

Abstract
The prognosis for children with recurrent and/or refractory neuroblastoma (NB) is dismal. The receptor tyrosine kinase-like orphan receptor 1 (ROR1), which is highly expressed on the surface of NB cells, provides a potential target for novel immunotherapeutics. Anti-ROR1 chimeric antigen receptor engineered ex vivo expanded peripheral blood natural killer (anti-ROR1 CAR exPBNK) cells represent this approach. N-803 is an IL-15 superagonist with enhanced biological activity. In this study, we investigated the in vitro and in vivo anti-tumor effects of anti-ROR1 CAR exPBNK cells with or without N-803 against ROR1+ NB models. Compared to mock exPBNK cells, anti-ROR1 CAR exPBNK cells had significantly enhanced cytotoxicity against ROR1+ NB cells, and N-803 further increased cytotoxicity. High-dimensional analysis revealed that N-803 enhanced Stat5 phosphorylation and Ki67 levels in both exPBNK and anti-ROR1 CAR exPBNK cells with or without NB cells. In vivo, anti-ROR1 CAR exPBNK plus N-803 significantly (p < 0.05) enhanced survival in human ROR1+ NB xenografted NSG mice compared to anti-ROR1 CAR exPBNK alone. Our results provide the rationale for further development of anti-ROR1 CAR exPBNK cells plus N-803 as a novel combination immunotherapeutic for patients with recurrent and/or refractory ROR1+ NB.
Keywords: MT: Regular Issue, neuroblastoma, chimerical antigen receptor, ROR1, expanded natural killer cells, targeted immunotherapy, cytotoxicity, IL-15 superagonist
Graphical abstract
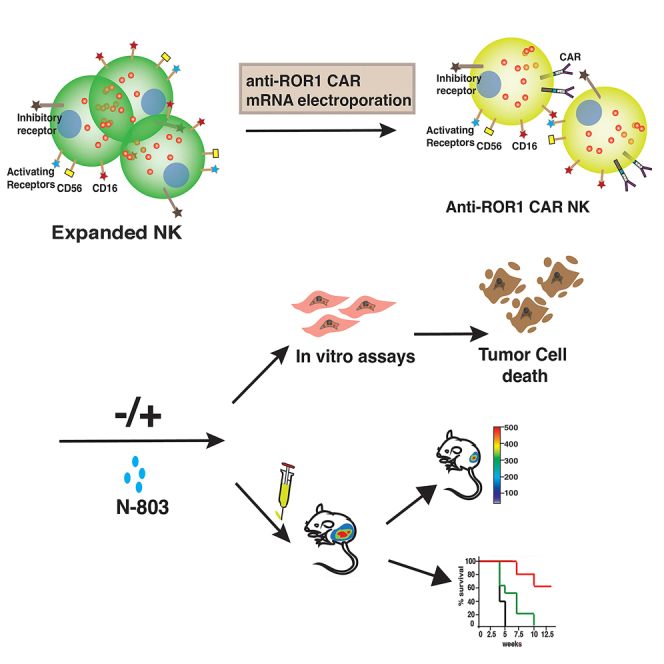
Cairo and colleagues present compelling preclinical data, offering a proof of concept for the combination of N-803 and anti-ROR1 CAR NK cells. This combination emerges as a promising strategy for future clinical studies targeting R/R ROR1+ NB tumors.
Introduction
Neuroblastoma (NB) is the third most common malignancy in childhood and the most common malignancy in infancy.1 The factors associated with response to treatment and survival in NB include disease stage at diagnosis and risk category.2 The survival of patients with advanced stage and high-risk disease has been improved by adoption of an aggressive multimodal treatment approach involving multiagent chemotherapy, local regional control with radiation and surgery, and autologous peripheral blood stem cell transplantation with subsequent targeted immunotherapy using anti-GD2 antibodies.3,4,5 Unfortunately, about half of these high-risk patients will experience a relapse or become refractory despite an initial good response to the therapy.6 Several factors have been implicated in the dismal outcome following treatments in advanced stage NB including but not limited to difference in biological profile of tumor cells due to presence of segmental chromosomal aberrations, poor infiltration of chemotherapeutic agents into the tumor space, chemotherapy-induced immune escape,7 upregulation of tumor growth factors, decreased apoptosis due to reactive oxygen species generation, and poor immunogenicity of tumor cells due to presence of inhibitory signaling molecules in the tumor microenvironment.8,9,10
Multiple immunotherapeutic approaches and targets to improve outcomes in patients with relapsed and/or refractory (R/R) disease are being investigated. For example, autologous, third-generation GD2-chimeric antigen receptor (CAR) T cells expressing the inducible caspase 9 suicide gene (GD2-CART01) were investigated to treat patients with R/R high-risk NB (NCT03373097).11 Receptor tyrosine kinase-like orphan receptor 1 (ROR1) is a transmembrane protein that has been shown to play a key role in embryonal development, particularly of neuronal and muscular tissues.12 Several studies have reported aberrant expression of ROR1 in different hematologic malignancies including acute lymphocytic leukemia and B cell chronic lymphocytic leukemia, as well as solid tumors including NB, breast cancer, and renal cell carcinoma.13,14 Dave et al. thoroughly examined ROR1 expression in both NB cell lines and patients primary NB cells.14 They found that ROR1 expressed on 13 NB cell lines with and without amplification of MYCN proto-oncogene (MYCN) by flow cytometry. ROR1 mRNA levels were significantly higher in NB patients with poor prognosis compared to those with good prognosis. Furthermore, ROR1 protein expression was detected across all stages of NB using primary patients’ tissues.14
Preclinical studies using immunotherapeutic approaches targeting ROR1 against a variety of malignancies have shown promising results.15 In addition, studies have demonstrated that ROR1 is overexpressed in all stages of NB.14 Overexpression of ROR1 in NB cells with minimal to limited expression in normal human tissues makes it an appealing candidate for development of targeted therapies. ROR1-targeted treatments are likely to have a low risk of on-target off-tumor (OTOT) side effects, frequently seen with widely used anti-GD2 antibodies and CAR T cells.16,17
To effectively leverage the potential of ROR1 as a target in NB, natural killer (NK) cells can be engineered to express anti-ROR1 CAR. NK cells are innate immune effector cells with a unique role in immune surveillance and tumor cells killing, making them an attractive effector cell population against a variety of malignancies, including NB, which is highly susceptible to NK cell-mediated cytotoxicity.18 We have previously demonstrated that peripheral blood NK cells can be significantly expanded ex vivo using genetically modified and irradiated feeder cells for use against different malignancies.19,20,21 We are currently evaluating the safety and efficacy of donor-derived ex vivo expanded NK cells in children with R/R NB through the New Approaches to Neuroblastoma Therapy (NANT) consortium (NCT02573896) in combination with irinotecan, temozolomide, and dinutuximab (NCT04211675).
We have also previously reported successful genetic modification of ex vivo expanded peripheral blood NK (exPBNK) cells by electroporating anti-CD20 CAR mRNA into exPBNK cells to express anti-CD20 CAR on the cell surface to enhance target recognition and increase cytotoxicity,22 suggesting the possibility of engineering exPBNK cells to express an anti-ROR1 CAR. Here, we present our findings from in vitro and in vivo assessment of the efficacy of anti-ROR1 CAR exPBNK cells against NB tumors. N-803 (previously known as ALT-803), an IL-15 super-agonist, is a known enhancer of NK cell effector function and has been demonstrated to have potent immune-stimulatory properties in multiple preclinical and clinical studies.8 To further enhance the potential for efficacy, we also investigated the effects of combining anti-ROR1 CAR exPBNK with N-803.
Results
Anti-ROR1 CAR expression on exPBNK cells following electroporation
Peripheral blood mononuclear cells (PBMNCs) from healthy donors were cultured with irradiated K562-mbIL21-41BBL cells as described21 in the presence of 50 IU/mL IL-2. The anti-ROR1 CAR fragment was excised with restriction enzymes from the lentiviral vector epHIV7-anti-ROR1-BB-ζ23 and ligated into a T7 promoter-driven pcDNA3.1 plasmid (Figure 1A). Anti-ROR1 CAR mRNA was in vitro transcribed as described previously.22 Expanded CD56+CD3− exPBNK cells were electroporated in the presence of anti-ROR1 CAR mRNA to generate anti-ROR1 CAR exPBNK cells or in nuclease-free H2O to generate mock exPBNK cells using Maxcyte electroporator. Flow cytometry was used to detect the expression of anti-ROR1 CAR in 67.35% of viable exPBNK cells after 48 h of electroporation (Figure 1B). Consistently with our previous report,22 the anti-ROR1 CAR expression utilizing CAR mRNA electroporation was transient and was reduced from 67.35% at 48 h to 36.3% at day 6 and 4.35% at day 10 after electroporation (Figure 1B).
Figure 1.

