

Abstract
INTRODUCTION:
Neuropeptides are small chains of amino acids that play vital roles in the endocrine and nervous systems, regulating diverse functions such as metabolism, pain perception, sleep and circadian rhythm, mood, and learning. Malfunctions in neuropeptide signaling have been implicated in many diseases, including insomnia, diabetes, and depression. Monitoring neuropeptides with high spatiotemporal resolution in vivo could provide insights into their functions in physiological conditions and disease pathophysiology and advance new drug development. Recent progress led to the development of fluorescent sensors for selected neuropeptides, including orexin and oxytocin, which helped reveal their in vivo release patterns and dynamics. However, given the large number of neuropeptides, the scalable development of neuropeptide sensors is appealing yet challenging.
RATIONALE:
Recent endeavors from our group and others in combining G protein-coupled receptors (GPCRs) with circularly permutated green fluorescent protein (cpGFP) have led to the development of several GPCR activation-based (GRAB) sensors that can detect neuromodulators with high spatiotemporal resolution. As most neuropeptide receptors belong to the GPCR superfamily, which endogenously has high affinity and specificity for their ligands, one could, in principle, develop genetically encoded fluorescence sensors for each neuropeptide. However, given the diversity of neuropeptides and their cognate GPCRs, developing and optimizing a GRAB sensor with each GPCR de novo could be labor-intensive. Despite variations in their sequences and structures, neuropeptide GPCRs do share a common structural change upon ligand-induced activation. Therefore, a streamlined approach by transplanting the entire cpGFP-containing intracellular loop 3 (ICL3) from existing GRAB sensors to new neuropeptide GRAB sensors could give the new sensors the ability to change fluorescence upon ligand binding without modifying and optimizing each sensor individually. This strategy could greatly accelerate the development of a wide variety of GRAB sensors tailored to detect the real-time dynamics of diverse endogenous neuropeptides with minimal optimization needed.
RESULTS:
Using this grafting strategy, we developed a series of highly selective and sensitive genetically encoded neuropeptide sensors for detecting somatostatin (SST), corticotropin-releasing factor (CRF), cholecystokinin (CCK), neuropeptide Y (NPY), neurotensin (NTS), and vasoactive intestinal peptide (VIP). These sensors are engineered by replacing the ICL3 in each new peptide GPCR with the entire cpGFP-containing ICL3 from previously optimized norepinephrine (NE) sensor GRABNE. This array of peptide sensors enables the detection of specific neuropeptides at nanomolar concentrations with minimal disruptions to neuronal activity, transcriptional profiling, and animal behaviors. We demonstrated the utilities of SST and CRF sensors in detail. Specifically, we used the SST1.0 sensor to detect activity-dependent SST release in cultured rat cortical neurons and mice pancreatic islets and to reveal the SST dynamics during conditioned learning in mice. Moreover, the CRF1.0 sensor reliably reported the electrical stimulation evoked release of CRF in acute brain slices from mice, monitored in vivo changes in CRF levels in the hypothalamus, and visualized spatially resolved cortical CRF dynamics in response to stress-inducing stimuli in mice.
CONCLUSION:
To permit the fast and scalable development of neuropeptide sensors, we have developed an ICL3 grafting method that efficiently generates genetically encoded fluorescent sensors. Leveraging this method, we developed a panel of sensors to detect the real-time dynamics of six commonly studied neuropeptides. We demonstrated that our GRAB SST and CRF sensors can be used to monitor neuropeptide dynamics in vitro, ex vivo, and in vivo with good sensitivity, selectivity, and spatiotemporal resolution. This flexible engineering strategy and toolkit of optimized peptide sensors pave the way for studying the release, regulation, and functions of diverse neuropeptides under both physiological and pathophysiological states.
Neuropeptides are key signaling molecules in the endocrine and nervous systems that regulate many critical physiological processes. Understanding the functions of neuropeptides in vivo requires the ability to monitor their dynamics with high specificity, sensitivity, and spatiotemporal resolution. However, this has been hindered by the lack of direct, sensitive, and noninvasive tools. We developed a series of GRAB (G protein–coupled receptor activation‒based) sensors for detecting somatostatin (SST), corticotropin-releasing factor (CRF), cholecystokinin (CCK), neuropeptide Y (NPY), neurotensin (NTS), and vasoactive intestinal peptide (VIP). These fluorescent sensors, which enable detection of specific neuropeptide binding at nanomolar concentrations, establish a robust tool kit for studying the release, function, and regulation of neuropeptides under both physiological and pathophysiological conditions.
Graphical abstract
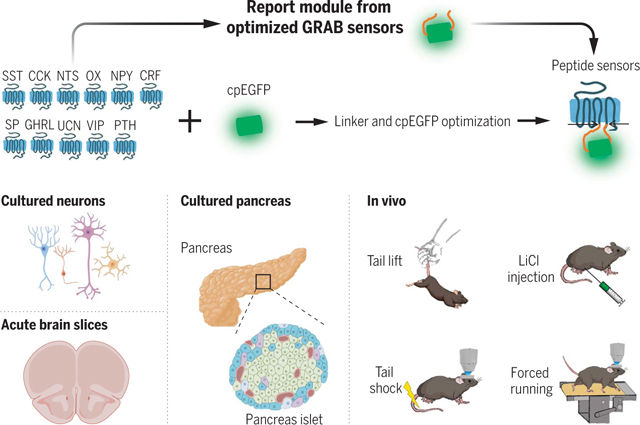
Development and applications of fluorescent sensors for detecting neuropeptides. OX, orexin; SP, substance P; GHRL, ghrelin; UCN, urocortin; PTH, parathyroid hormone-related peptide. [Created with BioRender.com]
Neuropeptides were first identified nearly seven decades ago as hormone regulators in the endocrine system and have since been recognized as highly effective signaling molecules in both central and peripheral tissues (1–4). In the brain, neuropeptides regulate many types of physiological functions, such as digestion, metabolism, sleep and circadian rhythm, reproduction, and higher cognitive processes (5–8). Thus, neuropeptide signaling—which is mediated primarily by G protein-coupled receptors (GPCRs)—provides a key site for drug targeting for a wide range of diseases and conditions such as insomnia, pain, obesity, and diabetes (9–11).
The ability to measure the spatial and temporal dynamics of neuropeptides in vivo is essential for understanding their functions and the mechanisms that regulate these key signaling molecules. However, current methods for detecting peptides in the brain either lack the necessary spatiotemporal resolution or are not suitable for in vivo application. Thus, the precise spatiotemporal dynamics and release patterns of endogenous peptides remain poorly understood.
Genetically encoded fluorescent indicators have proven suitable for measuring the dynamics of signaling molecules with high spatiotemporal resolution in vivo. For example, bacterial periplasmic binding protein (PBP) based sensors have been developed to detect neurotransmitters such as glutamate, acetylcholine, and serotonin (12–14). However, corresponding PBPs for peptides and proteins are unlikely to exist. Generating peptide-sensing PBPs with high affinity and selectivity will therefore require considerable bioengineering and screening. Notably, most neuropeptide receptors are GPCRs, and peptide or protein GPCR ligands make up 70% of all nonolfactory GPCR ligands in the human body (Fig. 1A) (15,16). Peptide GPCRs can provide a valuable opportunity for generating genetically encoded sensors with high sensitivity and selectivity. Previously, our group and others developed and characterized several GPCR activation-based (GRAB) intensiometric biosensors, using GPCRs as the ligand-sensing unit and circularly permutated green fluorescent protein (cpGFP) as the reporter module, for detecting small-molecule transmitters (17–21) and some peptides, including oxytocin and orexin (22–24). The strategy for developing these GRAB sensors includes screening for the optimal cpGFP placement site within the receptor’s third intracellular loop 3 (ICL3); however, given the large number of peptide and protein GPCRs (with 131 expressed in humans) and the high variability of ICL3 among GPCRs (ranging from 2 to 211 amino acids), developing and optimizing a GRAB sensor for each GPCR would be highly labor-intensive (25,26). Despite this structural variation in the ICL3, however, peptide GPCRs undergo a common structural change upon activation, with an outward movement of transmembrane 6 (TM6) observed in both class A and class B1 peptide GPCRs (Fig. 1B) (27–29). Thus, peptide GPCRs generated using the entire cpGFP-containing ICL3 in previously optimized GRAB sensors may retain the ability to couple the activation-induced conformational change with an increase in fluorescence, thereby accelerating the development of a wide variety of GRAB peptide sensors.
