

Abstract
Background
TNF (tumor necrosis factor)‐alpha inhibitors block a key protein in the inflammatory chain reaction responsible for joint inflammation, pain, and damage in ankylosing spondylitis.
Objectives
To assess the benefit and harms of adalimumab, etanercept, golimumab, and infliximab (TNF‐alpha inhibitors) in people with ankylosing spondylitis.
Search methods
We searched the following databases to January 26, 2009: MEDLINE (from 1966); EMBASE (from 1980); the Cochrane Central Register of Controlled Trials (CENTRAL; 2008, Issue 4); ACP Journal Club; CINAHL (from 1982); and ISI Web of Knowledge (from 1900). We ran updated searches in May 2012, October 2013, and in June 2014 for McMaster PLUS. We searched major regulatory agencies for safety warnings and clinicaltrials.gov for registered trials.
Selection criteria
Randomized controlled trials (RCTs) comparing adalimumab, etanercept, golimumab and infliximab to placebo, other drugs or usual care in patients with ankylosing spondylitis, reported in abstract or full‐text.
Data collection and analysis
Two authors independently assessed search results, risk of bias, and extracted data. We conducted Bayesian mixed treatment comparison (MTC) meta‐analyses using WinBUGS software. To investigate a class‐effect of harms across biologics, we pooled harms data using Review Manager 5.
Main results
We included twenty‐one, short‐term (24 weeks or less) RCTs with a total of 3308 participants; 18 contributed data to the MTC analysis: adalimumab (4 studies), etanercept (8 studies), golimumab (2 studies), infliximab (3 studies), and one head‐to‐head study (etanercept versus infliximab) which was unblinded and considered at a higher risk of bias. The risk of selection and detection bias was low or unclear for most of the studies. The risk of selective outcome reporting was low for most studies as they reported on outcomes recommended by the Assessment of SpondyloArthritis international Society. We found little heterogeneity and no significant inconsistency in the MTC analyses. The majority of the studies were funded by pharmaceutical companies. Most studies permitted concomitant therapy of stable doses of disease‐modifying anti‐rheumatic drugs, non‐steroidal anti‐inflammatory drugs, or corticosteroids, but allowances varied across studies.
Compared with placebo, there was high quality evidence that patients on an anti‐TNF agent were three to four times more likely to achieve an ASAS40 response (assessing spinal pain, function, and inflammation, as measured by the mean of intensity and duration of morning stiffness, and patient global assessment) by six months (adalimumab: risk ratio (RR) 3.53, 95% credible interval (Crl) 2.49 to 4.91; etanercept: RR 3.31, 95% Crl 2.38 to 4.53; golimumab: RR 2.90, 95% Crl 1.90 to 4.23; infliximab: RR 4.07, 95% Crl 2.80 to 5.74, with a 25% to 40% absolute difference between treatment and placebo groups. The number needed to treat (NNT) to achieve an ASAS 40 response ranged from 3 to 5.
There was high quality evidence of improvement in physical function on a 0 to 10 scale (adalimumab: mean difference (MD) ‐1.6, 95% Crl ‐2.2 to ‐0.9; etanercept: MD ‐1.1, 95% CrI ‐1.6 to ‐0.6; golimumab: MD ‐1.5, 95% Crl ‐2.3 to ‐0.7; infliximab: MD ‐2.1, 95% Crl ‐2.7 to ‐1.4, with an 11% to 21% absolute difference between treatment and placebo groups. The NNT to achieve the minimally clinically important difference of 0.7 points ranged from 2 to 4.
Compared with placebo, there was moderate quality evidence (downgraded for imprecision) that patients on an anti‐TNF agent were more likely to achieve an ASAS partial remission by six months (adalimumab: RR 6.28, 95% Crl 3.13 to 12.78; etanercept: RR 4.24, 95% Crl 2.31 to 8.09; golimumab: RR 5.18, 95% Crl 1.90 to 14.79; infliximab: RR 15.41, 95% Crl 5.09 to 47.98 with a 10% to 44% absolute difference between treatment and placebo groups. The NNT to achieve an ASAS partial remission response ranged from 3 to 11.
There was low to moderate level evidence of a greater reduction in spinal inflammation as measured by magnetic resonance imaging though the absolute differences were small and the clinical relevance of the difference was unclear: adalimumab (1 trial; ‐6% (95% confidence interval (CI) ‐12% to 0.05%); 1 trial: 53.6% mean decrease from baseline versus 9.4% mean increase in the placebo group), golimumab (1 trial; ‐2.5%, (95% CI ‐5.6% to ‐0.7%)), and infliximab (1 trial; ‐3% (95% CI ‐4% to ‐2.4%)).
Radiographic progression was measured in one trial (N = 60) of etanercept versus placebo and it found that radiologic changes were similar in both groups (detailed data not provided).
There were few events of withdrawals due to adverse events leading to imprecision around the estimates. When all the anti‐TNF agents were combined against placebo, there was moderate quality evidence from 16 studies of an increased risk of withdrawals due to adverse events in the anti‐TNF group (Peto odds ratio (OR) 2.44, 95% CI 1.26 to 4.72; total events: 38/1637 in biologic group; 7/986 in placebo) though the absolute increase in harm was small (1%; 95% CI 0% to 2%).
Due to low event rates, evidence of the effect of individual TNF‐inhibitors against placebo or for all four biologics pooled together versus placebo on serious adverse events is inconclusive (moderate quality; downgraded for imprecision). For all anti‐TNF pooled versus placebo based on 16 studies: Peto OR 1.45, 95% CI 0.85 to 2.48; 51/1530 in biologic group; 18/878 in placebo; absolute difference: 1% (95% CI 0% to 2%).
Using indirect comparison methodology, and one head‐to‐head study of etanercept versus infliximab, wide confidence intervals meant that results were inconclusive for evidence of differences in the major outcomes between different anti‐TNF agents. Regulatory agencies have published warnings about rare adverse events of serious infections, including tuberculosis, malignancies and lymphoma.
Authors' conclusions
There is moderate to high quality evidence that anti‐TNF agents improve clinical symptoms in the treatment of ankylosing spondylitis. More participants withdrew due to adverse events when on an anti‐TNF agent but we did not find evidence of an increase in serious adverse events, though event rates were low and trials had a short duration. The short‐term toxicity profile appears acceptable. Based on indirect comparison methodology, we are uncertain whether there are differences between anti‐TNF agents in terms of the key benefit or harm outcomes.
Keywords: Humans; Adalimumab; Anti‐Inflammatory Agents, Non‐Steroidal; Anti‐Inflammatory Agents, Non‐Steroidal/therapeutic use; Antibodies, Monoclonal; Antibodies, Monoclonal/therapeutic use; Antibodies, Monoclonal, Humanized; Antibodies, Monoclonal, Humanized/therapeutic use; Etanercept; Immunoglobulin G; Immunoglobulin G/therapeutic use; Infliximab; Randomized Controlled Trials as Topic; Receptors, Tumor Necrosis Factor; Receptors, Tumor Necrosis Factor/therapeutic use; Spondylitis, Ankylosing; Spondylitis, Ankylosing/drug therapy; Tumor Necrosis Factor‐alpha; Tumor Necrosis Factor‐alpha/antagonists & inhibitors
Plain language summary
Anti‐TNF‐alpha drugs for treating ankylosing spondylitis
Researchers looked at trials done up to June 2014 on the effect of anti‐TNF drugs (adalimumab (Humira®), etanercept (Enbrel®), golimumab (Simponi®), and infliximab (Remicade®)) on ankylosing spondylitis. They found 21 trials with 3308 participants. Most studies were funded by pharmaceutical companies.
What is ankylosing spondylitis and what are anti‐TNF drugs?
Ankylosing spondylitis is a type of arthritis, usually in the joints and ligaments of the spine, but it may also affect other joints. Pain and stiffness occurs and limits movement in the back and affected joints. It can come and go, last for long periods, and be quite severe.
Anti‐TNF drugs target a protein called 'tumor necrosis factor' that causes inflammation. These drugs suppress the immune system and reduce the inflammation in the joints, with the aim of preventing damage. Even though suppressing the immune system can make it slightly harder to fight off infections, it also helps to stabilize an overactive immune system.
The review shows that in people with ankylosing spondylitis, using anti‐TNF drugs for up to 24 weeks:
‐ improves pain, function and other symptoms of ankylosing spondylitis; ‐ may increase the chance of achieving partial remission of symptoms of ankylosing spondylitis; ‐ probably slightly improves spinal inflammation, as measured by magnetic resonance imaging (MRI); and ‐ probably causes slightly more people to drop out of studies because of side effects.
We do not have precise information about side effects and complications, but in these short‐term studies there was no evidence of an increase in serious adverse events. Possible side effects may include a serious infection (like tuberculosis) or upper respiratory infection. Rare complications may include certain types of cancer.
Best estimate of what happens to people with ankylosing spondylitis who take anti‐TNF drugs for up to 24 weeks:
ASAS40 (40% improvement in pain, function, and inflammation as measured by morning stiffness, and patient overall well‐being) Compared to 13 people out of 100 who experienced an improvement with a placebo, among people who took:
‐ adalimumab: 46 people out of 100 experienced improvement (33% improvement); ‐ etanercept: 43 people out of 100 experienced improvement (30% improvement); ‐ golimumab: 38 people out of 100 experienced improvement (25% improvement); and ‐ infliximab: 53 people out of 100 experienced improvement (40% improvement).
Partial remission (defined as a value of less than 2 on a 0 to 10 scale in each of pain, function, and inflammation as measured by morning stiffness, and patient overall well‐being) Compared to 3 people out of 100 who experienced an improvement with a placebo, among people who took:
‐ adalimumab: 19 people out of 100 experienced partial remission (16% improvement); ‐ etanercept: 13 people out of 100 experienced partial remission (10% improvement); ‐ golimumab: 16 people out of 100 experienced partial remission (13% improvement); and ‐ infliximab: 47 people out of 100 experienced partial remission (44% improvement).
Physical function (lower score means better function; 0 to 10 scale)
Compared to a score of 5 in people who took placebo, among people who took:
‐ adalimumab, they rated their function to be 3.4 (16% improvement); ‐ etanercept, they rated their function to be 3.9 (11% improvement); ‐ golimumab, they rated their function to be 3.5 (15% improvement); and ‐ infliximab, they rated their function to be 2.9 (21% improvement).
Spinal inflammation as measured by magnetic resonance imaging (MRI) Compared to people who took placebo, a small improvement in spinal inflammation was seen in:
‐ adalimumab (6% improvement); ‐ golimumab (2.5% improvement); and ‐ infliximab (3% improvement).
X‐rays of the joints
Only one study looked at x‐rays and found that joint changes were similar in both groups (detailed data not provided).
Side effects
When all the anti‐TNF drugs were combined, 16 people out of 1000 dropped out of the study because of side effects compared to 7 people out of 1000 who took placebo (absolute increase 1%).
There may be little or no difference in the number of people who have a serious side effect with an anti‐TNF drug compared to people who take a fake pill.
Summary of findings
Summary of findings for the main comparison. Summary of findings table.
| TNF‐alpha inhibitors for ankylosing spondylitis (short‐term results < 24 weeks) | |||||||
| Outcome |
Intervention and comparison |
Illustrative comparative risks |
Relative effect (95% CrI) |
Number of participants (studies) |
Quality of the evidence (GRADE) |
Comment | |
|
Assumed risk with comparator1 |
Corresponding risk with intervention (95% CI or Crl) |
||||||
| Placebo | TNF‐alpha inhibitor | ||||||
| ASAS40 | |||||||
| Adalimumab versus placebo | 13 per 100 | 46 per 100 (32 to 64) | RR 3.53 (2.49 to 4.91) | 659 (2 studies) | ⊕⊕⊕⊕ high | Absolute increased benefit % = 33% (95% Crl 19% to 51%) Relative % change= 253% (95% CI 149% to 391%) NNT = 4 (95% CI 2 to 6) |
|
| Etanercept (25 mg twice weekly or 50 mg once weekly) versus placebo | 13 per 100 | 43 per 100 (31 to 59) | RR 3.31 (2.38 to 4.53) | 584 (3 studies) | ⊕⊕⊕⊕ high | Absolute increased benefit % = 30% (95% Crl 18% to 46%) Relative % change = 231% (95% CI 138% to 353%) NNT = 4 (95% Cl 3 to 6) |
|
| Golimumab versus placebo | 13 per 100 | 38 per 100 (25 to 55) | RR 2.90 (1.90 to 4.23) | 429 (2 studies) | ⊕⊕⊕⊕ high | Absolute increased benefit % = 25% (95% Crl 12% to 42%) Relative % change = 190% (95% CI 90% to 323%) NNT = 5 (95% CI 3 to 9) |
|
| Infliximab versus placebo | 13 per 100 | 53 per 100 (36 to 75) | RR 4.07 (2.80 to 5.74) | 355 (2 studies) | ⊕⊕⊕⊕ high | Absolute increased benefit % = 40% (95% Crl 23% to 62%) Relative % change = 307% (95% CI 180% to 474%) NNT= 3 (95% CI 2 to 5) |
|
| BASFI (0 to 10 scale) | |||||||
| Adalimumab versus placebo | The mean BASFI in the control groups was 5 points | The mean BASFI in the intervention groups was 1.6 lower (2.2 to 0.9 lower) | 786 (4 studies) | ⊕⊕⊕⊕ high | Absolute increased benefit % = ‐16% (95% Crl ‐22% to ‐9%); Relative % change from baseline = ‐32% (‐44% to ‐18%); NNT to achieve the MCID of 0.7 points = 4 (95% CI 3 to 5) | ||
| Etanercept (25 mg twice weekly or 50 mg once weekly) versus placebo | The mean BASFI in the control groups was 5 points | The mean BASFI in the intervention groups was 1.1 lower (1.6 to 0.6 lower) | 553 (6 studies) | ⊕⊕⊕⊕ high | Absolute increased benefit % =‐11% (95% Crl ‐16% to ‐6%); Relative % change from baseline = ‐22% (‐32% to ‐12%); NNT to achieve the MCID of 0.7 points = 4 (4 to 6) |
||
| Golimumab versus placebo | The mean BASFI in the control groups was 5 points | The mean BASFI in the intervention groups was 1.5 lower (2.3 to 0.7 lower) | 429 (2 studies) | ⊕⊕⊕⊕ high | Absolute increased benefit % =‐15% (95% Crl ‐23% to ‐7%) Relative % change from baseline = ‐30% (‐46% to ‐14%) NNT to achieve the MCID of 0.7 points = 4 (3 to 5) |
||
| Infliximab versus placebo | The mean BASFI in the control groups was 5 points | The mean BASFI in the intervention groups was 2.1 lower (2.7 to 1.4 lower) | 348 (2 studies) | ⊕⊕⊕⊕ high | Absolute increased benefit % = ‐21% (95% Crl ‐27% to ‐14%) Relative % change from baseline = ‐42% (‐54% to ‐28%) NNT to achieve the MCID of 0.7 points = 2 (2 to 3) |
||
| ASAS partial remission | |||||||
| Adalimumab versus placebo | 3 per 100 | 19 per 100 (9 to 38) | RR 6.28 (3.13 to 12.78) | 659 (2 studies) | ⊕⊕⊕⊖ moderate2 | Absolute increased benefit % = 16% (95% Crl 6% to 35%) Relative % change = 528% (95% CI 213% to 1178%) NNT = 7 (95% CI 3 to 16) |
|
| Etanercept (25 mg twice weekly or 50 mg once weekly) versus placebo | 3 per 100 | 13 per 100 (7 to 24) | RR 4.24 (2.31 to 8.09) | 785 (3 studies) | ⊕⊕⊕⊖ moderate2 | Absolute increased benefit % = 10% (95% Crl 4% to 21%) Relative % change = 324% (95% CI 131% to 709%); NNT = 11 (95% CI 5 to 26) | |
| Golimumab versus placebo | 3 per 100 | 16 per 100 (6 to 44) | RR 5.18 (1.90 to 14.79) | 216 (1 study) | ⊕⊕⊕⊖ moderate2 | Absolute increased benefit % = 13% (95% Crl 3% to 41%) Relative % change = 418% (95% CI 90% to 1379%); NNT = 8 (95% CI 3 to 38) | |
| Infliximab versus placebo | 3 per 100 | 47 per 100 (16 to 90) | RR 15.41 (5.09 to 47.98) | 348 (2 studies) | ⊕⊕⊕⊖ moderate2 | Absolute increased benefit % = 44% (95% Crl 13% to 87%) Relative % change = 1441% (95% CI 409% to 4698%) NNT = 3 (95% CI 2 to 8) |
|
| MRI of spinal inflammation | |||||||
| Adalimumab versus placebo Lumbar spine MRI; SPARCC score (0 to 108) |
The mean SPARCC score in the control groups was 16.1 | The mean SPARCC score in the intervention groups was 6.5 lower (13.06 to 0.06 higher) | 46 (1 study) | ⊕⊕⊕⊖ moderate3 | Absolute increased benefit % = ‐6% (95% CI ‐12% to 0.05%) Relative % change = ‐33% (95% CI ‐66% to 0%) NNT = n/a 2nd study with MRI data: Lambert 2007 (N = 82): % change from baseline in SPARCC score, week 12 (no variance provided) 1. Spine: Adalimumab group = 53.6% mean decrease Placebo group = 9.4% mean increase Between group: P < 0.001 2. Sacroiliac joint, % mean decrease: Adalimumab group = 52.9%, Placebo group = 12.7% Between group: P = 0.017 |
||
| Etanercept (25 mg twice weekly or 50 mg once weekly) versus placebo | See comment | See comment | Not estimable | 0 (0) | See comment | No studies assessed this outcome | |
| Golimumab versus placebo Change from baseline in AS spine MRI activity score (0 to 138; lower means less erosions or edema) |
The mean change in the control group was ‐2.5 points | The mean change in the golimumab group was
3.4 points lower (7.7 to 0.90 points lower) |
60 (1 study) | ⊕⊕⊖⊖ low3,4 | Absolute increased benefit % = ‐2.5% (95% CI ‐5.6% to ‐0.7%) Relative % change = ‐35% (95% CI ‐80% to 9%) NNT = n/a |
||
| Infliximab versus placebo Change from baseline in AS spine MRI activity score (0 to 138; lower means less erosions or edema) |
The mean change in the control group was ‐0.6 points | The mean change in the infliximab group was
4.4 points lower (5.6 to 3.3 points lower) |
266 (1 study) | ⊕⊕⊕⊖ moderate5 | Absolute increased benefit % = ‐3% (95% CI ‐4% to ‐2.4%) Relative % change = ‐62% (95% CI ‐79% to ‐46%) NNT = 3 (95% CI 3 to 5) Inman 2010 assessed MRI in a substudy (N = 26): "when the evaluation was based on the entire spine (23 DVU score), the infliximab group had a mean reduction of 57.2% compared to 3.4% in the placebo group (P < 0.001)" |
||
| Radiographic progression | |||||||
| Adalimumab versus placebo | See comment | See comment | Not estimable | 0 (0) | See comment | No studies assessed this outcome | |
| Etanercept (25 mg twice weekly or 50 mg once weekly) versus placebo | See comment | See comment | Not estimable | 0 (0) | See comment | No studies measured this outcome | |
| Golimumab versus placebo | See comment | See comment | Not estimable | 0 (0) | See comment | No studies measured this outcome | |
| Infliximab versus placebo | See comment | See comment | Not estimable | 0 (0) | See comment | Braun 2002 (N = 60) used the Bath Ankylosing Spondylitis Radiology Index (BASRI) to measure radiographic progression but detailed data was not provided. The results stated the "initial degree of radiological axial changes assessed by the BASRIs was similar in both groups" | |
| Withdrawals due to adverse events | |||||||
| Adalimumab versus placebo | 7 per 1000 | 12 per 1000 (3 to 80) | RR 1.69 (0.35 to 10.84) | 659 (2 studies) | ⊕⊕⊕⊖ moderate2 | Absolute increased harm % = 0.6% (95% Crl ‐0.4% to 7%) Relative % change = 69% (95% CI ‐65% to 984%) NNT = n/a |
|
| Etanercept (25 mg twice weekly or 50 mg once weekly) versus placebo | 7 per 1000 | 26 per 1000 (9 to 83) | RR 3.65 (1.27 to 11.79) | 1061 (8 studies) | ⊕⊕⊕⊖ moderate2 | Absolute increased harm % = 2% (95% Crl 0.2% to 8%) Relative % change = 265% (95% CI 27% to 1079%) NNTH = 54 (95% CI 14 to 530) |
|
| Golimumab versus placebo | 7 per 1000 | 14 per 1000 (3 to 123) | RR 1.97 (0.36 to 17.51) | 429 (2 studies) | ⊕⊕⊕⊖ moderate2 | Absolute increased harm % = 1.6% (95% Crl ‐0.4% to 11.6%) Relative % change = 97% (95% CI ‐64% to 1651%) NNTH = n/a |
|
| Infliximab versus placebo | 7 per 1000 | 12 per 1000 (3 to 59) | RR 1.77 (0.43 to 8.46) | 424 (3 studies) | ⊕⊕⊕⊖ moderate2 | Absolute increased harm % = 0.5% (95% Crl ‐0.4% to 5.6%) Relative % change = 77% (95% CI ‐43% to 746%) NNT = n/a |
|
| All anti‐TNF agents versus placebo | 7 per 1000 | 16 per 1000 (8 to 33) | Peto OR 2.44 (1.26 to 4.72 | 2623 (16 studies) | ⊕⊕⊕⊖ moderate2 | Absolute increased harm % = 1% (95% CI 0% to 2%) Relative % change = 130% (95% CI 12% to 371%) NNTH = 101 (95% CI 555 to 40) |
|
| Serious adverse events | |||||||
| Adalimumab versus placebo | 15 per 1000 | 14 per 1000 (4 to 59) | RR 0.92 (0.26 to 3.93) | 659 (2 studies) | ⊕⊕⊕⊖ moderate2 | Absolute increased harm %= ‐0.2% (95% Crl ‐1.1% to 4.4%); Relative % change= ‐8% (95% CI ‐74% to 293%); NNT = n/a | |
| Etanercept (25 mg twice weekly or 50 mg once weekly) versus placebo | 15 per 1000 | 25 per 1000 (11 to 56) | RR 1.69 (0.76 to 3.72) | 1061 (8 studies) | ⊕⊕⊕⊖ moderate2 | Absolute increased harm % = 1% (95% Crl ‐0.4% to 4.1%) Relative % change = 67% (95% CI ‐27% to 282%) NNT = n/a |
|
| Golimumab versus placebo | 15 per 1000 | 10 per 1000 (2 to 50) | RR 0.69 (0.15 to 3.32) | 216 (1 study) | ⊕⊕⊕⊖ moderate2 | Absolute increased harm % = ‐0.5% (95% Crl ‐1.3% to 3.5%); Relative % change= ‐31% (95% CI ‐85% to 232%); NNT = n/a | |
| Infliximab versus placebo | 15 per 1000 | 38 per 1000 (11 to 166) | RR 2.53 (0.76 to 11.09) | 422 (3 studies) | ⊕⊕⊕⊖ moderate2 | Absolute increased harm % = 2.3% (95% Crl ‐0.4% to 15.1%) Relative % change = 153% (95% CI ‐24% to 1009%) NNT = n/a |
|
| All anti‐TNF agents versus placebo | 15 per 1000 | 22 per 1000 (13 to 36) | Peto OR 1.45 (0.85 to 2.48) | 2408 (15 studies) | ⊕⊕⊕⊖ moderate2 | Absolute increased harm % = 1% (95% CI 0% to 2%) Relative % change = 41% (95% CI ‐15% to 136%) NNTH = n/a |
|
95% CI = 95% confidence interval; 95% Crl = 95% credible interval; n/a = not applicable; NNTH = Number needed to treat for harm; RR = risk ratio; OR = odds ratio; SI = sacroiliac joint; SPARCC = Spondyloarthritis Research Consortium of Canada
NNT = not applicable for non‐statistically significant results
Note: Results for ASAS40, BASFI, ASAS partial remission, withdrawals due to adverse events, and serious adverse events are based on the mixed treatment comparison analyses. The 'All anti‐TNF agents versus placebo' results for withdrawals due to adverse events and serious adverse events are based on standard meta‐analyses in Review Manager 5.3.
1 Assumed risk based on the placebo event rate as calculated in the mixed treatment comparison analysis.
2 Downgraded for imprecision; fewer events than 300 (a threshold rule‐of‐thumb) and wide confidence interval.
3 Downgraded for imprecision: total population is < 400.
4 MRI substudy (N = 60 for placebo and 50 mg golimumab arms) conducted at 10/57 participating sites; N = 216 in full RCT; readers were blinded but concerns regarding only modest level of agreement. Downgraded for concerns regarding missing data (12% did not have baseline and follow‐up MRIs and imputed 7% of scores at week 14).
5 Downgraded for imprecision; total population is < 400. MRI data available for 194/201 in infliximab group; 72/78 in placebo group.
Background
Description of the condition
Ankylosing spondylitis is a chronic, inflammatory rheumatic disease characterized by inflammatory back pain due to sacroiliitis and spondylitis, enthesitis, and the formation of syndesmophytes (bony growths) leading to ankylosis. Extraspinal manifestations are common, including peripheral arthritis (25% to 50%), uveitis (eye inflammation) (25% to 40%), and inflammatory bowel disease (26%), and contribute to disease morbidity (Edmunds 1991; Inman 2011).
The etiology of the disease is not yet fully understood but there is a strong association with the HLA‐B27 gene (Inman 2011). Studies have shown the prevalence of ankylosing spondylitis in the adult general population to vary from 0.4% (Alaskan Inuit) to 1.4% (Northern Norway) (Khan 2002). A general rule is that the prevalence of ankylosing spondylitis is highest in HLA‐B27‐positive patients with a family member who also has the disease (20%), is least in the general population (0.2%), and is about 2% in those positive for HLA‐B27 (Inman 2011). The peak age of onset is in young adults between 20 and 30 years, although there is often a five to six year delay in diagnosis (Khan 2002).
Clinical symptoms usually begin with back pain and stiffness in adolescence and early adulthood which shows improvement with exercise and can lead to impaired spinal mobility, or chest expansion, or both. The disease course of ankylosing spondylitis is highly variable, with back pain and stiffness often the primary features early in the process, and chronic pain and joint changes later on (Inman 2011). The burden of disease in ankylosing spondylitis has been found to be similar to that of rheumatoid arthritis in terms of pain, disability and decreased well‐being (Zink 2000). Additionally, compared to the general population, those with ankylosing spondylitis experience higher work disability and absence from work, which can lead to substantial direct and indirect socioeconomic costs (Boonen 2001a; Boonen 2001b; Montacer 2009).
The goals of treatment of ankylosing spondylitis are to relieve symptoms (pain, stiffness, joint swelling), improve physical function, and delay or avoid structural damage which leads to physical impairments and deformities. Ankylosing spondylitis requires a multidisciplinary treatment approach and is usually managed with a combination of exercises, physiotherapy and drug therapy. Regular exercise is crucial for maintaining or improving spinal mobility and physical function (Dagfinrud 2008). Non‐steroidal anti‐inflammatory drugs (NSAIDs) are the mainstay of symptomatic drug therapy, reducing the pain and stiffness of inflammation. Although these interventions can alleviate the symptoms of the disease, it is not clear whether they are able to prevent or delay the structural damage leading to physical disability. Some evidence suggests continuous NSAID therapy may have an effect on the spinal radiographic changes seen in ankylosing spondylitis (Wanders 2005). At least one‐third of patients respond insufficiently to NSAID therapy or experience serious side effects from NSAIDs and thus require disease controlling drugs in addition to symptom‐modifying treatment. In contrast to rheumatoid arthritis, there are no established disease‐modifying anti‐rheumatic treatments in ankylosing spondylitis, although sulphasalazine may be effective for peripheral joint symptoms but not for axial disease (Dougados 2002; Chen 2014).
Description of the intervention
A major advance in treatment options for ankylosing spondylitis is the development of biologic therapies which target specific elements of the immune system. Tumor necrosis factor (TNF)‐alpha is a protein that the body produces during the inflammatory response. TNF‐alpha promotes inflammation and subsequent pain, tenderness, swelling and fever in several inflammatory conditions, including ankylosing spondylitis. Four anti‐TNF agents, also known as TNF‐inhibitors, have been developed to target the binding of this protein, thus reducing the pain, swelling, and inflammation associated with ankylosing spondylitis. The generic and trademark drug names are: adalimumab (Humira®), etanercept (Enbrel®), golimumab (Simponi®), and infliximab (Remicade®). Infliximab is given as an intravenous infusion over one to two hours while etanercept, adalimumab, and golimumab are given as subcutaneous injections. Etanercept and adalimumab are given as weekly or bi‐weekly injections, while golimumab is injected once a month. Recognized contraindications for treatment include tuberculosis, multiple sclerosis, lupus, malignancy, pregnant or lactating women, heart failure, hepatitis, and pneumonia.
How the intervention might work
As the result of research demonstrating that tumor necrosis factor‐alpha (TNF‐alpha) is present in inflamed sacroiliac joints (Braun 1995), treatments were developed to block TNF‐alpha. Etanercept is a receptor fusion protein that binds to TNF‐alpha, thus competitively inhibiting the binding of TNF‐alpha to the cell surface. Infliximab is a chimeric (mouse/human) monoclonal antibody of the IgG1κ isotype that binds with a high affinity to TNF‐alpha. Adalimumab is a recombinant human IgG1 monoclonal antibody specific for human TNF‐alpha, and golimumab is a human monoclonal antibody that binds to both soluble and transmembrane TNF‐alpha. These four agents prevent TNF‐alpha from promoting inflammation and therefore are thought to interrupt the processes responsible for the pain, tenderness, and swelling of joints in patients with ankylosing spondylitis.
Why it is important to do this review
Early open label studies demonstrated that biologics are efficacious in ankylosing spondylitis (Brandt 2000; Haibel 2004; Maksymowych 2002; Marzo‐Ortega 2001; Stone 2001) and RCTs showed them to be effective in improving disease activity, spinal mobility, function, and pain (Braun 2002; Gorman 2002; Van Den Bosch 2002). Recognized adverse effects of anti‐TNF‐alpha therapy include serious infections such as tuberculosis, allergic reactions and autoimmune reactions.
The relatively high cost of treatment and possible serious side effects of anti‐TNF‐alpha therapy led the Assessment of SpondyloArthritis international Society (ASAS) (Braun 2003; van der Heijde 2011) and the Spondyloarthritis Research Consortium of Canada (Maksymowych 2003) to develop recommendations for the use of TNF‐alpha inhibitors in ankylosing spondylitis.
While these biologics offer an important therapeutic advance by appearing to reduce disease activity and improve function and well‐being of patients, it is important to understand and try to quantify not only the potential benefits of this treatment, but also the potential harms. Clinicians and patients need this information in order to make an informed decision about the trade‐offs of using this treatment option. The evidence base for the individual biologics compared to each other is of interest to patients and other healthcare decision makers. We will include certolizumab in an update of this review. Head‐to‐head studies (i.e. one biologic versus another) are usually rare so we will undertake indirect comparisons using network meta‐analysis methodology to address this question.
Objectives
To assess the benefits and harms of adalimumab, etanercept, golimumab, and infliximab (TNF‐alpha inhibitors) in people with ankylosing spondylitis.
Methods
Criteria for considering studies for this review
Types of studies
We included randomized controlled trials (RCTs) and controlled clinical trials (CCTs). We defined 'short‐term' benefit and harms as those with equal to or less than six months duration and 'long‐term' benefit and harms as longer than six months.
Types of participants
We included studies of patients meeting the following ankylosing spondylitis classification criteria: 1961 Rome, 1966 New York, or modified 1984 New York. We did not apply any additional restrictions in studies with regard to age of patients, past or present (co‐)medication or ankylosing spondylitis‐related comorbidity. We included studies on spondyloarthropathies that mentioned ankylosing spondylitis patients as a subgroup, as far as the subgroup was properly randomized and outcome measures were available, specifically for the ankylosing spondylitis subgroup. We included patients on other medications, and with or without ankylosing spondylitis‐related comorbidity (e.g. peripheral joint impairment, inflammatory bowel disease, psoriasis). We did not impose restrictions on age or disease duration. We did not include diagnoses of axial spondyloarthritis, though we may consider this for an update of this review based on the classification criteria developed by ASAS (Rudwaleit 2009a; Rudwaleit 2009b).
Types of interventions
Adalimumab versus placebo, other medications, or usual care.
Etanercept versus placebo, other medications, or usual care.
Golimumab versus placebo, other medications, or usual care.
Infliximab versus placebo, other medications, or usual care.
Note that we added golimumab after the protocol for this review (Zochling 2005).
We did not impose any restrictions with regard to dose or concomitant treatments in the placebo group (for example, physical exercises, or NSAIDs, or both).
Types of outcome measures
The primary and secondary outcomes defined in the protocol and listed in the Differences between protocol and review section were chosen in 2005 when the protocol for this review was published (Zochling 2005). Since then, the Cochrane Collaboration has developed Summary of Findings tables which require choosing a maximum of seven major outcomes for presentation in the table. The Assessment of SpondyloArthritis international Society (formerly ASsessment in Ankylosing Spondylitis) (ASAS) Working Group) (http://www.asas‐group.org) has developed core sets of standardized outcome measures for use in clinical practice and trial settings. This work has been undertaken in conjunction with the OMERACT (Outcome Measures in Rheumatology, www.omeract.org) initiative which aims to establish standardized, validated outcome measures for use in clinical trials in the field of rheumatology. Following discussion with experts from ASAS, the following outcomes were chosen to be the major outcomes for this review:
ASAS40 (Brandt 2004)
BASFI (Bath Ankylosing Spondylitis Functional Index) (Calin 1994)
ASAS partial remission (Anderson 2001)
Magnetic resonance imaging (MRI) for evidence of inflammation
Radiographic progression
Withdrawals due to adverse events
Serious adverse events
The criteria for an ASAS40 response is: at least a 40% improvement with a minimum of 20 units (0 to 100 scale) improvement compared with baseline in at least three of four domains (spinal pain, function (BASFI), inflammation as measured by the mean of intensity and duration of morning stiffness in the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI), and patient global assessment), and with no worsening in the fourth domain. Partial remission is defined as a value of less than 2 on a 0 to 10 scale in each of the four domains as described above for the ASAS40.
Search methods for identification of studies
The Cochrane Musculoskeletal Group's Trial Search Co‐ordinators developed the search strategies. In the original search in January 2009, we searched the following electronic databases: Cochrane Library (2008, Issue 4) including the following databases: Cochrane Central Register of Controlled Trials (CENTRAL), Cochrane Database of Systematic Reviews (CDSR), Database of Abstracts of Reviews of Effects (DARE), Cochrane Library Health Technology Assessment Database (CLHTA), and NHS Economic Evaluation Database (NHS EED); MEDLINE (1966 to January 26, 2009); EMBASE (1980 to January 26, 2009); CINAHL (1982 to January 26, 2009); ISI Web of Knowledge (1900 to January 2009).
We reviewed the reference section of retrieved articles. We contacted authors of relevant papers and experts in the field regarding any further published or unpublished work. One review author (LM) handsearched conference proceedings from the American College of Rheumatology (ACR) and European League Against Rheumatism (EULAR, http://www.abstracts2view.com/eular/ textword search: ankylosing AND etanercept OR infliximab OR adalimumab) from 2005 to 2009, for both benefit and harms.
We conducted an updated search in May 2012. In October 2013, we conducted another updated search of all databases and also included a search for 'golimumab' from database inception. From September 2013 to June 2014 we received alerts of potential new studies identified by the McMaster PLUS database (McMaster PLUS evidence updates) through a service provided for Cochrane Musculoskeletal Group authors.
In October 2014 a search of clinicaltrials.gov was conducted for any completed trials meeting the review's inclusion criteria using 'ankylosing spondylitis' in condition and Phase 3 and 4 trials.
For safety assessments, we searched the websites of the regulatory agencies (US Food and Drug Administration‐MedWatch (http://www.fda.gov/Safety/MedWatch/default.htm), European Medicines Evaluation Agency (http://www.emea.europa.eu), Australian Adverse Drug Reactions Bulletin (http://www.tga.gov.au/adr/aadrb.htm), and UK Medicines and Healthcare products Regulatory Agency (MHRA) pharmacovigilance and drug safety updates (http://www.mhra.gov.uk); note 'Current Problems in Pharmacovigilance' was superseded by 'Drug Safety Update' in July 2007) using the terms “ankylosing spondylitis,” “adalimumab,” "humira", "etanercept", "enbrel", "infliximab", and "remicade” on April 1, 2010. We updated this search and included "golimumab"/"simponi" in November 2014.
We did not impose any language restrictions.
There were some abstracts from conference proceedings that were later published as full‐text articles; in this case, we only included the full‐text article, however, if the abstract provided additional important information that was not provided in the full‐text article, then we also included the data from the abstract. Some trials had more than one publication with the secondary publications reporting on other outcomes such as health‐related quality of life, patient‐reported outcomes, or magnetic resonance imaging (MRI) data.
The original MEDLINE search strategy is in Appendix 1. The other search strategies are available in Appendix 2 and Appendix 3.
Data collection and analysis
Selection of studies
Two people independently reviewed the results of the search strategy. The authors involved in screening the different search results were: JZ, JS, LM, MBJ, MV. We reviewed abstracts and if more information was required to determine whether the trial met the inclusion criteria, we obtained the full text. Disagreement was resolved by a third author (AB, GW).
Data extraction and management
Four review authors (JZ, LM, JS, MVS) independently extracted data from the included trials and entered the data into Review Manager 5 (RevMan 2014). We pilot‐tested data extraction forms on a selection of trials.
We extracted the following data.
General study information such as title, authors, contact address, publication source, publication year, country, study sponsor.
Characteristics of the study: design, study setting, inclusion/exclusion criteria, risk of bias criteria (e.g. randomisation method, allocation procedure, blinding of patients, caregivers and outcome assessors).
Characteris`tics of the study population and baseline characteristics of the intervention and control groups (age, sex, type of classification criteria, duration of disease, presence of comorbidity and peripheral disease, concomitant treatments, Bath Ankylosing Spondylitis Disease Activity Index (BASDAI), Bath Ankylosing Spondylitis Functional Index (BASFI), patient global assessment) and numbers in each group.
Characteristics of the intervention, such as treatment comparators, dose, method of administration, frequency of administration and duration of treatment.
Outcomes measures as noted above.
Results for the intention‐to‐treat population (where possible), summary measures with standard deviations, confidence intervals and P values where given, dropout rate and reasons for withdrawal.
When data for more than one time point was provided, we used the longest time point for the blinded phase and prior to any early‐escape option for the meta‐analysis. However, for some of the harms data, only results for the double‐blind period was reported, without providing data prior to the early‐escape option. In this case, we used the double‐blind period data with data for those that did not crossover.
