

Abstract
Herein we describe a dual photocatalytic system to synthesize phenol–pyridinium salts using visible light. Utilizing both electron donor–acceptor (EDA) complex and iridium(III) photocatalytic cycles, the C─N cross-coupling of unprotected phenols and pyridines proceeds in the presence of oxygen to furnish pyridinium salts. Photocatalytic generation of phenoxyl radical cations also enabled a nucleophilic aromatic substitution (SNAr) of a fluorophenol with an electron-poor pyridine. Spectroscopic experiments were conducted to probe the mechanism and reaction selectivity. The unique reactivity of these phenol–pyridinium salts were displayed in several derivatization reactions, providing rapid access to a diverse chemical space.
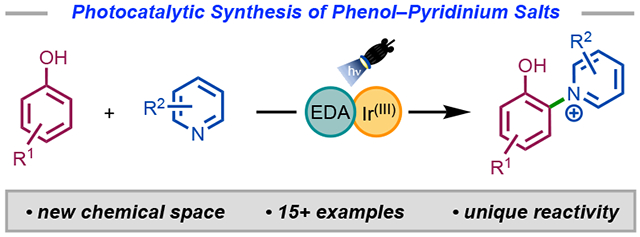
Graphical Abstract
INTRODUCTION
Pyridinium salts have emerged as a promising class of reactive intermediates to access uncharted chemical space and rapidly build molecular complexity.1,2 These salts have been utilized in two-electron processes such as pyridine functionalization, dearomatization reactions, as well as one-electron reactions for the transfer of functional groups (Figure 1).
Figure 1.

Synthetic utility of pyridinium salts for late-stage pyridine functionalization, dearomatization, and functional group transfer.
The ring-opening of pyridinium salts (Zincke imine) has been pivotal in synthesis of heterocyclic natural products3 and in late-stage drug scaffolds, especially for challenging regioselective pyridine functionalizations (e.g. loratadine, Figure 1).4,5 The design and synthesis of bespoke pyridinium activating groups has also allowed selective C─H functionalizations.6-10
Dearomatization of heterocyclic compounds has propelled the field of total synthesis into new horizons, granting synthetic chemists an unprecedented gateway to access a variety of saturated and partially unsaturated ring systems.11,12 Pyridines, in their free base form, do not readily undergo dearomatization and must be activated first by substitution on the nitrogen. From there, dearomatization reactions can be affected to access piperidines, which stand out as a particularly pertinent cyclic backbone in medicinal chemistry.13 Several research groups have successfully utilized such pyridinium dearomatizations in the total synthesis of natural products, particularly matrine-type lupin alkaloids,14 altemicidin alkaloids (Figure 1),15,16 and nuphar alkaloids.17
N-Functionalized pyridinium salts can engage in rich one-electron chemistries via photoredox and electrochemical catalysis to transfer functional groups via heterolytic (N-pyridyl radicals) or homolytic (generating C, N, or O radicals) cleavage (Figure 1).18,19,20 For example, Musacchio and co-workers recently implemented a photoreduction of N-alkoxy pyridinium salts as a means to generate alkoxyl radicals that are hydrogen atom transfer (HAT) reagents for benzylic amination.21 Watson and co-workers pioneered N-alkyl 2,4,6-triphenylpyridinium (Katritzky) salts to derivatize alkyl amines via pyridinium C─N bond cleavage.22
The formation of pyridinium salts is essential to access the chemistries described above. Generation of N-alkyl congeners is straightforward via N-alkylation23 or via Katritzky condensation with pyriliums.24 The latter method can also be employed to generate N-aryl pyridiniums from anilines, which are typically more difficult to generate.2 Alternate routes include nucleophilic aromatic substitution (SNAr) reaction of electron-poor haloarenes with pyridines to form Zincke salts.25 However, the Zincke salts are highly electrophilic and prone to ring-opening reactions resulting in a variety of rearrangement products.3 Direct C─H pyridination of arenes is possible by means of electrochemical oxidation of an arene to its radical cation and subsequent nucleophilic addition of the pyridine nitrogen; however, the pyridinium hydrolyzes to provide the corresponding aniline.26
Recent advances have leveraged photoredox catalysis and hypervalent iodine reagents to afford isolable N-aryl pyridinium salts of more electron rich arenes via C─H functionalization and SNAr. These approaches largely fall into two categories. In Scheme 1A, the pyridine component is the electrophile and the arene acts as the nucleophile. In Scheme 1B and Scheme 1C, the pyridine is the nucleophile and the arene serves as the electrophile. Notably, both approaches result in a C─H functionalization.
Scheme 1. Previous and Current Approach to N-Aryl Pyridinium Salts.

Photochemical generation of electrophilic N-pyridyl radical cations from the pyridinium N-OTf furnishes N-aryl pyridinium salts in high yields across a broad range of functional groups and structural complexity (Scheme 1A).27,28 The N-OTf reagent was prepared immediately prior to reaction from pyridine N-oxide under inert conditions, due to its limited stability under air. Addition of the arene nucleophile to the highly electrophilic N-pyridyl radical cation results in a mixture of regioisomers, which required reverse-phase column chromatography for isolation of pure products. A variety of 5- and 6-membered arenes nucleophiles with diverse electronic character were utilized; however, no phenols were described. Ritter and co-workers did describe the C(sp2)─H pyridination of phenyl acetate, with in situ deprotection giving rise to the phenol–pyridinium adducts, but three regioisomers were obtained in ~20% yields.27
Nucleophilic activation of the pyridinium can also be achieved by generation of in situ hypervalent iodine (N-HVI) reagents (Scheme 1B).29 With a broad tolerance to electronic nature of the pyridine (electron-poor and -rich), a range of aryl ethers were coupled to form N-aryl ether pyridinium salts in good yields. Methyl substituents were incompatible in this reaction as benzylic functionalization occurred. Furthermore, phenols are incompatible with this functionalization because they coordination with hypervalent iodine, which interferes with reactivity.
Following the seminal findings of Yoshida et al.,26 another report using an arene electrophile in pyridine N-arylation was disclosed by Sanford and co-workers (Scheme 1C). Two photocatalytic methods under anaerobic or aerobic conditions allowed either SNAr or oxidative C(sp2)─H pyridination, respectively.30 A plethora of electron-rich arenes, particularly aryl ethers proved effective, providing the products in good yields, but with varying levels of regioselectivity. Several reports of insertion of diarylamines into the C─H bonds of electron-rich arenes using both photochemical31 and electrochemical methods32 have proposed similar reaction pathways involving an arene radical cation. Notably, phenol and heterocyclic arenes were found to be incompatible as were substrates containing thiols or alcohols. In further work, the Sanford group found that the photocatalyst was not needed if UVA/B irradiation was used, which activated the substrates via an electron donor–acceptor (EDA) complex.33 This finding permitted the use of electron-rich and neutral pyridine nucleophiles, surpassing the limitations of previous research that solely focused on pyridine. However, electron-poor pyridines and phenols could not be utilized.
Reasoning that phenoxyl radical cation electrophiles could be generated photocatalytically, we sought to explore the functionalization with pyridine nucleophiles. The coupling of phenols with electron-poor species has been an underexplored area in the literature. In this study, we illustrate that a combined photocatalytic and EDA system is superior in the C(sp2)─H pyridination of unprotected phenols using visible light (Scheme 1D). With an iridium photocatalyst and under aerobic conditions, electron-poor/neutral pyridines are coupled with electron-rich phenols in moderate to good yields. This transformation allows for facile C─N bond formation without the need for any halides, intense UV light, in situ preparation of sensitive reagents, or column chromatography. Spectroscopic experiments support a mechanism in which a phenol is oxidized by the photocatalyst to a phenoxyl radical cation and subsequently intercepted by a nucleophilic pyridine. The concurrent formation of an EDA complex serves as an additional source of phenoxyl radical cation through SET. The practical significance of these findings becomes apparent through their application in a number of late-stage functionalizations.
RESULTS AND DISCUSSION
Reaction Optimization.
Our initial efforts focused on using a phenoxyl radical in a Minisci-type radical addition to forge a new C─C bond with pyridine. Previous work from our group detailed the photocatalytic oxidative coupling of phenols, thus supporting generation of phenoxyl radicals.34 Starting with an electron-poor pyridine 1a and an electron-rich phenol 2a, we envisioned Brønsted acid (trifluoroacetic acid) activation of 1a to facilitate the addition of 2a under oxidative photochemical conditions (Table 1).
Table 1.
Reaction Development and Optimizationa

| ||
|---|---|---|
| entry | change to conditions | yield 3a (%)b |
| 1 | none | 37 |
| 2 | Ru(bpz)PF6 instead of Ir(ppy)3 | 19 |
| 3 | MesAcrBF4 instead of Ir(ppy)3 | 10 |
| 4 | MeOH instead of HFIP | 7 |
| 5 | MeCN instead of HFIP | 34 |
| 6 | HBF4 (aq.) instead of TFA | 0 |
| 7 | AcOH instead of TFA | 9.5 |
| 8 | LiClO4 instead of TFA | 16 |
| 9 | 1a (2 equiv), O2 instead of air | 57 (54) |
| 10 | 2a (2 equiv), O2 instead of air | 56 |
Reaction conditions: 1a (0.25 mmol, 1.0 equiv), 2a (0.25 mmol), Ir(ppy)3 (0.0075 mmol, 3.0 mol%), TFA (0.25 mmol, 1.0 equiv), HFIP (2.0 mL, 0.10 M), air, 427 nm Kessil, 35 °C, 16 h.
