

Abstract
Chronic obstructive pulmonary disease (COPD) is a deadly and highly morbid disease. COPD susceptibility and heterogeneity are incompletely explained by environmental factors such as cigarette smoking. Studies in families and populations have demonstrated that a substantial proportion of COPD risk is related to genetic variation. Genetic association studies have identified hundreds of genetic variants that affect risk of COPD, lung function, and other COPD-related traits. These genetic variants are associated with other pulmonary and non-pulmonary traits, demonstrate a genetic basis for at least part of COPD heterogeneity, have a substantial impact on COPD risk in aggregate, implicate early life events in COPD pathogenesis, and often involve genes not previously suspected to have a role in COPD. Additional progress will require larger genetic studies with more ancestral diversity, improved profiling of rare variants, and better statistical methods. Through integration of genetic data with other omics data and comprehensive COPD phenotypes as well as functional description of genetic risk variant causal mechanisms, COPD genetics will continue to inform novel approaches to understanding the pathobiology, management, and treatment of COPD.
Introduction
Chronic obstructive pulmonary disease (COPD) is a leading and rising cause of morbidity and mortality worldwide (WHO). While usually diagnosed in the setting of exposure to noxious particles or gases, these exposures incompletely explain disease susceptibility. Cigarette smoking, the most common exposure worldwide, accounts for only a portion of the observed risk(1,2). While the health risks of cigarette smoking are incontrovertible, and mitigation of other environmental exposures such as outdoor pollution and indoor biomass fuel use should also be a top priority for public health, other factors are likely involved in COPD susceptibility. In fact, genetics explains a significant proportion of the phenotypic variability of COPD(3,4). Identifying these genetic factors may help explain COPD heterogeneity, estimate individual susceptibility, assess prognosis, and identify novel and personalized therapeutics.
In this review, we provide background of studies of COPD genetics and an overview of the past several decades of research. We review genome-wide association analyses and other genetic studies, and what these studies tell us about the genetic risk of COPD. We discuss the role of phenotypes and disease heterogeneity, the ability of genetics to predict COPD risk, the potential role of early life events in COPD pathogenesis, and specific molecular pathways and targets implicated by genetics studies. We also discuss the state of clinical translation and key elements needed to further the impact of genetic studies in COPD.
Genetic epidemiology and COPD
Human DNA consists of 23 pairs of chromosomes, totaling approximately two copies of 3 billion base pairs. The differences between any two individuals are small – on the order of 1 difference per 1000 base pairs, with most of this variation being due to single nucleotide variants (SNVs). This genetic variation has an impact on many human diseases and traits(5). Technological advances have made assessment of most SNVs in the human genome straightforward and inexpensive, available from a cheek swab or a blood sample, in an assay that only needs to be obtained once in a person’s lifetime. In contrast to other biomarkers such as proteins or expressed genes, which may be consequences and not causes of the disease process, genetic variants are randomly assigned at the time of conception and thus allow an investigation of potentially causal disease pathobiology. For drug development, human genetics support increases the likelihood of successful clinical trials(6).
Substantial evidence indicates that genetics are important in COPD susceptibility. Dozens of studies have shown significant heritability for lung function and COPD. For example, in one of the largest studies including > 50,000 twin pairs, the heritability of COPD was estimated at > 50%(7). In the COPDGene study of unrelated individuals, heritability was estimated at ~40%(8). Together, these studies strongly indicate that genetic factors are an important and substantial contributor to COPD susceptibility.
Approaches to Identify Genetic Variants in COPD
Initial studies in COPD tested only a few SNPs in or near candidate genes thought to play a role in COPD pathogenesis. To a large extent, however, these findings were not well replicated(9). Genome-wide association studies (GWAS), using larger sample sizes with greater statistical power - currently able to detect variants with odds ratios of < 1.2 using strict criteria for genome-wide significance - have failed to confirm nearly all of these previous candidates, and instead have found dozens to hundreds of susceptibility variants, near genes that largely had no previously suspected role in COPD pathogenesis. While there may still be a role for studying candidate genes with substantial prior evidence, publicly available genome-wide association studies that cover the variants of interest often can provide evidence that these candidate associations do not replicate in larger cohorts. The first GWAS of COPD, defined using post-bronchodilator spirometry, identified a locus near the CHRNA3/CHRNA5/IREB2 genes, as well as a region near HHIP(10). Subsequent larger studies identified genomic regions near FAM13A, RIN3, CYP2A6, and DSP. The most recent GWAS COPD including 35,735 cases and 222,076 controls identified 82 loci (defined using a distance around the top variant) at genome-wide significance. Of these, 50 had evidence for a second independent association, 60 had clear evidence of replication, and the remainder had either nominal or directionally consistent associations in population-based studies of lung function(11). As the largest COPD studies to date have relied on the Global Initiative for Chronic Obstructive Lung Disease (GOLD) definition of COPD based on lung function, analysis of spirometry – by virtue of being a quantitative trait and available in large numbers of subjects – may result in greater power to detect variants associated with COPD. Indeed large-scale studies of quantitative spirometry identified 279 replicated loci for forced expiratory volume in 1 second (FEV1), forced vital capacity (FVC), and FEV1/FVC ratio; most of which have some effect on COPD risk(12) (Figure 1).
Figure 1:

Karyogram of GWAS associations for COPD and related lung function phenotypes (FEV1 and FEV1/FVC)(11,12,105). Regions within 1 megabase were combined, and one gene name chosen to represent the region.
While the genetic architecture of COPD is still being learned, studies of heritability of traits such as height, and diseases such as schizophrenia and diabetes(13–15) as well as COPD(3) support a model where the largest contribution to the phenotype arises not from rare variants with large effect sizes, but from the combined effect of many common variants of small effect size. This common disease - common variant model lends strong support to the GWAS approach.
Rare variants, typically not found by standard GWAS approaches, clearly have a contribution to COPD risk, as demonstrated by severe alpha-1 antitrypsin (AAT) deficiency. The SERPINA1 gene encodes the AAT protein. The most common form of severe AAT deficiency is due to homozygosity at rs28929474, also known as the SERPINA1 Z allele, which occurs in approximately 1 in 3,000 individuals(16). Other Mendelian syndromes include severe emphysema as a prominent feature, such as cutis laxa(17), Ehlers Danlos syndrome(18), and telomeropathies(19). However, identification of other rare variants, especially outside of known GWAS loci, has been challenging(20), though some promising candidates, such as TBC1D10A and PTPN6, have been identified through genome-wide unrelated or family-based approaches(21–24).
COPD Phenotypes and Heterogeneity
In addition to discovering new genomic regions associated with COPD, some important lessons have been learned from GWAS. One important lesson relates to phenotypes of COPD. While post-bronchodilator lung function and cigarette smoke exposure are considered key factors for the diagnosis of COPD, most of the genetic variants identified do not seem to be substantially affected by the use pre- or post-bronchodilator lung function, the inclusion of adult asthmatics, or non-smokers(11,25). These data suggest that genetic factors leading chronic airflow limitation and COPD in asthmatics and non-smokers are shared with those of smokers, and helps explain why there is a high degree of overlap between discoveries using only smokers with clinical diagnoses of COPD, and population-based studies of spirometry. Similarly, despite a strong rationale for gene-by-environment interaction, evidence for interaction of SNVs with cigarette smoking is generally weak, in part due to power that is orders of magnitude lower than that to detect genetic main effects, measurement error, use of cross-sectional rather than longitudinal lung function, and likely other issues. Exceptions where differential effects by smoking status have been identified include the CHRNA5, CYP2A6, SERPINA1, and MECOM loci; however, weak interaction effects of other variants may be detectable in aggregate(26–29).
Studies of phenotypes related to COPD may provide additional insight into disease pathogenesis. Emphysema, airway wall thickening, and expiratory gas trapping assessed by chest CT may reflect different pathophysiologic processes leading to airflow obstruction and thus may be important endotypes of COPD. Results from these studies are mixed, with most studies identifying the same signals from COPD case-control studies (e.g., AGER and HHIP) with some exceptions (e.g. DLC1, SERPINA1, MAN2B1)(30,31). Similar findings have also been demonstrated in other COPD phenotypes such as chronic bronchitis and pulmonary artery enlargement(32,33). While new signals are intriguing, most of them have not been well replicated.
An alternative approach to genetic studies of novel phenotypes is to instead examine the phenotypic associations for a given genetic marker. One of the first examples of this approach was at the FAM13A locus, where genome-wide associations were found for both COPD and for pulmonary fibrosis; here, the FAM13A genotype associated with increased risk for COPD was associated with decreased risk for pulmonary fibrosis(25). Subsequent studies identified several additional overlapping and opposite risk loci for COPD and pulmonary fibrosis. Similar approaches identified loci shared between asthma and COPD, though most of the asthma and COPD risk variants have a concordant direction of effect(11). Causal modeling and mediation approaches can identify intermediate phenotypes, such as structural features of the lung that may mediate the relationship between genetics and airflow limitation(34,35). In addition to respiratory diseases, COPD and lung function risk loci are shared with autoimmune diseases including inflammatory bowel disease, celiac disease, height, cardiovascular disease, and eosinophil counts(11,12). This phenomenon, called pleiotropy, appears to be common in GWAS and may lead to further insights about the functional impact of COPD-associated genetic variants (Figure 2).