We extracted both change and final values, as reported in the publication. The network meta‐analysis required only end of study values so we calculated those when only a change value had been provided and used the standard deviation at baseline for the end of study standard deviation.
We used Plot Digitizer software to estimate results from five graphs (Plot Digitizer 2014) .
Assessment of risk of bias in included studies
Two independent reviewers (LM, JZ, MV, CM, MB) assessed risk of bias in the included RCTs. As recommended by the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), we assessed the following methodological domains.
Random sequence generation ‐ was the method used to generate the allocation sequence appropriate to produce comparable groups?
Allocation sequence concealment ‐ was the method used to conceal the allocation sequence appropriate to prevent the allocation being known in advance of, or during, enrolment?
Blinding of participants, personnel ‐ were measures used to blind study participants and personnel from knowledge of which intervention a participant received? We assessed patient‐ and physician‐assessed outcomes separately.
Blinding of outcome assessors ‐ were measures used to blind outcome assessors from knowledge of which intervention a participant received? We assessed patient‐ and physician‐assessed outcomes separately.
Incomplete outcome data ‐ how complete was the outcome data for the primary outcomes? Were dropout rates and reasons for withdrawal reported? Was missing data imputed appropriately? We considered an overall completion rate of 80% or higher as a low risk of bias. If completion rates were only provided by group, a less than 80% completion rate in the treatment group was considered a high risk of bias.
Selective outcome reporting ‐ were appropriate outcomes reported and were any key outcomes missing?
Ascertainment of outcome ‐ did the researchers actively monitor for adverse events (low risk of bias) or did they simply provide spontaneous reporting of adverse events that arise (high risk of bias)?
Definition of adverse outcomes‐ were definitions provided for general 'adverse event' or 'serious adverse event'?
We explicitly judged each of these criteria using: low risk of bias; high risk of bias; or unclear, meaning either lack of information or uncertainty over the potential for bias. We provided a reason for each judgement in the 'Risk of bias' table.
Measures of treatment effect
Methods for indirect and mixed treatment comparisons
For dichotomous outcome, we derived point estimates and 95% credible intervals for odds ratios (ORs), risk ratios (RR) and risk differences (RD). For continuous outcomes, we derived mean differences (MD) and 95% credible intervals.
Methods for direct treatment comparisons
In addition to the mixed treatment comparisons, we pooled data for serious adverse events and withdrawals due to adverse events to investigate a class‐effect of harms of biologics. We analysed this using Peto odds ratio (Peto OR) with 95% CIs given that the Peto OR is recommended when the outcome is a rare event (approximately less than 10%).
Unit of analysis issues
For studies with more than two arms, we halved the number of events and patients in the placebo arm to avoid double‐counting the placebo participants in the meta‐analysis. We only assessed the standard doses for each of the biologics for inclusion in the network meta‐analysis when multiple trial arms with different dosages were reported.
Dealing with missing data
We performed the following calculations for the purpose of entering data into Review Manager 5 when the mean and standard deviation was not provided in the published article. When the median change from baseline and interquartile range (IQR) change from baseline were reported (as in Inman 2008 and van der Heijde 2005), we assumed the median change to be the mean change and calculated the standard deviation as the IQR at baseline divided by 1.35 and this standard deviation (SD) assumed for the end of study score, as per the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
Where no SD at end of study was reported or the variance for the change from baseline was provided in the study report as the standard error of the change, the baseline SD was assumed for the end of study SD (Braun 2011; van der Heijde 2006a).
In Davis 2003, the standard error of the mean (SEM) was transformed to SD by the calculation SD = SEM *sqrt(N). In Barkham 2010 the 95% CI about the mean change was converted to SD using the formula in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). In Inman 2010, the SD was calculated from the P value for continuous outcomes. In Gorman 2002, the median was assumed for the mean for continuous beneficial outcomes.
We conducted sensitivity analyses to check the robustness of the estimates to these imputations.
For continuous outcomes, we calculated the mean difference (MD) based on the number of patients analysed at that time point. When the number of patients analysed was not presented for each time point, we used the number of randomized patients in each group at baseline.
Assessment of heterogeneity
Both network meta‐analysis and traditional meta‐analysis require studies to be sufficiently similar in order to pool their results. As a result, we carefully assessed heterogeneity across trials in terms of patient characteristics, trial methodologies, and treatment protocols across trials.
As outlined in the papers by Bucher 1997 and Song 2009, there are several key assumptions that must be met when undertaking indirect comparisons. Song breaks these assumptions into three components: i.homogeneity; ii. similarity of trial; iii. consistency of evidence. Homogeneity refers to the standard assumptions used for pooling studies in a meta‐analysis; ie. trials comparing two treatments must be both clinically and methodologically similar to be combined. Trial similarity is comprised of clinical similarity and methodological similarity and the similarity of the bridging treatment. By 'bridging treatment' we mean the common comparator (ie. in a trial of A versus C and B versus C, the bridging treatment is 'C'). The assumption of consistency means that the results of direct and indirect evidence should not be heterogeneous and that there is a consistent effect across the direct comparisons.
To ensure that the consistency assumption is valid, we formally assessed inconsistency by comparing the deviance and deviance information criterion statistics of the consistency and inconsistency models (Dias 2010; Dias 2011c). To help identify the loops in which inconsistency was present, we plotted the posterior mean deviance of the individual data points in the inconsistency model against their posterior mean deviance in the consistency model (Dias 2010). Using the plots, loops in which inconsistency is present could be identified.
In the standard Review Manager 5 meta‐analyses, we tested heterogeneity of the data by visual inspection of the forest plots and by using the I2 statistic (Higgins 2003). A value greater than 50% may be considered substantial heterogeneity.
Assessment of reporting biases
We visually inspected funnel plots to assess publication bias when there were more than 10 included studies for an outcome; however, this applied to only two outcomes: the pooled results for all anti‐TNF agents versus placebo for withdrawals due to adverse events and serious adverse events.
Data synthesis
We combined data in a meta‐analyses only when we decided it was meaningful to do so, i.e. when the treatments, participants and the underlying clinical question were similar enough for pooling to make sense. We planned to analyze and present separately 'short‐term' outcomes ( less than or equal to 6 months duration) and 'long‐term' outcomes ( greater than 6 months). However, all included studies were short‐term.
We made a post‐hoc decision to present the results combined for etanercept trials which used either 25 mg administered twice a week or 50 mg administered once a week. The results from the van der Heijde 2006b study showed these dosing regimens to be equivalent in both benefit and safety. We also pooled infliximab doses of 3 mg/kg and 5 mg/kg together as there was very little heterogeneity when the major outcomes for these two doses were pooled together in a standard meta‐analyses.
Methods for direct treatment comparisons
We conducted a pooled analyses in RevMan5 for all the included anti‐TNF inhibitors to assess for a class‐effect of harms of anti‐TNF agents. We used the Peto OR statistic which uses a fixed‐effect model because the data consisted of rare events (< 10%). Although not specified a priori, we decided to perform a sensitivity analysis using the Mantel‐Haenszel OR method with a standard continuity correction of 0.5 on those meta‐analyses in which we had used the Peto OR to check the robustness of our results (as recommended by Sweeting 2004).
Methods for indirect and mixed comparisons
Our primary analysis presents refined placebo estimates for the major outcomes ASAS40, partial remission, withdrawals due to AE, SAE and BASFI for each of the biologics using the network meta‐analysis methods described below. Using a Bayesian framework, the mixed treatment comparison (MTC) method provides a refined estimate of the treatment effect by combining the information from the direct and indirect data to strengthen the precision of the estimate of effect. This methodology utilizes techniques to preserve the randomisation inherent in the RCTs. It avoids the “naive” method of pooling the results across trials from the different treatment arms of interest and then comparing the results of treatment A versus treatment B versus treatment C. This naive method ignores the randomisation that was present in the original RCTs and introduces biases expected in an observational cohort (i.e. potential confounders are no longer likely to be randomly distributed between the treatment groups) (Bucher 1997; Wells 2009).
We used WinBUGS software (MRC Biostatistics Unit, Cambridge, UK) to conduct the Bayesian mixed treatment comparison meta‐analysis using a binomial likelihood model for dichotomous outcomes or a normal likelihood model for continuous outcomes which allows for the use of multi‐arm trials (Dias 2011a; Dias 2011b; Spiegelhalter 2003) and used the placebo as the reference group. We assigned vague priors, such as N(0, 1002), for basic parameters of the treatment effects in the model (Dias 2011b) and considered informative priors for the variance parameter in the random‐effects model (Turner 2005). To ensure convergence was reached, we assessed trace plots and the Brooks‐Gelman‐Rubin statistic (Spiegelhalter 2003). Three chains were fit in WinBUGS for each analysis, with at least 10,000 iterations, and a burn‐in of at least 10,000 iterations (Ades 2008; Spiegelhalter 2003).
We conducted both fixed‐effect and random‐effects network meta‐analyses; we assessed the deviance information criterion and compared the residual deviance to the number of unconstrained data points to assess model fit and determine the choice of model (Dias 2011a; Dias 2011b; Spiegelhalter 2003). For all analyses, the deviance information criterion and residual deviance for both models were close to each other. We used the random‐effects model as the primary results, as this model takes into consideration between‐study variation, whereas fixed‐effect models assume all the trials are estimating the same treatment effect (Cooper 2009; Dias 2011b).
For the continuous outcome, we derived mean and standard deviation for mean difference (MD) using Markov Chain Monte Carlo methods. We assigned vague priors for both basic parameters and the variance parameter in the models.
Summary of Findings table
We compiled 'Summary of findings' tables using GRADEpro (GRADEpro 2014) to improve the readability of the review. The outcomes included in the 'Summary of findings' table are: ASAS40, ASAS partial remission, BASFI, MRI, radiographic progression, withdrawals due to adverse events, and serious adverse events.
For dichotomous outcomes, we calculated the number needed to treat (NNT) from the control group event rate and the risk ratio using the Visual Rx NNT calculator (Cates 2008). We calculated the corresponding risk as per the GRADEPro Help file (Schünemann 2009): Risk (per 1000 people) = 1000 × assumed control risk × risk ratio (RR). We obtained the assumed control risk from the placebo estimate in the network meta‐analysis. The absolute increased benefit or harm and 95% CI was calculated as the corresponding risk minus the assumed control risk. The relative percentage change was calculated as the RR‐1.
For continuous outcomes, we calculated the NNT for the continuous measures of BASFI using the Wells calculator (available at the Cochrane Musculoskeletal Group editorial office). We used a minimally clinically important improvement of 0.7 points on a 0 to 10 scale as per the findings of Pavy 2005. We calculated the absolute benefit as the improvement in the intervention group minus the improvement in the control group, in the original units. We calculated relative percentage change as the absolute benefit divided by the control event rate.
We used the GRADE considerations (study limitations, consistency of effect, imprecision, indirectness, and publication bias) to assess the quality of a body of evidence as it relates to the studies which contribute data to the meta‐analyses for the prespecified outcomes. We used the methods and recommendations described in Section 8.5, 8.7, Chapter 11, and Section 13.5 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011; Schünemann 2011) using GRADEpro software. We provided footnotes to justify all decisions to down‐ or up‐grade the quality of studies.
Subgroup analysis and investigation of heterogeneity
We planned the following subgroup analyses a priori in order to explore possible effect size differences.
Intervention ‐ different dose; trial duration.
Characteristics of participants ‐ different ankylosing spondylitis classification criteria; severity of baseline disease (based on BASDAI, BASFI); age; disease duration; sex; with or without peripheral joint involvement.
Sensitivity analysis
We prespecified sensitivity analysis to assess the effect of study quality (proper generation of randomisation sequence, and adequate allocation concealment and blinding) on the overall estimates of effect.
Results
Description of studies
Results of the search
January 2009: The search of the electronic databases listed in the methods section for RCTs resulted in 2445 records. After de‐duplication, there were 1644 records left to screen. We assessed a total of 60 records in depth to see if they met the inclusion criteria. We included two additional articles from handsearching European League Against Rheumatism (EULAR) abstracts on their website.
After assessing all the records, we included 14 trials, with 24 published articles (either abstracts or full‐text articles) related to those trials. The additional articles related to a trial are listed as secondary references in the reference section.
Updated search May 2012: The search of the electronic databases listed in the methods section for RCTs resulted in 1686 records. After de‐duplication, there were 1483 records left to screen. We assessed seven records to see if they met the inclusion criteria. We did not conduct any handsearching since American College of Rheumatology (ACR) and EULAR conference abstracts were indexed electronically. We added three new studies.
Updated search October 2013: This search also included a search for golimumab from database inception as well as an update of the original search. After de‐duplication, we screened 499 records. We assessed four articles in depth and identified three new studies.
An update from the McMaster PLUS database in June 2014 alerted us to one new study and the full‐text publication of a previously included abstract.
A search of clinicaltrials.gov (https://clinicaltrials.gov/) in October 2014 found 89 records, but we did not identify any new completed trials.
A flow chart of the search results is provided in Figure 1.
1.
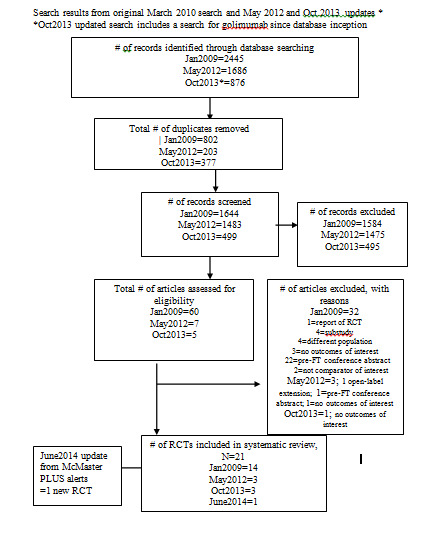
Study flow chart Note: October 2013 search included retrospective search for golimumab from database inception
Included studies
Further details on each included study are available in the Characteristics of included studies table.
Twenty‐one RCTs met the inclusion criteria with a total of 3308 participants. One thousand and twelve people received etanercept in 10 studies (Barkham 2010; Brandt 2003; Braun 2011; Calin 2004; Davis 2003; Dougados 2011; Gorman 2002; Giardina 2009, Huang 2008; Navarro‐Sarabia 2011; van der Heijde 2006b); 327 people received infliximab in 5 studies (Braun 2002;Giardina 2009; Inman 2010; van der Heijde 2005) (28 in combination with methotrexate (Marzo‐Ortega 2005); 501 received adalimumab in 4 studies (Hu 2012; Huang 2014; Lambert 2007; van der Heijde 2006a); 246 received golimumab in 2 studies (Bao 2014; Inman 2008). One study was an open‐label head‐to‐head study of etanercept (N = 25) and infliximab (N = 25) (Giardina 2009).
Eighteen RCTs contributed data to the mixed treatment comparison analysis: adalimumab (4 studies; Hu 2012; Huang 2014; Lambert 2007; van der Heijde 2006a), etanercept (8 studies; Barkham 2010; Brandt 2003; Calin 2004; Davis 2003; Dougados 2011; Gorman 2002; Huang 2008; van der Heijde 2006b), golimumab; 2 studies (Bao 2014; Inman 2008), infliximab (3 studies; Braun 2002; Inman 2010; van der Heijde 2005)) and one head‐to‐head study of etanercept to infliximab (Giardina 2009).
Additional data
We received additional data from the trial authors for the following studies: Brandt 2003, Braun 2002, Calin 2004, and Davis 2003 (though we were unable to use data from Davis 2003 since variance was not provided in the additional information received). This was mainly to obtain data on clinical endpoints where the published results for continuous outcomes had been reported as a statistic different from the mean and SD which is required for entry into Review Manager 5. We also sought additional details to clarify risk of bias items for some studies (Davis 2003; van der Heijde 2006a).
Participants
The majority of participants were Caucasian males in their early forties. The percentage of male participants in the treatment groups ranged between 65% to 80%, and 74% to 100% in the control groups. The mean age ranged from 38 to 45 years in the treatment groups and 39 to 47 years in the control groups. Between 75% and 98% of the participants in the treatment groups were Causasian with a similar distribution in the control groups (70% to 97%).
The mean disease duration in the treatment groups ranged from 8 to 16 years, and 10 to 17 years in the control groups.
Interventions
Table 2 summarizes the concomitant therapy permitted in each study.
1. Concomitant permitted therapy by study.
| Study ID | Concomitant/Background treatment |
| Adalimumab | |
| Lambert 2007 | Not reported |
| van der Heijde 2006a | Allowed to continue sulphasalazine (3 g/day), methotrexate (25 mg/week), hydroxychloroquine (400 mg/day), prednisone or prednisone equivalent (10 mg/day), and NSAIDs, if the dose had remained stable for at least 4 weeks before the baseline visit |
| Hu 2012, Huang 2014 | Concomitant use of methotrexate (≦ 25 mg/week), sulphasalazine (≦ 3 g/day), prednisone (≦ 10 mg/day), NSAIDs and/or analgesics was allowed but dose adjustments, induction and/or discontinuation of these therapies was not permitted |
| Etanercept | |
| Brandt 2003 | Allowed NSAIDs at the same or less dose at baseline |
| Calin 2004 | Allowed prestudy physiotherapy |
| Davis 2003, Gorman 2002, van der Heijde 2006b | Allowed stable doses of DMARDs, NSAIDs, and oral corticosteroids |
| Huang 2008 | Allowed stable DMARDs doses |
| Barkham 2010 | Allowed stable doses of DMARDs (sulphasalazine or methotrexate) and/or a NSAID for the duration but not corticosteroids |
| Golimumab | |
| Bao 2014, Inman 2008 | Allowed to continue concurrent treatment with stable doses of methotrexate, sulphasalazine, and hydroxychloroquine |
| Inman 2008 | Allowed to continue concurrent treatment with stable doses of methotrexate, sulphasalazine, and hydroxychloroquine, corticosteroids, and NSAIDs |
| Infliximab | |
| Braun 2002, van der Heijde 2005 | Allowed to continue on stable doses of NSAIDs |
| Inman 2010 | Concomitant therapy of NSAIDs, corticosteroids, analgesics, and DMARDs were allowed as long as doses remained stable in the study |
| Marzo‐Ortega 2005 | Allowed concomitant use of NSAIDs or oral corticosteroids |
DMARD ‐ disease‐modifying anti‐rheumatic drug NSAID ‐ non‐steroidal anti‐inflammatory drug
Adalimumab
Four studies assessed adalimumab at a dose of 40 mg every other week subcutaneously. Lambert 2007 and van der Heijde 2006a at 40 mg every other week for a 24‐week double‐blind period, though an early escape option was available after week 12. Both Hu 2012 and Huang 2014 had a 12‐week double‐blind phase.
Concomitant therapy
Lambert 2007 did not mention concomitant therapy. In van der Heijde 2006a, patients were allowed to continue sulphasalazine (3 g/day), methotrexate (25 mg/week), hydroxychloroquine (400 mg/day), prednisone or prednisone equivalent (10 mg/day), and NSAIDs, if the dose had remained stable for at least 4 weeks before the baseline visit. In Hu 2012 and Huang 2014, concomitant use of methotrexate (≦ 25 mg/week), sulphasalazine (≦ 3 g/day), prednisone (≦ 10 mg/day), NSAIDs and/or analgesics was allowed but dose adjustments, induction and/or discontinuation of these therapies was not permitted.
Etanercept
Four RCTs assessed etanercept at a dose of 25 mg twice weekly, delivered subcutaneously against placebo (Barkham 2010; Brandt 2003; Calin 2004; Davis 2003; Gorman 2002). van der Heijde 2006b assessed 50 mg once weekly versus 25 mg twice weekly versus placebo. Huang 2008 used 50 mg once weekly versus placebo. Navarro‐Sarabia 2011 assessed a high dose, 50 mg twice weekly, against the standard dose of 50 mg once weekly. Dougados 2011 assessed the effect of etanercept 50 mg once weekly against placebo in participants with advanced ankylosing spondylitis. Braun 2011 compared 50 mg once weekly to 3 g daily of sulphasalazine. The length of treatment ranged from 6 weeks (Brandt 2003 and Huang 2008) to 24 weeks (Davis 2003).
Concomitant therapy
Brandt 2003 allowed NSAIDs at the same or less dose at baseline; Calin 2004 allowed pre‐study physiotherapy; Davis 2003; Gorman 2002 and van der Heijde 2006b allowed stable doses of disease‐modifying anti‐rheumatic drugs, NSAIDs, and oral corticosteroids; Huang 2008 allowed stable disease‐modifying anti‐rheumatic drug doses; Barkham 2010 allowed stable doses of disease‐modifying anti‐rheumatic drugs sulphasalazine or methotrexate and/or a NSAID for the duration, but not corticosteroids.
Golimumab
Two studies assessed subcutaneous golimumab at a dose of 50 mg every 4 weeks (Bao 2014; Inman 2008). Both had a 24‐week double‐blind phase and an early escape option after week 16.
Concomitant therapy
In Bao 2014 and Inman 2008, patients were allowed to continue concurrent treatment with stable doses of methotrexate, sulphasalazine, and hydroxychloroquine. In Inman 2008, stable doses of corticosteroids, and NSAIDs were also allowed.
Infliximab
Four RCTs assessed infliximab; Braun 2002 assessed infliximab at 5 mg/kg intravenously at weeks 0, 2, and 6. van der Heijde 2005 delivered this same dose of infliximab at weeks 0, 2, 6, 12 and 18 weeks. Inman 2010 evaluated infliximab at 3 mg/kg delivered at weeks 0, 2, and 6. Marzo‐Ortega 2005 assessed infliximab (5 mg/kg) in combination with methotrexate against placebo plus methotrexate.
Concomitant therapy
In both Braun 2002 and van der Heijde 2005, patients were allowed to continue on stable doses of NSAIDs. It appears concomitant therapy of NSAIDs, corticosteroids, analgesics, and disease‐modifying anti‐rheumatic drugs were allowed as long as doses remained stable in the Inman 2010 study. Marzo‐Ortega 2005 allowed concomitant use of NSAIDs or oral corticosteroids.
Outcomes
All studies used the outcomes recommended by the Assessment of SpondyloArthritis international Society. The primary outcome in two studies was the BASDAI ≥ 50% (Brandt 2003; Braun 2002) and the ASAS20 in 14 studies (Bao 2014; Braun 2011; Calin 2004; Davis 2003; Gorman 2002; Huang 2008; Huang 2014; Inman 2008; Inman 2010; Lambert 2007; Navarro‐Sarabia 2011; van der Heijde 2005; van der Heijde 2006a; van der Heijde 2006b). The change in BASDAI score was the primary outcome in Marzo‐Ortega 2005.
In Dougados 2011, the primary outcome was the area under the curve in the BASDAI between baseline and week 12.
In Barkham 2010, the primary outcome was a change in the work instability of patients after three months, as measured by the Ankylosing Spondylitis Work Instability Scale.
In the abstract of Giardina 2009, the primary outcome was stated to be the proportion of patients achieving a 50% BASDAI response at week 102; Secondary: ASAS50; BASFI, back pain, morning stiffness, C‐reactive protein, and spinal mobility. However, in the full‐text article, the outcome defined as primary is not stated, and the 50% BASDAI response is not reported. ASAS20, ASAS40, BASDAI, BASFI, and adverse events were reported.
Hu 2012 did not state a primary outcome. Clinical outcomes like BASDAI and BASFI were reported along with lab measures (C‐reactive protein and serum DKK‐1) and imaging (MRI of both the lumbar spine and sacroiliac joints).
Source of funding
A total of 17 studies reported some type of industry sponsorship.
van der Heijde 2005 was supported by Centocor. Braun 2002 was funded by a grant from the German Minstry of Research and by Essex Pharma who provided the study drug. Inman 2010 did not report the funding source in the abstracts but the trial protocol states the study was sponsored by Schering‐Plough. Marzo‐Ortega 2005 reported that the study was supported by a grant in aid from Schering‐Plough, UK.
Brandt 2003 was supported by a grant from the German Minstry of Research and by Wyeth Pharma who provided the study drug. Calin 2004 was funded by Wyeth Research. Gorman 2002 was funded by the National Institute of Arthritis and Musculoskeletal and Skin Diseases and Immunex. The trial report states that Immunex was "not involved in the study design, data collection, statistical analysis, or manuscript preparation". Davis 2003 was supported by Immunex Corporation. van der Heijde 2006b was supported by Wyeth Pharmaceuticals (study drug and grants to investigational sites) Braun 2011 and Dougados 2011 were also supported by Wyeth, which was acquired by Pfzier in 2009. Navarro‐Sarabia 2011 was supported by Pfizer.
van der Heijde 2006a and Lambert 2007 were sponsored by Abbott Laboratories. Huang 2014 was sponsored by AbbVie.
Bao 2014 was funded by Janssen Research and Development. Inman 2008 was supported by Centocor Research and Development, Inc. and the Schering‐Plough Research Institute, Inc.
Barkham 2010, Giardina 2009, Hu 2012, and Huang 2008 did not list any source of funding.
Excluded studies
We excluded 6 studies after assessing the full‐text articles. The Characteristics of excluded studies table provides more details for the exclusions. Briefly, the participants in three studies (Barkham 2008b;Breban 2008; Haibel 2008) did not meet the review's inclusion criteria; the intervention in Li 2008 assesses the effect of methotrexate, not infliximab; and there is no separate information provided for ankylosing spondylitis patients in Van den Bosch 2002 (and we were unable to obtain this from the author). Morency 2011 provided data on the open‐label extension results of an included study.
Risk of bias in included studies
Figure 2 provides a graphical summary of the risk of bias of the included studies.
2.
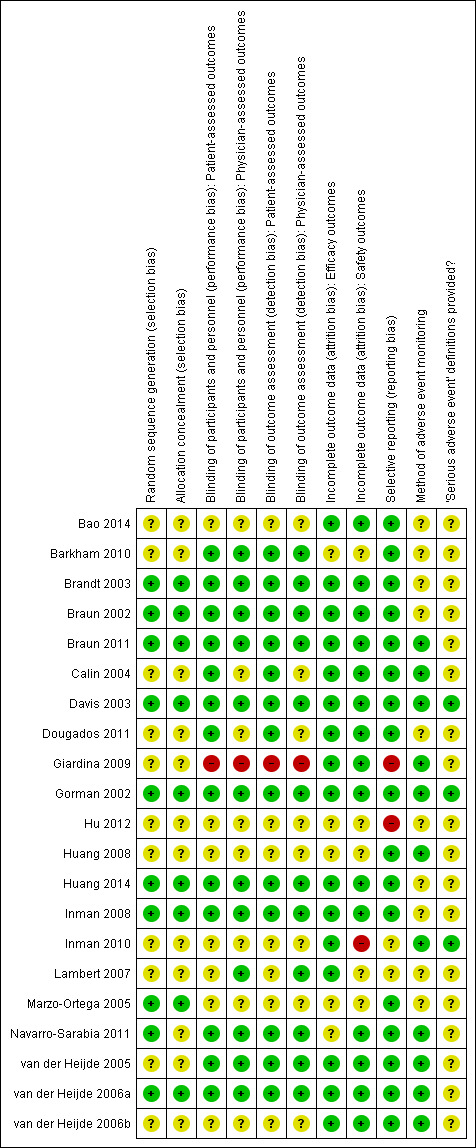
Methodological quality summary: review authors' judgements about each methodological quality item for each included study.
We received additional information from study authors on methodology and data for Davis 2003, Gorman 2002, and van der Heijde 2006a.
Huang 2008 was reported as an abstract, and did not provide enough information to make a judgement about risk of bias, and so was judged as 'unclear'.
Sequence generation
Bao 2014, Barkham 2010, Calin 2004, Dougados 2011, Giardina 2009, Hu 2012, Inman 2010, Lambert 2007, van der Heijde 2005, and van der Heijde 2006b did not provide any information regarding sequence generation, and so the judgement was 'unclear'. The 10 other studies provided evidence of appropriate generation of the randomisation sequence.
Allocation
Bao 2014, Barkham 2010, Calin 2004, Dougados 2011, Giardina 2009, Inman 2010, Hu 2012, Lambert 2007, Navarro‐Sarabia 2011, van der Heijde 2005, and van der Heijde 2006b did not provide information regarding the method of allocation concealment. The nine other studies provided evidence of appropriate concealment of allocation of the randomisation sequence.
Blinding of patient assessed outcomes
Barkham 2010, Braun 2002, Braun 2011, Brandt 2003, Calin 2004, Davis 2003, Dougados 2011, Gorman 2002, Huang 2014, Inman 2008, and van der Heijde 2005 reported the patient was blinded. We were unclear about the methods of blinding in Bao 2014, Hu 2012, Inman 2010, Lambert 2007, Marzo‐Ortega 2005, van der Heijde 2006a, and van der Heijde 2006b which were reported only as "double blind". There was no blinding in Giardina 2009, which places it at a high risk of bias.
Blinding of physician reported outcomes
Barkham 2010, Braun 2002, Brandt 2003, Davis 2003, Gorman 2002, Hu 2012, Huang 2014, Inman 2008, Lambert 2007, van der Heijde 2005, and van der Heijde 2006a reported that the investigator was blinded. Calin 2004 did not specify who other than the patient was blinded and physician/investigator blinding was unclear in Dougados 2011, Marzo‐Ortega 2005, and van der Heijde 2006b. There was no blinding in Giardina 2009 which places it at a high risk of bias.
Incomplete outcome data
We judged all trials but five trials to be at low risk of incomplete outcome data bias for beneficial outcomes as there was a low rate of missing data and most conducted an intention‐to‐treat analysis. Five were judged as unclear.
Selective outcome reporting
We judged most of the trials to be at low risk of selective outcome reporting bias as they reported on outcomes recommended by the Assessment of SpondyloArthritis international Society, with the exception of Giardina 2009 and Hu 2012 which we judged as 'high risk'. In Giardina 2009, the abstract we found first for this trial had the primary outcome listed as the proportion of people achieving a 50% response in BASDAI. However, the full‐text article did not report this outcome. We could not find a protocol for this trial. Hu 2012 did not state their primary outcome nor any adverse event data. In terms of risk of bias for selective adverse event reporting, we judged Inman 2010 and Lambert 2007 as ‘unclear’ given the lack of specifics provided on harms data. In Lambert 2007, the primary outcome was reported in an abstract but not in the full‐text article.
Method of adverse event monitoring
The following studies stated that the patients were actively monitored (though few details were provided on the specifics of the monitoring) for adverse events. These were judged to be at low risk of bias: Calin 2004, Davis 2003, Giardina 2009, Gorman 2002, Huang 2008, Navarro‐Sarabia 2011, van der Heijde 2005, van der Heijde 2006a, and van der Heijde 2006b. The rest of the studies did not mention how the patients were monitored for adverse events and were judged as 'unclear' risk of bias.
Definition of serious adverse event provided
The following studies used a common grading system, though the specific definition of serious adverse events was not provided in the articles: Davis 2003, Gorman 2002, Inman 2010; we judged these to be at low risk of bias. van der Heijde 2005 and van der Heijde 2006a did not provide general serious adverse events definitions, but each serious adverse event was clearly explained in the published report. The other studies did not report their definition of 'serious adverse events' and we judged them to be at unclear risk of bias.
Effects of interventions
See: Table 1
The review prespecified that outcomes measured at six months or less would assess short‐term results and greater than six months would assess long‐term results; however, all outcomes in the placebo‐controlled trials were reported at six months or less.
Table 1 provides an overview of the mixed treatment comparison refined placebo estimates for the major outcomes of ASAS40, physical function, ASAS partial remission, withdrawals due to adverse events and serious adverse events for the individual biologics and for the class‐effect analysis for the two adverse event outcomes. We did not pool data from magnetic resonance imaging and radiographic progression outcomes. Figure 3 shows the network diagram for ASAS40. Figure 4 and Figure 5 show the forest plots for the biologic versus placebo and head‐to‐head mixed treatment comparison estimates for the outcomes ASAS40 and withdrawals due to adverse events, respectively. The pairwise data for the individual trials that was used in the mixed treatment comparison analysis is available in the Data and Analyses section. Trace plots and Brooks–Gelman–Rubin statistic indicated convergence of the model in all analyses.
3.
ASAS40 Evidence Diagram
4.
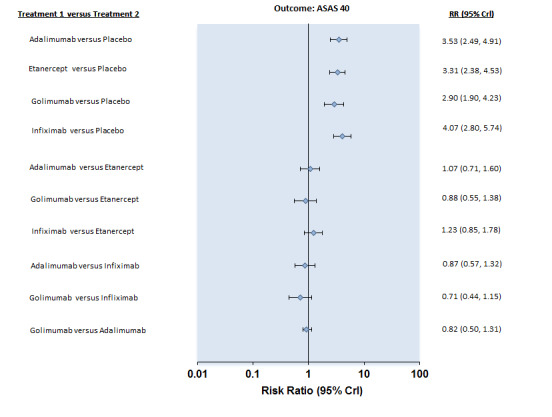
Forest Plot: ASAS40
5.
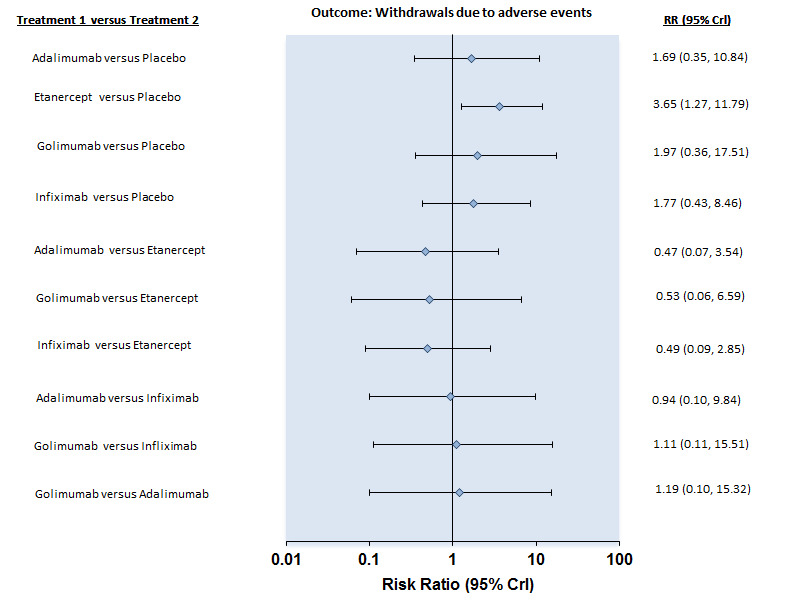
Forest Plot: Withdrawals due to adverse events
Individual biologics
Adalimumab (40 mg every other week) versus placebo
Four studies assessed the effect of adalimumab versus placebo (Hu 2012; Huang 2014; Lambert 2007; van der Heijde 2006a). Lambert 2007 did not report all benefits and adverse events in the published article but they were reported in an abstract and other publications of the same trial (Maksymowych 2005; Maksymowych 2008). Hu 2012 did not report on any adverse outcomes.
Major outcomes
ASAS40: There was high quality evidence that the adalimumab group was more likely than placebo to achieve the ASAS40 criteria (risk ratio (RR) 3.53, 95% credible interval (Crl) 2.49 to 4.91, with an absolute improvement of 33% (95% Crl 19% to 51%) and a NNT = 4 (95% confidence interval (CI) 2 to 6)).
Physical function (BASFI 0 to 10 scale, lower is better): There was high quality evidence of a clinically important improvement in physical function (mean difference (MD) ‐1.6, 95% CrI ‐2.2 to ‐0.9), with an absolute increased benefit of ‐16% (95% Crl ‐22% to ‐9%)); relative percentage change from baseline = ‐32% (95% CrI ‐44% to ‐18%); and NNT to achieve the minimally important difference (MCID) of 0.7 points = 4 (95% CI 3 to 5).
ASAS partial remission: There was moderate quality evidence (downgraded for imprecision) that the adalimumab group was more likely than placebo to meet the criteria for partial remission (RR 6.28, 95% Crl 3.13 to 12.78), with an absolute improvement of 16% (95% Crl 6% to 35%) and a NNT = 7 (95% CI 3 to 16).
Magnetic resonance imaging (MRI): There was moderate quality evidence of a small absolute improvement on spinal inflammation with unclear clinical relevance. Hu 2012 (N = 46) assessed spinal inflammation using the Spondyloarthritis Research Consortium of Canada (SPARCC) scoring method. SPARCC scores for the spine can range from 0 to 108 and SPARCC sacroiliac joint scores can range from 0 to 72. The MD for the lumbar spine was ‐6.5 (95% CI ‐13.06 to 0.06), with a small absolute benefit of ‐6% (95% CI ‐12% to 0.05%) and relative percentage change = ‐33% (95% CI ‐66% to 0%). The MD for the sacroiliac joint was ‐3.00 (95% CI ‐7.46 to 1.46).
Lambert 2007 used MRI to assess the effect of adalimumab compared to placebo in reducing spinal and sacroiliac joint inflammation using the SPARCC scoring method. MRIs were obtained for all participants (N = 82) at baseline and week 12. There was high quality evidence of a statistically significantly greater reduction in the mean spine SPARCC score of adalimumab‐treated patients (median change 6.3, range 34.0 to 2.0) compared with placebo‐treated patients (median change 0.5, range 26.0 to 13.5) (P < 0.001). The mean sacroiliac joint SPARCC score also decreased significantly between the adalimumab (median change 0.5, range 22.5 to 2.5) and placebo groups (median change 0.0, range 13.5 to 16.0) (P < 0.001). In terms of percentage change from baseline, placebo‐treated patients had a 9.4% mean increase in spine SPARCC scores compared to a 53.6% mean reduction in scores in adalimumab‐treated patients (P < 0.001). There was also a significant difference in the mean percentage reduction in adalimumab (52.9%) and placebo‐treated (12.7%) patients in the sacroiliac joint SPARCC score (P = 0.017).
Radiographic progression: Not reported.
Withdrawals due to adverse events: Based on moderate quality evidence, we are uncertain of the effect on withdrawals due to adverse events (RR 1.69, 95% CI 0.35 to 10.84) but the absolute numbers were low: 6/437 in the adalimumab group versus 2/222 in the placebo (absolute increased harm = 0.6% (95% Crl ‐0.4% to 7%); relative percentage change = 69% (95% CI ‐65% to 984%). We downgraded the evidence due to the low event rates and resulting imprecision.
Serious adverse events: Based moderate quality evidence, we are uncertain of the effect on serious adverse events (RR 0.92, 95% CI 0.26 to 3.93) but the absolute numbers were low: 7/437 in the adalimumab group versus 4/222 in the placebo (absolute increased harm = ‐0.2% (95% Crl ‐1.1% to 4.4%); relative percentage change = ‐8% (95% CI ‐74% to 293%)). We downgraded the evidence due to the low event rates and resulting imprecision.