Fig. 1. A general method for engineering fluorescent indicators for neuropeptides.

(A) (Left) Illustration of peptide-containing large dense-core vesicles (LDCVs), neurotransmitter-containing synaptic vesicles (SVs), and their receptors in a synapse. (Right) Proportion and number of peptide or protein GPCR ligands and nonpeptidergic GPCRs in humans, with corresponding examples. (B) Superposition of active (blue) and inactive (gray) structures of class A SSTR2 [Protein Data Bank (PDB) IDs 7XMR and 7XN9] and class B1CRF1R (PDB IDs 4K5Y and 6PB0). The dashed arrows indicate the movement of the sixth transmembrane domain (TM6). (C) Schematic diagram depicting the ICL3 transplantation strategy for developing GRAB sensors. (D) Fluorescence responses (ΔF/F0) of peptide GPCR chimeras with ICL3 transplanted from the indicated sensors. The amino acid (AA) numbers flanking cpGFP are labeled. The number and percentage of GPCRs with a maximum response exceeding 0.3 (dashed horizontal line) are shown, and these sensors are shaded in yellow. Each data point represents the average of between 100 and 300 cells measured in one well. (E) Schematic diagram showing the steps for GRAB sensor optimization, including the ICL3 replacement site, linker optimization, and cpEGFP optimization. Black triangles indicate the optimization sites. (F) Optimization of the SST, CCK, CRF, NTS, NPY, and VIP sensors. In each plot, the black dot indicates the initial version after ICL3 transplantation. After optimization, candidates with the highest ΔF/F0 were selected as the first-generation (1.0) sensors (blue dots). (G) Summary of the peak fluorescence response (top) and maximum brightness (bottom) of the indicated peptide sensors developed by transplanting ICL3 into the indicated class A and class B1 GPCRs (n = 4 wells containing 100 to 300 cells per well).
In this study, we used this strategy to develop a series of GRAB sensors for detecting neuropeptides with nanomolar affinity. These sensors are able to reveal peptide dynamics with single-cell spatial resolution and subsecond temporal resolution.
Developing a generalized method for engineering fluorescent sensors to detect neuropeptides
Neuropeptides and peptide receptors were widely expressed in the brain (30–32), and we chose these highly expressed peptide GPCRs as scaffolds for sensor development (Fig. 1C). To develop a scalable method for generating a series of genetically encoded peptide sensors, we replaced the ICL3 domains in various peptide GPCRs with the ICL3 in several existing sensors, including GRABNE1m, GRABDA2m, GRAB5-HT1.0, GRABACh3.0, and dLight1.3b (19–21,33,34). These sensor-derived ICL3s vary in length with respect to the number of amino acids that flank the cpGFP module (table S1); thus, GRAB peptide sensors were generated by replacing the ICL3 in the GPCR with the linker sequences and cpGFP derived from the inner membrane regions of TM5 and TM6, located at sites around 5.70 and 6.28, respectively (Fig. 1C). Each newly generated candidate peptide sensor was then expressed in human embryonic kidney (HEK) 293T (HEK293T) cells together with a plasma membrane-targeted mCherry (as a marker of surface expression) (fig. S1A). Each candidate’s performance was measured with respect to trafficking to the plasma membrane and the change in the sensor’s fluorescence in response to the appropriate ligand (Fig. 1D and fig. S1, B and C). Candidates with a trafficking index of >80% (measured as the Pearson correlation coefficient between the expression of a candidate and mCherry) and a fluorescence increase of >30% upon ligand application were considered as responsive peptide sensors.
We found that peptide sensors containing the ICL3s derived from GRABNE1m and GRABDA2m—both of which contain relatively long linkers (table S1)—had a higher trafficking index and a larger response than did sensors with ICL3s derived from GRAB5-HT10, GRABACh3.0, or dLight1.3b (Fig. 1D). For further development, we chose the more generally applicable ICL3 in GRABNE1m and optimized the peptide sensor prototypes in four steps, including modifying the ICL3 replacing sites, modifying the linker sequences, and modifying critical residues both in the cpGFP module and in the GPCR (fig. S2). The critical residues were chosen from the cpGFP alignment of different cpGFP-based sensors (20) and from the potential interface with cpGFP in the GPCRs (fig. S2). The first three steps are depicted schematically in Fig. 1E, and the complete optimization processes are shown for six peptide GRAB sensors in Fig. 1F, in which the optimal version of the CRF sensor yielded a >10-fold increase in fluorescence upon CRF binding compared with the original candidate (Fig. 1, E and F, and fig. S2). Using the same strategy for both class A and class B1 peptide GPCRs—including the receptors SSTR5 (somatostatin receptor type 5), CCKBR (gastrin/cholecystokinin type B receptor), NTSR1 (neurotensin receptor type 1), HCRTR2/OX2R [hypocretin (orexin) receptor 2], NPY1R (neuropeptide Y receptor type 1), TACR1 (tachykinin receptor 1), GHS-R (growth hormone secretagogue receptor type 1), CRF1R (corticotropin releasing factor type 1 receptor), CRF2R, VIPR2 (vasoactive intestinal polypeptide receptor 2), and PTH1R (parathyroid hormone 1 receptor)—we then developed and optimized a series of GRAB peptide sensors for detecting somatostatin (SST), cholecystokinin (CCK), neurotensin (NTS), orexin/hypocretin (OX), neuropeptide Y (NPY), substance P (SP), ghrelin (GHRL), corticotropin-releasing factor (CRF), urocortin (UCN), vasoactive intestinal peptide (VIP), and parathyroid hormone-related peptide (PTH) (Fig. 1G).
Characterization of GRAB peptide sensors in cultured cells
Next, we characterized the properties of the SST1.0, CCK1.0, CRF1.0, NPY1.0, NTS1.0, and VIP1.0 sensors (table S2); the full amino acid sequences of these sensors are shown in fig. S3. When expressed in HEK293T cells, all six GRAB sensors localized primarily to the plasma membrane and produced a robust change in fluorescence (ranging from a 2.5- to 12-fold increase in fluorescence) in response to their respective ligand (table S2), and each response was blocked by the corresponding GPCR antagonist (fig. S4, A and B). These sensors also retained the ligand selectivity of their respective GPCR scaffolds and had high sensitivity, with apparent half-maximum effective concentrations (EC50) of approximately 10 to 100 nM (fig. S4C and table S2). For example, the CRF1.0 sensor was based on CRF1R, which has a higher affinity for CRF than for UCN (35). As expected, the CRF1.0 sensor’s EC50 for CRF was 33 nM, compared with 69 nM for urocortin 1 (UCN1), whereas the peptides UCN2 and UCN3 had no effect on the CRF1.0 sensor (fig. S4C2). We then tested the ligand specificity of CRF1.0 and SST1.0 sensors and found that none of the sensors responded to glutamate (Glu), γ-aminobutyric acid (GABA), dopamine (DA), or any other neuropeptides tested (Fig. 2E).
Fig. 2. Characterization of SST1.0 and CRF1.0 sensors in vitro.