Figure 2:
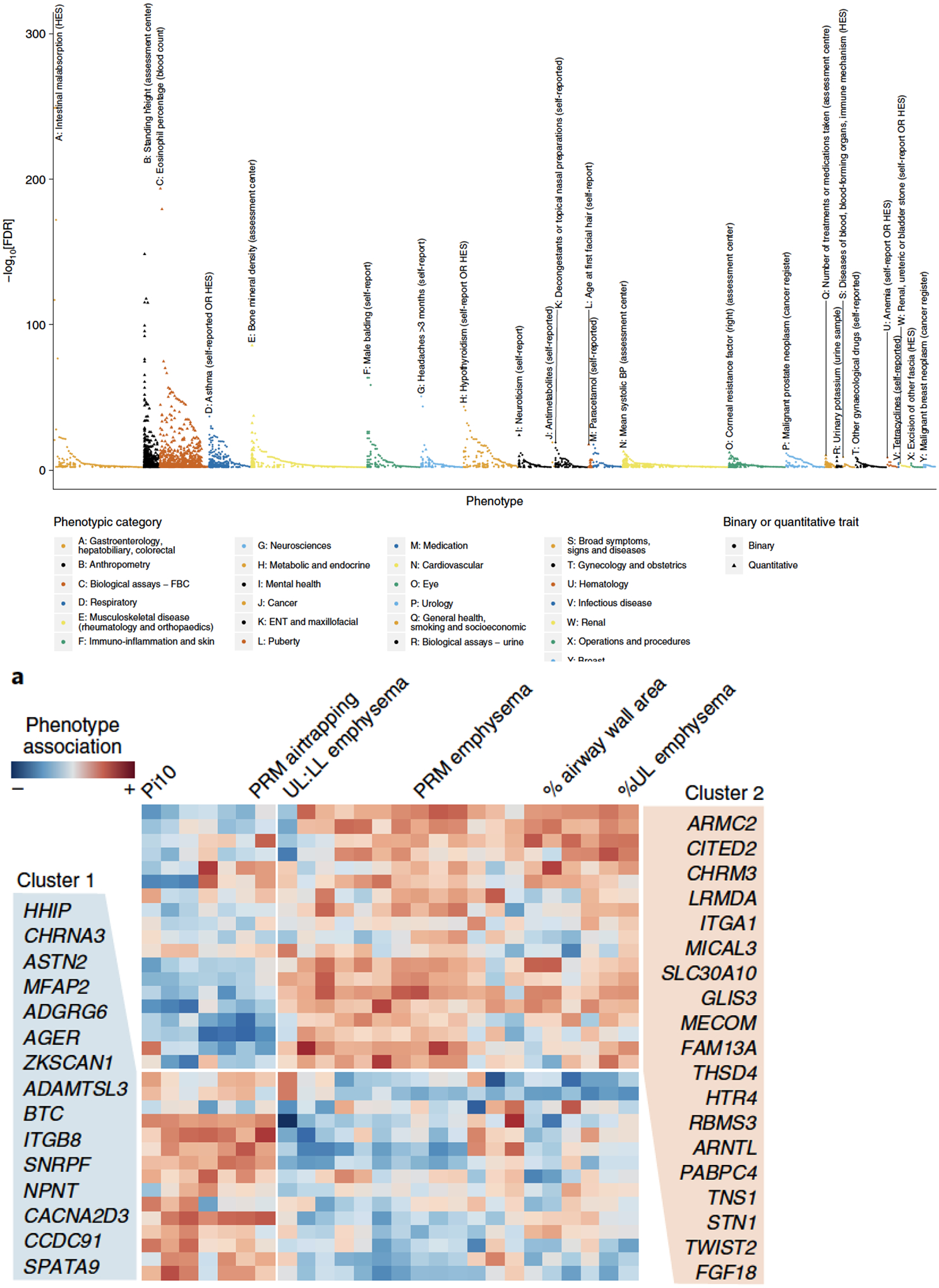
Multiple phenotypic effects identified at COPD and lung function genetic loci. A) Association of 279 lung function variants with 2,411 traits in UK Biobank(12), demonstrating extensive pleiotropy B) Hierarchical clustering of COPD genetic loci with chest imaging phenotypes in COPDGene(11). Variants clustered by association with airway features or emphysema features, suggesting that different groups of genetic variants may explain imaging heterogeneity in COPD.
These studies also highlight the relationship between genetics and COPD heterogeneity. Subjects with COPD vary not only in the degree of airflow limitation but also in the extent of emphysema and other imaging and pathologic characteristics, frequency and type of exacerbations, and presence of co-morbid conditions. Genetic variants may explain COPD heterogeneity. Severe alpha-1 antitrypsin deficiency is a paradigm of a COPD genetic subtype with specific phenotypic characteristics such as emphysema and liver disease that are a functional consequence of the underlying genetic mutation. Other individual rare variants of large effect, or groups of common variants, are likely to also have an impact on heterogeneity commonly observed in COPD(36,37) (Figure 2).
The impact of genetic risk
A second important insight from COPD genetic studies is the effect of genetics on risk. For Mendelian diseases such as severe alpha-1 antitrypsin deficiency and cystic fibrosis, a single genetic variant is responsible for a markedly elevated risk of disease. Genome-wide association studies in COPD have identified risk variants with odds ratios of ~1.3 or less, and thus individual variants are quite poor for risk prediction. Combining multiple genome-wide significant variants, and more recently, applying approaches that can leverage all variants and combine them into a single score per individual, demonstrates a substantial improvement in risk prediction. In COPD risk prediction, these advances have resulted in an increase of area under the curve (AUC) from 0.58 to an AUC of 0.68, and an increase in COPD risk in the top versus bottom decile of the risk score from 3.7, to 4.7, to 8.0 with more comprehensive risk scores(11,12,38–40) (Figure 3). These genetic risk scores (also termed polygenic risk scores, or PRS) also associate with important COPD characteristics, such as quantitative computed tomography emphysema and airway features, and lung growth patterns in a cohort of childhood asthmatics followed through to adulthood(40,41), raising the possibility that polygenic risk scores may not only predict overall risk, but may reflect risk of specific features of COPD.
Figure 3:
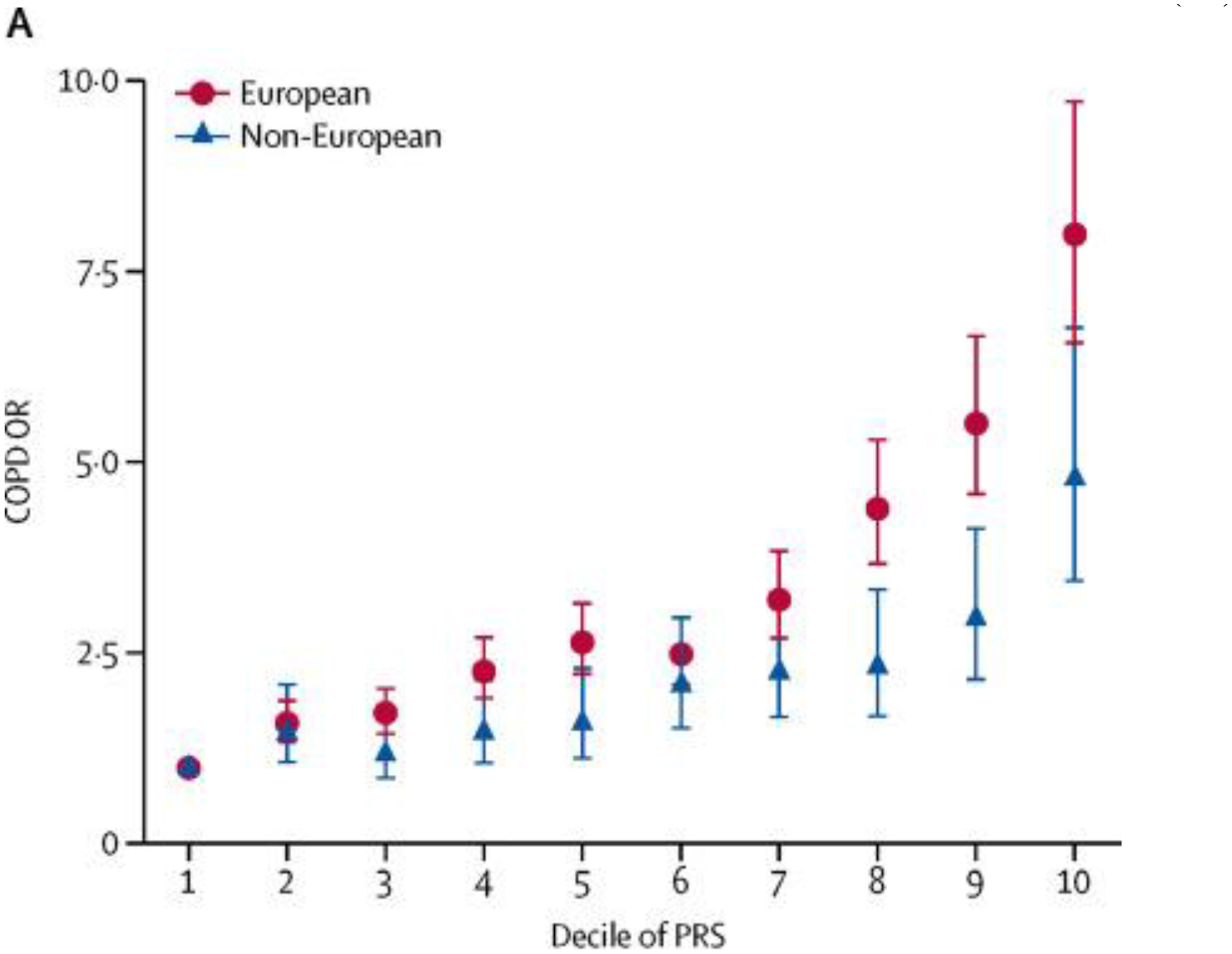
Odds ratios for COPD by decile of a polygenetic risk score(40).