Etanercept (25 mg twice weekly or 50 mg once weekly) versus placebo
Five studies assessed the effect of etanercept 25 mg twice weekly versus placebo (Barkham 2010; Brandt 2003; Calin 2004; Davis 2003; Gorman 2002). Two studies assessed the effect of 50 mg of etanercept once weekly versus placebo (Dougados 2011; Huang 2008).
van der Heijde 2006b performed a double‐blind, placebo‐controlled non‐inferiority trial with 356 patients to compare the benefit of 25 mg twice weekly and 50 mg once weekly. Both dosing regimens were found to be statistically significantly better than placebo in terms of ASAS20, ASAS5/6, ASAS40, BASDAI, BASFI, and other clinical measures. The study also showed that 50 mg once weekly was not inferior to the usual standard of 25 mg twice weekly in terms of the primary outcome of ASAS20 response at 12 weeks. As well, patient‐reported outcomes such as fatigue, EuroQOL‐5D, and SF‐36 scores were similar between the two doses. Injection site reactions were similar in the 50 mg once weekly and 25 mg twice weekly groups (20.7% versus 22.7%). Infections were also similar between the two groups (22.6% and 22.0%). The percentage of non‐infectious serious adverse events was 5.2% and 4.0% in the 50 mg once‐ and 25 mg twice‐per‐week groups, respectively. One serious infection occurred in each group.The authors concluded that "the efficacy and safety of etanercept 50 mg once weekly was comparable with that of the standard regimen of 25 mg twice weekly in patients with ankylosing spondylitis." Based on this result, we combined studies with the two dosing regimens in the standard and network meta‐analyses.
Dougados 2011 assessed the effect of 50 mg of etanercept once weekly versus placebo in a population with advanced and active ankylosing spondylitis. We pooled this results of this study with the ones above given the lack of heterogeneity of the results, even though the population had a longer disease duration than the other included studies. The majority of clinical outcomes showed statistically and clinically important improvements in this population.
Major outcomes
ASAS40: There was high quality evidence that the etanercept group participants were more likely than placebo to achieve the ASAS40 criteria (RR 3.31, 95% Crl 2.38 to 4.53), with an absolute improvement of 30% (95% CI 18% to 46%) and a NNT = 4 (95% CI 2 to 6).
Physical function (BASFI 0 to 10 scale, lower is better): There was high quality evidence of a clinically important improvement in physical function (MD ‐1.1, 95% CI ‐1.6 to ‐0.6), with an absolute increased benefit percentage = ‐11% (95% Crl ‐16% to ‐6%); relative percentage change from baseline = ‐22% (95% CI ‐32% to ‐12%); and NNT to achieve the MCID of 0.7 points = 4 (95% CI 4 to 6).
ASAS partial remission: There was moderate quality evidence (downgraded for imprecision) that the etanercept group participants were more likely than placebo to meet the criteria for partial remission (RR 4.24, 95% Crl 2.31 to 8.09), with an absolute improvement of 10% (95% Crl 4% to 21%) and a NNT = 11 (95% CI 5 to 26).
MRI and radiographic outcomes: Not assessed in these studies.
Withdrawals due to adverse events: There was moderate quality evidence of increased withdrawals due to adverse events in the etanercept versus placebo group (RR 3.65, 95% CI 1.27 to 11.79) although the absolute numbers were low: 22/655 in the etanercept group versus 1/402 in the placebo (absolute increased harm: 2% (95% Crl 0.2% to 8%; NNTH: 54 (95% CI 14 to 530). The evidence was downgraded due to the low event rates and resulting imprecision.
Serious adverse events: Based on moderate quality evidence, we are uncertain of the effect of etanercept on serious adverse events (RR 1.69, 95% CI 0.76 to 3.72) but the absolute numbers were low: 28/655 in the etanercept group versus 8/406 in the placebo (absolute increased harm = 1% (95% Crl ‐0.4% to 4.1%); relative percentage change = 67% (95% CI ‐27% to 282%)). The evidence was downgraded due to the low event rates and resulting imprecision.
Etanercept (50 mg twice weekly) versus Etanercept (50 mg once weekly)
Navarro‐Sarabia 2011 investigated a high‐dose (100 mg per week) versus a standard‐dose (50 mg per week) of etanercept. There was no evidence of a difference between the two doses in the major clinical beneficial outcomes and most results were precise in the estimate around a null effect. The evidence for the harms outcomes was inconclusive due to the low number of events and the wide confidence intervals. The authors concluded that the higher dose does not significantly increase the benefit of etanercept.
Etanercept 50 mg weekly versus sulphasalazine (3 g) daily
Braun 2011 (ASCEND study) found a statistically and clinically important improvement for ASAS40 (RR 1.84, 95% CI 1.47 to 2.29) (Analysis 6.1) and ASAS Partial Remission RR 2.14 (1.49 to 3.08) (Analysis 6.3) at 16 weeks in the etanercept compared to sulphasalazine group. BASFI was statistically significantly improved in the etanercept versus the sulphasalazine group (MD ‐10.63, 95% CI ‐15.22 to ‐6.04) . There was no evidence of a difference in the risk of serious adverse events between etanercept and sulphasalazine (Peto OR 0.86, 95% CI 0.24 to 3.05) Analysis 6.5).
6.1. Analysis.
Comparison 6 Etanercept versus sulphasalazine, Outcome 1 ASAS40 ‐ 16‐week.
6.3. Analysis.
Comparison 6 Etanercept versus sulphasalazine, Outcome 3 ASAS Partial remission.
6.5. Analysis.
Comparison 6 Etanercept versus sulphasalazine, Outcome 5 Serious adverse events.
A subanalysis of the ASCEND study was undertaken to investigate the benefit of etanercept versus sulphasalazine in patients with peripheral joint involvement (Braun 2012a). Of 566 subjects included in original study, 181 (etanercept 121; sulphasalazine 60) had ≥ 1 swollen peripheral joint and 364 (etanercept 250; sulphasalazine 124) had none at baseline. Ankylosing spondylitis patients treated with etanercept showed significantly greater improvement than those treated with sulphasalazine in all joint assessments regardless of swollen joint involvement. The authors noted that,"These findings support the role of etanercept as a key therapy for the management of subjects with ankylosing spondylitis regardless of peripheral joint involvement."
Golimumab (50 mg every 4 weeks) versus placebo
Major outcomes
ASAS40: There was high quality evidence that the golimumab group participants were more likely than placebo to achieve the ASAS40 criteria (RR 2.90, 95% Crl 1.90 to 4.23), with an absolute improvement of 25% (95% Crl 12% to 42%) and a NNT = 5 (95% CI 3 to 9).
Physical function (BASFI 0 to 10 scale, lower is better): There was high quality evidence of a clinically important improvement in physical function (MD ‐1.5, 95% Crl ‐2.3 to ‐0.7), with an absolute increased benefit percentage of ‐15% (95% Crl ‐23% to ‐7%); relative percentage change from baseline of ‐30% (95% CI ‐46% to ‐14%); and NNT to achieve the MCID of 0.7 points = 4 (95% CI 3 to 5).
ASAS partial remission: There was moderate quality evidence (downgraded for imprecision) that people in the golimumab group were more likely than those in the placebo group to meet the criteria for partial remission (RR 5.18, 95% Crl 1.90 to 14.79), with an absolute improvement of 13% (95% Crl 3% to 41%) and a NNT = 8 (95% CI 3 to 38).
MRI: A MRI substudy conducted at 10/57 participating sites (N = 60 for placebo and 50 mg golimumab arms) of Inman 2008 was reported in Braun 2012b. Evidence was downgraded to low quality due to concerns about missing data and the modest level of agreement between outcome assessors. The mean change from baseline in the golimumab group was 3.4 points lower (95% CI 7.7 to 0.90 points lower) than placebo, as measured by the ankylosing spondylitis spine MRI activity score (0 to 138; lower means less erosions or edema). This translated to a small absolute increased benefit of 2.5% (95% CI ‐5.6% to 0.7%) and a relative percentage change of ‐35% (95% CI ‐80% to 9%).
Radiographic progression: Not reported.
Withdrawals due to adverse events: Based on moderate quality evidence, we are uncertain of the effect of golimumab on withdrawals due to adverse events (RR 1.97, 95% CI 0.36 to 17.51) but the absolute numbers were low: 5/246 in the golimumab group versus 2/183 in the placebo (absolute increased harm = 1.6% (95% Crl ‐0.4% to 11.6%); relative percentage change of 97% (95% CI ‐64% to 1651%)). The evidence was downgraded due to the low event rates and resulting imprecision.
Serious adverse events: Based on moderate quality evidence, we are uncertain of the effect of golimumab on serious adverse events (RR 0.69, 95% CI 0.15 to 3.32) but the absolute numbers were low: 5/138 in the golimumab group versus 4/78 in the placebo group (absolute increased harm was ‐0.5% (95% Crl ‐1.3% to 3.5%); relative percentage change was ‐31% (95% CI ‐85% to 232%). The evidence was downgraded due to the low event rates and resulting imprecision. The total number of serious adverse events were not clearly reported in Bao 2014.
Infliximab versus placebo (pooled 3 mg/kg and 5 mg/kg)
Two studies assessed the effect of infliximab 5 mg/kg versus placebo; Braun 2002 at 12 weeks and van der Heijde 2005 at 24 weeks. Inman 2010 evaluated a 12‐week RCT of a lower dose of 3 mg/kg against placebo. There was no significant heterogeneity of including the 3 mg/kg dose with the 5 mg/kg dose results, so they were pooled together. Separate details for the lower dose are also reported below.
Major outcomes
ASAS40: There was high quality evidence that the infliximab group participants were more likely than placebo to achieve the ASAS40 criteria (RR 4.07, 95% Crl 2.80 to 5.74), with an absolute improvement of 40% (95% Crl 23% to 62%) and a NNT = 3 (95% CI 2 to 5).
Physical function (BASFI 0 to 10 scale, lower is better): There was high quality evidence of a clinically important improvement in physical function (MD ‐2.1, 95% CrI ‐2.7 to ‐1.4), with an absolute increased benefit percentage of ‐21% (95% Crl ‐27% to ‐14%); relative percentage change from baseline was ‐42% (95% CI ‐54% to ‐28%); and NNT to achieve the MCID of 0.7 points = 2 (95% CI 2 to 3).
ASAS Partial remission: There was moderate quality evidence (downgraded for imprecision) that participants in the infliximab group were more likely than placebo to meet the criteria for partial remission (RR 15.41, 95% Crl 5.09 to 47.98), with an absolute improvement of 44% (95% Crl 13% to 87%) and a NNT = 3 (95% CI 2 to 8).
Withdrawals due to adverse events: Based on moderate quality evidence, we are uncertain of the effect of infliximab on withdrawals due to adverse events (RR 1.77, 95% CI 0.43 to 8.46) but the absolute numbers were low: 5/274 in the infliximab group versus 2/150 in the placebo (absolute increased harm was 0.5% (95% Crl ‐0.4% to 5.6%); relative percentage change was 77% (95% CI ‐43% to 746%). The evidence was downgraded due to the low event rates and resulting imprecision.
Serious adverse events: Based on moderate quality evidence, we are uncertain of the effect of infliximab on serious adverse events (RR 2.53, 95% CI 0.76 to 11.09) but the absolute numbers were low: 11/275 in the infliximab group versus 2/147 in the placebo (absolute increased harm: 2.3% (95% Crl ‐0.4% to 15.1%); relative percentage change was 153% (95% CI ‐24% to 1009%). The evidence was downgraded due to the low event rates and resulting imprecision.
MRI: Data was reported in a secondary publication of van der Heijde 2005 (Braun 2006). The MRI Activity Score was used to assess spinal inflammation as detected by MRI. There was high quality evidence of a greater reduction in MRI Activity Score from baseline to week 24 in the infliximab‐treated group (MD ‐4.42, 95% CI ‐5.59 to ‐3.25 on a 0 to 138 scale, lower means less erosions or edema), though the absolute increase in benefit was small: ‐3% (95% CI ‐4% to ‐2.4%); NNT = 3 (95% CI 3 to 5) with a relative percentage change of ‐62% (95% CI ‐79% to ‐46%). As well, evidence of some ("some" defined as MRI Activity score > 1 where each vertebral unit is scored from 0 to 6 with 0 = no inflammation) spinal inflammation by week 24 was 37% in the infliximab group compared to 73.6% in the placebo group (P < 0.001).
A separate publication (Maksymowych 2010) reported on spinal inflammation as measured by the SPARCC MRI method in a subset of the Inman 2010 trial participants. Thirty‐six patients at two of the sites involved in the trial participated in the MRI investigation. They found a large treatment effect in favor of infliximab (mean percentage change based on evaluation of the most severely affected discovertebral units (6 DVU score) in the infliximab group was ‐55.1% compared to +5.8% in the placebo group (P < 0.001). When the evaluation was based on the entire spine (23 DVU score), the infliximab group had a mean reduction of 57.2% compared to 3.4% in the placebo group (P < 0.001).
Radiographic progression: Radiological change was assessed using the Bath ankylosing spondylitis radiology index (BASRI) in Braun 2002 though data were not shown. They found that the "initial degree of radiological axial changes assessed by the BASRIs was similar in both groups." Interestingly, the study stated that there was "no less benefit in patients with higher BASRI scores than those with lower scores"; indicating that the amount of ankylosis did not impact the benefit of infliximab. Radiographic outcome data was not reported in van der Heijde 2005 or Inman 2010.
Low dose infliximab (3 mg/kg) versus placebo
Inman 2010 conducted a study on 76 patients to compare the benefit of infliximab at 3 mg/kg versus placebo. The first 12 weeks were a double‐blind placebo phase and then there was an open‐label phase where placebo patients switched to 3 mg/kg infliximab and received infusions at weeks 14, 16, 22 and every 8 weeks afterwards. Patients were eligible for a dose escalation to 5 mg/kg at weeks 22 or 38 if they were not responding adequately.
Significantly more participants in the infliximab group than placebo achieved an ASAS40 response at 12 weeks (RR 5.69 (1.83 to 17.74). However, after the 12‐week double‐blind placebo phase, 68% of patients in the 3 mg/kg infliximab group switched to the 5 mg/kg by 38 weeks because of lack of benefit.
Adverse event data were not presented separately for the placebo‐controlled, 12‐week phase, with the exception of one serious adverse event of arthralgia in an infliximab‐treated patient.
Infliximab + methotrexate versus placebo + methotrexate
Marzo‐Ortega 2005 assessed the benefit and harms of adding infliximab (5 mg/kg) or placebo to methotrexate therapy. The final infliximab or placebo treatment was given at 22 weeks. At 30 weeks, neither the change in BASDAI (MD ‐1.14, 95% CI ‐2.76 to 0.48) (Analysis 7.1), nor the ASAS20 response (RR 2.33, 95% CI 0.80 to 6.80) (Analysis 7.2) or a 50% improvement in BASDAI (RR 2.50, 95% CI 0.87 to 7.22) (Analysis 7.3) were statistically significantly different between the two groups. There was a statistically significant difference at 10 weeks, but this did not extend to 30 weeks. The last dosing of infliximab was at 22 weeks, so the authors concluded that the addition of methotrexate to the treatment regimen did not lengthen the beneficial period of infliximab. There was a significantly greater reduction in the number of lesions in the sacroiliac joints and spine resolving completely in the combination group versus the methotrexate monotherapy group, as assessed by MRI. No serious adverse events were seen in either group.
7.1. Analysis.
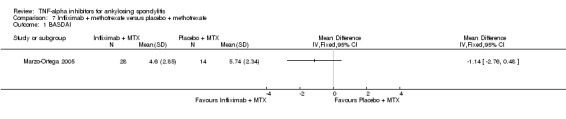
Comparison 7 Infliximab + methotrexate versus placebo + methotrexate, Outcome 1 BASDAI.
7.2. Analysis.
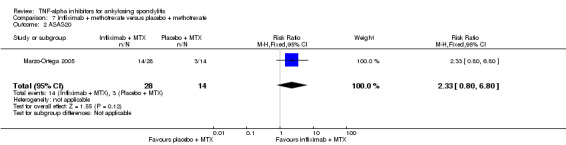
Comparison 7 Infliximab + methotrexate versus placebo + methotrexate, Outcome 2 ASAS20.
7.3. Analysis.
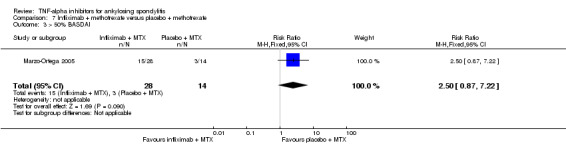
Comparison 7 Infliximab + methotrexate versus placebo + methotrexate, Outcome 3 > 50% BASDAI.
Adverse events ‐ pooled results from all four anti‐TNF agents versus placebo
We pooled the results from adalimumab, etanercept, golimumab, and infliximab to assess for a class‐effect of adverse effects of TNF‐inhibitors. Given our interest in adverse events was prespecified in short‐term (less than or equal to 6 months) and long‐term (greater than 6 months) periods, and that all time points reported were six months or less, we pooled all trials together for an assessment of short‐term effects. The adverse events reported for the 50 mg once weekly and 25 mg twice weekly groups in the trial of etanercept (van der Heijde 2006b) and infliximab 3mg/kg and 5mg/kg doses were pooled together for this analysis .
Withdrawals due to adverse events: Based on 16 studies (N = 2623 participants), there is moderate quality evidence (downgraded for imprecision) of an increase in withdrawals due to adverse events in the anti‐TNF group versus placebo (Peto OR 2.44, 95% CI 1.26 to 4.72) though the absolute increase was small (1%, 95% CI 0% to 2%) with 38/1637 in the biologic group and 7/986 in the placebo group (Analysis 8.1).
8.1. Analysis.
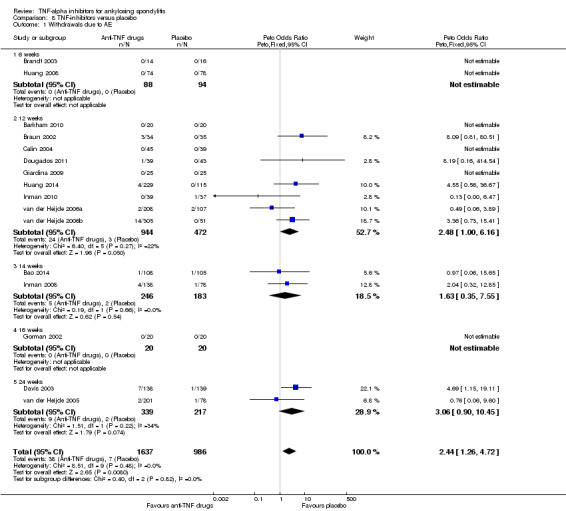
Comparison 8 TNF‐inhibitors versus placebo, Outcome 1 Withdrawals due to AE.
Serious adverse events: Based on 15 studies (N = 2408 participants) and moderate quality evidence, results were inconclusive for evidence of a difference in serious adverse events in the anti‐TNF group versus placebo (Peto OR 1.45, 95% CI 0.85 to 2.48) though the absolute increase was small (1%, 95% CI 0% to 2%) with 51/1530 in the biologic group and 18/878 in the placebo group (Analysis 8.2).
8.2. Analysis.
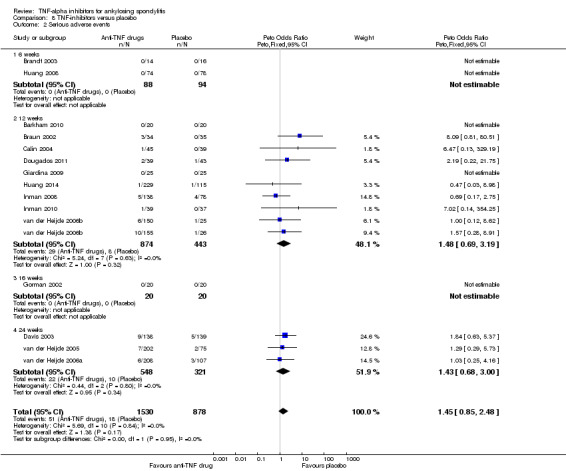
Comparison 8 TNF‐inhibitors versus placebo, Outcome 2 Serious adverse events.
Most studies did not provide a definition of a 'serious adverse event'. Two studies (Davis 2003; Gorman 2002) stated that the National Cancer Institute Common Toxicity criteria scale was used to grade adverse events and abnormal laboratory values and Inman 2010 used MedDRA ver9.
Head‐to‐head comparisons
Direct evidence: Etanercept versus infliximab
One RCT (Giardina 2009) assessed the benefit and harm of etanercept compared to infliximab over a two‐year period. The trial was reported first in a conference abstract and later a full‐text article was published. The risk of bias for this study is high given the lack of blinding of participants and outcome assessors and there were no details provided about the method of sequence generation and allocation concealment.
Fifty patients were enrolled in the trial. To calculate the proportion of responders reported in the abstract for entry into RevMan, the number of participants in each treatment group was assumed to be the denominators at end of study. However, there is a slight discrepancy in the number of people in each group; the full‐text article states that there were 25 people in each group, while the abstract states there were 26 people in the etanercept group and 24 people in the infliximab group. We used the numbers from the full‐text article.
Major outcomes
ASAS40: We are uncertain whether there is a difference between etanercept and infliximab at 12 weeks (RR 0.79, 95% CI 0.45 to 1.38) (Analysis 5.1).
5.1. Analysis.
Comparison 5 Etanercept versus infliximab, Outcome 1 ASAS40 ‐ 12 weeks.
BASFI (0 to 10 scale): A statistically significant MD of 1.50 (95% CI 0.94 to 2.06) in favour of infliximab was found.
Partial remission: Not reported.
MRI and radiographic data: Not reported.
Withdrawals due to adverse events: The abstract states that "no patients discontinued therapy".
Serious adverse events: Although two "severe infections" were reported in the infliximab group and one in the etanercept group, the text stated that there were no discontinuations due to adverse events, so we considered there to be no serious adverse events.
Indirect evidence from mixed treatment comparison (MTC) analysis
Indirect comparisons of one treatment versus another are useful when there is no, or limited, direct evidence from head‐to‐head RCTs comparing treatments of interest to practitioners who must make choices as to which treatment to prescribe to their patient. In the case of the four anti‐TNF agents for use in ankylosing spondylitis, the majority of RCTs assess each of adalimumab, etanercept, golimumab and infliximab against placebo and we also found one open‐label RCT comparing etanercept to infliximab over a two year period (Giardina 2009). The lack of direct comparisons between anti‐TNF agents provides a compelling reason to undertake a mixed treatment comparison analysis. Figure 3 describes the relationship of trials in this systematic review. It is a network with one closed loop (Wells 2009).
We used the direct evidence from the Giardina 2009 study in the mixed treatment comparison analysis to give refined estimates of the comparison of etanercept versus infliximab and refined estimates of the four anti‐TNF agents versus placebo. As well, the mixed treatment comparison analysis provided us with new indirect estimates of adalimumab versus etanercept, golimumab versus etanercept, adalimumab versus infliximab, golimumab versus infliximab, and adalimumab versus golimumab. We performed indirect comparisons on the following outcomes: ASAS40, ASAS partial remission, withdrawals due to adverse events, and serious adverse events. Giardina 2009 was able to contribute data to all the outcomes except ASAS partial remission. The outcomes of withdrawals due to adverse events and serious adverse events had rare events, so in the mixed treatment comparison analysis, a random‐effects model was used and the zero events were adjusted for by adding 0.5.
Table 3, Table 4, Table 5, Table 6 and Table 7 provide results for the mixed treatment comparison placebo estimates and also show the new estimates for the different biologics against each other. For the mixed treatment comparison‐derived head‐to‐head estimates, there were wide confidence intervals and no consistency as to which biologic was favored in terms of ASAS40, partial remission, BASFI, withdrawals due to adverse events, and serious adverse events.
2. ASAS40: odds ratios, risk ratios and risk differences for all treatment comparisons (random‐effects model).
| Treatment | Reference | OR (95% CrI) | RR (95% CrI) | RD% (95% CrI) |
| Etanercept | Placebo | 5.16 (3.14 to 8.62) | 3.31 (2.38 to 4.53) | 30.98 (20.08 to 42.47) |
| Infliximab | 7.75 (4.11 to 15.45) | 4.07 (2.80 to 5.74) | 41.08 (26.62 to 55.88) | |
| Adalimumab | 5.84 (3.33 to 10.68) | 3.53 (2.49 to 4.91) | 34.00 (21.03 to 48.00) | |
| Golimumab | 4.12 (2.23 to 7.74) | 2.90 (1.90 to 4.23) | 25.50 (12.66 to 40.31) | |
| Infliximab | Etanercept | 1.50 (0.73 to 3.23) | 1.23 (0.85 to 1.78) | 10.01 (‐7.67 to 28.21) |
| Adalimumab | 1.13 (0.53 to 2.46) | 1.07 (0.71 to 1.60) | 3.09 (‐15.40 to 21.89) | |
| Golimumab | 0.80 (0.36 to 1.78) | 0.88 (0.55 to 1.38) | ‐5.45 (‐23.60 to 13.99) | |
| Adalimumab | Infliximab | 0.76 (0.31 to 1.76) | 0.87 (0.57 to 1.32) | ‐6.89 (‐27.99 to 13.79) |
| Golimumab | 0.53 (0.21 to 1.30) | 0.71 (0.44 to 1.15) | ‐15.62 (‐36.44 to 6.40) | |
| Golimumab | Adalimumab | 0.71 (0.30 to 1.62) | 0.82 (0.50 to 1.31) | ‐8.45 (‐28.45 to 11.78) |
| Random‐effects model | Residual deviance | 18.86 versus 20 data points | ||
| Deviance information criteria | 124.187 | |||
| Fixed‐effect model | Residual deviance | 19.51 versus 20 data points | ||
| Deviance information criteria | 123.44 |
CrL ‐ credible interval OR ‐odds ratio RD ‐risk difference RR ‐risk ratio
3. BASFI: mean difference for all treatment comparisons (random‐effects model).
| Treatment | Reference | Mean (SD) |
| Etanercept | Placebo | ‐1.09 (‐1.60 to ‐0.56) |
| Infliximab | ‐2.07 (‐2.71 to ‐1.35) | |
| Adalimumab | ‐1.57 (‐2.21 to ‐0.89) | |
| Golimumab | ‐1.49 (‐2.27 to ‐0.69) | |
| Infliximab | Etanercept | ‐0.98 (‐1.69 to ‐0.23) |
| Adalimumab | ‐0.48 (‐1.32 to 0.36) | |
| Golimumab | ‐0.40 (‐1.36 to 0.54) | |
| Adalimumab | Infliximab | 0.51 (‐0.46 to 1.43) |
| Golimumab | 0.59 (‐0.49 to 1.61) | |
| Golimumab | Adalimumab | 0.08 (‐0.95 to 1.10) |
| Random‐effects model | Residual deviance | 27.04 versus 28 data points |
| Deviance information criteria | 31.943 | |
| Fixed‐effect model | Residual deviance | 34.09 versus 28 data points |
| Deviance information criteria | 34.13 |
SD ‐ standard deviation
4. Partial remission: odds ratios, risk ratios and risk differences for all treatment comparisons (random‐effects model).
| Treatment | Reference | OR (95% CrI) | RR (95% CrI) | RD% (95% CrI) |
| Etanercept | Placebo | 4.72 (2.43 to 9.72) | 4.24 (2.31 to 8.09) | 9.66 (3.79 to 19.10) |
| Infliximab | 28.18 (6.25to 284.40) | 15.41 (5.09 to 47.98) | 43.61 (16.89 to 82.38) | |
| Adalimumab | 7.53 (3.39 to 18.33) | 6.28 (3.13 to 12.78) | 15.74 (6.11 to 32.38) | |
| Golimumab | 5.96 (1.97 to 23.86) | 5.18 (1.90 to 14.79) | 12.39 (2.69 to 38.31) | |
| Infliximab | Etanercept | 5.94 (1.12 to 65.22) | 3.60 (1.09 to 12.19) | 33.62 (1.91 to 76.29) |
| Adalimumab | 1.59 (0.53 to 4.93) | 1.47 (0.58 to 3.67) | 5.83 (‐7.73 to 24.11) | |
| Golimumab | 1.26 (0.34 to 5.71) | 1.22 (0.38 to 4.00) | 2.68 (‐11.26 to 28.71) | |
| Adalimumab | Infliximab | 0.27 (0.02 to 1.52) | 0.41 (0.12 to 1.35) | ‐26.97 (‐72.15 to 7.93) |
| Golimumab | 0.21 (0.02 to 1.56) | 0.34 (0.08 to 1.35) | ‐29.87 (‐74.54 to 9.13) | |
| Golimumab | Adalimumab | 0.78 (0.19 to 4.08) | 0.82 (0.25 to 2.92) | ‐3.23 (‐23.14 to 24.98) |
| Random‐effects model | Residual deviance | 12.93 versus 16 data points | ||
| Deviance information criteria | 88.137 | |||
| Fixed‐effect model | Residual deviance | 12.56 versus 16 data points | ||
| Deviance information criteria | 87.316 |
CrL ‐ credible interval OR ‐ odds ratio RD ‐ risk difference RR ‐ risk ratio
5. Serious adverse events: odds ratios, risk ratios and risk differences for all treatment comparisons (random‐effects model).
| Treatment | Reference | OR (95% CrI) | RR (95% CrI) | RD% (95% CrI) |
| Etanercept | Placebo | 1.70 (0.76 to 3.84) | 1.69 (0.76 to 3.72) | 1.03 (‐0.48 to 3.24) |
| Infliximab | 2.60 (0.75 to 12.62) | 2.53 (0.76 to 11.09) | 2.34 (‐0.45 to 12.84) | |
| Adalimumab | 0.92 (0.25 to 4.08) | 0.92 (0.26 to 3.93) | ‐0.11 (‐1.48 to 3.85) | |
| Golimumab | 0.69 (0.15 to 3.44) | 0.69 (0.15 to 3.32) | ‐0.44 (‐1.67 to 3.47) | |
| Infliximab | Etanercept | 1.53 (0.38 to 9.81) | 1.51 (0.39 to 8.49) | 1.27 (‐2.48 to 12.27) |
| Adalimumab | 0.54 (0.14 to 2.67) | 0.55 (0.14 to 2.59) | ‐1.09 (‐3.67 to 3.03) | |
| Golimumab | 0.40 (0.07 to 2.47) | 0.41 (0.08 to 2.40) | ‐1.41 (‐3.93 to 2.78) | |
| Adalimumab | Infliximab | 0.35 (0.05 to 2.28) | 0.36 (0.05 to 2.22) | ‐2.33 (‐13.00 to 2.47) |
| Golimumab | 0.25 (0.03 to 2.03) | 0.26 (0.03 to 1.99) | ‐2.68 (‐13.32 to 2.04) | |
| Golimumab | Adalimumab | 0.74 (0.09 to 6.02) | 0.75 (0.09 to 5.78) | ‐0.31 (‐4.40 to 3.85) |
| Random‐effect model | Residual deviance | 21.86 versus 30 data points | ||
| Deviance information criteria | 109.922 | |||
| Fixed‐effect model | Residual deviance | 21.57 versus 30 data points | ||
| Deviance information criteria | 109.171 |
CrL ‐ credible interval OR ‐ odds ratio RD ‐ risk difference RR ‐ risk ratio
6. Withdrawal due to adverse events: odds ratios, risk ratios and risk differences for all treatment comparisons (random‐effects model).
| Treatment | Reference | OR (95% CrI) | RR (95% CrI) | RD% (95% CrI) |
| Etanercept | Placebo | 3.73 (1.27 to 12.40) | 3.65 (1.27 to 11.79) | 1.94 (0.27 to 5.35) |
| Infliximab | 1.78 (0.43 to 8.77) | 1.77 (0.43 to 8.46) | 0.55 (‐0.55 to 4.45) | |
| Adalimumab | 1.70 (0.35 to 11.56) | 1.69 (0.35 to 10.84) | 0.49 (‐0.61 to 6.14) | |
| Golimumab | 1.98 (0.36 to 19.49) | 1.97 (0.36 to 17.51) | 0.70 (‐0.59 to 10.58) | |
| Infliximab | Etanercept | 0.49 (0.08 to 2.94) | 0.49 (0.09 to 2.85) | ‐1.28 (‐5.08 to 3.07) |
| Adalimumab | 0.46 (0.07 to 3.72) | 0.47 (0.07 to 3.54) | ‐1.33 (‐5.10 to 4.63) | |
| Golimumab | 0.52 (0.06 to 7.31) | 0.53 (0.06 to 6.59) | ‐1.16 (‐5.16 to 9.25) | |
| Adalimumab | Infliximab | 0.94 (0.10 to 10.34) | 0.94 (0.10 to 9.84) | ‐0.07 (‐4.00 to 5.61) |
| Golimumab | 1.11 (0.11 to 17.16) | 1.11 (0.11 to 15.51) | 0.12 (‐3.92 to 10.05) | |
| Golimumab | Adalimumab | 1.19 (0.10 to 16.81) | 1.19 (0.10 to 15.32) | 0.19 (‐5.13 to 9.86) |
| Random‐effects model | Residual deviance | 28.38 versus 32 data points | ||
| Deviance information criteria | 112.54 | |||
| Fixed‐effect model | Residual deviance | 28.92 versus 32 data points | ||
| Deviance information criteria | 112.474 |
CrL ‐ credible interval OR ‐ odds ratio RD ‐ risk difference RR ‐ risk ratio
As already noted, we had direct estimates of the benefit and harm of etanercept versus infliximab from the Giardina 2009 study. We found the indirect mixed treatment comparison estimate and direct estimates to be similar in terms of magnitude and direction of effect.
With respect to the homogeneity assumption, statistical heterogeneity (as assessed by I2 values) for the individual pairwise meta‐analyses (e.g. adalimumab versus placebo, etanercept versus placebo, golimumab versus placebo and infliximab versus placebo) used in the indirect comparison meta‐analyses were all very low (see Data and analyses 1 to 4).
For the assumption of trial similarity, Table 8 shows key potential effect modifiers for the individual anti‐TNFs versus placebo comparisons included in the network meta‐analysis. We compared the distribution of mean age, disease duration, baseline BASDAI and BASFI for trials of adalimumab versus placebo to etanercept versus placebo to golimumab versus placebo and to infliximab versus placebo. The results for the modifiers are similar and overlap across comparisons.
7. Demographic and clinical characteristics of studies included in network meta‐analysis.
| Study ID | # of patients | Duration, weeks | Age (yrs, SD) | % male | Disease duration (yrs, SD) | Baseline BASDAI (SD) | Baseline BASFI (SD) | |
| Tx | Control | |||||||
| Etanercept versus placebo | ||||||||
| Gorman 2002 | 20 | 20 | 16 | 38 (10) | 65 | 15 (10) | Not reported | 4.5 (2.1) |
| Brandt 2003 | 14 | 16 | 6 | 39.8 (9.1) | 71.4 | 14.9 (8.3) | 6.5 (1.2) | 6.2 (1.8) |
| Davis 2003 | 138 | 139 | 24 | 42.1 | 76 | 10.1 | 58.1 | 51.7 |
| Calin 2004 | 45 | 39 | 12 | 45.3 (9.5) | 80 | 15 (8.8) | 61 | 60.2 |
| van der Heijde 2006b | 150 | 51 | 12 | 39.8 (10.7) | 76 | 10 (9.1) | 59.4 (16.7) | 57.7 (20.1) |
| Huang 2008 | 74 | 78 | 6 | abstract; no details reported | ||||
| Barkham 2010 | 20 | 20 | 12 | 40.8 (9.7) | 75 | 11 (2–45)# | 6.05 (1.71) | 5.60 (1.98) |
| Dougados 2011 | 39 | 43 | 12 | 46 (11) | 95 | 19 | 64 (12) | 63 (20) |
| Infliximab versus placebo | ||||||||
| Braun 2002 | 34 | 35 | 12 | 40.6 (8) | 68 | 16.4 (8.3) | 6.5(1.2) | 5.1 (2.2) |
| van der Heijde 2005 | 201 | 78 | 24 | 40 (32,47)* | 78 | 10.1 | 6.6 (5.3, 7.6)* | 5.7 (4.5, 7.1)* |
| Inman 2010 | 39 | 37 | 12 | 42.9 (10.4) | 82 | 11.7 (10.6) | Not reported | Not reported |
| Adalimumab versus placebo | ||||||||
| van der Heijde 2006a | 208 | 107 | 12 | 41.7 (11.69) | 75.5 | 11.3 (10) | 6.3 (1.7) | 5.2 (2.2) |
| Lambert 2007 | 38 | 40 | 12 | 41.9 (11.1) | 76 | 14.5 (9) | 6.2 (1.7) | 5.3 (2.0) |
| Hu 2012 | 26 | 20 | 12 | 28.2 (6.9) | 92 | 7.4 (5.7) | 5.9 (1.4) | 3.7 (2.1) |
| Huang 2014 | 229 | 115 | 12 | 30.1 (8.1) | 80.8 | 8.1 (6.0)+ | 6.0 (1.4) | 4.3 (2.3) |
| Golimumab versus placebo | ||||||||
| Inman 2008 | 138 | 78 | 14 | 38 (30, 47)* | 73.9 | 11 (6, 18)*+ | 6.6 (5.6, 7.6)* | 5.0 (3.2, 6.7)* |
| Bao 2014 | 108 | 105 | 14 | 30.5 (10.3) | 83.3 | 6.1 (5.9)+ | 6.6 (1.3) | 5.0 (2.4) |
| Etanercept versus infliximab | ||||||||
| Giardina 2009 | 25 | 25 | 12** | 32.6 (6.8) | 80 | 15.7 (6.5) | 6.6 (1.1) | 6.5 (1.1) |
Note: data from the treatment group provided in this table; for Giardina 2009, the etanercept baseline data is provided (very similar to the infliximab group) Giardina 2009 is the only unblinded study. We did not include Braun 2011 or Marzo‐Ortega 2005 *median (IQR) # median (range) + years since symptoms occurred **study ran for 104 weeks, but week 12 data used in the network meta‐analysis BASDAI: Bath Ankylosing Spondylitis Disease Activity Index BASFI: Bath Ankylosing Spondylitis Functional Index
As noted in the paper by Hochberg 2003 et al, “another method of assessing the validity of this assumption is to compare the proportion of patients randomly allocated to receive placebo who develop the study outcome”. For ASAS40, the range of responses was 12% to 21%; for partial remission the range was 1% to 6%. Thus, it seems that it is likely that the patients in these trials are drawn from similar patient populations.
With respect to methodological risk of bias, most of the studies are similar in terms of sequence generation and allocation concealment, being either at low or unclear risk of bias due to lack of details in the published reports. However, all but the etanercept versus infliximab Giardina 2009 study were reported to be “double‐blind”, though details of the methods of blinding were not always clearly reported in the other studies. Giardina 2009 was an unblinded study, presumably because it was difficult to blind treatments with different routes of administration; i.e. subcutaneous versus infusion. This puts it at a higher risk of bias than the other studies. As well, the length of outcome assessment was quite different in the Giardina 2009 study as the full duration of the trial was 104 weeks. As all other studies reported results at 24 weeks or less, and we used the 12 week data from Giardina 2009.
