

Abstract
The common fruit fly Drosophila melanogaster provides a powerful platform to investigate the genetic, molecular, cellular, and neural circuit mechanisms of behavior. Research in this model system has shed light on multiple aspects of brain physiology and behavior, from fundamental neuronal function to complex behaviors. A major anatomical region that modulates complex behaviors is the mushroom body (MB). The MB integrates multimodal sensory information and is involved in behaviors ranging from sensory processing/responses to learning and memory. Many genes that underlie brain disorders are conserved, from flies to humans, and studies in Drosophila have contributed significantly to our understanding of the mechanisms of brain disorders. Genetic mutations that mimic human diseases—such as Fragile X syndrome, neurofibromatosis type 1, Parkinson's disease, and Alzheimer's disease—affect MB structure and function, altering behavior. Studies dissecting the effects of disease-causing mutations in the MB have identified key pathological mechanisms, and the development of a complete connectome promises to add a comprehensive anatomical framework for disease modeling. Here, we review Drosophila models of human neurodevelopmental and neurodegenerative disorders via the effects of their underlying mutations on MB structure, function, and the resulting behavioral alterations.
Brain disorders affect a large percentage of the human population—current estimates suggest that ∼15% of people suffer from neurological disorders (Feigin et al. 2020) and 49.5% are affected by at least one class of mental health disorder (Merikangas et al. 2010). Two major classes of brain disorders are neurodevelopmental disorders and neurodegenerative disorders. Neurodevelopmental disorders alter biological processes during development, often impacting cognitive function and behavior. Such disorders affect 1%–3% of the world population (Kochinke et al. 2016). In addition, neurodegenerative disorders, characterized by progressive loss of neurons, affect 12%–14% of the population (Feigin et al. 2020). In some cases, genetic mutations are the root cause of the disorder. Such mutations exert complex effects on cellular signaling pathways, circuit function, and systemic physiology. Given this complexity, powerful genetic models such as Drosophila facilitate mechanistic dissection of disease pathophysiology. There is significant conservation of genes and cellular functions between flies and humans (Adams et al. 2000; Mohr and Perrimon 2019); ∼75% of known human disease-causing genes have orthologs in Drosophila (Rubin et al. 2000). To facilitate investigation of these conserved genes and signaling pathways, databases of human diseases have been generated for Drosophila (Millburn et al. 2016). Genetic screens and high-throughput phenotypic analyses in Drosophila have advanced the understanding of human diseases that result from a variety of genetic mutations.
The Drosophila mushroom body (MB) provides a robust anatomical platform to dissect the mechanisms underlying sensory integration/processing, complex behaviors such as learning and memory, and the effects of human disease-causing mutations on these processes. The MB consists of approximately 2000 intrinsic neurons in each hemisphere, which are called Kenyon cells (KCs). KC somata and dendrites are in the posterior brain and project fasciculated axon bundles anteriorly. As these axon bundles approach the front of the brain, they diverge, sending collaterals dorsally and medially into five lobes: the α, α′, β, β′, and γ lobes (Fig. 1A,B; Crittenden et al. 1998; Aso et al. 2014a). Along the longitudinal length of each lobe, there are multiple anatomically and functionally distinct compartments (Fig. 1B; Tanaka et al. 2008; Mao and Davis 2009). Each compartment receives input from discrete sets of modulatory afferent neurons (such as dopaminergic neurons) and sends their cholinergic output to discrete downstream output neurons (MBONs) that have different behavioral roles (Aso et al. 2014b; Barnstedt et al. 2016). This allows sensory information to drive different behavioral responses in a context-dependent manner (Aso et al. 2014b). Multimodal sensory signals are processed in the MB including visual, olfactory, gustatory, tactile, and auditory stimuli (Wolf et al. 1998; Liu et al. 1999; Popov et al. 2003; Kirkhart and Scott 2015; Vogt et al. 2016). Information is processed and modified by experience to alter learned behaviors via plasticity in the MB (Yu et al. 2005, 2006; Séjourné et al. 2011; Perisse et al. 2013; Tomchik and Davis 2013; Boto et al. 2014, 2019; Cohn et al. 2015; Hige et al. 2015; Yamagata et al. 2015; Berry et al. 2018; Louis et al. 2018; Handler et al. 2019; Phan et al. 2019; Zhang et al. 2019; Bilz et al. 2020; Baltruschat et al. 2021; Stahl et al. 2022). Along with associative learning, the MB modulates state-dependent behaviors such as sleep and hunger (Pitman et al. 2006; Sitaraman et al. 2015; Tsao et al. 2018).
Figure 1.
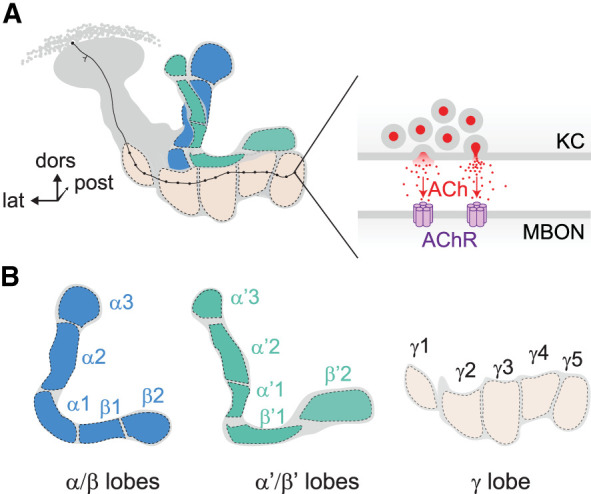
Diagram of mushroom body (MB) anatomy. (A) Frontal view of the MB lobes and their compartments. A single KC innervating the γ lobe is shown in black. The KC axon travels through multiple compartments, making en passant synapses with monoaminergic (e.g., dopaminergic) neurons (not shown) and downstream mushroom body output neurons (MBONs). (Inset) Expanded synaptic-scale view showing the presynaptic release of acetylcholine (ACh) onto a downstream MBON, activating ACh receptors (AChRs). (dors) Dorsal, (post) posterior, (lat) lateral. (B) Separate views of the α, β, α′, β′, and γ lobes. Each KC innervates either the α/β lobe, the α′/β′ lobe, or the γ lobe, passing through multiple synaptic compartments. Each numbered compartment is labeled.
In this review, we will highlight major findings in two broad categories of neurological diseases that are modeled in the Drosophila MB: neurodegenerative and neurodevelopmental disorders. Multiple genetic mutations that drive brain disorders in humans affect MB structure and function. The thoroughly characterized anatomy of the MB, along with its role in mediating complex behaviors, makes it an outstanding platform to dissect the mechanisms of these disorders. Complete coverage of the literature in this area would be too vast for a single review, and readers are directed to additional reviews for more information (Jaiswal et al. 2012; Şentürk and Bellen 2018; Mariano et al. 2020). Drosophila have been used in genetic research for more than 100 y, and a vast genetic toolkit has been developed over this time. For further information, readers are directed to technical reviews describing the Gal4/UAS system (and numerous derivatives/alternatives) (Luan et al. 2020), the types of genetic screens used (Yoon 2023), and current catalogs of RNAi and CRISPR mutations (Adams et al. 2000; Port et al. 2020; Hu et al. 2021; Zirin et al. 2022). The first sections of this review focus on major findings from studies on neurodegenerative disorders, defined by a progressive loss of susceptible neuronal populations (Dugger and Dickson 2017). In the neurodegenerative realm, we will focus on modeling Alzheimer's disease (AD) and Parkinson's disease (PD). Next, we will cover neurodevelopmental disorders, which are defined by their onset during a developmental period (Morris-Rosendahl and Crocq 2020). In this realm, we will highlight research into Fragile X syndrome (FXS) and neurofibromatosis type 1 (NF1). Finally, we briefly discuss several other disorders modeled in Drosophila. Throughout the review, we emphasize research that involves the MB.
Alzheimer's disease
AD is a neurodegenerative disease that typically appears late in adulthood. The disease results in neurodegeneration, with severe cases causing cortical shrinkage and ventricle enlargement (Fig. 2A,B). The first clue into the molecular nature of AD came from the discovery that the amyloid beta (Aβ) peptide accumulates in the brains of AD patients (Yoshikai et al. 1990). Aβ is generated via cleavage of the amyloid precursor protein (APP) (Drosophila express an APP-like protein, APPL). Under nonpathological conditions, this cleavage is catalyzed by α-secretase; however, under disease-state conditions, γ-secretase/β-site APP cleaving enzyme-1 (BACE1) produces a less soluble, neurotoxic Aβ peptide: Aβ42 peptide (De Strooper and Annaert 2000; O'Brien and Wong 2011; Liu et al. 2019). Pathophysiology of AD may involve altered neuronal excitability; accumulation of Aβ42 increases the excitability of neuronal circuits associated with neuronal degeneration (Ping et al. 2015; Tabuchi et al. 2015; Kaldun et al. 2021). Loss of synaptic inhibition leads to the neuronal excitation of cells localized near plaque formations (Busche et al. 2008). This potential pathophysiological mechanism has been modeled extensively in the MB; excess Aβ42 in the MB impairs courtship memory (Feng et al. 2018a), aversive short-/middle-term memory (Martin-Peña et al. 2017; Kaldun et al. 2021), forgetting (Kaldun et al. 2021), and middle-term appetitive memory (Kaldun et al. 2021). Mouse models have shown both increases in neuronal activity in some brain regions (e.g., the hippocampus) and decreases in activity of others (e.g., the amygdala, which exhibits increased inhibitory transmission) (Busche et al. 2008; Klein et al. 2014; Šišková et al. 2014). Notably, accumulation of Aβ42 in the MB α, β, and γ lobes decreases neuronal activity due to age-dependent loss of the A-type K+ channel Kv4 (Aso et al. 2009; Feng et al. 2018a). Behavioral alterations, such as impaired courtship memory, could result from a decrease in the odor-evoked activity of the MB (Feng et al. 2018a), where Aβ42 has aggregated (Higham et al. 2019). The loss of Kv4 activity also affects circuits outside of the MB, including those mediating circadian rhythms and flight maintenance (Ryglewski and Duch 2009; Feng et al. 2018b; Smith et al. 2019).
Figure 2.
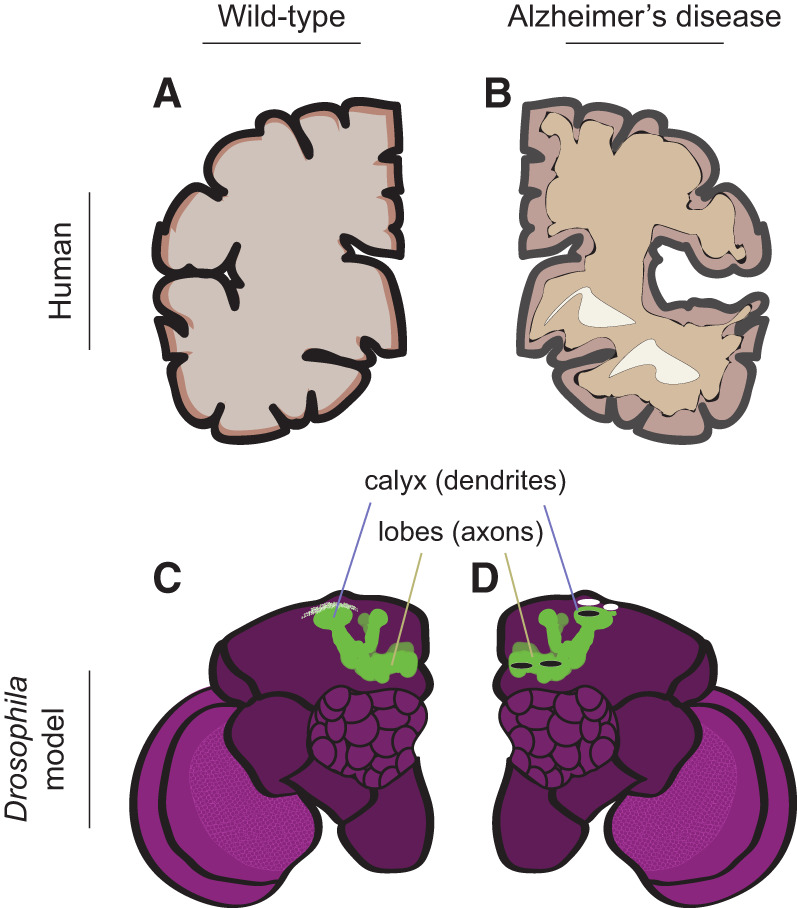
Alzheimer's disease (AD) pathology in humans and in the Drosophila AD model. (A) Diagram of a transverse section of healthy human brain. (B) AD pathology in the human brain (contralateral view relative to A), showing atrophied cerebral cortex and enlarged ventricles (white patches). (C) Diagram of a wild-type Drosophila brain, showing the MBs in green. (D) Pathology in the Drosophila R406W AD model (contralateral view relative to C). White ellipses represent vacuoles in the MB calyx (dendritic) regions, and black ellipses represent tau accumulation of R406W in the MB γ-lobe (axonal) and calyx (dendritic) regions.
Aggregation of Aβ42 is highly dependent on the specific mutation(s) in the precursor peptide. Sequencing of patient-derived mutations has pinpointed mutations responsible for early onset AD (EOAD). EOAD is characterized by a severe amalgamation of misfolded amyloid structures identified in individuals before the age of 65 (Nilsberth et al. 2001). One model of EOAD involves expressing mutant Aβ containing the Arctic mutation (Aβ42Arc). This mutation (APP E693G) is one of several that produces an autosomal dominant form of AD. When Aβ42Arc is expressed in the MB, the flies exhibit neuronal cell loss, visible in the dendritic region of the MB (calyx) as well as its axonal lobes (Fig. 2C,D; Iijima et al. 2008). Further, Drosophila models of EOAD cause progressive aggregation of Aβ42, and learning and memory deficits appear 7–9 d posteclosion. Other pathologies, such as mitochondrial perturbations, are observed even earlier (Kaldun et al. 2021; Wang and Davis 2021).