Early life and genetic risk
The association of a COPD polygenic risk score with lung growth patterns begs the question of when genetic risk to COPD occurs. Early efforts at identifying COPD genetic susceptibility in the candidate gene era focused on the idea that individuals started with similar degrees of normal lung function and had different rates of lung function decline due to varied responses to cigarette exposure, for example through detoxifying enzymes such as EPHX1 and GSTM1, or genes related to inflammatory pathways such as TNF. However, COPD genome-wide association studies have generally not found enrichment in either inflammatory or detoxifying enzyme pathways. Instead, several lines of evidence suggest that genetic susceptibility begins early in life. COPD-associated genetic loci are largely contained within loci discovered in general populations; several of these loci are shared with height, and COPD and lung function loci are significantly more likely than chance to overlap with regions of the genome that are regulatory in fetal lung, and lung developmental genes(11,39,42). A second piece of evidence comes from looking at studies of childhood lung function or lung function decline. Overall, studies of lung function loci identified in adults appear to have similar effects in children, but most may not have an effect on lung function decline in adults(39,43). Supporting these data are reports of the decreasing heritability of lung function and other phenotypes with age(44). Together, these data are consistent with recent reports in longitudinal cohorts demonstrating that a subset of individuals who have fixed airflow obstruction as adults have reduced lung function in earlier life(45,46). Thus, some genetic risk factors for COPD, and similarly, the currently developed polygenic risk score may reflect – at least in part – the effects of respiratory system development and other events that result in lower lung function that precede later adulthood.
While genetic studies strengthen the case for the importance of early life events in COPD, these findings do not mean that these genetic risk factors cannot have influences later in life. In one of the most extensively studied genetic associations, Zhou et al described how a COPD-associated region could affect expression of the HHIP gene, and how haploinsufficiency of this gene in a murine model led to age-related emphysema that was attenuated with antioxidant treatment(47,48). Thus, HHIP, despite its roles in lung development may also have a role in regulating adult lung function.
While lung function decline appears to have a genetic component(49), identifying the specific genetic factors associated with decline has been challenging, with no genome-wide significant and independently replicated findings(50,51). Reasons for these findings include a smaller relative effect of genetics on decline versus environmental factors; decreased sample size relative to cross-sectional cohorts; and variability and relatively short-term follow-up available in longitudinal lung function studies. Addressing these issues could identify novel variants and clarify the role of currently identified variants on the cycle of chronic inflammation and progressive lung function decline that can persist years after smoking cessation in COPD patients(52).
Functional Genetics
The HHIP example also illustrates one of the main misconceptions regarding genome-wide association studies. Arguably, the main goal of genetic association studies is to discover new disease genes. For therapeutics, having genetic support for a drug target increases the likelihood of success in clinical trials and subsequent drug approval(6). However, GWAS themselves do not implicate a specific gene, and are better understood as implicating a specific region of the genome. Although the functions of most GWAS loci are unknown, by convention loci are often named for their closest gene. This may lead to the incorrect assumption that the named closest gene is causal. In fact, the effector gene for a GWAS risk locus is usually not known, and may be several hundred thousand base pairs distant(53). Similarly, the specific causal variant(s), cell type(s), and environment(s) leading to disease are generally unknown. Identifying a COPD-associated variant that is also an expression quantitative trait locus (eQTL) – i.e., a variant that also affects gene expression – can identify a causal gene, but most GWAS loci are likely not explained by existing eQTLs, particularly when the pervasiveness of eQTLs is considered(54). Previous studies have used multiple methods to identify the potential causal gene at a given locus(11,12), but of the dozens to hundreds of loci in COPD, only a small fraction of implicated loci have been investigated for functional effects(55–60). However, despite these challenges, these investigations have often led to important discoveries regarding disease mechanisms (Table 1). For example, FAM13A appears to promote β-catenin degradation, inhibiting Wnt activation relevant for alveolar type 2 cell repair and regeneration; as well as modulating airway TGF- β1 signaling(24,29,61). AGER and SFTPD both harbor coding variants that increase susceptibility to COPD and encode proteins that are biomarkers for COPD(62–65). In some cases, finding a COPD risk variant that also alters a molecular phenotype such as gene expression or alternative splicing can implicate a causal gene, such as in TGFB2 and FBXO38(66,67). For other regions, genes in or near the associated region offer intriguing hypotheses that require follow-up testing: ADGRG6 (previously GPR126), a G protein-coupled receptor which may have roles in airway remodeling(24); CHRM3, a cholinergic muscarinic receptor; genes in the MAPK pathway; IL17RD, which can regulate pathways of IL-17; CHIA, which encodes a protein that degrades chitin; and several genes involved in extracellular matrix, cell adhesion, cell-cell interactions, and elastin-associated microfibrils, including ITGA1, NPNT, MFAP2, and ADAMTSL3(11,25).
Table 1:
Selected genetic associations with evidence linking variants to genes and supportive functional data. eQTL = expression quantitative trait locus, pQTL = protein quantitative trait locus
| Gene name | Evidence for gene | Implications |
|---|---|---|
| AGER | Nonsynonymous / coding variant (pQTL, rs2070600) | sRAGE, encoded by AGER, associated with emphysema and lung function decline(69) |
| HHIP | Gene expression, chromosomal conformation capture, functional variants and eQTL, (e.g. rs6537296, rs1542725), murine model | Increased susceptibility in haploinsufficient mouse to cigarette smoke, age-related emphysema, and lymphocytic inflammation(47,48,100) |
| FAM13A | Gene expression, functional variants and eQTL (e.g. rs2013701), murine model | Effects on Wnt/B-catenin pathway, induces reactive oxygen species(61) |
| SFTPD | Nonsynonymous / coding variant (pQTL, rs721917) | Surfactant protein with immunomodulatory role; biomarker for COPD(25,62,64,65) |
| TGFB2 | Gene expression, functional variant identification in fibroblasts and eQTL (rs1690789) | With other associations, implicating members of the TGFB pathway in COPD(101) |
| SERPINA1 | Nonsynonymous variant, protein levels (pQTL rs28929474), familial segregation | Discovery in 1960’s led to protease-antiprotease hypothesis in COPD, AAT augmentation therapy(16,102) |
| TERT | Rare variants affect telomere length (e.g., rs372511089), segregation in families | Telomere pathway mutations can predispose to COPD and pulmonary fibrosis(103,104) |
Clinical and translational implications
What are the clinical and translational implications of the reported findings in COPD genetics? Simply understanding that a substantial portion of COPD susceptibility comes from genetic factors and early life events may help mitigate the detrimental effects of self-blame common in COPD(68). Our understanding of the genetic impact on specific phenotypes has also changed. Most identified COPD genetic susceptibility loci overlap with genetic susceptibility to reduced pre-bronchodilator spirometry in the general population. Analyses of other phenotypes or subsets of COPD has to date also found a high degree of overlap; whether discovery of novel loci not related to population-based lung function is due to a lack of sufficient sample size or specificity for these other phenotypes is not known. Genetic risk factors are complex and the genetic risk for COPD partially overlaps with asthma, pulmonary fibrosis, and many other phenotypes. Genetic susceptibility to COPD explained by currently available GWAS is due to many common variants of small effect. Thus, while pulmonologists traditionally consider genetic risk in settings such as alpha-1 antitrypsin deficiency, from a population standpoint a larger number of subjects at markedly elevated genetic risk are likely to be identified by aggregation of these common variants using approaches such as polygenic risk scores. High genetic risk individuals identified by common variant aggregation could be targeted for counseling on avoidance of harmful occupational exposures and smoking; or could be identified for clinical trials. Perhaps most importantly, the discovery of a large number of genetic loci relevant for human disease offers opportunities to investigate new pathobiology, link existing biological hypotheses to human disease, or develop biomarkers, such as sRAGE and SFTPD(29,62,69).
Future directions in COPD Genetics
Despite tremendous advances in COPD genetics over the last several decades, the field is still in its relative infancy. The first genome-wide association study was reported just over a decade ago, and whole genome sequencing data has only recently become available. The fraction of genetic risk (or heritability) that is explained by replicated COPD genetic loci is only ~10% in those of European ancestry and even less so in others. This ‘missing heritability’ is likely due to other, mostly common variants but also rare variants yet to be discovered(15,52). While a major goal of genetics is to develop therapeutics, this translational impact is most immediately apparent when the effects of the drug target are already well understood, and genetics is used to provide the link to human disease. Of the COPD loci identified by GWAS, the underlying pathobiology is understood partially for a handful, and not at all for the vast majority. A limited number of rare variants have been associated with COPD, including alpha-1 antitrypsin deficiency and telomeropathies. Finding additional rare variants in COPD, based on experience with rare variants in other complex diseases(70–73) suggests that these variants may be very rare, have more modest effect sizes, or are associated with specific phenotypes, and thus will require sample sizes at least as large as those for traditional genome-wide association studies to reach the same statistical rigor. While most studies have focused on the COPD case-control phenotype defined by spirometry, COPD is a heterogeneous disease, and phenotypes are much more nuanced than the presence or absence of airflow limitation. There is an increasing appreciation for the trajectory in early life and young adulthood that leads to COPD(74,75). While genetic factors solely affecting developmental processes may not be amenable to therapeutic intervention, some genetic loci associated with growth and development likely overlap with processes involved in injury, repair, and inflammation. For those with disease, there are marked differences in lung parenchyma, vasculature, and structure(76,77); and comorbidities such as asthma, cardiovascular disease, and lung cancer share some of the same genetic risk factors(11,78). Genetics also likely underlie differences in rates of lung function decline, emphysema progression, and rates and types of exacerbations(49,79,80).