Yields as judged by 1H-NMR spectroscopy with CH2Br2 as internal standard. Yield in parentheses corresponds to the isolated yield (IY) of the optimized condition after trituration with Et2O.
The reaction yielded the desired coupled product mass by LCMS, but NMR analysis did not support the desired C─C bond formation. Instead, the phenol–pyridinium salt 3a was obtained in a 37% assay yield (AY) (Table 1, entry 1). While trifluoromethyl radicals (ω = 1.672 eV)35 can be engaged in nucleophilic radical addition to pyridinium,36 the phenoxyl radical is not sufficiently nucleophilic (ω = 2.236 eV).35 Therefore, we discovered that upon oxidation, the phenol undergoes a polarity reversal, transforming into an electrophilic radical cation which can undergo nucleophilic addition of pyridine to form a new C─N bond.
This led us to screen a series of photocatalysts, solvents, additives, and oxidants to optimize the transformation (full optimization results found in Supporting Information). A screening of photocatalysts revealed that Ir(ppy)3 (Ir(III)*/Ir(II) = +0.31 V vs. SCE)37 was the most successful (Table 1, entries 2–3). Photocatalysts with lower oxidation potentials (Eox) were found to perform better in the reaction (see the Supporting Information). Replacing hexafluoroisopropanol (HFIP) with an alternative alcoholic solvent, MeOH, resulted in diminished yields (entry 4). While MeCN produced yields similar to HFIP (entry 5), it proved to be less effective when considering the entire substrate scope. An evaluation of reaction additives was undertaken to probe the activation of the pyridine and counterion stability. Other Brønsted acids, AcOH and HBF4, along with counterion sources, LiClO4, led to inferior yields (entries 6–8). Product conversion was only detected for acid additives with pKa, values ranging from 0 to ~5 (Figure 2). The use of strong acids or increased amounts of TFA eroded the reactivity due to the complete protonation of the pyridine (see Mechanism section). Optimal conditions were achieved when using 2 equiv of 1a under a headspace of O2 to afford 3a in 57% assay yield and 54% isolated yield after trituration with Et2O (entry 9). With an excess of pyridine, stronger acids like HBF4 can be employed to obtain comparable yields. Although achieving a similar assay yield, employing an excess of phenol 2a posed challenges during purification as phenol coupling products were observed (entry 10).
Figure 2.

1H-NMR assay yield 3a (%) vs. pKa acid additive (H2O) (see Supporting Information for full table).
Reaction Controls.
In order to probe the specific role of each reagent and bolster our understanding of the mechanism, we embarked on a series of control reactions with our optimized conditions (Table 2).
Table 2.
Reaction Control and Mechanistic Screena

| ||
|---|---|---|
| entry | change to conditions | yield 3a (%)b |
| 1 | none | 57 |
| 2 | Ar instead of O2 | 4 |
| 3 | No 427 nm Kessil | 0 |
| 4 | No Ir(ppy)3 | 15 |
| 5 | No TFA | trace |
| 6 | TEMPO (5 equiv) | trace |
| 7 | BHT (5 equiv) | 11 |
Reaction conditions: 1a (0.50 mmol, 2.0 equiv), 2a (0.25 mmol), Ir(ppy)3 (0.0075 mmol, 3.0 mol%), TFA (0.25 mmol, 1.0 equiv), HFIP (2.0 mL, 0.10 M), O2 (1 atm), 427 nm Kessil, 35 °C, 16 h.
yields as judged by 1H-NMR spectroscopy with CH2Br2 as internal standard.
When the reaction was conducted under an argon atmosphere, poor conversion was observed, providing evidence for an oxidative quenching photocatalytic process (entry 2). For our iridium-catalyzed system, we would expect a Ir(III)/Ir(III)*/Ir(IV) cycle, where O2 is needed to reoxidize the excited photocatalyst. In the absence of light, no coupling product was generated (entry 3). Without any photocatalyst present, a low level of background reactivity (15% assay yield) was obtained, providing initial evidence toward an electron donor–acceptor complex in the catalytic cycle (entry 4) (see Mechanism section). Eliminating TFA from the reaction mixture resulted in only minimal product conversion (entry 5). The presence of the acid additive is imperative for both pyridine protonation and the generation of the necessary salt counterion, thereby driving the equilibrium toward the desired product. The addition of common radical inhibitors, specifically 2,2,6,6-tetramethylpiperidinooxy (TEMPO) and butylated hydroxytoluene (BHT), into the reaction mixture significantly decreased the product yield (entries 6–7). Even though no TEMPO-trapped adducts were detected in the HRMS analysis, the observed inhibition in product conversion provides support for the involvement of a radical-driven chemical process.
Pyridine Scope.
Equipped with these optimized conditions, we investigated the chemical scope of this reaction using various pyridine derivatives (Scheme 2).
Scheme 2. Substrate Scope – Pyridine Explorationa.

aReaction conditions: 1a–j (0.50 mmol, 2.0 equiv), 2a (0.25 mmol), Ir(ppy)3 (0.0075 mmol, 3.0 mol%), TFA (0.25 mmol, 1.0 equiv), HFIP (2.0 mL, 0.10 M), O2 (1 atm), 427 nm Kessil, 35 °C, 16 h. Isolated yields shown. Yield in parentheses was performed on a 2.6 mmol scale. NR = no reaction.
Electron-deficient and neutral pyridines displayed good reactivity, resulting in the formation of phenol–pyridinium salts in isolated yields reaching up to 66%. Hammett sigma constants of monosubstituted pyridines correlated well with pyridinium yield, favoring more withdrawing substituents (see the Supporting Information). The reaction demonstrated scalability, resulting in a 41% isolated yield of 3a when conducted on a 2.6 mmol scale. Pyridines bearing electron-withdrawing groups, including halides, at the C3, C4, or C5 positions, readily generated the coupled products in good yields (3a–3f). These less nucleophilic substrates were unreactive in other photochemical SNAr methods,33 although recently enabled by hypervalent iodine.29 Unsubstituted azines exhibited limited compatibility, with pyridine (3g) and pyrazine (3h) affording only 19% and 18% isolated yields, respectively. C2-substituted pyridines (3i) were unreactive to our conditions, as similarly observed by Wengryniuk and co-workers.29 With their EDA system, Sanford et al. were successfully able to couple 2-methylpyridine, albeit a para-substituted electrophile (4-phenylbenzene) was utilized.33 Thus, steric hinderance appears to be a significant limiting factor. Despite their increased nucleophilicity, electron-rich pyridines (3j) were unsuccessful. (see the Supporting Information for other failed pyridines and Mechanism section for further scope discussion).
Phenol Scope.
The substitution pattern and electronic nature of the phenol had a significant impact on the product formation (Scheme 3). 2,4-Disubstituted phenols were effective when the para-position contained an electron-donating (OMe) group (3a, 3k, 3l, 3m, 3n). Under these constraints, electron-poor aryls (3n) were even tolerated, albeit in lower yield (31%). Tetrasubstituted 2,6-dimethylhydroquinone (2o) underwent pyridination to form 3o in a 39% isolated yield. Reaction with 2-tert-butylhydroquinone (2p) resulted in both pyridination and intramolecular cyclization to afford 3p in a 53% yield. Surprisingly, hydroquinone (2q) did not undergo competitive oxidation to benzoquinone, instead forming desired 3q in a 72% isolated yield. In prior C─H aminations of phenols, hydroquinone (2q) failed to produce product.38 Aryl ethers (3t) and halides (3u) were unreactive under our conditions. Electron-neutral 2,4-disubstituted phenols underwent competitive oxidative dimerization and did not afford product (3s).
Scheme 3. Substrate Scope – Phenol Explorationa.

aReaction conditions: 1e (0.50 mmol, 2.0 equiv), 2a–u (0.25 mmol), Ir(ppy)3 (0.0075 mmol, 3.0 mol%), TFA (0.25 mmol, 1.0 equiv), HFIP (2.0 mL, 0.10 M), O2 (1 atm), 427 nm Kessil, 35 °C, 16 h. Isolated yields shown. Yield in parentheses was performed on a 2.6 mmol scale. NR = no reaction.
The use of 2,6-disubstituted phenols (3r) of varying electronic character only formed trace amounts of the desired product (see the Supporting Information for rest of failed phenol substrates). In our previous studies on phenolic couplings, we calculated that open para-positions are much more nucleophilic than ortho-positions.39 The ortho-positions are therefore more electrophilic and more susceptible to attack by a nucleophile. Recent work on copper-catalyzed,40 photochemical,38,41 and electrochemical.42 C─H amination of phenols has reported exclusive ortho selectivity. In these works, the authors invoke aminyl and phenoxy radical intermediates and bond formation mediated by radical-radical cross-coupling. Interestingly, for Xia et al. the addition of TEMPO did not decrease cross-coupling yields and molecular oxygen was an insufficient oxidant.41 In this work, Stern-Volmer analysis revealed that diarylamines were more efficient quenchers than phenols. In a photoelectrocatalytic approach aimed at C─H amination of aryl ethers, Hu and colleagues highlighted an extensive hydrogen-bonding network formed with HFIP, aryl ether, and nitrogen nucleophile as the dominant factor influencing their ortho selectivity.43 A similar phenomenon could explain the observed regioselectivity in our study.