A second hallmark of human AD is the presence of neurofibrillary tangles composed of aggregated tau, a microtubule-binding protein. This aggregation occurs due to tau hyperphosphorylation and subsequent dissociation of the protein from microtubules (Hutton and Hardy 1997). Tau is critical for microtubule stability and for proper axonal trafficking (Cowan et al. 2010). Overexpression of wild-type tau in the Drosophila MB does not produce a degenerative phenotype (and Drosophila do not exhibit neurofibrillary tangles). Nonetheless, tau hyperphosphorylation recapitulates some features of human AD, including neuronal degradation (Wittmann et al. 2001). Modeling a form of EOAD, expression of the R406W mutation in Drosophila drives axonal decay and vacuole formation through the accumulation of hyperphosphorylated tau in cholinergic neurons (Fig. 2; Wittmann et al. 2001; Mershin et al. 2004). Comparing the pathology of human AD and the R406W model, Drosophila exhibit brain vacuoles but not neurofibrillary tangles, and AD patients exhibit neurofibrillary tangles, but not vacuoles. Despite these differences, a common feature is neurodegeneration, and neurodegeneration in the R406W model is progressive with age (Wittmann et al. 2001; Ali et al. 2012; Passarella and Goedert 2018). Further, there is impairment of learning and memory in Drosophila—accumulation of tau in the MB reduces short-term olfactory aversive memory (Mershin et al. 2004). Although overexpression assays identified tau as a target for neurodegenerative alterations, the molecular mechanisms were initially unclear. Genetic modifier screens targeting tauopathies subsequently identified a group of proteins that alter tau phosphorylation (Shulman and Feany 2003). The PAR-1 kinase kicks off a cascade of downstream phosphorylation events following the initial phosphorylation of Drosophila tau (Nishimura et al. 2004). Further, the key neurotoxic effects are localized to the phosphorylation sites at Ser238 and Thr245 (Kosmidis et al. 2010).
A common feature of many neurodegenerative diseases is misfolding and accumulation of proteins (Colla 2019). The unfolded protein response (UPR) is activated to restore proper endoplasmic reticulum (ER) homeostasis by increasing folding capacity (Walter and Ron 2011). This response is primarily due to a loss of Ca2+ homeostasis within the cell (Torres et al. 2010). There are three downstream components of the UPR pathway, inositol-requiring enzyme 1 (IRE1), PPKR-like endoplasmic reticulum kinase (PERK), and activating transcription factor (ATF6) (Mou et al. 2020) The UPR stress response of both IRE1 and PERK increase autophagy by increasing expression of autophagy receptors SQSTM1/p62, NBR1, and BNIP3L/NIX levels (Adolph et al. 2013; Deegan et al. 2015). Because of ER stress, PERK increases apoptotic response and translational arrest by activating eLF2α (Lin et al. 2007; Urra et al. 2013).
As understanding of AD pathology has increased, the range of potential therapeutic targets has expanded. Genetic screens in Drosophila have uncovered a range of mechanisms underlying AD dysfunction, involving oxidative stress, JNK signaling, and apolipoprotein D (Bowers et al. 2011; Hopkins 2013; Briston and Hicks 2018). These findings point to oxidative stress as a major contributor to AD pathology. Relatedly, metabolic changes, including changes in glucose metabolism through the TCA cycle, are potential therapeutic targets for AD (Lin and Beal 2006; Mattson et al. 2008; Reddy 2009; Wang et al. 2009). In Drosophila, expression of human tau drives mitochondrial elongation (along with mitochondrial dysfunction and cell death), implicating altered mitochondrial fusion/fission dynamics (DuBoff et al. 2012). Further, aggregation of Aβ42 alters the localization of mitochondria, increasing soma localization and decreasing mitochondrial localization to dendritic and axonal regions. This process involves a cAMP/PKA-dependent mechanism (Iijima-Ando et al. 2009). Translocation of mitochondria away from the axons and dendrites depletes local ATP sources that are used to generate cAMP, limits the mobilization of synaptic vesicles to the cleft for neurotransmission, and reduces synaptic strength (Verstreken et al. 2005). This neuropathological response is already present on the first day after eclosion, when mitochondria exhibit an increased number and altered morphology (Wang and Davis 2021).
An array of potential therapeutic treatments to rescue physiological, morphological, and behavioral responses have been explored using the MB as a test bed. Initial studies focused on the inhibition of the cleavage pathway for Aβ42 through the utilization of γ-secretase inhibitors (Chakraborty et al. 2011). Recent studies have focused on decreasing neuronal excitability induced by AD. Aβ42Arc expression induces sleep fragmentation, implicating sleep disruption and alteration of excitability in sleep-regulating circuits as a potential AD mechanism (Tabuchi et al. 2015; Gerstner et al. 2017). To ameliorate the alteration in neuronal excitability, gaboxadol and levetiracetam have been tested. Application of these drugs in flies expressing Aβ42Arc rescues memory deficits through reduction of circuitry excitability (Kaldun et al. 2021). Most recently, clinical trials have been initiated within individuals that suffer from Down syndrome, a population with the greatest risk of EOAD. The triplicate copy of Chromosome 21 may increase AD risk due to an extra copy of the DYRK1A kinase, which hyperphosphorylates tau (Ryoo et al. 2007). Administration of a DYRK1A inhibitor decreases the amount of phosphorylated tau, while rescuing both sleep and memory deficits in Drosophila (Zhu et al. 2022). Although a number of pathological mechanisms have been identified in AD, the genetic architecture underlying the disease is complex (Andrews et al. 2023). Future research will likely include more genetic sequencing of patients, as well as mechanistic studies and screens in animal models.
Parkinson's disease
PD is the most common movement disorder, characterized by the progressive loss of dopaminergic neurons in the nigrostriatal pathway (Fig. 3A,B) and concomitant motor dysfunction (Schrag et al. 2015). Locomotor deficits in PD include resting tremors, impaired movement initiation, and general instability (Whitworth 2011). The disease often drives accumulation of α-synuclein (α-syn) into globular structures referred to as Lewy bodies (Spillantini et al. 1997). Some cases of PD result from mutations in a single gene, though most result from a complex interaction of genetic and environmental factors (Selvaraj and Piramanayagam 2019). PD is modeled in Drosophila by depleting dopaminergic neurons via several approaches: inhibition of the mitochondrial respiratory transport chain (using the drug rotenone) (Betarbet et al. 2000), inducing oxidative stress (using paraquat) (Przedborski and Ischiropoulos 2005), or transgenically expressing α-syn (Feany and Bender 2000). Although Drosophila do not express an endogenous α-syn gene, transgenic expression of human α-syn recapitulates the central features of the disorder (progressive loss of dopaminergic neurons and locomotor dysfunction) (Feany and Bender 2000).
Figure 3.
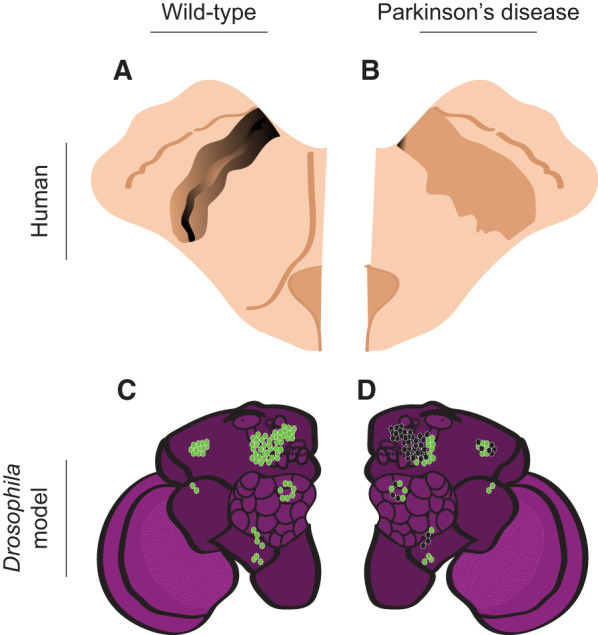
Parkinson's disease (PD) pathology in humans and in the Drosophila PD model. (A) Diagram of the human midbrain in transverse section, with the substantia nigra in black. (B) PD diminishes the substantia nigra (light brown) (contralateral view relative to A). (C) Diagram of dopaminergic neurons in wild-type Drosophila. Green circles represent dopaminergic neurons. (D) In the Pink1 Drosophila model of PD, dopaminergic neurons are lost in the PPL1, PPL2, and PPM1,2,3 clusters (contralateral view relative to C). Healthy neurons are shown in green and black circles represent neurons lost in the PD model.
Several subsets of dopaminergic neurons exhibit degeneration or morphological alterations in Drosophila PD models, including PPL1, PPL2, PAM, PPM1/2, PPM3, and PAL dopaminergic neurons (Fig. 3C,D). Among these, the PPL1, PPL2, and PAM neurons innervate the MB, modulating locomotion and learning (Claridge-Chang et al. 2009; Liu et al. 2012; Aso et al. 2014a; Galili et al. 2014; Cassari et al. 2015; Boto et al. 2019). To examine motor aberrations induced by α-syn aggregation and identify key dopaminergic clusters, a range of behavioral assays have been used, including startle-induced negative geotaxis (Riemensperger et al. 2013). This approach identified 15 protocerebral anterior medial (PAM) neurons, which directly innervate the β′ lobe, as crucial for locomotor alterations (Riemensperger et al. 2013). Pathophysiological changes to this circuitry involve neuronal hyperexcitability that progresses in an age-dependent manner; this diminishes connectivity between PAM neurons and KCs (Riemensperger et al. 2013). Correspondingly, α-syn aggregation causes the loss of neurons in the PPL1 and PPM1/2 clusters (Narwal et al. 2024).
The most common form of heritable PD results from mutations in the leucine-rich repeat kinase (LRRK2) (Paisán-Ruíz et al. 2004; Zimprich et al. 2004; Rajput et al. 2006; Ross et al. 2006). A hallmark of this form of PD is sleep deficiency (Berg et al. 2005). Expression of human LRRK2 (hLLRK2) in the MBs leads to a fragmentation in sleep that can be attenuated by melatonin (Sun et al. 2016). Furthermore, melatonin can also rescue long-term memory deficits induced by hLRRk2 expression (Ran et al. 2018). The pathological changes that produce PD-like behavioral outcomes likely involve changes in multiple signaling cascades including AKT, PTEN, and JNK (Mehdi et al. 2016). Expression of hLLRK2 leads to the degeneration of dopaminergic neurons localized to the PPL1 and PPM1/2 clusters (Islam et al. 2016).
The second most common form of inherited PD is due to a mutation in the PTEN-induced putative kinase (PINK1) (Kasten et al. 2018). In mammals, the hallmark of PD caused by PINK1 mutations is increased energetic demand within the dopaminergic neurons of the substantial nigra (Bolam and Pissadaki 2012). The increased energetic demand suggests that PINK1 mutations affect mitochondrial function (Pilsl and Winklhofer 2012). Neurons are lost in the PPL1, PPL2, PPM 1/2, and PPM3 clusters (Pirooznia et al. 2020; Zárate et al. 2022). In Drosophila, PINK1 binds directly to the mitochondrial protein PGAM5 to activate mitochondrial degradation followed by cell loss (Imai et al. 2010). PINK1 mutations can be exacerbated with the loss of PGAM5 (Ishida et al. 2012). PGAM5 increases mitochondrial turnover and fission events (Yu et al. 2020). Although there are additional Drosophila models of PD, we focus here on those that affect the MB. Mutations in hLRRK2, PINK1, α-syn, and Parkin also result in the loss of dopaminergic neurons that do not innervate the MB (MacLeod et al. 2013; Fellgett et al. 2021; Narwal et al. 2024). Examples of affected areas include dopaminergic neurons innervating the central complex (Dumitrescu et al. 2023) and subesophageal ganglion (Cording et al. 2017).
Emerging research has begun to unravel the roles of apoptosis and the UPR (Martinez et al. 2019). Accumulation of α-syn within the ER creates a stress-like response, activating the UPR (Walter and Ron 2011). In Drosophila, the response to the accumulation of α-syn involves hyperactivation of the IRE1 (Yan et al. 2019). Alternatively, the ATF6 pathway, which plays a role in the liberation of proteases, is inhibited through α-syn accumulation, causing dopaminergic neuronal death (Egawa et al. 2011). Unlike ATF6, the attenuation of PERK, which has been shown to cause apoptosis, can rescue the loss of PPL1 neurons in the Pink1 background (Popovic et al. 2023).
The primary treatment for PD, the administration of levodopa (l-DOPA)—a precursor to dopamine—has been used since the early 1970s (Tolosa et al. 1998). However, long-term exposure to high doses of l-DOPA can lead to a drug-induced dyskinesia (Parkinson Study Group 2000). Importantly, the effect of PD on monoaminergic pathways involves more than the loss of dopaminergic neurons. In Drosophila, l-DOPA decreases the innervation of the α/α′ lobes by serotonergic dorsal paired medial (DPM) neurons (Niens et al. 2017). Furthermore, the loss of PPL2 neurons that innervate the MB dendrites (among other brain regions) can be rescued by increasing serotonergic activity (Zárate et al. 2022). Overall, research in Drosophila has shed light on the molecular mechanisms that potentially drive the pathophysiology of PD (Shukla et al. 2014; Maitra et al. 2019; Ma et al. 2022; Zhang et al. 2023). Examining these molecular mechanisms within the well-defined anatomical context of the MB and its associated dopaminergic circuitry provides a platform to test novel therapeutic strategies for PD. Moving forward, these will likely include targeting α-syn receptors, autophagy-mediated pathways, and/or niacin targets (Rai et al. 2021).
Fragile X syndrome
FXS is a monogenetic inherited disorder (affecting ∼1 in 6000 births) caused by an expansion in the CGG trinucleotide repeat in the 5′-untranslated region of the FMR1 gene. Up to 55 repeats are present in normal individuals, and this expands to more than 200 in severely affected individuals (Willemsen et al. 2011). The number of CGG repeats affects FMR1 transcription, with longer repeats decreasing the amount of FMR protein (FMRP) (Schwemmle et al. 1997). FMRP is a major regulator of mRNA, modulating mRNA transport, stabilization, and translation (Bagni and Greenough 2005; Bassell and Warren 2008). FXS causes a range of symptoms, including developmental delays, learning disabilities, social and behavioral problems, impaired executive function, attention-deficit/hyperactivity disorder, sleep disturbances, intellectual disability, and anxiety (Crowe and Hay 1990; Fisch et al. 2002; Loesch et al. 2004; Scharf et al. 2015). Morphological alterations in the brain include aberrant dendritic spine morphology (Fig. 4A,B; Irwin et al. 2000).
Figure 4.