The field of COPD genetics is rising to meet these challenges. Studies of more diverse ancestries and rarer variants are essential both for discovery of new genetic risk factors but also understanding the biology of discovered loci(81,82). Phenotypes continue to be refined, with attention to longitudinal studies(83,84), via more advanced imaging algorithms such as vascular segmentation and application of deep learning(85). Studies of both the pleiotropic effects of individual associated variants, and studies of specific phenotypes and subtypes will answer questions about the role of genetics in disease heterogeneity. Recently, loss of function variants in IL33 initially associated with asthma were also found to be associated with COPD, leading to a clinical trial of an IL33 inhibitor in COPD(24). Environmental exposures are clearly critical to COPD, and some genetic risk factors may be involved in gene-by-environment interactions(26,27), though power to detect such interactions is an important limitation. The growth of large epidemiologic studies and health systems in biobanks, and the decreasing cost of genotyping and DNA sequencing - in the UK Biobank, Kadoorie, and the Trans-Omics in Precision Medicine (TOPMed) program(23,86,87) will enable larger studies to increase the number of associated loci, leading to synergistic understanding of disease; refine understanding of associated phenotypes and disease risk in different ancestries; and lead to better and more useful polygenic risk prediction and stratification, including identification of disease subtypes. Most studies examine germline genetic variation, which is present in every cell, versus somatic mutations, which occur after conception, particularly in tumors; recently, somatic mutations related to clonal hematopoiesis in blood cells were associated with COPD (Qiao et al, accepted). Genetic variants cause their effect through other molecules, and do not act alone. Biochemical alterations to DNA, and molecules that bind to DNA, can affect the expression of genes. These epigenetic alterations are also associated with COPD susceptibility, severity, and mortality(88–92). Integrative omics and networks and systems approaches pair genetic variation with other omics data and examine relationships between genes and omics types. Combining genetic variation with gene expression; methylation and other epigenetic measures (such as microRNA); proteomics; and metabolomics from blood, lung, and respiratory and immune cell types from samples of different ages and clinical conditions can identify new effector genes, biomarkers, and disease pathobiology(92–97). The process of identifying the effect of a given associated locus - functional genetics, is perhaps the most challenging and immediate problem in human complex disease genetics. In addition to integrative omics methods, advances in high throughput functional studies such as massively parallel reporter assays (MPRA) and CRISPR editing techniques - combined, for example, with single-cell methods(98,99) allow high-throughput testing to identify causal variants. Resources such as TOPMed, LungMAP, ENCODE, and the Human Cell Atlas provide resources to identify relevant genetic regions and cell types.
COPD genetics has, to date, identified dozens of genetic loci, leading to an understanding of novel disease pathobiology. It has also confirmed a strong component of genetic risk for disease, and identified an effect early in life, and can quantify risk in an individual. Future studies will continue to advance our understanding of disease pathobiology; allow better risk stratification, disease classification, and subtyping across different ancestries; and ultimately result in information to better manage patients and develop new therapies for this deadly disease.
Figure 4:
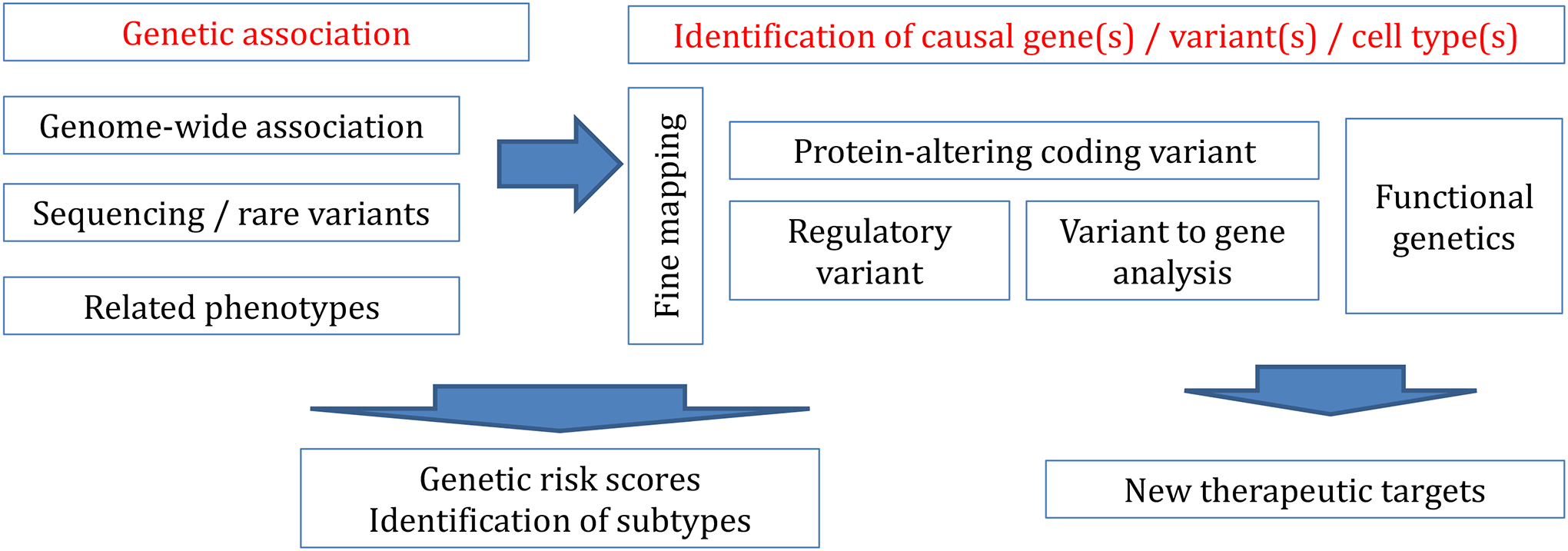
Genetic association with COPD or a COPD related phenotype, ideally with replication, can be done through standard genome-wide association studies for common variants, sequencing and rare variant based studies.
Once an association is identified, efforts should be made to identify the likely causal variant(s) through a procedure called ‘fine mapping’, since linkage disequilibrium results in extensive correlation of variants through the genome. For rare protein altering variants, there may be no other likely causal variants; for intragenic regions with high linkage disequilibrium, there may be hundreds or more potential causal variants.
Identifying a causal gene for regulatory variants (the majority of variants found by GWAS) is challenging: variant (or SNP) to gene strategies include molecular QTL (e.g. expression QTL, or eQTL) analysis ideally with colocalization, identification of open chromatin regions that correlate with gene expression, usually in specific cell types. ‘Functional genetics’ encompasses a set of experimental approaches to test the perturbation of an individual variant, regulatory region, or gene, in a specific cell type, organoid, or organism model, consistent with a hypothesis generated from downstream genetic analyses.
GWAS or association studies alone are sufficient to generate genetic risk scores, or in the case of variant(s) of large effect or affecting one pathway, a disease subtype (e.g. alpha-1 antitrypsin deficiency), though fine mapping and other methods may help improve these scores. To identify novel drug pathways and therapeutic targets, there should be an understanding of the basic effector gene and cell types, ideally after functional genetics assays.
Table 2:
Ongoing and Future Approaches in COPD Genetics
| Approach | Description |
|---|---|
| Biobanks | Large population- or healthcare system-based collections with a range of phenotyping, enabling an increase in sample size and examination of pleiotropic or genotype-first effects |
| Populations of diverse ancestry | Initiatives will enable and enhance discoveries and mitigate health inequalities |
| Phenomics | Comprehensive and detailed measures of phenotypes to identify and elucidate genetic mechanisms and disease heterogeneity |
| Whole-genome sequencing | Comprehensive analysis of the genome allowing identification and discovery of rare variants associated with disease |
| Integrative genomics | Incorporation of genetics with gene expression, epigenetics, proteins, and metabolites to identify genetic effects; systems biology and network methods to understand gene regulation |
| Single-cell sequencing | Identification of genetics, gene regulation, and gene expression in individual lung cells, rather than heterogeneous bulk tissue |
| High-throughput functional genomics | Methods to identify functional variants and effector genes at large scale; to be followed by detailed experimental approaches |
Key messages.
Genetics accounts for a substantial fraction of risk to develop COPD
Genetic variants, assigned at birth, can lead to important insight into causal pathobiology
While alpha-1 antitrypsin deficiency remains the most important genetic risk factor for COPD, most genetic risk appears to come from common variants, dozens to hundreds of which have been identified by GWAS
Each of these associations can lead to new discoveries about disease pathobiology
Though the key gene and functional variants in most COPD GWAS are unknown, at least some appear to be related to lung growth and development
A score comprised of many genetic variants can identify a group at high risk for COPD
Larger studies of more diverse phenotypes, ethnicities, rare variants, and integrative and network approaches incorporating other omics data are key future directions to understanding COPD genetics
Funding:
No funding was provided specifically for this Series paper.
The investigators are supported by R01HL137927, R01 HL152728, and P01 HL114501(EKS), R01 HL089856, R01 HL133135, P01 HL132825, and R01HL147148 (EKS and MHC), R01HL149861 and R01HL135142 (MHC); K08HL136928 (BDH).