(A) Primary cultured rat cortical neurons expressing SST1.0 (A1) or CRF1.0 (A2), showing sensor expression (left), pseudocolor responses (middle), and cell membrane localization (right). Scale bars, 100 μm (left) and 20 μm (right). (B) Example fluorescence traces (left) and summary data (right) of neurons expressing SST1.0 (B1) or CRF1.0 (B2); where indicated, peptides and antagonists were applied (n = 66 to 115 ROIs from three or four coverslips). (C) Normalized dose-response curves of neurons expressing SST1.0 (C1) or CRF1.0 (C2) in response to the indicated ligands; n = 3 cultures each with 20 to 40 ROIs. (D) Summary of the fluorescence change measured in neurons expressing SST1.0 (D1) or CRF1.0 (D2) in response to a 2-hour continuous application of 1 μM SST-14 or 300 nM CRF, respectively; n = 4 cultures each with 20 to 40 ROIs. (E) Summary of normalized ΔF/F0 in HEK293T cells expressing SST1.0 (left) or CRF1.0 (right) in response to the indicated compounds applied: SST-14, CRF, UCN2, UCN3, CCK-8s, NPY, NTS, VIP, pituitary adenylate cyclase-activating polypeptide (PACAP), adrenocorticotropic hormone (ACTH), β-melanocyte-stimulating hormone (β-MSH), γ-MSH, growth hormone-releasing hormone (GHRH), oxytocin (OT), vasopressin (AVP), vosotocin, isotocin, glucagon, glucagon-like peptide (GLP-1), DAMGO, dynorphin A (DynA), enkephalin (Enk), goserelin, ghrelin, galanin, orexin A (OX-A), SP, neurokinin B (NKB), and bombesin (BB) were applied at 1 μM, while Glu, GABA, DA, norepinephrine (NE), serotonin (5-HT), histamine (HA), ATP, ADP, adenosine (Ado), teriparatide (TPTD), and taltirelin were applied at 10 μM (n = 4 wells containing 100 to 300 cells per well). (F and G) G protein and β-arrestin coupling were measured using the split-luciferase complementation assay (F1), a cyclic adenosine monophosphate (cAMP) reporter (F2), and the Tango assay (G1 and G2) in cells expressing either the wild-type peptide receptor (red), sensor (green), or no receptor (Ctrl; gray) in the presence of the indicated concentrations of the ligand; n = 3 wells each. AU, arbitrary units. (H) RNA-seq analysis shows that GRAB peptide sensors did not alter the cellular transcriptome. Comparisons of transcriptomes between cortex tissue expressing SST1.0 or EGFP-CAAX (H1) and between CRF1.0 or EGFP-CAAX (H2). Pearson's correlation coefficient analysis was used to evaluate the differential RNA expression.
We also measured the single-photon spectra of these six peptide sensors and found a common excitation peak at ~500 nm and a common emission peak at ~520 nm, with an isosbestic point of the excitation wavelength at ~420 nm (fig. S4E and table S2). The two-photon excitation cross section of the SST and CRF sensors showed excitation peaks at 920 to 930 nm in the presence of the respective ligands (fig. S4F). The kinetics of the peptide sensors’ responses were also measured by locally applying the corresponding peptide ligands and antagonists and then recording the change in fluorescence using line-scan confocal microscopy. The resulting time constants of the rise in the signal (τon) ranged from approximately 0.3 to 0.9 s, and the time constants of the signal decay (τoff) ranged from approximately 3 to 12 s (fig. S4D and table S2).
Next, we measured the properties of our GRAB peptide sensors expressed in cultured rat cortical neurons. All six neuropeptide sensors, including the SST1.0 and CRF1.0 sensors, localized to the neuronal membrane both at the cell body and in extended ramified neurites and also responded robustly to ligand application (Fig. 2, A and B, and fig. S5). Moreover, when expressed in cultured neurons, the peptide sensors’ responses and apparent EC50 values were measured, and the responses were again blocked by the respective GPCR antagonists (Fig. 2, B and C, and table S3). Finally, for most of the peptide sensors, the ligand-induced change in fluorescence remained stable for up to 120 min in neurons exposed to a saturated ligand concentration (Fig. 2D), indicating minimal internalization of the peptide sensors.
We then tested whether our GRAB peptide sensors couple to downstream signaling pathways by measuring G protein-mediated signaling and β-arrestin recruitment. Although wild-type peptide receptors activated both signaling pathways, their corresponding GRAB sensors elicited significantly reduced or virtually no downstream signaling (Fig. 2, F and G). Coexpression of peptide sensors showed no detectable alteration to the affinity and efficacy of corresponding wild-type receptors in response to their ligands (fig. S6, A and B), and no significant differences were observed in neuronal Ca2+ response (fig. S6, C to G). Additionally, RNA sequencing (RNA-seq) analysis shows that GRAB peptide sensors did not alter the cellular transcriptome in either cultured neurons or the mouse cortex (Fig. 2H and fig. S7). Furthermore, expressing GRAB peptide sensors exhibits no detectable change in the expression level and localization of native GPCRs (figs. S8 and S9), highlighting that overexpression of peptide sensors does not disrupt endogenous signaling.
Our SST, CCK, CRF, NPY, NTS, and VIP sensors were all highly sensitive, specific, and produced a robust real-time increase in fluorescence in response to their corresponding ligands, without activating downstream signaling pathways. We chose the SST and CRF sensors for further study.
The SST1.0 sensor can be used to detect the release of endogenous SST in cortical neurons
Neuropeptides are widely used as markers to categorize various types of neurons, with SST-expressing neurons representing subsets of interneurons in the cerebral cortex (31, 36). Although used as a marker for neuronal subpopulations, whether SST is actually released from cortex neurons—and the spatiotemporal pattern of its potential release—has not been well investigated. Applying trains of electrical field stimuli to cultured mouse hippocampal neurons can induce the fusion of peptide-containing dense-core vesicles (37, 38). To detect SST release from these neurons, we expressed the SST1.0 sensor in cultured primary rat cortical neurons. Applying increasing numbers of pulse trains elicited increasingly strong responses then reached a plateau (Fig. 3, A to C, and table S4). Application of 75 mM K+ to depolarize the neurons also induced a robust increase in SST1.0 fluorescence that was blocked by the SST receptor antagonist BIM 23056; moreover, no increase in response was measured in neurons expressing the membrane-targeted EGFP-CAAX (Fig. 3, A to C, and fig. S10A). The signal was reversible, and the rise and decay half-times of the SST1.0 signal induced by stimulation and K+ application are summarized in fig. S10B. Furthermore, the SST1.0 response was directly correlated with the corresponding increase in cytosolic Ca2+ levels measured using the fluorescent Ca2+ indicator Calbryte-590 (fig. S10C).
Fig. 3. Imaging SST release in cultured neurons and pancreatic islets.

(A) (Left) Schematic diagram depicting the experimental strategy. (Middle) Fluorescence image of rat cortical neurons expressing SST1.0. (Right) Pseudocolor images of zoom-in boutons or soma areas bounded by yellow rectangles in the middle image; where indicated, train electrical stimuli (50 pulses delivered at 50 Hz, 0.5-s intertrain interval) or 75 mM K+ were applied. The white arrow indicates soma regions. Scale bars, 100 μm (middle) and 50 μm (right). (B) Example traces of the change in SST1.0 (green) and Calbryte-590 (red) in response to electric stimuli; SST1.0 (with or without antagonist BIM) and EGFP-CAAX fluorescence in response to 75 mM K+; yellow shading indicates the 75 mM KCl perfusion time. (C) Summary of the peak change in fluorescence measured in neurons expressing SST1.0 or EGFP-CAAX in response to burst stimuli or 75 mM K+. (D) (Left) Schematic diagram depicting the experimental strategy in which pancreatic islets were isolated, infected with adenoviruses expressing SST1.0, and treated with high (20 mM) glucose. (Right) Example fluorescence images of an SST1.0-expressing pancreatic islet before and after application of 20 mM glucose. Scale bar, 50 μm. (E) SST1.0 fluorescence was measured at the indicated ROIs in the same pancreatic islet shown in (D). On the basis of the response patterns (right panel), ROI1 and ROI2 are classified as nonburst regions (blue), whereas ROI3 is classified as a burst region (red). Scale bar, 50 μm. (F and G) Summary of the burst frequency (F) and peak response (G) measured for nonburst and burst regions; n = 30 to 55 ROIs from three islets. (H) Representative spatial-temporal profile of the SST1.0 fluorescence response measured during a single burst. (I) Example time-lapse pseudocolor images of SST1.0 fluorescence measured in the burst region. The white arrow indicates the location from which the signal originates. Scale bar, 10 μm. (J) Summary of the effective diffusion coefficient (D); note that the y axis is a log scale (n = 12 burst events from three islets).