Construction and anti-ROR1 CAR expression on expanded NK cells
(A) Schema of anti-ROR1 CAR construct in pcDNA3.1 consisting of anti-ROR1 single chain fragment variable (scFv), 41BB, CD3zeta, and truncated CD19. (B) The anti-ROR1 CAR exPBNK cells were generated by electroporating exPBNK with anti-ROR1 CAR mRNA using Maxcyte electroporator. Mock exPBNK cells were generated by electroporating exPBNK cells with RNase-free H2O. Anti-ROR1 CAR expression was evaluated by flow cytometry analysis using an FITC-conjugated goat anti-mouse IgG, F(ab′)2 fragment-specific antibody at days 1, 2, 4, 6, and 10. The top panel shows the representative flow cytometry dot plots of forward scatter (FSC) vs. CAR. The bottom panel summarizes the percentage of anti-ROR1 CAR expression on exPBNK cells. Data are presented as mean ± SEM, n = 3. ∗∗∗p < 0.001.
Anti-ROR1 CAR exPBNK cells significantly enhanced in vitro cytotoxicity against ROR1+ NB cells with significantly enhanced intracellular expression of CD107a, IFN-γ, and granzyme B
We further confirmed ROR1 expression on the surface of NB cells using NB cell lines (Figure S1). To investigate the in vitro cytotoxicity of anti-ROR1 CAR exPBNK cells, we co-cultured anti-ROR1 CAR exPBNK cells with carboxyfluorescein diacetate succinimidyl ester (CFSE)-labeled ROR1+ NB cells: SH-SY5Y (Figure 2A) and SKNFI (Figure 2B) and SKNBE(2)N (Figure 2C). ROR1+ osteosarcoma HOS cells (Figure 2D) were used as positive control, and ROR1− MCF-7 cells were used as negative control (Figure 2E). The Cayman’s 7-AAD/CFSE Cell-Mediated Cytotoxicity Assay was used to measure the in vitro cytotoxicity. Anti-ROR1 CAR exPBNK cells showed significantly enhanced in vitro cytotoxicity against CFSE-labeled ROR1+ SH-SY5Y (Figure 2A), SKNFI (Figure 2B), and SKNBE(2)N (Figure 2C) and the ROR1+ osteosarcoma HOS cells (Figure 2D) used as the positive control at effector to target (E:T) ratios of 5:1, 10:1, or 40:1 compared to mock exPBNK cells. There was no significant difference of in vitro cytotoxicity between anti-ROR1 CAR exPBNK cells and mock exPBNK cells when targeting ROR1− MCF-7 cells used as a negative control (Figure 2E).
Figure 2.

Anti-ROR1 CAR enhances exPBNK cell in vitro cytolytic activity against ROR1+ NB cells
The exPBNK cells electroporated with anti-ROR1 CAR mRNA (anti-ROR1-CAR) or H2O (mock) were incubated for 4 h with ROR1+ NB cell lines: SH-SY5Y (A), SKNFI (B), SKNBE(2)N (C), ROR1+ osteosarcoma cell line HOS (D), and ROR1− MCF-7 (E) cell line. In vitro cytotoxicity was measured by 7-AAD/CFSE Cell-Mediated Cytotoxicity Assay. The representative flow cytometry contour plots are shown in the left panel of (A). At indicated by E:T ratios, the cytotoxicity of anti-ROR1 CAR exPBNK cells was significantly higher than mock NK cells against ROR1+ NB and osteosarcoma cells but not ROR1− MCF-7 cells. Data are presented as mean ± SEM, n = 3. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Consistent with the enhanced in vitro cytotoxicity, anti-ROR1 CAR exPBNK cells had significantly enhanced intracellular expression of CD107a (Figure 3A) (SH-SY5Y p < 0.001, SKNFI p = 0.004, SKNBE(2)N p = 0.003), interferon-γ (IFN-γ) (Figure 3B) (SH-SY5Y p = 0.002, SKNFI p = 0.005, SKNBE(2)N p = 0.004), granzyme B (Figure 3C) (SH-SY5Y p = 0.007, SKNFI p = 0.002, SKNBE(2)N p = 0.003), and perforin (Figure 3D) (SH-SY5Y p = 0.001, SKNFI p < 0.001, SKNBE(2)N p < 0.001) compared to the mock exPBNK cells.
Figure 3.

Anti-ROR1 CAR NK cells had significantly enhanced intracellular expression of CD107a, IFN-γ, granzyme B targeting ROR1+ NB cells
The exPBNK cells electroporated with anti-ROR1 CAR mRNA (anti-ROR1-CAR) or H2O (mock) were incubated with SH-SY5Y, SKNFI, or SKNBE(2)N at an E:T = 10:1 ratio for 4 h. (A) CD107a expression in exPBNK or anti-ROR1 CAR NK cells was detected with anti-human CD107a–APC antibody gated on CD56 cells (labeled with an anti-CD56–FITC antibody) by flow cytometry and significantly increased (p < 0.001, p = 0.004, p = 0.003, respectively). (B) IFN-γ expression in exPBNK or anti-ROR1 CAR NK cells was detected with anti-human IFN-γ-Alexa Fluor 647 antibody gated on CD56 cells (labeled with an anti-CD56-FITC antibody) by flow cytometry and significantly increased (p = 0.002, p = 0.005, p = 0.004, respectively). The same cells stained with isotype-matched controls were used for gating. (C) Granzyme B expression in exPBNK or anti-ROR1 CAR NK cells was detected with anti-human granzyme B-APC antibody gated on CD56 cells (labeled with an anti-CD56-FITC antibody) by flow cytometry and significantly increased (p = 0.007, p = 0.002, p = 0.003, respectively). (D) Perforin expression in exPBNK or anti-ROR1 CAR NK cells was detected with anti-human perforin-APC antibody gated on CD56 cells (labeled with an anti-CD56-FITC antibody) by flow cytometry and significantly increased (p = 0.001, p < 0.001, p < 0.001, respectively). The same cells stained with isotype-matched controls were used for gating. Data are presented as mean ± SEM, n = 3.
N-803 significantly enhanced anti-ROR1 CAR exPBNK cells in vitro cytotoxicity against NB cells and significantly increased granzyme B, IFN-γ, and perforin secretion
We previously demonstrated that N-803 significantly enhanced the antibody-dependent cell-mediated cytotoxicity of expanded NK cells combined with dinutuximab when targeting GD2+ solid tumors.19 To investigate if N-803 enhances the in vitro cytotoxicity of anti-ROR1 CAR exPBNK cells, anti-ROR1 CAR exPBNK cells and exPBNK cells were cultured in RPMI1640 medium + 10% fetal bovine serum (FBS) with or without 3.5 ng/mL N-803 for 48 h. We found that N-803 significantly enhanced the in vitro cytotoxicity of anti-ROR1 CAR exPBNK cells against SKNFI-Luc cells at an E:T ratio of 3:1 (Figure 4A) (p = 0.0001) with significantly enhanced secretion of granzyme B (Figure 4B) (p = 0.0009), IFN-γ (Figure 4C) (p = 0.0147), and perforin (Figure 4D) (p = 0.0166) compared to anti-ROR1 CAR exPBNK cells in the absence of N-803. Similarly, N-803 significantly enhanced the in vitro cytotoxicity of anti-ROR1 CAR exPBNK cells against CHLA-255-Luc cells at an E:T ratio of 1:1 (Figure 4E) (p = 0.0004) with significantly enhanced secretion of granzyme B (Figure 4F) (p = 0.0028), IFN-γ (Figure 4G) (p = 0.0004), and perforin (Figure 4H) (p = 0.0140) compared to anti-ROR1 CAR exPBNK cells.
Figure 4.

N-803 significantly enhanced the in vitro cytotoxicity with the enhanced release of granzyme B, IFN-γ, and perforin of anti-ROR1 CAR NK against NB cells
(A) Mock NK or anti-ROR1 CAR NK cells were incubated with or without 3.5 ng/mL N-803 for 48 h and then used for in vitro cytotoxicity assays against SKNFI at an E:T = 3:1. N-803 significantly enhanced the in vitro cytotoxicity of anti-ROR1 CAR NK cells against SKNFI-Luc (p = 0.0001) compared to anti-ROR1 CAR NK cells. (B) After 24 h co-culture under the condition as described in (A), the supernatants were collected for ELISA assays to determine the released granzyme B level. Granzyme B level was significantly enhanced (p = 0.0009). (C) After 24 h co-culture under the condition as described in (A), the supernatants were collected for ELISA assays to determine the released IFN-γ level. IFN-γ level was significantly enhanced (p = 0.0147). (D) After 24 h co-culture under the condition as described in (A), the supernatants were collected for ELISA assays to determine the released perforin level. Perforin level was significantly enhanced (p = 0.0166). (E) Mock NK or anti-ROR1 CAR NK cells were incubated with or without 3.5 ng/mL N-803 for 48 h and then used for in vitro cytotoxicity assays against CHLA-255 at an E:T = 1:1. N-803 significantly enhanced the in vitro cytotoxicity of anti-ROR1 CAR NK cells against CHLA-255 (p = 0.0004) compared to anti-ROR1 CAR NK cells. (F) After 24 h co-culture under the condition as described in (E), the supernatants were collected for ELISA assays to determine the released granzyme B level. Granzyme B level was significantly enhanced (p = 0.0028). (G) After 24 h co-culture under the condition as described in (E), the supernatants were collected for ELISA assays to determine the released IFN-γ level. IFN-γ level was significantly enhanced (p = 0.0004). (H) After 24 h co-culture under the condition as described in (E), the supernatants were collected for ELISA assays to determine the released perforin level. Perforin level was significantly enhanced (p = 0.0140). Data are presented as mean ± SEM, n = 3–4.