The funders had no role in study design, data collection, data analysis, data interpretation, writing, or the decision to publish. The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute or the National Institutes of Health.
Conflicts of interest:
Edwin K. Silverman and Michael H. Cho have received grant support from GlaxoSmithKline and Bayer. Michael H. Cho has received consulting and speaking fees from Illumina and AstraZeneca.
Glossary
- Single nucleotide polymorphism / single nucleotide variant
The majority of human genetic variation consists of single nucleotide polymorphisms (SNPs); though the term SNV (single nucleotide variant) is more inclusive of the increasingly rare variation being discovered. The term ‘genetic variation’ includes these and other types of variants, such as insertion/deletion (indel), and copy number and structural variants of larger size
- Genome-wide association study (GWAS)
A GWAS is a method to test for an association of a genetic variant with disease or a specific trait. In contrast to a candidate gene study which only tests a few variants, a GWAS tests millions of SNVs across the genome, and includes extensive quality control, accounts for population substructure, adjusts for multiple testing, and usually includes replication
- Heritability
Heritability is a measure of the degree of variation in a trait due to genetic factors versus other (e.g., environmental, stochastic) factors, and can range from 0–100%. Heritability estimates can vary depending on the relative contribution of genetics to the phenotype in the population studied
- Genetic architecture
A concept encompassing the number, allele frequency, and magnitude of genetic variants (and their interactions with other genetic variants and environmental factors) associated with a given trait
- Pleiotropy
the ability of one gene (or genetic variant) to influence two or more seemingly unrelated traits
- Polygenic Risk Score
a numeric value generally created by aggregating the contribution of thousands or more genetic variants from genome-wide association studies. The resulting score can be used to assess the relative risk of disease
- Epigenetics
the study of mitotically heritable or other stable effects that alter the transcriptional potential of the cell not immediately due to the underlying DNA sequence
- Search strategy and selection criteria
We searched for COPD [MeSH] and “genetic association study” [MeSH] through October 9, 2021; and “genetic*” [MeSH] in PubMed from Jan 1, 2015 through October 9, 2021
References
- 1.Burrows B, Knudson RJ, Cline MG, Lebowitz MD. Quantitative relationships between cigarette smoking and ventilatory function. Am Rev Respir Dis [Internet]. Am Rev Respir Dis; 1977. Feb [cited 2021 Feb 22];115(2):195–205. Available from: http://www.ncbi.nlm.nih.gov/pubmed/842934 [DOI] [PubMed] [Google Scholar]
- 2.Marsh S, Aldington S, Shirtcliffe P, Weatherall M, Beasley R. Smoking and COPD: What really are the risks? Eur Respir J [Internet]. Eur Respir J; 2006. Oct [cited 2021 Feb 22];28(4):883–4. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17012635 [DOI] [PubMed] [Google Scholar]
- 3.Zhou JJ, Cho MH, Castaldi PJ, Hersh CP, Silverman EK, Laird NM. Heritability of Chronic Obstructive Pulmonary Disease and Related Phenotypes in Smokers. Am J Respir Crit Care Med [Internet]. 2013. Aug 23 [cited 2013 Aug 27];188(8):941–7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23972146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silverman EK, Chapman H a, Drazen JM, Weiss ST, Rosner B, Campbell EJ, et al. Genetic epidemiology of severe, early-onset chronic obstructive pulmonary disease. Risk to relatives for airflow obstruction and chronic bronchitis. Am J Respir Crit Care Med [Internet]. 1998. Jun;157(6 Pt 1):1770–8. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9620904 [DOI] [PubMed] [Google Scholar]
- 5.Polderman TJC, Benyamin B, de Leeuw CA, Sullivan PF, van Bochoven A, Visscher PM, et al. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat Genet [Internet]. 2015. Jul 18 [cited 2019 Jul 25];47(7):702–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25985137 [DOI] [PubMed] [Google Scholar]
- 6.Nelson MR, Tipney H, Painter JL, Shen J, Nicoletti P, Shen Y, et al. The support of human genetic evidence for approved drug indications. Nat Genet [Internet]. 2015. Jun 29 [cited 2015 Jun 30];47(8):856–60. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26121088 [DOI] [PubMed] [Google Scholar]
- 7.Ingebrigtsen T, Thomsen SF, Vestbo J, van der Sluis S, Kyvik KO, Silverman EK, et al. Genetic influences on Chronic Obstructive Pulmonary Disease - a twin study. Respir Med [Internet]. 2010. Dec [cited 2011 Mar 2];104(12):1890–5. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20541380 [DOI] [PubMed] [Google Scholar]
- 8.Zhou JJ, Cho MH, Castaldi PJ, Hersh CP, Silverman EK, Laird NM. Heritability of COPD and Related Phenotypes in Smokers. Am J Respir Crit Care Med [Internet]. 2013. Aug 23 [cited 2013 Aug 27]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/23972146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Castaldi PJ, Cho MH, Cohn M, Langerman F, Moran S, Tarragona N, et al. The COPD genetic association compendium: a comprehensive online database of COPD genetic associations. Hum Mol Genet [Internet]. 2009/11/26. 2010. Feb 1 [cited 2014 Sep 17];19(3):526–34. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2798725&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pillai SG, Ge D, Zhu G, Kong X, Shianna KV, Need AC, et al. A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet [Internet]. 2009/03/21. 2009;5(3):e1000421. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=19300482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakornsakolpat P, Prokopenko D, Lamontagne M, Reeve NF, Guyatt AL, Jackson VE, et al. Genetic landscape of chronic obstructive pulmonary disease identifies heterogeneous cell-type and phenotype associations. Nat Genet [Internet]. 2019. Mar 25 [cited 2019 Jun 28];51(3):494–505. Available from: http://www.nature.com/articles/s41588-018-0342-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shrine N, Guyatt AL, Erzurumluoglu AM, Jackson VE, Hobbs BD, Melbourne CA, et al. New genetic signals for lung function highlight pathways and chronic obstructive pulmonary disease associations across multiple ancestries. Nat Genet [Internet]. 2019. Mar 25 [cited 2019 Dec 1];51(3):481–93. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30804560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fuchsberger C, Flannick J, Teslovich TM, Mahajan A, Agarwala V, Gaulton KJ, et al. The genetic architecture of type 2 diabetes. Nature [Internet]. Nature; 2016. [cited 2021 Feb 21];536(7614):41–7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27398621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature [Internet]. Nature; 2014. Jul 24 [cited 2021 Feb 21];511(7510):421–7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25056061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang J, Bakshi A, Zhu Z, Hemani G, Vinkhuyzen AAE, Lee SH, et al. Genetic variance estimation with imputed variants finds negligible missing heritability for human height and body mass index. Nat Genet [Internet]. 2015. Oct 31 [cited 2019 Sep 5];47(10):1114–20. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26323059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Silverman EK, Sandhaus RA. Clinical practice. Alpha1-antitrypsin deficiency. N Engl J Med [Internet]. 2009/06/26. 2009. Jun 25 [cited 2015 Feb 16];360(26):2749–57. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19553648 [DOI] [PubMed] [Google Scholar]
- 17.Callewaert B, Renard M, Hucthagowder V, Albrecht B, Hausser I, Blair E, et al. New insights into the pathogenesis of autosomal dominant cutis laxa with report of five ELN mutations. Hum Mutat [Internet]. 2011. Feb [cited 2011 Feb 23]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/21309044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ayres JG, Pope FM, Reidy JF, Clark TJ. Abnormalities of the lungs and thoracic cage in the Ehlers-Danlos syndrome. Thorax [Internet]. Thorax; 1985. Apr [cited 2021 Feb 21];40(4):300–5. Available from: http://www.ncbi.nlm.nih.gov/pubmed/4023980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stanley SE, Merck SJ, Armanios M. Telomerase and the Genetics of Emphysema Susceptibility. Implications for Pathogenesis Paradigms and Patient Care. Ann Am Thorac Soc [Internet]. Ann Am Thorac Soc; 2016. [cited 2021 Feb 21];13 Suppl 5(Suppl 5):S447–51. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28005428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moll M, Jackson VE, Yu B, Grove ML, London SJ, Gharib SA, et al. A systematic analysis of protein-altering exonic variants in chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol. 2021. Jul;321(1):L130–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bossé Y, Lamontagne M, Gaudreault N, Racine C, Levesque M-H, Smith BM, et al. Early-onset emphysema in a large French-Canadian family: a genetic investigation. Lancet Respir Med [Internet]. Lancet Respir Med; 2019. [cited 2021 Feb 21];7(5):427–36. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31000475 [DOI] [PubMed] [Google Scholar]