The SST1.0 sensor can be used to detect glucose-stimulated SST release in isolated pancreatic islets
SST plays an essential role in feeding and energy expenditure by affecting central and peripheral tissues (39). In pancreatic islets, the release of SST from delta (δ) cells is critical for regulating the activity of glucagon-releasing α cells and insulin-releasing β cells (40, 41). However, the spatiotemporal pattern of SST release in individual islets has not been investigated. To measure SST release in islets, we expressed SST1.0 under the control of a cytomegalovirus (CMV) promoter in mouse pancreatic islets cultures using adenovirus infection. Application of the peptide SST-14—but not CCK—caused a robust increase in SST1.0 fluorescence, and this response was blocked by the SST receptor antagonist BIM 23056 but not by the CCK receptor antagonist YM 022 (fig. S10, D to G). We then examined whether SST1.0 can detect the release of endogenous SST in islets in response to high glucose stimulation (42,43). Application of 20 mM glucose caused a progressive increase in SST1.0 fluorescence (Fig. 3, D and E, and fig. S10, H to J). Moreover, the increase in SST1.0 fluorescence had a distinct spatial pattern within the islet, with regions that could be classified as either nonburst or burst regions (Fig. 3E and movie S1). Analyzing these regions separately revealed that burst regions exhibited a phasic SST1.0 response in the presence of 20 mM glucose, with a higher burst rate and larger peak responses compared with nonburst regions (Fig. 3, F and G). During a single burst event, the SST1.0 signal first increased at a focal hotspot and then propagated over time to neighboring cells (Fig. 3, H and I, and fig. S10K). The response measured near the initial hotspot was more rapid and robust than the responses measured farther away from the hotspot (fig. S10L). Moreover, at the 10 s time point, this propagation of the SST1.0 signal had an average half-width of ~6 μm (fig. S10, M to O), and this half-width increased over time, with an average diffusion coefficient of ~0.4 μm2/s (Fig. 3J).
Detection of endogenous SST release in a Pavlovian conditioning process
SST is widely distributed throughout the central nervous system (44). Previous studies indicate a correlation between endogenous SST levels and learning and memory performance. Mice with SST deficiencies exhibit impaired associative learning (45–47). The basolateral amygdala (BLA) is known as the hub for converging inputs of different modalities during associative learning, with BLA SST interneurons involved in these learning processes (48–50). However, the SST dynamics during the learning process remains unknown. We recorded the SST1.0 signal in mouse BLA during olfactory Pavlovian learning (Fig. 4, A and B). In the early training phase, reward but not punishment unconditional stimulus (US) evoked a transient increase in SST1.0 signals (Fig. 4C). After conditioning by pairing the conditional stimulus (CS; odor) with the US (reward), a fluorescence response developed to the reward-predictive CS, lasting until reward delivery and then decreasing after the US. In contrast, no observable SST1.0 response was found during “nothing” or “punishment” trials (Fig. 4, C to E). To evaluate whether expression of SST1.0 would affect normal animal behaviors, we recorded body weight and food and water consumption and performed open field test and elevated plus maze test. We found no significant behavioral difference between mice expressing enhanced yellow fluorescent protein (EYFP; control) or SST1.0 at BLA (fig. S14, A to D).
Fig. 4. Detection of endogenous SST release in a Pavlovian conditioning process.

(A) Schematic for fiber photometry recording of SST1.0-expressing neurons from the BLA of a mouse. (B) Schematic for olfactory Pavlovian conditioning tasks. (C) Exemplar time-aligned lick trials, pseudocolor images, and averaged traces from a mouse in early-training and well-learned sessions. (D) Averaged traces of SST1.0 signals in early-training and well-learned sessions (n = 5 mice). (E) Group analysis of the peak ΔF/F0 of SST1.0 signals to US and CS in different sessions. Each trace represents data from one animal (n = 5 mice); Studenťs t test, n.s., not significant; *P < 0.05. (US responses between early-training and well-learned: P = 0.6336 in nothing trial, P = 0.0184 in reward trial, and P = 0.8859 in punishment trial; CS responses between early-training and well-learned: P = 0.6517 in nothing trial, P = 0.0900 in reward trial, and P = 0.3499 in punishment trial.) Values with error bars indicate mean ± SEM.
Characterization of the CRF1.0 sensor expressed in acute brain slices
CRF is an anxiogenic neuropeptide, and CRF neurons in the central amygdala (CeA) play an important role in several conditions related to fear, anxiety, and alcohol addiction (51–54). To test whether the CRF1.0 sensor can be used to measure the release of endogenous CRF in the CeA, we expressed the CRF1.0 sensor in the CeA and then recorded the response in acute brain slices using two-photon fluorescence microscopy (Fig. 5A). Electric stimuli delivered at 20 Hz induced a robust increase in CRF1.0 fluorescence, with larger responses induced by increased numbers of pulses, and this response was significantly blocked by treating the slices with the CRF receptor antagonist AHCRF (α-helical CRF) (Fig. 5, B and C); in contrast, no response was measured in slices expressing EGFP-CAAX (Fig. 5C). Additionally, slices expressing the CRF-insensitive mutant (CRFmut), which harbors a point mutation at the ligand-binding pocket (28) (see fig. S13, A to D, for expression in HEK293T cells), showed no observable response compared with CRF1.0 (fig. S11, A to D), and CRISPR-mediated knockout of the Crh gene at CeA lead to significantly reduced CRF1.0 response to electrical stimulation (fig. S11, E to H). To examine whether expression of GRAB peptide sensors would alter the physiological properties of neurons, we compared the calcium signals and the GIRK (G protein-coupled inwardly rectifying potassium) channel currents between sensor or control fluorescent protein-expressing neurons in acute slices (fig. S12). There was no significant difference in the electric stimuli- or high potassium-induced calcium signals (fig. S12, A to G) nor significant alterations to the current-voltage curves of GABABR agonist baclofen-induced GIRK currents (fig. S12, H to L). The rise and decay half-times increased with increasing pulse numbers, with on and off t50 values of approximately 0.6 to 1.8 s and 3.5 to 6.4 s, respectively (Fig. 5D). Finally, the CRF1.0 signal propagated during electrical stimulation (Fig. 5, E to H) with an average diffusion coefficient of 3.5 × 103 μm2/s (Fig. 5, I and J).
Fig. 5. Detection of endogenous CRF release in acute brain slices using CRF1.0.