High-dimensional analysis of N-803 activated anti-ROR1 CAR exPBNK cells with/without NB
To evaluate the phenotypic and functional effects of N-803 activation on anti-ROR1 CAR exPBNK cells, we utilized a custom, 34 parameter, CyTOF (mass cytometry) NK cell targeted panel as previously described.24 Anti-ROR1 CAR exPBNK cells and exPBNK cells were co-cultured with or without SKNFI and N-803 for 2 days. After the cells were stained and fixed, the samples were run on a CyTOF2. Samples (three biologic replicates per condition) were concatenated, and data were visualized with viSNE. We found that N-803 enhanced Stat5 phosphorylation (pStat5) in exPBNK and anti-ROR1 CAR exPBNK cells (Figure 5A) in the absence of NB cells compared to corresponding controls. N-803 also enhanced the levels of pStat5 in anti-ROR1 CAR exPBNK cells in the presence of SKNFI cells compared to the level of pStat5 in exPBNK cells or in anti-ROR1 CAR exPBNK cells in the presence of SKNFI cells without N-803 (Figure 5A), indicating both N-803 and the interaction of CAR and NB target cells contribute the enhanced pStat5 levels. Consistently with our previous report that N-803 significantly enhanced exPBNK cell proliferation, N-803 enhanced Ki67 levels in both mock exPBNK and anti-ROR1 CAR exPBNK cells with or without SKNFI (Figure 5B) compared to controls.
Figure 5.

High-dimensional analysis of N-803 activated anti-ROR1 CAR NK cells with/without NB by mass cytometry
Anti-ROR1 CAR NK cells or NK cells were co-cultured with or without SKNFI and N-803 for 2 days. After the cells were stained and fixed, the samples were run on a CyTOF2. The experiment was repeated 3 times using NK cells from 3 different donors. (A) The representative viSNE plots show pStat5 in anti-ROR1 CAR NK cells or NK cells under the indicated conditions by mass cytometry. (B) The representative viSNE plots show Ki67 levels in anti-ROR1 CAR NK cells or NK cells under the indicated conditions by mass cytometry.
Because the functions of NK cells are highly regulated by the signals from the NK repertoire of receptors,25 we further examined the activating receptors NKG2D and NKp30 and the inhibitory receptors NKG2A and CD94. Consistently with our previous report, N-803 enhanced the expression of NKG2D (Figure S2A) and NKp30 (Figure S2B) on both exPBNK cells and anti-ROR1 CAR exPBNK cells. However, levels of NKG2D (Figure S2A) and NKp30 (Figure S2B) were less enhanced by N-803 on mock exPBNK or anti-ROR1 CAR exPBNK cells in the presence compared to the absence of target NB cells, suggesting tumor cell inhibition of these NK-activating receptors.
Interestingly, N-803 did not enhance the expression of inhibitory receptor NKG2A (Figure S2C) or CD94 (Figure S2D) on either mock exPBNK cells or anti-ROR1 CAR exPBNK cells. C-X-C motif chemokine receptor 3 (CXCR3) plays a vital role in NK migration and tumor infiltration.26 We found that N-803 enhanced the expression of CXCR3 on mock exPBNK and anti-ROR1 CAR exPBNK cells (Figure S2E). Compared to CXCR3 (median on exPBNK: 239.9 ± 74.8, median on CAR exPBNK: 216.3 ± 51.8), the expression of CXCR4 on the mock exPBNK and anti-ROR1 CAR exPBNK cells was relatively low (median on exPBNK: 11.1 ± 1.1, median on CAR NK: 11.9 ± 1.4). Furthermore, N-803 did not enhance CXCR4 expression on mock exPBNK cells nor on anti-ROR1 CAR exPBNK cells (Figure S2F).
Anti-ROR1 CAR exPBNK+N-803 cells significantly enhanced mice survival in NB xenografted NSG mice
To investigate if anti-ROR1 CAR exPBNK cells were significantly more effective compared to mock exPBNK cells in limiting NB growth and improving mice survival in NB xenografts, we xenografted SKNFI-Luc to immunodeficient NSG mice using a semi-disseminated mouse model. In this model, SKNFI-Luc cells were injected into the peritoneal cavity of NSG mice, and after tumor cells were established in mice at day 7, anti-ROR1 CAR exPBNK cells, mock exPBNK cells, or PBS was administered to mice once a week for 6 weeks through intraperitoneal injection. We found that with low tumor burden, the mice treated with anti-ROR1 CAR exPBNK cells (n = 8) significantly extended the survival of SKNFI xenografted NSG mice compared to the PBS (n = 8, p = 0.0019) or mock exPBNK cell (n = 8, p = 0.0073) -treated control groups (Figure 6A), an effect associated with reduced tumor burden. However, all mice eventually died, likely due to tumor migration and growth, including the group treated with anti-ROR1 CAR exPBNK cells, indicating NB resistance to or escape from anti-ROR1 CAR exPBNK cell treatment.
Figure 6.

The combination of anti-ROR1 CAR NK + N-803 significantly extended the survival of NB xenografted NSG mice
(A) 1 × 106 of SKNFI-Luc cells were intraperitoneally injected in NSG mice on day 0. After confirming the tumor engraftment at day 7, 5 × 106 exPBNK cells, 5 × 106 anti-ROR1 CAR exPBNK cells, or PBS was intraperitoneally injected to each mouse once a week for 6 weeks. The mice treated with anti-ROR1 CAR NK cells (n = 8) significantly extended the survival of SKNFI xenografted mice compared to the control groups that were treated with PBS (n = 8, p = 0.0019) or mock NK cells (n = 8, p = 0.0073). (B) 1 × 106 of SKNFI-Luc cells was intraperitoneally injected in NSG mice on day 0. After confirming the tumor engraftment at day 7, 5 × 106 exPBNK cells, 5 × 106 anti-ROR1 CAR exPBNK cells, 5 × 106 exPBNK cells + 0.2 mg/kg N-803, 5 × 106 anti-ROR1 CAR exPBNK cells + 0.2 mg/kg N-803, or PBS was intraperitoneally injected into each mouse once a week for 6 weeks. The mice treated with anti-ROR1 CAR NK cells + N-803 (n = 8) significantly extended the survival of SKNFI xenografted mice compared to the control groups that were treated with NK + N-803 (n = 9, p = 0.018) or anti-ROR1 CAR NK cells alone (n = 7, p = 0.055). (C) Representative bioluminescence images of mice of each group of (B) are shown at day 7, day 81, and day 137. (D) 1 × 106 of CHLA255-Luc cells was intraperitoneally injected in NSG mice on day 0. After confirming the tumor engraftment at day 7, 5 × 106 exPBNK cells, 5 × 106 anti-ROR1 CAR exPBNK cells, 5 × 106 exPBNK cells + 0.2 mg/kg N-803, 5 × 106 anti-ROR1 CAR exPBNK cells + 0.2 mg/kg N-803, or PBS was intraperitoneally injected into each mouse once a week for 6 weeks. The mice treated with anti-ROR1 CAR NK cells + N-803 (n = 9) significantly extended the survival of CHLA255 xenografted mice compared to the control groups that were treated with mock NK + N-803 (n = 9, p = 0.032) or anti-ROR1 CAR NK cells alone (n = 10, p < 0.021).
To investigate if N-803 enhances the therapeutic efficacy of anti-ROR1 CAR exPBNK cells against NB in vivo using human NB xenografted NSG mice, the same SKNFI-Luc mouse model was used. After tumor cells were established in mice at day 7, anti-ROR1 CAR exPBNK cells, mock exPBNK cells, anti-ROR1 CAR exPBNK+N-803, exPBNK+N-803, or PBS was administered to mice once a week for 6 weeks through intraperitoneal injection. We found that with low tumor burden, the survival of SKNFI xenografted mice was further prolonged by addition of N-803 to anti-ROR1 CAR exPBNK cell treatment (n = 8) compared to either ROR1 CAR exPBNK cells alone (n = 7; p = 0.055) or mock exPBNK+N-803 (n = 9; p = 0.018) (Figure 6B). The median survival time was 139 days for the PBS-treated group, 138 days for the exPBNK-treated group, 172 days for the anti-ROR1 CAR exPBNK-treated group, 249 days for the anti-ROR1 CAR exPBNK+N-803-treated group, and 139 days for the exPBNK+N-803-treated group. As observed in bioluminescence images, the anti-ROR1 CAR exPBNK+N-803-treated mice consistently had lower tumor burdens than controls groups (Figure 6C).