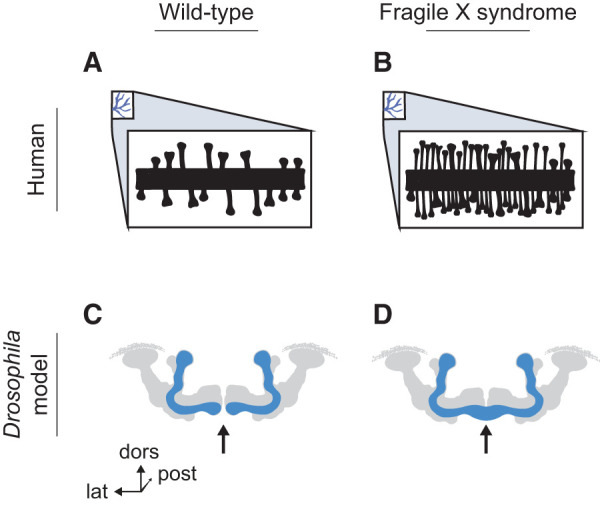
Fragile X syndrome (FXS) pathology in humans and in the Drosophila FXS model. (A) Diagram of dendritic spines in healthy humans. (B) Effects of FXS on dendritic spine morphology in humans. (C) Diagram of wild-type Drosophila MBs, showing the α/β lobes (in blue). The midline of the brain is marked with an arrow—note that the β lobes do not cross the midline. (dors) Dorsal, (post) posterior, (lat) lateral. (D) Pathology in the Drosophila FXS model. The β lobes overgrow, crossing the midline and infiltrating the contralateral lobe (arrow).
The Drosophila FMR1 gene (dFMR1) has high sequence homology with the human variant, including a conserved pair of KH domains (Wan et al. 2000). Loss-of-function mutations in dFMR1 alter circadian rhythms, courtship behaviors, and synaptic branching (Dockendorff et al. 2002). These mutations also alter MB morphology. In dFMR1 mutants, the KC axons that form the MB β-lobe aberrantly cross the midline of the brain (Fig. 4C,D), merging with the β lobe in the contralateral hemisphere (Michel et al. 2004). The dendritic branching pattern of the KCs is also altered by the addition of higher-order branches and supernumerary dendritic process formation (Pan et al. 2004). Long-term memory is dependent on dFMR1 expression (Dockendorff et al. 2002; McBride et al. 2005; Kanellopoulos et al. 2012). These morphological aberrations likely contribute to the deficits in learning. dFMR1 mutants also exhibit increases in synaptic boutons along the axons with increases in synaptic vesicle area. These phenotypes are likely due to either hyperactivity or inhibition of exocytotic events (Pan et al. 2004). Interestingly, dFMR1 heterozygotes have normal MBs but still show impaired long-term memory (Kanellopoulos et al. 2012).
Some FXS phenotypes may result from alterations in cAMP/PKA signaling. Human blood cell samples from FXS patients provided an early indication that FMRP could regulate the cAMP/PKA pathway (Berry-Kravis and Huttenlocher 1992). cAMP/PKA signaling plays a role in both memory acquisition and consolidation (Zars et al. 2000; Blum et al. 2009), and FMRP decreases cAMP generation following stimulation of adenylyl cyclases (Kelley et al. 2007). To examine the role of cAMP/PKA in a developmental/anatomical context, several studies investigated the role of FMRP in PKA regulation in the MB. PKA dynamically regulates the actin cytoskeleton to ensure proper neuronal growth (Lin et al. 2005; Cingolani and Goda 2008; Zhu et al. 2015). Localization of PKA is an important factor in its developmental effects; A-kinase anchor proteins (AKAPs) localize and regulate PKA activity (Smith et al. 2017; Wild and Dell'Acqua 2018). The Drosophila AKAP homolog, Rugose, interacts with PKA in the MB γ lobe and modulates short-term memory (Zhao et al. 2013). In FXS models, loss of FMRP down-regulates the expression of Rugose. This decreases PKA activity and alters F-actin assembly in the MB γ lobe (Sears et al. 2019). Patient-derived mutations have revealed other previously undefined roles of the arginine–glycine–glycine (RGG) domain of FMRP. For instance, a negative self-regulatory feedback loop suppresses FMRP levels due to PKA activation (Sears and Broadie 2020).
The role of FMRP in the maturation and pruning of dendrites was first identified in a mouse model, where the loss of FMRP led to increases in spine lengths and decreases in pruning identified in pyramidal neurons (Comery et al. 1997). Similarly, in dendritic arborization (multidendritic) neurons of Drosophila larvae, loss of FMR1 increases higher-order dendritic arborizations (Lee et al. 2003). One factor associated with increases in spine density is the activation of metabotropic glutamate receptors (mGluRs), which leads to the increase of FMRP in postsynaptic dendrites. Loss of FMRP generates an excessive number of long and thin dendrites (Fig. 4; Weiler and Greenough 1999). Similar to the mouse model, the MB possesses mGluRs localized to the dendrites within the calyces (Devaud et al. 2008). dFMR1 mutants exhibit increased mGluR expression, which leads to learning deficits that can be rescued through knockdown or pharmacological inhibition of mGluR (Kanellopoulos et al. 2012). Interestingly, increasing cAMP levels by inhibiting the phosphodiesterase also rescues mGluR-mediated learning deficits (Kanellopoulos et al. 2012; Choi et al. 2016).
Although therapeutic targeting of mGluR5 has shown promising results in rodents, clinical trials in humans have yet to produce an FDA-approved therapy. This is likely a consequence of the multitude of FXS symptoms (Scharf et al. 2015). Currently, symptoms are individually targeted, increasing the risk of side effects. Because the syndrome is monogenetic, much current research on potential therapeutics focuses on methods to increase endogenous FMR1 expression.
Neurofibromatosis type 1
NF1 results from loss-of-function mutations in the NF1 gene in humans, which encodes a protein called neurofibromin (Nf1). This disorder affects ∼1 in 3500 individuals (Evans et al. 2010; Hirbe and Gutmann 2014; Uusitalo et al. 2015). Although it is monogenetic in origin, genetic modifiers influence NF1 symptoms (Easton et al. 1993). Symptoms include the formation of tumors/cancers as well as an increased incidence of brain disorders. These include attention-deficit/hyperactivity disorder, autism spectrum disorder (ASD), learning disabilities, sleep disturbances, and others (North et al. 1994, 1995; Ferner et al. 1996; Wang et al. 2021). The Nf1 protein contains a central Ras-GTPase-activating protein-related domain (GRD), which negatively regulates Ras activity (Martin et al. 1990). In addition to modulating Ras signaling, loss of Nf1 also reduces cAMP/PKA levels and impacts G protein–coupled receptor signal transduction (Guo et al. 1997; Hannan et al. 2006; Ho et al. 2007; Anastasaki and Gutmann 2014; Xie et al. 2016). Importantly, Drosophila express an Nf1 ortholog, which shares 60% amino acid homology and conserved Ras GAP functionality with humans (The et al. 1997; Williams et al. 2001; Walker et al. 2006). The Drosophila NF1 model mimics a range of morphological and behavioral features of NF1, including increased metabolic rate, reduced body size, altered circadian rhythms, decreased sleep, changes in social behavior, increased grooming, and altered synaptic transmission (The et al. 1997; Williams et al. 2001; Bai and Sehgal 2015; King et al. 2016, 2020; Bai et al. 2018; Moscato et al. 2020; Botero et al. 2021; Dyson et al. 2022; Brown et al. 2023). Nf1 deficiency alters behaviors through effects on different sets of neurons (Guo et al. 2000; Buchanan and Davis 2010; King et al. 2020; Moscato et al. 2020; Georganta et al. 2021; Dyson et al. 2022), which are detailed further below.
Drosophila nf1 mutants show impaired learning and memory, particularly in olfactory classical conditioning (a commonly used associative learning paradigm) (Guo et al. 2000; Ho et al. 2007; Buchanan and Davis 2010; Gouzi et al. 2011; Qin et al. 2012; Georganta et al. 2021). This is reminiscent of NF1 in humans, which increases the incidence of learning disabilities (North et al. 1995). Learning and memory deficits in Drosophila result from alterations in MB function, with contributions from several cell types. In KCs, Nf1 is required for the acquisition of olfactory associative memory (Buchanan and Davis 2010). Rescue of wild-type Nf1 protein in a subset of MB neurons—the α/β neurons—restores normal memory (Buchanan and Davis 2010). Further, Nf1 interacts with cAMP/PKA signaling in the MB (Buchanan and Davis 2010). In addition to the KCs, Nf1 modulates memory via actions in a set of inhibitory GABAergic neurons that innervate the MB. Loss of Nf1 increases GABAergic circuits innervating the MB, contributing to learning deficits (Georganta et al. 2021). Rescuing Nf1 expression in these circuits restores normal memory (Georganta et al. 2021). This occurs via regulation of Ras in the GABAergic neurons, and signaling upstream of Ras contributes as well. The receptor tyrosine kinase anaplastic lymphoma kinase (Alk) is an upstream regulator of Ras that colocalizes with Nf1 in the nervous system and modulates the Nf1 learning phenotype (Gouzi et al. 2011; Bai and Sehgal 2015; Georganta et al. 2021). Thus, learning and memory are modulated by both cAMP and Ras signaling (Guo et al. 1997; Ho et al. 2007), with cAMP-dependent effects in KCs (Buchanan and Davis 2010) and Ras-dependent effects in the GABAergic neurons that innervate the MB (Georganta et al. 2021). This represents one example in which Nf1 deficiency in one set of cells can act via Ras and potentially affect another set of cells via cAMP/PKA signaling to produce a phenotype (Georganta et al. 2021). A similar interaction of different signaling pathways across cell types regulates the growth phenotype in Drosophila nf1 mutants (Walker et al. 2006) and optic glioma growth in mice (Pan et al. 2021).
Nf1 regulates circadian rhythms and sleep via its function in the MB. Loss of Nf1 disrupts the normal circadian rhythms of locomotor activity in Drosophila, along with reducing sleep (Williams et al. 2001; Bai and Sehgal 2015; Bai et al. 2018). Mutations in Alk also alter sleep because of effects in the MB, where Alk and Nf1 interact (Bai and Sehgal 2015). Circadian rhythms drive oscillations in gene expression in the MB, which are dependent on Nf1 and cAMP/PKA signaling (Almeida et al. 2021). Nf1 also acts downstream from the circadian clock and outside the MB, with the pars intercerebralis being one major site of action (Bai et al. 2018). In addition to its effect on sleep quantity, Nf1 modulates sleep quality (sleep depth) and the interaction of sleep with metabolic state (Brown et al. 2023). Like many other animals, flies exhibit a set of sleep states (Hendricks et al. 2000; Shaw et al. 2000). Loss of Nf1 fragments sleep and prevents flies from entering deep sleep (Brown et al. 2023). Further, it alters the interaction of sleep with metabolic state. When animals sleep, they normally suppress their metabolic rate. Yet flies with Nf1 mutations do not exhibit this suppression of metabolism during sleep (Brown et al. 2023). The sleep–metabolism interaction is not known to map to the MB. Yet reminiscent of the learning deficits, it relies on GABAergic circuitry. Nf1 is required in neurons that express the GABAA receptor Rdl, suggesting that circuitry immediately postsynaptic to GABAergic circuits regulates sleep–metabolism interactions (Brown et al. 2023).
Related to the learning and sleep phenotypes described above, loss of Nf1 in flies drives ASD-like behavioral changes, such as increased grooming (King et al. 2016, 2020) and altered social behavior (Moscato et al. 2020). Although the focus of this review is on MB effects, it is noteworthy that Nf1 modulates ASD-like phenotypes via actions outside the MB. In addition to those noted above, the grooming phenotype maps to cholinergic, Oct–Tyr receptor-expressing neurons in the ventral nerve cord (King et al. 2020). This effect is Ras-dependent and includes a developmental contribution (King et al. 2020). Additionally, social behavior alterations result from Nf1 function in adult chemosensory cells (ppk23+ neurons) and are also Ras-dependent (Moscato et al. 2020). Thus, the loss of Nf1 alters different behaviors via actions on different circuits. In some cases, the effects are likely additive across multiple neurons/circuits (King et al. 2020).
Therapeutic development to date has focused intensively on the mitogen-activated protein kinase (MAPK) signaling pathway (Ras/MEK/ERK). This pathway is important for some phenotypes in the Drosophila NF1 model. Increases in phospho-ERK accompany Nf1-dependent changes in body size, synaptic growth, and learning (Walker and Bernards 2014; Georganta et al. 2021). Metabolic alterations in nf1 mutants are dependent on Ras and likely involve ERK (Botero et al. 2021). In addition to MEK/ERK, other signaling pathways downstream from Ras are dysregulated in NF1 (Anastasaki et al. 2022). Among these, mTor has been implicated in other animal models of NF1, including modulating memory (in mammals) via effects on presynaptic neurotransmitter release (Asati et al. 2016; Choi et al. 2016). Whether/how this pathway contributes to Drosophila phenotypes is currently unknown. Future studies will be needed to understand how signaling pathways such as cAMP/PKA and PI3K/AKT/mTor modulate NF1 phenotypes, as well as how these pathways interact with MAPK signaling (such interactions occur in other model systems/phenotypes) (Anastasaki and Gutmann 2014). Ras signaling alterations also contribute to the circadian rhythm, grooming, and social behavior phenotypes (Williams et al. 2001; King et al. 2020; Moscato et al. 2020).
Molecular dissection of the signaling pathways downstream from Ras and cAMP will likely aid the development of therapeutic strategies. Similar to FXS, current treatments for NF1 are palliative and symptom-specific. Some patients experience multiple symptoms, and no single treatment addresses all of them (Walker and Upadhyaya 2018). A pharmacological inhibitor of MEK, selumetinib, is used to treat pediatric plexiform neurofibromas (effects of this pharmacological intervention have not been tested in Drosophila) (Gross et al. 2020). Another strategy involves the use of statins to inhibit Ras. Lovastatin, an HMG-CoA reductase, improves learning and attention in mice (Li et al. 2005). Another HMG-CoA reductase, simvastatin, rescues quantal size deficits at the Drosophila neuromuscular junction (Dyson et al. 2023). However, statins have not shown efficacy in clinical trials with children suffering from NF1 (Li et al. 2005; Payne et al. 2016). New approaches to treating NF1 may involve small molecules focusing on the regulation of Ras and/or cAMP signaling pathways (Walker and Upadhyaya 2018), as well as molecules targeting downstream effects on neuronal excitability (Pan et al. 2021; Dyson et al. 2023).