Photocatalytic SNAr with para-Fluorophenol.
Notably, the limitation of ortho-selectivity could be overcome with para-fluoro phenol 2v, which underwent selective SNAr with 4-(trifluoromethyl)pyridine (1a) to afford phenol–pyridinium salt 3v in a 46% yield after trituration (Scheme 4). Under an argon atmosphere, this reactivity was significantly hindered, leading to only a trace amount of product detected. This finding stands in contrast to work from Sanford et al. where SNAr reactivity required argon atmospheres as C─H pyridination occurred under aerobic conditions.30 Without O2, the IrIII* is not oxidizing enough (IrIII* → IrII = +0.31 V), but under O2, IrIV → IrIII = +0.77 V, it is.37 In a departure from the C─H pyridination described above where no reaction occurred without TFA, here reaction still proceeded albeit in lower yield (23%). We hypothesize that fluoride ions formed during the reaction serve as the counterion source in the absence of an acid. Addition of TFA promotes EDA complexation which aids in the oxidation of the phenol (to radical cation) to increase the product yield (46%).
Scheme 4. SNAr of Fluorophenol with Electron-Poor Nucleophilesa.

aReaction conditions: 1a (0.50 mmol, 2.0 equiv), 2v (0.25 mmol), Ir(ppy)3 (0.0075 mmol, 3.0 mol%), TFA (0.25 mmol, 1.0 equiv), HFIP (2.0 mL, 0.10 M), O2 (1 atm), 427 nm Kessil, 35 °C, 16 h. Isolated yield shown. *Yield judged by 1H-NMR spectroscopy with CH2Br2 as internal standard. F− pyridinium salt formed.
In our conditions, the photogenerated phenoxyl radical cation is hypothesized to undergo nucleophilic substitution ipso to fluorine even with very weak nucleophiles (see the Supporting Information for plausible mechanism). We have thus discovered an inverse electron-demand SNAr reaction, in which an electron-rich phenol functions as the electrophilic arene, while an electron-poor pyridine acts as the nucleophile, all under acidic conditions. This result allows the use of electron-neutral phenols and 2,6-disubstitution patterns, both of which were unreactive in the direct oxidative C(sp2)─H pyridination described above. This approach is reminiscent of recent work by Knowles and co-workers, who leveraged the electrophilic nature of the phenoxyl radical to facilitate challenging SNAr reactions involving carboxylates and halophenols.44 Their investigation, supported by transient absorption spectroscopy, elucidated the long-lived lifetime of the phenoxyl radical and its electron-with-drawing properties, suggesting an alkali metal-mediated concerted mechanism following stepwise proton-transfer/electron-transfer (PT/ET) of the phenol. While electron-deficient carboxylates/benzoates exhibited competence in the Knowles work (N ~ 7–9 eV), electron-poor pyridines possess lower nucleophilicity (N ~ 2 eV) and are thus more difficult to engage in such reactivity (see the Supporting Information).
Mechanistic Studies.
To gain insight into the mechanism and the reaction limitations, spectroscopic experiments were conducted. Stern-Volmer fluorescence quenching analysis (Figure 3A) was undertaken to investigate the extent of electron transfer between Ir(ppy)3 and each reactant of a C(sp2)─H pyridination. Phenol 2a was found to most effectively quench the excited state photocatalyst, Ir(ppy)3*, with a Stern-Volmer constant (kSV) of 18.138 M−1 in HFIP. Quenching was nearly absent for the pyridine and pyridinium 1a species. As a result, it is likely that the photocatalyst selectively oxidizes the phenol within the reaction mixture.
Figure 3.

A) Steady-state Stern-Volmer quenching experiments with the excited state of Ir(ppy)3 in HFIP (λex = 320 nm, λmax ~ 362 nm). kSV refers to the Stern-Volmer constant. B) UV-Vis analysis of each respective starting material and a 1:1 mixture (λrange = 275-600 nm). C) Calculated phenol nucleophilicity parameters (site nucleophilicity in brackets in eV, N = global nucleophilicity) from ref 34. Measured oxidation potentials (measured in HFIP, vs. Ag/AgNO3) were sourced from ref 45. Product isolated yields shown in parentheses.
Given the electronic bias of the reaction for electron-poor pyridines and electron-rich phenols, we conducted UV-Vis analysis to investigate the potential formation of an EDA complex46 (Figure 3B). For a mixture of 2,4-dimethoxyphenol (2a) and 4-trifluoro-methylpyridine (1a) in the presence of TFA, a distinctive bathochromic shift was observed – indicative of the formation of an EDA complex, as highlighted by the green line.33 The observed 15% assay yield in the absence of Ir(ppy)3 is most likely attributed to this exciplex formation. In the case of the unreactive substrate 4-methoxypyridine (1j), the lack of a bathochromic shift suggests that EDA complexation is presumably a crucial factor for the success of the reaction. As such, the pyridine serves two separate roles: as a nucleophile for which electron-rich pyridines should be favorable and as an EDA activator for which electron-poor pyridines are required. These two trends counter each other with the experimental results indicating that EDA complexation is more important for net reactivity. However, for phenols that resulted in trace product formation (2r, 2s), EDA complex formation was still observed, indicating that another factor governs the phenol reactivity (see the Supporting Information).
Phenols with high site-nucleophilicity (2r) (0.64 eV) are less electrophilic and therefore less susceptible to nucleophilic addition from pyridine (Figure 3C). Also, the elevated oxidation potential (Eox = 0.64 V) of 2r is not expected to influence the reactivity significantly, given that Ir(ppy)3 is sufficiently oxidizing (Ir(IV)/Ir(III) = +0.77 V).37 While possessing a similar site nucleophilicity (0.31 eV) and oxidation potential (0.47 V) to 2a, phenol 2s has a notably lower global nucleophilicity (3.48 eV).34,45 Despite EDA formation occurring, the SET process may be more inefficient with a less electron-rich donor, especially given the lower energy light (427 nm) employed.
Based upon the above experimental results alongside the control experiments and reaction limitations, we propose a dual-catalytic cycle involving both an iridium-based photocatalyst and EDA complex (Scheme 5). Following a typical oxidative quenching photocatalytic cycle, Ir(III) is excited by blue light (427 nm) to form the excited Ir(III)* which is oxidized by oxygen to form the potent oxidant Ir(IV) (photocatalytic cycle). This species can oxidize phenol C to form Ir(III) and phenoxyl radical cation F which facilitates nucleophilic addition of pyridine A (F to G). Oxidation by the perhydroxyl radical (HOO•) affords the phenol–pyridinium salt H. Protonation of A to form pyridinium B enables EDA complexation with C (EDA complex cycle). Single-electron transfer of EDA complex D then serves as an additional source of radical cation F and regeneration of pyridine A after subsequent oxidation (E to A). Pyridyl and phenoxyl radical-radical coupling is unlikely due to the polarity mismatch (see Supporting Information for alternate mechanisms). In the literature there is also no evidence of pyridyl radical cation formation via HAT of pyridinium.
Scheme 5. Proposed Mechanism of Phenol Oxidative C─H Pyridination.

Phenoxyl radical cation intermediates have previously been proposed en route to unsymmetrical phenol dimers using stoichiometric persulfate. Phenol radical cations [pKa(C6H5OH•+) ~ −2.0]47 are thermodynamically prone to deprotonation by the superoxide anion (O2•−)) [pKa(HOO•) = 4.9].48 Unproductive deprotonation would result in the formation of neutral phenoxyl radicals, which would likely undergo dimerization (a main reaction byproduct).50 4-Trifluoromethyl pyridinium [pKa(4-CF3pyr-H+) = 2.26]51 could serve as an acidic scavenger of O2•− to avoid phenoxyl deprotonation. Disproportionation with HOO• can also consume the superoxide anion.52,53 Additionally, HFIP has been found to have a unique ability to stabilize radical cation intermediates.54 With strong hydrogen bond interactions, HFIP has been shown to form a negatively charged [HFIP•O2]− adduct after single electron transfer of molecular oxygen to produce singlet oxygen.55 The presence of singlet oxygen was supported by a trapping experiment with 9,10-dimethylanthracene and the generation of H2O2 was substantiated with a KI peroxide test of the aqueous layer of a successful reaction (see the Supporting Information).
Derivatization.
We next sought to explore the synthetic utility of these phenol–pyridinium salts (Scheme 6). Serving as a synthon for piperidine derivatives, we were able to fully hydrogenate 3a with Pd/C under atmospheric pressure of hydrogen in a 54% yield (Scheme 6A). With the employment of a mild base and heat, hydrolysis provided N-phenol 2-pyridone 4b in a 39% yield (Scheme 6B). This method provides an additional pathway to the N-arylation of pyridones, complementing established Chan-Lam, Buchwald-Hartwig, and other cross-couplings. Previously, Ritter and co-workers reported photochemical SNAr to access 2-pyridones from 2-halo pyridinium salts.56 Semi-reduced 2-pyridone 4c was obtained in a 50% yield with NaBH4/NaOH/H2O (Scheme 6C).