Furthermore, the mice treated with anti-ROR1 CAR exPBNK cells and N-803 (n = 9) significantly extended the survival of CHLA-255 xenografted mice compared to the control groups that were treated with mock exPBNK+N-803 (n = 9, p = 0.032) or anti-ROR1 CAR exPBNK cells only (n = 10, p = 0.021) (Figure 6D).
Discussion
In this study, the anti-ROR1 CAR exPBNK cells that we generated as a potential novel immunotherapeutic for NB displayed significantly enhanced in vitro cytotoxicity against ROR1+ NB cells and significantly extended in vivo survival in NB xenografted NSG mice. When combined with the IL-15 superagonist N-803, the prolongation of survival conferred by anti-ROR1 CAR exPBNK cells was significantly enhanced in vivo using NB xenografted NSG mice. To our knowledge, this is the first preclinical study investigating the therapeutic potential of targeted anti-ROR1 CAR exPBNK cells in combination with an IL-15 superagonist in treating NB.
Despite the advances in the molecular phenotyping of NB and the introduction of highly intensive multimodality treatment regimens, the prognosis of R/R NB remains dismal and represents an urgent and unmet need.6,9,10 Therapeutic options for patients with R/R NB are limited; therefore, new treatment strategies are desperately needed for this poor risk population.9,10 ROR1, due to its high and consistent expression by NB cells, is an appealing target for new therapeutic development. ROR1 plays an essential role in normal embryogenesis and an important role in oncogenesis by activating several cell survival signaling events, including the non-canonical WNT signaling pathway, PI3K/AKT pathway, the NF-κB pathway, and the STAT3 pathway.27 ROR1 is overexpressed in many types of malignant tumors such as chronic lymphocytic leukemia, some types of non-Hodgkin lymphoma, lung adenocarcinoma, and NB, etc.28 With the development of a new anti-ROR1 detection antibody, ROR1 expression was also detected in normal adult tissues, including parathyroid, pancreatic islets, which raised the potential OTOT toxicity of anti-ROR1 CAR T therapy.29
Others have targeted ROR1 for the treatment of NB. At the time this manuscript was written, there were 22 clinical trials (3 completed, 6 active not recruiting, and 13 active recruiting) utilizing anti-ROR1 monoclonal antibodies, anti-ROR1 antibody drug conjugates, or anti-ROR1 CAR T cells cellular or immunotherapies for treating patients with a variety of cancers (Table S1). None of these studies have resulted in a novel approved therapeutic for this indication to date.
NB is widely regarded as among the most sensitive tumor to direct killing by NK cells because of reduced class one major histocompatibility complex expression.30 However, NK cell number and function are decreased in most cancer patients at diagnosis and are further reduced by radiation, chemotherapy, and following high-dose conditioning therapy and autologous stem cell transplantation.31,32 To address the problem of NK cell number and function, Denman et al. developed a method expanding and activating NK cells using feeder cells that expresses membrane-bound IL-21.21 This process inhibits apoptosis and promotes telomere extension, enabling robust expansion of NK cells without replicative senescence.21 An NANT trial (NCT02573896) is ongoing to determine the maximum tolerated dose of autologous ex vivo expanded NK cells utilizing this feeder cell line when combined with dinutuximab and to assess the feasibility of adding lenalidomide at the recommended phase II dose of the ex vivo expanded NK cells with dinutuximab for treatment of children with R/R NB.
MYCN is an immunosuppressive oncogene and plays a central role in maintaining the malignant potential of high-risk NB tumors.33 MYCN amplification is one of the most critical prognostic factors in high-risk NB and strongly associated with unfavorable outcome in high-risk NB.34 Possible strategies to treat MYCN-amplified NB have been proposed and applied such as blocking MYCN-dependent transcription, HDAC inhibitors, inducing differentiation, or suppressing MDM2.34 Our in vitro data showed that anti-ROR1 CAR exPBNK cells killed both MYCN nonamplified NB cells (SH-SY5Y and SKNFI) and MYCN-amplified NB cells (SKNBE(2)N) (Figure 2) in an E:T ratio-dependent manner, suggesting a novel therapeutic strategy to treat MYCN-amplified NB by adoptive anti-ROR1 CAR exPBNK cell therapy.
Anti-ROR1 CAR exPBNK cell-based therapy has advantages over anti-ROR1 CAR T therapy. Firstly, due to the transient expression of anti-ROR1 CAR on NK cells (Figure 1B), the risk of OTOT is relatively low. In our in vivo murine experiments, we did not observe any adverse effects on mice following the anti-ROR1 CAR exPBNK cell treatment even though the scFv of the anti-ROR1 CAR was derived from a monoclonal antibody 2A2, which recognizes both human and murine ROR1.23 Secondly, the allogeneic CAR exPBNK cells are not predicted to increase graft vs. host disease that is associated with allogeneic anti-ROR1 CAR T cells.35,36 Third, anti-ROR1 CAR exPBNK cells can be manufactured as an “off-the-shelf” product37 to eliminate the need for a patient-specific product.
IL-15 clinical application has been limited by its short half-life, tight posttranslational regulation, and high-dose associated toxicity.38,39 Extensive efforts have been made to overcome these limitations by increasing the molecular size, forming IL-15/IL-15Ra complexes, or by overexpressing soluble IL-15 (sIL15), IL-15/IL-15Ra complex through IL-15-armored T cells or NK cells. N-803 comprises an interleukin-15 superagonist mutein (IL-15N72D) and a dimeric IL-15 receptor alpha (IL-15Rα)/Fc fusion protein.40 Important for any novel therapeutic, N-803 has been recognized to be a safe, well-tolerated agent in several phase 1 human clinical trials when given subcutaneously.41,42,43,44 N-803 significantly increased NK and CD8+ T cell numbers and function with minimal toxicities.41,42,43 The combination of N-803 with NK-based cell therapy showed promising preclinical results. N-803 combined with dinutuximab and exPBNK cells significantly enhanced in vitro cytotoxicity of exPBNK cells and significantly extended the survival of immunodeficient mice xenografted with human solid tumors.19 As demonstrated in the present studies, the addition of the IL-15 superagonist N-803 further increases the potential of ROR1-CAR exPBNK cell therapy. Clinical studies of N-803 demonstrated that N-803 was well tolerated via both the intravenous (i.v.) and subcutaneous (s.c.) routes, but s.c. administration of N-803 resulted in more sustained drug levels and better biologic activity on blood NK and CD8+ T cells while avoiding the constitutional toxicities seen with i.v. administration.43,44 Other IL-15-based superagonists with different structures are also in preclinical and clinical development, such as SOT101 (SO-C101), a human fusion protein formed by linking IL-15 to the IL-15Rα sushi domain.45,46 Similar to N-803, SOT101 prolongs the half-life of IL-15 and promotes the development and differentiation of NK cell.45 In the phase I/Ib AURELIO-03 trial, SOT101 exhibited a favorable safety profile and promising efficacy, both as a monotherapy and in combination with pembrolizumab (NCT04234113).47 But it will be interesting to investigate the clinical outcomes of SOT101 in combination with NK-based cell therapies. This will emphasize the pivotal role of combining IL-15 superagonists with NK-based cell therapy for further clinical success. Armoring CAR NK with IL-15 or IL-15/IL-15Rα significantly enhanced CAR NK cell proliferation and long-term persistence and increased CAR NK metabolic fitness and effector function.48,49,50 The phase I/II clinical trial of iC9/CAR.19/IL15 CB-NK cells in treating refractory B cell lymphoma or leukemia showed promising clinical outcome without developing cytokine release syndrome (CRS), neurotoxicity, or graft vs. host disease (NCT03056339).35 And, the infused iC9/CAR.19/IL15 CB-NK cells persisted for at least 12 months.35 Even though only picogram quantities of IL-15 were produced from iC9/CAR.19/IL15 CB-NK cells,49 safety has become a concern in IL-15 or IL-15/IL-15Rα-armored NK cell therapy due to several reasons. Enhancing inflammatory cytokines, impeding NK activation, inducing NK exhaustion, and manipulating the tumor microenvironment to facilitate tumor evasion through prolonged stimulation with IL-15 or IL-15/IL-15Rα complexes are all critical mechanisms to consider.51,52,53 Incorporating a safety switch in IL-15-armored CAR NK cells should be considered to eliminate the potential unwanted and uncontrolled adverse effects.