However, there was no significant inconsistency evident in the consistency plot for ASAS40 (Figure 6). The other major outcomes had similar consistency plots.
6.
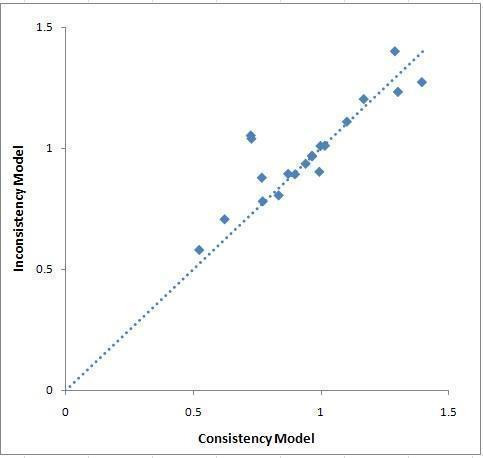
ASAS40: Plot of Posterior Mean Deviance of the Individual Data Points in the Inconsistency Model against Their Posterior Mean Deviance in the Consistency Model
Adverse effect warnings from regulatory websites
Table 9 summarizes the warnings on the use of TNF‐inhibitors from the websites of FDA MedWatch, European Medicines Evaluation Agency, Australian Adverse Drug Reactions Bulletin, and UK Medicines and Healthcare products Regulatory Agency (MHRA) Drug Safety Updates. Most warnings concerned tuberculosis and other serious infections as well as lymphoma and other malignancies, and recommended avoiding the concomitant use of biologics.
8. Summary of warnings on the TNF‐inhibitors from regulatory agencies.
| Summary of warning and conclusions | Date warning posted |
| MedWatch: The US Food and Drug Administration (FDA) Safety Information and Adverse Event Reporting Program | |
| Humira (adalimumab) injection: Postmarketing Experience: Hepato‐biliary disorders: … added … hepatitis | May 2014 |
| Humira (adalimumab)
5 WARNINGS AND PRECAUTIONS 5.1 Serious infections
5.2 Malignancies
5.3 Hypersensitivity reactions
5.10 Immunizations
6 ADVERSE REACTIONS 6.2 Postmarketing experience
7 DRUG INTERACTIONS 7.2 Biologic products
|
May 2013 |
Humira (adalimumab) 6.2 Postmarketing experience
|
May 2012 |
| Humira (adalimumab) BOXED WARNING Malignancy
WARNINGS and PRECAUTIONS
ADVERSE REACTIONS Postmarketing experience
|
Nov 2009 |
| Infliximab (Remicade) WARNINGS AND PRECAUTIONS Skin cancer
Concurrent administration with other biological therapeutics
ADVERSE REACTIONS Postmarketing experience
|
Mar 2013 |
| Enbrel (Etanercept) Injection: ADVERSE REACTIONS‐ Postmarketing Experience: Sarcoidosis | Dec 2012 |
| Warning of ongoing safety review of TNF blockers and malignancy in children, adolescents, and young adults (30 years of age or younger). FDA is requiring the manufacturers of TNF blockers to perform enhanced safety surveillance for these products | 03 Nov 2011 |
| FDA notified healthcare professionals that the Boxed Warning for the entire class of Tumor Necrosis Factor‐alpha (TNFα) blockers has been updated to include the risk of infection from two bacterial pathogens, Legionella and Listeria. In addition, the Boxed Warning and Warnings and Precautions sections of the labels for all of the TNFα blockers have been revised so that they contain consistent information about the risk for serious infections and the associated disease‐causing pathogens Patients treated with TNFα blockers are at increased risk for developing serious infections involving multiple organ systems and sites that may lead to hospitalizations or death due to bacterial, mycobacterial, fungal, viral, parasitic, and other opportunistic pathogens |
07 Sep 2011 |
| FDA continues to receive reports of a rare cancer of white blood cells (known as Hepatosplenic T‐Cell Lymphoma or HSTCL, primarily in adolescents and young adults being treated for Crohn’s disease and ulcerative colitis with medicines known as tumor necrosis factors (TNF) blockers, as well as with azathioprine, and/or mercaptopurine. TNF blockers include Remicade (infliximab), Enbrel (etanercept), Humira (adalimumab), Cimzia (certolizumab pegol) and Simponi (golimumab) | 04 Nov 2011 and 14 Apr 2011 |
| Label warnings added since 2000 for infliximab: hepatotoxicity; infections (pneumonia specifically added), lymphoma, tuberculosis, and other serious opportunistic infections including histoplasmosis, listeriosis, and pneumocystosis, malignancies Label warnings added since 2000 for etanercept: serious infections leading to hospitalizations or death, including bacterial sepsis and tuberculosis; recommendation to screen for latent tuberculosis infection before beginning Enbrel; lymphoma and other malignancies, including acute and chronic leukemia Label warnings added since 2000 for adalimumab: lymphoma and other malignancies; skin reactions: new or worsening psoriasis (all sub‐types including pustular and palmoplantar) ; serious infections with the combined use of Humira (adalimumab) and anakinra, hypersensitivity reactions, including anaphylaxis, and hematologic events, including pancytopenia and aplastic anemia |
|
| European Medicines Evaluation Agency (EMEA) | |
|
EPAR summary for the public: Adalimumab: Humira must not be used in patients with active tuberculosis, other severe infections, or moderate to severe heart failure (an inability of the heart to pump enough blood around the body) EPAR summary for the public: Etanercept: Enbrel must not be used in patients who have or are at risk of sepsis (when bacteria and toxins circulate in the blood and start to damage the organs), or in patients with infections. Patients developing a serious infection should stop Enbrel treatment Revised public statement on Enbrel (etanercept) ‐ Serious haematological reactions EPAR summary for the public: Golimumab: Simponi must not be used in people who are hypersensitive (allergic) to golimumab or any of the other ingredients. It must not be used in patients with tuberculosis, other severe infections, or moderate or severe heart failure (an inability of the heart to pump enough blood around the body). Due to an increased risk of infection, patients taking Simponi must be monitored closely for infections, including tuberculosis, during and for up to five months after treatment The most serious side effects include serious infections, such as sepsis (blood infection), pneumonia (lung infection), tuberculosis and infections due to fungi or yeasts, demyelinating disorders (disorders suggesting damage to the protective sheath around nerves, such as changes to vision and weak arms or legs), lymphoma (a type of cancer of the white blood cells), re‐activation of hepatitis B (a liver disease), congestive heart failure (a heart disease), lupus‐like syndrome and blood reactions EPAR summary for the public: Infliximab: Remicade must not be used in patients who have experienced hypersensitivity (allergy) to infliximab in the past, or who are hypersensitive (allergic) to mouse proteins or any of the other ingredients of Remicade. Remicade must not be used in patients with tuberculosis, other severe infections, or moderate or severe heart failure (an inability of the heart to pump enough blood around the body) |
14 Nov 2014 03 Sep 2014 03 Nov 2000 23 Oct 2013 1 Mar 2012 |
| Australian Adverse Drug Reactions | |
| Bulletin Golimumab: Infections Serious and sometimes fatal infections due to bacterial (including sepsis and pneumonia), mycobacterial, invasive fungal, viral, protozoal, or other opportunistic pathogens have been reported in patients receiving TNF‐blockers including SIMPONI. Among opportunistic infections, tuberculosis, histoplasmosis, aspergillosis, candidiasis, coccidioidomycosis, listeriosis, and pneumocystosis were the most commonly reported with TNF‐blockers. Patients have frequently presented with disseminated rather than localized disease, and were often taking concomitant immunosuppressants such as methotrexate (MTX) or corticosteroids. The concomitant use of a TNF‐blocker and abatacept or anakinra was associated with a higher risk of serious infections; therefore, the concomitant use of SIMPONI and these biologic products is not recommended Tuberculosis Cases of reactivation of tuberculosis or new tuberculosis infections have been observed in patients receiving TNF‐blockers, including Simponi Hepatitis B virus reactivation The use of TNF‐blockers including SIMPONI has been associated with reactivation of hepatitis B virus (HBV) in patients who are chronic hepatitis B carriers (i.e. surface antigen positive). Patients should be tested for Hepatitis B virus (HBV) infection before initiating treatment with immunosuppressants, including Simponi Malignancies The potential role of TNF‐blocking therapy in the development of malignancies is not known. Caution should be exercised when considering TNF‐blocking therapy for patients with a history of malignancy or when considering continuing treatment in patients who develop malignancy Paediatric Malignancy Post‐marketing cases of malignancies, some fatal, have been reported among children, adolescents and young adults (up to 22 years of age) who received TNF‐blocking agents (initiation of therapy ≤ 18 years of age) to treat Juvenile Idiopathic Arthritis (JIA), Crohn’s disease or other conditions Lymphoma In the controlled portions of clinical trials of all the TNF‐blocking agents including Simponi, more cases of lymphoma have been observed among patients receiving anti‐TNF treatment compared with control patients Leukaemia Cases of acute and chronic leukaemia have been reported with postmarketing TNF‐blocker use in rheumatoid arthritis and other indications Skin cancers Melanoma has been reported in patients treated with TNF blocking agents, including Simponi. Merkel cell carcinoma has been reported in patients treated with other TNF‐blocking agents Congestive Heart Failure Cases of worsening congestive heart failure (CHF) and new onset CHF have been reported with TNF‐blockers including SIMPONI. Cases of CHF in patients with known cardiovascular risk factors have been observed with Simponi Neurological events Use of TNF‐blocking agents has been associated with cases of new onset or exacerbation of clinical symptoms and/or radiographic evidence of central nervous system demyelinating disorders, including multiple sclerosis (MS) and peripheral demyelinating disorders, including Guillain‐Barré syndrome Haematological cytopaenias There have been post‐marketing reports of pancytopaenia, leukopaenia, neutropaenia, aplastic anaemia, and thrombocytopaenia in patients receiving TNF‐blockers. Cytopaenias including pancytopaenia, have been infrequently reported with Simponi in clinical trials |
16 Aug 2013 |
| Drug‐induced lupus erythematosus (June 2009): An emerging association with TNF inhibitors TNA‐alpha inhibitors (Dec. 2006): While extremely effective, TNF inhibitors are associated with several serious reactions. These include: · Hypersensitivity reactions ‐ immediately post‐injection or delayed · Serious and life‐threatening infection and sepsis · Recrudescence of tuberculosis and other granulomatous diseases · Reactivation of hepatitis B · Malignancy, including lymphoma · Haematological reactions such as pancytopenia and aplastic anaemia · Autoimmunity ‐ e.g. drug‐induced lupus · CNS reactions, including demyelinating disorders and seizures · New‐onset heart failure or worsening of advanced heart failure |
June 2009 Dec 2006 |
| UK MHRA Medicines and Healthcare products Regulatory Agency: Drug Safety Updates (formerly Current Problems in Pharmacovigilance) | |
| ‐ letter to healthcare providers re: reports of hepatosplenic T‐cell lymphoma in patients treated with Humira® (adalimumab) ‐ congestive cardiac failure, cardiomyopathy, the frequency of blood dyscrasias, demyelination, infections, adult respiratory distress syndrome and TB should be kept under close monitoring by the MA (marketing authorization) holder Highlight of the European Agency for the Evaluation of Medicinal Products (EMEA) public statement regarding tuberculosis (TB) or other opportunistic infections following infliximab (Remicade) therapy. “Prescribers and patients who are receiving Remicade need to be aware of the risk of developing infections upon starting therapy and to be especially vigilant for signs of infection throughout treatment. If active TB is suspected (persistent cough, wasting/weight loss, low grade fever), Remicade treatment should be withheld until the infection has been treated” |
July 2008 20 Dec 2000 |
DMARDs: Disease‐modifying anti‐rheumatic drugs JIA: Juvenile idiopathic arthritis TB: tuberculosis
Sensitivity analyses
As Figure 2 shows, there were few concerns about studies with high risk of bias, with the exception of Giardina 2009 which did not blind patients or investigators. The results of the network meta‐analyses showed good consistency so we did not perform a sensitivity analysis around study quality.
Mantel‐Haenzsal versus Peto OR for rare events
Although using the Mantel‐Haenszel method changed the width of some confidence intervals, none of the point estimates changed significantly, nor did any estimates change in their statistical significance. Therefore, we feel our estimates of outcomes with rare events using the Peto OR are robust.
Subgroup analyses
We explored the characteristics of the studies as described below and found them sufficiently similar and did not conduct subgroup analyses.
Dose
We pooled the 5 mg/kg dose and 3 mg/kg dose of infliximab for major outcomes in a standard meta‐analysis. There was very little heterogeneity when these doses were pooled. We investigated the effect estimates in the low‐dose and high‐dose groups separately and found them to be quite similar.
Trial duration
All RCT portions of the studies were six months or less duration and we had specified a priori that trials shorter than or equal to six months would be used to assess short‐term benefits and harms.
Patient characteristics
Overall, the studies were quite homogeneous (Table 8). Barkham 2010 and Huang 2008 were reported in abstracts, so details of patient characteristics are not available. All studies included patients meeting the modified New York classification criteria. Patients were similar across trials in terms of age (range of mean age in treatment group 30 to 45 years). Calin 2004 included the oldest patients but the effect estimates were similar to the other studies for BASFI. All studies included a majority of men (% male in treatment group range: 65% to 83%, with the exception of Dougados 2011 which focused on people with advanced ankylosing spondylitis. We included and excluded Dougados 2011 from a standard meta‐analysis of the major outcomes for trials of etanercept and found that the study introduced very little heterogeneity and the effect estimates were very similar: BASFI went from I2 = 24% to 0% and both ASAS40 and withdrawals due to adverse events stayed at I2 = 0% when Dougados 2011 was removed. Therefore we decided it was appropriate to pool it with the other studies.
BASDAI and BASFI baseline scores were similar across studies (BASDAI range: 5.8 to 6.6 and BASFI range: 4.5 to 6.5). Disease duration was similar, with almost all ranging from 10 to 15 years in the treatment groups across studies. The two exceptions were Dougados 2011 at 19 years but as explained above, we felt it was appropriate to use it in the analyses with the other studies. Bao 2014 had the shortest disease duration at 6.1 years, but its results were very similar to the other golimumab study for the major outcomes (ASAS40 and withdrawals due to adverse events both had I2 = 0%).
Only Brandt 2003 did not allow concomitant disease‐modifying anti‐rheumatic drug therapy but in terms of the BASDAI, the result from this study was very similar to the other studies of etanercept.
Almost all included studies excluded patients with complete ankylosis (fusion) (exclusion criteria not reported in Lambert 2007 and van der Heijde 2006a restricted a priori to < 10% of recruited patients could have complete ankylosis).
Assessment of publication bias
We had planned to assess publication bias by visual inspection of funnel plots. However, it was very difficult to assess plots with few studies in them. According to the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), when assessing publication bias using statistical tests for funnel plot asymmetry, at least 10 studies should be included in the meta‐analysis, otherwise the power of the test is too low. There is no guidance regarding the minimum number of studies needed for visual inspection. We decided to look at plots with more than 10 studies and there were two analyses that met this criteria. Both appear to have a funnel shape (and it is more apparent when the treatment effect is a risk ratio rather than Peto OR) with the studies distributed fairly symmetrically around the mean effect size. There is not strong evidence of small‐study bias when assessing these plots.
Discussion
Summary of main results
Twenty‐one RCTs with a total of 3308 participants met the inclusion criteria for this review. The major beneficial outcomes were based on those recommended by the Assessment of SpondyloArthritis international Society (ASAS), and the primary analysis results were based on mixed treatment comparison refined placebo estimates.
High quality evidence showed that in the short‐term (less than 24 weeks), each biologic ‐ adalimumab, etanercept, golimumab, and infliximab ‐ when compared to placebo, improved ankylosing spondylitis symptoms of pain, function, stiffness and global well‐being. Moderate quality evidence showed a greater number of participants met the partial responder criteria. There was low to moderate quality evidence for less erosions and edema in spine and sacroiliac joints as measured by magnetic resonance imaging (MRI) in trials of adalimumab, golimumab, and infliximab, though the absolute improvement was small and we are not sure of the clinical relevance.
Radiological progression was measured in only one study of etanercept versus placebo and changes were similar in both groups. Given that all trials were 24 weeks or less, it is not surprising that most did not measure radiographic progression.
We pooled adverse event data from the four anti‐TNF agents to investigate a class effect of the TNF‐inhibitors. When all the anti‐TNF agents were combined against placebo, there was moderate quality evidence from 16 studies of an increased risk of withdrawals due to adverse events in the anti‐TNF group, but results were inconclusive for evidence of a difference between groups in terms of risk of serious adverse events in the anti‐TNF group versus placebo. We downgraded the evidence to moderate, given the low number of events.
Our search of major regulatory agency websites for warnings of adverse effects highlighted concerns of serious infections, including tuberculosis, as well as lymphoma, and malignancies associated with anti‐TNF use.
Indirect comparisons can be useful when there is limited direct evidence for a clinically‐important question. Appropriate methods should be employed to undertake indirect comparisons; for example, “naive” pooling methods should be avoided, and in this analysis, we have undertaken a suitable methodology, mixed treatment comparisons, for our indirect comparisons. The direct estimates from the one head‐to‐head study of etanercept versus infliximab (Giardina 2009) were comparable in terms of the magnitude and direction of effect with the refined estimates obtained from the MTC analysis, thereby increasing our confidence in the indirect comparison estimates. For the mixed treatment comparison‐derived head‐to‐head estimates, there were wide confidence intervals and no consistency as to which biologic was favored in terms of the major outcomes. Therefore, we do not have evidence that one anti‐TNF agent appears to confer more benefit or harm than another.
One RCT (Braun 2011) assessed etanercept (50 mg once a week) compared to sulphasalazine. Etanercept was found to have a statistically significantly better response in terms of ASAS20, ASAS40, and ASAS5/6 at 16 weeks. As well, Bath Ankylosing Spondylitis Disease Activity Index (BASDAI), Bath Ankylosing Spondylitis Functional Index (BASFI), Bath Ankylosing Spondylitis Metrology Index (BASMI), nocturnal back pain, and Modified Schobers response were all greater in the etanercept versus the sulphasalazine groups.
Infliximab in combination with methotrexate versus placebo plus methotrexate was assessed in Marzo‐Ortega 2005. At 30 weeks, there was no statistically significant difference between the two groups in terms of ASAS20, > 50% BASDAI response, or change in BASDAI, though there had been at 10 weeks. The assessment at 30 weeks was 8 weeks after the last dose of infliximab had been given and the authors concluded that the addition of methotrexate did not confer a longer benefit of infliximab. Caution is needed though, as this is based on only one small study.
Overall completeness and applicability of evidence
In total, twenty‐one trials addressed the use of anti‐TNF agents: adalimumab (three trials), etanercept (eight trials versus placebo; one versus sulphasalazine, one a high versus low dose), golimumab (2 trials), infliximab (4 trials, one assessed the benefit of infliximab in combination with methotrexate), and one trial of etanercept versus infliximab, for the treatment of ankylosing spondylitis. The RCTs were between six and 24 weeks duration, so all data about benefit and harm is based on short‐term studies. We assessed appropriate outcomes based on Assessment of SpondyloArthritis international Society recommendations to establish short‐term benefit of the anti‐TNF agents against placebo.
Participants in the included studies had high disease activity (entry criteria was a BASDAI ≥ 4). The high levels of disease activity seen in the patients included in these trials may not be typical of patients seen in daily clinical practice. In addition, patients selected for RCTs generally have few major comorbidities. Almost all studies excluded patients with complete ankylosis of the spine, and many excluded patients with conditions related to the concerns of potential harms with biologics, i.e. recent serious infections, history of infectious diseases or malignancies in last five years, and signs of severe renal, cardiac, hepatic, demyelinating, or other diseases. This may impact the generalizability of these results to clinical practice.
As disease duration of the participants enrolled in these studies was mainly 10 years and longer, the applicability of this evidence to those with shorter disease duration is unclear. However, in Rudwaleit 2004, a shorter disease duration (≤ 10 years) and a lower BASFI (< 4.5) were both found to be strong predictors of a major clinical response (as assessed by > 50% BASDAI response). Therefore, it is of interest to conduct trials of anti‐TNF agents in populations with early disease to determine if there is indeed a better response and any effect on the progression of ankylosis spondylitis in this population and to determine when it is appropriate to start anti‐TNF therapy (Haibel 2010).
Most trials used a standard dose for each of the biologics. One trial (van der Heijde 2006b) assessed the benefit of giving 50 mg of etanercept once a week versus 25 mg twice a week and concluded that the benefit and safety outcomes were similar between the two groups. Another trial (Inman 2010) evaluated the effect of a lower dose of infliximab (3 mg/kg) compared to the standard dose of 5 mg/kg. Although a previous open‐label study of lower dose infliximab showed sustained benefit in the majority of patients, in the extension phase of this RCT (the double‐blind phase was to 12 weeks), by week 38, most participants (68%) required an increase to 5 mg/kg to maintain benefit.
A limitation of this review is that we did not include non‐RCT data to assess changes in the radiological progression of patients exposed to anti‐TNF therapy. It is unlikely that changes in radiological progression will be seen in RCTs of six months or less duration. In addition, the lack of including non‐RCT data limits the assessment of rare or delayed harms which are unlikely to be found in short‐term RCTs.
An editorial on the study of abatacept in rheumatoid arthritis (Boers 2006) highlights the desire of clinicians for active comparator trials, once the benefit of a treatment has been established against placebo. As described above in the summary of results section, only one head‐to‐head study of anti‐TNF agents (Giardina 2009) was found in our systematic search. We therefore performed indirect comparisons using mixed treatment comparison methodology to explore the evidence base and test the robustness of the results. However, additional large, well conducted, head‐to‐head studies would be helpful to provide clinicians with a stronger evidence base regarding differences between the various biologics.
Certolizumab pegol, another TNF‐inhibitor, has been approved for use in ankylosing spondylitis and it will be included in our update of this review. As well, we may need to consider whether a new disease activity measure, the Ankylosing Spondylitis Disease Activity Score (ASDAS) should be included as a major outcome. A further consideration for future updates is that new studies also apply the new ASAS axial spondyloarthropathy (axSpA) criteria and we will need discussion on including more heterogeneous populations of radiographic axSpA (ankylosing spondylitis) and non‐radiographic axSpA.
Quality of the evidence
Adequate allocation concealment can avoid selection bias in controlled trials and there is evidence that inadequate allocation concealment leads to an overestimation of the treatment effect (Schultz 1995). We obtained additional information from the authors for some studies to clarify the method of allocation. Still, there were eleven studies which did not provide enough information on this domain and we had to mark it as unclear.
Blinding of participants was not clearly reported in seven studies. Giardina 2009 was clearly not blinded; this two‐year study compared etanercept (given subcutaneously) with infliximab (given as an infusion), so it is evident why the patients were not blinded. Given that the primary outcome measured in most trials was the BASDAI or ASAS20, both self reported measures, it is necessary for patients to be blinded to ensure detection bias has not been introduced in these studies.
Risk of bias due to attrition bias was unclear in five studies, and completion rates were greater than 80% in all but one study, Marzo‐Ortega 2005, which had a large imbalance in withdrawals in the treatment and placebo arms, mostly due to lack of benefit in the placebo group (93% versus 64%). All the trials reported the numbers of patients who dropped out in the treatment and placebo groups. The drop‐out rates were generally higher in the placebo group than the treatment group in all trials and there was a much higher rate of withdrawal due to lack of benefit in the placebo groups. In most trials the missing data were imputed using last observation carried forward analysis for continuous data and 'non‐responders' for dichotomous outcomes like ASAS20. Most trials reported a proper intention‐to‐treat analysis. The other trials (Braun 2002; Inman 2010; van der Heijde 2006b) reported a 'modified' intention‐to‐treat analysis as one defined by those subjects who received at least one infusion of study medication. Although fewer than 3% of participants were affected, it is of interest to know why patients who were randomized did not receive the study drug.
The Assessment of SpondyloArthritis international Society (ASAS) and OMERACT groups have had great success in standardizing outcomes that should be measured in trials of interventions for ankylosing spondylitis. The ASAS response criteria were developed for use in clinical trials and the ASAS40 and ASAS5/6 response criteria have both shown good discrimination in anti‐TNF studies (Brandt 2004). The trials included in this systematic review usually reported outcome measures as recommended by ASAS and OMERACT for trials on ankylosing spondylitis patients. There is little risk of bias due to selective reporting in these trials in terms of beneficial outcomes; however, adverse events were less clearly reported.
Heterogeneity was low in pooled analyses of the major outcomes. The low event rates in the outcomes of partial remission, withdrawals due to side effects, and serious adverse events led to downgrading the quality of evidence for the resulting imprecise estimates.
When combining studies it is important that the outcome measures are comparable. Of note, different definitions of serious adverse events were used in the assessment of these events across the trials. Most trials did not provide a specific definition, two stated they used the National Cancer Institute Common Toxicity Criteria, and another MedDRA version 9. We assumed for the purpose of this review that the definitions were similar enough to warrant combining.
Only two outcomes had enough studies in which to visually inspect a funnel plot for publication bias. These plots did not indicate a potential lack of publication of smaller, 'negative' studies but overall we do not have much evidence whether publication bias is an issue in this systematic review.
With regards to detecting adverse events in RCTs, it was noted in Yazici 2008 that an inadequate sample size (Type II error) is a possible reason that a significant difference in the number of adverse events between treatment and placebo groups is often not observed. All the included studies termed themselves 'efficacy and safety' studies. But in none of the trials was there a discussion of necessary sample sizes to detect adverse events.
We concluded that there is moderate (downgraded for concerns about imprecision due to low event rates) to high level evidence for the short‐term outcomes of ASAS40 response, achievement of partial remission, physical function, and disease activity. Evidence for improvement in inflammation as measured by MRI results was low to moderate due to small population and concerns about missing data in one study. Harms outcomes were downgraded to moderate due to low event rates and resulting imprecise estimates.
Potential biases in the review process
We undertook a systematic, thorough search of the electronic literature and searched key conference proceedings to identify all studies meeting the inclusion criteria for this review. However, we did not approach pharmaceutical companies for additional data and it is possible that additional data from this source could contribute to this review. Study selection, data extraction, and risk of bias assessments were done in duplicate and independently and we reached consensus by discussing any discrepancies.
Published trial reports did not provide enough details to adequately assess risk of bias and some variance measures necessary for meta‐analysis were missing from the report. We contacted some authors for further information and while some of the requested data was provided, it is a limitation of this review that not all the data were available. For some trials we had to undertake transformations and assumptions in order to enter continuous data into our mixed treatment comparison spreadsheets and this may reduce the accuracy of our estimates.
Some adverse event outcomes consisted of sparse data, with few events in the groups. In cases where there were no events in either study arm (zero total event studies), the study did not contribute to the meta‐analysis. The rationale for this method is that no information on the magnitude of the treatment effect can be obtained from these studies. An investigation by Sweeting 2004 confirmed that "zero total event studies do not contribute to a fixed meta‐analysis" and so we felt it appropriate not to include these in our meta‐analysis. The Peto OR was shown to be one of the least biased estimators of a treatment effect when using sparse data. However, in the case of unbalanced study arms with four times as many participants in one arm as another, there is concern that the estimate will be biased (Bradburn 2007). Given that none of our included studies were unbalanced by more than 4:1, we decided the Peto OR was a suitable method. When analyzing rare data in a meta‐analysis, it is recommended that various methods are used to determine the treatment effect estimate and sensitivity analyses are undertaken (Sweeting 2004). For those analyses in which studies had zero events in one arm, or an event rate < 10%, we used the Peto OR. We also performed the Mantel‐Haenszel method with the standard continuity correction of 0.5 to check the robustness of our results and found that results did not change significantly. We did not perform other sensitivity analyses with other continuity corrections, but given that there was consistency of results with the standard continuity correction, we are confident that our estimates are robust.
For the indirect comparison results to be considered robust, it must be investigated whether the trials included in each standard meta‐analysis comparison (e.g. A‐B and C‐B) are homogeneous; whether the effect of the linking treatment is similar across comparisons; and whether there is consistency of evidence. To judge the likelihood of these assumptions being true, the comparability of the linking treatment, the patient population, the methodological risk of bias, the study design, and the date of publication must be assessed (Song 2009; Wells 2009). We did assess the studies included in terms of the criteria noted above. As noted in Table 8 and the Results section, the patient populations were fairly similar in terms of length of disease duration, severity of disease, inclusion/exclusion trial criteria, and trial duration. The one head‐to head study of etanercept versus infliximab (Giardina 2009) did not blind the participants (given the difference in route of administration of the two biologics) and it was measured at a longer follow‐up time than the other included studies (two years versus six months, though interim results were available). However, since the treatment effect observed for etanercept and infliximab was similar to that seen in other placebo‐controlled studies, and including this study allowed us to have a closed loop in our network meta‐analysis for evaluating the consistency of the direct and indirect evidence, we decided it was appropriate to include this in our indirect comparison analysis.
However, it must be cautioned that while we assessed the necessary assumptions for undertaking indirect comparisons, indirect comparisons may not provide the same strength of validity of results that a well conducted, head‐to‐head, RCT may.
A protocol was published for this review (Zochling 2005) and outcomes and analyses were specified a priori. However, since this time, there have been significant changes to the methodology recommended in the Cocharane Handbook for Systematic Reviews of Interventions (Higgins 2011), specifically around assessing the risk of bias, grading the quality of evidence, and providing 'Summary of findings' tables. Our protocol listed outcomes recommended by the Assessment of SpondyloArthritis international Society (ASAS) but for the 'Summary of findings' table, we had to choose a maximum of seven outcomes, representing both benefit and harm. In discussion with ASAS members we decided on our seven major outcomes and we do not think that any selection bias was introduced when making this choice.
Agreements and disagreements with other studies or reviews
The National Institute for Health and Clinical Excellence (NICE) undertook a technology appraisal report to provide guidance on the use of adalimumab, etanercept and infliximab for ankylosing spondylitis for the UK National Health Service (NICE 2008). The report was issued in May 2008 and the results are very similar to this review. This Cochrane review includes some newer information on the benefit of low‐dose infliximab and use of 50 mg once per week of etanercept. The NICE report also conducted indirect comparisons of the anti‐TNF inhibitors and as this review did, found no statistically significant difference between the three biologics in terms of ASAS response rates. The NICE 2008 review assessed cost‐effectiveness of three biologics in detail and recommended that the high cost of infliximab precluded its recommendation for use in people with ankylosing spondylitis. Both adalimumab and etanercept were recommended for use subject to the conditions outlined in the report. Golimumab was recommended for use in the NICE 2011 report.
A recent meta‐analysis (Callhoff 2014) of RCTs assessed the benefit of anti‐TNF agents adalimumab, certolizumab, etanercept, golimumab or infliximab against placebo for both ankylosing spondylitis and non‐radiographic axial spondyloarthritis. Like this review, they found that anti‐TNF agents showed improved benefit compared to placebo. Effect sizes were greater in the ankylosing spondylitis population compared to the non‐radiographic axial spondyloarthritis trials, though when they adjusted the estimates for the year of publication as a proxy for disease severity, there was no longer a difference between the two populations.
While the data for this systematic review is based on RCTs, a study based on the British Society for Rheumatology Biologic Register assessed the use of anti‐TNF therapy for ankylosing spondylitis in the UK in routine care. They found the mean improvement in BASDAI was 3.6 (0 to 10 scale), 52% of patients achieved a BASDAI > 50 and concluded that "routine clinical use improves disease activity and functional impairment in patients with ankylosing spondylitis" (Lord 2010).
Authors' conclusions
Implications for practice.
There is moderate to high level quality of evidence for a clinically important benefit of adalimumab, etanercept, golimumab, and infliximab compared to placebo in improving disease activity and function, and achieving partial remission in ankylosing spondylitis in the short‐term. Reduction in spinal inflammation, as measured by magnetic resonance imaging (MRI), was based on low to moderate quality evidence and the clinical relevance of the small absolute changes is unclear. There is little evidence on radiographic progression from these short‐term RCTs. There is moderate quality evidence for a small increased risk of withdrawals due to adverse events for the anti‐tumor necrosis factor (TNF) agents as a group against placebo, but results were inconclusive for evidence of a difference between groups in terms of risk of serious adverse events. We downgraded evidence given the low number of events in the harm outcomes. For the indirect head‐to‐head estimates, there were wide confidence intervals and no consistency as to which biologic was favored in terms of the major outcomes. Therefore, we do not find evidence that one treatment appears to confer more benefit than another. Given that the included studies were all six months or less in duration, we did not assess evidence on the long‐term benefit or effectiveness of anti‐TNF therapy for ankylosing spondylitis.
Implications for research.
Future trials of anti‐TNF agents should focus on populations with early disease to determine if there is indeed a better response, the effect on the progression of ankylosis in this population, and to determine when it is appropriate to start anti‐TNF therapy. Given the paucity of head‐to‐head study data, we performed a network meta‐analysis to estimate effects of one anti‐TNF agent versus another. However, we recommend that large, well‐conducted randomized trials of anti‐TNF agents versus each other are needed to clearly address issues of effectiveness among these agents. Studies based on biologic registry and other observational data will be useful to assess rare and delayed adverse effects.
History
Protocol first published: Issue 3, 2005 Review first published: Issue 4, 2015
| Date | Event | Description |
|---|---|---|
| 11 November 2008 | Amended | Converted to new review format |
| 20 May 2005 | New citation required and conclusions have changed | Substantive amendment |
Acknowledgements
Many thanks to Tamara Rader, Louise Falzon, Jessie McGowan and Nancy Santesso for their assistance with the literature searching. Thanks to Bharbhoor Dhaliwal for assistance with screening the searches. Colin McCann assisted with screening search results and data extraction. Thanks to Lily Chen for her statistical expertise in running the mixed treatment comparison analyses. Thank you to the authors of primary trials who provided us with additional information.