Scheme 6. Pyridinium Salt Derivatizationa.

aReaction conditions: A) 3a (0.042 mmol), Pd/C (0.0042 mmol, 10 mol%), H2 (1 atm), EtOAc (2.0 mL, 20 mM), rt. B) 3a (0.12 mmol), K2CO3 (0.90 mmol, 7.5 equiv), MeCN (1.50 mL, 80 mM), 50 °C. C) 3a (0.050 mmol), NaBH4 (0.20 mmol, 4.0 equiv), NaOH (0.20 mmol, 4.0 equiv), H2O (1.3 mL, 35 mM), rt.
CONCLUDING REMARKS
In summary, this report enables the synthesis of previously unreported phenol–pyridinium salts utilizing low-energy visible light redox catalysis. By harnessing both EDA complexation and transition-metal photocatalysts, phenols and pyridines are selectively coupled to yield pyridinium salts with moderate to satisfactory yields. A fluorophenol is also engaged in photochemical SNAr allowing the addition of pyridine derivatives that are weakly nucleophilic due to the presence of electron-withdrawing groups. Mechanistic and spectroscopic experiments offer substantial support for the proposed mechanism of C(sp2)─H and SNAr pyridination, which revolves around the photochemical generation of phenoxyl radical cations. These mild reaction conditions facilitate the construction of valuable synthetic intermediates, as exemplified in several functionalization strategies. Building upon established findings in pyridinium redox chemistry, our ongoing research efforts are focused on using these phenol–pyridinium salts to generate challenging phenoxyl radicals and expanding the scope of SNAr-type reactions.
EXPERIMENTAL SECTION
General Information.
NMR spectra (1H, 13C{1H}, 19F) were recorded on a Fourier transform NMR spectrometer at 298 K at 400, 500, or 600 MHz (for 1H), 101, 126, or 151 MHz (for 13C{1H}), and 376 MHz (for 19F). Chemical shifts are reported relative to the solvent resonance peak (CDCl3: δ = 7.26 ppm, DMSO-d6: δ = 2.50 ppm, CD3CN: δ = 1.94 ppm) for 1H NMR spectra and (CDCl3: δ = 77.16 ppm, DMSO-d6: δ = 39.52 ppm, CD3CN: δ = 1.32 ppm) for 13C{1H} spectra. NMR spectra multiplicities are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br s = broad singlet, dd = doublet of doublets, did = doublet of doublet of doublets, dt = doublet of triplets, td = triplet of doublets, m = multiple, coupling constants (J, reported in Hz), and number of protons. 13C{1H} and 19F spectra were both fully proton decoupled. 19F splitting was observed in the 13C{1H} spectra: ─CF3 (quartets), ─F (doublets). Accurate mass measurement analyses were conducted on either a Waters GCT Premier, time-of-flight, GCMS with electron ionization (EI), or an LCT Premier XE, time-of-flight, LCMS with electrospray ionization (ESI). Unless otherwise noted, yields refer to isolated material based on product purity (≥95%) determined by 1H-NMR spectroscopy following purification (trituration or column chromatography). For light irradiation, a 427 nm Kessil PR160L lamp (100% intensity, max 45W, 7 cm from wall of flask, no filters) was employed with a commercial blade fan for cooling. Borosilicate glass vials were used for reaction vessels.
General Procedure for the Synthesis of 3a–v.
An 8 mL scintillation vial equipped with a stir bar was charged with phenol derivative (0.25 mmol, 1.0 equiv), HFIP (2.0 mL, 0.10 M), pyridine derivative (0.50 mmol, 2.0 equiv), TFA (0.25 mmol, 1.0 equiv), and Ir(ppy)3 (0.0075 mmol, 3.0 mol%). The vial was then capped, sealed with parafilm, and sparged by bubbling oxygen through the solvent via a needle for 10 min prior to irradiation with a 427 nm Kessil lamp (~35 °C) for 16 h (see Figure S2). After 16 h, the reaction was filtered through a Celite™ plug into a pre-weighed 20 mL scintillation vial and concentrated in vacuo until a minimal amount of solvent remained. To this mixture was then slowly added Et2O to precipitate the pyridinium salt product and the liquid was then decanted (trituration). This process was repeated 3-5 more times until the liquid was mostly clear to afford pure (>95%) pyridinium salt product as determined by 1H-, 13C{1H}-, and 19F-NMR spectroscopy.
4-Trifluoromethyl-1-(2-hydroxy-3,5-dimethoxyphenyl)pyridin-1-ium trifluoroacetate (3a).
The product 3a was obtained as an amorphous brown solid (56 mg, 54% yield); 1H-NMR (500 MHz, DMSO-d6) δ 10.00 (s, 1H), 9.56 (d, J = 6.2 Hz, 2H), 8.82 (d, J = 6.2 Hz, 2H), 6.97 – 6.92 (m, 2H), 3.92 (s, 3H), 3.77 (s, 3H); 13C{1H}-NMR (101 MHz, CD3CN) δ 160.8 (q, J = 33.4 Hz), 154.3, 152.2, 149.6, 145.9 (q, J = 36.6 Hz), 135.9, 131.5, 125.8 (q, J = 3.1 Hz), 122.3 (q, J = 282.5 Hz), 118.3 (q, J = 296.3 Hz), 104.1, 101.7, 57.3, 56.8; 19F-NMR (376 MHz, CD3CN) δ −65.9, −75.7; HRMS (ESI-TOF) m/z = 300.0848 calcd for C14H13F3NO3+ [M-CO2CF3]+, found 300.0845.
4-Cyano-1-(2-hydroxy-3,5-dimethoxyphenyl)pyridin-1-ium trifluoroacetate (3b).
The product 3b was obtained as an amorphous dark brown solid (61 mg, 66% yield); 1H-NMR (500 MHz, DMSO-d6) δ 10.00 (s, 1H), 9.51 (d, J = 6.9 Hz, 2H), 8.86 (d, J = 6.8 Hz, 2H), 6.97 – 6.88 (m, 2H), 3.91 (s, 3H), 3.77 (s, 3H); 13C{1H}-NMR (151 MHz, DMSO-d6) δ 157.7 (q, J = 32.0 Hz), 152.3, 149.6, 147.9, 134.1, 130.7, 130.0, 128.0, 117.2 (q, J = 259.0 Hz), 114.8, 103.1, 101.5, 56.6, 56.0; 19F-NMR (376 MHz, DMSO-d6) δ −73.4; HRMS (ESI-TOF) m/z = 257.0926 calcd for C14H13N2O3+ [M-CO2CF3]+, found 257.0942.
4-Acetyl-1-(2-hydroxy-3,5-dimethoxyphenyl)pyridin-1-ium trifluoroacetate (3c).
The product 3c was obtained as an amorphous dark brown solid (46 mg, 47% yield); 1H-NMR (400 MHz, CD3CN) δ 9.10 (d, J = 6.3 Hz, 2H), 8.45 (d, J = 6.3 Hz, 2H), 6.79 (d, J = 2.7 Hz, 1H), 6.66 (d, J = 2.6 Hz, 1H), 3.87 (s, 3H), 3.78 (s, 3H), 2.75 (s, 3H); 13C{1H}-NMR (101 MHz, CD3CN) δ 196.0, 160.7 (q, J = 33.6 Hz), 154.3, 151.9, 150.3, 148.9, 135.8, 131.4, 126.7, 118.1 (q, J = 294.9 Hz), 103.8, 101.7, 57.3, 56.8, 27.7; 19F-NMR (376 MHz, CD3CN) δ −75.8; HRMS (ESI-TOF) m/z = 274.1079 calcd for C15H16NO4+ [M-CO2CF3]+, found 274.1093.
3-Methoxycarbonyl-1-(2-hydroxy-3,5-dimethoxyphenyl)pyridin-1-ium trifluoroacetate (3d).
The product 3d was obtained as an amorphous dark brown solid (53 mg, 53% yield); 1H-NMR (500 MHz, DMSO-d6) δ 9.87 (s, 1H), 9.57 (s, 1H), 9.39 (d, J = 5.9 Hz, 1H), 9.15 (d, J = 8.2 Hz, 1H), 8.42 (t, J = 6.9 Hz, 1H), 6.96 (d, J = 2.6 Hz, 1H), 6.92 (d, J = 2.6 Hz, 1H), 3.99 (s, 3H), 3.91 (s, 3H), 3.77 (s, 3H); 13C{1H}-NMR (126 MHz, DMSO-d6) δ 162.1, 152.3, 149.8, 149.7, 147.3, 146.3, 134.3, 130.2, 129.7, 128.2, 102.8, 101.6, 56.5, 55.9, 53.6; Note: Trifluoroacetate 13C{1H} signals were not observed but are seen in the 19F-NMR; 19F-NMR (376 MHz, DMSO-d6) δ −73.7; HRMS (ESI-TOF) m/z = 290.1028 calcd for C15H16NO5+ [M-CO2CF3]+, found 290.1041.
3-Fluoro-1-(2-hydroxy-3,5-dimethoxyphenyl)pyridin-1-ium trifluoroacetate (3e).