Consistent with our previous studies,19 our mass cytometry data show that N-803 significantly stimulated the pStat5 signal transduction pathway and the proliferation of exPBNK and anti-ROR1 CAR exPBNK cells (Figure 5). NK cells express diverse activating and inhibitory receptors, and the balance of signals between these receptors determines the net functionality of NK cells.54 Anti-ROR1 CAR exPBNK cells express the activating receptors such as NKG2D and NKp30 (Figure S2), which can potentially eliminate NB cells in a CAR-independent manner. Decreased expression of the activating receptors such as NKp30 and NKG2D on the NK cells was associated with poorer prognosis.55 Our current study showed that N-803 was able to enhance the expression of NKG2D and NKp30 in both exPBNK and anti-ROR1 CAR exPBNK cells without NB cell co-culture (Figure S2). However, NB cells reduced the expression of NKG2D and NKp30 on exPBNK and anti-ROR1 CAR exPBNK cells, partially contributing to the NB relapse from the treatment of exPBNK and anti-ROR1 CAR exPBNK cells (Figure S2).
A limitation of the in vivo studies presented herein may be that the mice were immunodeficient. A recent clinical trial showed that systematic N-803 reduced the clinical activity of the adoptive haploidentical NK cell cells due to the accelerated host T cell alloreactivity.56,57 Since our mouse model lacks host T cells, we were not able to observe host T cell alloreactivity by N-803 to the adoptive anti-ROR1 CAR exPBNK cells. However, N-803 doses to be administered, the types of allogeneic PBMNC donors to derive anti-ROR1 CAR exPBNK cells, and the intensity of lymphodepletion need to be tailored to improve the potential efficacy of N-803 and adoptive anti-ROR1 exPBNK cell therapy with a low risk of any effects on T cell function in the future clinical trials for patients with R/R NB tumors.
In summary, our findings demonstrate that anti-ROR1 CAR exPBNK cells exhibit significantly enhanced in vitro and in vivo anti-tumor effects compared to mock exPBNK cells against NB and that N-803 further increases anti-ROR1 CAR exPBNK anti-NB activity both in vitro and in vivo. Our preclinical data provide a compelling proof of concept that the combination of N-803 and anti-ROR1 CAR exPBNK cells would be a promising strategy for future clinical studies against R/R ROR1+ NB tumors.
Materials and methods
Cell lines and reagents
SH-SY5Y, SKNFI, and SKNBE(2)N were generously provided by Nai-Kong Cheung from Memorial Sloan Kettering Cancer Center, New York, NY, USA, and HOS cells were purchased from the American Type Culture Collection, Gaithersburg, MD, USA. CHLA-255 cells were generously provided by Robert Seeger from Children’s Hospital Los Angeles, Los Angeles, CA, USA. K526-mbIL21-41BBL cells were generously provided by Dean A. Lee, MD/PhD, from Nationwide Children’s Hospital, Columbus, OH, USA.21 N-803 was generously provided by Patrick Soon-Shiong, MD, from ImmunityBio, Culver City, CA, USA. Anti-ROR1 scFv was generously provided by Stanley Riddell, MD, from Fred Hutchinson Cancer Center, Seattle, WA, USA. Leukocytes were obtained after informed consent from healthy donors at the New York Blood Center, New York, NY, USA. PBMNCs were obtained by Ficoll gradient (Cytiva, Marlborough, MA, USA) separation as we previously described.22
NK cell expansion
PBMNCs were stimulated with irradiated genetically modified K562-mbIL21-41BBL cells as we previously described.21 ExPBNK cells were isolated by negative selection using Miltenyi NK cell isolation kit (Miltenyi Biotec, Cambridge, MA, USA) as we have previously described.22 Expanded purified NK cells were cultured in Gibco RPMI 1640 medium (Thermo Fisher) supplemented with 10% heat-inactivated FBS (Thermo Fisher), 100 U/mL penicillin, 100 μg/mL streptomycin (Thermo Fisher), 4 mmol/L glutamine (Thermo Fisher), and 50 IU/mL IL-2.
Anti-ROR1 CAR exPBNK cell generation
The anti-ROR1 CAR mRNA was synthesized in vitro using the mMESSAGE mMACHINE T7 Ultra kit as we previously described.22 ExPBNK cells were electroporated with anti-ROR1 CAR mRNA using the MaxCyte GT electroporation system (Maxcyte, Rockville, MD, USA). Anti-ROR1 CAR mRNA electroporation efficacy was evaluated by flow cytometry analysis using an FITC-conjugated goat anti-mouse IgG, F(ab′)2 fragment-specific antibody. Expanded NK cells electroporated without H2O (mock exPBNK cells) were used as control. Anti-ROR1 CAR expression was determined by flow cytometry analysis on days 1, 2, 4, 6, and 10 post electroporation.
7-AAD/CFSE cell-mediated cytotoxicity
Target tumor cells (ROR1+) and control MCF-7 (ROR1−) cells were labeled with CFSE (Cayman) according to the manufacturer’s instructions and co-cultured with anti-ROR1 CAR exPBNK cells at different E:T ratios for 4–6 h. The target cells only were used as background. After washing with PBS, the cells were stained with 7-AAD viability dye. The dead tumor cells (7-AAD+ CFSE+) were monitored by flow cytometry. The percentage of cytotoxicity was calculated as follows: [100% × dead targets/(dead targets + live targets)] (experiment) − [100% × dead targets/(dead targets + live targets)] (background).
Intracellular expression of CD107a, IFN-γ, and granzyme B assays
Intracellular expressions of CD107a, IFN-γ, and granzyme B were determined as we previously described.22 Briefly, anti-ROR1 CAR exPBNK cells or mock exPBNK cells were mixed with NB cells at a ratio of 10:1 in the absence of exogenous cytokines in RPMI1640 medium. 5 μL anti-CD107a-APC (BD Biosciences, Franklin Lakes, NJ, USA) was added to each well and incubated for 1 h at 37°C. After 1 h, brefeldin A (Golgi Plug; BD Biosciences) was added to each well, and the cells were incubated for an additional 3 h. Cells were then washed, fixed, permeabilized using Cytofix/Cytoperm reagent kit (BD Biosciences, Franklin Lakes, NJ, USA), and resuspended in staining buffer containing anti-CD56-FITC (BD Biosciences).
Intracellular IFN-γ and granzyme B were analyzed after anti-ROR1 CAR exPBNK cells or mock exPBNK cells were mixed with NB cells, at a ratio of 10:1 in the absence of exogenous cytokines in RPMI1640 medium with brefeldin A for 4 h (Golgi Plug; BD Biosciences). Cells were then washed, fixed, permeabilized using Cytofix/Cytoperm reagent kit (BD Biosciences, Franklin Lakes, NJ, USA), and resuspended in staining buffer containing anti-CD56-FITC and anti-IFN-γ-Alexa Fluor 647 (BD Biosciences) or anti-human granzyme B APC antibody (Biolegend, San Diego, CA, USA).
All experiments included appropriate isotype controls. medium alone served as a control, and a stained control sample with target cells was included to detect spontaneous degranulation. Samples were analyzed on an MACs.c.UANT flow cytometer (Miltenyi Biotec, Cambridge, MA, USA), and a minimum of 10,000 events was collected.
Luciferase-based in vitro cytotoxicity
Anti-ROR1 CAR exPBNK cells were cultured in RPMI1640 + 10% FBS with 3.5 ng/mL N-803 for 48 h, followed by adding SKNFI-Luc at E:T ratio of 3:1 or CHLA-255-Luc at 1:1, respectively. Cytotoxicity was determined by luciferase-based cytotoxicity assay 24 h after co-culture with NB cells with Britelite plus reporter gene assay (PerkinElmer, 6066761). After adding luciferin, the luminescence emission of viable cells was measured by a plate reader (Molecular devices, Filter max F5 microplate reader) within 15 min. Cytotoxicity was calculated as follows: percent cytotoxicity = (luminescence release (untreated tumor cells) − luminescence release (treated tumor cells))/luminescence release (untreated tumor cells)) ×100.
Enzyme-linked immunosorbent assay
IFN-γ (Thermo Fisher, cat# EHIFNG), granzyme B (Thermo Fisher, cat# BMS2027-2), and perforin (Thermo Fisher, cat# BMS2306) concentrations were analyzed by enzyme-linked immunosorbent assay (ELISA) as we previously described according to the manufacturers’ instructions.15 Briefly, recombinant standards were run with serial dilutions. Cell culture supernatants were diluted at 1:1 or 1:4 with assay diluent. 100 uL of diluted samples and standard were added to microwells simultaneously and incubated for 2–2.5 h at room temperature. Biotin conjugated anti-human IFN-γ, granzyme B, or perforin antibody was used and incubated for 1 h at room temperature. After washing, streptavidin-HRP solution was added for 30 min at room temperature. ELISA plates were developed with 100 μL TMB substrate reagents. TMB Stop Solution was added to halt the reaction. The absorbance at 450 nm was measured on a Molecular Devices Multifilter F5 plate reader.