- 22.Qiao D, Lange C, Beaty TH, Crapo JD, Barnes KC, Bamshad M, et al. Exome Sequencing Analysis in Severe, Early-Onset Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med [Internet]. 2016. Jun 15 [cited 2021 Feb 21];193(12):1353–63. Available from: http://www.atsjournals.org/doi/10.1164/rccm.201506-1223OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao X, Qiao D, Yang C, Kasela S, Kim W, Ma Y, et al. Whole genome sequence analysis of pulmonary function and COPD in 19,996 multi-ethnic participants. Nat Commun [Internet]. 2020. Dec 14 [cited 2021 Feb 21];11(1):5182. Available from: http://www.nature.com/articles/s41467-020-18334-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hall RJ, O’Loughlin J, Billington CK, Thakker D, Hall IP, Sayers I. Functional genomics of GPR126 in airway smooth muscle and bronchial epithelial cells. FASEB J Off Publ Fed Am Soc Exp Biol. United States; 2021. Jul;35(7):e21300. [DOI] [PubMed] [Google Scholar]
- 25.Hobbs BD, de Jong K, Lamontagne M, Bossé Y, Shrine N, Artigas MS, et al. Genetic loci associated with chronic obstructive pulmonary disease overlap with loci for lung function and pulmonary fibrosis. Nat Genet [Internet]. 2017. Feb 6 [cited 2017 Jul 5];49(3):426–32. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28166215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aschard H, Tobin MD, Hancock DB, Skurnik D, Sood A, James A, et al. Evidence for large-scale gene-by-smoking interaction effects on pulmonary function. Int J Epidemiol [Internet]. 2017. Jan 12 [cited 2017 Jan 18];dyw318. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28082375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Silverman EK, Province M a, Campbell EJ, Pierce J a, Rao DC. Family study of alpha 1-antitrypsin deficiency: effects of cigarette smoking, measured genotype, and their interaction on pulmonary function and biochemical traits. Genet Epidemiol [Internet]. 1992. Jan;9(5):317–31. Available from: http://www.ncbi.nlm.nih.gov/pubmed/1427021 [DOI] [PubMed] [Google Scholar]
- 28.Kim W, Prokopenko D, Sakornsakolpat P, Hobbs BD, Lutz SM, Hokanson JE, et al. Genome-Wide Gene-by-Smoking Interaction Study of Chronic Obstructive Pulmonary Disease. Am J Epidemiol [Internet]. 2021. May 4;190(5):875–85. Available from: 10.1093/aje/kwaa227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Milne S, Li X, Hernandez Cordero AI, Yang CX, Cho MH, Beaty TH, et al. Protective effect of club cell secretory protein (CC-16) on COPD risk and progression: a Mendelian randomisation study. Thorax. England; 2020. Nov;75(11):934–43. [DOI] [PubMed] [Google Scholar]
- 30.Manichaikul A, Hoffman EA, Smolonska J, Gao W, Cho MH, Baumhauer H, et al. Genome-wide study of percent emphysema on computed tomography in the general population. The Multi-Ethnic Study of Atherosclerosis Lung/SNP Health Association Resource Study. Am J Respir Crit Care Med [Internet]. 2014. Feb 15 [cited 2014 Jul 19];189(4):408–18. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24383474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cho MH, Castaldi PJ, Hersh CP, Hobbs BD, Barr RG, Tal-Singer R, et al. A Genome-Wide Association Study of Emphysema and Airway Quantitative Imaging Phenotypes. Am J Respir Crit Care Med [Internet]. 2015. Sep 1 [cited 2019 Dec 1];192(5):559–69. Available from: http://www.atsjournals.org/doi/10.1164/rccm.201501-0148OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee JH, Cho MH, Hersh CP, McDonald M-LN, Crapo JD, Bakke PS, et al. Genetic susceptibility for chronic bronchitis in chronic obstructive pulmonary disease. Respir Res [Internet]. 2014. Sep 21 [cited 2021 Feb 21];15(1):113. Available from: https://respiratory-research.biomedcentral.com/articles/10.1186/s12931-014-0113-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee JH, Cho MH, Hersh CP, McDonald M-LN, Wells JM, Dransfield MT, et al. IREB2 and GALC are associated with pulmonary artery enlargement in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol [Internet]. 2015. Mar [cited 2021 Feb 21];52(3):365–76. Available from: http://www.atsjournals.org/doi/10.1165/rcmb.2014-0210OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oelsner EC, Ortega VE, Smith BM, Nguyen JN, Manichaikul AW, Hoffman EA, et al. A Genetic Risk Score Associated with Chronic Obstructive Pulmonary Disease Susceptibility and Lung Structure on Computed Tomography. Am J Respir Crit Care Med [Internet]. 2019. Sep 15 [cited 2021 Feb 21];200(6):721–31. Available from: https://www.atsjournals.org/doi/10.1164/rccm.201812-2355OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parker MM, Lutz SM, Hobbs BD, Busch R, McDonald MN, Castaldi PJ, et al. Assessing pleiotropy and mediation in genetic loci associated with chronic obstructive pulmonary disease. Genet Epidemiol [Internet]. 2019. Apr [cited 2021 Feb 21];43(3):318–29. Available from: http://doi.wiley.com/10.1002/gepi.22192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Udler MS, Kim J, von Grotthuss M, Bonàs-Guarch S, Cole JB, Chiou J, et al. Type 2 diabetes genetic loci informed by multi-trait associations point to disease mechanisms and subtypes: A soft clustering analysis. Langenberg C, editor. PLOS Med [Internet]. 2018. Sep 21 [cited 2019 Jul 25];15(9):e1002654. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30240442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Udler MS, McCarthy MI, Florez JC, Mahajan A. Genetic Risk Scores for Diabetes Diagnosis and Precision Medicine. Endocr Rev [Internet]. The Endocrine Society; 2019. Dec 1 [cited 2019 Oct 16];40(6):1500–20. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31322649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Busch R, Hobbs BD, Zhou J, Castaldi PJ, McGeachie MJ, Hardin ME, et al. Genetic association and risk scores in a chronic obstructive pulmonary disease meta-analysis of 16,707 subjects. Am J Respir Cell Mol Biol. 2017;57(1):35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wain LV, Shrine N, Artigas MS, Erzurumluoglu AM, Noyvert B, Bossini-Castillo L, et al. Genome-wide association analyses for lung function and chronic obstructive pulmonary disease identify new loci and potential druggable targets. Nat Genet [Internet]. 2017. Mar 6 [cited 2019 Dec 1];49(3):416–25. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28166213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moll M, Sakornsakolpat P, Shrine N, Hobbs BD, DeMeo DL, John C, et al. Chronic obstructive pulmonary disease and related phenotypes: polygenic risk scores in population-based and case-control cohorts. Lancet Respir Med [Internet]. Lancet Respir Med; 2020. [cited 2021 Feb 21];8(7):696–708. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32649918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oelsner EC, Ortega VE, Smith BM, Nguyen JN, Manichaikul AW, Hoffman EA, et al. A Genetic Risk Score Associated with Chronic Obstructive Pulmonary Disease Susceptibility and Lung Structure on Computed Tomography. Am J Respir Crit Care Med [Internet]. 2019. Sep 15 [cited 2019 Oct 16];200(6):721–31. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30925230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Portas L, Pereira M, Shaheen SO, Wyss AB, London SJ, Burney PGJ, et al. Lung Development Genes and Adult Lung Function. Am J Respir Crit Care Med [Internet]. Am J Respir Crit Care Med; 2020. [cited 2021 Feb 21];202(6):853–65. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32392078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.John C, Soler Artigas M, Hui J, Nielsen SF, Rafaels N, Pare PD, et al. Genetic variants affecting cross-sectional lung function in adults show little or no effect on longitudinal lung function decline. Thorax. England; 2017. May;72(5):400–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ge T, Chen C-Y, Neale BM, Sabuncu MR, Smoller JW. Phenome-wide heritability analysis of the UK Biobank. PLoS Genet [Internet]. PLoS Genet; 2017. [cited 2021 Feb 21];13(4):e1006711. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28388634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McGeachie MJ, Yates KP, Zhou X, Guo F, Sternberg AL, Van Natta ML, et al. Patterns of Growth and Decline in Lung Function in Persistent Childhood Asthma. N Engl J Med [Internet]. 2016. May 12 [cited 2021 Feb 21];374(19):1842–52. Available from: http://www.nejm.org/doi/10.1056/NEJMoa1513737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lange P, Celli B, Agustí A, Boje Jensen G, Divo M, Faner R, et al. Lung-Function Trajectories Leading to Chronic Obstructive Pulmonary Disease. N Engl J Med [Internet]. 2015. Jul 9 [cited 2021 Feb 21];373(2):111–22. Available from: http://www.nejm.org/doi/10.1056/NEJMoa1411532 [DOI] [PubMed] [Google Scholar]
- 47.Zhou X, Baron RM, Hardin M, Cho MH, Zielinski J, Hawrylkiewicz I, et al. Identification of a chronic obstructive pulmonary disease genetic determinant that regulates HHIP. Hum Mol Genet [Internet]. 2011/12/06. 2011; Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=22140090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lao T, Jiang Z, Yun J, Qiu W, Guo F, Huang C, et al. Hhip haploinsufficiency sensitizes mice to age-related emphysema. Proc Natl Acad Sci [Internet]. 2016. Aug 9 [cited 2017 Jan 20];113(32):E4681–7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27444019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gottlieb DJ, Wilk JB, Harmon M, Evans JC, Joost O, Levy D, et al. Heritability of longitudinal change in lung function. The Framingham study. Am J Respir Crit Care Med [Internet]. 2001;164(9):1655–9. Available from: http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=11719305 [DOI] [PubMed] [Google Scholar]