The product 3e was obtained as an amorphous light brown solid (51 mg, 56% yield); 1H-NMR (500 MHz, DMSO-d6) δ 9.93 (s, 1H), 9.70 – 9.65 (m, 1H), 9.14 (d, J = 6.0 Hz, 1H), 8.85 (t, J = 7.5 Hz, 1H), 8.44 – 8.36 (m, 1H), 6.97 – 6.91 (m, 2H), 3.91 (s, 3H), 3.78 (s, 3H); 13C{1H}-NMR (126 MHz, DMSO-d6) δ 158.7 (q, J = 250.6 Hz), 157.8 (d, J = 29.6 Hz), 152.2, 149.6, 144.1 (d, J = 3.1 Hz), 137.1, 136.7 (d, J = 17.7 Hz), 134.3 (d, J = 20.3 Hz), 130.0, 129.1 (d, J = 8.7 Hz), 120.9 (q, J = 252.4 Hz), 102.8, 101.6, 56.6, 56.0; 19F-NMR (376 MHz, DMSO-d6) δ −73.4, −117.1; HRMS (ESI-TOF) m/z = 250.0879 calcd for C13H13FNO3+ [M-CO2CF3]+, found 250.0876.
3-Bromo-5-(methoxycarbonyl)-1-(2-hydroxy-3,5-dimethoxyphenyl)pyridin-1-ium trifluoroacetate (3f).
The product 3f was obtained as an amorphous light brown solid (36 mg, 30% yield); 1H-NMR (600 MHz, DMSO-d6) δ 9.92 (s, 1H), 9.85 (s, 1H), 9.60 (s, 1H), 9.37 (s, 1H), 6.97 (d, J = 2.8 Hz, 1H), 6.92 (d, J = 2.8 Hz, 1H), 3.98 (s, 3H), 3.91 (s, 3H), 3.77 (s, 3H); 13C{1H}-NMR (151 MHz, DMSO-d6) δ 161.2, 152.2, 150.7, 149.5, 148.5, 146.3, 134.3, 130.2, 129.6, 122.3, 103.1, 101.6, 56.6, 55.9, 53.8; Note: Trifluoroacetate 13C{1H} signals were not observed but are seen in the 19F-NMR; 19F-NMR (376 MHz, DMSO-d6) δ −73.4; HRMS (ESI-TOF) m/z = 368.0134 calcd for C15H15BrNO5+ [M-CO2CF3]+, found 368.0127.
1-(2-Hydroxy-3,5-dimethoxyphenyl)pyridin-1-ium trifluoroacetate (3g).
The product 3g was obtained as an amorphous light brown solid (16 mg, 19% yield); 1H-NMR (500 MHz, CD3CN) δ 8.87 (d, J = 5.8 Hz, 2H), 8.63 (t, J = 7.7 Hz, 1H), 8.13 (t, J = 6.9 Hz, 2H), 6.79 (d, J = 2.5 Hz, 1H), 6.64 (d, J = 2.8 Hz, 1H), 3.89 (s, 3H), 3.78 (s, 3H); 13C{1H}-NMR (126 MHz, CD3CN) δ 160.7 (q, J = 34.3 Hz), 154.2, 151.4, 147.7, 147.2, 135.6, 131.3, 128.8, 118.0 (q, J = 293.1 Hz), 103.3, 101.9, 57.2, 56.7; 19F-NMR (376 MHz, CD3CN) δ −75.9; HRMS (ESI-TOF) m/z = 232.0974 calcd for C13H14NO3+ [M-CO2CF3]+, found 232.0966.
1-(2-Hydroxy-3,5-dimethoxyphenyl)pyrazin-1-ium trifluoroacetate (3h).
The product 3h was obtained as an amorphous brown solid (16 mg, 18% yield); 1H-NMR (400 MHz, CD3CN) δ 9.51 (br s, 2H), 9.00 (br s, 2H), 6.87 – 6.82 (m, 1H), 6.65 (s, 1H), 3.90 (s, 3H), 3.80 (s, 3H); 13C{1H}-NMR (126 MHz, CD3CN) δ 160.7 (q, J = 22.2 Hz), 154.5, 152.0, 151.9, 138.7 (2 carbons), 135.6, 120.5 (q, J = 247.0 Hz), 104.5, 101.1, 57.3, 56.8; 19F-NMR (376 MHz, CD3CN) δ −75.8; HRMS (ESI-TOF) m/z = 233.0926 calcd for C12H13N2O3+ [M-CO2CF3]+, found 233.0937.
4-Trifluoromethyl-1-(3-(tert-butyl)-2-hydroxy-5-methoxyphenyl)pyridin-1-ium trifluoroacetate (3k).
The product 3k was obtained as an amorphous light brown solid (56 mg, 51% yield); 1H-NMR (400 MHz, CD3CN) δ 9.13 (d, J = 6.4 Hz, 2H), 8.40 (d, J = Hz, 2H), 7.15 (d, J = 3.1 Hz, 1H), 6.89 (d, J = 3.2 Hz, 1H), 3.78 (s, 3H), 1.43 (s, 9H); 13C{1H}-NMR (151 MHz, DMSO-d6) δ 157.9 (q, J = 30.9 Hz), 152.3, 149.7, 143.5 (q, J = 35.0 Hz), 142.6, 142.1, 133.4, 124.9 (q, J = 3.7 Hz), 121.5 (q, J = 279.2 Hz), 117.2 (q, J = 302.3 Hz), 117.0, 108.5, 55.7, 35.4, 29.4; 19F-NMR (376 MHz, CD3CN) δ −66.0, −75.8; HRMS (ESI-TOF) m/z = 326.1368 calcd for C17H19F3NO2+ [M-CO2CF3]+, found 326.1357.
4-Trifluoromethyl-1-(2-hydroxy-5-methoxy-3-methylphenyl)pyridin-1-ium trifluoroacetate (3l).
The product 3l was obtained as an amorphous yellow solid (48 mg, 48% yield); 1H-NMR (400 MHz, CD3CN) δ 10.96 (s, 1H), 9.23 (d, J = 6.6 Hz, 2H), 8.42 (d, J = 6.5 Hz, 2H), 7.08 (d, J = 2.7 Hz, 1H), 6.89 (d, J = 3.0 Hz, 1H), 3.78 (s, 3H), 2.34 (s, 3H); 13C{1H}-NMR (151 MHz, CD3CN) δ 160.8 (q, J = 32.3 Hz), 153.9, 149.7, 145.8 (q, J = 36.0 Hz), 143.6, 133.1, 133.0, 125.7 (q, J = 3.3 Hz), 122.4 (q, J = 275.1 Hz), 121.3, 118.1 (q, J = 295.4 Hz), 109.0, 56.7, 17.2; 19F-NMR (376 MHZ, CD3CN) δ −65.9, −75.8; HRMS (ESI-TOF) m/z = 284.0898 calcd for C14H13F3NO2+ [M-CO2CF3]+, found 284.0885.
4-Trifluoromethyl-1-(2-hydroxy-5-methoxy-[1,1'-biphenyl]-3-yl)pyridin-1-ium trifluoroacetate (3m).
The product 3m was obtained as an amorphous light brown solid (56 mg, 49% yield); 1H-NMR (400 MHz, CD3CN) δ 9.21 (d, J = 6.6 Hz, 2H), 8.41 (d, J = 6.6 Hz, 2H), 7.63 – 7.58 (m, 2H), 7.48 – 7.42 (m, 2H), 7.41 – 7.36 (m, 1H), 7.20 (d, J = 3.1 Hz, 1H), 7.04 (d, J = 3.1 Hz, 1H), 3.84 (s, 3H); 13C{1H}-NMR (151 MHz, CD3CN) δ 160.6 (q, J = 32.0 Hz), 154.2, 149.8, 146.0 (q, J = 35.3 Hz), 142.4, 138.0, 136.1, 134.0, 130.5, 129.4, 129.0, 125.8 (q, J = 3.1 Hz), 122.3 (q, J = 274.2 Hz), 120.8, 118.1 (q, J = 285.7 Hz), 111.3, 56.9; 19F-NMR (376 MHz, CD3CN) δ −65.9, −75.8; HRMS (ESI-TOF) m/z = 346.1055 calcd for C19H15F3NO2+ [M-CO2CF3]+, found 346.1050.
4-Trifluoromethyl-1-(4’-cyano-2-hydroxy-5-methoxy-[1,1'-biphenyl]-3-yl)pyridin-1-ium trifluoroacetate (3n).
The product 3n was obtained as an amorphous beige solid (38 mg, 31% yield); 1H-NMR (400 MHz, CD3CN) δ 9.17 (d, J = 6.5 Hz, 2H), 8.35 (d, J = 6.5 Hz, 2H), 7.78 – 7.72 (m, 4H), 7.17 (d, J = 3.1 Hz, 1H), 7.04 (d, J = 3.1 Hz, 1H), 3.82 (s, 3H); Note: minor impurity at 8.01 ppm (integrates to <5%); 13C{1H}-NMR (151 MHz, CD3CN) δ 152.2, 149.2, 145.3 (q, J = 29.2 Hz), 143.7, 135.1, 133.9, 133.8, 132.9, 131.1, 125.4 (q, J = 3.2 Hz), 122.4 (q, J = 273.9 Hz), 120.9, 119.8, 112.0, 111.5, 57.0; Note: Trifluoroacetate 13C{1H} signals were not observed but are seen in the 19F-NMR; 19F-NMR (376 MHz, CD3CN) δ −65.8, −75.7; HRMS (ESI-TOF) m/z = 371.1007 calcd for C20H14F3N2O2+ [M-CO2CF3]+, found 371.1003.