Other diseases modeled in the MB
Although this review focuses on four commonly studied diseases that alter the MB circuit, other diseases have been modeled as well. For instance, Angelman syndrome is a rare neurodevelopmental disorder that is characterized by delayed development, seizures, and intellectual disabilities. The disease is caused by mutations in Ube3a, which encodes a ubiquitin ligase (Kishino et al. 1997). Although the loss of the Drosophila homolog, dube3a, does not induce seizures, significant deficits were identified in long-term memory, climbing, and circadian rhythms (Wu et al. 2008). Similar to FXS, the fusing of β lobes is present along with the loss of α lobes in dube3a mutants (Chakraborty et al. 2015). Outside of the MB, mutant larvae exhibit morphological characteristics that are shared with mammals, including alterations in dendritic arborizations (Dindot et al. 2008; Lu et al. 2009). These dendritic alterations appear to be cell autonomous, and are dependent on expression level (Lu et al. 2009). In larval dendritic arborization (multidendritic) sensory neurons, Ube3a is responsible for proper pruning (Furusawa et al. 2023). The transport of Ube3a to presynaptic dendritic terminals is dependent on the kinesin motor and functions by maintaining the BMP signaling (Furusawa et al. 2023). For further information on Angelman syndrome, see Maranga et al. (2020).
Another well-defined class of neurodegenerative disorders that has been modeled includes the polyglutamine (polyQ) diseases. There are nine different polyQ diseases, including Huntington's, spinocerebellar ataxia types 1, 2, 3, 6, 7, and 17, spinobulbar muscular atrophy, and dentatorubral–pallidoluysian atrophy (Xu et al. 2015). Each of these is caused by a CAG repeat leading to a glutamine track forming somewhere in the protein-coding region of the gene (Macdonald et al. 1993). The number of these repeats varies significantly across the different diseases (Koide et al. 1994; Deka et al. 1995; Ikeuchi et al. 1995a,b; Komure et al. 1995). These polyQ repeats lead to a protein aggregation formation that precedes neurodegeneration. A multitude of transgenic models corresponding to each of the nine polyQ diseases have been generated as tools to identify the underlying pathophysiology (Fernandez-Funez et al. 2000; Chan et al. 2002; Takeyama et al. 2002; Warrick et al. 2005; Pandey et al. 2007; Nedelsky et al. 2010; Nisoli et al. 2010; Napoletano et al. 2011). Although many screens use S2 cells, or the eye, to investigate aggregation/neurodegeneration, significant loss in the α, β, and γ lobes of the MB has also been observed (Agrawal et al. 2005; Zhang et al. 2010; Song et al. 2013; Tandon and Sarkar 2023). This selective degradation is also found in the human neuropathology of each of the nine diseases (Tandon et al. 2024). For an extensive review on polyQ current research and therapeutics, see Tandon et al. (2024).
Overview and outlook
The Drosophila MB has functioned as a key source to identify the fundamental mechanisms of several distinct neurological disorders. The insights garnered from the model have shed light on the etiological identification of aberrated molecular mechanisms underlying multiple aspects of disease states. Although current genetic sequencing has identified many monogenetic mutations that cause disorders, many associated risk factors have yet to be discovered. Advancements in patient-derived sequencing will further personalize the therapeutic potential associated with many risk factors. These advancements, coupled with a comprehensive Drosophila connectome (Scheffer et al. 2020), will enable further dissection of how diseases alter biology (Scheffer et al. 2020). Further genetic studies are necessary to identify and characterize the full breadth of pathways that underlie and modulate brain disorders, and the MB provides an anatomical node to examine their effects on anatomy and complex behavior. From the forward genetic approaches that have identified foundational gene function to developmental and behavioral studies and detailed circuit studies, significant contributions have been made into disease mechanisms at the genetic, molecular, cellular, circuit, and behavioral levels. These discoveries are likely to continue and accelerate as fundamental nervous system function is more thoroughly dissected.
Acknowledgments
We thank Drs. Karla Kaun and André Fiala for organizing the Special Issue of Learning & Memory on the mushroom body, as well as Drs. André Fiala, Ilona Grunwald-Kadow, and Yoshinori Aso for organizing the 2023 mushroom body meeting that provided inspiration for these articles. Research in our laboratory on the mechanisms of learning and memory and NF1 is supported by the National Institutes of Health (NIH)/National Institute of Neurological Disorders and Stroke (NINDS) R01 NS114403, R01 NS124176, R01 NS126361, R01 NS097237, and R21 NS124198.
Footnotes
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.053816.123.
Freely available online through the Learning & Memory Open Access option.
References
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, et al. 2000. The genome sequence of Drosophila melanogaster. Science 287: 2185–2195. 10.1126/science.287.5461.2185 [DOI] [PubMed] [Google Scholar]
- Adolph TE, Tomczak MF, Niederreiter L, Ko HJ, Böck J, Martinez-Naves E, Glickman JN, Tschurtschenthaler M, Hartwig J, Hosomi S, et al. 2013. Paneth cells as a site of origin for intestinal inflammation. Nature 503: 272. 10.1038/nature12599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal N, Pallos J, Slepko N, Apostol BL, Bodai L, Chang LW, Chiang AS, Thompson LM, Marsh JL. 2005. Identification of combinatorial drug regimens for treatment of Huntington's disease using Drosophila. Proc Natl Acad Sci 102: 3777–3781. 10.1073/pnas.0500055102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali YO, Ruan K, Zhai RG. 2012. NMNAT suppresses tau-induced neurodegeneration by promoting clearance of hyperphosphorylated tau oligomers in a model of tauopathy. Hum Mol Genet 21: 237–250. 10.1093/hmg/ddr449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida PM, Solis BL, Feidler A, Nagoshi E, Stickley L. 2021. Neurofibromin 1 in mushroom body neurons mediates circadian wake drive through activating cAMP–PKA signaling. Nat Commun 12: 5758. 10.1038/s41467-021-26031-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasaki C, Gutmann DH. 2014. Neuronal NF1/RAS regulation of cyclic AMP requires atypical PKC activation. Hum Mol Genet 23: 6712–6721. 10.1093/hmg/ddu389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anastasaki C, Orozco P, Gutmann DH. 2022. RAS and beyond: the many faces of the neurofibromatosis type 1 protein. Dis Model Mech 15: dmm049362. 10.1242/dmm.049362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews SJ, Renton AE, Fulton-Howard B, Podlesny-Drabiniok A, Marcora E, Goate AM. 2023. The complex genetic architecture of Alzheimer's disease: novel insights and future directions. EBioMedicine 90: 104511. 10.1016/j.ebiom.2023.104511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asati V, Mahapatra DK, Bharti SK. 2016. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: structural and pharmacological perspectives. Eur J Med Chem 109: 314–341. 10.1016/j.ejmech.2016.01.012 [DOI] [PubMed] [Google Scholar]
- Aso Y, Grübel K, Busch S, Friedrich AB, Siwanowicz I, Tanimoto H. 2009. The mushroom body of adult Drosophila characterized by GAL4 drivers. J Neurogenet 23: 156–172. 10.1080/01677060802471718 [DOI] [PubMed] [Google Scholar]
- Aso Y, Hattori D, Yu Y, Johnston RM, Iyer NA, Ngo TTB, Dionne H, Abbott LF, Axel R, Tanimoto H, et al. 2014a. The neuronal architecture of the mushroom body provides a logic for associative learning. Elife 3: e04577. 10.7554/eLife.04577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aso Y, Sitaraman D, Ichinose T, Kaun KR, Vogt K, Belliart-Guerin G, Placais PY, Robie AA, Yamagata N, Schnaitmann C, et al. 2014b. Mushroom body output neurons encode valence and guide memory-based action selection in Drosophila. Elife 3: e04580. 10.7554/eLife.04580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagni C, Greenough WT. 2005. From mRNP trafficking to spine dysmorphogenesis: the roots of Fragile X syndrome. Nat Rev Neurosci 6: 376–387. 10.1038/nrn1667 [DOI] [PubMed] [Google Scholar]
- Bai L, Sehgal A. 2015. Anaplastic lymphoma kinase acts in the Drosophila mushroom body to negatively regulate sleep. PLoS Genet 11: e1005611. 10.1371/journal.pgen.1005611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai L, Lee Y, Hsu CT, Williams JA, Cavanaugh D, Zheng X, Stein C, Haynes P, Wang H, Gutmann DH, et al. 2018. A conserved circadian function for the neurofibromatosis 1 gene. Cell Rep 22: 3416–3426. 10.1016/j.celrep.2018.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltruschat L, Prisco L, Ranft P, Lauritzen JS, Fiala A, Bock DD, Tavosanis G. 2021. Circuit reorganization in the Drosophila mushroom body calyx accompanies memory consolidation. Cell Rep 34: 108871. 10.1016/j.celrep.2021.108871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnstedt O, Owald D, Felsenberg J, Brain R, Moszynski JP, Talbot CB, Perrat PN, Waddell S. 2016. Memory-relevant mushroom body output synapses are cholinergic. Neuron 89: 1237–1247. 10.1016/j.neuron.2016.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassell GJ, Warren ST. 2008. Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron 60: 201–214. 10.1016/j.neuron.2008.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg D, Schweitzer KJ, Leitner P, Zimprich A, Lichtner P, Belcredi P, Brüssel T, Schulte C, Maass S, Nägele T, et al. 2005. Type and frequency of mutations in the LRRK2 gene in familial and sporadic Parkinson's disease. Brain 128: 3000–3011. 10.1093/brain/awh666 [DOI] [PubMed] [Google Scholar]
- Berry JA, Phan A, Davis RL. 2018. Dopamine neurons mediate learning and forgetting through bidirectional modulation of a memory trace. Cell Rep 25: 651–662.e5. 10.1016/j.celrep.2018.09.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry-Kravis E, Huttenlocher PR. 1992. Cyclic-AMP metabolism in Fragile-X syndrome. Ann Neurol 31: 22–26. 10.1002/ana.410310105 [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. 2000. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci 3: 1301–1306. 10.1038/81834 [DOI] [PubMed] [Google Scholar]
- Bilz F, Geurten BRH, Hancock CE, Widmann A, Fiala A. 2020. Visualization of a distributed synaptic memory code in the Drosophila brain. Neuron 106: 963–976.e4. 10.1016/j.neuron.2020.03.010 [DOI] [PubMed] [Google Scholar]
- Blum AL, Li WH, Cressy M, Dubnau J. 2009. Short- and long-term memory in Drosophila require cAMP signaling in distinct neuron types. Curr Biol 19: 1341–1350. 10.1016/j.cub.2009.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolam JP, Pissadaki EK. 2012. Living on the edge with too many mouths to feed: why dopamine neurons die. Mov Disord 27: 1478–1483. 10.1002/mds.25135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botero V, Stanhope BA, Brown EB, Grenci EC, Boto T, Park SJ, King LB, Murphy KR, Colodner KJ, Walker JA, et al. 2021. Neurofibromin regulates metabolic rate via neuronal mechanisms in Drosophila. Nat Commun 12: 4285. 10.1038/s41467-021-24505-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boto T, Louis T, Jindachomthong K, Jalink K, Tomchik SM. 2014. Dopaminergic modulation of cAMP drives nonlinear plasticity across the Drosophila mushroom body lobes. Curr Biol 24: 822–831. 10.1016/j.cub.2014.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boto T, Stahl A, Zhang XF, Louis T, Tomchik SM. 2019. Independent contributions of discrete dopaminergic circuits to cellular plasticity, memory strength, and valence in Drosophila. Cell Rep 27: 2014–2021.e2. 10.1016/j.celrep.2019.04.069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers S, Truong AP, Neitz RJ, Hom RK, Sealy JM, Probst GD, Quincy D, Peterson B, Chan WM, Galemmo RA, et al. 2011. Design and synthesis of brain penetrant selective JNK inhibitors with improved pharmacokinetic properties for the prevention of neurodegeneration. Bioorg Med Chem Lett 21: 5521–5527. 10.1016/j.bmcl.2011.06.100 [DOI] [PubMed] [Google Scholar]
- Briston T, Hicks AR. 2018. Mitochondrial dysfunction and neurodegenerative proteinopathies: mechanisms and prospects for therapeutic intervention. Biochem Soc Trans 46: 829–842. 10.1042/BST20180025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EB, Zhang J, Lloyd E, Lanzon E, Botero V, Tomchik S, Keene AC. 2023. Neurofibromin 1 mediates sleep depth in Drosophila. PLoS Genet 19: e1011049. 10.1371/journal.pgen.1011049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan ME, Davis RL. 2010. A distinct set of brain neurons required for neurofibromatosis type 1–dependent learning and memory. J Neurosci 30: 10135–10143. 10.1523/JNEUROSCI.0283-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, Garaschuk O. 2008. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science 321: 1686–1689. 10.1126/science.1162844 [DOI] [PubMed] [Google Scholar]