(A) Schematic illustration depicting the experimental design in which CRF1.0 or EGFP-CAAX was expressed virally in the CeA; after 3 weeks, acute slices were prepared. (B) Two-photon fluorescence images of acute slices, showing expression of CRF1.0 (left). Example pseudocolor images of acute slices expressing CRF1.0 at baseline and in response to 1, 5, 20, and 100 electric stimuli [i.e., pulses (P)] delivered at 20 Hz, and the response to 100 pulses measured in the presence of 100 nM AHCRF. The dashed white circles indicate the ROI used to calculate the response, and the approximate position of the stimulating electrode is indicated. Scale bars, 100 μm. (C) Representative traces (left) and summary (right) of the change in CRF1.0 fluorescence in response to electric stimuli delivered at 20 Hz in ACSF and 100 pulses delivered in the presence of AHCRF; also shown is the response measured in slices expressing EGFP-CAAX (n = 3 to 6 slices from one to three mice). (D) Fitted curves (top) and summary (bottom) of on and off t50 of the change in CRF1.0 fluorescence; n = 2 to 6 slices. (E) Example time-lapse pseudocolor images of CRF1.0 expressed in the CeA; during the first 5 s, 100 pulses were delivered at 20 Hz. Scale bar, 100 μm. (F to H) Spatial-temporal profile (F), temporal dynamics (G), and spatial dynamics (H) of the fluorescence change shown in (E). The profile in (F) shows the average response of three trials conducted in one slice. The traces in (G) and (H) correspond to the indicated distances and times, respectively, and the data in (H) were fitted with a Gaussian function. (I) Square of the full width at half maximum (FWHM2) plotted against time on the basis of the data shown in (H); the diffusion coefficient (D) was measured as the slope of a line fitted to the data. (J) Summary of the diffusion coefficient (D) measured CRF in the CeA; n = 6 slices from three mice.
The CRF1.0 sensor can be used to measure CRF release in vivo
CRF neurons in the paraventricular nucleus of the hypothalamus (PVN) play an essential role in regulating the stress response via the endocrine axis (8). In addition, these neurons also respond rapidly to both aversive and appetitive stimuli (55–57). To investigate the specificity of our CRF1.0 sensor in vivo, we expressed CRF1.0 or a CRFmut in the mouse PVN. We recorded the signal using fiber photometry while infusing CRF and/or AHCRF through an intracerebroventricular cannula (Fig. 6A). CRF1.0 fluorescence increased in a dose-dependent manner after CRF infusion (Fig. 6B), and the increase was blocked by coadministration of AHCRF (Fig. 6D); in contrast, CRFmut expressed in the PVN showed virtually no response to CRF, even at the highest concentration (Fig. 6C).
Fig. 6. Using fiber photometry to measure endogenous CRF release in vivo.

(A) (Left) Schematic diagrams depicting the strategy for virus injection, fiber and cannula implantation, and measurement of CFR1.0 or CRFmut in the PVN. (Right) image showing the expression of CRF1.0 (green) in the PVN and the approximate location of the optic fiber above the PVN; the nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). Scale bar, 200 μm. (B to D) Traces (left panels) and summary of the response (right panels) measured in mice expressing CRF1.0 [(B) and (D)] or CRFmut (C); the indicated concentrations of CRF and α-helical CRF 9–41 (AHCRF) were infused through the cannula. (E) Schematic diagram depicting the strategy for virus injection and fiber photometry recording. (F) Image showing the expression of CRF1.0 (green) and the approximate location of the imaging fiber; the nuclei were counterstained with DAPI (blue). Scale bars, 300 μm (left) and 40 μm (right). (G to J) Illustration (G), representative traces (H), average traces per stimulus-response (I), and summary data (J) of the change in CRF1.0 and CRFmut fluorescence measured before and during a 30-s tail lift (G1 to J1) and before and after an intraperitoneal injection of LiCl or saline (G2 to J2); n = 3 to 6 animals.
Next, we measured the dynamics of CRF release in the PVN during stressful experiences in mice expressing CRF1.0 (Fig. 6, E and F). Suspending the mouse by the tail for 30 s induced a robust time-locked increase in CRF1.0 fluorescence, whereas mice expressing CRFmut or EGFP-CAAX in the PVN showed no visible response (Fig. 6, G1 to J1, and fig. S13, E to H). Similarly, an intraperitoneal injection of lithium chloride (LiCl), an abdominal malaise-inducing stimulus, but not saline, elicited a long-lasting increase in CRF1.0 fluorescence, whereas no response was measured in mice expressing CRFmut or EGFP-CAAX (Fig. 6, G2 to J2, and fig. S13, E to H; the rise half-times are shown in fig. S13I). We also observed no significant alteration to animal behaviors, including metabolism, performance in open field test, immobility time in tail suspension and forced swimming tests, and sucrose preference (fig. S14, E to I), indicating that expression of CRF1.0 had no negative effects on animal behaviors.
CRF is expressed abundantly in neocortical interneurons, and CRF receptors are present in pyramidal cells (58, 59). In the frontal cortex, CRF mediates stress-induced executive dysfunction (60, 61). We therefore investigated the role of CRF in the mouse cortex during various behavioral paradigms. We injected virus expressing CRF1.0 into the motor cortex and prefrontal cortex (PFC) and then performed two-photon imaging of CRF1.0-expressing layer 2/3 neurons in head-fixed mice (Fig. 7A). We observed a transient reversible increase in CRF1.0 fluorescence in both the motor cortex and PFC in response to tail shocks; in contrast, no response was detected in mice expressing CRFmut or EGFP-CAAX (Fig. 7, B1 to D1 and G, and fig. S15, A to E; the kinetics and time constants are shown in Fig. 7, E and F).
Fig. 7. Spatially resolved measurements of CRF in vivo using two-photon imaging.
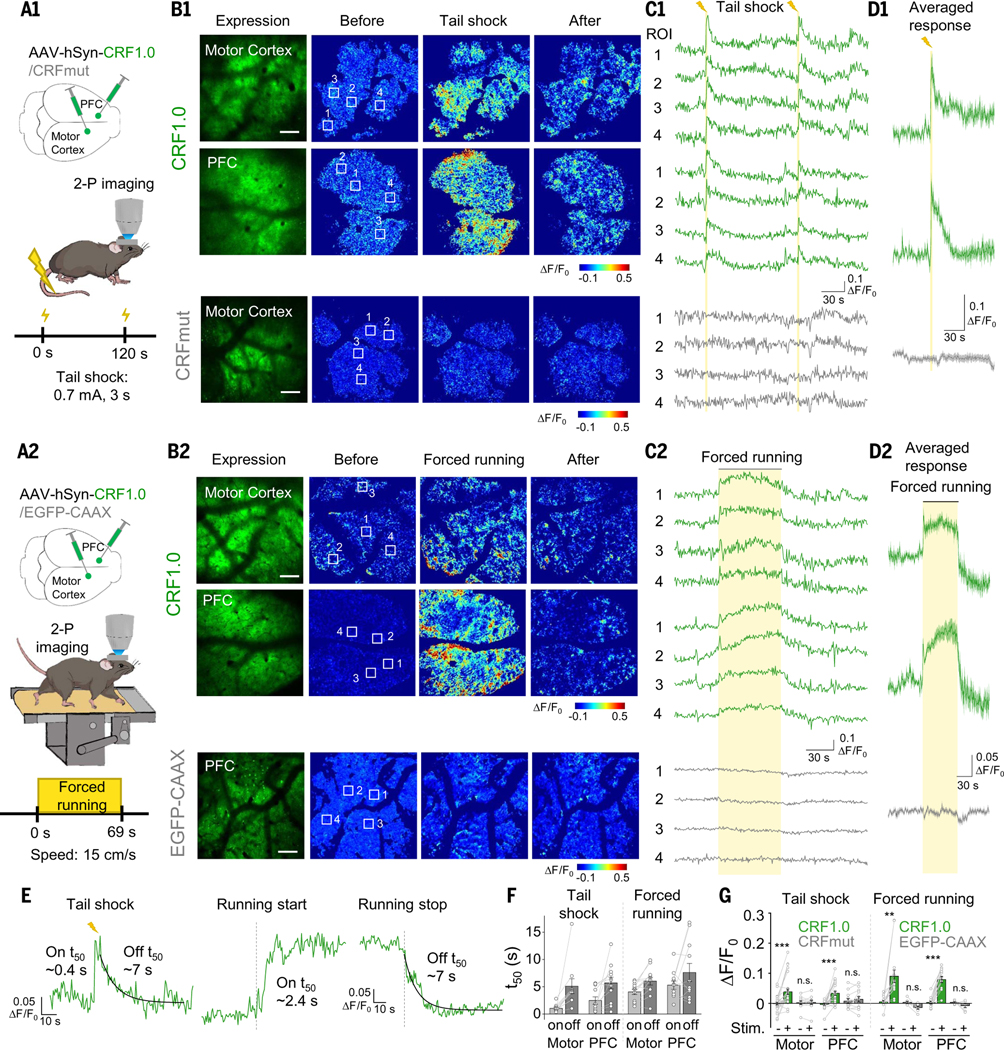
(A to D) Schematic diagrams (A), representative expression and pseudocolor response images (B), representative traces measured at the indicated ROIs (C), and average traces per stimulus (D) measured in the head-fixed mice expressing CRF1.0 or CRFmut (B1 to D1, top and bottom panels, respectively). The mice were subjected to tail shock (A1 to D1) and forced running on a treadmill (A2 to D2), and two-photon imaging was performed in the motor cortex and PFC. Scale bars, 100 μm. (E and F) Representative traces (E) and summary (F) of the rise and decay t50 values of the CRF1.0 signal in response to tail shock and forced running; n = 10 to 12 trials from three mice. (G) Summary of the peak fluorescence response measured in the motor cortex and PFC in mice expressing CRF1.0, CRFmut, or EGFP-CAAX in response to tail shock and forced running; n = 3 to 7 mice each.