Mass cytometry analysis
The expanded NK cells from 3 donors were set up for co-culture under 8 conditions: (1) exPBNK, (2) anti-ROR1 CAR exPBNK, (3) exPBNK + N-803, (4) anti-ROR1 exPBCAR NK + N-803, (5) exPBNK + SKNFI cells, (6) anti-ROR1 CAR exPBNK + SKNFI, (7) exPBNK + SKNFI cells + N-803, (8) anti-ROR1 CAR exPBNK + SKNFI cells + N-803. After 2 days of co-culture, the cells were collected, washed, and cryopreserved in 10% DMSO in FBS. The batched thawed cells were stained with 34 specific antibodies according to the manufacturer’s protocol.58,59 Immunophenotypic assignments were performed as we previously described.60 The multiparametric analysis of immune markers as well as viSNE analysis were performed on Cytobank (Cytobank, Santa Clara, CA, USA) as we previously described.61
Xenograft models
6- to 8-week-old NOD/SCID/γ-chain−/− (NSG) mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Mice were bred, treated, and maintained under pathogen-free conditions in-house under New York Medical College Institutional Animal Care and Use Committee-approved protocols and as mandated by federal law and regulations. The experimental protocols were conducted in accordance with the recommendations of the Guide for Care and Use of Laboratory Animals with respect to restraint, husbandry, surgical procedures, feed and fluid regulation, and veterinary care. The animal care and use program at New York Medical College is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International.
Luciferase-expressing NB cells (SKNFI-Luc cells) were generated as we have previously described.22 1 × 106 of SKNFI-Luc cells or CHLA-255-Luc cells were intraperitoneally injected in NSG mice on day 0. After confirming the tumor engraftment at day 7, 5 × 106 exPBNK cells, 5 × 106 anti-ROR1 CAR exPBNK cells, 5 × 106 exPBNK cells + 0.2 mg/kg N-803, 5 × 106 anti-ROR1 CAR exPBNK cells + 0.2 mg/kg N-803, or PBS was intraperitoneally injected into each mouse. ExPBNK cells and N-803 were administered once a week for 6 weeks. Tumor engraftment and progression were evaluated using the Xenogen IVIS-200 system (PerkinElmer, Shelton, Connecticut) as we have previously described.22 Tumor size was estimated according to the following formula: tumor size (cm3) = length (cm) × width2 (cm) × 0.5. Mice were followed until death or sacrificed if any tumor size reached 2 cm3 or larger.
Statistical analysis
Statistical analyses were performed using the Prism program 10.0 (GraphPad Software). Average values are reported as the mean ± SEM. Results were compared using one-way ANOVA with p < 0.05 considered as significant. Probability of survival in animal studies was determined by the Kaplan-Meier method and comparison of survival curves of experimental groups by log rank test using the Prism program 10.0 (GraphPad Software).
Data and code availability
Data are available upon reasonable request.
Acknowledgments
The authors would like to thank Erin Morris, BSN, Virginia Davenport, RN, and Patricia Spilman for their excellent assistance with the preparation of this manuscript. The authors would like to thank Hing Wong, Peter R. Rhode, Patrick Soon-Shiong, John H. Lee, and Jeffrey Safrit from ImmunityBio, Culver City, CA, USA, for providing N-803. The research for this study was primarily supported by the moonshot and PIDDN NIH 1U54 CA232561-01A1 grant (M.S.C., D.A.L., T.P.C.) with additional support from Pediatric Cancer Research Foundation (PI: M.S.C.), New York Medical College School of Medicine Translational Science Institute Children Health Translational Research Award (Co-PI: Y.C.), Children Cancer Foundation (PI: M.S.C.), and Department of Defense W81XWH-16-PRCRP-TTSA #CA160461 (partnering PI: M.S.C.).
Author contributions
Y.C. and M.S.C. conceived and designed the study. Y.C., G.N., M.T., and D.A.L. developed the methodology, performed the analysis, and interpreted the data. Y.C., G.N., and M.S.C. wrote the manuscript. Y.C., G.N., M.T., D.A.L., P.T., J.A.-C., K.K., K.F., A.S.M., Y.L., W.L., J.A., G.K.B., S.R., T.P.C., and M.S.C. reviewed and revised the manuscript. D.A.L., S.R., G.K.B., T.P.C., and M.S.C. provided administrative, technical, and material support. All authors approved the final manuscript for submission.
Declaration of interests
This was presented in part at Transplantation & Cellular Therapy Meetings of the American Society for Transplantation and Cellular Therapy (2019). M.S.C. has served as a consultant for Jazz Pharmaceuticals, Omeros Pharmaceuticals, Servier Pharmaceuticals, Abbvie, and Novartis Pharmaceuticals; Speakers Bureau for Jazz Pharmaceuticals, Servier Pharmaceuticals, Amgen, Inc., Sanofi, and Sobi; Advisory Board for Astra Zeneca; and research funding from Celularity, Merck, Miltenyi Biotec, Servier, Omeros, Jazz, and Janssen. D.A.L. reports personal fees and other from Kiadis Pharma, CytoSen Therapeutics, Courier Therapeutics, and Caribou Biosciences outside the submitted work. In addition, D.A.L. has a patent broadly related to NK cell therapy of cancer with royalties paid to Kiadis Pharma. T.P.C. recently served as a one-time consultant to Blueprint, Incyte, Oncopeptides, DSMB chair for SpringWorks, and is a cofounder of Vironexis Biotherapeutics, Inc.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omton.2024.200820.
Contributor Information
Yaya Chu, Email: yayachu.nymc@gmail.com.
Mitchell S. Cairo, Email: mitchell_cairo@nymc.edu.
Supplemental information
References
- 1.Yan P., Qi F., Bian L., Xu Y., Zhou J., Hu J., Ren L., Li M., Tang W. Comparison of Incidence and Outcomes of Neuroblastoma in Children, Adolescents, and Adults in the United States: A Surveillance, Epidemiology, and End Results (SEER) Program Population Study. Med. Sci. Mon. Int. Med. J. Exp. Clin. Res. 2020;26 doi: 10.12659/MSM.927218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perel Y., Valteau-Couanet D., Michon J., Lavrand F., Coze C., Bergeron C., Notz A., Plantaz D., Chastagner P., Bernard F., et al. [Prognosis of neuroblastoma in childhood. Methods of assessment and clinical use] Arch. Pediatr. 2004;11:834–842. doi: 10.1016/j.arcped.2004.02.022. [DOI] [PubMed] [Google Scholar]
- 3.Park J.R., Kreissman S.G., London W.B., Naranjo A., Cohn S.L., Hogarty M.D., Tenney S.C., Haas-Kogan D., Shaw P.J., Kraveka J.M., et al. Effect of Tandem Autologous Stem Cell Transplant vs Single Transplant on Event-Free Survival in Patients With High-Risk Neuroblastoma: A Randomized Clinical Trial. JAMA. 2019;322:746–755. doi: 10.1001/jama.2019.11642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ozkaynak M.F., Gilman A.L., London W.B., Naranjo A., Diccianni M.B., Tenney S.C., Smith M., Messer K.S., Seeger R., Reynolds C.P., et al. A Comprehensive Safety Trial of Chimeric Antibody 14.18 With GM-CSF, IL-2, and Isotretinoin in High-Risk Neuroblastoma Patients Following Myeloablative Therapy: Children's Oncology Group Study ANBL0931. Front. Immunol. 2018;9:1355. doi: 10.3389/fimmu.2018.01355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu A.L., Gilman A.L., Ozkaynak M.F., London W.B., Kreissman S.G., Chen H.X., Smith M., Anderson B., Villablanca J.G., Matthay K.K., et al. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N. Engl. J. Med. 2010;363:1324–1334. doi: 10.1056/NEJMoa0911123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maris J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010;362:2202–2211. doi: 10.1056/NEJMra0804577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y., Hu R., Xi B., Nie D., Xu H., Liu A. Mechanisms of Senescence-Related NKG2D Ligands Release and Immune Escape Induced by Chemotherapy in Neuroblastoma Cells. Front. Cell Dev. Biol. 2022;10 doi: 10.3389/fcell.2022.829404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nayyar G., Chu Y., Cairo M.S. Overcoming Resistance to Natural Killer Cell Based Immunotherapies for Solid Tumors. Front. Oncol. 2019;9:51. doi: 10.3389/fonc.2019.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qiu B., Matthay K.K. Advancing therapy for neuroblastoma. Nat. Rev. Clin. Oncol. 2022;19:515–533. doi: 10.1038/s41571-022-00643-z. [DOI] [PubMed] [Google Scholar]