- Cassari M, Issa AR, Riemensperger T, Petitgas C, Rival T, Coulom H, Iché-Torres M, Han KA, Birman S. 2015. A dopamine receptor contributes to paraquat-induced neurotoxicity in Drosophila. Hum Mol Genet 24: 197–212. 10.1093/hmg/ddu430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty R, Vepuri V, Mhatre SD, Paddock BE, Miller S, Michelson SJ, Delvadia R, Desai A, Vinokur M, Melicharek DJ, et al. 2011. Characterization of a Drosophila Alzheimer's disease model: pharmacological rescue of cognitive defects. PLoS ONE 6: e20799. 10.1371/journal.pone.0020799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty M, Paul BK, Nayak T, Das A, Jana NR, Bhutani S. 2015. The E3 ligase ube3a is required for learning in Drosophila melanogaster. Biochem Biophys Res Commun 462: 71–77. 10.1016/j.bbrc.2015.04.110 [DOI] [PubMed] [Google Scholar]
- Chan HYE, Warrick JM, Andriola I, Merry D, Bonini NM. 2002. Genetic modulation of polyglutamine toxicity by protein conjugation pathways in Drosophila. Hum Mol Genet 11: 2895–2904. 10.1093/hmg/11.23.2895 [DOI] [PubMed] [Google Scholar]
- Choi CH, Schoenfeld BP, Bell AJ, Hinchey J, Rosenfelt C, Gertner MJ, Campbell SR, Emerson D, Hinchey P, Kollaros M, et al. 2016. Multiple drug treatments that increase cAMP signaling restore long-term memory and aberrant signaling in Fragile X syndrome models. Front Behav Neurosci 10: 136. 10.3389/fnbeh.2016.00136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani LA, Goda Y. 2008. Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci 9: 494. 10.1038/nrn2410 [DOI] [PubMed] [Google Scholar]
- Claridge-Chang A, Roorda RD, Vrontou E, Sjulson L, Li HY, Hirsh J, Miesenböck G. 2009. Writing memories with light-addressable reinforcement circuitry. Cell 139: 405–415. 10.1016/j.cell.2009.08.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn R, Morantte I, Ruta V. 2015. Coordinated and compartmentalized neuromodulation shapes sensory processing in Drosophila. Cell 163: 1742–1755. 10.1016/j.cell.2015.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colla E. 2019. Linking the endoplasmic reticulum to Parkinson's disease and α-synucleinopathy. Front Neurosci 13: 560. 10.3389/fnins.2019.00560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comery TA, Harris JB, Willems PJ, Oostra BA, Irwin SA, Weiler IJ, Greenough WT. 1997. Abnormal dendritic spines in Fragile X knockout mice: maturation and pruning deficits. Proc Natl Acad Sci 94: 5401–5404. 10.1073/pnas.94.10.5401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cording AC, Shiaelis N, Petridi S, Middleton CA, Wilson LG, Elliott CJH. 2017. Targeted kinase inhibition relieves slowness and tremor in a Drosophila model of LRRK2 Parkinson's disease. NPJ Parkinsons Dis 3: 34. 10.1038/s41531-017-0036-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan CM, Bossing T, Page A, Shepherd D, Mudher A. 2010. Soluble hyper-phosphorylated tau causes microtubule breakdown and functionally compromises normal tau in vivo. Acta Neuropathol 120: 593–604. 10.1007/s00401-010-0716-8 [DOI] [PubMed] [Google Scholar]
- Crittenden JR, Skoulakis EMC, Han KA, Kalderon D, Davis RL. 1998. Tripartite mushroom body architecture revealed by antigenic markers. Learn Mem 5: 38–51. 10.1101/lm.5.1.38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe SF, Hay DA. 1990. Neuropsychological dimensions of the Fragile X syndrome: support for a non-dominant hemisphere dysfunction hypothesis. Neuropsychologia 28: 9–16. 10.1016/0028-3932(90)90082-Y [DOI] [PubMed] [Google Scholar]
- Deegan S, Koryga I, Glynn SA, Gupta S, Gorman AM, Samali A. 2015. A close connection between the PERK and IRE arms of the UPR and the transcriptional regulation of autophagy. Biochem Biophys Res Commun 456: 305–311. 10.1016/j.bbrc.2014.11.076 [DOI] [PubMed] [Google Scholar]
- Deka R, Miki T, Yin SJ, Mcgarvey ST, Shriver MD, Bunker CH, Raskin S, Hundrieser J, Ferrell RE, Chakraborty R. 1995. Normal CAG repeat variation at the DRPLA locus in world populations. Am J Hum Genet 57: 508–511. [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, Annaert W. 2000. Proteolytic processing and cell biological functions of the amyloid precursor protein. J Cell Sci 113: 1857–1870. 10.1242/jcs.113.11.1857 [DOI] [PubMed] [Google Scholar]
- Devaud JM, Couet-Redt C, Bockaert J, Grau Y, Parmentier ML. 2008. Widespread brain distribution of the Drosophila metabotropic glutamate receptor. Neuroreport 19: 367–371. 10.1097/WNR.0b013e3282f524c7 [DOI] [PubMed] [Google Scholar]
- Dindot SV, Antalffy BA, Bhattacharjee MB, Beaudet AL. 2008. The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum Mol Genet 17: 111–118. 10.1093/hmg/ddm288 [DOI] [PubMed] [Google Scholar]
- Dockendorff TC, Su HS, McBride SMJ, Yang ZH, Choi CH, Siwicki KK, Sehgal A, Jongens TA. 2002. Drosophila lacking dFMR1 activity show defects in circadian output and fail to maintain courtship interest. Neuron 34: 973–984. 10.1016/S0896-6273(02)00724-9 [DOI] [PubMed] [Google Scholar]
- DuBoff B, Götz J, Feany MB. 2012. Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron 75: 618–632. 10.1016/j.neuron.2012.06.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugger BN, Dickson DW. 2017. Pathology of neurodegenerative diseases. Cold Spring Harb Perspect Biol 9: a028035. 10.1101/cshperspect.a028035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumitrescu E, Copeland JM, Venton BJ. 2023. Parkin knockdown modulates dopamine release in the central complex, but not the mushroom body heel, of aging Drosophila. Acs Chem Neurosci 14: 198–208. 10.1021/acschemneuro.2c00277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson A, Ryan M, Garg S, Evans G, Baines RA. 2022. Loss of NF1 in Drosophila larvae causes tactile hypersensitivity and impaired synaptic transmission at the neuromuscular junction. J Neurosci 42: 9450–9472. 10.1523/JNEUROSCI.0562-22.2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyson A, Ryan M, Garg S, Evans G, Baines RA. 2023. A targeted, low-throughput compound screen in a Drosophila model of neurofibromatosis type 1 identifies simvastatin and BMS-204352 as potential therapies for autism spectrum disorder (ASD). eNeuro 10: ENEURO.0461-22.2023. 10.1523/ENEURO.0461-22.2023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton DF, Ponder MA, Huson SM, Ponder BAJ. 1993. An analysis of variation in expression of neurofibromatosis (Nf) type-1 (Nf1)—evidence for modifying genes. Am J Hum Genet 53: 305–313. [PMC free article] [PubMed] [Google Scholar]
- Egawa N, Yamamoto K, Inoue H, Hikawa R, Nishi K, Mori K, Takahashi R. 2011. The endoplasmic reticulum stress sensor, ATF6α, protects against neurotoxin-induced dopaminergic neuronal death. J Biol Chem 286: 7947–7957. 10.1074/jbc.M110.156430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans DG, Howard E, Giblin C, Clancy T, Spencer H, Huson SM, Lalloo F. 2010. Birth incidence and prevalence of tumor-prone syndromes: estimates from a UK family genetic register service. Am J Med Genet A 152a: 327–332. 10.1002/ajmg.a.33139 [DOI] [PubMed] [Google Scholar]
- Feany MB, Bender WW. 2000. A Drosophila model of Parkinson's disease. Nature 404: 394–398. 10.1038/35006074 [DOI] [PubMed] [Google Scholar]
- Feigin VL, Vos T, Nichols E, Owolabi MO, Carroll WM, Dichgans M, Deuschl G, Parmar P, Brainin M, Murray C. 2020. The global burden of neurological disorders: translating evidence into policy. Lancet Neurol 19: 255–265. 10.1016/S1474-4422(19)30411-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellgett A, Middleton CA, Munns J, Ugbode C, Jaciuch D, Wilson LG, Chawla S, Elliott CJH. 2021. Multiple pathways of LRRK2-G2019S/Rab10 interaction in dopaminergic neurons. J Parkinsons Dis 11: 1805–1820. 10.3233/JPD-202421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G, Pang J, Yi X, Song Q, Zhang JX, Li C, He G, Ping Y. 2018a. Down-regulation of KV4 channel in Drosophila mushroom body neurons contributes to Aβ42-induced courtship memory deficits. Neuroscience 370: 236–245. 10.1016/j.neuroscience.2017.06.008 [DOI] [PubMed] [Google Scholar]
- Feng G, Zhang JX, Li MZ, Shao LZ, Yang LN, Song Q, Ping Y. 2018b. Control of sleep onset by Shal/Kv4 channels in Drosophila circadian neurons. J Neurosci 38: 9059–9071. 10.1523/JNEUROSCI.0777-18.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Funez P, Nino-Rosales ML, de Gouyon B, She WC, Luchak JM, Martinez P, Turiegano E, Benito J, Capovilla M, Skinner PJ, et al. 2000. Identification of genes that modify ataxin-1-induced neurodegeneration. Nature 408: 101–106. 10.1038/35040584 [DOI] [PubMed] [Google Scholar]
- Ferner RE, Hughes RA, Weinman J. 1996. Intellectual impairment in neurofibromatosis 1. J Neurol Sci 138: 125–133. 10.1016/0022-510X(96)00022-6 [DOI] [PubMed] [Google Scholar]
- Fisch GS, Simensen RJ, Schroer RJ. 2002. Longitudinal changes in cognitive and adaptive behavior scores in children and adolescents with the Fragile X mutation or autism. J Autism Dev Disord 32: 107–114. 10.1023/A:1014888505185 [DOI] [PubMed] [Google Scholar]
- Furusawa K, Ishii K, Tsuji M, Tokumitsu N, Hasegawa E, Emoto K. 2023. Presynaptic Ube3a E3 ligase promotes synapse elimination through down-regulation of BMP signaling. Science 381: 1197–1205. 10.1126/science.ade8978 [DOI] [PubMed] [Google Scholar]
- Galili DS, Dylla KV, Lüdke A, Friedrich AB, Yamagata N, Wong JYH, Ho CH, Szyszka P, Tanimoto H. 2014. Converging circuits mediate temperature and shock aversive olfactory conditioning in Drosophila. Curr Biol 24: 1712–1722. 10.1016/j.cub.2014.06.062 [DOI] [PubMed] [Google Scholar]
- Georganta EM, Moressis A, Skoulakis EMC. 2021. Associative learning requires neurofibromin to modulate GABAergic inputs to Drosophila mushroom bodies. J Neurosci 41: 5274–5286. 10.1523/JNEUROSCI.1605-20.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstner JR, Lenz O, Vanderheyden WM, Chan MT, Pfeiffenberger C, Pack AI. 2017. Amyloid-β induces sleep fragmentation that is rescued by fatty acid binding proteins in Drosophila. J Neurosci Res 95: 1548–1564. 10.1002/jnr.23778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouzi JY, Moressis A, Walker JA, Apostolopoulou AA, Palmer RH, Bernards A, Skoulakis EMC. 2011. The receptor tyrosine kinase Alk controls neurofibromin functions in Drosophila growth and learning. PLoS Genet 7: e1002281. 10.1371/journal.pgen.1002281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross AM, Wolters PL, Dombi E, Baldwin A, Whitcomb P, Fisher MJ, Weiss B, Kim A, Bornhorst M, Shah AC, et al. 2020. Selumetinib in children with inoperable plexiform neurofibromas. N Engl J Med 383: 1290. 10.1056/NEJMx200013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo HF, The I, Hannan F, Bernards A, Zhong Y. 1997. Requirement of Drosophila NF1 for activation of adenylyl cyclase by PACAP38-like neuropeptides. Science 276: 795–798. 10.1126/science.276.5313.795 [DOI] [PubMed] [Google Scholar]
- Guo HF, Tong JY, Hannan F, Luo L, Zhong Y. 2000. A neurofibromatosis-1-regulated pathway is required for learning in Drosophila. Nature 403: 895–898. 10.1038/35002593 [DOI] [PubMed] [Google Scholar]
- Handler A, Graham TGW, Cohn R, Morantte I, Siliciano AF, Zeng JZ, Li YL, Ruta V. 2019. Distinct dopamine receptor pathways underlie the temporal sensitivity of associative learning. Cell 178: 60–75.e19. 10.1016/j.cell.2019.05.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannan F, Ho I, Tong JJY, Zhu YH, Nurnberg P, Zhong Y. 2006. Effect of neurofibromatosis type I mutations on a novel pathway for adenylyl cyclase activation requiring neurofibromin and Ras. Hum Mol Genet 15: 1087–1098. 10.1093/hmg/ddl023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendricks JC, Finn SM, Panckeri KA, Chavkin J, Williams JA, Sehgal A, Pack AI. 2000. Rest in Drosophila is a sleep-like state. Neuron 25: 129–138. 10.1016/S0896-6273(00)80877-6 [DOI] [PubMed] [Google Scholar]
- Hige T, Aso Y, Modi MN, Rubin GM, Turner GC. 2015. Heterosynaptic plasticity underlies aversive olfactory learning in Drosophila. Neuron 88: 985–998. 10.1016/j.neuron.2015.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higham JP, Malik BR, Buhl E, Dawson JM, Ogier AS, Lunnon K, Hodge JJL. 2019. Alzheimer's disease associated genes Ankyrin and tau cause shortened lifespan and memory loss in Drosphila. Front Cell Neurosci 13: 260. 10.3389/fncel.2019.00260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirbe AC, Gutmann DH. 2014. Neurofibromatosis type 1: a multidisciplinary approach to care. Lancet Neurol 13: 834–843. 10.1016/S1474-4422(14)70063-8 [DOI] [PubMed] [Google Scholar]
- Ho IS, Hannan F, Guo HF, Hakker I, Zhong Y. 2007. Distinct functional domains of neurofibromatosis type 1 regulate immediate versus long-term memory formation. J Neurosci 27: 6852–6857. 10.1523/JNEUROSCI.0933-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins PCR. 2013. Neurodegeneration in a Drosophila model for the function of TMCC2, an amyloid protein precursor-interacting and apolipoprotein E-binding protein. PLoS ONE 8: e55810. 10.1371/journal.pone.0055810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu YH, Comjean A, Rodiger J, Liu YF, Gao Y, Chung V, Zirin J, Perrimon N, Mohr SE. 2021. FlyRNAi.org-the database of the Drosophila RNAi screening center and transgenic RNAi project: 2021 update. Nucleic Acids Res 49: D908–D915. 10.1093/nar/gkaa936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton M, Hardy J. 1997. The presenilins and Alzheimer's disease. Hum Mol Genet 6: 1639–1646. 10.1093/hmg/6.10.1639 [DOI] [PubMed] [Google Scholar]