Finally, head-fixed mice were forced to run on a treadmill. In response to this stressful stimulus, CRF1.0 fluorescence was monitored using two-photon microscopy (Fig. 7A2). At the onset of forced running, CRF1.0 fluorescence first increased, then reached a plateau within ~5 s, and finally returned to baseline after the treadmill stopped; in contrast, no response was measured in mice expressing CRFmut or EGFP-CAAX (Fig. 7, B2 to D2 and G, and fig. S15, A to E; the kinetics and time constants are shown in Fig. 7, E and F).
Discussion
We developed and characterized a series of highly selective and sensitive genetically encoded neuropeptide sensors. Moreover, as proof-of-principle, we demonstrated that our SST and CRF sensors can be used to monitor their corresponding peptides in vitro, ex vivo, and in vivo. We used our SST sensor to monitor activity-dependent SST release in cultured cortical neurons as well as pancreatic islets. SST1.0 sensor also revealed the SST dynamic changes in the process of conditioned learning. In acute brain slices, our CRF sensors reliably reported the electrical stimulation evoked release of CRF in the central amygdala. Moreover, the CRF sensor was successfully used to measure in vivo changes in CRF levels in response to stress-inducing stimuli.
The ICL3 with relatively long linkers derived from GRABNE1m, GRABDA2m, empirically showed higher membrane trafficking index and fluorescence response, which may accommodate the folding of TM5, TM6, and cpGFP. Further detailed structural study could help to understand its mechanism. Using our peptide sensors, we observed electrically evoked CRF and CCK (fig. S16) release in acute brain slices and measured their average apparent diffusion coefficients during signal propagation. This signal spread may derive both from the increase of peptide release and from the diffusion of released peptides. Our calculated diffusion coefficients are relatively higher than those of glutamate in the synaptic cleft (~330 μm2/s) (62), dopamine in the rat brain (~68 μm2/s) (63), and GFP-tagged tissue plasminogen activator (~0.02 μm2/s) (64) estimated by other methods. Further studies could apply optogenetic and chemogenic tools to drive the release from peptidergic neurons. The development and optimization of red sensors for neuropeptides will facilitate research of this field, which could be applied in multiplexed dual-color recording with green sensors in the future. By combining these GRAB peptide sensors with neurotransmitter sensors, it may be possible to monitor the real-time release of both neuropeptides and neurotransmitters, providing new insights into the mechanisms and functions of neuropeptide corelease.
In addition to its use in vitro in cultured neurons, we also measured the endogenous SST release in isolated pancreatic islets, consisting of cell types that secrete glucagon and insulin to maintain blood glucose levels (40). The finest temporal resolution of pulsatile SST release measured in previous studies was on the order of 30 s (42, 43). Using our SST sensor, we measured changes in SST levels in response to high glucose at the single-cell level with high temporal resolution on the order of seconds. SST released from 5 cells functions as a paracrine regulator to integrate signals from ghrelin, dopamine, acetylcholine, and leptin (65). Moreover, pancreatic islets receive regulatory input that affects Ca2+ fluctuations in α and β cells. These fluctuations are subsequently translated into the appropriate release of glucagon and insulin (66). Thus, our SST sensor and other hormone and/or transmitter sensors, such as ghrelin, UCN3, DA, and adenosine triphosphate (ATP) sensors, can be combined with Ca2+ indicators to study pancreatic islets in healthy conditions and in diabetic animal models.
Finally, our in vivo experiments show that these sensors can be used to directly monitor neuropeptide release within specific brain regions during behaviors, supporting their utility in freely moving animals. Although the peptide-expressing cortical neurons are well established (67), it remains unclear whether these peptides are released in a behaviorally relevant manner. In addition to the axonal release, neuropeptides can also be released from large dense-core vesicles in the somatodendritic compartment, likely contributing to volume transmission and exerting their function through paracrine modulation (37, 68, 69). The CRF1.0 sensor exhibits similar signals when expressed in different cell types in brain slices (fig. S17). Different cell types may have different regulations of neuropeptide release. These peptide sensors may be helpful to shed light on the understanding of the peptide release mechanism in the future. Although sensor fluorescence does not directly represent endogenous receptor activation, when and where these neuropeptides are released can nonetheless be examined using these sensors, thus helping elucidate their regulatory role on neural circuits.
Our peptide GRAB sensors were designed to take advantage of the native peptide receptors, inheriting their high selectivity and sensitivity. For example, CRF1.0 and SST1.0 sensors showed selectivity profiles similar to those of their native receptors, CRF1R and SSTR5 (Fig. 2E and fig. S4C). We further validated the selectivity of CRF1.0 by CRISPR-mediated knockout of the corresponding Crh gene in mice (fig. S11, E to H), and the provisional selectivity of SST1.0 could be validated by SST deletion experiment. Peptides bind to receptors with high affinity and potency, with the median inhibition constant (Ki) and EC50 at nanomolar range, targeting class A GPCRs (10, 70). The difference in sensor EC50 values for HEK cells and neurons maybe due to the differences in lipid and cholesterol compositions of cell membranes. Although the sensors’ affinity remains lower than that of native receptors at present (table S3), it is sufficient to detect endogenous neuropeptide changes ex vivo and in vivo (Figs. 4 to 7). Our series dilution experiment further suggests that the virus titer is not a major factor in determining the sensor fluorescence change upon neuropeptide binding given sufficient incubation time, and the titer is within a reasonable range (3 × 1012 to 1013 genome copies per milliliter in our case) (fig. S13, J to M). Improving neuropeptide sensors to achieve picomolar sensitivity in the future will be the key to capture neuropeptide release under diverse natural conditions.
The “on” kinetics of GRAB peptide sensors are 300 to 400 ms and “off ” kinetics are 3 to 12 s, which is in a similar range to that of naïve peptide receptors. There is relatively limited information for the kinetics of native peptide GPCRs (71, 72). The τon and τoff time constants of the CRF receptor are 0.1 s (at 1 μM concentration) and 142 min, respectively (73). A time constant of ~1 s for the parathyroid hormone receptor was measured by intramolecular fluorescence resonance energy transfer in vivo (74). The τon and τoff time constants of gonadotropin-releasing hormone receptor are 1 to 20 s (at 1 μM concentration) and 2 to 6 min, respectively (75). Given the lack of direct kinetics data of native peptide GPCRs, we also compared the reported equilibrium dissociation constant (Kd) of native peptide receptors with that of sensors. The Kd values of high- and low-affinity CRF binding sites are approximately 0.2 and 20 nM, respectively (73). The EC50 of CRF1.0 sensor is 18.6 ± 1.6 nM, indicating that there is not much difference in sensors’ EC50 with native receptors’ Kd values. The kinetics of GRAB peptide sensor’s signal reflect the GPCR structure change upon ligand binding, which is at the first level of signal transduction. It would be even slower if the measurement was at secondary effector levels that require activation of cellular signaling cascades.