Appendices
Appendix 1. MEDLINE search
Database: Ovid MEDLINE(R) In‐Process & Other Non‐Indexed Citations and Ovid MEDLINE(R)
1 exp spondylitis, ankylosing/ 2 exp spondylarthropathies/ 3 (ankylosing or spondyl$).tw. 4 (bekhterev or bechterew).tw. 5 4 or 1 or 3 or 2 6 exp Receptors, Tumor Necrosis Factor/ 7 exp tumor necrosis factor/ 8 exp Antibodies, Monoclonal/ 9 anti‐tumo?r necrosis factor$.sh,rn,tw. 10 antitumo?r necrosis factor$.sh,rn,tw. 11 anti‐tnf.sh,rn,tw. 12 antitnf.sh,rn,tw. 13 etanercept.sh,rn,tw. 14 enbrel.sh,rn,tw. 15 infliximab.sh,rn,tw. 16 remicade.sh,rn,tw. 17 adalimumab.sh,rn,tw. 18 humira.sh,rn,tw. 19 or/6‐18 20 5 and 19 21 randomized controlled trial.pt. 22 controlled clinical trial.pt. 23 randomized.ab. 24 placebo.ab. 25 drug therapy.fs. 26 randomly.ab. 27 trial.ab. 28 groups.ab. 29 27 or 25 or 28 or 21 or 26 or 22 or 24 or 23 30 (animals not (humans and animals)).sh. 31 29 not 30 32 20 and 31 33 from 32 keep 1‐10
Appendix 2. Additional search strategies (January 2009 search)
Database: EMBASE <1980 to 2009 Week 04> Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 exp Spondylitis/ (10156) 2 (ankylosing or spondyl$).mp. (21570) 3 (bekhterev or bechterew).mp. (201) 4 or/1‐3 (21620) 5 exp Tumor Necrosis Factor Receptor/ (5586) 6 exp Tumor Necrosis Factor/ (19676) 7 exp Monoclonal Antibody/ (170176) 8 anti‐tumo?r necrosis factor$.mp. (1439) 9 anti‐tnf.mp. (3283) 10 etanercept.mp. (7693) 11 enbrel.mp. (1723) 12 infliximab.mp. (11164) 13 remicade.mp. (2218) 14 exp adalimumab/ (3580) 15 humira.sh,rn,tw. (966) 16 trudexa.sh,rn,tw. (10) 17 Monoclonal antibody D2e7.rn,tw. (4) 18 or/5‐17 (195408) 19 4 and 18 (1592) 20 random$.ti,ab. (388596) 21 factorial$.ti,ab. (8106) 22 (crossover$ or cross over$ or cross‐over$).ti,ab. (39123) 23 placebo$.ti,ab. (108834) 24 (doubl$ adj blind$).ti,ab. (84015) 25 (singl$ adj blind$).ti,ab. (7379) 26 assign$.ti,ab. (107363) 27 allocat$.ti,ab. (33957) 28 volunteer$.ti,ab. (98245) 29 crossover procedure.sh. (20893) 30 double blind procedure.sh. (71118) 31 randomized controlled trial.sh. (164870) 32 single blind procedure.sh. (7912) 33 or/20‐32 (652278) 34 exp animal/ or nonhuman/ or exp animal experiment/ (3407483) 35 exp Spondylitis/ (10156) 36 (ankylosing or spondyl$).mp. (21570) 37 (bekhterev or bechterew).mp. (201) 38 or/35‐37 (21620) 39 exp Tumor Necrosis Factor Receptor/ (5586) 40 exp Tumor Necrosis Factor/ (19676) 41 exp Monoclonal Antibody/ (170176) 42 anti‐tumo?r necrosis factor$.mp. (1439) 43 anti‐tnf.mp. (3283) 44 etanercept.mp. (7693) 45 enbrel.mp. (1723) 46 infliximab.mp. (11164) 47 remicade.mp. (2218) 48 exp adalimumab/ (3580) 49 humira.sh,rn,tw. (966) 50 trudexa.sh,rn,tw. (10) 51 Monoclonal antibody D2e7.rn,tw. (4) 52 or/39‐51 (195408) 53 38 and 52 (1592) 54 random$.ti,ab. (388596) 55 factorial$.ti,ab. (8106) 56 (crossover$ or cross over$ or cross‐over$).ti,ab. (39123) 57 placebo$.ti,ab. (108834) 58 (doubl$ adj blind$).ti,ab. (84015) 59 (singl$ adj blind$).ti,ab. (7379) 60 assign$.ti,ab. (107363) 61 allocat$.ti,ab. (33957) 62 volunteer$.ti,ab. (98245) 63 crossover procedure.sh. (20893) 64 double blind procedure.sh. (71118) 65 randomized controlled trial.sh. (164870) 66 single blind procedure.sh. (7912) 67 or/54‐66 (652278) 68 53 and 67 (230) 69 from 68 keep 1 (1)
Database: EBM Reviews ‐ Cochrane Central Register of Controlled Trials <4th Quarter 2008> Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 exp Spondylarthropathies/ (363) 2 (ankylosing or spondyl$).ti,ab. (668) 3 (bekhterev or bechterew).ti,ab. (4) 4 or/1‐3 (790) 5 exp Receptors, Tumor Necrosis Factor/ (417) 6 exp Tumor Necrosis Factors/ (1445) 7 exp Antibodies, Monoclonal/ (1920) 8 anti‐tumor necrosis factor$.ti,ab. (88) 9 antitumor necrosis factor$.ti,ab. (2) 10 anti‐tnf.ti,ab. (121) 11 antitnf.ti,ab. (1) 12 etanercept.ti,ab. (224) 13 enbrel.ti,ab. (25) 14 infliximab.ti,ab. (256) 15 remicade.ti,ab. (10) 16 adalimumab.ti,ab. (73) 17 humira.ti,ab. (4) 18 6 or 11 or 7 or 9 or 17 or 12 or 15 or 14 or 8 or 16 or 10 or 13 or 5 (3597) 19 4 and 18 (89) 20 from 19 keep 1 (1)
Database: CLEED, CDSR, ACP Journal Club, DARE, CLHTA Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 exp Spondylarthropathies/ (40) 2 (ankylosing or spondyl$).ti,ab. (61) 3 (bekhterev or bechterew).ti,ab. (0) 4 or/1‐3 (74) 5 exp Receptors, Tumor Necrosis Factor/ (43) 6 exp Tumor Necrosis Factors/ (57) 7 exp Antibodies, Monoclonal/ (237) 8 anti‐tumor necrosis factor$.ti,ab. (4) 9 antitumor necrosis factor$.ti,ab. (1) 10 anti‐tnf.ti,ab. (9) 11 antitnf.ti,ab. (0) 12 etanercept.ti,ab. (70) 13 enbrel.ti,ab. (2) 14 infliximab.ti,ab. (91) 15 remicade.ti,ab. (5) 16 adalimumab.ti,ab. (30) 17 humira.ti,ab. (4) 18 6 or 11 or 7 or 9 or 17 or 12 or 15 or 14 or 8 or 16 or 10 or 13 or 5 (363) 19 4 and 18 (26) 20 from 19 keep 1‐26 (26) 21 from 20 keep 1 (1)
Web of Knowledge 1900‐2008
#5 #4 AND #3
#4 TS=(trial* or random* or placebo* or control* or double or treble or triple or blind* or mask* or allocat* or prospective* or volunteer*or comparative or evaluation or follow‐up or followup)
#3 #2 AND #1
#2 TS=(tumor necrosis factor* or tumour necrosis factor* or monoclonal antibod* or anti‐tnf or antitnf or etanercept or enbrel or infliximab or remicade)
#1 TS=(ankylos* or spondyl*)
CINAHL
| S12 | S4 and S11 |
| S11 | S5 or S6 or S7 or S8 or S9 or S10 |
| S10 | TX etanercept or TX enbrel or TX infliximab or TX remicade or TX adalimumab or TX humira |
| S9 | (MH "etanercept+") |
| S8 | TX "anti‐tnf" or TX antitnf |
| S7 | TX "anti‐tumor necrosis factor*" or TX "anti‐tumour necrosis factor*" or TX "antitumor necrosis factor*" or TX "antitumour necrosis factor*" |
| S6 | (MH "Antibodies, Monoclonal") |
| S5 | (MH "Tumor Necrosis Factor") |
| S4 | (S1 or S2 or S3) |
| S3 | TX (bekhterev or bechterew) |
| S2 | TX (ankylosing or spondyl*) |
| S1 | (MH "Spondylarthropathies+") |
Appendix 3. Updated search strategy (October 2013, including golimumab)
Database: Ovid MEDLINE(R) In‐Process & Other Non‐Indexed Citations and Ovid MEDLINE(R) <1946 to Present>
Search Strategy:
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
1 Spondylitis, Ankylosing/ (11743)
2 (bekhterev* or bechterew*).tw. (669)
3 (ankylosing adj (spondyl$ or rheumatoid)).tw. (9939)
4 or/1‐3 (14314)
5 etanercept.sh,rn,tw. (4782)
6 enbrel.sh,rn,tw. (219)
7 infliximab.sh,rn,tw. (10011)
8 remicade.sh,rn,tw. (227)
9 adalimumab.sh,rn,tw. (5447)
10 humira.sh,rn,tw. (186)
11 Simponi.sh,rn,tw. (9)
12 Golimumab.sh,rn,tw. (335)
13 or/5‐12 (15002)
14 4 and 13 (1224)
15 meta‐analysis.mp,pt. (80626)
16 search.tw. (181139)
17 systematic review.tw. (46201)
18 medline.tw. (63445)
19 cochrane database of systematic reviews.jn. (18098)
20 or/15‐19 (273222)
21 randomized controlled trial.pt. (388158)
22 controlled clinical trial.pt. (89760)
23 randomized.ab. (303172)
24 placebo.ab. (163015)
25 clinical trials as topic.sh. (175009)
26 randomly.ab. (214577)
27 trial.ti. (130732)
28 or/21‐27 (928846)
29 exp animals/ not humans.sh. (4050087)
30 28 not 29 (858635)
31 14 and 30 (319)
32 14 and 20 (58)
33 11 or 12 (335)
Database: Embase Classic+Embase <1947 to 2013 October 09>
Search Strategy:
‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐
1 (bekhterev* or bechterew*).tw. (1228)
2 (ankylosing adj (spondyl$ or rheumatoid)).tw. (14716)
3 ankylosing spondylitis/ (20512)
4 or/1‐3 (22655)
5 etanercept/ (16881)
6 (enbrel or etanercept).tw. (8641)
7 infliximab/ (26746)
8 (remicade or infliximab).tw. (13987)
9 adalimumab/ (13106)
10 (adalimumab or humira).tw. (7067)
11 golimumab/ (1576)
12 (Golimumab or Simponi).tw. (830)
13 or/5‐12 (36950)
14 4 and 13 (3140)
15 randomized.tw. (399359)
16 meta‐analysis.tw. (60911)
17 systematic review.tw. (52090)
18 or/15‐17 (473294)
19 random$.tw. (872666)
20 factorial$.tw. (22787)
21 crossover$.tw. (49676)
22 cross over.tw. (22540)
23 cross‐over.tw. (22540)
24 placebo$.tw. (205650)
25 (doubl$ adj blind$).tw. (150905)
26 (singl$ adj blind$).tw. (14326)
27 assign$.tw. (239903)
28 allocat$.tw. (82346)
29 volunteer$.tw. (184925)
30 crossover procedure/ (38960)
31 double blind procedure/ (122751)
32 randomized controlled trial/ (360167)
33 single blind procedure/ (18364)
34 or/19‐33 (1431673)
35 14 and 18 (288)
36 14 and 34 (501)
37 11 or 12 (1626)
38 4 and 37 (423)
39 34 and 38 (67)
40 18 and 38 (53)
41 39 or 40 (74)
Cochrane Library 2013 Issue 9 ID Search
#1 MeSH descriptor: [Spondylarthropathies] this term only
#2 ankylosing or spondyl:ti,ab
#3 bekhterev or bechterew:ti,ab
#4 MeSH descriptor: [Receptors, Tumor Necrosis Factor] this term only
#5 MeSH descriptor: [Tumor Necrosis Factors] this term only
#6 MeSH descriptor: [Antibodies, Monoclonal] this term only
#7 anti‐tumor necrosis factor:ti,ab
#8 anti‐tnf:ti,ab
#9 antitnf:ti,ab
#10 etanercept:ti,ab
#11 enbrel:ti,ab
#12 infliximab:ti,ab
#13 remicade:ti,ab
#14 adalimumab:ti,ab
#15 humira:ti,ab
#16 (golimumab or simponi):ti,ab
#17 #1 or #2 or #3
#18 #4 or #5 or #6 or #7 or #8 or #9 or #10 or #11 or #12 or #13 or #14 or #15
#19 #17 and #18
CINAHL via Ebscohost
2012‐2013
| S14 | S1 OR S2 |
| S13 | S3 OR S4 OR S5 OR S6 OR S7 OR S8 OR S9 OR S10 OR S11 or S12 |
| S12 | "simponi" |
| S11 | "golimumab" |
| S10 | "humira" |
| S9 | "humira" |
| S8 | "adalimumab" |
| S7 | (MH "Adalimumab") |
| S6 | "remicade" |
| S5 | (MH "Infliximab") |
| S4 | "enbrel" |
| S3 | (MH "Etanercept") |
| S2 | "(bekhterev or bechterew)" |
| S1 | (MH "Spondylitis, Ankylosing") |
Database: Web of Science – Oct 09, 2013 Search Strategy
Topic=(ankylosing spondylitis) AND Topic=(( anti‐tnf or etanercept or enbrel or infliximab or remicade or adalimumab or humira or golimumab or simponi))
Timespan= Limited to 2012‐2013
Databases=SCI‐EXPANDED, SSCI, A&HCI, CPCI‐S, CPCI‐SSH.
Refined by document type (Meeting or Abstract)
Data and analyses
Comparison 1. Adalimumab versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 BASFI (0‐10 VAS) | 4 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.1 12 weeks | 4 | 786 | Mean Difference (IV, Fixed, 95% CI) | ‐1.56 [‐1.89, ‐1.23] |
| 2 ASAS 40 | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.1 12 weeks | 2 | 659 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.76 [2.56, 5.53] |
| 3 ASAS partial remission | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 3.1 12 weeks | 2 | 659 | Risk Ratio (M‐H, Fixed, 95% CI) | 5.91 [2.92, 11.94] |
| 4 MRI SPARCC score (lumbar spine; scale 0‐108) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5 MRI SPARCC score (sacroiliac joint; scale 0‐72)) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 6 Withdrawals due to adverse events | 2 | 659 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.48 [0.34, 6.46] |
| 6.1 12 weeks | 2 | 659 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.48 [0.34, 6.46] |
| 7 Serious adverse events | 2 | 659 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.89 [0.25, 3.15] |
| 7.1 12 weeks | 2 | 659 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.89 [0.25, 3.15] |
1.1. Analysis.
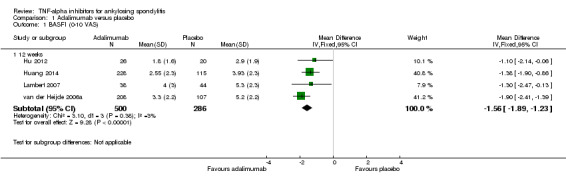
Comparison 1 Adalimumab versus placebo, Outcome 1 BASFI (0‐10 VAS).
1.2. Analysis.
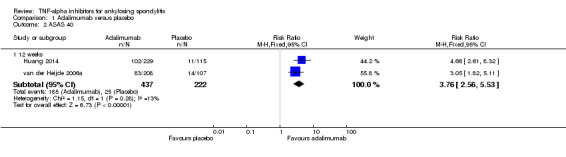
Comparison 1 Adalimumab versus placebo, Outcome 2 ASAS 40.
1.3. Analysis.
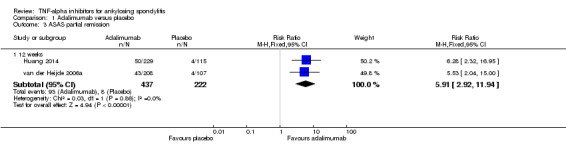
Comparison 1 Adalimumab versus placebo, Outcome 3 ASAS partial remission.
1.4. Analysis.
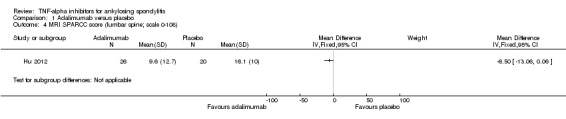
Comparison 1 Adalimumab versus placebo, Outcome 4 MRI SPARCC score (lumbar spine; scale 0‐108).
1.5. Analysis.
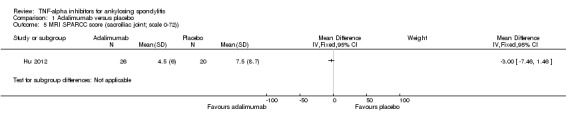
Comparison 1 Adalimumab versus placebo, Outcome 5 MRI SPARCC score (sacroiliac joint; scale 0‐72)).
1.6. Analysis.
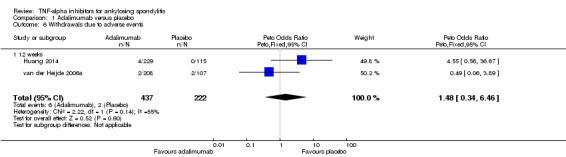
Comparison 1 Adalimumab versus placebo, Outcome 6 Withdrawals due to adverse events.
1.7. Analysis.
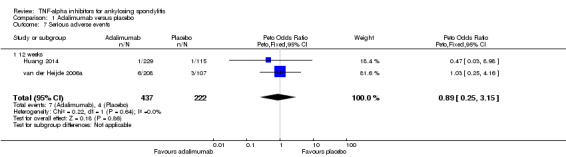
Comparison 1 Adalimumab versus placebo, Outcome 7 Serious adverse events.
Comparison 2. Infliximab versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 ASAS 40 | 2 | 355 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.84 [2.28, 6.46] |
| 1.1 12 weeks | 1 | 76 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.42 [1.41, 8.26] |
| 1.2 24 weeks | 1 | 279 | Risk Ratio (M‐H, Fixed, 95% CI) | 4.01 [2.13, 7.55] |
| 2 BASFI | 3 | 424 | Mean Difference (IV, Fixed, 95% CI) | ‐1.84 [‐2.18, ‐1.49] |
| 2.1 12 weeks | 2 | 145 | Mean Difference (IV, Fixed, 95% CI) | ‐1.34 [‐2.04, ‐0.64] |
| 2.2 24 weeks | 1 | 279 | Mean Difference (IV, Fixed, 95% CI) | ‐2.0 [‐2.40, ‐1.60] |
| 3 ASAS partial remission | 2 | 348 | Risk Ratio (M‐H, Fixed, 95% CI) | 17.47 [3.42, 89.14] |
| 4 Serious adverse events | 3 | 422 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 2.47 [0.75, 8.14] |
| 4.1 12 weeks | 2 | 145 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 7.80 [1.07, 56.65] |
| 4.2 24 weeks | 1 | 277 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.29 [0.29, 5.73] |
| 5 Withdrawals due to adverse events | 3 | 424 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.48 [0.39, 5.62] |
| 5.1 12 weeks | 2 | 145 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.99 [0.38, 10.42] |
| 5.2 24 weeks | 1 | 279 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.78 [0.07, 8.44] |
| 6 Spinal inflammation (MRI activity score (0‐138)) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 7 Spinal inflammation (MRI Activity score >1) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only |
2.1. Analysis.
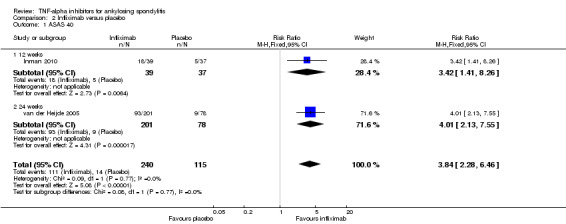
Comparison 2 Infliximab versus placebo, Outcome 1 ASAS 40.
2.2. Analysis.
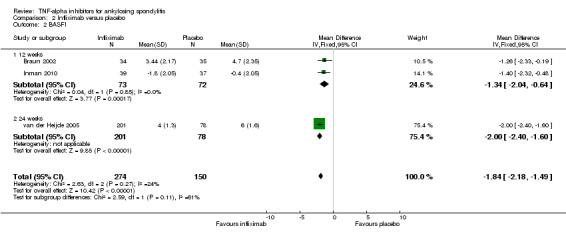
Comparison 2 Infliximab versus placebo, Outcome 2 BASFI.
2.3. Analysis.
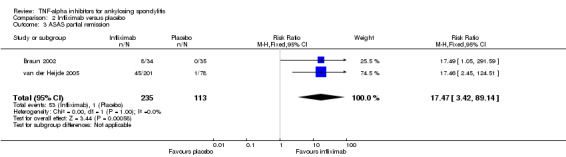
Comparison 2 Infliximab versus placebo, Outcome 3 ASAS partial remission.
2.4. Analysis.
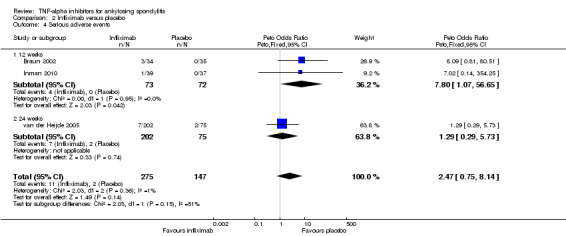
Comparison 2 Infliximab versus placebo, Outcome 4 Serious adverse events.
2.5. Analysis.
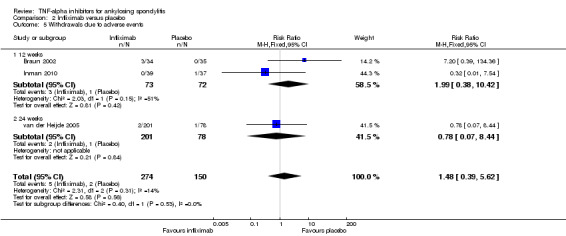
Comparison 2 Infliximab versus placebo, Outcome 5 Withdrawals due to adverse events.
2.6. Analysis.
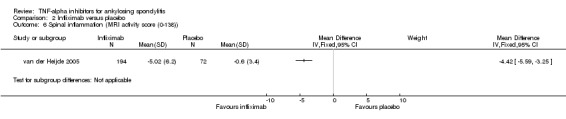
Comparison 2 Infliximab versus placebo, Outcome 6 Spinal inflammation (MRI activity score (0‐138)).
2.7. Analysis.
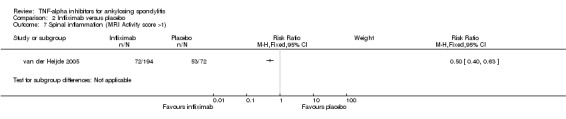
Comparison 2 Infliximab versus placebo, Outcome 7 Spinal inflammation (MRI Activity score >1).
Comparison 3. Golimumab versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 ASAS40 | 2 | 429 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.87 [1.89, 4.35] |
| 2 BASFI | 2 | 429 | Mean Difference (IV, Random, 95% CI) | ‐1.48 [‐1.95, ‐1.02] |
| 3 ASAS partial remission | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4 ASspiMRI‐a change from baseline (spinal inflammation, score 0‐138)) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 5 Withdrawals due to adverse events | 2 | 429 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.63 [0.35, 7.55] |
| 6 Serious adverse event | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Subtotals only |
3.1. Analysis.
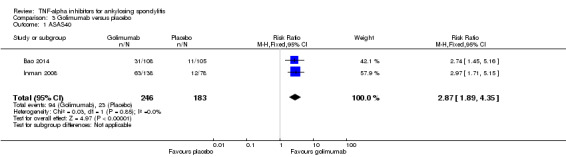
Comparison 3 Golimumab versus placebo, Outcome 1 ASAS40.
3.2. Analysis.
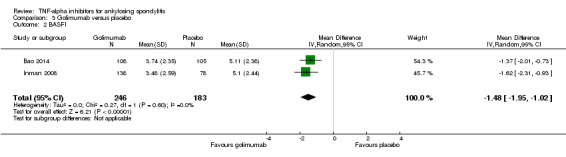
Comparison 3 Golimumab versus placebo, Outcome 2 BASFI.
3.3. Analysis.
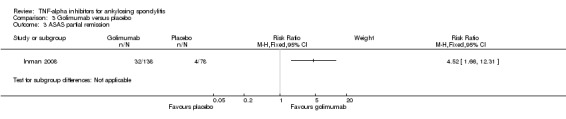
Comparison 3 Golimumab versus placebo, Outcome 3 ASAS partial remission.
3.4. Analysis.
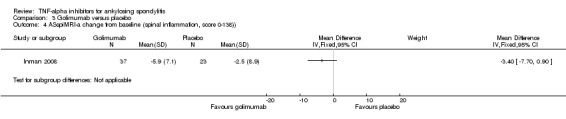
Comparison 3 Golimumab versus placebo, Outcome 4 ASspiMRI‐a change from baseline (spinal inflammation, score 0‐138)).
3.5. Analysis.
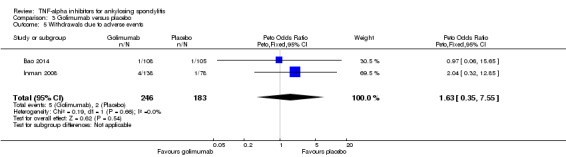
Comparison 3 Golimumab versus placebo, Outcome 5 Withdrawals due to adverse events.
3.6. Analysis.
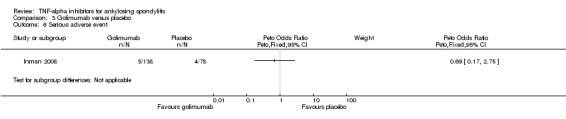
Comparison 3 Golimumab versus placebo, Outcome 6 Serious adverse event.
Comparison 4. Etanercept (25 mg twice weekly or 50mg once weekly) versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 ASAS 40 | 3 | 590 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.82 [2.04, 3.91] |
| 1.1 6‐12 weeks | 2 | 508 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.08 [2.11, 4.48] |
| 1.2 Advanced AS | 1 | 82 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.98 [1.05, 3.76] |
| 2 BASFI (0‐10 scale, none to severe limitations) | 6 | 553 | Mean Difference (IV, Fixed, 95% CI) | ‐1.35 [‐1.75, ‐0.95] |
| 2.1 6 weeks | 1 | 30 | Mean Difference (IV, Fixed, 95% CI) | ‐0.80 [‐2.48, 0.88] |
| 2.2 12 weeks | 2 | 124 | Mean Difference (IV, Fixed, 95% CI) | ‐1.35 [‐2.16, ‐0.54] |
| 2.3 16 weeks | 1 | 40 | Mean Difference (IV, Fixed, 95% CI) | ‐0.90 [‐2.50, 0.70] |
| 2.4 24 weeks | 1 | 277 | Mean Difference (IV, Fixed, 95% CI) | ‐1.87 [‐2.48, ‐1.26] |
| 2.5 Advanced AS | 1 | 82 | Mean Difference (IV, Fixed, 95% CI) | ‐0.60 [‐1.45, 0.25] |
| 3 ASAS Partial remission | 3 | 785 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.99 [2.21, 7.20] |
| 4 Withdrawals due to adverse events | 8 | 1061 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 4.21 [1.55, 11.44] |
| 4.1 6 weeks | 2 | 182 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.2 12 weeks | 3 | 480 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 3.36 [0.73, 15.41] |
| 4.3 16 weeks | 1 | 40 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 4.4 24 weeks | 1 | 277 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 4.69 [1.15, 19.11] |
| 4.5 Advanced AS | 1 | 82 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 8.19 [0.16, 414.54] |
| 5 Serious adverse events | 8 | 1061 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.76 [0.81, 3.82] |
| 5.1 6 weeks | 2 | 182 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 5.2 12 weeks | 3 | 480 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.56 [0.43, 5.59] |
| 5.3 16 weeks | 1 | 40 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 5.4 24 weeks | 1 | 277 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.84 [0.63, 5.37] |
| 5.5 Advanced AS | 1 | 82 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 2.19 [0.22, 21.75] |
4.1. Analysis.
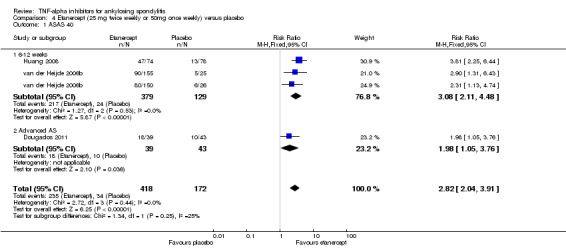
Comparison 4 Etanercept (25 mg twice weekly or 50mg once weekly) versus placebo, Outcome 1 ASAS 40.
4.2. Analysis.
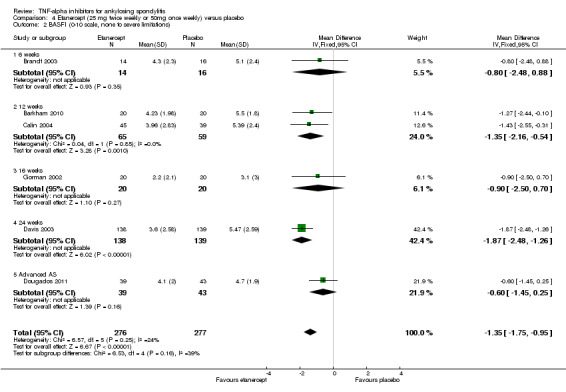
Comparison 4 Etanercept (25 mg twice weekly or 50mg once weekly) versus placebo, Outcome 2 BASFI (0‐10 scale, none to severe limitations).
4.3. Analysis.
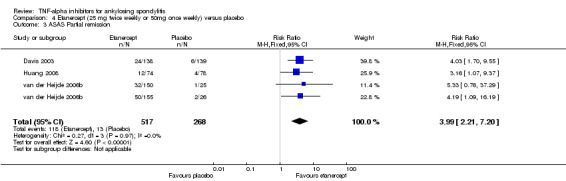
Comparison 4 Etanercept (25 mg twice weekly or 50mg once weekly) versus placebo, Outcome 3 ASAS Partial remission.
4.4. Analysis.
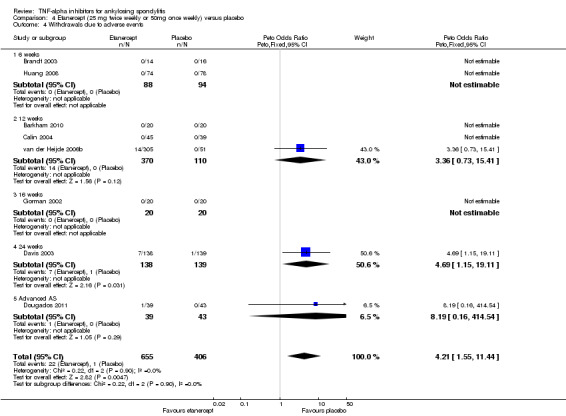
Comparison 4 Etanercept (25 mg twice weekly or 50mg once weekly) versus placebo, Outcome 4 Withdrawals due to adverse events.
4.5. Analysis.
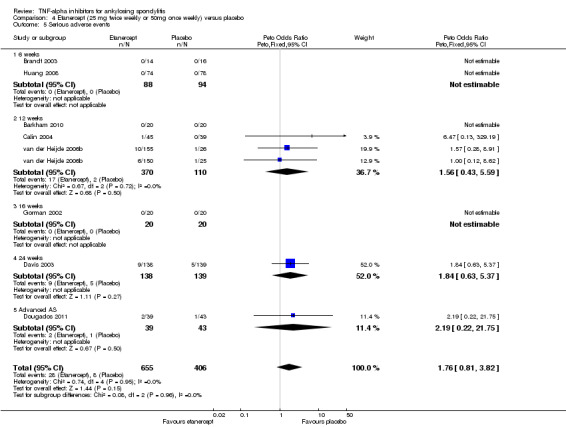
Comparison 4 Etanercept (25 mg twice weekly or 50mg once weekly) versus placebo, Outcome 5 Serious adverse events.
Comparison 5. Etanercept versus infliximab.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 ASAS40 ‐ 12 weeks | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2 BASFI ‐ 12 weeks | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only |
5.2. Analysis.
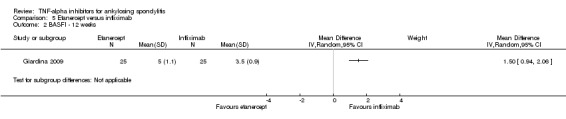
Comparison 5 Etanercept versus infliximab, Outcome 2 BASFI ‐ 12 weeks.
Comparison 6. Etanercept versus sulphasalazine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 ASAS40 ‐ 16‐week | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 2 BASFI | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 3 ASAS Partial remission | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4 Withdrawals due to adverse events | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Totals not selected | |
| 5 Serious adverse events | 1 | Peto Odds Ratio (Peto, Fixed, 95% CI) | Totals not selected |
6.2. Analysis.
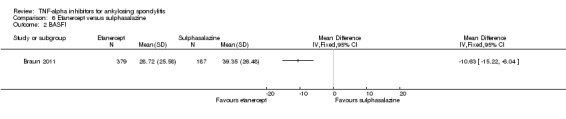
Comparison 6 Etanercept versus sulphasalazine, Outcome 2 BASFI.
6.4. Analysis.
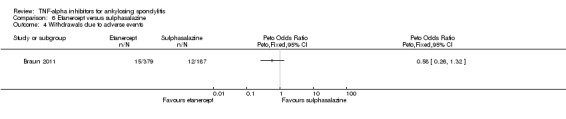
Comparison 6 Etanercept versus sulphasalazine, Outcome 4 Withdrawals due to adverse events.
Comparison 7. Infliximab + methotrexate versus placebo + methotrexate.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 BASDAI | 1 | Mean Difference (IV, Fixed, 95% CI) | Totals not selected | |
| 2 ASAS20 | 1 | 42 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.33 [0.80, 6.80] |
| 3 > 50% BASDAI | 1 | 42 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.5 [0.87, 7.22] |
Comparison 8. TNF‐inhibitors versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Withdrawals due to AE | 16 | 2623 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 2.44 [1.26, 4.72] |
| 1.1 6 weeks | 2 | 182 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 1.2 12 weeks | 9 | 1416 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 2.48 [1.00, 6.16] |
| 1.3 14 weeks | 2 | 429 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.63 [0.35, 7.55] |
| 1.4 16 weeks | 1 | 40 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 1.5 24 weeks | 2 | 556 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 3.06 [0.90, 10.45] |
| 2 Serious adverse events | 15 | 2408 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.45 [0.85, 2.48] |
| 2.1 6 weeks | 2 | 182 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2.2 12 weeks | 9 | 1317 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.48 [0.69, 3.19] |
| 2.3 16 weeks | 1 | 40 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 0.0 [0.0, 0.0] |
| 2.4 24 weeks | 3 | 869 | Peto Odds Ratio (Peto, Fixed, 95% CI) | 1.43 [0.68, 3.00] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Bao 2014.
| Methods | Multicenter, 24‐week, randomized, double‐blind, placebo‐controlled phase III trial followed by subcutaneous administration of golimumab 50mg to all patients from week 24 forward | |
| Participants | Golimumab 50 mg (N = 108) Placebo N = (105) Age (mean (SD), years): Treatment group ‐ 30.5 (10.27); Control group ‐ 30.6 (8.60) N (%) male: Treatment group ‐ 90 (83.3); Control group ‐ 87 (82.9) Ethnicity: Chinese Duration of symptoms (mean (SD), years): Treatment group ‐ 6.1 (5.93); Control group ‐6.6 (5.67) Duration since ankylosing spondylitis diagnosis: Treatment group 4.2 (5.22); control group 3.7 (3.88) Inclusion: Diagnosis of ankylosing spondylitis for at least 3 months defined as definite by the 1984 modified New York criteria; BASDAI of >=4 (0‐10cm scale) and visual analogue scale score for total back pain of >=4 (0‐10cm scale) Exclusion: prior biologic anti‐TNF therapy; complete ankylosis of the spine, failed tuberculosis screening Patients were allowed to continue receiving stable doses of MTX, SSZ and HCQ during study participation |
|
| Interventions | Subcutaneous injections of placebo (group 1) or golimumab 50 mg (group 2) every 4 weeks. At week 16, patients with < 20% improvement from baseline in both total back pain and morning stiffness measures entered double‐blind early escape, whereby those in group 1 were started on golimumab 50 mg and those in group 2 continued to receive golimumab 50 mg. At week 24, all patients still receiving placebo crossed over to golimumab 50 mg subcutaneous injections | |
| Outcomes | The primary study endpoint was the proportion of patients achieving ASAS20 at week 14. Other clinical assessments: BASFI, Bath Ankylosing Spondylitis Disease Activity Index (BASDAI), Bath Ankylosing Spondylitis Metrology Index (BASMI), ASAS40 response, ASAS5/6 response, ASAS partial remission, SF‐36 health‐related quality of life (HRQoL) questionnaire, Jenkins Sleep Evaluation Questionnaire (JSEQ) | |
| Notes |
NCT01248793. " This study was funded by Janssen Research & Development, LLC, Spring House, PA, USA. Janssen statisticians and programmers performed the analyses. All authors reviewed and approved the content of the manuscript before submission and jointly agreed to submit the final version of the manuscript. The manuscript was prepared by C.B., F.H., M.A.K., K.F., Z.W., C.H., E.C.H., Michelle Perate (non‐author; a paid consultant for Janssen Scientific Affairs), and Mary Whitman (non‐author; Janssen Biotech)." Four authors are employees of Janssen Pharmaceuticals. MTX = methotrexate, SSZ = sulphasalazine and HCQ = hydroxychloroquine |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details provided |
| Allocation concealment (selection bias) | Unclear risk | No details provided |
| Blinding of participants and personnel (performance bias) Patient‐assessed outcomes | Unclear risk | "Double‐blind", no details provided |
| Blinding of participants and personnel (performance bias) Physician‐assessed outcomes | Unclear risk | "Double‐blind", no details provided |
| Blinding of outcome assessment (detection bias) Patient‐assessed outcomes | Unclear risk | "Double‐blind", no details provided |
| Blinding of outcome assessment (detection bias) Physician‐assessed outcomes | Unclear risk | "Double‐blind", no details provided |
| Incomplete outcome data (attrition bias) Efficacy outcomes | Low risk | 101/105 in golimumab group and 102/108 in placebo group completed 52 weeks; Intention‐to‐treat analyses |
| Incomplete outcome data (attrition bias) Safety outcomes | Low risk | 101/105 in golimumab group and 102/108 in placebo group completed 52 weeks; Intention‐to‐treat analyses |
| Selective reporting (reporting bias) | Low risk | Reported on primary outcome as defined in protocol on clinicaltrials.gov and on Assessment of SpondyloArthritis international Society (ASAS)‐recommended outcomes |
| Method of adverse event monitoring Safety outcomes | Unclear risk | No details |
| 'Serious adverse event' definitions provided? Safety outcomes | Unclear risk | No details |
Barkham 2010.
| Methods | "Double blind, placebo controlled trial". States that participants were randomized to etanercept or placebo in a 1 to 1 ratio | |
| Participants | N = 40, 20 per group. 32 (80%) male; mean age 40.1 years (range 20‐61 years); mean duration of symptoms 17 years | |
| Interventions | Etanercept (25 mg) or placebo twice weekly for 3 months | |
| Outcomes | Primary outcome was a change in the work instability of patients after 3 months as measured by the Ankylosing Spondylitis Work Instability Scale (AS WIS) scale. Secondary outcomes included changes in AS WIS score after 6 months, clinical response (BASDAI, BASFI, Ankylosing Spondylitis Quality of Life (ASQoL)) and gait analysis at 3 and 6 months | |
| Notes | Funding source not reported. First reference reported as an abstract from a conference proceeding | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) Patient‐assessed outcomes | Low risk | "The site monitor, investigators, and patients remained blinded until after the data through week 12 had been finalised" ; although details not provided, probably done adequately |
| Blinding of participants and personnel (performance bias) Physician‐assessed outcomes | Low risk | "The site monitor, investigators, and patients remained blinded until after the data through week 12 had been finalised" ; although details not provided, probably done adequately |
| Blinding of outcome assessment (detection bias) Patient‐assessed outcomes | Low risk | "The site monitor, investigators, and patients remained blinded until after the data through week 12 had been finalised" ; although details not provided, probably done adequately |
| Blinding of outcome assessment (detection bias) Physician‐assessed outcomes | Low risk | "The site monitor, investigators, and patients remained blinded until after the data through week 12 had been finalised" ; although details not provided, probably done adequately |
| Incomplete outcome data (attrition bias) Efficacy outcomes | Unclear risk | Unclear if all completed trial |
| Incomplete outcome data (attrition bias) Safety outcomes | Unclear risk | unclear if all completed trial |
| Selective reporting (reporting bias) | Low risk | Assessment of SpondyloArthritis international Society (ASAS)‐recommended clinical outcomes reported in addition to primary outcome |
| Method of adverse event monitoring Safety outcomes | Unclear risk | Not reported |
| 'Serious adverse event' definitions provided? Safety outcomes | Unclear risk | Not reported |
Brandt 2003.
| Methods | Multicenter, randomized, placebo‐controlled trial | |
| Participants | N = 14 etanercept ; N = 16 placebo
Age (mean years): Treatment group ‐ 40; Control group ‐ 32
% male: Treatment group ‐ 71%; Control group ‐ 75%
% white: Not reported
Disease duration (years): Treatment group ‐ 15; Control group ‐11
Patients fulfilled the modified NY criteria for ankylosing spondylitis and had active disease defined by BASDAI >=4 and spinal pain of >=4 on 0‐10 scale Excluded: active tuberculosis in past 3 years, serious infection in past 2 months, malignancies in past 5 years, multiple sclerosis or related disorder, current signs of severe disease. Disease‐modifying, anti‐rheumatic drugs (DMARDs) and corticosteroids withdrawn at least 4 weeks prior to screening. Also, widespread ankylosis. Non‐steroidal, anti‐inflammatory drugs (NSAIDs) at same or less dose at baseline were allowed |
|
| Interventions | Etanercept 25 mg twice weekly subcutaneously vs placebo for 6 weeks | |
| Outcomes | Primary: BASDAI >= 50% by week 6. Others: BASDAI, BASFI, BASMI, ASAS20%, SF‐36, Bath Ankylosing Spondylitis Radiology Index ‐spine (BASRI‐s), adverse events | |
| Notes | "Supported by a grant (Kompentenznetz Rheuma) from the German MInistry of Research and by Wyeth Pharma who provided the study drug" | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Initials and sex of the 33 remaining patients were reported to a central independent registration office by fax. Patients were randomly allocated to one of the treatment groups" |
| Allocation concealment (selection bias) | Low risk | "Initials and sex of the 33 remaining patients were reported to a central independent registration office by fax. Patients were randomly allocated to one of the treatment groups" |
| Blinding of participants and personnel (performance bias) Patient‐assessed outcomes | Low risk | "The pharmacist at each center prepared the medication, which was delivered in a blinded manner." "The placebo solution containing bacteriostatic water was supplied and administered identically." "Investigators and patients remained blinded until week 12, 6 weeks after the placebo controlled phase had finished" |
| Blinding of participants and personnel (performance bias) Physician‐assessed outcomes | Low risk | "Investigators and patients remained blinded until week 12, 6 weeks after the placebo controlled phase had finished" |
| Blinding of outcome assessment (detection bias) Patient‐assessed outcomes | Low risk | "The pharmacist at each center prepared the medication, which was delivered in a blinded manner." "The placebo solution containing bacteriostatic water was supplied and administered identically." "Investigators and patients remained blinded until week 12, 6 weeks after the placebo controlled phase had finished" |
| Blinding of outcome assessment (detection bias) Physician‐assessed outcomes | Low risk | Investigators and patients remained blinded until week 12, 6 weeks after the placebo controlled phase had finished" |
| Incomplete outcome data (attrition bias) Efficacy outcomes | Low risk | Reasons for withdrawal provided. > 80% follow‐up in both treatment and placebo groups |
| Incomplete outcome data (attrition bias) Safety outcomes | Low risk | Reasons for withdrawal provided. > 80% follow‐up in both treatment and placebo groups |
| Selective reporting (reporting bias) | Low risk | Assessment of SpondyloArthritis international Society (ASAS)‐recommended outcomes reported. "As the primary end point of the study, an improvement in disease activity of 50% between baseline and week 6, measured by the BASDAI, was chosen. The secondary outcome parameters analyzed were improvements in numeric rating scale for spinal pain, BASFI, Bath Anyklosing Spondylitis Metrology Index (BASMI), SF‐36, the ASAS response criteria, serum C‐reactive protein (CRP) level, and erythrocyte sedimentation rate (ESR) |
| Method of adverse event monitoring Safety outcomes | Unclear risk | Method not reported; or efficacy, clinical questionnaires filled out every 3 weeks |
| 'Serious adverse event' definitions provided? Safety outcomes | Unclear risk | Serious adverse event definition not provided |
Braun 2002.
