized bacterial outer membrane vesicles potentiate safe and efficient tumor microenvironment reprogramming for anticancer therapy. Adv. Mater. 2020;32(47) doi: 10.1002/adma.202002085. [DOI] [PubMed] [Google Scholar]
- 18.Dudziak D., Kamphorst A.O., Heidkamp G.F., et al. Differential antigen processing by dendritic cell subsets in vivo. Science. 2007;315(5808):107–111. doi: 10.1126/science.1136080. [DOI] [PubMed] [Google Scholar]
- 19.Shakhar G., Lindquist R.L., Skokos D., et al. Stable T cell-dendritic cell interactions precede the development of both tolerance and immunity in vivo. Nat. Immunol. 2005;6(7):707–714. doi: 10.1038/ni1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Birkholz K., Schwenkert M., Kellner C., et al. Targeting of DEC-205 on human dendritic cells results in efficient MHC class II-restricted antigen presentation. Blood. 2010;116(13):2277–2285. doi: 10.1182/blood-2010-02-268425. [DOI] [PubMed] [Google Scholar]
- 21.Sartorius R., D'Apice L., Trovato M., et al. Antigen delivery by filamentous bacteriophage fd displaying an anti-DEC-205 single-chain variable fragment confers adjuvanticity by triggering a TLR9-mediated immune response. EMBO Mol. Med. 2015;7(7):973–988. doi: 10.15252/emmm.201404525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iijima M., Kadoya H., Hatahira S., et al. Nanocapsules incorporating IgG Fc-binding domain derived from Staphylococcus aureus protein A for displaying IgGs on immunosensor chips. Biomaterials. 2011;32(6):1455. doi: 10.1016/j.biomaterials.2010.10.057. -1164. [DOI] [PubMed] [Google Scholar]
- 23.Achmuller C., Kaar W., Ahrer K., et al. N(pro) fusion technology to produce proteins with authentic N termini in E. coli. Nat. Methods. 2007;4(12):1037–1043. doi: 10.1038/nmeth1116. [DOI] [PubMed] [Google Scholar]
- 24.Vila J.A., Ripoll D.R., Scheraga H.A. Atomically detailed folding simulation of the B domain of staphylococcal protein A from random structures. Proc. Natl. Acad. Sci. U. S. A. 2003;100(25):14812–14816. doi: 10.1073/pnas.2436463100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iijima M., Matsuzaki T., Yoshimoto N., et al. Fluorophore-labeled nanocapsules displaying IgG Fc-binding domains for the simultaneous detection of multiple antigens. Biomaterials. 2011;32(34):9011–9020. doi: 10.1016/j.biomaterials.2011.08.012. [DOI] [PubMed] [Google Scholar]
- 26.Carreno B.M., Magrini V., Becker-Hapak M., et al. Cancer immunotherapy. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science. 2015;348(6236):803–808. doi: 10.1126/science.aaa3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stronen E., Toebes M., Kelderman S., et al. Targeting of cancer neoantigens with donor-derived T cell receptor repertoires. Science. 2016;352(6291):1337–1341. doi: 10.1126/science.aaf2288. [DOI] [PubMed] [Google Scholar]
- 28.Akazawa Y., Saito Y., Yoshikawa T., et al. Efficacy of immunotherapy targeting the neoantigen derived from epidermal growth factor receptor T790M/C797S mutation in non-small cell lung cancer. Cancer Sci. 2020;111(8):2736–2746. doi: 10.1111/cas.14451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hsiue E.H., Wright K.M., Douglass J., et al. Targeting a neoantigen derived from a common TP53 mutation. Science. 2021;371(6533) doi: 10.1126/science.abc8697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen F., Zou Z., Du J., et al. Neoantigen identification strategies enable personalized immunotherapy in refractory solid tumors. J. Clin. Investig. 2019;129(5):2056–2070. doi: 10.1172/JCI99538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gerritzen M.J.H., Martens D.E., Wijffels R.H., et al. Bioengineering bacterial outer membrane vesicles as vaccine platform. Biotechnol. Adv. 2017;35(5):565–574. doi: 10.1016/j.biotechadv.2017.05.003. [DOI] [PubMed] [Google Scholar]