- 10.DuBois S.G., Macy M.E., Henderson T.O. High-Risk and Relapsed Neuroblastoma: Toward More Cures and Better Outcomes. Am. Soc. Clin. Oncol. Educ. Book. 2022;42:1–13. doi: 10.1200/EDBK_349783. [DOI] [PubMed] [Google Scholar]
- 11.Del Bufalo F., De Angelis B., Caruana I., Del Baldo G., De Ioris M.A., Serra A., Mastronuzzi A., Cefalo M.G., Pagliara D., Amicucci M., et al. GD2-CART01 for Relapsed or Refractory High-Risk Neuroblastoma. N. Engl. J. Med. 2023;388:1284–1295. doi: 10.1056/NEJMoa2210859. [DOI] [PubMed] [Google Scholar]
- 12.Yoda A., Oishi I., Minami Y. Expression and function of the Ror-family receptor tyrosine kinases during development: lessons from genetic analyses of nematodes, mice, and humans. J. Recept. Signal Transduct. Res. 2003;23:1–15. doi: 10.1081/rrs-120018757. [DOI] [PubMed] [Google Scholar]
- 13.Rebagay G., Yan S., Liu C., Cheung N.K. ROR1 and ROR2 in Human Malignancies: Potentials for Targeted Therapy. Front. Oncol. 2012;2:34. doi: 10.3389/fonc.2012.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dave H., Butcher D., Anver M., Bollard C.M. ROR1 and ROR2-novel targets for neuroblastoma. Pediatr. Hematol. Oncol. 2019;36:352–364. doi: 10.1080/08880018.2019.1646365. [DOI] [PubMed] [Google Scholar]
- 15.Gohil S.H., Paredes-Moscosso S.R., Harrasser M., Vezzalini M., Scarpa A., Morris E., Davidoff A.M., Sorio C., Nathwani A.C., Della Peruta M. An ROR1 bi-specific T-cell engager provides effective targeting and cytotoxicity against a range of solid tumors. OncoImmunology. 2017;6 doi: 10.1080/2162402X.2017.1326437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blom T., Lurvink R., Aleven L., Mensink M., Wolfs T., Dierselhuis M., van Eijkelenburg N., Kraal K., van Noesel M., van Grotel M., Tytgat G. Treatment-Related Toxicities During Anti-GD2 Immunotherapy in High-Risk Neuroblastoma Patients. Front. Oncol. 2020;10 doi: 10.3389/fonc.2020.601076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Richman S.A., Nunez-Cruz S., Moghimi B., Li L.Z., Gershenson Z.T., Mourelatos Z., Barrett D.M., Grupp S.A., Milone M.C. High-Affinity GD2-Specific CAR T Cells Induce Fatal Encephalitis in a Preclinical Neuroblastoma Model. Cancer Immunol. Res. 2018;6:36–46. doi: 10.1158/2326-6066.CIR-17-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cho D., Shook D.R., Shimasaki N., Chang Y.H., Fujisaki H., Campana D. Cytotoxicity of activated natural killer cells against pediatric solid tumors. Clin. Cancer Res. 2010;16:3901–3909. doi: 10.1158/1078-0432.CCR-10-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chu Y., Nayyar G., Jiang S., Rosenblum J.M., Soon-Shiong P., Safrit J.T., Lee D.A., Cairo M.S. Combinatorial immunotherapy of N-803 (IL-15 superagonist) and dinutuximab with ex vivo expanded natural killer cells significantly enhances in vitro cytotoxicity against GD2(+) pediatric solid tumors and in vivo survival of xenografted immunodeficient NSG mice. J. Immunother. Cancer. 2021;9 doi: 10.1136/jitc-2020-002267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chu Y., Nayyar G., Kham Su N., Rosenblum J.M., Soon-Shiong P., Lee J., Safrit J.T., Barth M., Lee D., Cairo M.S. Novel cytokine-antibody fusion protein, N-820, to enhance the functions of ex vivo expanded natural killer cells against Burkitt lymphoma. J. Immunother. Cancer. 2020;8 doi: 10.1136/jitc-2020-001238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Denman C.J., Senyukov V.V., Somanchi S.S., Phatarpekar P.V., Kopp L.M., Johnson J.L., Singh H., Hurton L., Maiti S.N., Huls M.H., et al. Membrane-bound IL-21 promotes sustained ex vivo proliferation of human natural killer cells. PLoS One. 2012;7 doi: 10.1371/journal.pone.0030264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chu Y., Hochberg J., Yahr A., Ayello J., van de Ven C., Barth M., Czuczman M., Cairo M.S. Targeting CD20+ Aggressive B-cell Non-Hodgkin Lymphoma by Anti-CD20 CAR mRNA-Modified Expanded Natural Killer Cells In Vitro and in NSG Mice. Cancer Immunol. Res. 2015;3:333–344. doi: 10.1158/2326-6066.CIR-14-0114. [DOI] [PubMed] [Google Scholar]
- 23.Hudecek M., Schmitt T.M., Baskar S., Lupo-Stanghellini M.T., Nishida T., Yamamoto T.N., Bleakley M., Turtle C.J., Chang W.C., Greisman H.A., et al. The B-cell tumor-associated antigen ROR1 can be targeted with T cells modified to express a ROR1-specific chimeric antigen receptor. Blood. 2010;116:4532–4541. doi: 10.1182/blood-2010-05-283309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ciurea S.O., Schafer J.R., Bassett R., Denman C.J., Cao K., Willis D., Rondon G., Chen J., Soebbing D., Kaur I., et al. Phase 1 clinical trial using mbIL21 ex vivo-expanded donor-derived NK cells after haploidentical transplantation. Blood. 2017;130:1857–1868. doi: 10.1182/blood-2017-05-785659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lanier L.L. NK cell recognition. Annu. Rev. Immunol. 2005;23:225–274. doi: 10.1146/annurev.immunol.23.021704.115526. [DOI] [PubMed] [Google Scholar]
- 26.Wendel M., Galani I.E., Suri-Payer E., Cerwenka A. Natural killer cell accumulation in tumors is dependent on IFN-gamma and CXCR3 ligands. Cancer Res. 2008;68:8437–8445. doi: 10.1158/0008-5472.CAN-08-1440. [DOI] [PubMed] [Google Scholar]
- 27.Quezada M.J., Lopez-Bergami P. The signaling pathways activated by ROR1 in cancer. Cell. Signal. 2023;104 doi: 10.1016/j.cellsig.2023.110588. [DOI] [PubMed] [Google Scholar]
- 28.Zhao Y., Zhang D., Guo Y., Lu B., Zhao Z.J., Xu X., Chen Y. Tyrosine Kinase ROR1 as a Target for Anti-Cancer Therapies. Front. Oncol. 2021;11 doi: 10.3389/fonc.2021.680834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Balakrishnan A., Goodpaster T., Randolph-Habecker J., Hoffstrom B.G., Jalikis F.G., Koch L.K., Berger C., Kosasih P.L., Rajan A., Sommermeyer D., et al. Analysis of ROR1 Protein Expression in Human Cancer and Normal Tissues. Clin. Cancer Res. 2017;23:3061–3071. doi: 10.1158/1078-0432.CCR-16-2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wolfl M., Jungbluth A.A., Garrido F., Cabrera T., Meyen-Southard S., Spitz R., Ernestus K., Berthold F. Expression of MHC class I, MHC class II, and cancer germline antigens in neuroblastoma. Cancer Immunol. Immunother. 2005;54:400–406. doi: 10.1007/s00262-004-0603-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Farhan S., Lee D.A., Champlin R.E., Ciurea S.O. NK cell therapy: targeting disease relapse after hematopoietic stem cell transplantation. Immunotherapy. 2012;4:305–313. doi: 10.2217/imt.11.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Waldhauer I., Steinle A. NK cells and cancer immunosurveillance. Oncogene. 2008;27:5932–5943. doi: 10.1038/onc.2008.267. [DOI] [PubMed] [Google Scholar]
- 33.Raieli S., Di Renzo D., Lampis S., Amadesi C., Montemurro L., Pession A., Hrelia P., Fischer M., Tonelli R. MYCN Drives a Tumor Immunosuppressive Environment Which Impacts Survival in Neuroblastoma. Front. Oncol. 2021;11 doi: 10.3389/fonc.2021.625207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang M., Weiss W.A. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 2013;3:a014415. doi: 10.1101/cshperspect.a014415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu E., Marin D., Banerjee P., Macapinlac H.A., Thompson P., Basar R., Nassif Kerbauy L., Overman B., Thall P., Kaplan M., et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020;382:545–553. doi: 10.1056/NEJMoa1910607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanber K., Savani B., Jain T. Graft-versus-host disease risk after chimeric antigen receptor T-cell therapy: the diametric opposition of T cells. Br. J. Haematol. 2021;195:660–668. doi: 10.1111/bjh.17544. [DOI] [PubMed] [Google Scholar]