| Methods | Multicenter, randomized placebo‐controlled trial | |
| Participants | N = 34 infliximab; N = 35 placebo
Age (mean years): Treatment group ‐ 41; Control group ‐ 39
% male: Treatment group ‐ 68%; Control group ‐ 63%
% white: Treatment group ‐ not reported
Disease duration (years): Treatment group ‐ 16; Control group ‐15
Patients fulfilled the modified NY criteria for AS and had active disease defined by BASDAI >= 4 and spinal pain of >= 4 on 0‐10 scale Excluded: The main reasons for exclusion were severe comorbidity, insufficient disease activity, complete ankylosis, incorrect diagnosis, and disease‐modifying, anti‐rheumatic drugs therapy. Also, active tuberculosis in past 3 years, specific changes in radiograph of chest at baseline, serious infection in past 2 months, malignancies in past 5 years, signs of severe renal, hepatic, haematological, gastrointestinal, endocrine, pulmonary, cardiac, neurological, cerebral disease. Disease‐modifying, anti‐rheumatic drugs and corticosteroids withdrawn at least 4 weeks prior to screening. Patients allowed non‐steroidal, anti‐inflammatory drugs but dose could not increase from baseline dose |
|
| Interventions | Infliximab 5 mg/kg intravenous vs placebo administered at 0, 2, 6 weeks for 12 weeks | |
| Outcomes | Primary: BASDAI >= 50% by week 12. Others BASDAI, BASFI, BASMI, ASAS20%, SF‐36, spinal pain, serum C‐reactive protein (CRP) level, erythrocyte sedimentation rate (ESR), adverse events | |
| Notes | "Funded by a grant (Kompentenznetz Rheuma) from the German MInistry of Research and by Essex Pharma, Munich, who provided the study drug" | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The allocation schedule was generated by computer‐generated random numbers, done in blocks of four for every centre. Thus, within each group of patients enrolled by a single centre, two were randomly assigned to placebo, and two to infliximab" |
| Allocation concealment (selection bias) | Low risk | "The allocation schedule was generated by computer‐generated random numbers, done in blocks of four for every centre. Thus, within each group of patients enrolled by a single centre, two were randomly assigned to placebo, and two to infliximab." "Investigators were informed by fax about the randomisation, and were provided with the trial number of the patient." This information was kept in a sealed envelope that was only opened in case of a serious adverse event |
| Blinding of participants and personnel (performance bias) Patient‐assessed outcomes | Low risk | "Investigators and patients were unaware of treatment status until all case report forms had been completed" |
| Blinding of participants and personnel (performance bias) Physician‐assessed outcomes | Low risk | "Investigators and patients were unaware of treatment status until all case report forms had been completed." "The information had to be sent back once the patient had completed the trial." (referring to the sealed envelope with the group assignment) |
| Blinding of outcome assessment (detection bias) Patient‐assessed outcomes | Low risk | "Investigators and patients were unaware of treatment status until all case report forms had been completed" |
| Blinding of outcome assessment (detection bias) Physician‐assessed outcomes | Low risk | "Investigators and patients were unaware of treatment status until all case report forms had been completed." "The information had to be sent back once the patient had completed the trial."(referring to the sealed envelope with the group assignment) |
| Incomplete outcome data (attrition bias) Efficacy outcomes | Low risk | Greater than 80% follow‐up. Reasons for withdrawal were provided. The last observation carried forward method was applied to the four infliximab group members that withdrew |
| Incomplete outcome data (attrition bias) Safety outcomes | Low risk | Greater than 80% follow‐up. Reasons for withdrawal were provided. The last observation carried forward method was applied to the four infliximab group members that withdrew |
| Selective reporting (reporting bias) | Low risk | Assessment of SpondyloArthritis international Society (ASAS) recommended outcomes were reported. "The primary endpoint was improvement of disease activity by 50% between baseline and week 12, measured by BASDAI" |
| Method of adverse event monitoring Safety outcomes | Unclear risk | Not reported |
| 'Serious adverse event' definitions provided? Safety outcomes | Unclear risk | Not provided |
Braun 2011.
| Methods | RCT. Double‐blind | |
| Participants | N = 566. AS patients had active disease based on BASDAI VAS>=30; morning stiffness visual analogue scale (VAS)>=30; VAS>=30 for 2 of the following:patient global assessment of disease activity, pain, BASFI, and be a candidate for sulphasalazine or etanercept. All patients had failed >= 1 non‐steroidal, anti‐inflammatory drug for >= 3 months Exclusion: complete ankylosis of the spine; previous etanercept treatment; sulphasalazine treatment within 6 months of screening Mean age = 41 years; 74% male; average disease duration = 7.5 years |
|
| Interventions | Etanercept 50 mg once weekly (N = 379). Sulfasalzine 3 g daily (N =187) | |
| Outcomes | Primary outcome: proportion of patients achieving ASAS20 at 16 weeks | |
| Notes | ASCEND (Ankylosing Spondylitis Study Comparing ENbrel with sulphasalazine Dosed Weekly) (NCT00247962). Funding source:"Supported by Wyeth Pharmaceuticals, which was acquired by Pfizer Inc. in October 2009" | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computerized randomisation/enrolment system |
| Allocation concealment (selection bias) | Low risk | Computerized randomisation/enrolment system |
| Blinding of participants and personnel (performance bias) Patient‐assessed outcomes | Low risk | Double‐blind; All patients treated with visually identical injections and tablets |
| Blinding of participants and personnel (performance bias) Physician‐assessed outcomes | Low risk | Double‐blind; All patients treated with visually identical injections and tablets; probably investigator blinded |
| Blinding of outcome assessment (detection bias) Patient‐assessed outcomes | Low risk | Double‐blind; All patients treated with visually identical injections and tablets |
| Blinding of outcome assessment (detection bias) Physician‐assessed outcomes | Low risk | Double‐blind; All patients treated with visually identical injections and tablets; probably investigator blinded |
| Incomplete outcome data (attrition bias) Efficacy outcomes | Low risk | < 10% withdrawals in each group; reasons provided; modified intention‐to‐treat analysis reported |
| Incomplete outcome data (attrition bias) Safety outcomes | Low risk | < 10% withdrawals in each group; reasons provided; modified intention‐to‐treat analysis reported |
| Selective reporting (reporting bias) | Low risk | Assessment of SpondyloArthritis international Society (ASAS)‐recommended outcomes reported |
| Method of adverse event monitoring Safety outcomes | Low risk | Safety was assessed by noting the rate of reported AEs and performing routine physical examinations and laboratory tests. Data on the occurrence of AEs were collected at each study visit and via telephone contact with patients 15 days after the 16‐week treatment period |
| 'Serious adverse event' definitions provided? Safety outcomes | Unclear risk | Not reported |
Calin 2004.
| Methods | Multicenter, randomized, placebo‐controlled trial | |
| Participants | N = 45 etanercept; N = 39 placebo
Age (mean, years): Treatment group ‐ 45; Control group ‐ 41
% male: Treatment group ‐ 80%; Control group ‐ 77%
% white: Treatment group ‐ 93%; Control group ‐ 95%
Disease duration (years): Treatment group ‐ 15; Control group ‐10 Patients fulfilled the modified NY criteria for AS and had active disease defined by score >= 30 on visual analogue scale 0‐100 for spinal inflammation and a score of >= 30 on at least 2 of the 3 domains: back pain, patient global assessment and physical function. Patients were excluded if they had complete ankylosis (fusion) of the spine; previously used TNFa inhibitors, including etanercept; used disease‐modifying anti‐rheumatic drugs other than hydroxychloroquine, sulphasalazine, or methotrexate within 4 weeks of baseline; used multiple non‐steroidal anti‐inflammatory drugs (NSAIDs); used > 10 mg prednisone daily; or changed doses of NSAIDs or prednisone within 2 weeks of baseline. Patients were permitted to continue prestudy physiotherapy |
|
| Interventions | Etanercept 25 mg twice weekly subcutaneously vs placebo for 12 weeks | |
| Outcomes | Primary: ASAS20 by week 12. Others: BASDAI, BASFI, ASAS50, ASAS70, spinal inflammation, nocturnal and total pain, spinal mobility, serum C‐reactive protein (CRP) level, erythrocyte sedimentation rate (ESR), adverse events | |
| Notes | NCT00421915. "Trial was funded by Wyeth Research" | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No details provided on sequence generation |
| Allocation concealment (selection bias) | Unclear risk | No details provided on allocation concealment |
| Blinding of participants and personnel (performance bias) Patient‐assessed outcomes | Low risk | "To preserve the integrity of the blind study, placebo and etanercept supplies were similar in appearance" |
| Blinding of participants and personnel (performance bias) Physician‐assessed outcomes | Unclear risk | No details provided other than 'double‐blind' |
| Blinding of outcome assessment (detection bias) Patient‐assessed outcomes | Low risk | "To preserve the integrity of the blind study, placebo and etanercept supplies were similar in appearance" |
| Blinding of outcome assessment (detection bias) Physician‐assessed outcomes | Unclear risk | No details provided other than 'double‐blind' |
| Incomplete outcome data (attrition bias) Efficacy outcomes | Low risk | Reasons for withdrawal reported. Follow‐up was greater than 80%. "Disease activity and safety analyses were based on the intention to treat population and included all patients who received at least one dose of the ‘‘blinded’’ test article. The last observation carried forward technique was used to handle missing data for continuous and ordinal end points" |
| Incomplete outcome data (attrition bias) Safety outcomes | Low risk | Reasons for withdrawal reported. Follow‐up was greater than 80%. "Disease activity and safety analyses were based on the intention to treat population and included all patients who received at least one dose of the ‘‘blinded’’ test article. The last observation carried forward technique was used to handle missing data for continuous and ordinal end points" |
| Selective reporting (reporting bias) | Low risk | Assessment of SpondyloArthritis international Society (ASAS)‐recommended outcomes reported. "The primary efficacy end point was the percentage of ASAS 20 responders after 12 weeks of treatment" |
| Method of adverse event monitoring Safety outcomes | Low risk | "Patients were monitored for adverse events and abnormal laboratory tests" over the course of the study |
| 'Serious adverse event' definitions provided? Safety outcomes | Unclear risk | Serious adverse event definition not provided |
Davis 2003.
| Methods | Multicenter, randomized, placebo‐controlled trial | |
| Participants | N = 138 etanercept; N = 139 placebo
Age (mean, years): Treatment group ‐ 42; Control group ‐ 42
% male: Treatment group ‐ 76%; Control group ‐ 76%
% white: Treatment group ‐ 94%; Control group ‐ 91%
Disease duration (years): Treatment group ‐ 10; Control group ‐10 Patients fulfilled the modified NY criteria for AS and had active disease defined by score >=30 on visual analogue scale 0‐100 for morning stiffness and a score of >= 30 on at least 2 of the 3 domains: back pain, patient global assessment and BASFI. Patients were excluded if they had complete ankylosis (fusion) of the spine based on radiographic assessment; previously used TNFa inhibitors, had a serious infection (requiring hospitalizations or IV antibiotics) within 4 weeks of screening or were pregnant. Patients were allowed to continue receiving hydroxychloroquine, sulphasalazine, or methotrexate at stable doses during the study but were excluded if they had received any other disease‐modifying, anti‐rheumatic drug within 4 weeks of baseline. Also allowed to continue on stable non‐steroidal, anti‐inflammatory drug, prednisone, and analgesics |
|
| Interventions | Etanercept 25 mg twice weekly subcutaneously vs placebo for 24 weeks | |
| Outcomes | Primary: ASAS20 by week 12 and 24. Others: ASAS50, ASAS70, partial remission (defined as value < 20 mm (0‐100mm scale) in each of 4 ASAS domains (patient global assessment, pain, BASFI, inflammation)). BASDAI, spinal mobility, peripheral joint count, serum C‐reactive protein (CRP) level, erythrocyte sedimentation rate (ESR), assessor global assessment, adverse events | |
| Notes | "Supported by Immunex Corporation" NCT00356356 for long‐term extension |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Patients, investigators, assessors, other study site personnel, and representatives of the sponsor were blinded to the randomisation schedule and to treatment assignment until completion of the trial" |
| Allocation concealment (selection bias) | Low risk | "Patients, investigators, assessors, other study site personnel, and representatives of the sponsor were blinded to the randomisation schedule and to treatment assignment until completion of the trial" |
| Blinding of participants and personnel (performance bias) Patient‐assessed outcomes | Low risk | "Patients, investigators, assessors, other study site personnel, and representatives of the sponsor were blinded to the randomisation schedule and to treatment assignment until completion of the trial" |
| Blinding of participants and personnel (performance bias) Physician‐assessed outcomes | Low risk | "Patients, investigators, assessors, other study site personnel, and representatives of the sponsor were blinded to the randomisation schedule and to treatment assignment until completion of the trial" |
| Blinding of outcome assessment (detection bias) Patient‐assessed outcomes | Low risk | "Patients, investigators, assessors, other study site personnel, and representatives of the sponsor were blinded to the randomisation schedule and to treatment assignment until completion of the trial" |
| Blinding of outcome assessment (detection bias) Physician‐assessed outcomes | Low risk | "Patients, investigators, assessors, other study site personnel, and representatives of the sponsor were blinded to the randomisation schedule and to treatment assignment until completion of the trial" |
| Incomplete outcome data (attrition bias) Efficacy outcomes | Low risk | Completed study: 86% in placebo and 91% in treatment; 1 loss to follow‐up in placebo and 2 loss to follow‐up in treatment. Last observation carried forward (LOCF) used for missing data |
| Incomplete outcome data (attrition bias) Safety outcomes | Low risk | Completed study: 86% in placebo and 91% in treatment; 1 loss to follow‐up in placebo and 2 loss to follow‐up in treatment. LOCF used for missing data |
| Selective reporting (reporting bias) | Low risk | ASAS20 was primary outcome; not stated that it was prespecified in the protocol, but it is an appropriate outcome |
| Method of adverse event monitoring Safety outcomes | Low risk | Patients used a diary to record presence of adverse events |
| 'Serious adverse event' definitions provided? Safety outcomes | Low risk | Adverse events graded on a scale derived from the National Cancer Institute Common Toxicity criteria |
Dougados 2011.
| Methods | 12‐week randomized double‐blind placebo‐controlled multicenter study conducted in 21 centers in four European countries (France, Germany, The Netherlands and Hungary) | |
| Participants | Etanercept (N = 39) Placebo (N = 43) Inclusion criteria was patients with advanced and active ankylosing spondylitis Age (mean, years): Treatment group ‐ 46; Control group ‐ 48 % male: Treatment group ‐ 95%; Control group ‐ 91% Disease duration (years): Treatment group ‐ 19; Control group ‐23 Inclusion criteria: Men and women aged 18–70 years were eligible if they had a current diagnosis of ankylosing spondylitis as defined by the modified New York criteria. Advanced ankylosing spondylitis and severe spinal ankylosis defined by having one of the following three criteria: (1) two intervertebral adjacent bridges and/or fusion at the lumbar spine; (2) three intervertebral adjacent bridges and/or fusion at the thoracic spine; or (3) two intervertebral adjacent bridges and/or fusion at the cervical spine. As well: ‐ radiologic evidence of spine and hip ‐ baseline pain with axial involvement of the overall level of ankylosing spondylitis neck, back or hip for a score ≧ 30 on a 0‐100 mm visual analogue scale (VAS). patients had to have an active refractory disease defined by a score ≧ 40 on the BASDAI (0‐100) despite optimal non‐steroidal, antif‐inflammatory drug treatment |
|
| Interventions | Etanercept 50 mg subcutaneous injection once weekly, with placebo (1:1 ratio) | |
| Outcomes | The primary end point was the normalized net incremental area under the curve in the BASDAI between randomisation (baseline) and week 12 and was calculated as the area between baseline and the patient global assessment curve as a function of time, using the linear trapezoidal method, divided by the number of days the patient remained in the study Secondary outcomes: ASAS20, ASAS40, ASAS5/6, ASAS partial remission, and improvement in BASDAI of at least 50% (BASDAI50), improvement in Ankylosing Spondylitis‐Disease Activity State (AS‐DAS) and AS‐DAS status, BASFI, BASMI, Minimum Clinically Important Improvement (MCII) and Patient Acceptable Symptom State (PASS) |
|
| Notes | NCT00420238; "This study was sponsored by Wyeth Pharmaceuticals (Wyeth was acquired by Pfizer in October 2009)" | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomised double‐blind", no further details provided |
| Allocation concealment (selection bias) | Unclear risk | "Randomised double‐blind", no further details provided |
| Blinding of participants and personnel (performance bias) Patient‐assessed outcomes | Low risk | "Matching placebo" |
| Blinding of participants and personnel (performance bias) Physician‐assessed outcomes | Unclear risk | "Matching placebo" ; unclear if investigators were properly blinded and no details provided on allocation concealment |
| Blinding of outcome assessment (detection bias) Patient‐assessed outcomes | Low risk | "Matching placebo" |
| Blinding of outcome assessment (detection bias) Physician‐assessed outcomes | Unclear risk | "Matching placebo" ; unclear if investigators were properly blinded and no details provided on allocation concealment |
| Incomplete outcome data (attrition bias) Efficacy outcomes | Low risk | < 10% drop out rate; Intention‐to‐treat analysis conducted |
| Incomplete outcome data (attrition bias) Safety outcomes | Low risk | < 10% drop out rate; Intention‐to‐treat analysis conducted |
| Selective reporting (reporting bias) | Low risk | Important Assessment of SpondyloArthritis international Society (ASAS)‐recommended outcomes reported |
| Method of adverse event monitoring Safety outcomes | Unclear risk | Not provided |
| 'Serious adverse event' definitions provided? Safety outcomes | Unclear risk | Not provided |
Giardina 2009.
| Methods | "Two year randomized study" | |
| Participants | N = 25 (abstract had N = 26) etanercept; N = 25 (abstract had N = 24) infliximab Inclusion criteria: active disease for > 3 months; BASDAI > 4; visual analogue scale for spinal pain > 4 Age (mean, years): Etanercept group ‐ 32.6 SD 6.8; Infliximab group ‐ 31.9 SD 9.2 % male: Etanercept group ‐ 80%; Infliximab group ‐ 76% Disease duration (years): Etanercept group ‐ 15.7 SD 6.5; Infliximab group ‐15.4 SD 10.6 |
|
| Interventions | Etanercept 25 mg twice weekly or 5 mg/kg infliximab at week 0, 2, 6, and then every 6 weeks for a period of 102 weeks | |
| Outcomes | In the abstract, the primary outcome was stated to be the proportion of patients achieving a 50% BASDAI response at week 102; Secondary: ASAS50; BASFI, back pain, morning stiffness, C‐reactive protein (CRP), spinal mobility. However, in the full‐text article, the outcome defined as primary is not stated, and the 50% BASDAI response is not reported. ASAS20 and 40, BASDAI, BASFI and adverse events were reported. | |
| Notes | Reported as a full‐text and an abstract from a conference. Funding source not reported. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Patients were randomised to receive alternatively etanercept or inXiximab with a ratio of 1:1" |
| Allocation concealment (selection bias) | Unclear risk | No details on concealment of allocation |
| Blinding of participants and personnel (performance bias) Patient‐assessed outcomes | High risk | "Open‐label" |
| Blinding of participants and personnel (performance bias) Physician‐assessed outcomes | High risk | "Open‐label" |
| Blinding of outcome assessment (detection bias) Patient‐assessed outcomes | High risk | "Open‐label" |
| Blinding of outcome assessment (detection bias) Physician‐assessed outcomes | High risk | "Open‐label" |
| Incomplete outcome data (attrition bias) Efficacy outcomes | Low risk | "No patients discontinued therapy" |
| Incomplete outcome data (attrition bias) Safety outcomes | Low risk | "No patients discontinued therapy" |
| Selective reporting (reporting bias) | High risk | Primary outcome listed in abstract: proportion of people achieving a 50% response in BASDAI; full‐text article does not state the primary outcome, but 50% BASDAI response not reported |
| Method of adverse event monitoring Safety outcomes | Low risk | Patients were monitored for adverse events and abnormal lab values over the course of the study |
| 'Serious adverse event' definitions provided? Safety outcomes | Unclear risk | No serious adverse event definition given |
Gorman 2002.
| Methods | Randomized, placebo‐controlled trial | |
| Participants | N = 20 (etanercept); N = 20 (placebo)
Age (median, years): Treatment group ‐ 38; Control group ‐ 39
% male: Treatment group ‐ 65%; Control group ‐ 90%
% white: Treatment group ‐ 75%; Control group ‐ 70%
Disease duration (years): Treatment group ‐ 15; Control group ‐12 Patients >= 18 years of age and classified as having definite ankylosing spondylitis based on the modified New York criteria. Active spondylitis was defined as the presence of inflammatory back pain (stiffness and pain that worsened with rest and improved with exercise), morning stiffness for at least 45 minutes, and at least moderate disease activity as assessed by the patient and the physician. The patient’s global assessment of disease activity was based on a five‐point scale (1, none; 2, mild; 3, moderate; 4, severe; and 5, very severe). The physician’s assessment was measured with the use of a visual analogue scale (0 mm absence of disease activity and 100 mm very severe activity); a moderate or higher level of disease activity was defined by the placement of a vertical line at 40 mm or higher. Patients were excluded if they had a spondylitis other than ankylosing spondylitis, clinical or radiographic evidence of complete spinal ankylosis, a history of recurrent infections or cancer, or a serious liver, renal, hematologic, or neurologic disorder. Patients continued to take drugs that had already been prescribed for ankylosing spondylitis if the doses had not been changed for at least four weeks before randomisation and if they remained unchanged throughout the trial. Acceptable medications included non‐steroidal anti‐inflammatory drugs,oral corticosteroids (« 10 mg per day), gold injections («50 mg per month), methotrexate («20 mg per week), and sulphasalazine(« 3 g per day) |
|
| Interventions | Twice‐weekly subcutaneous injections of etanercept (25 mg) versus placebo for four months | |
| Outcomes | Primary: ASAS20 Secondary outcomes: physician’s global assessment of disease activity, measures of spinal mobility, the scores for enthesitis and peripheral‐joint tenderness, erythrocyte sedimentation rate, C‐reactive protein, adverse events | |
| Notes | NCT00000433. "The majority of funding for the study was provided by the National Institute of Arthritis and Musculoskeletal and Skin Diseases. Immunex, the pharmaceutical funding source, supplied etanercept and placebo and provided partial funding. Immunex was not involved in the study design, data collection, statistical analysis, or manuscript preparation; these tasks were performed by the authors" | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "A statistician not otherwise involved with the study randomly assigned patients to the study groups, using computer‐generated, random blocks of two and four" |
| Allocation concealment (selection bias) | Low risk | "Cards with the group assignments were placed in sequentially numbered envelopes that were opened by the study pharmacist as each patient was enrolled" |
| Blinding of participants and personnel (performance bias) Patient‐assessed outcomes | Low risk | "The patients and study investigators were unaware of the group assignments" |
| Blinding of participants and personnel (performance bias) Physician‐assessed outcomes | Low risk | "The patients and study investigators were unaware of the group assignments" |
| Blinding of outcome assessment (detection bias) Patient‐assessed outcomes | Low risk | "The patients and study investigators were unaware of the group assignments" |
| Blinding of outcome assessment (detection bias) Physician‐assessed outcomes | Low risk | "The patients and study investigators were unaware of the group assignments" |
| Incomplete outcome data (attrition bias) Efficacy outcomes | Low risk | Reasons for withdrawal were provided. Follow‐up was greater than 80% in both groups. Intention‐to‐treat analyses were performed |
| Incomplete outcome data (attrition bias) Safety outcomes | Low risk | Reasons for withdrawal were provided. Follow‐up was greater than 80% in both groups |
| Selective reporting (reporting bias) | Low risk | "The primary outcome measure was a prespecified, composite treatment response, defined as 20 percent or greater improvement in at least three of five measures of disease activity, as recommended by the ASsessments in Ankylosing Spondylitis Working Group". Corresponds to the protocol on clinicaltrials.gov. |
| Method of adverse event monitoring Safety outcomes | Low risk | "Side effects monitored at each clinic visit by means of open ended questions..." |
| 'Serious adverse event' definitions provided? Safety outcomes | Low risk | Adverse events graded on a scale derived from the National Cancer Institute Common Toxicity criteria |
Hu 2012.
| Methods | This was a randomized, double‐blind, placebo‐controlled study | |
| Participants | Patients were adults (18 and 65 years) diagnosed as having AS defined by the modified New York criteria who had been treated unsuccessfully (nonresponsive or lack of tolerance) with ≥ 1 non‐steroidal anti‐inflammatory drug (NSAIDs). Active ankylosing spondylitis at baseline was defined by fulfilment of at least two of the following three criteria: a BASDAI score ≥ 4, total back pain visual analog scale score ≥ 40, or morning stiffness of ≥ 1 h in duration. Patients could continue taking sulphasalazine ( ≤ 3 g/day), methotrexate ( ≤ 25 mg/week), prednisone and/or prednisone equivalents ( ≤ 10 mg/day), and/or NSAIDs as long as these doses had remained stable for 4 weeks before | |
| Interventions | 40 mg adalimumab (n = 26) or placebo (n = 20) every other week during an initial 12‐week double‐blind period, and all switched to adalimumab treatment for another 12 weeks | |
| Outcomes | "Bath Ankylosing Spondylitis Disease Activity Index (BASDAI), Bath Ankylosing Spondylitis Function Index (BASFI), C‐reactive protein (CRP), Ankylosing Spondylitis Disease Activity Scores (ASDAS) and serum DKK‐1 levels were measured and magnetic resonance imaging (MRI) of both the lumbar spine and sacroiliac joints were obtained at baseline, week 12 and week 24. Spinal and sacroiliac joint inflammations were evaluated using the Spondyloarthritis Research Consortium of Canada (SPARCC) MRI index, and fatty deposition lesions (FDL) were assessed in a dichotomous manner" | |
| Notes | No source of funding reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomized, double‐blind, placebo‐controlled comparison..." No further details provided |
| Allocation concealment (selection bias) | Unclear risk | "Randomized, double‐blind, placebo‐controlled comparison..." No further details provided |
| Blinding of participants and personnel (performance bias) Patient‐assessed outcomes | Unclear risk | "Randomized, double‐blind, placebo‐controlled comparison..."No further details provided |
| Blinding of participants and personnel (performance bias) Physician‐assessed outcomes | Unclear risk | "Randomized, double‐blind, placebo‐controlled comparison..." No further details provided |
| Blinding of outcome assessment (detection bias) Patient‐assessed outcomes | Unclear risk | "Randomized, double‐blind, placebo‐controlled comparison..."No further details provided |
| Blinding of outcome assessment (detection bias) Physician‐assessed outcomes | Unclear risk | "Randomized, double‐blind, placebo‐controlled comparison..." "Each image was rated by two independent readers (XHD and ZYH) who were blinded to the patients’ identities," Unclear if readers were blinded to the treatment group assignment |
| Incomplete outcome data (attrition bias) Efficacy outcomes | Unclear risk | No details provided on number of patients completing the trial |
| Incomplete outcome data (attrition bias) Safety outcomes | Unclear risk | No details provided on number of patients completing the trial |
| Selective reporting (reporting bias) | High risk | While some recommended efficacy outcomes were reported (e.g. BASDAI, BASFI, ASDAS), no adverse outcomes were reported. No primary outcome stated |
| Method of adverse event monitoring Safety outcomes | Unclear risk | No adverse event data reported |
| 'Serious adverse event' definitions provided? Safety outcomes | Unclear risk | No adverse event data reported |
Huang 2008.
| Methods | Randomized, double‐blind, placebo‐controlled study for 6 weeks with 6 week open label afterwards | |
| Participants | N = 74 etanercept; N = 78 placebo. Adult patients with ankylosing spondylitis | |
| Interventions | Etanercept 50 mg once weekly for 6 weeks or placebo subcutaneously. Patients receiving hydrochloroquine, sulphasalazine, or methotrexate at screening continued on the medication | |
| Outcomes | Primary endpoint: ASAS20 week 6. Secondary ASAS40, ASAS5/6, adverse events | |
| Notes | Abstract from conference proceeding. Funding source not reported. NCT00434044 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) Patient‐assessed outcomes | Unclear risk | "Double blind" |
| Blinding of participants and personnel (performance bias) Physician‐assessed outcomes | Unclear risk | "Double blind" |
| Blinding of outcome assessment (detection bias) Patient‐assessed outcomes | Unclear risk | "Double blind" |
| Blinding of outcome assessment (detection bias) Physician‐assessed outcomes | Unclear risk | "Double blind" |
| Incomplete outcome data (attrition bias) Efficacy outcomes | Unclear risk | 2/152 withdrew, no group or reason given |
| Incomplete outcome data (attrition bias) Safety outcomes | Unclear risk | 2/152 withdrew, no group or reason given |
| Selective reporting (reporting bias) | Low risk | Primary outcome ASAS20 reported and appropriate |
| Method of adverse event monitoring Safety outcomes | Low risk | "Safety evaluation included adverse event and routine lab monitoring" |
| 'Serious adverse event' definitions provided? Safety outcomes | Unclear risk | No serious adverse event definition provided |
Huang 2014.
| Methods | Placebo‐controlled, double‐blind, randomized, phase III trial conducted between January 2010 and February 2011 at nine study sites in the People’s Republic of China. 12‐week double‐blind phase was followed by a 12‐week open‐label phase, during which all patients received open‐label adalimumab 40 mg every other week. | |
| Participants | 344 patients ; N = 115 placebo; N = 229 adalimumab Age (mean (standard deviation (SD), years): Treatment group ‐ 30.1 (8.7); Control group ‐ 29.6 (7.5) N (%) male: Treatment group ‐ 185 (80.8); Control group ‐ 95 (82.6) Ethnicity: Chinese Duration of AS symptoms (mean (SD), years): Treatment group ‐ 8.1 (6.0); Control group ‐7.7 (4.7) Duration since AS diagnosis, mean (SD), years: Treatment group 3.0 (3.2) ; Control group 3.0 (3.8) Inclusion criteria: Adults (18 to 65 years) fulfilling modified New York Criteria for ankylosing spondylitis, active disease (as defined by >= 2 of the following: BASDAI >= 4; total back pain on a visual analogue scale (VAS) >= 4 cm; and >= 1 hour of morning stiffness; and had an inadequate response or were intolerant to >=1 non‐steroidal anti‐inflammatory drug (NSAID). Exclusion: Patients with latent or active tuberculosis; total spinal ankylosis; unstable extra‐articular manifestations (e.g. psoriasis, uveitis, inflammatory bowel disease); surgery involving the spine or joints within the previous 2 months; intra‐articular or spinal/paraspinal corticosteroid injections within the previous 28 days; positive serology for HIV antibody, hepatitis B surface antibody or hepatitis C virus antibody; recent infection requiring anti‐infectives; listeriosis; histoplasmosis; immunodeficiency syndrome; or chronic recurring infections. Patients with moderate to severe congestive heart failure, recent cerebrovascular accident, central nervous system demyelinating disease, or history of malignancy (except for successfully treated non‐metastatic non‐melanoma skin cancer or localized cervical carcinoma in situ) were also excluded. Prior exposure to TNF inhibitors, natalizumab or efalizumab at any time, or use of traditional Chinese medicines within 28 days of baseline was not allowed |
|
| Interventions | Adalimumab 40 mg or matching placebo subcutaneously every other week (EOW) Concomitant use of methotrexate (<= 25 mg/week), sulphasalazine (<= 3 g/day), prednisone (<= 10 mg/day), NSAIDs and/or analgesics was allowed but dose adjustments, induction and/or discontinuation of these therapies were only permitted during the open‐label period. Other pharmacological therapies for ankylosing spondylitis except for those listed above could not be initiated at any time during the study |
|
| Outcomes | The primary efficacy endpoint was the percentage of patients achieving the ASAS20 response criteria at week 12. Secondary endpoints were ASAS40 and ASAS5/6 response, ASAS partial remission; BASDAI50; Ankylosing Spondylitis Disease Activity Score (ASDAS); disease activity, pain and spinal mobility by measuring changes from baseline in patient global assessment (VAS), total back pain (VAS), inflammation/morning stiffness,BASDAI, physician’s global assessment of disease activity (VAS),nocturnal pain (VAS), patient’s global assessment of pain (VAS), tender joint count, swollen joint count, Maastricht AS Enthesitis Score (MASES), BASMI‐linear and chest expansion. All measures recorded on a VAS were reported on a 0–10 cm scale. Health‐related quality of life, physical function and work productivity measures included the Health Assessment Questionnaire modified for spondyloarthropathies (HAQ‐S), 36‐Item Short‐Form Health Survey, V.2 (SF‐36v2), BASFI, Bath AS Patient Global Index (BAS‐G) and Work Productivity and Activity Impairment‐Specific Health Problem Questionnaire (WPAI‐SHP) Note: BASFI was measured on a 0‐100 scale as per the result on clinicaltrials.gov. Converted to 0‐10 scale for meta‐analysis |
|
| Notes | (NCT01114880). "This study was sponsored by AbbVie". FH is a consultant and has served on speakers bureaus for AbbVie China. Data extracted for efficacy from 12‐week placebo‐controlled phase | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Participants were centrally randomised using an interactive voice response or web‐based system in a 2:1 ratio" |
| Allocation concealment (selection bias) | Low risk | "Participants were centrally randomised using an interactive voice response or web‐based system in a 2:1 ratio" |
| Blinding of participants and personnel (performance bias) Patient‐assessed outcomes | Low risk | "Study overseers, investigators, study site personnel and patients remained blinded to treatment during this phase, which was followed by a 12‐week open‐label phase, during which all patients received open‐label adalimumab 40 mg EOW" |
| Blinding of participants and personnel (performance bias) Physician‐assessed outcomes | Low risk | "Study overseers, investigators, study site personnel and patients remained blinded to treatment during this phase, which was followed by a 12‐week open‐label phase, during which all patients received open‐label adalimumab 40 mg EOW" |
| Blinding of outcome assessment (detection bias) Patient‐assessed outcomes | Low risk | "Study overseers, investigators, study site personnel and patients remained blinded to treatment during this phase, which was followed by a 12‐week open‐label phase, during which all patients received open‐label adalimumab 40 mg EOW" |
| Blinding of outcome assessment (detection bias) Physician‐assessed outcomes | Low risk | "Study overseers, investigators, study site personnel and patients remained blinded to treatment during this phase, which was followed by a 12‐week open‐label phase, during which all patients received open‐label adalimumab 40 mg EOW" |
| Incomplete outcome data (attrition bias) Efficacy outcomes | Low risk | Completer of 12 week double‐blind period: 113/115 placebo; 224/229 adalimumab; Intention‐to‐treat analysis |
| Incomplete outcome data (attrition bias) Safety outcomes | Low risk | Completer of 12 week double‐blind period: 113/115 placebo; 224/229 adalimumab; Intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Low risk | Same primary outcome as listed on clinicaltrials.gov. Assessment of SpondyloArthritis international Society (ASAS)‐recommended outcomes reported |
| Method of adverse event monitoring Safety outcomes | Unclear risk | "Safety evaluations were conducted at every study visit and included adverse event (AE) monitoring and assessments of clinical laboratory and vital signs."‐from published article. "Events were collected by non‐systematic assessment" ‐ from clinicaltrials.gov |
| 'Serious adverse event' definitions provided? Safety outcomes | Unclear risk | Not provided |
Inman 2008.