4-Trifluoromethyl-1-(3,6-dihydroxy-2,4-dimethylphenyl)pyridin-1-ium trifluoroacetate (3o).
The product 3o was obtained as an amorphous brown solid (38 mg, 39% yield); 1H-NMR (400 MHz, CD3CN) δ 8.98 (br s, 2H), 8.47 (br s, 2H), 6.99 (s, 1H), 2.24 (s, 3H), 1.93 (s, 3H); Note: 1.93 ppm signal overlaps with the solvent signal; 13C{1H}-NMR (151 MHz, CD3CN) δ 160.9 (q, J = 32.7 Hz), 150.3, 146.7, 146.1 (q, J = 36.8 Hz), 145.5, 131.4, 129.2, 126.4 (q, J = 3.3 Hz), 122.3 (q, J = 274.7 Hz), 121.2, 118.4 (q, J = 271.5 Hz), 117.3, 17.4, 11.7; 19F-NMR (376 MHz, CD3CN) δ −66.0, −75.8; HRMS (ESI-TOF) m/z = 284.0898 calcd for C14H13F3NO2+ [M-CO2CF3]+, found 284.0906.
4-Trifluoromethyl-1-(5-hydroxy-3,3-dimethyl-2,3-dihydrobenzoiuran-6-yl)pyridin-1-ium trifluoroacetate (3p).
The product 3p was obtained as an amorphous brown solid (56 mg, 53% yield); 1H-NMR (600 MHz, CD3CN) δ 9.14 (d, J = 6.5 Hz, 2H), 8.41 (d, J = 6.4 Hz, 2H), 7.27 (s, 1H), 6.79 (s, 1H), 3.09 (s, 2H), 1.46 (s, 6H); 13C{1H}-NMR (151 MHz, CD3CN) δ 161.0 (q, J = 33.2 Hz), 152.8, 149.4, 146.1, 145.4 (q, J = 36.2 Hz), 135.1, 129.4, 125.6 (q, J = 3.7 Hz), 122.4 (q, J = 275.4 Hz), 118.3 (q, J = 296.3 Hz), 116.5, 106.2, 89.0, 43.5, 28.1; 19F-NMR (376 MHz, CD3CN) δ −65.9, −75.7; HRMS (ESI-TOF) m/z = 310.1055 calcd for C16H15F3NO2+ [M-CO2CF3]+, found 310.1041.
4-Trifluoromethyl-1-(2,5-dihydroxyphenyl)pyridin-1-ium trifluoroacetate (3q).
The product 3q was obtained as an amorphous dark brown solid (66 mg, 72% yield); 1H-NMR (400 MHz, DMSO-d6) δ 10.63 (s, 1H), 9.75 (s, 1H), 9.54 (d, J = 6.6 Hz, 2H), 8.77 (d, J = 6.7 Hz, 2H), 7.11 – 7.04 (m, 2H), 7.00 (dd, J = 8.9, 2.8 Hz, 1H); Note: minor impurity peak at 6.55 pm (integrates to <5%) (Dearomatized quinone isomer); 13C{1H}-NMR (101 MHz, DMSO-d6) δ 157.9 (q, J = 31.2 Hz), 150.3, 148.8, 143.4 (d, J = 35.6 Hz), 142.5, 129.8, 124.7 (q, J = 4.0 Hz), 122.7 (q, J = 282.1 Hz), 120.0, 118.2, 117.3 (q, J = 300.1 Hz), 112.7; Note: minor impurity at 115.6 ppm (Dearomatized quinone isomer); 19F-NMR (376 MHz, DMSO-d6) δ −63.9, −73.6; HRMS (ESI-TOF) m/z = 256.0585 calcd for C12H9F3NO2+ [M-CO2CF3]+, found 256.0592.
4-Trifluoromethyl-1-(4-Hydroxy-3,5-dimethylphenyl)pyridin-1-ium trifluoroacetate (3v).
The product 3v was obtained as an amorphous dark brown solid (44 mg, 46% yield); 1H-NMR (400 MHz, CD3CN) δ 9.17 (d, J = 6.6 Hz, 2H), 8.44 (d, J = 6.4 Hz, 2H), 7.35 (s, 2H), 2.33 (s, 6H); 13C{1H}-NMR (151 MHz, CD3CN) δ 160.5 (q, J = 26.2 Hz), 158.4, 147.2, 144.9 (q, J = 36.0 Hz), 134.9, 128.1, 126.1 (q, J = 3.5 Hz), 124.9, 122.4 (q, J = 275.1 Hz), 117.4 (q, J = 292.8 Hz), 17.1; 19F-NMR (376 MHz, CD3CN) δ −65.7, −75.6; HRMS (ESI-TOF) m/z = 268.0949 calcd for C14H13F3NO+ [M-CO2CF3]+, found 268.0923.
Synthesis of Derivatized Products 4a–c.
2,4-Dimethoxy-6-(4-(trifluoromethyl)piperidin-1-yl)phenol (4a).
An 8 mL scintillation vial equipped with a stirbar was charged with 4-trifluoromethyl-1-(2-hydroxy-3,5-dimethoxyphenyl)pyridin-1-ium trifluoroacetate (3a, 18 mg, 0.042 mmol, 1.0 equiv), Pd/C (9.0 mg, 0.0085 mmol, 10 mol%, 10% w/w), and EtOAc (2.0 mL, 0.020 M). The vial was capped and then sparged with hydrogen for 10 min and then stirred at 25 °C for 72 h under a hydrogen-filled balloon. (Note: The balloon was refilled every 16 h.) After completion, the reaction was subject to vacuum and backfilled with argon. The resultant suspension was filtered through Celite™. This solution was then dry loaded onto silica and purified by normal phase column chromatography (25:65 n-hexanes:EtOAc) to afford the title compound 4a as an amorphous orange solid (7.0 mg, 54% yield); 1H-NMR (400 MHz, CDCl3) δ 6.33 (d, J = 2.7 Hz, 1H), 6.27 (d, J = 2.7 Hz, 1H), 3.87 (s, 3H), 3.76 (s, 3H), 3.28 (d, J = 11.8 Hz, 2H), 2.66 (td, J = 12.0, 2.1 Hz, 2H), 2.22 – 2.12 (m, 1H), 2.04 – 1.95 (m, 2H), 1.82 (qd, J = 12.6, 4.0 Hz, 2H); 13C{1H}-NMR (151 MHz, CDCl3) δ 153.2, 147.6, 139.4, 134.1, 127.6 (q, J = 278.4 Hz), 97.9, 95.9, 56.3, 55.9, 51.3, 40.2 (q, J = 28.0 Hz), 25.4 (q, J = 2.6 Hz); 19F-NMR (376 MHz, CDCl3) δ −73.7; HRMS (ESI-TOF) m/z = 306.1317 calcd for C14H19F3NO3 [M+H]+, found 306.1309.
1-(2-Hydroxy-3,5-dimethoxyphenyl)-4-(trifluoromethyl)pyridin-2(1H)-one (4b).
An 8 mL scintillation vial equipped with a stirbar was charged with 4-trifluoromethyl-1-(2-hydroxy-3,5-dimethoxyphenyl)pyridin-1-ium trifluoroacetate (3a, 55 mg, 0.13 mmol, 1.0 equiv), K2CO3 (140 mg, 1.0 mmol, 7.5 equiv) , and anhydrous MeCN (2.0 mL, 70 mM). The vial was capped, parafilmed, and then stirred at 50 °C in an oil bath for 24 h. The reaction mixture was diluted with CH2Cl2, washed with H2O, extracted with CH2Cl2 (3x), dried over sodium sulfate, and concentrated in vacuo. This material was then dry loaded onto silica and purified by normal phase column chromatography (30:70 n-hexanes:EtOAc) to afford the title compound 4b as an amorphous orange solid (16 mg, 39% yield); 1H-NMR (400 MHz, CDCl3) δ 7.49 (d, J = 7.2 Hz, 1H), 6.99 (s, 1H), 6.59 (d, J = 2.7 Hz, 1H), 6.43 (dd, J = 7.2, 1.9 Hz, 1H), 6.34 (d, J = 2.7 Hz, 1H), 5.83 (s, 1H), 3.91 (s, 3H), 3.77 (s, 3H); 13C{1H}-NMR (151 MHz, CDCl3) δ 161.4, 153.7, 149.5, 141.9 (q, J = 34.2 Hz), 140.6, 135.6, 127.4, 122.2 (q, J = 273.6 Hz), 119.5 (q, J = 4.4 Hz), 102.5, 101.9 (q, J = 2.4 Hz), 100.9, 56.5, 56.0; 19F-NMR (376 MHz, CDCl3) δ −66.8; HRMS (ESI-TOF) m/z = 316.0797 calcd for C14H13F3NO4 [M+H]+, found 316.0775.