Neuropeptides bind to endogenous GPCRs and transduce signals. If peptide sensors recapitulate the signal transduction or compete the ligand binding with native receptors, the endogenous signal fidelity and normal animal behavior could be affected when using peptide sensors. Most of our peptide sensors show minimal downstream coupling; for example, the SST1.0 sensors exhibit virtually no coupling (Fig. 2, F and G), suggesting that the expression of these peptide sensors will not affect the normal functions of cells. However, the CRF1.0 sensor still shows significant coupling, albeit with orders of magnitude lower affinity and 60% reduced efficacy (Fig. 2, F and G). The structures of peptide GPCRs bound to G proteins and β-arrestin have been solved, and the interaction sites have been identified (28, 76); altering these sites in the CRF1.0 sensor will allow future modifications to further reduce downstream coupling. At cellular signaling levels, expressing peptide sensors showed no obvious alterations to the cellular transcriptome (Fig. 2H and fig. S7), neural calcium activity in culture neurons or acute slices (figs. S6 and S11). At behavior outcome levels, we found no significant changes between mice expressing peptide sensors or control fluorescence proteins (fig. S14).
In summary, this series of newly generated GRAB peptide sensors can be used both in vitro and in vivo to monitor the rate and range of peptide release with a high spatiotemporal resolution. These tools have the potential to advance our understanding of the roles of neuropeptides in health and disease.
Materials and methods
Cell lines
HEK293T cells (CRL-3216, ATCC) were used to generate cell lines stably expressing the CRF1.0, SST1.0, CCK1.0, NPY1.0, NTS1.0, and VIP1.0 sensors. These stable cell lines were generated by transfecting cells with pCS7-PiggyBAC (S103P, S509G) (77) together with vectors containing a 5′ PiggyBac inverted terminal repeat sequence (ITR), CAG promoter, the GRAB peptide sensor coding region, internal ribosomal entry site (IRES) sequence, a puromycin-encoding gene, and a 3′ PiggyBac ITR; 24 hours after transfection, the cells were selected by culturing in 1 μg/ml puromycin. The HTLA cell line for the Tango assay was a gift from B. L. Roth (78). All cell lines were cultured in Dulbecco’s modified Eagle’s medium (Biological Industries, 06–1055-57–1ACS) supplemented with 10% (v/v) fetal bovine serum (FBS; CellMax, SA301.02) and 1% (v/v) penicillinstreptomycin (Gibco, 15140122) at 37°C in humidified air containing 5% CO2.
Cultured rat primary cortical neurons
Rat cortical neurons were obtained from postnatal day 0 (P0) Sprague-Dawley rat pups of both sexes (Beijing Vital River Laboratory Animal Technology Co., Ltd.). In brief, the brain was removed, and the cortex was dissected, dissociated in 0.25% trypsin-EDTA (Gibco, 25200–056), and plated on glass coverslips precoated with poly-D-lysine hydrobromide (Sigma, P7280). The neurons were cultured in Neurobasal medium (Gibco, 21103049) supplemented with 2% B-27 (Gibco, A3582801), 1% GlutaMAX (Gibco, 35050061), and 1% penicillin-streptomycin (Gibco, 15140122) at 37°C in humidified air containing 5% CO2.
Mice
C57BL/6N mice of both sexes (6 to 8 weeks of age and 10 to 12 weeks of age) were obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd., and group-housed (up to five mice per cage) under a 12-hours/12-hours light/dark cycle with the ambient temperature maintained at 25°C. CaMKIIα-Cre mice (JAX Strain 005359), Vglut2-Cre mice (JAX Strain 028863), and Gad2-Cre mice (JAX Strain 019022) were obtained from Jackson Laboratory. All surgical and experimental protocols were approved by the Animal Care and Use Committee at Peking University, the University of Science and Technology of China, New York University, the Institute of Neuroscience, and the Chinese Academy of Sciences and were performed in accordance with the standards established by the Association for the Assessment and Accreditation of Laboratory Animal Care. Detailed information about the sex and littermate status of the mice used is given below, in the section pertaining to each experiment.
Molecular biology
Molecular cloning was conducted using the Gibson assembly method. Primers for Gibson assembly were synthesized by Tsingke Biotechnology Co., Ltd., with 30-base pair overlap. The coding sequences for the GPCRs were polymerase chain reaction (PCR)-amplified from the corresponding full-length human GPCR cDNAs (hORFeome database 8.1) using GoldenStar T6 DNA Polymerase (Tsingke, TSE102). The ICL3 from the GRAB-NE (19), GRAB-DA (33), GRAB-ACh (20), GRAB-5-HT (34), and dLight (21) sensors were PCR-amplified from the corresponding sensors. Chimeric GPCRs and GRAB sensors were cloned into the modified pDisplay vector (Invitrogen) with an upstream immunoglobulin K-chain leader sequence and followed by an IRES sequence and mCherry-CAAX. Sanger sequencing was performed to verify the sequence of all clones. GPCR/Sensor-SmBit was constructed from β2AR-SmBit, and LgBit-mGs/mGsi/mGsq was a gift from N. A. Lambert (79). The GRAB peptide sensors were cloned into the pAAV vector under the control of the human Synapsin promoter and used for AAV packing.
Transfection of cell lines and virus infection of primary cultures
HEK293T cells and HTLA cells at 50 to 60% confluency were transfected with a mixture of polyethylenimine (PEI) and plasmid DNA at a 3:1 (w/w) ratio; after 6 to 8 hours, the transfection reagent was replaced with standard culture medium, and the cells were cultured for an additional 24 to 36 hours for expression of the transfected plasmids.
AAV9 viruses expressing the indicated GRAB peptide sensors were packaged at WZ Biosciences and BrainVTA (Wuhan) Co., Ltd. Each virus [at a titer of 3 × 1013 to 5 × 1013 viral genomes per milliliter (vg/ml) was added to cultured rat cortical neurons at DIV5–7, and the neurons were imaged 7 to 10 days later.
Fluorescence imaging of cultured cells and primary neurons
HEK293T cells and primary neurons were imaged using a Ti-E A1 inverted confocal microscope (Nikon) and an Opera Phenix High-Content Screening System (PerkinElmer). The confocal microscope was equipped with a 10×/0.45 numerical aperture (NA) objective, a 20×/0.75 NA objective, and a 40×/1.35 NA oil-immersion objective. A 488-nm laser and 525/50-nm emission filter were used to image green fluorescence, and a 561-nm laser and 595/50-nm emission filter were used to image red fluorescence. Cells were cultured on glass coverslips in 24-well plates and imaged in a custom-made chamber. The Opera Phenix system was equipped with 20×/1.0 NA and 40×/1.15 NA water-immersion objectives. A 488-nm laser and 525/50-nm emission filter were used to image green fluorescence, and a 561-nm laser and 600/30-nm emission filter were used to image red fluorescence. Cells were cultured and imaged in CellCarrier Ultra 96-well plates (PerkinElmer).