| Methods | 24‐week, double‐blind, placebo‐controlled study | |
| Participants | Placebo (N = 78) Golimumab 50 mg (N = 138) Golimumab 100 mg (N = 140) Age (median (interquartile range, years) 41.0 (31.0–50.0) 38.0 (30.0–47.0) 38.0 (29.0–46.0) Male, No (%) 55 (70.5) 102 (73.9) 98 (70.0) White, No (%) 57 (73.1) 103 (74.6) 102 (72.9) Years since symptoms occurred, median (interquartile range) 16 (5–25) 11( 6–18) 9.5 (14‐18) Inclusion criteria: Adult patients who had AS (diagnosed according to the modified New York Criteria for 3 months before the first administration of the study agent, a Bath AS Disease Activity Index (BASDAI) score of 4 (0–10‐point scale), a spinal pain assessment score of 4 on a visual analogue scale (VAS); 0–10‐cm scale), and an inadequate response to current or previous non‐steroidal anti‐inflammatory drugs (NSAIDs) or disease‐modifying anti‐rheumatic drugs (DMARDs). Failure to DMARDs for at least 3 months. Normal chest radiograph within 3 months before randomisation and to have undergone screening for latent tuberculosis (TB) Exclusion criteria: any of the following: complete ankylosis of the spine, any other inflammatory rheumatic disease, a serious infection within 2 months before randomisation, active or latent TB or positive results of a tuberculin skin test before screening or recent contact with a person with active TB, an opportunistic infection within 6 months of screening, hepatitis, human immunodeficiency virus, a transplanted organ, malignancy, multiple sclerosis, or congestive heart failure |
|
| Interventions | 3‐arm study. Patients were randomly assigned in a 1:1.8:1.8 ratio to receive placebo or golimumab at a dose of 50 mg or 100 mg every 4 weeks. Patients were allowed to continue concurrent treatment with methotrexate (MTX), sulphasalazine, hydroxychloroquine, corticosteroids, and NSAIDs at stable doses during the study. At week 16 there was an early‐escape option for those patients who had not responded. Note ‐only extracted 50 mg data (100 mg only indicated for patients > 100 kg and who fail to achieve response to 50mg – not many in the trial and not standard dose) at week 14 |
|
| Outcomes | The primary end point was the proportion of patients who achieved ASAS20 at week 14. Secondary end points included ASAS40, ASAS partial remission, ASAS5/6. Disease activity was evaluated using the BASDAI, the back pain VAS, the night pain VAS, the patient’s global assessment, and the C‐reactive protein level. Physical function was evaluated using the BASFI. Range of motion was assessed using the BASMI (3‐point scale), and chest expansion. Health‐related quality of life was measured using the Short Form 36 (SF‐36) Health Survey. Sleep disturbance was assessed using the Jenkins Sleep Evaluation Questionnaire(JSEQ). Presence of antibodies to golimumab | |
| Notes | NCT00265083. Supported by Centocor Research and Development, Inc. and the Schering‐Plough Research Institute, Inc | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "An interactive voice‐response system with adaptive treatment allocation was used to assign patients to treatment" |
| Allocation concealment (selection bias) | Low risk | "An interactive voice‐response system with adaptive treatment allocation was used to assign patients to treatment" |
| Blinding of participants and personnel (performance bias) Patient‐assessed outcomes | Low risk | "To maintain blinding, patients in the 50‐mg group received active golimumab in the 0.5‐ml syringe and placebo in the 1.0‐ml syringe; patients in the 100‐mg group received placebo in the 0.5‐ml syringe and active golimumab in the 1.0‐ml syringe; and patients in the placebo group received placebo in both syringes" |
| Blinding of participants and personnel (performance bias) Physician‐assessed outcomes | Low risk | Matching placebo and adequate allocation concealment; investigators probably blinded adequately |
| Blinding of outcome assessment (detection bias) Patient‐assessed outcomes | Low risk | "To maintain blinding, patients in the 50‐mg group received active golimumab in the 0.5‐ml syringe and placebo in the 1.0‐ml syringe; patients in the 100‐mg group received placebo in the 0.5‐ml syringe and active golimumab in the 1.0‐ml syringe; and patients in the placebo group received placebo in both syringes" |
| Blinding of outcome assessment (detection bias) Physician‐assessed outcomes | Low risk | matching placebo and adequate allocation concealment; investigators probably blinded adequately |
| Incomplete outcome data (attrition bias) Efficacy outcomes | Low risk | < 10% withdrawal; participants who entered early escape option were considered non‐responders at week 24; "data from all randomized patients were analyzed according to their assigned treatment group" |
| Incomplete outcome data (attrition bias) Safety outcomes | Low risk | < 10% withdrawal;all participants who received study drug were assessed in the safety analyses ; "data from all randomized patients were analyzed according to their assigned treatment group" |
| Selective reporting (reporting bias) | Low risk | Outcomes recommended by Assessment of SpondyloArthritis international Society (ASAS) were reported |
| Method of adverse event monitoring Safety outcomes | Unclear risk | Not reported |
| 'Serious adverse event' definitions provided? Safety outcomes | Unclear risk | Not reported |
Inman 2010.
| Methods | Randomized, double‐blind, placebo‐controlled, multi‐center trial | |
| Participants | N = 76; N = 39 infliximab, N = 37 placebo Note: one abstract assessing spinal inflammation with magnetic resonance imaging (MRI) indicates N=32 (16 infliximab and 16 placebo). The study protocol from clinicaltrials.gov indicates the study is complete and the sample is 76 participants Age (mean, years): Treatment group ‐ 42.9 (10.4); Control group ‐ 39.3 (9) % male: Treatment group ‐ 82%; Control group ‐ 78% % white: Treatment group ‐ 87%; Control group ‐ 89% Disease duration (years): Treatment group ‐ 11.7 (10.6); Control group ‐11.1 (10.3) Positive for HLA‐B27: infliximab = 72%; placebo = 73% Inclusion: Adults (> 18 years) with active ankylosing spondylitis (BASDAI >= 4) In those patients taking non‐steroidal anti‐inflammatory drugs (NSAIDs), disease‐modifying anti‐rheumatic drugs (DMARDs), analgesics, or corticosteroids, the dose must have been stable for at least 14 days (30 days for DMARD) prior to the first infusion of study drug. Patients were excluded from the study if they had a history of chronic/recurrent infectious disease, including tuberculosis, hepatitis B, or HIV, and/or a diagnosis of malignancy or lymphoproliferative disease currently or within the past 5 years |
|
| Interventions | Infliximab (IFX) 3 mg/kg or placebo intravenously at weeks 0, 2, and 6. An open‐label phase followed after week 12 which lasted 46 weeks and the placebo group crossed over to receive infusions of IFX 3 mg/kg at Weeks 14, 16, and 22, and every 8 weeks thereafter. "All patients could receive dose‐escalation of IFX to 5 mg/kg at Weeks 22 or 38 if the patient had an absolute BASDAI score > 3 and a relative decrease of < 50% in BASDAI from baseline". Follow‐up was for 52 weeks | |
| Outcomes | Primary outcome listed in study protocol was ASAS 20 at week 12. Other clinical outcomes in the protocol: BASDAI, BASFI, BASGI, BASMI, ASAS 40/50/70 and ASAS 5/6 and MRI at week 12 were reported in the abstracts (though the number of participants is unclear) | |
| Notes |
NCT00202865. Study known as "CANaDian evaluation of Low DosE infliximab (CANDLE)." Reported in one full‐text article and 3 abstracts from conferences. "Supported by Schering‐Plough, Canada" |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) Patient‐assessed outcomes | Unclear risk | "Double‐blind"; no further details reported |
| Blinding of participants and personnel (performance bias) Physician‐assessed outcomes | Unclear risk | "Double‐blind"; no further details reported |
| Blinding of outcome assessment (detection bias) Patient‐assessed outcomes | Unclear risk | "Double‐blind"; no further details reported |
| Blinding of outcome assessment (detection bias) Physician‐assessed outcomes | Unclear risk | "Double‐blind"; no further details reported |
| Incomplete outcome data (attrition bias) Efficacy outcomes | Low risk | Fig.1 shows the details of patient flow throughout the study. The number of people completing the study at week 12 (RCT phase) was not clearly reported, but the number of drop outs was low. ITT analysis performed, defined as those who received one dose of the study drug |
| Incomplete outcome data (attrition bias) Safety outcomes | High risk | Safety outcomes reported for the combined RCT and open‐label phase; not for RCT phase separately |
| Selective reporting (reporting bias) | Unclear risk | Harms data for RCT phase not reported separately |
| Method of adverse event monitoring Safety outcomes | Low risk | "Safety and tolerability were assessed by the incidence of treatment emergent adverse events" |
| 'Serious adverse event' definitions provided? Safety outcomes | Low risk | All adverse events were coded using the MedDRA dictionary of terms (version 9.0) |
Lambert 2007.
| Methods | Randomized, multicenter, double‐blind, placebo‐controlled study | |
| Participants | N = 38 adalimumab, N = 44 placebo Age (mean (SD), years): Treatment group ‐ 41.9(11.1); Control group ‐ 40 (10.9) % male: Treatment group ‐ 76.3%; Control group ‐ 81.8% % white: Treatment group ‐ not reported Disease duration (years (SD)): Treatment group ‐ 14.5 (9); Control group ‐12.1 (8.7) Inclusion:Patients were adults (18 years of age) diagnosed as having ankylosing spondylitis as defined by the modified New York criteria, who had been treated unsuccessfully (nonresponse or lack of tolerance) with 1 non‐steroidal anti‐inflammatory drug (NSAIDs). Patients who had failed to respond to 1 disease‐modifying anti‐rheumatic drug (DMARD) (e.g., methotrexate,sulphasalazine) were also allowed to enrol. Active ankylosing spondylitis at baseline was defined by fulfilment of 2 of the following 3 criteria: a BASDAI score greater than or equal to 4, total back pain visual analog scale score greater than or equal to 40, or morning stiffness of 1 hour in duration. Patients could continue taking sulphasalazine (3 g/day), methotrexate (25 mg/week), hydroxychloroquine (400 mg/day), prednisone and/or prednisone equivalents (10 mg/day), and/or NSAIDs as long as these doses had remained stable for 4 weeks before baseline |
|
| Interventions | 40 mg adalimumab or placebo every other week for 24 weeks (double‐blind phase) but an early escape option to non‐responders was available after week 12. Study visits occurred at baseline, week 2, week 4, every 4 weeks through week 24 | |
| Outcomes | Primary endpoint was ASAS20 at 12 weeks but results were not provided in full‐text Lambert article, but were reported in a conference abstract (Maksymowych 2005) Secondary outcomes of MRI of spine and SI joints scored using SPARCC methodology at week 12 were reported in Lambert2007. Another publication from this trial, Maksymowych 2008, which was focused on biomarkers for structural damage, also reported BASDAI, total back pain, patient global, BASFI, BASMI |
|
| Notes |
NCT00195819; M03‐606 study group "ROLE OF THE STUDY SPONSOR An advisory committee, including authors from academic institutions and Abbott Laboratories, and members of the Abbott Laboratories clinical trial team designed the study, which was conducted at 11 centers in Canada. Clinical data were collected and analyzed by Abbott Laboratories. Data analyses were reviewed by members of the advisory committee. All authors reviewed and assisted in the manuscript preparation during its development, agreed to submit the manuscript, and approved the content of the submitted manuscript" |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported |
| Allocation concealment (selection bias) | Unclear risk | Not reported |
| Blinding of participants and personnel (performance bias) Patient‐assessed outcomes | Unclear risk | Trial protocol states "Double Blind (Subject, Investigator)" but no details provided |
| Blinding of participants and personnel (performance bias) Physician‐assessed outcomes | Low risk | "Readers were qualified, trained radiologists who were blinded to the patients' identities, treatments, and imaging time points" |
| Blinding of outcome assessment (detection bias) Patient‐assessed outcomes | Unclear risk | Trial protocol states "Double Blind (Subject, Investigator)" but no details provided |
| Blinding of outcome assessment (detection bias) Physician‐assessed outcomes | Low risk | "Readers were qualified, trained radiologists who were blinded to the patients' identities, treatments, and imaging time points" |
| Incomplete outcome data (attrition bias) Efficacy outcomes | Low risk | "At baseline and week 12, all 44 patients in the placebo group and 38 in the adalimumab group had evaluable MRIs" |
| Incomplete outcome data (attrition bias) Safety outcomes | Unclear risk | Safety data not reported in primary publication |
| Selective reporting (reporting bias) | Unclear risk | Primary outcome and adverse events reported only in abstract. |
| Method of adverse event monitoring Safety outcomes | Unclear risk | Not reported |
| 'Serious adverse event' definitions provided? Safety outcomes | Unclear risk | Not reported |
Marzo‐Ortega 2005.
| Methods | 30 week, single‐center, randomized, double‐blind placebo controlled trial | |
| Participants | N = 28 infliximab + methotrexate (MTX), N = 14 placebo + MTX Age (mean (range), years): Treatment group ‐ 41 (28–74); Control group ‐ 39 (30–56) % male: Treatment group ‐ 82%; Control group ‐ 79% % white: ‐ not reported Disease duration (median years (range)): Treatment group ‐ 8 (0‐41) ; Control group ‐ 10 (0‐35) Inclusion: fulfil the modified New York criteria for ankylosing spondylitis,14 be older than 18 years of age, and have active spinal disease. This was defined as persistent inflammatory back pain (defined as 3 cm or more on a 10 cm visual analogue scale (VAS)) and a raised inflammatory response in serum as shown by a C reactive protein (CRP) value of more than 10 mg/L despite treatment with conventional agents such as an optimal dose of non‐steroidal anti‐inflammatory drugs (NSAIDs) or disease‐modifying anti‐rheumatic drugs (DMARDs) Exclusion: any history of tuberculosis, active infection, demyelinating disease, previous lymphoproliferative or malignant disorder, pregnancy, breast feeding, or uncontrolled concomitant disease in the opinion of the investigator. |
|
| Interventions | Infusions of infliximab (5 mg/kg in 250 ml 0.9% NaCl) + MTX or placebo + MTX. The infusion regimen was weeks 0, 2, 6, 14, and 22. All subjects also received a dose of 7.5 mg with folic acid cover (5 mg twice a week), which was eventually increased to 10 mg a week | |
| Outcomes | The primary outcome was evaluation of change in the BASDAI score at weeks 4, 10, and 30. Secondary outcomes were ASAS 20 and BASDAI 50% response. Magnetic resonance imaging (MRI) also assessed | |
| Notes | This study was supported by a grant in aid from Schering‐Plough, UK | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "A 2:1 randomisation list was generated by a statistician (who was unconnected with the final analysis of results)" |
| Allocation concealment (selection bias) | Low risk | "Study participants, clinical observers, and metrologists were unaware of the randomisation code, which was kept in the hospital pharmacy" |
| Blinding of participants and personnel (performance bias) Patient‐assessed outcomes | Unclear risk | "Double‐blind" |
| Blinding of participants and personnel (performance bias) Physician‐assessed outcomes | Unclear risk | "Double‐blind" |
| Blinding of outcome assessment (detection bias) Patient‐assessed outcomes | Unclear risk | "Double‐blind" |
| Blinding of outcome assessment (detection bias) Physician‐assessed outcomes | Unclear risk | "Double‐blind" |
| Incomplete outcome data (attrition bias) Efficacy outcomes | Unclear risk | 93% completed treatment, 64% completed in control group; an intention‐to‐treat analysis performed with last observation carried forward imputation for missing data |
| Incomplete outcome data (attrition bias) Safety outcomes | Unclear risk | 93% completed treatment, 64% completed in control group; an intention‐to‐treat analysis performed with last observation carried forward imputation for missing data |
| Selective reporting (reporting bias) | Low risk | Appropriate outcomes measured |
| Method of adverse event monitoring Safety outcomes | Unclear risk | Not reported |
| 'Serious adverse event' definitions provided? Safety outcomes | Unclear risk | Not reported |
Navarro‐Sarabia 2011.
| Methods | 12‐week double‐blind, placebo‐controlled randomized pilot study to evaluate the effect of etanercept 100 vs 50 mg/week to treat ankylosing spondylitis | |
| Participants | N = 54 etanercept 100 mg/week; N = 54 etancercept 50 mg/week Age (mean (SD), years): Treatment group ‐ 40.22 (10.36); Control group ‐ 42.63 (10.66) % male: Treatment group ‐ 79.6%; Control group ‐ 79.6% % white: ‐ not reported Disease duration (years since diagnosis, mean years (standard deviation (SD)): Treatment group ‐ 7.03 (6.83) ; Control group ‐ 7.28 (7.06) Inclusion criteria: Adult outpatients with ankylosing spondylitis diagnosis as defined by the modified New York criteria for ankylosing spondylitis and with inflammatory activity maintained for >12 weeks, who had failed treatment with at least two non‐steroidal anti‐inflammatory drugs (NSAIDs) at maximum recommended doses during at least 3 months Exclusion criteria: Complete ankylosis, contraindications for anti‐TNF alpha treatment, treatment with more than 10mg/day of disease‐modifying anti‐rheumatic drugs (DMARDs) or prednisone, NSAID use within 2 weeks of baseline, previous TNF inhibitors or biologics use, abnormal haematological profiles, psychiatric disease, history of alcohol or drug abuse |
|
| Interventions | Etanercept 50 mg twice a week or 50 mg once a week plus a second injection of placebo | |
| Outcomes | The primary efficacy endpoint was the proportion of subjects who achieved ASAS20 response at Week 12. Patients’ global assessment of disease activity and pain were measured using a visual analogue scale (VAS), physical function was assessed using the BASFI score and inflammation was measured using the score of the morning‐stiffness items of the BASDAI. Secondary endpoints were the proportion of subjects who achieved ASAS40, ASAS50, ASAS70, ASAS5/6 response and partial remission at Week 12, nocturnal and overall spine pain, physician global assessment of disease activity, activity index (BASDAI), spinal mobility (BASMI) score , complete peripheral joint count (ACR64/66 index), tenderness of enthesis [Maastricht Ankylosing Spondylitis Enthesis Score (MASES) index], C‐reactive protein and erythrocyte sedimentation rate. Quality of life was assessed by the European Quality of Life Scale. (EuroQoL) and 36‐item Short‐Form Health Survey (SF‐36) questionnaires |
|
| Notes | NCT00873730; LOADET. This work was supported by Pfizer S.A. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Patients were sequentially numbered at the screening visit |
| Allocation concealment (selection bias) | Unclear risk | "Upon completion of the baseline evaluation, eligible subjects were randomly allocated to a treatment group." Method of concealment of allocation not reported |
| Blinding of participants and personnel (performance bias) Patient‐assessed outcomes | Low risk | All study personnel and participants, including statisticians, were blinded to treatment assignment for the whole duration of the study |
| Blinding of participants and personnel (performance bias) Physician‐assessed outcomes | Low risk | All study personnel and participants, including statisticians, were blinded to treatment assignment for the whole duration of the study |
| Blinding of outcome assessment (detection bias) Patient‐assessed outcomes | Low risk | All study personnel and participants, including statisticians, were blinded to treatment assignment for the whole duration of the study |
| Blinding of outcome assessment (detection bias) Physician‐assessed outcomes | Low risk | All study personnel and participants, including statisticians, were blinded to treatment assignment for the whole duration of the study |
| Incomplete outcome data (attrition bias) Efficacy outcomes | Unclear risk | The intent‐to‐treat (ITT) population was defined as 'All randomized patients who had received at least one treatment dose and who had undergone at least one therapy evaluation'. This resulted in 48/54 of etanercept 100 mg/week and 49/54 etanercept 50 mg/week. Unclear what effect the 10% missing may have made to the estimate |
| Incomplete outcome data (attrition bias) Safety outcomes | Low risk | Analysis done on the number randomized |
| Selective reporting (reporting bias) | Low risk | ASAS‐recommended outcomes reported |
| Method of adverse event monitoring Safety outcomes | Low risk | Safety was assessed by the evaluation of the percentage and type of adverse events and serious adverse events, vital signs, physical examination, early withdrawals and laboratory results. Safety was assessed during all the study, until 15 days after the last study visit |
| 'Serious adverse event' definitions provided? Safety outcomes | Unclear risk | Not reported |
van der Heijde 2005.
| Methods | Multicenter, randomized, placebo‐controlled study for 24 weeks | |
| Participants | N = 201 infliximab N = 78 placebo
Age (mean, years): Treatment group ‐ 40; Control group ‐ 41
% male: Treatment group ‐ 78%; Control group ‐ 87%
% white: Treatment group ‐ 98%; Control group ‐ 97% Disease duration (years): Treatment group ‐ 8; Control group ‐13 Patient fulfilling the modified New York criteria for at least 3 months prior to screening, with BASDAI >= 4 (range 0–10) and spinal pain assessment score >= 4 on a visual analogue scale (0–10 cm) were eligible for the study. Patients were also required to have a normal chest radiograph within 3 months prior to randomisation and either a negative tuberculosis (TB) test. Exclusion: total ankylosis of the spine (defined by syndesmophytes present on the lateral views of spinal radiographs at all intervertebral levels from T6 through S1), any other inflammatory rheumatic disease, fibromyalgia, a serious infection within 2 months prior to randomisation, TB (active or latent) or recent contact with a person with active TB, an opportunistic infection within 6 months of screening, hepatitis, human immunodeficiency virus, a transplanted organ, malignancy, multiple sclerosis, or congestive heart failure. Patients were allowed to receive concurrent stable doses of non‐steroidal anti‐inflammatory drugs, acetaminophen (paracetamol), or tramadol during the study. Patients were not permitted to receive sulphasalazine or methotrexate within 2 weeks prior to screening, systemic corticosteroids within 1 month prior to screening, anti‐TNF therapy other than infliximab within 3 months prior to screening, infliximab at any time prior to screening, Disease‐modifying anti‐rheumatic drugs other than sulphasalazine or methotrexate within 6 months prior to screening, or cytotoxic drugs within 12 months prior to screening |
|
| Interventions | 5 mg/kg infliximab at weeks 0, 2, 6, 12, and 18 vs placebo | |
| Outcomes | Primary end point: ASAS20 responders at week 24. Other: BASDAI, night pain, patient’s global assessment, BASF, BASMI, chest expansion, the Mander enthesis index, the total swollen joint index, the C‐reactive protein level, SF‐36, adverse events Magnetic resonance imaging (MRI) reported in another publication (Braun 2006): MRI Activity Score based on the amount of bone marrow edema or erosions,as follows: 0 no erosions or bone marrow edema, 1 minor bone marrow edema involving 25% of the vertebral unit, 2 moderate bone marrow edema involving 20% but 50% of the vertebral unit, 3 major bone marrow edema involving 50% of the vertebral unit, 4 bone marrow edema and minor erosion involving 25% of the vertebral unit, 5 bone marrow edema and moderate erosion involving 20% but 50% of the vertebral unit, and 6 bone marrow edema and major erosion involving 50% of the vertebral unit. Thus, the MRI Activity Score for each vertebral unit ranged from 0 to 6. With 23 vertebral units assessed (from C2 to S1), the total MRI Activity Score for the spine ranged from 0 to 138 |
|
| Notes | NCT00207701 'Ankylosing Spondylitis Study for the Evaluation of Recombinant Infliximab Therapy (ASSERT)' trial. "Supported by Centocor Inc" | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomly assigned in 3:8 ratio" |
| Allocation concealment (selection bias) | Unclear risk | Unclear "Patients were allocated to treatment groups using an adaptive treatment allocation stratified by investigational site and C‐reactive protein level..." |
| Blinding of participants and personnel (performance bias) Patient‐assessed outcomes | Low risk | "Double blind"; "both infliximab and placebo were supplied as sterile, white, lyophilized powders in single‐use 20‐ml vials" |
| Blinding of participants and personnel (performance bias) Physician‐assessed outcomes | Low risk | "Double blind"; probably yes given the blinding of the study drug |
| Blinding of outcome assessment (detection bias) Patient‐assessed outcomes | Low risk | "Double blind"; "both infliximab and placebo were supplied as sterile, white, lyophilized powders in single‐use 20‐ml vials" |
| Blinding of outcome assessment (detection bias) Physician‐assessed outcomes | Low risk | "Double blind"; probably yes given the blinding of the study drug |
| Incomplete outcome data (attrition bias) Efficacy outcomes | Low risk | Fairly complete follow up; 75/78 completed in placebo and 199/201 completed in infliximab group. Intention‐to‐treat analysis |
| Incomplete outcome data (attrition bias) Safety outcomes | Low risk | Fairly complete follow up; 75/78 completed in placebo and 199/201 completed in infliximab group. Intention‐to‐treat analysis |
| Selective reporting (reporting bias) | Low risk | ASAS‐recommended outcomes reported |
| Method of adverse event monitoring Safety outcomes | Low risk | "Safety assessments included adverse events, infections, infusion reactions, premature discontinuations, and lab tests" |
| 'Serious adverse event' definitions provided? Safety outcomes | Unclear risk | No general serious adverse event (SAE) definition provided; but all SAEs that occurred were explained |
van der Heijde 2006a.
| Methods | Multicenter, randomized (2:1 ratio) placebo‐controlled study | |
| Participants | N = 208 adalimumab; N = 107 placebo
Age (mean, years): Treatment group ‐ 42; Control group ‐ 43
% male: Treatment group ‐ 76%; Control group ‐ 74%
% white: Treatment group ‐ 97%; Control group ‐ 93% Disease duration (years): Treatment group ‐ 11; Control group ‐10 Patients >= 18 years of age and classified as having definite ankylosing spondylitis based on the modified New York criteria. All had active disease, defined as fulfilment of at least 2 of the following 3 criteria: BASDAI >= 4, a total back pain score >4 (visual analogue scale 0–10 cm), or a duration of morning stiffness > 1 hour Patients with stable and well‐controlled psoriasis, uveitis, inflammatory bowel disease (i.e., ulcerative colitis,Crohn’s disease), and reactive arthritis were allowed to participate. Inadequate response or intolerance to 1 or more non‐steroidal anti‐inflammatory (NSAIDs) was defined by the investigators. Patients in whom 1 or more disease‐modifying anti‐rheumatic drugs (DMARDs) had failed were also allowed to participate. Patients were allowed to continue any of the following medications if the dose had remained stable for at least 4 weeks before the baseline visit: sulphasalazine (<=3gm/day), methotrexate (<= 25 mg/week), hydroxychloroquine(<= 400 mg/day), prednisone or prednisone equivalent (<=10mg/day), and NSAIDs Exclusions: "previously received anti‐TNF therapy, cyclosporine, azathioprine, or DMARDs (other than the medications and doses listed above) at any time and patients who had received intraarticular injection(s) with corticosteroids within 4 weeks prior to baseline. Patients with latent tuberculosis (TB) were allowed to participate in the study if a documented history of treatment was available or if treatment for latent TB was initiated before the first dose of study medication. Patients with clinically active TB were excluded from the study. History of any recent infections requiring antibiotic treatment; hepatitis or human immunodeficiency virus; a significant history of cardiac, renal, neurologic, psychiatric, endocrinologic, metabolic, or hepatic disease; and a history of demyelinating disease or multiple sclerosis. History of cancer or lymphoproliferative disease other than a successfully treated nonmetastatic squamous cell or basal cell carcinoma and/or localized carcinoma in situ of the cervix" |
|
| Interventions | Adalimumab 40 mg every other week or placebo for a 24‐week period. Patients who did not achieve a 20% response according to the ASsessment in Ankylosing Spondylitis International Working Group criteria for improvement (ASAS20) at weeks 12, 16, or 20 were eligible for “early‐escape” open label treatment with adalimumab 40 mg every other week | |
| Outcomes | Primary efficacy outcome: percentage of ASAS20 responders at week 12. Secondary: ASAS5/6, ASAS40 | |
| Notes | NCT00085644. 'Adalimumab Trial Evaluating Long‐term Efficacy and Safety for Ankylosing Spondylitis (ATLAS)'. "Supported by Abbott Laboratories" | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | From study author: "A random number generator was used to generate the randomisation numbers. All patients were centrally randomized using an Interactive Voice Response System (IVRS). Randomization occurred within each site" |
| Allocation concealment (selection bias) | Low risk | From study author: "The patient, sponsor and the study sites were blinded to treatment allocation." "The treatment allocation for each patient was provided to the site in a sealed envelope, to be opened in the case of an emergency in which the investigator believed that knowledge of study drug treatment was required. However, no patient was unblinded during the course of the double blind period" |
| Blinding of participants and personnel (performance bias) Patient‐assessed outcomes | Low risk | From study author: "The patient, sponsor and the study sites were blinded to treatment allocation." "The treatment allocation for each patient was provided to the site in a sealed envelope, to be opened in the case of an emergency in which the investigator believed that knowledge of study drug treatment was required. However, no patient was unblinded during the course of the double blind period" |
| Blinding of participants and personnel (performance bias) Physician‐assessed outcomes | Low risk | From study author: "The patient, sponsor and the study sites were blinded to treatment allocation." "The treatment allocation for each patient was provided to the site in a sealed envelope, to be opened in the case of an emergency in which the investigator believed that knowledge of study drug treatment was required. However, no patient was unblinded during the course of the double blind period" |
| Blinding of outcome assessment (detection bias) Patient‐assessed outcomes | Low risk | From study author: "Yes, patients and assessors were blinded. In particular, the assessor who performed the tender and swollen joint counts, MASES, and the physical examination was blinded to information from the patient reported questionnaires at all visits" |
| Blinding of outcome assessment (detection bias) Physician‐assessed outcomes | Low risk | From study author: "Yes, patients and assessors were blinded. In particular, the assessor who performed the tender and swollen joint counts, MASES, and the physical examination was blinded to information from the patient reported questionnaires at all visits" |
| Incomplete outcome data (attrition bias) Efficacy outcomes | Low risk | 95% of participants completed 24 weeks. Intention‐to‐treat analysis performed using non‐responder imputation |
| Incomplete outcome data (attrition bias) Safety outcomes | Low risk | 95% of participants completed 24 weeks |
| Selective reporting (reporting bias) | Low risk | Outcomes reported as prespecified in trial protocol (ASAS20 at week 12). Appropriate outcomes were reported |
| Method of adverse event monitoring Safety outcomes | Low risk | "Adverse events and other safety assessments were completed throughout the study" |
| 'Serious adverse event' definitions provided? Safety outcomes | Unclear risk | No general serious adverse event (SAE) definition provided; but each SAE that occurred was detailed in the report |
van der Heijde 2006b.
| Methods | 12‐week, randomized, placebo‐controlled, double‐blind, multicenter study with three treatment groups in a 3:3:1 ratio (etanercept 50 mg once weekly: etanercept 25 mg twice weekly: placebo) Study carried out in 38 centers in 11 European countries |
|
| Participants | Etanercept 50mg once weekly:N=155; mean age (SD)=41.5 (11.0); 69.7% male; disease duration, years (SD) = 9.0 (8.7) Etanercept 25mg twice weekly:N=150; mean age (SD)=39.8 (10.7); 76% male; disease duration, years (SD) = 10.0 (9.1) Placebo: N = 51; mean age (SD) = 40.1 (10.9); 78.4% male; disease duration, years (SD) = 8.5 (6.8) Inclusion ‐ age 18 to 70 years with active AS based on the Modified New York Criteria for ankylosing spondylitis. Active ankylosing spondylitis defined by visual analogue scale (VAS) >= 30 for duration and intensity of morning stiffness and two or more of the following:patient global assessment of disease activity VAS >=30; mean of nocturnal and total pain VAS scores >=30 "Concomitant oral non‐steroidal anti‐inflammatory drugs and oral corticosteroids ((10 mg/day), if stable for >2 weeks before randomisation, and disease‐modifying antirheumatic drugs (hydroxychloroquine, sulphasalazine and methotrexate), if stable for >4 weeks before randomisation, were permitted" Exclusion: "Patients previously treated with TNFa inhibitors, including etanercept or other biological agents, or disease‐modifying antirheumatic drugs (other than hydrochloroquine, sulphasalazine and methotrexate) less than 4 weeks before baseline, were not eligible. Other important exclusion criteria included complete ankylosis (fusion) of the spine based on radiographic assessment and concurrent medical events, such as uncontrolled hypertension, unstable angina pectoris, congestive heart failure, severe pulmonary disease, cancer, demyelinating diseases of the central nervous system and serious infections" |
|
| Interventions | Etanercept 50 mg once weekly versus etanercept 25 mg twice weekly versus placebo | |
| Outcomes | Non‐inferiority design to compare etanercept 50 mg once weekly to 25 mg twice weekly. Primary outcome: ASAS20 at week 12. Secondary outcomes: Secondary outcomes: ASAS40 and ASAS5/6 criteria at all time points | |
| Notes | NCT00418548. "Study was supported by Wyeth Pharmaceuticals, Collegeville, Pennsylvania, USA (study drug and grants to investigational sites) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not reported in article |
| Allocation concealment (selection bias) | Unclear risk | Not reported in article |
| Blinding of participants and personnel (performance bias) Patient‐assessed outcomes | Unclear risk | "Double‐blind", no further details reported |
| Blinding of participants and personnel (performance bias) Physician‐assessed outcomes | Unclear risk | "Double‐blind", no further details reported |
| Blinding of outcome assessment (detection bias) Patient‐assessed outcomes | Unclear risk | "Double‐blind", no further details reported |
| Blinding of outcome assessment (detection bias) Physician‐assessed outcomes | Unclear risk | "Double‐blind", no further details reported |
| Incomplete outcome data (attrition bias) Efficacy outcomes | Low risk | Reasons for withdrawal were provided. Follow‐up was greater than 80% in all groups. Modified intention‐to‐treat analyses were performed in which "all participants who received at least one dose of the test drugs" were included in the analyses (356/361 randomized). "A last‐observation‐carried‐forward approach was used to impute missing data in the modified intent‐to‐treat population analysis" |
| Incomplete outcome data (attrition bias) Safety outcomes | Low risk | Same as efficacy outcomes |
| Selective reporting (reporting bias) | Low risk | All appropriate outcomes were assessed |
| Method of adverse event monitoring Safety outcomes | Low risk | "Safety assessments were based on reports of adverse events, routine physical examinations and laboratory test results" |
| 'Serious adverse event' definitions provided? Safety outcomes | Unclear risk | Refers to "non‐infectious serious adverse events" but no definition provided |
AID: articular index according to Dougados AS: Ankylosing Spondylitis ASAS20 response: defined as 20% or greater improvement in at least three of five measures of disease activity, as recommended by the Assessment of SpondyloArthritis international Society (ASAS) (duration of morning stiffness, degree of nocturnal spinal pain, the BASFI, the patient’s global assessment of disease activity, and the score for joint swelling), one of which was required to be duration of morning stiffness or degree of nocturnal spinal pain, with no worsening in any of the measures. If the swollen‐joint score was zero throughout the study, improvement was required in at least two of the four other outcome measures, with the aforementioned restrictions. Twenty per cent improvement is defined as improvement of at least 20% and absolute improvement of at least 10 units (on a scale of 0–100) in three or more of the following domains: patient global assessment, pain, function (from the BASFI score), and inflammation (measured by the mean of the two morning stiffness‐related BASDAI and visual analogue scores). Furthermore, deterioration in the potentially remaining domain has to be absent, defined as a change for the worse of 20% or more, and net worsening of 10 units or more (on a scale of 0–100) ASAS40 response: at least a 40% improvement with a minimum of 20 units (0 to 100 scale) improvement compared with baseline in at least three of four domains (spinal pain, function (BASFI), inflammation as measured by the mean of intensity and duration of morning stiffness in the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI), and patient global assessment), and with no worsening in the fourth domain. ASAS50 response: at least a 50% with a minimum of 20 units (0 to 100 scale) improvement compared with baseline in at least three of four domains (spinal pain, function (BASFI), inflammation as measured by the mean of intensity and duration of morning stiffness in the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI), and patient global assessment), and with no worsening in the fourth domain. ASAS70 response: at least a 70% with a minimum of 20 units (0 to 100 scale) improvement compared with baseline in at least three of four domains (spinal pain, function (BASFI), inflammation as measured by the mean of intensity and duration of morning stiffness in the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI), and patient global assessment), and with no worsening in the fourth domain. ASAS Partial remission: a value of less than 2 on a 0 to 10 scale in each of the four domains as described above for the ASAS40. ASAS 5/6 response criteria require at least 20% improvement in 5 of 6 domains: spinal mobility (according to the Bath Ankylosing Spondylitis Metrology Index [BASMI];other instruments may be used) and acute‐phase reactants (the CRP concentration) in addition to the 4 domains included in the ASAS 20 response criteria. BASDAI: Bath Ankylosing Spondylitis Disease Activity Index BASDAI 50: 50% improvement of the initial BASDAI BASFI: Bath Ankylosing Spondylitis Functional Index BASMI: Bath Ankylosing Spondylitis Metrology index BASRI‐s: Bath Ankylosing Spondylitis Radiology Index for the spine CRP: C‐reactive protein level DFI: Dougados Functional Index DMARD: disease‐modifying anti‐rheumatic drug ESR: erythrocyte sedimentation rate NSAID: non‐steroidal anti‐inflammatory drug SF‐36: short form 36. A health‐related assessment of quality of life VAS: Visual analogue scale
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Barkham 2008b | Trial participant inclusion criteria does not meet review participant inclusion criteria (inflammatory back pain by Calin criteria < 3yrs) |
| Breban 2008 | Trial participants did not meet complete modified NY criteria for ankylosing spondylitis and the intervention was systematic versus on‐demand treatment using infliximab |
| Haibel 2008 | The population included in this study "axial spondylarthritis without radiographically defined sacroiliitis" does not meet the review's inclusion criteria. This study is in early ankylosing spondylitis patients |
| Li 2008 | The intervention of infliximab + methotrexate vs infliximab + placebo assesses effect of methotrexate and does not meet the review's intervention inclusion criteria |
| Morency 2011 | Open‐label extension of Lambert 2007 |
| Van den Bosch 2002 | Patients included ankylosing spondylitis, reactive arthritis, psoriatic arthritis, and undifferentiated spondyloarthritis. Results for ankylosing spondylitis patients only were not available |
Characteristics of studies awaiting assessment [ordered by study ID]
Zhang 2009.
| Methods | Randomized, placebo‐controlled trial |
| Participants | 86 Chinese patients with AS |
| Interventions | Etanercept or placebo for 6 weeks followed by 6 week open‐label extension |
| Outcomes | ASAS20 and other ASAS responses, changes in BASDAI, BASFI, morning stiffness, nocturnal spinal pain |
| Notes | Chinese paper; abstract in English. Same authors as Huang 2008 and same intervention so it might be a report from the same study, though only reports on 86 participants as opposed to 152 in Huang 2008. Awaiting translation and confirmation with authors |
Differences between protocol and review
For the primary analysis of the major outcomes in this review, we conducted Bayesian mixed treatment comparison meta‐analyses using WinBUGS software. We added golimumab to the included interventions. We selected seven major outcomes for inclusion in the 'Summary of findings' table after discussions with ankylosing spondylitis experts in the Assessment of SpondyloArthritis international Society and reported on only these seven. The original protocol outcomes are listed below.
Primary and secondary outcome measures will be based on the DC‐ART Ankylosing Spondylitis Working Group Core Set (van der Heijde 1997) and the International ASAS consensus statement for the use of TNF‐alpha inhibitors in patients with AS (Braun 2003). Secondary outcomes will be proportion of responders based on either the ASAS measures (ASAS20 (Anderson 2001), ASAS40 and/or ASAS 5 of 6 (Brandt 2004)), or on any alternative response criteria formulated by the authors. Additional outcomes including quality of life measures, other imaging outcomes and reduction of other medications will also be recorded. Finally, adverse events will be reported separately, distinguishing between withdrawal due to adverse events and withdrawal due to inefficacy of therapy.
Primary outcomes: Physical function Spinal Pain Spinal stiffness (duration of morning stiffness) Spinal mobility Patient global assessment Peripheral joint/enthesis inflammation Changes in spine radiographs Fatigue Acute phase reactants (erthyrocyte sedimentation rate (ESR), C‐reactive protein (CRP)) Disease activity (BASDAI)Secondary outcomes: ASAS20 response ASAS40 response ASAS 5 out of 6 responseOther: Changes in hip radiograph Physician global assessement Quality of life MRI evidence of suppression of inflammation Reduction of steroid or NSAID use
Adverse effects: Data will be collected on: a) Total withdrawals b) Withdrawals due to adverse effects c) Withdrawals due to inefficacy d) Any reported adverse effects such as infections, allergic reactions
We followed the latest methods as outlined in the Cochrane Handbook (Higgins 2011) and therefore used the GRADE criteria to assess the quality of the evidence rather than the grading system described in the 2004 book Evidence‐based Rheumatology (Tugwell 2004) as stated in the protocol.
Contributions of authors
LM: conceptualized the idea, performed screening and data extraction, entered data into Review Manager and Excel, performed analyses, and wrote the review.
JZ: conceptualized the idea, performed screening and data extraction, entered data into Review Manager, and wrote and commented on drafts of the review.
AB: conceptualized the idea, provided clinical expertise, and commented on drafts of the review.
JAS: performed screening and data extraction, and commented on drafts of the review.
MV: performed screening and data extraction, and commented on drafts of the review.
MJB: performed screening and data extraction, and commented on drafts of the review.
ETG: performed screening, risk of bias and GRADE assessments, and commented on drafts of the review.