- Iijima K, Chiang HC, Hearn SA, Hakker I, Gatt A, Shenton C, Granger L, Leung A, Iijima-Ando K, Zhong Y. 2008. Aβ42 mutants with different aggregation profiles induce distinct pathologies in Drosophila. PLoS ONE 3: e1703. 10.1371/journal.pone.0001703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iijima-Ando K, Hearn SA, Shenton C, Gatt A, Zhao LJ, Iijima K. 2009. Mitochondrial mislocalization underlies Aβ42-induced neuronal dysfunction in a Drosophila model of Alzheimer's disease. PLoS ONE 4: e8310. 10.1371/journal.pone.0008310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeuchi T, Koide R, Tanaka H, Onodera O, Igarashi S, Takahashi H, Kondo R, Ishikawa A, Tomoda A, Miike T, et al. 1995a. Dentatorubral–pallidoluysian atrophy: clinical features are closely related to unstable expansions of trinucleotide (CAG) repeat. Ann Neurol 37: 769–775. 10.1002/ana.410370610 [DOI] [PubMed] [Google Scholar]
- Ikeuchi T, Onodera O, Oyake M, Koide R, Tanaka H, Tsuji S. 1995b. Dentatorubral–pallidoluysian atrophy (DRPLA): close correlation of CAG repeat expansions with the wide spectrum of clinical presentations and prominent anticipation. Semin Cell Biol 6: 37–44. 10.1016/1043-4682(95)90013-6 [DOI] [PubMed] [Google Scholar]
- Imai Y, Kanao T, Sawada T, Kobayashi Y, Moriwaki Y, Ishida Y, Takeda K, Ichijo H, Lu BW, Takahashi R. 2010. The loss of PGAM5 suppresses the mitochondrial degeneration caused by inactivation of PINK1 in Drosophila. PLoS Genet 6: e1001229. 10.1371/journal.pgen.1001229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin SA, Galvez R, Greenough WT. 2000. Dendritic spine structural anomalies in Fragile-X mental retardation syndrome. Cereb Cortex 10: 1038–1044. 10.1093/cercor/10.10.1038 [DOI] [PubMed] [Google Scholar]
- Ishida Y, Sekine Y, Oguchi H, Chihara T, Miura M, Ichijo H, Takeda K. 2012. Prevention of apoptosis by mitochondrial phosphatase PGAM5 in the mushroom body is crucial for heat shock resistance in Drosophila melanogaster. PLoS ONE 7: e30265. 10.1371/journal.pone.0030265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam MS, Nolte H, Jacob W, Ziegler AB, Pütz S, Grosjean Y, Szczepanowska K, Trifunovic A, Braun T, Heumann H, et al. 2016. Human R1441C LRRK2 regulates the synaptic vesicle proteome and phosphoproteome in a Drosophila model of Parkinson's disease. Hum Mol Genet 25: 5365–5382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaiswal M, Sandoval H, Zhang K, Bayat V, Bellen HJ. 2012. Probing mechanisms that underlie human neurodegenerative diseases in Drosophila. Annu Rev Genet 46: 371–396. 10.1146/annurev-genet-110711-155456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaldun JC, Lone SR, Camps AMH, Fritsch C, Widmer YF, Stein JV, Tomchik SM, Sprecher SG. 2021. Dopamine, sleep, and neuronal excitability modulate amyloid-β-mediated forgetting in Drosophila. PLoS Biol 19: e3001412. 10.1371/journal.pbio.3001412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanellopoulos AK, Semelidou O, Kotini AG, Anezaki M, Skoulakis EMC. 2012. Learning and memory deficits consequent to reduction of the Fragile X mental retardation protein result from metabotropic glutamate receptor-mediated inhibition of cAMP signaling in Drosophila. J Neurosci 32: 13111–13124. 10.1523/JNEUROSCI.1347-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasten M, Hartmann C, Hampf J, Schaake S, Westenberger A, Vollstedt EJ, Balck A, Domingo A, Vulinovic F, Dulovic M, et al. 2018. Genotype–phenotype relations for the Parkinson's disease genes Parkin, PINK1, DJ1: MDSGene systematic review. Mov Disord 33: 730–741. 10.1002/mds.27352 [DOI] [PubMed] [Google Scholar]
- Kelley DJ, Davidson RJ, Elliott JL, Lahvis GP, Yin JCP, Bhattacharyya A. 2007. The cyclic AMP cascade is altered in the Fragile X nervous system. PLoS ONE 2: e931. 10.1371/journal.pone.0000931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King LB, Koch M, Murphy KR, Velazquez Y, Ja WW, Tomchik SM. 2016. Neurofibromin loss of function drives excessive grooming in Drosophila. G3 (Bethesda) 6: 1083–1093. 10.1534/g3.115.026484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King LB, Boto T, Botero V, Aviles AM, Jomsky BM, Joseph C, Walker JA, Tomchik SM. 2020. Developmental loss of neurofibromin across distributed neuronal circuits drives excessive grooming in Drosophila. PLoS Genet 16: e1008920. 10.1371/journal.pgen.1008920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkhart C, Scott K. 2015. Gustatory learning and processing in the Drosophila mushroom bodies. J Neurosci 35: 5950–5958. 10.1523/JNEUROSCI.3930-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishino T, Lalande M, Wagstaff J. 1997. Erratum: UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet 15: 411. 10.1038/ng0497-411d [DOI] [PubMed] [Google Scholar]
- Klein RC, Acheson SK, Mace BE, Sullivan PM, Moore SD. 2014. Altered neurotransmission in the lateral amygdala in aged human apoE4 targeted replacement mice. Neurobiol Aging 35: 2046–2052. 10.1016/j.neurobiolaging.2014.02.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochinke K, Zweier C, Nijhof B, Fenckova M, Cizek P, Honti F, Keerthikumar S, Oortveld MA, Kleefstra T, Kramer JM, et al. 2016. Systematic phenomics analysis deconvolutes genes mutated in intellectual disability into biologically coherent modules. Am J Hum Genet 98: 149–164. 10.1016/j.ajhg.2015.11.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koide R, Ikeuchi T, Onodera O, Tanaka H, Igarashi S, Endo K, Takahashi H, Kondo R, Ishikawa A, Hayashi T, et al. 1994. Unstable expansion of CAG repeat in hereditary dentatorubral–pallidoluysian atrophy (DRPLA). Nat Genet 6: 9–13. 10.1038/ng0194-9 [DOI] [PubMed] [Google Scholar]
- Komure O, Sano A, Nishino N, Yamauchi N, Ueno S, Kondoh K, Sano N, Takahashi M, Murayama N, Kondo I, et al. 1995. DNA analysis in hereditary dentatorubral–pallidoluysian atrophy—correlation between CAG repeat length and phenotypic variation and the molecular-basis of anticipation. Neurology 45: 143–149. 10.1212/WNL.45.1.143 [DOI] [PubMed] [Google Scholar]
- Kosmidis S, Grammenoudi S, Papanikolopoulou K, Skoulakis EMC. 2010. Differential effects of τ on the integrity and function of neurons essential for learning in Drosophila. J Neurosci 30: 464–477. 10.1523/JNEUROSCI.1490-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A, Li WJ, Xu KY, Bogert BA, Su K, Gao FB. 2003. Control of dendritic development by the Drosophila Fragile X–related gene involves the small GTPase Rac1. Development 130: 5543–5552. 10.1242/dev.00792 [DOI] [PubMed] [Google Scholar]
- Li WD, Cui YJ, Kushner SA, Brown RAM, Jentsch JD, Frankland PW, Cannon TD, Silva AJ. 2005. The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Curr Biol 15: 1961–1967. 10.1016/j.cub.2005.09.043 [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. 2006. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443: 787–795. 10.1038/nature05292 [DOI] [PubMed] [Google Scholar]
- Lin B, Kramár EA, Bi XN, Brucher FA, Gall CM, Lynch G. 2005. Theta stimulation polymerizes actin in dendritic spines of hippocampus. J Neurosci 25: 2062–2069. 10.1523/JNEUROSCI.4283-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, LaVail MM, Walter P. 2007. IRE1 signaling affects cell fate during the unfolded protein response. Science 318: 944–949. 10.1126/science.1146361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Wolf R, Ernst R, Heisenberg M. 1999. Context generalization in Drosophila visual learning requires the mushroom bodies. Nature 400: 753–756. 10.1038/23456 [DOI] [PubMed] [Google Scholar]
- Liu C, Plaçais PY, Yamagata N, Pfeiffer BD, Aso Y, Friedrich AB, Siwanowicz I, Rubin GM, Preat T, Tanimoto H. 2012. A subset of dopamine neurons signals reward for odour memory in Drosophila. Nature 488: 512–516. 10.1038/nature11304 [DOI] [PubMed] [Google Scholar]
- Liu PP, Xie Y, Meng XY, Kang JS. 2019. History and progress of hypotheses and clinical trials for Alzheimer's disease. Signal Transduct Target Ther 4: 29. 10.1038/s41392-019-0063-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loesch DZ, Huggins RM, Hagerman RJ. 2004. Phenotypic variation and FMRP levels in Fragile X. Ment Retard Dev Disabil Res Rev 10: 31–41. 10.1002/mrdd.20006 [DOI] [PubMed] [Google Scholar]
- Louis T, Stahl A, Boto T, Tomchik SM. 2018. Cyclic AMP–dependent plasticity underlies rapid changes in odor coding associated with reward learning. Proc Natl Acad Sci 115: E448–E457. 10.1073/pnas.1709037115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YB, Wang F, Li Y, Ferris J, Lee JA, Gao FB. 2009. The Drosophila homologue of the Angelman syndrome ubiquitin ligase regulates the formation of terminal dendritic branches. Hum Mol Genet 18: 454–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan HJ, Diao FQ, Scott RL, White BH. 2020. The Drosophila Split Gal4 system for neural circuit mapping. Front Neural Circuits 14: 603397. 10.3389/fncir.2020.603397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma MQ, Moulton MJ, Lu SZ, Bellen HJ. 2022. ‘Fly-ing’ from rare to common neurodegenerative disease mechanisms. Trends Genet 38: 972–984. 10.1016/j.tig.2022.03.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, Srinidhi L, Barnes G, Taylor SA, James M, Groot N, et al. 1993. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 72: 971–983. 10.1016/0092-8674(93)90585-E [DOI] [PubMed] [Google Scholar]
- MacLeod DA, Rhinn H, Kuwahara T, Zolin A, Di Paolo G, McCabe BD, Marder KS, Honig LS, Clark LN, Small SA, et al. 2013. RAB7L1 interacts with LRRK2 to modify intraneuronal protein sorting and Parkinson's disease risk. Neuron 79: 202–203. 10.1016/j.neuron.2013.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra U, Scaglione MN, Chtarbanova S, O'Donnell JM. 2019. Innate immune responses to paraquat exposure in a Drosophila model of Parkinson's disease. Sci Rep 9: 12714. 10.1038/s41598-019-48977-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao ZM, Davis RL. 2009. Eight different types of dopaminergic neurons innervate the Drosophila mushroom body neuropil: anatomical and physiological heterogeneity. Front Neural Circuits 3: 5. 10.3389/neuro.04.005.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maranga C, Fernandes TG, Bekman E, da Rocha ST. 2020. Angelman syndrome: a journey through the brain. FEBS J 287: 2154–2175. 10.1111/febs.15258 [DOI] [PubMed] [Google Scholar]
- Mariano V, Achsel T, Bagni C, Kanellopoulos AK. 2020. Modelling learning and memory in Drosophila to understand intellectual disabilities. Neuroscience 445: 12–30. 10.1016/j.neuroscience.2020.07.034 [DOI] [PubMed] [Google Scholar]
- Martin GA, Viskoohil D, Bollag G, McCabe PC, Crosier WJ, Haubruck H, Conroy L, Clark R, O'Connell P, Cawthon RM, et al. 1990. The gap-related domain of the neurofibromatosis type-1 gene-product interacts with Ras P21. Cell 63: 843–849. 10.1016/0092-8674(90)90150-D [DOI] [PubMed] [Google Scholar]
- Martinez A, Lopez N, Gonzalez C, Hetz C. 2019. Targeting of the unfolded protein response (UPR) as therapy for Parkinson's disease. Biol Cell 111: 161–168. 10.1111/boc.201800068 [DOI] [PubMed] [Google Scholar]
- Martin-Peña A, Rincon-Limas DE, Fernandez-Funez P. 2017. Anti-Aβ single-chain variable fragment antibodies restore memory acquisition in a Drosophila model of Alzheimer's disease. Sci Rep 7: 11268. 10.1038/s41598-017-11594-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Gleichmann M, Cheng A. 2008. Mitochondria in neuroplasticity and neurological disorders. Neuron 60: 748–766. 10.1016/j.neuron.2008.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride SM, Choi CH, Wang Y, Liebelt D, Braunstein E, Ferreiro D, Sehgal A, Siwicki KK, Dockendorff TC, Nguyen HT, et al. 2005. Pharmacological rescue of synaptic plasticity, courtship behavior, and mushroom body defects in a Drosophila model of Fragile X syndrome. Neuron 45: 753–764. 10.1016/j.neuron.2005.01.038 [DOI] [PubMed] [Google Scholar]
- Mehdi SJ, Rosas-Hernandez H, Cuevas E, Lantz SM, Barger SW, Sarkar S, Paule MG, Ali SF, Imam SZ. 2016. Protein kinases and Parkinson's disease. Int J Mol Sci 17: 1585. 10.3390/ijms17091585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merikangas KR, He JP, Burstein M, Swanson SA, Avenevoli S, Cui LH, Benjet C, Georgiades K, Swendsen J. 2010. Lifetime prevalence of mental disorders in U.S. adolescents: results from the National Comorbidity Survey Replication-Adolescent Supplement (NCS-A). J Am Acad Child Adolesc Psychiatry 49: 980–989. 10.1016/j.jaac.2010.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mershin A, Pavlopoulos E, Fitch O, Braden BC, Nanopoulos DV, Skoulakis EMC. 2004. Learning and memory deficits upon TAU accumulation in Drosophila mushroom body neurons. Learn Mem 11: 277–287. 10.1101/lm.70804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel CI, Kraft R, Restifo LL. 2004. Defective neuronal development in the mushroom bodies of Drosophila Fragile X mental retardation 1 mutants. J Neurosci 24: 5798–5809. 10.1523/JNEUROSCI.1102-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millburn GH, Crosby MA, Gramates LS, Tweedie S, Consortium F. 2016. FlyBase portals to human disease research using Drosophila models. Dis Model Mech 9: 245–252. 10.1242/dmm.023317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr SE, Perrimon N. 2019. Drosophila melanogaster: a simple system for understanding complexity. Dis Model Mech 12: dmm041871. 10.1242/dmm.041871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris-Rosendahl DJ, Crocq MA. 2020. Neurodevelopmental disorders-the history and future of a diagnostic concept. Dialogues Clin Neurosci 22: 65–72. 10.31887/DCNS.2020.22.1/macrocq [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscato EH, Dubowy C, Walker JA, Kayser MS. 2020. Social behavioral deficits with loss of neurofibromin emerge from peripheral chemosensory neuron dysfunction. Cell Rep 32: 107856. 10.1016/j.celrep.2020.107856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mou Z, Yuan YH, Zhang Z, Song LK, Chen NH. 2020. Endoplasmic reticulum stress, an important factor in the development of Parkinson's disease. Toxicol Lett 324: 20–29. 10.1016/j.toxlet.2020.01.019 [DOI] [PubMed] [Google Scholar]