- 50.Tang W, Kowgier M, Loth DW, Soler Artigas M, Joubert BR, Hodge E, et al. Large-Scale Genome-Wide Association Studies and Meta-Analyses of Longitudinal Change in Adult Lung Function. Chen L, editor. PLoS One [Internet]. 2014. Jul 1 [cited 2017 Jan 19];9(7):e100776. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24983941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hansel NN, Ruczinski I, Rafaels N, Sin DD, Daley D, Malinina A, et al. Genome-wide study identifies two loci associated with lung function decline in mild to moderate COPD. Hum Genet [Internet]. 2013. Jan [cited 2013 Jun 6];132(1):79–90. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22986903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wainschtein P, Jain D, Zheng Z, Group TopmAW, Consortium NT-O for PM (TOPMed), Cupples LA, et al. Recovery of trait heritability from whole genome sequence data. bioRxiv [Internet]. 2021. Jan 1;588020. Available from: http://biorxiv.org/content/early/2021/06/11/588020.abstract [Google Scholar]
- 53.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science [Internet]. 2012. Sep 7 [cited 2013 Mar 3];337(6099):1190–5. Available from: http://www.sciencemag.org/cgi/doi/10.1126/science.1222794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chun S, Casparino A, Patsopoulos NA, Croteau-Chonka DC, Raby BA, De Jager PL, et al. Limited statistical evidence for shared genetic effects of eQTLs and autoimmune-disease-associated loci in three major immune-cell types. Nat Genet [Internet]. 2017/02/20. 2017. Apr;49(4):600–5. Available from: https://pubmed.ncbi.nlm.nih.gov/28218759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou X, Baron RM, Hardin M, Cho MH, Zielinski J, Hawrylkiewicz I, et al. Identification of a chronic obstructive pulmonary disease genetic determinant that regulates HHIP. Hum Mol Genet [Internet]. 2012. Mar 15 [cited 2021 Feb 21];21(6):1325–35. Available from: https://academic.oup.com/hmg/article-lookup/doi/10.1093/hmg/ddr569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jiang Z, Lao T, Qiu W, Polverino F, Gupta K, Guo F, et al. A Chronic Obstructive Pulmonary Disease Susceptibility Gene, FAM13A, Regulates Protein Stability of β-Catenin. Am J Respir Crit Care Med [Internet]. 2016. Jul 15 [cited 2021 Feb 21];194(2):185–97. Available from: http://www.atsjournals.org/doi/10.1164/rccm.201505-0999OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.House JS, Li H, DeGraff LM, Flake G, Zeldin DC, London SJ. Genetic variation in HTR4 and lung function: GWAS follow-up in mouse. FASEB J [Internet]. 2015. Jan 23 [cited 2021 Feb 21];29(1):323–35. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1096/fj.14-253898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cloonan SM, Glass K, Laucho-Contreras ME, Bhashyam AR, Cervo M, Pabón MA, et al. Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice. Nat Med [Internet]. 2016. Feb 11 [cited 2021 Feb 21];22(2):163–74. Available from: http://www.nature.com/articles/nm.4021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang X, Guo Y, Yang J, Niu J, Du L, Li H, et al. A functional variant alters binding of activating protein 1 regulating expression of FGF7 gene associated with chronic obstructive pulmonary disease. BMC Med Genet [Internet]. 2019. Dec 18 [cited 2021 Feb 21];20(1):33. Available from: https://bmcmedgenet.biomedcentral.com/articles/10.1186/s12881-019-0761-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Silverman EK. Applying Functional Genomics to Chronic Obstructive Pulmonary Disease. Ann Am Thorac Soc [Internet]. 2018. Dec [cited 2021 Feb 21];15(Suppl 4):S239–42. Available from: https://www.atsjournals.org/doi/10.1513/AnnalsATS.201808-530MG [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jiang Z, Lao T, Qiu W, Polverino F, Gupta K, Guo F, et al. A Chronic Obstructive Pulmonary Disease Susceptibility Gene, FAM13A, Regulates Protein Stability of β-Catenin. Am J Respir Crit Care Med [Internet]. 2016. Jul 15 [cited 2017 Jan 20];194(2):185–97. Available from: http://www.atsjournals.org/doi/10.1164/rccm.201505-0999OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Obeidat M, Li X, Burgess S, Zhou G, Fishbane N, Hansel NN, et al. Surfactant protein D is a causal risk factor for COPD: results of Mendelian randomisation. Eur Respir J [Internet]. 2017. Nov 30 [cited 2021 Feb 22];50(5):1700657. Available from: http://erj.ersjournals.com/lookup/doi/10.1183/13993003.00657-2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yonchuk JG, Silverman EK, Bowler RP, Agustí A, Lomas DA, Miller BE, et al. Circulating soluble receptor for advanced glycation end products (sRAGE) as a biomarker of emphysema and the RAGE axis in the lung. Am J Respir Crit Care Med [Internet]. 2015. Oct 1 [cited 2021 Feb 22];192(7):785–92. Available from: http://www.atsjournals.org/doi/10.1164/rccm.201501-0137PP [DOI] [PubMed] [Google Scholar]
- 64.Sun W, Kechris K, Jacobson S, Drummond MB, Hawkins GA, Yang J, et al. Common Genetic Polymorphisms Influence Blood Biomarker Measurements in COPD. Gibson G, editor. PLoS Genet [Internet]. 2016. Aug 17 [cited 2017 Jun 22];12(8):e1006011. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27532455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Foreman MG, Demeo DL, Hersh CP, Reilly JJ, Carey VJ, Litonjua A a, et al. Polymorphisms in surfactant protein-D are associated with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol [Internet]. 2011. May [cited 2011 Jan 15];44(3):316–22. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20448057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Saferali A, Yun JH, Parker MM, Sakornsakolpat P, Chase RP, Lamb A, et al. Analysis of genetically driven alternative splicing identifies FBXO38 as a novel COPD susceptibility gene. Barsh GS, editor. PLoS Genet [Internet]. 2019. Jul 3 [cited 2021 Feb 22];15(7):e1008229. Available from: https://dx.plos.org/10.1371/journal.pgen.1008229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Parker MM, Hao Y, Guo F, Pham B, Chase R, Platig J, et al. Identification of an emphysema-associated genetic variant near TGFB2 with regulatory effects in lung fibroblasts. Elife [Internet]. 2019. Jul 25 [cited 2021 Feb 22];8. Available from: https://elifesciences.org/articles/42720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Plaufcan MR, Wamboldt FS, Holm KE. Behavioral and characterological self-blame in chronic obstructive pulmonary disease. J Psychosom Res [Internet]. J Psychosom Res; 2012. Jan [cited 2021 Feb 22];72(1):78–83. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22200527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yonchuk JG, Silverman EK, Bowler RP, Agustí A, Lomas DA, Miller BE, et al. Circulating Soluble Receptor for Advanced Glycation End Products (sRAGE) as a Biomarker of Emphysema and the RAGE Axis in the Lung. Am J Respir Crit Care Med [Internet]. 2015. Oct 1 [cited 2017 Jun 27];192(7):785–92. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26132989 [DOI] [PubMed] [Google Scholar]
- 70.Flannick J, Thorleifsson G, Beer NL, Jacobs SBR, Grarup N, Burtt NP, et al. Loss-of-function mutations in SLC30A8 protect against type 2 diabetes. Nat Genet [Internet]. 2014. Apr [cited 2015 Aug 29];46(4):357–63. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4051628&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Momozawa Y, Mni M, Nakamura K, Coppieters W, Almer S, Amininejad L, et al. Resequencing of positional candidates identifies low frequency IL23R coding variants protecting against inflammatory bowel disease. Nat Genet [Internet]. 2010. Dec [cited 2010 Dec 15];43(1):43–7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21151126 [DOI] [PubMed] [Google Scholar]
- 72.Do R, Stitziel NO, Won H-H, Jørgensen AB, Duga S, Angelica Merlini P, et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature [Internet]. 2014. Dec 10 [cited 2014 Dec 10];518(7537):102–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25487149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kiezun A, Garimella K, Do R, Stitziel NO, Neale BM, McLaren PJ, et al. Exome sequencing and the genetic basis of complex traits. Nat Genet [Internet]. 2012. Jun [cited 2013 Mar 1];44(6):623–30. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22641211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ross JC, Castaldi PJ, Cho MH, Hersh CP, Rahaghi FN, Sánchez-Ferrero GV, et al. Longitudinal Modeling of Lung Function Trajectories in Smokers with and without Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med [Internet]. 2018. Oct 15 [cited 2021 Feb 22];198(8):1033–42. Available from: https://www.atsjournals.org/doi/10.1164/rccm.201707-1405OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Agusti A, Faner R. Lung function trajectories in health and disease. Lancet Respir Med [Internet]. 2019. Apr [cited 2021 Feb 22];7(4):358–64. Available from: https://linkinghub.elsevier.com/retrieve/pii/S2213260018305290 [DOI] [PubMed] [Google Scholar]
- 76.Smith BM, Traboulsi H, Austin JHM, Manichaikul A, Hoffman EA, Bleecker ER, et al. Human airway branch variation and chronic obstructive pulmonary disease. Proc Natl Acad Sci [Internet]. 2018. Jan 30 [cited 2019 Oct 15];115(5):E974–81. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29339516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Castaldi PJ, Cho MH, Estépar RSJ, McDonald M-LN, Laird N, Beaty TH, et al. Genome-Wide Association Identifies Regulatory Loci Associated with Distinct Local Histogram Emphysema Patterns. Am J Respir Crit Care Med [Internet]. 2014. Jul 9 [cited 2014 Jul 18]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/25006744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McKay JD, Hung RJ, Han Y, Zong X, Carreras-Torres R, Christiani DC, et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat Genet [Internet]. Nat Genet; 2017. Jul [cited 2021 Feb 22];49(7):1126–32. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28604730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim W, Cho MH, Sakornsakolpat P, Lynch DA, Coxson HO, Tal-Singer R, et al. DSP variants may be associated with longitudinal change in quantitative emphysema. Respir Res [Internet]. Respir Res; 2019. Jul 19 [cited 2021 Feb 22];20(1):160. Available from: http://www.ncbi.nlm.nih.gov/pubmed/31324189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pavord ID, Chanez P, Criner GJ, Kerstjens HAM, Korn S, Lugogo N, et al. Mepolizumab for Eosinophilic Chronic Obstructive Pulmonary Disease. N Engl J Med [Internet]. Massachusetts Medical Society; 2017. Oct 26 [cited 2017 Nov 18];377(17):1613–29. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28893134 [DOI] [PubMed] [Google Scholar]