- 37.Heipertz E.L., Zynda E.R., Stav-Noraas T.E., Hungler A.D., Boucher S.E., Kaur N., Vemuri M.C. Current Perspectives on "Off-The-Shelf" Allogeneic NK and CAR-NK Cell Therapies. Front. Immunol. 2021;12 doi: 10.3389/fimmu.2021.732135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Conlon K.C., Lugli E., Welles H.C., Rosenberg S.A., Fojo A.T., Morris J.C., Fleisher T.A., Dubois S.P., Perera L.P., Stewart D.M., et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J. Clin. Oncol. 2015;33:74–82. doi: 10.1200/JCO.2014.57.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Van den Bergh J.M.J., Smits E.L.J.M., Versteven M., De Reu H., Berneman Z.N., Van Tendeloo V.F.I., Lion E. Characterization of Interleukin-15-Transpresenting Dendritic Cells for Clinical Use. J. Immunol. Res. 2017;2017 doi: 10.1155/2017/1975902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu X., Marcus W.D., Xu W., Lee H.I., Han K., Egan J.O., Yovandich J.L., Rhode P.R., Wong H.C. Novel human interleukin-15 agonists. J. Immunol. 2009;183:3598–3607. doi: 10.4049/jimmunol.0901244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Margolin K., Morishima C., Velcheti V., Miller J.S., Lee S.M., Silk A.W., Holtan S.G., Lacroix A.M., Fling S.P., Kaiser J.C., et al. Phase I Trial of ALT-803, A Novel Recombinant IL15 Complex, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2018;24:5552–5561. doi: 10.1158/1078-0432.CCR-18-0945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wrangle J.M., Velcheti V., Patel M.R., Garrett-Mayer E., Hill E.G., Ravenel J.G., Miller J.S., Farhad M., Anderton K., Lindsey K., et al. ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: a non-randomised, open-label, phase 1b trial. Lancet Oncol. 2018;19:694–704. doi: 10.1016/S1470-2045(18)30148-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Romee R., Cooley S., Berrien-Elliott M.M., Westervelt P., Verneris M.R., Wagner J.E., Weisdorf D.J., Blazar B.R., Ustun C., DeFor T.E., et al. First-in-human phase 1 clinical study of the IL-15 superagonist complex ALT-803 to treat relapse after transplantation. Blood. 2018;131:2515–2527. doi: 10.1182/blood-2017-12-823757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Foltz J.A., Hess B.T., Bachanova V., Bartlett N.L., Berrien-Elliott M.M., McClain E., Becker-Hapak M., Foster M., Schappe T., Kahl B., et al. Phase I Trial of N-803, an IL15 Receptor Agonist, with Rituximab in Patients with Indolent Non-Hodgkin Lymphoma. Clin. Cancer Res. 2021;27:3339–3350. doi: 10.1158/1078-0432.CCR-20-4575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Desbois M., Beal C., Charrier M., Besse B., Meurice G., Cagnard N., Jacques Y., Bechard D., Cassard L., Chaput N. IL-15 superagonist RLI has potent immunostimulatory properties on NK cells: implications for antimetastatic treatment. J Immunother Cancer. 2020;8:e000632. doi: 10.1136/jitc-2020-000632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mortier E., Quéméner A., Vusio P., Lorenzen I., Boublik Y., Grötzinger J., Plet A., Jacques Y. Soluble interleukin-15 receptor alpha (IL-15R alpha)-sushi as a selective and potent agonist of IL-15 action through IL-15R beta/gamma. Hyperagonist IL-15 x IL-15R alpha fusion proteins. J. Biol. Chem. 2006;281:1612–1619. doi: 10.1074/jbc.M508624200. [DOI] [PubMed] [Google Scholar]
- 47.Garralda E., Naing A., Galvao V., LoRusso P., Grell P., Cassier P.A., Gomez-Roca C.A., Korakis I., Bechard D., Palova Jelinkova L., et al. Interim safety and efficacy results from AURELIO-03: A phase 1 dose escalation study of the IL-2/IL-15 receptor ßγ superagonist SOT101 as a single agent and in combination with pembrolizumab in patients with advanced solid tumors. J. Clin. Oncol. 2022;40:2502. [Google Scholar]
- 48.Silvestre R.N., Eitler J., de Azevedo J.T.C., Tirapelle M.C., Fantacini D.M.C., de Souza L.E.B., Swiech K., Covas D.T., Calado R.T., Montero P.O., et al. Engineering NK-CAR.19 cells with the IL-15/IL-15Rα complex improved proliferation and anti-tumor effect in vivo. Front. Immunol. 2023;14 doi: 10.3389/fimmu.2023.1226518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu E., Tong Y., Dotti G., Shaim H., Savoldo B., Mukherjee M., Orange J., Wan X., Lu X., Reynolds A., et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32:520–531. doi: 10.1038/leu.2017.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li L., Mohanty V., Dou J., Huang Y., Banerjee P.P., Miao Q., Lohr J.G., Vijaykumar T., Frede J., Knoechel B., et al. Loss of metabolic fitness drives tumor resistance after CAR-NK cell therapy and can be overcome by cytokine engineering. Sci. Adv. 2023;9 doi: 10.1126/sciadv.add6997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Christodoulou I., Ho W.J., Marple A., Ravich J.W., Tam A., Rahnama R., Fearnow A., Rietberg C., Yanik S., Solomou E.E., et al. Engineering CAR-NK cells to secrete IL-15 sustains their anti-AML functionality but is associated with systemic toxicities. J. Immunother. Cancer. 2021;9 doi: 10.1136/jitc-2021-003894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fiore P.F., Di Matteo S., Tumino N., Mariotti F.R., Pietra G., Ottonello S., Negrini S., Bottazzi B., Moretta L., Mortier E., Azzarone B. Interleukin-15 and cancer: some solved and many unsolved questions. J. Immunother. Cancer. 2020;8 doi: 10.1136/jitc-2020-001428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Felices M., Lenvik A.J., McElmurry R., Chu S., Hinderlie P., Bendzick L., Geller M.A., Tolar J., Blazar B.R., Miller J.S. Continuous treatment with IL-15 exhausts human NK cells via a metabolic defect. JCI Insight. 2018;3 doi: 10.1172/jci.insight.96219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vivier E., Nunès J.A., Vély F. Natural killer cell signaling pathways. Science. 2004;306:1517–1519. doi: 10.1126/science.1103478. [DOI] [PubMed] [Google Scholar]
- 55.Cozar B., Greppi M., Carpentier S., Narni-Mancinelli E., Chiossone L., Vivier E. Tumor-Infiltrating Natural Killer Cells. Cancer Discov. 2021;11:34–44. doi: 10.1158/2159-8290.CD-20-0655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pende D., Meazza R. N-803: a double-edged sword in haplo-NK therapy. Blood. 2022;139:1122–1124. doi: 10.1182/blood.2021014789. [DOI] [PubMed] [Google Scholar]
- 57.Berrien-Elliott M.M., Becker-Hapak M., Cashen A.F., Jacobs M., Wong P., Foster M., McClain E., Desai S., Pence P., Cooley S., et al. Systemic IL-15 promotes allogeneic cell rejection in patients treated with natural killer cell adoptive therapy. Blood. 2022;139:1177–1183. doi: 10.1182/blood.2021011532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bagwell C.B., Hunsberger B., Hill B., Herbert D., Bray C., Selvanantham T., Li S., Villasboas J.C., Pavelko K., Strausbauch M., et al. Multi-site reproducibility of a human immunophenotyping assay in whole blood and peripheral blood mononuclear cells preparations using CyTOF technology coupled with Maxpar Pathsetter, an automated data analysis system. Cytometry B Clin. Cytom. 2020;98:146–160. doi: 10.1002/cyto.b.21858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hadjadj J., Yatim N., Barnabei L., Corneau A., Boussier J., Smith N., Péré H., Charbit B., Bondet V., Chenevier-Gobeaux C., et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science. 2020;369:718–724. doi: 10.1126/science.abc6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Behbehani G.K., Bendall S.C., Clutter M.R., Fantl W.J., Nolan G.P. Single-cell mass cytometry adapted to measurements of the cell cycle. Cytometry A. 2012;81:552–566. doi: 10.1002/cyto.a.22075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chu Y., Milner J., Lamb M., Maryamchik E., Rigot O., Ayello J., Harrison L., Shaw R., Behbehani G.K., Mardis E.R., et al. Manufacture and Characterization of Good Manufacturing Practice-Compliant SARS-COV-2 Cytotoxic T Lymphocytes. J. Infect. Dis. 2023;227:788–799. doi: 10.1093/infdis/jiac500. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available upon reasonable request.