- Napoletano F, Occhi S, Calamita P, Volpi V, Blanc E, Charroux B, Royet J, Fanto M. 2011. Polyglutamine Atrophin provokes neurodegeneration in Drosophila by repressing fat. EMBO J 30: 945–958. 10.1038/emboj.2011.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narwal S, Singh A, Tare M. 2024. Analysis of α-syn and parkin interaction in mediating neuronal death in Drosophila model of Parkinson's disease. Front Cell Neurosci 17: 1295805. 10.3389/fncel.2023.1295805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedelsky NB, Pennuto M, Smith RB, Palazzolo I, Moore J, Nie ZP, Neale G, Taylor JP. 2010. Native functions of the androgen receptor are essential to pathogenesis in a Drosophila model of spinobulbar muscular atrophy. Neuron 67: 936–952. 10.1016/j.neuron.2010.08.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niens J, Reh F, Çoban B, Cichewicz K, Eckardt J, Liu YT, Hirsh J, Riemensperger TD. 2017. Dopamine modulates serotonin innervation in the Drosophila brain. Front Syst Neurosci 11: 76. 10.3389/fnsys.2017.00076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsberth C, Westlind-Danielsson A, Eckman CB, Condron MM, Axelman K, Forsell C, Stenh C, Luthman J, Teplow DB, Younkin SG, et al. 2001. The ‘Arctic’ APP mutation (E693G) causes Alzheimer's disease by enhanced Aβ protofibril formation. Nat Neurosci 4: 887–893. 10.1038/nn0901-887 [DOI] [PubMed] [Google Scholar]
- Nishimura I, Yang YF, Lu BW. 2004. PAR-1 kinase plays an initiator role in a temporally ordered phosphorylation process that confers tau toxicity in Drosophila. Cell 116: 671–682. 10.1016/S0092-8674(04)00170-9 [DOI] [PubMed] [Google Scholar]
- Nisoli I, Chauvin JP, Napoletano F, Calamita P, Zanin V, Fanto M, Charroux B. 2010. Neurodegeneration by polyglutamine Atrophin is not rescued by induction of autophagy. Cell Death Differ 17: 1577–1587. 10.1038/cdd.2010.31 [DOI] [PubMed] [Google Scholar]
- North K, Joy P, Yuille D, Cocks N, Mobbs E, Hutchins P, McHugh K, de Silva M. 1994. Specific learning disability in children with neurofibromatosis type 1: significance of MRI abnormalities. Neurology 44: 878–883. 10.1212/WNL.44.5.878 [DOI] [PubMed] [Google Scholar]
- North K, Joy P, Yuille D, Cocks N, Hutchins P. 1995. Cognitive function and academic performance in children with neurofibromatosis type 1. Dev Med Child Neurol 37: 427–436. 10.1111/j.1469-8749.1995.tb12026.x [DOI] [PubMed] [Google Scholar]
- O'Brien RJ, Wong PC. 2011. Amyloid precursor protein processing and Alzheimer's disease. Annu Rev Neurosci 34: 185–204. 10.1146/annurev-neuro-061010-113613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paisán-Ruíz C, Jain S, Evans EW, Gilks WP, Simón J, van der Brug M, de Munain AL, Aparicio S, Gil AM, Khan N, et al. 2004. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron 44: 595–600. 10.1016/j.neuron.2004.10.023 [DOI] [PubMed] [Google Scholar]
- Pan LY, Zhang YQ, Woodruff E, Broadie K. 2004. The Drosophila Fragile X gene negatively regulates neuronal elaboration and synaptic differentiation. Curr Biol 14: 1863–1870. 10.1016/j.cub.2004.09.085 [DOI] [PubMed] [Google Scholar]
- Pan Y, Hysinger JD, Barron T, Schindler NF, Cobb O, Guo X, Yalçın B, Anastasaki C, Mulinyawe SB, Ponnuswami A, et al. 2021. NF1 mutation drives neuronal activity-dependent initiation of optic glioma. Nature 594: 277–282. 10.1038/s41586-021-03580-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey UB, Nie ZP, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, DiProspero NA, Knight MA, Schuldiner O, et al. 2007. HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447: 859–863. 10.1038/nature05853 [DOI] [PubMed] [Google Scholar]
- Parkinson Study Group. 2000. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. J Am Med Assoc 284: 1931–1938. 10.1001/jama.284.15.1931 [DOI] [PubMed] [Google Scholar]
- Passarella D, Goedert M. 2018. β-Sheet assembly of Tau and neurodegeneration in Drosophila melanogaster. Neurobiol Aging 72: 98–105. 10.1016/j.neurobiolaging.2018.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne JM, Barton B, Ullrich NJ, Cantor A, Hearps SJC, Cutter G, Rosser T, Walsh KS, Gioia GA, Wolters PL, et al. 2016. Randomized placebo-controlled study of lovastatin in children with neurofibromatosis type 1. Neurology 87: 2575–2584. 10.1212/WNL.0000000000003435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perisse E, Yin Y, Lin AC, Lin SW, Huetteroth W, Waddell S. 2013. Different Kenyon cell populations drive learned approach and avoidance in Drosophila. Neuron 79: 945–956. 10.1016/j.neuron.2013.07.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan A, Thomas CI, Chakraborty M, Berry JA, Kamasawa N, Davis RL. 2019. Stromalin constrains memory acquisition by developmentally limiting synaptic vesicle pool size. Neuron 101: 103–118.e5. 10.1016/j.neuron.2018.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilsl A, Winklhofer KF. 2012. Parkin, PINK1 and mitochondrial integrity: emerging concepts of mitochondrial dysfunction in Parkinson's disease. Acta Neuropathol 123: 173–188. 10.1007/s00401-011-0902-3 [DOI] [PubMed] [Google Scholar]
- Ping Y, Hahm ET, Waro G, Song Q, Vo-Ba DA, Licursi A, Bao H, Ganoe L, Finch K, Tsunoda S. 2015. Linking Aβ42-induced hyperexcitability to neurodegeneration, learning and motor deficits, and a shorter lifespan in an Alzheimer's model. PLoS Genet 11: e1005025. 10.1371/journal.pgen.1005025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirooznia SK, Yuan CQ, Khan MR, Karuppagounder SS, Wang L, Xiong YL, Kang SU, Lee Y, Dawson VL, Dawson TM. 2020. PARIS induced defects in mitochondrial biogenesis drive dopamine neuron loss under conditions of parkin or PINK1 deficiency. Mol Neurodegener 15: 17. 10.1186/s13024-020-00363-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitman JL, McGill JJ, Keegan KP, Allada R. 2006. A dynamic role for the mushroom bodies in promoting sleep in Drosophila. Nature 441: 753–756. 10.1038/nature04739 [DOI] [PubMed] [Google Scholar]
- Popov AV, Sitnik NA, Savvateeva-Popova EV, Wolf R, Heisenberg M. 2003. The role of central parts of the brain in the control of sound production during courtship in Drosophila melanogaster. Neurosci Behav Physiol 33: 53–65. 10.1023/A:1021179331583 [DOI] [PubMed] [Google Scholar]
- Popovic R, Mukherjee A, Leal NS, Morris L, Yu YZ, Loh SHY, Martins LM. 2023. Blocking dPerk in the intestine suppresses neurodegeneration in a Drosophila model of Parkinson's disease. Cell Death Dis 14: 206. 10.1038/s41419-023-05729-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Port F, Strein C, Stricker M, Rauscher B, Heigwer F, Zhou J, Beyersdörffer C, Frei J, Hess A, Kern K, et al. 2020. A large-scale resource for tissue-specific CRISPR mutagenesis in Drosophila. Elife 9: e53865. 10.7554/eLife.53865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przedborski S, Ischiropoulos H. 2005. Reactive oxygen and nitrogen species: weapons of neuronal destruction in models of Parkinson's disease. Antioxid Redox Signal 7: 685–693. 10.1089/ars.2005.7.685 [DOI] [PubMed] [Google Scholar]
- Qin HT, Cressy M, Li WH, Coravos JS, Izzi SA, Dubnau J. 2012. γ Neurons mediate dopaminergic input during aversive olfactory memory formation in Drosophila. Curr Biol 22: 608–614. 10.1016/j.cub.2012.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai SN, Singh P, Varshney R, Chaturvedi VK, Vamanu E, Singh MP, Singh BK. 2021. Promising drug targets and associated therapeutic interventions in Parkinson's disease. Neural Regen Res 16: 1730–1739. 10.4103/1673-5374.306066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajput A, Dickson DW, Robinson CA, Ross OA, Dächsel JC, Lincoln SJ, Cobb SA, Rajput ML, Farrer MJ. 2006. Parkinsonism, Lrrk2 G2019S, and tau neuropathology. Neurology 67: 1506–1508. 10.1212/01.wnl.0000240220.33950.0c [DOI] [PubMed] [Google Scholar]
- Ran DZ, Xie BG, Gan ZJ, Sun XC, Gu HY, Yang JQ. 2018. Melatonin attenuates hLRRK2-induced long-term memory deficit in a Drosophila model of Parkinson's disease. Biomed Rep 9: 221–226. 10.3892/br.2018.1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy PH. 2009. Amyloid β, mitochondrial structural and functional dynamics in Alzheimer's disease. Exp Neurol 218: 286–292. 10.1016/j.expneurol.2009.03.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riemensperger T, Issa AR, Pech U, Coulom H, Nguyen MV, Cassar M, Jacquet M, Fiala A, Birman S. 2013. A single dopamine pathway underlies progressive locomotor deficits in a Drosophila model of Parkinson disease. Cell Rep 5: 952–960. 10.1016/j.celrep.2013.10.032 [DOI] [PubMed] [Google Scholar]
- Ross OA, Toft M, Whittle AJ, Johnson JL, Papapetropoulos S, Mash DC, Litvan I, Gordon MF, Wszolek ZK, Farrer MJ, et al. 2006. Lrrk2 and Lewy body disease. Ann Neurol 59: 388–393. 10.1002/ana.20731 [DOI] [PubMed] [Google Scholar]
- Rubin GM, Yandell MD, Wortman JR, Miklos GLG, Nelson CR, Hariharan IK, Fortini ME, Li PW, Apweiler R, Fleischmann W, et al. 2000. Comparative genomics of the eukaryotes. Science 287: 2204–2215. 10.1126/science.287.5461.2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryglewski S, Duch C. 2009. Shaker and Shal mediate transient calcium-independent potassium current in a Drosophila flight motoneuron. J Neurophysiol 102: 3673–3688. 10.1152/jn.00693.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryoo SR, Jeong HK, Radnaabazar C, Yoo JJ, Cho HJ, Lee HW, Kim IS, Cheon YH, Ahn YS, Chung SH, et al. 2007. DYRK1A-mediated hyperphosphorylation of tau—a functional link between Down syndrome and Alzheimer disease. J Biol Chem 282: 34850–34857. 10.1074/jbc.M707358200 [DOI] [PubMed] [Google Scholar]
- Scharf SH, Jaeschke G, Wettstein JG, Lindemann L. 2015. Metabotropic glutamate receptor 5 as drug target for Fragile X syndrome. Curr Opin Pharmacol 20: 124–134. 10.1016/j.coph.2014.11.004 [DOI] [PubMed] [Google Scholar]
- Scheffer LK, Xu CS, Januszewski M, Lu ZY, Takemura SY, Hayworth KJ, Huang GB, Shinomiya K, Maitlin-Shepard J, Berg S, et al. 2020. A connectome and analysis of the adult Drosophila central brain. Elife 9: e57443. 10.7554/eLife.57443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrag A, Horsfall L, Walters K, Noyce A, Petersen I. 2015. Prediagnostic presentations of Parkinson's disease in primary care: a case-control study. Lancet Neurol 14: 57–64. 10.1016/S1474-4422(14)70287-X [DOI] [PubMed] [Google Scholar]
- Schwemmle S, de Graaff E, Deissler H, Gläser D, Wöhrle D, Kennerknecht I, Just W, Oostra BA, Döerfler W, Vogel W, et al. 1997. Characterization of FMR1 promoter elements by in vivo-footprinting analysis. Am J Hum Genet 60: 1354–1362. 10.1086/515456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears JC, Broadie K. 2020. FMRP-PKA activity negative feedback regulates RNA binding-dependent fibrillation in brain learning and memory circuitry. Cell Rep 33: 108266. 10.1016/j.celrep.2020.108266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears JC, Choi WJ, Broadie K. 2019. Fragile X mental retardation protein positively regulates PKA anchor Rugose and PKA activity to control actin assembly in learning/memory circuitry. Neurobiol Dis 127: 53–64. 10.1016/j.nbd.2019.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Séjourné J, Plaçais PY, Aso Y, Siwanowicz I, Trannoy S, Thoma V, Tedjakumala SR, Rubin GM, Tchénio P, Ito K, et al. 2011. Mushroom body efferent neurons responsible for aversive olfactory memory retrieval in Drosophila. Nat Neurosci 14: 903–910. 10.1038/nn.2846 [DOI] [PubMed] [Google Scholar]
- Selvaraj S, Piramanayagam S. 2019. Impact of gene mutation in the development of Parkinson's disease. Genes Dis 6: 120–128. 10.1016/j.gendis.2019.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Şentürk M, Bellen HJ. 2018. Genetic strategies to tackle neurological diseases in fruit flies. Curr Opin Neurobiol 50: 24–32. 10.1016/j.conb.2017.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw PJ, Cirelli C, Greenspan RJ, Tononi G. 2000. Correlates of sleep and waking in Drosophila melanogaster. Science 287: 1834–1837. 10.1126/science.287.5459.1834 [DOI] [PubMed] [Google Scholar]
- Shukla AK, Pragya P, Chaouhan HS, Tiwari AK, Patel DK, Abdin MZ, Chowdhuri DK. 2014. Heat shock protein-70 (Hsp-70) suppresses paraquat-induced neurodegeneration by inhibiting JNK and caspase-3 activation in Drosophila model of Parkinson's disease. PLoS ONE 9: e98886. 10.1371/journal.pone.0098886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulman JM, Feany MB. 2003. Genetic modifiers of tauopathy in Drosophila. Genetics 165: 1233–1242. 10.1093/genetics/165.3.1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šišková Z, Justus D, Kaneko H, Friedrichs D, Henneberg N, Beutel T, Pitsch J, Schoch S, Becker A, von der Kammer H, et al. 2014. Dendritic structural degeneration is functionally linked to cellular hyperexcitability in a mouse model of Alzheimer's disease. Neuron 84: 1023–1033. 10.1016/j.neuron.2014.10.024 [DOI] [PubMed] [Google Scholar]