- 81.Wyss AB, Sofer T, Lee MK, Terzikhan N, Nguyen JN, Lahousse L, et al. Multiethnic meta-analysis identifies ancestry-specific and cross-ancestry loci for pulmonary function. Nat Commun [Internet]. 2018. Dec 30 [cited 2021 Feb 22];9(1):2976. Available from: http://www.nature.com/articles/s41467-018-05369-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Burkart KM, Sofer T, London SJ, Manichaikul A, Hartwig FP, Yan Q, et al. A Genome-wide Association Study in Hispanics/Latinos Identifies Novel Signals for Lung Function. The Hispanic Community Health Study/Study of Latinos. Am J Respir Crit Care Med [Internet]. 2018. Feb 2 [cited 2018 Jul 4];rccm.201707–1493OC. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29394082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bui DS, Perret JL, Walters EH, Abramson MJ, Burgess JA, Bui MQ, et al. Lifetime Risk Factors for Pre- and Post-Bronchodilator Lung Function Decline. A Population-based Study. Ann Am Thorac Soc [Internet]. 2020. Mar [cited 2021 Feb 22];17(3):302–12. Available from: https://www.atsjournals.org/doi/10.1513/AnnalsATS.201904-329OC [DOI] [PubMed] [Google Scholar]
- 84.Washko G, Colangelo L, Estépar RSJ, Ash S, Bhatt S, Okajima Y, et al. Adult Life-Course Trajectories of Lung Function and the Development of Emphysema: The CARDIA Lung Study. Am J Med [Internet]. 2019. Jul 29 [cited 2019 Sep 16]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/31369720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.González G, Ash SY, Vegas-Sánchez-Ferrero G, Onieva Onieva J, Rahaghi FN, Ross JC, et al. Disease Staging and Prognosis in Smokers Using Deep Learning in Chest Computed Tomography. Am J Respir Crit Care Med [Internet]. 2018. Jan 15 [cited 2021 Feb 22];197(2):193–203. Available from: http://www.atsjournals.org/doi/10.1164/rccm.201705-0860OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Taliun D, Harris DN, Kessler MD, Carlson J, Szpiech ZA, Torres R, et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature [Internet]. Nature; 2021. [cited 2021 Feb 22];590(7845):290–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/33568819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen Z, Chen J, Collins R, Guo Y, Peto R, Wu F, et al. China Kadoorie Biobank of 0.5 million people: survey methods, baseline characteristics and long-term follow-up. Int J Epidemiol [Internet]. Int J Epidemiol; 2011. Dec [cited 2021 Feb 22];40(6):1652–66. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22158673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vucic EA, Chari R, Thu KL, Wilson IM, Cotton AM, Kennett JY, et al. DNA methylation is globally disrupted and associated with expression changes in chronic obstructive pulmonary disease small airways. Am J Respir Cell Mol Biol [Internet]. 2014. May [cited 2015 Sep 2];50(5):912–22. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4068945&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Morrow JD, Cho MH, Hersh CP, Pinto-Plata V, Celli B, Marchetti N, et al. DNA methylation profiling in human lung tissue identifies genes associated with COPD. Epigenetics [Internet]. 2016. Oct 2 [cited 2020 Feb 8];11(10):730–9. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27564456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yoo S, Takikawa S, Geraghty P, Argmann C, Campbell J, Lin L, et al. Integrative analysis of DNA methylation and gene expression data identifies EPAS1 as a key regulator of COPD. PLoS Genet [Internet]. 2015. Jan [cited 2015 Aug 17];11(1):e1004898. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4287352&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Qiu W, Baccarelli A, Carey VJ, Boutaoui N, Bacherman H, Klanderman B, et al. Variable DNA methylation is associated with chronic obstructive pulmonary disease and lung function. Am J Respir Crit Care Med [Internet]. 2012. Feb 15 [cited 2015 Jul 9];185(4):373–81. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3297093&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Morrow JD, Glass K, Cho MH, Hersh CP, Pinto-Plata V, Celli B, et al. Human Lung DNA Methylation Quantitative Trait Loci Colocalize with Chronic Obstructive Pulmonary Disease Genome-Wide Association Loci. Am J Respir Crit Care Med [Internet]. 2018. May 15 [cited 2021 Feb 22];197(10):1275–84. Available from: https://www.atsjournals.org/doi/10.1164/rccm.201707-1434OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lamontagne M, Bérubé J-C, Obeidat M, Cho MH, Hobbs BD, Sakornsakolpat P, et al. Leveraging lung tissue transcriptome to uncover candidate causal genes in COPD genetic associations. Hum Mol Genet [Internet]. 2018. May 15 [cited 2021 Feb 22];27(10):1819–29. Available from: https://academic.oup.com/hmg/article/27/10/1819/4935076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Morrow JD, Zhou X, Lao T, Jiang Z, DeMeo DL, Cho MH, et al. Functional interactors of three genome-wide association study genes are differentially expressed in severe chronic obstructive pulmonary disease lung tissue. Sci Rep [Internet]. 2017. May 13 [cited 2021 Feb 22];7(1):44232. Available from: http://www.nature.com/articles/srep44232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sharma A, Kitsak M, Cho MH, Ameli A, Zhou X, Jiang Z, et al. Integration of Molecular Interactome and Targeted Interaction Analysis to Identify a COPD Disease Network Module. Sci Rep [Internet]. 2018. Dec 27 [cited 2021 Feb 22];8(1):14439. Available from: http://www.nature.com/articles/s41598-018-32173-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Halu A, Liu S, Baek SH, Hobbs BD, Hunninghake GM, Cho MH, et al. Exploring the cross-phenotype network region of disease modules reveals concordant and discordant pathways between chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Hum Mol Genet [Internet]. 2019. Jul 15 [cited 2021 Feb 22];28(14):2352–64. Available from: https://academic.oup.com/hmg/article/28/14/2352/5424998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Benway CJ, Liu J, Guo F, Du F, Randell SH, Cho MH, et al. Chromatin Landscapes of Human Lung Cells Predict Potentially Functional Chronic Obstructive Pulmonary Disease Genome-Wide Association Study Variants. Am J Respir Cell Mol Biol. 2021. Jul;65(1):92–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Patwardhan RP, Lee C, Litvin O, Young DL, Pe’er D, Shendure J. High-resolution analysis of DNA regulatory elements by synthetic saturation mutagenesis. Nat Biotechnol [Internet]. Nat Biotechnol; 2009. Dec [cited 2021 Feb 22];27(12):1173–5. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19915551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Gasperini M, Hill AJ, McFaline-Figueroa JL, Martin B, Kim S, Zhang MD, et al. A Genome-wide Framework for Mapping Gene Regulation via Cellular Genetic Screens. Cell [Internet]. 2019. Jan 10 [cited 2019 Jul 25];176(1–2):377–390.e19. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30612741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yun JH, Lee C, Liu T, Liu S, Kim EY, Xu S, et al. Hedgehog interacting protein-expressing lung fibroblasts suppress lymphocytic inflammation in mice. JCI Insight [Internet]. The American Society for Clinical Investigation; 2021. Aug 10; Available from: 10.1172/jci.insight.144575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Parker MM, Lutz SM, Hobbs BD, Busch R, McDonald MN, Castaldi PJ, et al. Assessing pleiotropy and mediation in genetic loci associated with chronic obstructive pulmonary disease. Genet Epidemiol [Internet]. 2019. Apr [cited 2019 Sep 14];43(3):318–29. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30740764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Laurell C-B, Eriksson A The electrophoretic alpha-1 globulin pattern of serum in alpha 1-antitrypsin deficiency. 1963;15:132–40. [DOI] [PubMed] [Google Scholar]
- 103.Stanley SE, Chen JJL, Podlevsky JD, Alder JK, Hansel NN, Mathias RA, et al. Telomerase mutations in smokers with severe emphysema. J Clin Invest [Internet]. American Society for Clinical Investigation; 2014. Dec 22 [cited 2015 Jan 7];125(125(2)):563–70. Available from: http://www.jci.org/articles/view/78554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Nunes H, Monnet I, Kannengiesser C, Uzunhan Y, Valeyre D, Kambouchner M, et al. Is telomeropathy the explanation for combined pulmonary fibrosis and emphysema syndrome?: report of a family with TERT mutation. Am J Respir Crit Care Med [Internet]. 2014. Mar 15 [cited 2014 Sep 10];189(6):753–4. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24628319 [DOI] [PubMed] [Google Scholar]
- 105.Wyss AB, Sofer T, Lee MK, Terzikhan N, Nguyen JN, Lahousse L, et al. Multiethnic meta-analysis identifies ancestry-specific and cross-ancestry loci for pulmonary function. Nat Commun [Internet]. 2018. Dec 30 [cited 2018 Oct 4];9(1):2976. Available from: http://www.nature.com/articles/s41467-018-05369-0 [DOI] [PMC free article] [PubMed] [Google Scholar]