- 32.Sinha G. Listeria vaccines join the checkpoint frenzy. Nat. Biotechnol. 2014;32(12):1176–1177. doi: 10.1038/nbt1214-1176d. [DOI] [PubMed] [Google Scholar]
- 33.MacDiarmid J.A., Mugridge N.B., Weiss J.C., et al. Bacterially derived 400 nm particles for encapsulation and cancer cell targeting of chemotherapeutics. Cancer Cell. 2007;11(5):431–445. doi: 10.1016/j.ccr.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 34.Savas J.N., De Wit J., Comoletti D., et al. Ecto-Fc MS identifies ligand-receptor interactions through extracellular domain Fc fusion protein baits and shotgun proteomic analysis. Nat. Protoc. 2014;9(9):2061–2074. doi: 10.1038/nprot.2014.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feng B., Huang S., Ge F., et al. 3D antibody immobilization on a planar matrix surface. Biosens. Bioelectron. 2011;28(1):91–96. doi: 10.1016/j.bios.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 36.Hughes A., Clarson J., Tang C., et al. CML patients with deep molecular responses to TKI have restored immune effectors and decreased PD-1 and immune suppressors. Blood. 2017;129(9):1166–1176. doi: 10.1182/blood-2016-10-745992. [DOI] [PubMed] [Google Scholar]
- 37.Chen B., Khodadoust M.S., Olsson N., et al. Predicting HLA class II antigen presentation through integrated deep learning. Nat. Biotechnol. 2019;37(11):1332–1343. doi: 10.1038/s41587-019-0280-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bonsack M., Hoppe S., Winter J., et al. Performance evaluation of MHC class-I binding prediction tools based on an experimentally validated MHC-peptide binding data set. Cancer Immunol. Res. 2019;7(5):719–736. doi: 10.1158/2326-6066.CIR-18-0584. [DOI] [PubMed] [Google Scholar]
- 39.Delamarre L., Mellman I., Yadav M. Cancer immunotherapy. Neo approaches to cancer vaccines. Science. 2015;348(6236):760–761. doi: 10.1126/science.aab3465. [DOI] [PubMed] [Google Scholar]
- 40.Hollingsworth R.E., Jansen K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines. 2019;4:7. doi: 10.1038/s41541-019-0103-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tran E., Robbins P.F., Lu Y.C., et al. T-cell transfer therapy targeting mutant KRAS in cancer. N. Engl. J. Med. 2016;375(23):2255–2262. doi: 10.1056/NEJMoa1609279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bejarano L., Jordao M.J.C., Joyce J.A. Therapeutic targeting of the tumor microenvironment. Cancer Discov. 2021;11(4):933–959. doi: 10.1158/2159-8290.CD-20-1808. [DOI] [PubMed] [Google Scholar]
- 43.Aldea M., Andre F., Marabelle A., et al. Overcoming resistance to tumor-targeted and immune-targeted therapies. Cancer Discov. 2021;11(4):874–899. doi: 10.1158/2159-8290.CD-20-1638. [DOI] [PubMed] [Google Scholar]
- 44.Lin L., Kane N., Kobayashi N., et al. High-dose per fraction radiotherapy induces both antitumor immunity and immunosuppressive responses in prostate tumors. Clin. Cancer Res. 2021;27(5):1505–1515. doi: 10.1158/1078-0432.CCR-20-2293. [DOI] [PubMed] [Google Scholar]
- 45.Lu X., Horner J.W., Paul E., et al. Effective combinatorial immunotherapy for castration-resistant prostate cancer. Nature. 2017;543(7647):728–732. doi: 10.1038/nature21676. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.