1-(2-Hydroxy-3,5-dimethoxyphenyl)-4-(trifluoromethyl)-5,6-dihydropyridin-2(1H)-one (4c).
An 8 mL scintillation vial equipped with a stirbar was charged with 4-trifluoromethyl-1-(2-hydroxy-3,5-dimethoxyphenyl)pyridin-1-ium trifluoroacetate (3a, 19 mg, 0.046 mmol, 1.0 equiv), NaBH4 (7.0 mg, 0.18 mmol, 4.0 equiv), NaOH (7.4 mg, 0.18 mmol, 4.0 equiv), and H2O (1.3 mL, 0.035 M). The vial was capped and stirred at 25 °C for 16 h. The reaction mixture was concentrated in vacuo, dry loaded onto silica, and purified by normal phase column chromatography (30:70 n-hexanes:EtOAc) to afford the title compound 4c as an amorphous light brown solid (7.3 mg, 50% yield); 1H-NMR (400 MHz, CDCl3) δ 6.53 – 6.51 (m, 1H), 6.48 (d, J = 2.7 Hz, 1H), 6.33 (d, J = 2.7 Hz, 1H), 5.61 (s, 1H), 3.91 (t, J = 6.9 Hz, 2H), 3.88 (s, 3H), 3.76 (s, 3H), 2.70 (t, J = 6.9 Hz, 2H); 13C{1H}-NMR (151 MHz, CDCl3) δ 162.4, 153.4, 149.3, 138.1 (q, J = 32.0 Hz), 135.4, 128.7, 125.8 (q, J = 4.7 Hz), 122.6 (q, J = 255.3 Hz), 102.2, 99.3, 56.4, 55.9, 48.3, 22.5 (q, J = 3.4 Hz); 19F-NMR (376 MHz, CDCl3) δ −70.4; HRMS (ESI-TOF) m/z = 318.0953 calcd for C14H15F3NO4 [M+H]+, found 318.0965.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the NSF (CHE2102626) and the NIH (R35 GM131902) for financial support of this research. Partial instrumentation support was provided by the NIH and NSF (1S10RR023444, CHE-1827457, 3R01GM118510-03S1, 3R01GM087605-06S1), as well as the Vagelos Institute for Energy Science and Technology. We thank Dr. Charles W. Ross III (UPenn) for chromatographic and mass spectral method development, training, analyses, and data interpretation. We are also grateful to Kyle Shaffer (UPenn) for fluorescence and UV-Vis analytical help.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Experimental procedures, product characterization, and NMR spectral data (PDF).
FAIR data, includes the primary NMR FID files, for compounds [2p, 2n, 3a–h, 3k–q, 3v, 4a–c] (ZIP).
The authors declare no competing financial interest.
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.
REFERENCES
- (1).He FS; Ye SQ; Wu J Recent Advances in Pyridinium Salts as Radical Reservoirs in Organic Synthesis. ACS Catal. 2019, 9 (10), 8943–8960. [Google Scholar]
- (2).Sowmiah S; Esperanca J; Rebelo LPN; Afonso CAM Pyridinium salts: from synthesis to reactivity and applications. Org. Chem. Front 2018, 5 (3), 453–493. [Google Scholar]
- (3).Vanderwal CD Reactivity and Synthesis Inspired by the Zincke Ring-Opening of Pyridines. J Org. Chem 2011, 76 (23), 9555–9567. [DOI] [PubMed] [Google Scholar]
- (4).Boyle BT; Levy JN; de Lescure L; Paton RS; McNally A Halogenation of the 3-position of pyridines through Zincke imine intermediates. Science 2022, 378 (6621), 773–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Joo JM Adding functions to pyridines. Science 2022, 378 (6621), 710–711. [DOI] [PubMed] [Google Scholar]
- (6).Fier PS A Bifunctional Reagent Designed for the Mild, Nucleophilic Functionalization of Pyridines. J Am. Chem. Soc 2017, 139 (28), 9499–9502. [DOI] [PubMed] [Google Scholar]
- (7).Fier PS; Kim S; Cohen RD A Multifunctional Reagent Designed for the Site-Selective Amination of Pyridines. J. Am. Chem. Soc 2020, 142 (19), 8614–8618. [DOI] [PubMed] [Google Scholar]
- (8).Cao H; Cheng Q; Studer A Radical and ionic meta-C─H functionalization of pyridines, quinolines, and isoquinolines. Science 2022, 378 (6621), 779–785. [DOI] [PubMed] [Google Scholar]
- (9).Josephitis CM; Nguyen HMH; McNally A Late-Stage C─H Functionalization of Azines. Chem. Rev 2023, 123 (12), 7655–7691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kim M; Koo Y; Hong SW N-Functionalized Pyridinium Salts: A New Chapter for Site-Selective Pyridine C-H Functionalization via Radical-Based Processes under Visible Light Irradiation. Acc. Chem. Res 2022, 55 (20), 3043–3056. [DOI] [PubMed] [Google Scholar]
- (11).Lovering F; Bikker J; Humblet C Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J Med. Chem 2009, 52 (21), 6752–6756. [DOI] [PubMed] [Google Scholar]
- (12).a) Roche SP; Porco JA Dearomatization Strategies in the Synthesis of Complex Natural Products. Angew. Chem. Int. Ed 2011, 50 (18), 4068–4093. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Escolano M; Gaviña D; Alzuet-Piña G; Díaz-Oltra S; Sánchez-Roselló M; Pozo C Recent Strategies in the Nucleophilic Dearomatization of Pyridines, Quinolines, and Isoquinolines. Chem. Rev 2024, 10.1021/acs.chemrev.3c00625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Frolov NA; Vereshchagin AN Piperidine Derivatives: Recent Advances in Synthesis and Pharmacological Applications. Int. J Mol. Sci 2023, 24 (3), 2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kerkovius JK; Stegner A; Turlik A; Lam PH; Houk KN; Reisman SE A Pyridine Dearomatization Approach to the Matrine-Type Lupin Alkaloids. J. Am. Chem. Soc 2022, 144 (35), 15938–15943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Magnani CSH; Maimone TJ Dearomative Synthetic Entry into the Altemicidin Alkaloids. J. Am. Chem. Soc 2021, 143 (21), 7935–7939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Magnani CSH; Hernández-Meléndez JR; Tantillo DJ; Maimone TJ Total Synthesis of Altemicidin: A Surprise Ending for a Monoterpene Alkaloid. JACS Au 2023, 3 (10), 2883–2893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Robinson DJ; Ortiz KG; O'Hare NP; Karimov RR Dearomatization of Heteroarenium Salts with ArBpin Reagents. Application to the Total Synthesis of a Nuphar Alkaloid. Org. Let 2022, 24 (19), 3445–3449. [DOI] [PubMed] [Google Scholar]
- (18).Rössler SL; Jelier BJ; Magnier E; Dagousset G; Carreira EM; Togni A Pyridinium Salts as Redox-Active Functional Group Transfer Reagents. Angew. Chem. Int. Ed 2020, 59 (24), 9264–9280. [DOI] [PubMed] [Google Scholar]
- (19).Urbiña-Alvarez J; Rincón-Carvajal S; Gamba-Sánchez D Ammonia surrogates in the synthesis of primary amines. Org. Biomol. Chem 2023, 21 (35), 7036–7051. [DOI] [PubMed] [Google Scholar]
- (20).Basch CH; Liao JN; Xu JY; Piane JJ; Watson MP Harnessing Alkyl Amines as Electrophiles for Nickel-Catalyzed Cross Couplings via C─N Bond Activation. J Am. Chem. Soc 2017, 139 (15), 5313–5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).a) Das M; Zamani L; Bratcher C; Musacchio PZ Azolation of Benzylic C─H Bonds via Photoredox-Catalyzed Carbocation Generation. J. Am. Chem. Soc 2023, 145 (7), 3861–3868. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wei N; Beil SB Light-driven benzylic azolation. Trends in Chemistry 2023, 5 (9), 711–712. [Google Scholar]
- (22).a) Hoerrner ME; Baker KM; Basch CH; Bampo EM; Watson MP Deaminative Arylation of Amino Acid-derived Pyridinium Salts. Org. Lett 2019, 21 (18), 7356–7360. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang J; Hoerrner ME; Watson MP; Weix DJ Nickel-Catalyzed Synthesis of Dialkyl Ketones from the Coupling of N-Alkyl Pyridinium Salts with Activated Carboxylic Acids. Angew. Chem. Int. Ed 2020, 59 (32), 13484–13489. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Tcyrulnikov S; Cai QQ; Twitty JC; Xu JY; Atifi A; Bercher OP; Yap GPA; Rosenthal J; Watson MP; Kozlowski MC Dissection of Alkylpyridinium Structures to Understand Deamination Reactions. ACS Catal. 2021, 11 (14), 8456–8466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Castejon H; Wiberg KB Solvent effects on methyl transfer reactions. 1. The Menshutkin Reaction. J Am. Chem. Soc 1999, 121 (10), 2139–2146. [DOI] [PubMed] [Google Scholar]