- Sitaraman D, Aso Y, Rubin GM, Nitabach MN. 2015. Control of sleep by dopaminergic inputs to the Drosophila mushroom body. Front Neural Circuits 9: 73. 10.3389/fncir.2015.00073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith FD, Esseltine JL, Nygren PJ, Veesler D, Byrne DP, Vonderach M, Strashnov I, Eyers CE, Eyers PA, Langeberg LK, et al. 2017. Local protein kinase A action proceeds through intact holoenzymes. Science 356: 1288–1293. 10.1126/science.aaj1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith P, Buhl E, Tsaneva-Atanasova K, Hodge JJL. 2019. Shaw and Shal voltage-gated potassium channels mediate circadian changes in Drosophila clock neuron excitability. J Physiol 597: 5707–5722. 10.1113/JP278826 [DOI] [PubMed] [Google Scholar]
- Song W, Smith MR, Syed A, Lukacsovich T, Barbaro BA, Purcell J, Bornemann DJ, Burke J, Marsh JL. 2013. Morphometric analysis of Huntington's disease neurodegeneration in Drosophila. Methods Mol Biol 1017: 41–57. 10.1007/978-1-62703-438-8_3 [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VMY, Trojanowski JQ, Jakes R, Goedert M. 1997. α-Synuclein in Lewy bodies. Nature 388: 839–840. 10.1038/42166 [DOI] [PubMed] [Google Scholar]
- Stahl A, Noyes NC, Boto T, Botero V, Broyles CN, Jing M, Zeng JZ, King LB, Li YL, Davis RL, et al. 2022. Associative learning drives longitudinally graded presynaptic plasticity of neurotransmitter release along axonal compartments. Elife 11: e76712. 10.7554/eLife.76712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun XC, Ran DZ, Zhao XF, Huang Y, Long SM, Liang FY, Guo WY, Nucifora FC, Gu HY, Lu XL, et al. 2016. Melatonin attenuates hLRRK2-induced sleep disturbances and synaptic dysfunction in a Drosophila model of Parkinson's disease. Mol Med Rep 13: 3936–3944. 10.3892/mmr.2016.4991 [DOI] [PubMed] [Google Scholar]
- Tabuchi M, Lone SR, Liu S, Liu QL, Zhang JL, Spira AP, Wu MN. 2015. Sleep interacts with Aβ to modulate intrinsic neuronal excitability. Curr Biol 25: 702–712. 10.1016/j.cub.2015.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeyama K, Ito S, Yamamoto A, Tanimoto H, Furutani T, Kanuka H, Miura M, Tabata T, Kato S. 2002. Androgen-dependent neurodegeneration by polyglutamine-expanded human androgen receptor in Drosophila. Neuron 35: 855–864. 10.1016/S0896-6273(02)00875-9 [DOI] [PubMed] [Google Scholar]
- Tanaka NK, Tanimoto H, Ito K. 2008. Neuronal assemblies of the Drosophila mushroom body. J Comp Neurol 508: 711–755. 10.1002/cne.21692 [DOI] [PubMed] [Google Scholar]
- Tandon S, Aggarwal P, Sarkar S. 2024. Polyglutamine disorders: pathogenesis and potential drug interventions. Life Sci 344: 122562. 10.1016/j.lfs.2024.122562 [DOI] [PubMed] [Google Scholar]
- Tandon S, Sarkar S. 2023. Glipizide ameliorates human poly(Q) mediated neurotoxicity by upregulating insulin signalling in Drosophila disease models. Biochem Biophys Res Commun 645: 88–96. 10.1016/j.bbrc.2023.01.022 [DOI] [PubMed] [Google Scholar]
- The I, Hannigan GE, Cowley GS, Reginald S, Zhong Y, Gusella JF, Hariharan IK, Bernards A. 1997. Rescue of a Drosophila NF1 mutant phenotype by protein kinase A. Science 276: 791–794. 10.1126/science.276.5313.791 [DOI] [PubMed] [Google Scholar]
- Tolosa E, Martí MJ, Valldeoriola F, Molinuevo JL. 1998. History of levodopa and dopamine agonists in Parkinson's disease treatment. Neurology 50: S2–S10. 10.1212/WNL.50.6_Suppl_6.S2 [DOI] [PubMed] [Google Scholar]
- Tomchik SM, Davis RL. 2013. Drosophila memory research through four eras: genetic, molecular biology, neuroanatomy, and systems neuroscience. Handb Behav Neurosci 22: 359–377. 10.1016/B978-0-12-415823-8.00027-7 [DOI] [Google Scholar]
- Torres M, Castillo K, Armisén R, Stutzin A, Soto C, Hetz C. 2010. Prion protein misfolding affects calcium homeostasis and sensitizes cells to endoplasmic reticulum stress. PLoS ONE 5: e15658. 10.1371/journal.pone.0015658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao CH, Chen CC, Lin CH, Yang HY, Lin S. 2018. Drosophila mushroom bodies integrate hunger and satiety signals to control innate food-seeking behavior. Elife 7: e35264. 10.7554/eLife.35264.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urra H, Dufey E, Lisbona F, Rojas-Rivera D, Hetz C. 2013. When ER stress reaches a dead end. Biochim Biophys Acta Mol Cell Res 1833: 3507–3517. 10.1016/j.bbamcr.2013.07.024 [DOI] [PubMed] [Google Scholar]
- Uusitalo E, Leppävirta J, Koffert A, Suominen S, Vahtera J, Vahlberg T, Pöyhönen M, Peltonen J, Peltonen S. 2015. Incidence and mortality of neurofibromatosis: a total population study in Finland. J Invest Dermatol 135: 904–906. 10.1038/jid.2014.465 [DOI] [PubMed] [Google Scholar]
- Verstreken P, Ly CV, Venken KJT, Koh TW, Zhou Y, Bellen HJ. 2005. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 47: 365–378. 10.1016/j.neuron.2005.06.018 [DOI] [PubMed] [Google Scholar]
- Vogt K, Aso Y, Hige T, Knapek S, Ichinose T, Friedrich AB, Turner GC, Rubin GM, Tanimoto H. 2016. Direct neural pathways convey distinct visual information to Drosophila mushroom bodies. Elife 5. 10.7554/eLife.14009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JA, Bernards A. 2014. A Drosophila screen identifies neurofibromatosis-1 genetic modifiers involved in systemic and synaptic growth. Rare Dis 2: e28341. 10.4161/rdis.28341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JA, Upadhyaya M. 2018. Emerging therapeutic targets for neurofibromatosis type 1. Expert Opin Ther Targets 22: 419–437. 10.1080/14728222.2018.1465931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JA, Tchoudakova AV, McKenney PT, Brill S, Wu DY, Cowley GS, Hariharan IK, Bernards A. 2006. Reduced growth of Drosophila neurofibromatosis 1 mutants reflects a non-cell-autonomous requirement for GTPase-activating protein activity in larval neurons. Genes Dev 20: 3311–3323. 10.1101/gad.1466806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, Ron D. 2011. The unfolded protein response: from stress pathway to homeostatic regulation. Science 334: 1081–1086. 10.1126/science.1209038 [DOI] [PubMed] [Google Scholar]
- Wan LL, Dockendorff TC, Jongens TA, Dreyfuss G. 2000. Characterization of dFMR1, a Drosophila melanogaster homolog of the Fragile X mental retardation protein. Mol Cell Biol 20: 8536–8547. 10.1128/MCB.20.22.8536-8547.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XJ, Davis RL. 2021. Early mitochondrial fragmentation and dysfunction in a Drosophila model for Alzheimer's disease. Mol Neurobiol 58: 143–155. 10.1007/s12035-020-02107-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XL, Su B, Zheng L, Perry G, Smith MA, Zhu XW. 2009. The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer's disease. J Neurochem 109: 153–159. 10.1111/j.1471-4159.2009.05867.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Wei CJ, Cui XW, Li YH, Gu YH, Gu B, Li QF, Wang ZC. 2021. Impacts of NF1 gene mutations and genetic modifiers in neurofibromatosis type 1. Front Neurol 12: 704639. 10.3389/fneur.2021.704639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrick JM, Morabito LM, Bilen J, Gordesky-Gold B, Faust LZ, Paulson HL, Bonini NM. 2005. Ataxin-3 suppresses polyglutamine neurodegeneration in Drosophila by a ubiquitin-associated mechanism. Mol Cell 18: 37–48. 10.1016/j.molcel.2005.02.030 [DOI] [PubMed] [Google Scholar]
- Weiler IJ, Greenough WT. 1999. Synaptic synthesis of the Fragile X protein: possible involvement in synapse maturation and elimination. Am J Med Genet 83: 248–252. [DOI] [PubMed] [Google Scholar]
- Whitworth AJ. 2011. Drosophila models of Parkinson's disease. Adv Genet 73: 1–50. 10.1016/B978-0-12-380860-8.00001-X [DOI] [PubMed] [Google Scholar]
- Wild AR, Dell'Acqua ML. 2018. Potential for therapeutic targeting of AKAP signaling complexes in nervous system disorders. Pharmacol Ther 185: 99–121. 10.1016/j.pharmthera.2017.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemsen R, Levenga J, Oostra BA. 2011. CGG repeat in the FMR1 gene: size matters. Clin Genet 80: 214–225. 10.1111/j.1399-0004.2011.01723.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JA, Su HS, Bernards A, Field J, Sehgal A. 2001. A circadian output in Drosophila mediated by neurofibromatosis-1 and Ras/MAPK. Science 293: 2251–2256. 10.1126/science.1063097 [DOI] [PubMed] [Google Scholar]
- Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB. 2001. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293: 711–714. 10.1126/science.1062382 [DOI] [PubMed] [Google Scholar]
- Wolf R, Wittig T, Liu L, Wustmann G, Eyding D, Heisenberg M. 1998. Drosophila mushroom bodies are dispensable for visual, tactile, and motor learning. Learn Mem 5: 166–178. 10.1101/lm.5.1.166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Bolduc FV, Bell K, Tully T, Fang Y, Sehgal A, Fischer JA. 2008. A Drosophila model for Angelman syndrome. Proc Natl Acad Sci 105: 12399–12404. 10.1073/pnas.0805291105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie KQ, Colgan LA, Dao MT, Muntean BS, Sutton LP, Orlandi C, Boye SL, Boye SE, Shih CC, Li YQ, et al. 2016. NF1 is a direct G protein effector essential for opioid signaling to Ras in the striatum. Curr Biol 26: 2992–3003. 10.1016/j.cub.2016.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Tito AJ, Rui YN, Zhang S. 2015. Studying polyglutamine diseases in Drosophila. Exp Neurol 274: 25–41. 10.1016/j.expneurol.2015.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata N, Ichinose T, Aso Y, Plaçais PY, Friedrich AB, Sima RJ, Preat T, Rubin GM, Tanimoto H. 2015. Distinct dopamine neurons mediate reward signals for short- and long-term memories. Proc Natl Acad Sci 112: 578–583. 10.1073/pnas.1421930112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C, Liu JQ, Gao JM, Sun Y, Zhang L, Song HY, Xue L, Zhan LX, Gao GJ, Ke ZJ, et al. 2019. IRE1 promotes neurodegeneration through autophagy-dependent neuron death in the Drosophila model of Parkinson's disease. Cell Death Dis 10: 800. 10.1038/s41419-019-2039-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon WH. 2023. Recent progress in using Drosophila as a human genetic disease research. J Genet Med 20: 39–45. 10.5734/JGM.2023.20.2.39 [DOI] [Google Scholar]
- Yoshikai S, Sasaki H, Dohura K, Furuya H, Sakaki Y. 1990. Genomic organization of the human amyloid β-protein precursor gene. Gene 87: 257–263. 10.1016/0378-1119(90)90310-N [DOI] [PubMed] [Google Scholar]
- Yu D, Keene AC, Srivatsan A, Waddell S, Davis RL. 2005. Drosophila DPM neurons form a delayed and branch-specific memory trace after olfactory classical conditioning. Cell 123: 945–957. 10.1016/j.cell.2005.09.037 [DOI] [PubMed] [Google Scholar]
- Yu D, Akalal DB, Davis RL. 2006. Drosophila α/β mushroom body neurons form a branch-specific, long-term cellular memory trace after spaced olfactory conditioning. Neuron 52: 845–855. 10.1016/j.neuron.2006.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B, Ma J, Wang DZ, Wang ZG, Wang SS. 2020. Mitochondrial phosphatase PGAM5 modulates cellular senescence by regulating mitochondrial dynamics. Nat Commun 11: 2549. 10.1038/s41467-020-16312-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zárate RV, Hidalgo S, Navarro N, Molina-Mateo D, Arancibia D, Rojo-Cortés F, Oliva C, Andrés ME, Zamorano P, Campusano JM. 2022. An early disturbance in serotonergic neurotransmission contributes to the onset of parkinsonian phenotypes in Drosophila melanogaster. Cells-Basel 11: 1544. 10.3390/cells11091544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zars T, Fischer M, Schulz R, Heisenberg M. 2000. Localization of a short-term memory in Drosophila. Science 288: 672–675. 10.1126/science.288.5466.672 [DOI] [PubMed] [Google Scholar]
- Zhang S, Binari R, Zhou R, Perrimon N. 2010. A genomewide RNA interference screen for modifiers of aggregates formation by mutant Huntingtin in Drosophila. Genetics 184: 1165–1179. 10.1534/genetics.109.112516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XF, Noyes NC, Zeng JZ, Li YL, Davis RL. 2019. Aversive training induces both presynaptic and postsynaptic suppression in Drosophila. J Neurosci 39: 9164–9172. 10.1523/JNEUROSCI.1420-19.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JJ, Lentz L, Goldammer J, Iliescu J, Tanimura J, Riemensperger TD. 2023. Asymmetric presynaptic depletion of dopamine neurons in a Drosophila model of Parkinson's disease. Int J Mol Sci 24: 8585. 10.3390/ijms24108585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Lu Y, Zhao X, Yao X, Shuai Y, Huang C, Wang L, Jeong SH, Zhong Y. 2013. Dissociation of rugose-dependent short-term memory component from memory consolidation in Drosophila. Genes Brain Behav 12: 626–632. 10.1111/gbb.12056 [DOI] [PubMed] [Google Scholar]
- Zhu GQ, Liu Y, Wang YB, Bi XN, Baudry M. 2015. Different patterns of electrical activity lead to long-term potentiation by activating different intracellular pathways. J Neurosci 35: 621–633. 10.1523/JNEUROSCI.2193-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu BF, Parsons T, Stensen W, Svendsen JMS, Fugelli A, Hodge JJL. 2022. DYRK1a inhibitor mediated rescue of Drosophila models of Alzheimer's disease–Down syndrome phenotypes. Front Pharmacol 13: 881385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, et al. 2004. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44: 601–607. 10.1016/j.neuron.2004.11.005 [DOI] [PubMed] [Google Scholar]
- Zirin J, Bosch J, Viswanatha R, Mohr SE, Perrimon N. 2022. State-of-the-art CRISPR for in vivo and cell-based studies in Drosophila. Trends Genet 38: 437–453. 10.1016/j.tig.2021.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]