GW: conceptualized the idea, provided statistical expertise, and commented on drafts of the review.
PT: conceptualized the idea, provided clinical expertise, and commented on drafts of the review.
Sources of support
Internal sources
Institute of Population Health, University of Ottawa, Canada.
Rheumazentrum Ruhrgebiet, Germany.
External sources
-
NIHR Cochrane Incentive Award 2013, UK.
Award to assist with completion of the review.
Declarations of interest
We received a National Institute for Health Research (NIHR) Cochrane Incentive Award to assist with the completion of this review.
LJM: was an associate member of the Assessment of SpondyloArthritis international Society from 2005 to 2014.
JZ: none known
AB: Grants: to department only: Abbvie, Pfizer, Merck, Amgen Travel support to department: Janssen‐Cilag; Honorarium: part to department, part personally: Pfizer, UCB, Janssen‐Cilag, Abbvie
JAS: received research grants from Takeda and Savient and consultant fees from Savient, Takeda, Regeneron and Allergan. JAS is a member of the executive of OMERACT, an organization that develops outcome measures in rheumatology and receives arms‐length funding from 36 companies; a member of the American College of Rheumatology's Guidelines Subcommittee of the Quality of Care Committee; and a member of the Veterans Affairs Rheumatology Field Advisory Committee.
ETG: none known.
MV: none known.
MBJ: none known.
PT: grants/honoraria from Bristol Myers, Chiltern International, and UCB.
GW: none known
New
References
References to studies included in this review
Bao 2014 {published data only}
- Bao C, Huang F, Khan MA, Fei K, Zhong W, Han C, et al. Safety and efficacy of golimumab in Chinese patients with active ankylosing spondylitis: 1‐year results of a multicentre, randomized, double‐blind, placebo‐controlled phase III trial. Rheumatology 2014;53:1654‐63. [DOI] [PubMed] [Google Scholar]
Barkham 2010 {published data only}
- Barkham N, Coates L, Keen H, Hensor E, Fraser A, Redmond A, et al. A double blind placebo controlled trial of etanercept in the prevention of work disability in ankylosing spondylitis. Rheumatology 2008;47:ii72. [DOI] [PubMed] [Google Scholar]
- Barkham N, Coates LC, Keen H, Hensor E, Fraser A, Redmond A, et al. Double‐blind placebo‐controlled trial of etanercept in the prevention of work disability in ankylosing spondylitis. Annals of the Rheumatic Diseases 2010;69(11):1926‐8. [DOI] [PubMed] [Google Scholar]
Brandt 2003 {published and unpublished data}
- Brandt J, Khariouzov A, Listing J, Haibel H, Sorensen H, Grassnickel L. Six‐month results of a double‐blind, placebo‐controlled trial of etanercept treatment in patients with active ankylosing spondylitis. Arthritis and Rheumatism 2003;48:1667‐75. [DOI] [PubMed] [Google Scholar]
Braun 2002 {published and unpublished data}
- Braun J, Brandt J, Listing J, Zink A, Alten R, Krause A, et al. Treatment of active ankylosing spondylitis with infliximab, a double‐blind placebo controlled multicenter trial. Lancet 2002;359:1187‐93. [DOI] [PubMed] [Google Scholar]
Braun 2011 {published data only}
- Braun J, Burgos‐Vargas R, Horst‐Bruinsma IE, Freundlich B, Vlahos B, Koenig AS, et al. Assessment of clinical efficacy in a randomized, double‐blind study of etanercept and sulphasalazine in patients with ankylosing spondylitis. Arthritis and Rheumatism. 2008; Vol. 58:S415. [DOI] [PubMed]
- Braun J, Pavelka K, Ramos‐Remus C, Dimic A, Vlahos B, Freundlich B, et al. Clinical efficacy of etanercept versus sulfasalazine in ankylosing spondylitis subjects with peripheral joint involvement. Journal of Rheumatology 2012;39(4):836‐40. [DOI] [PubMed] [Google Scholar]
- Braun J, Horst‐Bruinsma IE, Huang F, Burgos‐Vargas R, Vlahos B, Koenig AS, et al. Clinical efficacy and safety of etanercept versus sulfasalazine in patients with ankylosing spondylitis: A randomized, double‐blind trial. Arthritis and Rheumatism 2011;63(6):1543‐51. [DOI] [PubMed] [Google Scholar]
- Sieper J, Moots R, Taylor A, Sato R, Singh A, Roberson D, et al. Assessment of patient‐reported outcomes from a randomized, double‐blind study of etanercept and sulphasalazine in patients with ankylosing spondylitis. Arthritis and Rheumatism. 2008; Vol. 58:S580.
Calin 2004 {published and unpublished data}
- Calin A, Dijkmans B, Emery P, Hakala M, Kalden J, Leirsalo‐Repo M, et al. Outcomes of a multicentre randomised clinical trial of etanercept to treat ankylosing spondylitis. Annals of the Rheumatic Diseases 2004;63:1595‐1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Davis 2003 {published data only (unpublished sought but not used)}
- Davis J, Heijde D, Braun J, Dougados M, Cush J, Clegg D, et al. Recombinant Human Tumor Necrosis Factor Receptor (Etanercept) for Treating Ankylosing Spondylitis. Arthritis and Rheumatism 2003;48(11):3230‐6. [DOI] [PubMed] [Google Scholar]
- Davis J, Heijde, Braun J, Dougados M, Clegg DO, Kivits A, et al. Effiacy and safety of up to 192 weeks of etanercept therapy in patients with ankylosing spondylitis. Annals of the Rheumatic Diseases. 2008; Vol. 67:346‐52. [DOI] [PubMed]
- Davis JC, Heijde DM, Braun J, Dougados M, Cush J, Clegg D, et al. Sustained durability and tolerability of etanercept in ankylosing spondylitis for 96 weeks. Annals of the Rheumatic Diseases 2005;64:1557‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Dougados 2011 {published data only}
- Dougados M, Braun J, Szanto S, Combe B, Elbaz M, Geher P, et al. Efficacy of etanercept on rheumatic signs and pulmonary function tests in advanced ankylosing spondylitis: Results of a randomised double‐blind placebo‐controlled study (SPINE). Annals of the Rheumatic Diseases 2011;70:799‐804. [DOI: 10.1136/ard.2010.139261] [DOI] [PMC free article] [PubMed] [Google Scholar]
Giardina 2009 {published data only}
- Braun J, Baraliakos X, Hermann KG, Heijde D, Inman RD, Deodhar AA, et al. Golimumab reduces spinal inflammation in ankylosing spondylitis: MRI results of the randomised, placebo‐controlled GO‐RAISE study. Annals of the Rheumatic Diseases 2012;71(6):878‐84. [DOI: 10.1136/annrheumdis‐2011‐200308] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardina AR, Ferrante A, Ciccia F, Impastato R, Miceli M, Principato A, et al. A 2‐year comparative open label randomized study of efficacy and safety of etanercept and infliximab in patients with ankylosing spondylitis. Rheumatology International 2009 October 23 [Epub ahead of print]. [DOI: 10.1007/s00296-009-1157-3] [DOI] [PubMed]
- Impastato R, Ciccia F, Ferrante A, Principato A, Giardina A, Cadelo M, et al. Infliximab and etanercept are both effective and safe in patients with ankylosing spondylitis. A two‐year randomised study. Annals of the Rheumatic Diseases 2007;66(Suppl II):399. [Google Scholar]
Gorman 2002 {published data only}
- Gorman JD, Sack KE, Davis JC. Treatment of ankylosing spondylitis by inhibition of tumor necrosis factor alpha. New England Journal of Medicine 2002;346:1349‐56. [DOI] [PubMed] [Google Scholar]
Hu 2012 {published data only}
- Hu Z, Xu M, Qiuxia L, Zhiming L, Liao Z, Cao S, et al. Adalimumab significantly reduces inflammation and serumDKK‐1 level but increases fatty deposition in lumbar spine inactive ankylosing spondylitis. International Journal of Rheumatic Diseases 2012;15:358‐65. [DOI] [PubMed] [Google Scholar]
Huang 2008 {published data only}
- Huang F, MacPeek D, Shunle C, Li Z, Gu J, Wu D, et al. Etanercept is effective and well tolerated in Chinese patients with ankylosing spondylitis: 6‐week placebo‐controlled trial followed by 6‐week open‐label treatment. Annals of the Rheumatic Diseases. 2008; Vol. 67:Suppl II: 521.
- Huang F, Zhang J, Huang JL, Wu DH, Li ZG, Chen SL, et al. A multicenter, double‐blind, placebo‐controlled, randomized, phase III clinical study of etanercept in treatment of ankylosing spondylitis. Zhonghua Nei Ke Za Zhi (Chinese Journal of Internal Medicine) 2010;49:741–5. [PubMed] [Google Scholar]
Huang 2014 {published data only}
- Huang F, Gu J, Zhu P, Bao C, Xu, J, Xu H, et al. Efficacy and safety of adalimumab in Chinese adults with active ankylosing spondylitis: results of a randomised, controlled trial. Annals of the Rheumatic Diseases 2014;73:587‐94. [DOI: 10.1136/annrheumdis-2012-202533] [DOI] [PubMed] [Google Scholar]
Inman 2008 {published data only}
- Braun J, Baraliakos X, Hermann KG, Heijde D, Inman RD, Deodhar AA, et al. Golimumab reduces spinal inflammation in ankylosing spondylitis: MRI results of the randomised, placebo‐controlled GO‐RAISE study. Annals of the Rheumatic Diseases 2012;71(6):878‐84. [DOI: 10.1136/annrheumdis-2011-200308] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inman RD, Davis JC Jr, Heijde Dv, Diekman L, Sieper J, Kim SI, et al. Efficacy and safety of golimumab in patients with ankylosing spondylitis: results of a randomized, double‐blind, placebo‐controlled, phase III trial. Arthritis and Rheumatism 2008;58(11):3402‐12. [DOI] [PubMed] [Google Scholar]
Inman 2010 {published data only}
- Inman R, Maksymowych W, CANDLE group. A double‐blind, placebo‐controlled trial of low dose infliximab in ankylosing spondylitis. Journal of Rheumatology 2010;37(6):1‐8. [DOI: ] [DOI] [PubMed] [Google Scholar]
- Inman R, Shojania K, Keystone E, Haraoui B, Mitchell C, Maksymowych W. CANaDian evaluation of Low dosE infliximab in ankylosing spondylitis (CANDLE). Journal of Rheumatology 2008;35(6):1213‐14. [Google Scholar]
- Maksymowych W, Salonen D, Inman R, Paul FL, Lambert RG. CANaDian evaluation of Low dosE Infliximab in Ankylosing Spondylitis (CANDLE) ‐ 12 week magnetic resonance imaging evaluation of spinal inflammation with the SPARCC MRI method. Journal of Rheumatology 2008;35(6):1211‐12. [Google Scholar]
- Maksymowych WP, Salonen D, Inman RD, Rahman P, Lambert RGW. Low‐dose infliximab (3 mg/kg) significantly reduces spinal inflammation on magnetic resonance imaging in patients with ankylosing spondylitis: a randomized placebo‐controlled study. Journal of Rheumatology 2010;37(8):1728‐34. [DOI] [PubMed] [Google Scholar]
Lambert 2007 {published data only}
- Lambert RGW, Salonen D, Rahman P, Inman RD, Wong RL, Einstein SG, et al. Adalimumab significantly reduces both spinal and sacroiliac joint inflammation in patients with ankylosing spondylitis. Arthritis and Rheumatism 2007;56(12):4005‐14. [DOI] [PubMed] [Google Scholar]
- Maksymowych W, Rahman P, Keystone E, Wong R, Inman R, M03‐606 Study Group. Efficacy of adalimumab in active AS: Results from the Canadian AS study. Arthritis and Rheumatism. 2005:S217.
- Maksymowych W, Rahman P, Shojania K, Olszynski W, Thomson G, Ballal S, et al. Beneficial effects of adalimumab on biomarkers reflecting structural damage in patients with ankylosing spondylitis. Journal of Rheumatology 2008;35(10):2030‐7. [PubMed] [Google Scholar]
Marzo‐Ortega 2005 {published data only}
- Marzo‐Ortega H, McGonagle D, Jarrett S, Haugeberg G, Hensor E, O'Connor P, et al. Infliximab in combination with methotrexate in active ankylosing spondylitis: A clinical and imaging study. Annals of the Rheumatic Diseases 2005;64(11):1568‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Navarro‐Sarabia 2011 {published data only}
- Navarro‐Sarabia F, Fernandez‐Sueiro JL, Torre‐Alonso JC, Gratacos J, Queiro R, Gonzalez C, et al. High‐dose etanercept in ankylosing spondylitis: results of a 12‐week randomized, double blind, controlled multicentre study (LOADET study). Rheumatology 2011;50(10):1828‐37. [DOI] [PubMed] [Google Scholar]
van der Heijde 2005 {published data only}
- Braun J, Deodhar A, Dijkmans B, Geusens P, Sieper J, Williamson P, et al. Efficacy and safety of infliximab in patients with ankylosing spondylitis over a two‐year period. Arthritis and Rheumatism 2008;59:1270‐8. [DOI] [PubMed] [Google Scholar]
- Braun J, Landewe R, Hermann KGA, Han J, Yan S, Williamson P, et al. Major reduction in spinal inflammation in patients with ankylosing spondylitis after treatment with infliximab ‐ Results of a multicenter, randomized, double‐blind, placebo‐controlled magnetic resonance imaging study. Arthritis and Rheumatism 2006;54(5):1646‐52. [DOI] [PubMed] [Google Scholar]
- Heldmann F, Brandt J, Braun J, EASIC Study Group. European ankylosing spondylitis infliximab cohort (EASIC): safety of longterm therapy with infliximab in patients with ankylosing spondylitis. Annals of the Rheumatic Diseases. 2008; Vol. 67:Suppl II:386.
- Heldmann F, Brandt J, Horst‐Bruinsma, Landewe R, Sieper J, Burmester G, et al. European Ankylosing Spondylitis Infliximab Cohort (EASIC): Safety of longterm therapy in patients with ankylosing spondylitis. Annals of the Rheumatic Diseases. 2009; Vol. 68 (Suppl 3):626.
- Heijde D, Dijkmans B, Geusens P, Sieper J, DeWoody K, Williamson P, et al. Efficacy and Safety of Infliximab in Patients with Ankylosing Spondylitis. Results of a Randomized, Placebo‐Controlled Trial (ASSERT). Arthritis and Rheumatism 2005;52(2):582‐91. [DOI] [PubMed] [Google Scholar]
- Heijde D, Han C, Bala M, Williamson P, Han J, Braun J. Infliximab improves fatigue and pain in patients with ankylosing spondylitis: Results of a randomized, placebo‐controlled trial. Arthritis and Rheumatism. 2005; Vol. 52 (9):s35. [DOI] [PubMed]
- Heijde D, Han C, Bala M, Williamson P, Han J, Braun J. Infliximab improves fatigue and pain in patients with ankylosing spondylitis: Results of a randomized, placebo‐controlled trial (ASSERT). Annals of the Rheumatic Diseases. 2005; Vol. 64:318‐9.
van der Heijde 2006a {published and unpublished data}
- Davis Jr JC, Revicki D, Heijde DMF, Rentz AM, Wong RL, Kupper H, et al. Health‐related quality of life outcomes in patients with active ankylosing spondylitis treated with adalimumab: Results from a randomized controlled study. Arthritis Care and Research 2007; Vol. 57, issue 6:1050‐7. [DOI: 10.1002/art.22887] [DOI] [PubMed]
- Revicki DA, Luo MP, Wordsworth P, Wong RL, Chen N, Davis Jr J. Adalimumab reduces pain, fatigue, and stiffness in patients with ankylosing spondylitis: Results from the adalimumab trial evaluating long‐term safety and efficacy for ankylosing spondylitis (ATLAS). Journal of Rheumatology 2008;35(7):1346‐53. [PubMed] [Google Scholar]
- Heijde D, Dijkmans B, Schiff M, Kivitz A, Vlam K, Wordsworth P, et al. Maintenance of reduction in disease activity and partial remission in patients with ankylosing spondylitis (AS) ‐ 3 year results from the adalimumab (Humira) trial evaluating long‐term efficacy and safety in AS (ATLAS). Arthritis and Rheumatism. 2008:S579.
- Heijde D, Kivitz A, Schiff MH, Sieper J, Dijkmans BAC, Braun J, ATLAS study group. Efficacy and safety of adalimumab in patients with ankylosing spondylitis. Arthritis and Rheumatism 2006;54(7):2136‐46. [DOI] [PubMed] [Google Scholar]
- Heijde D, Schiff M, Sieper J, Kivitz A, Wong R, Kupper H, et al. Adalimumab effectiveness for the treatment of ankylosing spondylitis is maintained for up to 2 years: long‐term results from the ATLAS trial. Annals of the Rheumatic Diseases 2009;68:922‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
van der Heijde 2006b {published data only}
- Braun J, McHugh N, Singh A, Wajdula JS, Sato R. Improvement in patient‐reported outcomes for patients with ankylosing spondylitis treated with etanercept 50 mg once‐weekly and 25 mg twice‐weekly. Rheumatology 2007;46(6):999‐1004. [DOI] [PubMed] [Google Scholar]
- Heijde D, Silva JC, Dougados M, Geher P, Van DH‐B, Juanola X, et al. Etanercept 50 mg once weekly is as effective as 25 mg twice weekly in patients with ankylosing spondylitis. Annals of the Rheumatic Diseases 2006;65(12):1572‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Barkham 2008b {published data only}
- Barkham N, Keen HI, Coates P, O'Connor EMA, Hensor AD, Fraser LS, et al. A randomized controlled trial of infliximab shows clinical and MRI efficacy in HLA B27 positive very early ankylosing spondylitis. Annals of the Rheumatic Diseases. 2008; Vol. 67:Suppl II:382.
Breban 2008 {published data only}
- Breban M, Ravaud P, Claudepierre P, Baron G, Henry Y‐D, Hudry C, et al. Maintenance of infliximab treatment in ankylosing spondylitis: Results of a one‐year randomized controlled trial comparing systematic versus on‐demand treatment. Arthritis and Rheumatism 2008;58(1):88‐97. [DOI: 10.1002/art.23167] [DOI] [PubMed] [Google Scholar]
Haibel 2008 {published data only}
- Haibel H, Rudwaleit M, Listing J, Heldmann F, Wong RL, Kupper H, et al. Efficacy of adalimumab in the treatment of axial spondylarthritis without radiographically defined sacroiliitis: Results of a twelve‐week randomized, double‐blind, placebo‐controlled trial followed by an open‐label extension up to week fifty‐two. Arthritis and Rheumatism 2008; Vol. 58, issue 7:1981‐91. [DOI: 10.1002/art.23606] [DOI] [PubMed]
Li 2008 {published data only}
- Li EK, Griffth JF, Lee VW, Wang YX, Li TK, Lee KK, et al. Short‐term effiacy of combination methotrexate and infliximab in patients with ankylosing spondylitis: a clinical and magnetic resonance imaging correlation. Rheumatology 2008;47(9):1358‐63. [DOI] [PubMed] [Google Scholar]
Morency 2011 {published data only}
- Morency N, Lambert R, Maksymowych W. Direct imaging evidence that adalimumab induces resolution of inflammatory lesions in AS patients at sites of complete spinal ankylosis. Journal of Rheumatology 2011;38(6):1138. [Google Scholar]
Van den Bosch 2002 {published data only}
- Kruithof E, Bosch F, Baeten D, Herssens A, Keyser F, Mielants H, et al. Repeated infusions of infliximab, a chimeric anti‐TNF alpha monoclonal antibody, in patients with active spondyloarthropathy: one year follow up. Annals of Rheumatic Diseases 2002;61:207‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch F, Kruithof E, Baeten D, Herssens A, Keyser F, Mielants H, et al. Randomized double‐blind comparison of chimeric monoclonal antibody to tumor necrosis factor alpha (infliximab) versus placebo in active spondyloarthropathy. Arthritis and Rheumatism 2002;46(3):755‐65. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
Zhang 2009 {published data only}
- Zhang J, Zhang YM, Zhang J‐L, Deng X‐H, Huang F. Efficacy of etanercept in patients with ankylosing spondylitis: A double‐blind, randomized, placebo controlled trial. [Chinese]. Chinese Journal of New Drugs 2009;18(19):1846. [Google Scholar]
Additional references
Ades 2008
- Ades AE, Welton NJ, Caldwell D, Price M, Goubar A, Lu G. Multiparameter evidence synthesis in epidemiology and medical decision‐making. Journal of Health Services Research Policy 2008;13 Suppl 3:12‐22. [DOI] [PubMed] [Google Scholar]
Anderson 2001
- Anderson JJ, Baron G, Heijde D, Felson DT, Dougados M. Ankylosing Spondylitis Assessment group preliminary definition of short‐term improvement in ankylosing spondylitis. Arthritis and Rheumatism 2001;44(8):1876‐86. [DOI] [PubMed] [Google Scholar]
Boers 2006
- Boers M. Abatacept in rheumatoid arthritis: a new branch on the biologics tree (editorial). Annals of the Rheumatic Diseases 2006;144:933‐5. [DOI] [PubMed] [Google Scholar]
Boonen 2001a
- Boonen A, Chorus A, Miedema H, Heidje D, Temple H, Linden S. Employment, work disability, and work days lost in patients with ankylosing spondylitis: a cross sectional study of Dutch patients. Annals of Rheumatic Diseases 2001;60:353‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Boonen 2001b
- Boonen A, Chorus A, Miedema H, Heijde D, Landewé R, Schouten H, et al. Withdrawal from labour force due to work disability in patients with ankylosing spondylitis. Annals of Rheumatic Diseases 2001;60:1033‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bradburn 2007
- Bradburn MJ, Deeks JJ, Berlin JA, Russell Localio A. Much ado about nothing: a comparison of the performance of meta‐analytical methods with rare events. Statistics in Medicine 2007;26(1):53‐77. [DOI] [PubMed] [Google Scholar]
Brandt 2000
- Brandt J, Haibel H, Cornely D, Golder W, Gonzalez J, Reddig J, et al. Successful treatment of active ankylosing spondylitis with the anti‐tumor necrosis factor alpha monoclonal antibody infliximab. Arthritis and Rheumatism 2000;43(6):1346‐52. [DOI] [PubMed] [Google Scholar]
Brandt 2004
- Brandt J, Listing J, Sieper J, Rudwaleit M, Heijde D, Braun J. Development and preselection of criteria for short term improvement after anti‐TNF alpha treatment in ankylosing spondylitis. Annals of the Rheumatic Diseases 2004;63(11):1438‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Braun 1995
- Braun J, Bollow M, Neure L, Seipelt E, Seyrekbasan F, Herbst H, et al. Use of immunohistologic and in situ hybridization techniques in the examination of sacroiliac joint biopsy specimens from patients with ankylosing spondylitis. Arthritis and Rheumatism 1995;38(4):499‐505. [DOI] [PubMed] [Google Scholar]
Braun 2003
- Braun J, Pham T, Sieper J, Davis J, Linden S, Dougados M, et al. International ASAS consensus statement for the use of anti‐tumor necrosis factor agents in patients with ankylosing spondylitis. Annals of Rheumatic Diseases 2003;62:817‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Braun 2006
- Braun J, Landewe R, Hermann KGA, Han J, Yan S, Williamson P, et al. Major reduction in spinal inflammation in patients with ankylosing spondylitis after treatment with infliximab ‐ Results of a multicenter, randomized, double‐blind, placebo‐controlled magnetic resonance imaging study. Arthritis and Rheumatism 2006;54(5):1646‐52. [DOI] [PubMed] [Google Scholar]
Braun 2012a
- Braun J, Pavelka K, Ramos‐Remus C, Dimic A, Vlahos B, Freundlich B, et al. Clinical efficacy of etanercept versus sulfasalazine in ankylosing spondylitis subjects with peripheral joint involvement. Journal of Rheumatology 2012;39(4):836‐40. [DOI] [PubMed] [Google Scholar]
Braun 2012b
- Braun J, Baraliakos X, Hermann K‐G A, Heijde D, Inman RD, Deodhar AA, et al. Golimumab reduces spinal inflammation in ankylosing spondylitis: MRI results of the randomised, placebo‐ controlled GO‐RAISE study. Annals of the Rheumatic Diseases 2012;71:878‐84. [DOI: 10.1136/annrheumdis-2011-200308] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bucher 1997
- Bucher HC, Guyatt GH, Griffith LE, Walter SD. The results of direct and indirect treatment comparisons in meta‐analysis of randomized controlled trials. Journal of Clinical Epidemiology 1997;50(6):683‐91. [DOI] [PubMed] [Google Scholar]
Calin 1994
- Calin A, Garrett S, Whitelock H, Kennedy LG, O'Hea J, Mallorie P, et al. A new approach to defining functional ability in ankylosing spondylitis: the development of the Bath Ankylosing Spondylitis Functional Index. Journal of Rheumatology 1994;21(12):2281‐85. [PubMed] [Google Scholar]
Callhoff 2014
Cates 2008 [Computer program]
- Dr. Chris Cates. Visual Rx3. Dr Chris Cates' EBM website www.nntonline.com. Dr. Chris Cates, 2008.
Chen 2014
- Chen J, Lin S, Liu C. Sulfasalazine for ankylosing spondylitis. Cochrane Database of Systematic Reviews 2014, Issue 11. [DOI: 10.1002/14651858.CD004800] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cooper 2009
- Cooper NJ, Sutton AJ, Morris D, Ades AE, Welton NJ. Addressing between‐study heterogeneity and inconsistency in mixed treatment comparisons: application to stroke prevention treatments in individuals with non‐rheumatic atrial fibrillation. Statistics in Medicine 2009;28(14):1861‐81. [DOI] [PubMed] [Google Scholar]
Dagfinrud 2008
- Dagfinrud H, Kvien TK, Hagen KB. Physiotherapy interventions for ankylosing spondylitis. Cochrane Database of Systematic Reviews 2008, Issue 1. [DOI: 10.1002/14651858.CD002822] [DOI] [PMC free article] [PubMed] [Google Scholar]
Dias 2010
- Dias S, Welton NJ, Caldwell DM, Ades AE. Checking consistency in mixed treatment comparison meta‐analysis. Statistics in Medicine 2010;29:932‐44. [DOI: 10.1002/sim.3767] [DOI] [PubMed] [Google Scholar]
Dias 2011a
- Dias S, Welton NJ, Sutton AJ, Ades AE, National Institute for Health and Clinical Excellence, Decision Support Unit. NICE DSU technical support document 1: introduction to evidence synthesis for decision making. www.cc‐arcc.ca/common/pages/UserFile.aspx?fileId=282147 (accessed 22 Apr 2014).
Dias 2011b
- Dias S, Welton NJ, Sutton AJ, Ades AE, National Institute for Health and Clinical Excellence, Decision Support Unit. NICE DSU technical support document 2: a generalised linear modelling framework for pairwise and network meta‐analysis of randomised controlled trials. www.nicedsu.org.uk/TSD2%20General%20meta%20analysis%20corrected%2015April2014.pdf (accessed 22 Apr 2014). [PubMed]
Dias 2011c
- Dias S, Welton NJ, Sutton AJ, Caldwell DM, Lu G, Ades AE, National Institute for Health and Clinical Excellence, Decision Support Unit. NICE DSU technical support document 4: inconsistency in networks of evidence based on randomised controlled trials. www.nicedsu.org.uk/TSD4%20Inconsistency.final.15April2014.pdf (accessed 22 Apr 2014). [PubMed]
Dougados 2002
- Dougados M, Kijkmans B, Khan M, Maksymowych W, Linden S, Brandt J. Conventional treatments for ankylosing spondylitis. Annals of Rheumatic Diseases 2002;61(Suppl iii):iii40‐iii50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Edmunds 1991
- Edmunds L, Elswood J, Kennedy LG, Calin A. Primary ankylosing spondylitis, psoriatic and enteropathic spondyloarthropathy: a controlled analysis. Journal of Rheumatology 1991;18(5):696‐8. [PubMed] [Google Scholar]
GRADEpro 2014 [Computer program]
- McMaster University. GRADEpro. [www.gradepro.org]. McMaster University, Version 3.6 [2014].
Haibel 2004
- Haibel H, Brandt HC, Rudwaleit M, Listing J, Braun J, Kupper H, et al. Efficacy and safety of adalimumab in the treatment of active ankylosing spondylitis: Preliminary results of an open‐label, 20‐week trial. Arthritis and Rheumatism 2004;50(Suppl):S217. [Google Scholar]
Haibel 2010
- Haibel H, Sieper J. Editorial review: how early should ankylosing spondylitis be treated with a tumor necrosis factor‐blocker?. Current Opinion in Rheumatology 2010;22:388‐92. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins J, Thompson S, Deeks J, Altman D. Measuring inconsistency in meta‐analysis. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Hochberg 2003
- Hochberg MC, Tracy JK, Hawkins‐Holt M, Flores RH. Comparison of the efficacy of the tumour necrosis factor (alpha) blocking agents adalimumab, etanercept, and infliximab when added to methotrexate in patients with active rheumatoid arthritis. Annals of the Rheumatic Diseases 2003;62(Suppl 2):ii13‐ii16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Inman 2011
- Inman RD. The spondyloarthropathies. In: Goldman L, Schafer AI editor(s). Cecil Medicine. 24th Edition. Philadelphia, PA: Elsevier Saunders, 2011. [Google Scholar]
Khan 2002
- Khan M. Update on spondyloarthropathies. Annals of Internal Medicine 2002;136:896‐907. [DOI] [PubMed] [Google Scholar]
Lord 2010
- Lord PAC, Farragher TM, Lunt M, Watson KD, Symmons DPM, Hyrich KL. Predictors of response to anti‐TNF therapy in ankylosing spondylitis: results from the British Society for Rheumatology Biologics Register. Rheumatology 2010;49:563‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Maksymowych 2002
- Maksymowych W, Jhangri G, Lambert R, Mallon C, Buenviaje H, Pedryca E, et al. Infliximab in ankylosing spondylitis: a prospective observational inception cohort analysis of efficacy and safety. Journal of Rheumatology 2002;29:959‐65. [PubMed] [Google Scholar]
Maksymowych 2003
- Maksymowych W, Inman R, Gladman D, Thompson G, Stone M, Karsh J, et al. Canadian Rheumatolgy Association Consensus on the use of anti‐tumor necrosis factor‐alpha directed therapies in the treatment of spondyloarthritis. Journal of Rheumatology 2003;30:1356‐63. [PubMed] [Google Scholar]
Maksymowych 2005
- Maksymowych W, Rahman P, Keystone E, Wong R, Inman R, M03‐606 Study Group. Efficacy of adalimumab in active AS: Results from the Canadian AS study. Arthritis and Rheumatism. 2005:S217.
Maksymowych 2008
- Maksymowych W, Rahman P, Shojania K, Olszynski W, Thomson G, Ballal S, et al. Beneficial effects of adalimumab on biomarkers reflecting structural damage in patients with ankylosing spondylitis. Journal of Rheumatology 2008;35(10):2030‐7. [PubMed] [Google Scholar]
Maksymowych 2010
- Maksymowych WP, Salonen D, Inman RD, Rahman P, Lambert RGW. Low‐dose infliximab (3 mg/kg) significantly reduces spinal inflammation on magnetic resonance imaging in patients with ankylosing spondylitis: a randomized placebo‐controlled study. Journal of Rheumatology 2010;37(8):1728‐34. [DOI] [PubMed] [Google Scholar]
Marzo‐Ortega 2001
- Marzo‐Ortega H, McGonagle D, O'Connor P, Emery P. Efficacy of etanercept in the treatment of the entheseal pathology in resistant spondylarthropathy: a clinical and magnetic resonance imaging study. Arthritis and Rheumatism 2001;44(9):2112‐7. [DOI] [PubMed] [Google Scholar]
Montacer 2009
- Montacer KM, Mehdi Ghannouchi M, Hamdi W, Azzouz D, Kochbati S, Saadellaoui K, et al. Impact of the ankylosing spondylitis on the professional activity. Joint Bone Spine 2009;76(4):378‐82. [DOI: 10.1016/j.jbspin.2008.08.010] [DOI] [PubMed] [Google Scholar]
NICE 2008
- National Institute for Health and Clinical Excellence. Adalimumab, etanercept and infliximab for ankylosing spondylitis. NICE technology appraisal guidance 143. www.nice.org.uk/TA143 (accessed January 2010).
NICE 2011
- National Institute for Health and Clinical Excellence. Golimumab for the treatment of ankylosing spondylitis: NICE technology appraisal guidance 233. www.nice.org.uk/guidance/ta233/resources/guidance‐golimumab‐for‐the‐treatment‐of‐ankylosing‐spondylitis‐pdf 2011.
Pavy 2005
- Pavy S, Brophy S, Calin A. Establishment of the minimum clinically important difference for the bath ankylosing spondylitis indices: a prospective study. Journal of Rheumatology 2005;32(1):80‐5. [PubMed] [Google Scholar]
Plot Digitizer 2014 [Computer program]
- Huwaldt JA, Steinhorst S. Plot Digitizer. Open source; http://plotdigitizer.sourceforge.net/, 27 April 2014.
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Rudwaleit 2004
- Rudwaleit M, Listing J, Brandt J, Braun J, Sieper J. Prediction of a major clinical response (BASDAI 50) to TNF‐ blockers in ankylosing spondylitis?. Annals of the Rheumatic Diseases 2004;63:665‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Rudwaleit 2009a
- Rudwaleit M, Landewe R, Heijde D, Listing J, Brandt J, Braun J, et al. The development of Assessment of SpondyloArthritis international Society classification criteria for axial spondyloarthritis (part I): classification of paper patients by expert opinion including uncertainty appraisal. Annals of the Rheumatic Diseases 2009;68:770‐6. [DOI] [PubMed] [Google Scholar]
Rudwaleit 2009b
- Rudwaleit M, Heijde D, Landewé R, Listing J, Akkoc N, Brandt J, et al. The development of Assessment of SpondyloArthritis international Society classification criteria for axial spondyloarthritis (part II): validation and final selection. Annals of the Rheumatic Diseases 2009;68:777‐83. [DOI] [PubMed] [Google Scholar]
Schultz 1995
- Schultz K, Chalmers I, Hayes R, Altman D. Empirical evidence of bias. Dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273:408‐12. [DOI] [PubMed] [Google Scholar]
Schünemann 2009
- Schünemann H, Brożek J, Oxman A, editors. GRADE handbook for grading quality of evidence and strength of recommendation. Version 3.2. The GRADE Working Group. updated March 2009, issue Completing the Summary of Findings for an Outcome: Dichotomous outcomes ‐ Corresponding risk.
Schünemann 2011
- Schünemann HJ, Oxman AD, Higgins JPT, Vist GE, Glasziou P, Guyatt GH. Chapter 11: Presenting results and 'Summary of findings' tables. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Song 2009
- Song F, Loke YK, Walsh T, Glenny A‐M, Eastwood AJ, Altman DG. Methodological problems in the use of indirect comparisons for evaluating healthcare interventions: survey of published systematic reviews. BMJ 2009;338:b1147. [DOI: 10.1136/bmj.b1147] [DOI] [PMC free article] [PubMed] [Google Scholar]
Spiegelhalter 2003
- Spiegelhalter D, Thomas A, Best N, Lunn D. WinBUGS user manual [Internet]. Version 1.4. Cambridge, UK: MRC Biostatistics Unit, Institute of Public Health (accessed 21 Apr 2014).
Stone 2001
- Stone M, Salonen D, Lax M, Payne U, Lapp V, Inman R. Clinical and imaging correlates of response to treatment with infliximab in patients with ankylosing spondylitis. Journal of Rheumatology 2001;28:1605‐14. [PubMed] [Google Scholar]
Sweeting 2004
- Sweeting MJ, Sutton AJ, Lambert PC. What to add to nothing? Use and avoidance of continuity corrections in meta‐analysis of sparse data. Statistics in Medicine 2004;23:1351‐75. [DOI] [PubMed] [Google Scholar]
Turner 2005
- Turner RM, Thompson SG, Spiegelhalter DJ. Prior distributions for the intracluster correlation coefficient, based on multiple previous estimates, and their application in cluster randomized trials. Clinical Trials 2005;2(2):108‐18. [DOI] [PubMed] [Google Scholar]
Van Den Bosch 2002
- Bosch F, Kruithof E, Baeten D, Herssens A, Keyser F, Mielants H, et al. Randomized double‐blind comparison of chimeric monoclonal antibody to tumor necrosis factor alpha (infliximab) versus placebo in active spondylarthropathy. Arthritis and Rheumatism 2002;46:755‐65. [DOI] [PubMed] [Google Scholar]
van der Heijde 1997
- Heijde D, Bellamy N, Calin A, Dougados M, Khan MA, Linden S. Preliminary core sets for endpoints in ankylosing spondylitis. Journal of Rheumatology 1997;24:2225‐9. [PubMed] [Google Scholar]
van der Heijde 2011
- Heijde D, Sieper J, Maksymowych WP, Dougados M, Burgos‐Vargas R, Landewé R, et al. Assessment of SpondyloArthritis international Society. 2010 Update of the international ASAS recommendations for the use of anti‐TNF agents in patients with axial spondyloarthritis. Annals of the Rheumatic Diseases 2011;70(6):905‐8. [DOI: 10.1136/ard.2011.151563] [DOI] [PubMed] [Google Scholar]
Wanders 2005
- Wanders A, Heijde D, Landewe R, Behier JM, Calin A, Olivieri I, et al. Nonsteroidal antiinflammatory drugs reduce radiographic progression in patients with ankylosing spondylitis. Arthritis and Rheumatism 2005;52(6):1756‐65. [DOI] [PubMed] [Google Scholar]
Wells 2009
- Wells GA, Sultan SA, Chen L, Khan M, Coyle D, Canadian Agency for Drugs and Technologies in Health. Indirect Evidence: Indirect Treatment Comparisons in Meta‐Analysis. www.cadth.ca/media/pdf/H0462_itc_tr_e.pdf 2009.
Yazici 2008
- Yazici Y, Adler NM, Yazici H. Most tumour necrosis factor inhibitor trials in rheumatology are undeservedly called 'efficacy and safety' trials: a survey of power considerations. Rheumatology 2008;47(7):1054‐7. [DOI] [PubMed] [Google Scholar]
Zink 2000
- Zink A, Braun J, Listing J, Wollenhaupt J. Disability and handicap in rheumatoid arthritis and ankylosing spondylitis ‐ results from the German rheumatological database. Journal of Rheumatology 2000;27:613‐32. [PubMed] [Google Scholar]
References to other published versions of this review
Zochling 2005
- Zochling J, Maxwell L, Beardmore J, Boonen A. TNF‐alpha inhibitors for ankylosing spondylitis. Cochrane Database of Systematic Reviews 2005, Issue 3. [DOI: 10.1002/14651858.CD005468] [DOI] [PMC free article] [PubMed] [Google Scholar]