- (24).Sambiagio C; Ferrari M; van Beurden K; della Ca N; van Schijndel J; Noël T Continuous-Flow Synthesis of Pyrylium Tetrafluoroborates: Application to Synthesis of Katritzky Salts and Photoinduced Cationic RAFT Polymerization. Org. Lett 2021, 23 (6), 2042–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Cheng WC; Kurth MJ The Zincke reaction. A review. Org. Prep. Proced. Int 2002, 34 (6), 585–608. [Google Scholar]
- (26).Morofuji T; Shimizu A; Yoshida J Electrochemical C─H Amination: Synthesis of Aromatic Primary Amines via N-Arylpyridinium Ions. J. Am. Chem. Soc 2013, 135 (13), 5000–5003. [DOI] [PubMed] [Google Scholar]
- (27).Ham WS; Hillenbrand J; Jacq J; Genicot C; Ritter T Divergent Late-Stage (Hetero)aryl C─H Amination by the Pyridinium Radical Cation. Angew. Chem. Int. Ed 2019, 58 (2), 532–536. [DOI] [PubMed] [Google Scholar]
- (28).Rössler SL; Jelier BJ; Tripet PF; Shemet A; Jeschke G; Togni A; Carreira EM Pyridyl Radical Cation for C─H Amination of Arenes. Angew. Chem. Int. Ed 2019, 58 (2), 526–531. [DOI] [PubMed] [Google Scholar]
- (29).Motsch BJ; Kaur JY; Wengryniuk SE I(III)-Mediated Arene C─H Amination Using (Hetero)Aryl Nucleophiles. Org. Lett 2023, 25 (14), 2548–2553. [DOI] [PubMed] [Google Scholar]
- (30).Mantell MA; Lasky MR; Lee M; Remy M; Sanford MS SNAr and C─H Amination of Electron Rich Arenes with Pyridine as a Nucleophile Using Photoredox Catalysis. Org. Lett 2021, 23 (13), 5213–5217. [DOI] [PubMed] [Google Scholar]
- (31).a) Romero NA; Margrey KA; Tay NE; Nicewicz DA Site-selective arene C─H amination via photoredox catalysis. Science 2015, 349 (6254), 1326–1330. [DOI] [PubMed] [Google Scholar]; b) Tay NES; Nicewicz DA Cation Radical Accelerated Nucleophilic Aromatic Substitution via Organic Photoredox Catalysis. J. Am. Chem. Soc 2017, 139 (45), 16100–16104. [DOI] [PubMed] [Google Scholar]; c) Margrey KA; McManus JB; Bonazzi S; Zecri F; Nicewicz DA Predictive Model for Site-Selective Aryl and Heteroaryl C─H Functionalization via Organic Photoredox Catalysis. J. Am. Chem. Soc 2017, 139 (32), 11288–11299. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Pistritto VA; Liu SB; Nicewicz DA Mechanistic Investigations into Amination of Unactivated Arenes via Cation Radical Accelerated Nucleophilic Aromatic Substitution. J. Am. Chem. Soc 2022, 144 (33), 15118–15131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).a) Möhle S; Herold S; Richter F; Nefzger H; Waldvogel SR Twofold Electrochemical Amination of Naphthalene and Related Arenes. ChemElectroChem 2017, 4 (9), 2196–2210. [Google Scholar]; b) Magomedov A; Kasparavicius E; Rakstys K; Paek S; Gasilova N; Genevicius K; Juska G; Malinauskas T; Nazeeruddin MK; Getautis V Pyridination of hole transporting material in perovskite solar cells questions the long-term stability. J. Mater. Chem. C 2018, 6 (33), 8874–8878. [Google Scholar]
- (33).Lasky MR; Salvador TK; Mukhopadhyay S; Remy MS; Vaid TP; Sanford MS Photochemical C(sp2)─H Pyridination via Arene–Pyridinium Electron Donor–Acceptor Complexes. Angew. Chem. Int. Ed 2022, 61 (46), e202208741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Niederer KA; Gilmartin PH; Kozlowski MC Oxidative Photocatalytic Homo- and Cross-Coupling of Phenols: Nonenzymatic, Catalytic Method for Coupling Tyrosine. ACS Catal. 2020, 10 (24), 14615–14623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).De Vleeschouwer F; Van Speybroeck V; Waroquier M; Geerlings P; De Proft F Electrophilicity and nucleophilicity index for radicals. Org. Lett 2007, 9 (14), 2721–2724. [DOI] [PubMed] [Google Scholar]
- (36).Ji YN; Brueckl T; Baxter RD; Fujiwara Y; Seiple IB; Su S; Blackmond DG; Baran PS Innate C─H trifluoromethylation of heterocycles. PNAS 2011, 108 (35), 14411–14415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Prier CK; Rankic DA; MacMillan DWC Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev 2013, 113 (7), 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Zhao YT; Huang BB; Yang C; Xia WJ Visible-Light-Promoted Direct Amination of Phenols via Oxidative Cross-Dehydrogenative Coupling Reaction. Org. Lett 2016, 18 (14), 3326–3329. [DOI] [PubMed] [Google Scholar]
- (39).Nieves-Quinones Y; Paniak TJ; Lee YE; Kim SM; Tcyrulnikov S; Kozlowski MC Chromium-Salen Catalyzed Cross-Coupling of Phenols: Mechanism and Origin of the Selectivity. J Am. Chem. Soc 2019, 141 (25), 10016–10032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Shao NQ; Yao ZL; Wang DH Cu(II)-Catalyzed Ortho-Selective Amination of Simple Phenols with O-Benzoylhydroxylamines. Isr. J. Chem 2020, 60 (3-4), 429–432. [Google Scholar]
- (41).Zhao YT; Huang BB; Yang C; Li B; Gou BQ; Xia WJ Photocatalytic Cross-Dehydrogenative Amination Reactions between Phenols and Diarylamines. ACS Catal. 2017, 7 (4), 2446–2451. [Google Scholar]
- (42).Tang S; Wang SY; Liu YC; Cong HJ; Lei AW Electrochemical Oxidative C─H Amination of Phenols: Access to Triarylamine Derivatives. Angew. Chem. Int. Ed 2018, 57 (17), 4737–4741. [DOI] [PubMed] [Google Scholar]
- (43).Zhang L; Liardet L; Luo JS; Ren D; Grätzel M; Hu XL Photoelectrocatalytic arene C─H amination. Nat. Catal 2019, 2 (4), 366–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Shin NY; Tsui E; Reinhold A; Scholes GD; Bird MJ; Knowles RR Radicals as Exceptional Electron-Withdrawing Groups: Nucleophilic Aromatic Substitution of Halophenols Via Homolysis-Enabled Electronic Activation. J. Am. Chem. Soc 2022, 144 (47), 21783–21790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Libman A; Shalit H; Vainer Y; Narute S; Kozuch S; Pappo D Synthetic and Predictive Approach to Unsymmetrical Biphenols by Iron-Catalyzed Chelated Radical-Anion Oxidative Coupling. J Am. Chem. Soc 2015, 137 (35), 11453–11460. [DOI] [PubMed] [Google Scholar]
- (46).Wortman AK; Stephenson CRJ EDA photochemistry: Mechanistic investigations and future opportunities. Chem 2023, 9 (9), 2390–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Nicholas AMD; Arnold DR Thermochemical parameters for organic radicals and radical ions. Part 1. The estimation of the pKa of radical cations based on thermochemical calculations. Can.J. Chem 1982, 60 (17), 2165–2179. [Google Scholar]
- (48).Sawyer DT; Roberts JL; Calderwood TS; Sugimoto H; McDowell MS Reactivity and Activation of Dioxygen-Derived Species in Aprotic Media (A Model Matrix for Biomembranes). Philosophical Transactions of the Royal Society of London Series B, Biological Sciences 1985, 311 (1152), 483–503. [DOI] [PubMed] [Google Scholar]
- (50).Hayyan M; Hashim MA; AlNashef IM Superoxide Ion: Generation and Chemical Implications. Chem. Rev 2016, 116 (5), 3029–3085. [DOI] [PubMed] [Google Scholar]
- (51).Hajduk A; Ulrich N Determination of acidity constants of pyridines, imidazoles, and oximes by capillary electrophoresis. Electrophoresis 2023, 44 (17-18), 1353–1360. [DOI] [PubMed] [Google Scholar]
- (52).Lee J; von Gunten U; Kim JH Persulfate-Based Advanced Oxidation: Critical Assessment of Opportunities and Roadblocks. Environ Sci. Technol 2020, 54 (6), 3064–3081. [DOI] [PubMed] [Google Scholar]
- (53).Sawyer DT; Valentine JS How super is superoxide? Acc. Chem. Res 1981, 14 (12), 393–400. [Google Scholar]
- (54).Motiwala HF; Armaly AM; Cacioppo JG; Coombs TC; Koehn KRK; Iv VMN; Aube J HFIP in Organic Synthesis. Chem. Rev 2022, 122 (15), 12544–12747. [DOI] [PubMed] [Google Scholar]
- (55).Han J; Wang L; Cao WJ; Yuan QQ; Zhou XG; Liu SL; Wang XB Photogeneration of singlet oxygen catalyzed by hexafluoroisopropanol for selective degradation of dyes. iScience 2023, 26 (8), 107306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Hillenbrand J; Ham WS; Ritter T C─H Pyridonation of (Hetero-)Arenes by Pyridinium Radical Cations. Org. Lett 2019, 21 (13), 5363–5367. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.