The cells were imaged in Tyrode’s solutions containing (in millimolar concentrations): 150 NaCl, 4 KCl, 2 MgCl2, 2 CaCl2,10 HEPES, and 10 glucose (pH adjusted to between 7.35 and 7.45 with NaOH). Where indicated, the following compounds were applied to the cells in Tyrode’s solution by bath application or a custom-made perfusion system: SST-28 (Anaspec), SST-14 (Anaspec), CCK-8s (Abcam), CCK-4 (Abcam), CRF (Anaspec), UCNI (MedChemExpress), UCNII (MedChemExpress), UCNIII (Abcam), NTS (Anaspec), NPY (Abcam), VIP (Anaspec), PACAP(1–38) (MedChemExpress), PACAP(1–27) (MedChemExpress), Orexin-B (GL Biochem), Substance P (Tocris), Ghrelin (Tocris), teriparatide [human parathyroid hormone-(1–34)] (MedChemExpress), Glu (Sigma-Aldrich), GABA (Tocris), DA (Sigma-Aldrich), NE (Tocris), 5-HT (Tocris), HA (Tocris), ATP (Tocris), ADP (MedChemExpress), Ado (Tocris), ACTH (MedChemExpress), β-MSH (MedChemExpress), γ-MSH (MedChemExpress), GHRH (Anaspec), OT (Anaspec), AVP (Tocris), vasotocin (MedChem-Express), isotocin (KS-V peptide), glucagon (GLPBIO), GLP-1 (MedChemExpress), DAMGO (Tocris), dynorphin A (Tocris), [Leu5]-enkephalin (MedChemExpress), goserelin (MedChemExpress), galanin (Tocris), orexin-A (Tocris), NKB (Tocris), bombesin (MedChemExpress), taltirelin (MedChemExpress), BIM23056 (Abcam), YM 022 (Tocris), NBI 27914 (Santa Cruz), antalarmin (Cayman), α-helical CRF (Tocris), SR142948 (Tocris), BIBO 3304 (Tocris), and PACAP(6–38) (Tocris). For high K+ stimulation, Tyrode’s solution contained 79 mM NaCl and 75 mM KCl. For screening candidates using SSTR5, NPY1, NPY5, GHS-R, AVPR2, NTSR1, CCKBR, HCRTR2 (OX2), OPRM1, GRPR, TACRI (NK1), TRHR, VIPR1VIPR2, CRF1R, or PTH as scaffolds in Fig. 1, the following compounds were applied respectively (in micromolar concentrations): 1 SST-28,1 NPY, 1 NPY, 1 ghrelin, 5 desmopressin (Tocris), 1 NTS, 1 CCK-8s, 1 orexin-B, 1 DAMGO (Tocris), 10 bombesin (Tocris), 10 substance P, 20 taltirelin (Tocris), 1 VIP, 1 VIP, 1 CRF, and 1 teriparatide.
Neuron cultures were incubated in culture medium with 5 μMCalbryte590 AM (ATT Bioquest) at 37°C for 30 min before calcium imaging. Field electric stimuli were delivered by GRASS S88 stimulator. The pulse duration is 1 ms, 50 pulses were delivered at 50 Hz for 1 s, which are considered as 1× burst stimulation. The time interval between each burst is 0.5 s.
Spectra measurements
The linear optical properties of the GRAB peptide sensors expressed in HEK293T cells were measured using a Safire 2 plate reader (Tecan). Cells were harvested and transferred to blackwall 384-well plates containing either saline alone or saline containing the corresponding peptides. Emission spectra were measured using an excitation wavelength of 455 nm with a bandwidth of 20 nm, and emissions were collected using an emission wavelength step size of 5 nm. Excitation spectra were measured using excitation light ranging from 300 to 520 nm with a wavelength step size of 5 nm, and emission light was collected at 560 nm with a bandwidth of 20 nm.
The two-photon fluorescence spectra of the GRAB peptide sensors expressed in HEK293T cells were measured at 10-nm increments from 700 to 1050 nm using a Bruker Ultima Investigator two-photon microscope equipped with Spectra-Physics Insight X3. Cells were measured in Tyrode’s solutions or Tyrode’s solutions containing the corresponding peptides. The two-photon laser power at various wavelengths was calibrated, and the fluorescence measured in untransfected cells was subtracted as background.
Tango assay
HTLA cells were cultured and transfected in six-well plates and placed in 96-well plates (white with a clear flat bottom), and solutions containing various concentrations of peptides were applied; 12 hours after induction, the medium was discarded, and 40 μl of Bright-Glo Luciferase Assay Reagent (Promega) diluted 20-fold in phosphate-buffered saline (PBS) was added to each well at room temperature. After a 10-min reaction in the dark, luminescence was measured using a Victor X5 multi-label plate reader (PerkinElmer).
Mini G protein luciferase complementation assay
HEK293T cells were cultured and transfected in six-well plates and grown to between 80 and 90% confluency. The cells were then dissociated using a cell scraper, resuspended in PBS, and placed in 96-well plates (white with a clear flat bottom) containing Nano-Glo Luciferase Assay Reagent (Promega) diluted 1000-fold in PBS at room temperature. Solutions containing various concentrations of peptides were added to the wells. After a 10-min reaction in the dark, luminescence was measured using a Victor X5 multi-label plate reader (PerkinElmer).
Pancreatic islet isolation and imaging of SST1.0 sensor
Male C57BL/6N mice (10 weeks of age) were obtained from Beijing Vital River Laboratory Animal Technology Co., Ltd. The mice were sacrificed by cervical dislocation, and primary pancreatic islets were isolated using collagenase P digestion and purified by hand-picking under a dissecting microscope. After isolation, the islets were cultured overnight in RPMI-1640 medium containing 10% FBS (10099141C, Gibco), 8 mM D-glucose, 100 units/ml penicillin, and 100 μg/ml streptomycin for overnight culture at 37°C in a 5% CO2 humidified air atmosphere.
Adenovirus (ADV) expressing the SST1.0 sensor (pAdeno-MCMV-SST1.0) was prepared by OBiO Technology (Shanghai) Corp., Ltd. The islets were infected with pAdeno-MCMV-SST1.0 by 1 hour exposure in 200 μl culture medium (~4 × 106 plaque-forming units (PFU) per islet), followed by addition of regular medium and further culture for 16 to 20 hours before use.
All fluorescence images were acquired using Dragonfly 200 series (Andor) with a Zyla4.2 sCMOS camera (Andor) and the Fusion software. All channels were collected with a 40×/0.85 NA Microscope Objective (Warranty Leica HCX PL APO). The SST concentrations in culture medium were measured according to the protocol of the Somatostatin (Human, Rat, Mouse, Porcine)-EIA Kit (Phoenix Pharmaceuticals, EK-060–03). Briefly, batches of five islets were incubated in Krebs-Ringer buffer containing 3 or 20 mmol/liter glucose for 0.5 hours. At the end of incubation, the media were removed for enzyme-linked immunosorbent assay.
Fluorescence imaging of peptide sensors in acute brain slices
Male C57BL/6N mice (6 to 8 weeks of age) were anesthetized with an intraperitoneal injection of tribromoethanol (Avertin; 500 mg/kg body weight), and the AAV9-hSyn-CRF1.0, AAV9-hSyn-CCK1.0, AAV9-hSyn-EGFP-CAAX, or AAV9-CMV-saCAS9-sgRNA virus (300 nl, 3 × 1013 to 5 × 1013 vg/ml, WZ Biosciences), AAV9-hSyn-CRFmut, hsyn-NES-jRGECO1a (300 nl, 3 × 1012vg/ml, BrainVTA) virus was injected into the left CeA (AP: −1.2 mm relative to Bregma; ML: −2.5 mm relative to Bregma; DV: −4.4 mm from the dura) or the left CA1 (AP: −2.0 mm relative to Bregma; ML: −1.5 mm relative to Bregma; DV: −1.5 mm from the dura) at a rate of 30 nl/min. After 3 weeks, to allow for virus expression, the mice were anesthetized with Avertin and perfused with ice-cold oxygenated slicing buffer containing (in millimolar concentrations): 110 choline-Cl, 2.5 KCl, 7 MgCl2, 1 NaH2PO4,0.5 CaCl2,25 NaHCO3, and 25 glucose (pH 7.4). The brains were dissected, and 300-μm-thick coronal slices were cut in ice-cold oxygenated slicing buffer using a VT1200 vibratome (Leica). The slices were transferred and allowed to recover for at least 40 min at 34°C in oxygenated artificial cerebrospinal fluid (ACSF) containing (in millimolar concentrations): 125 NaCl, 2.5 KCl, 1.3 MgCl2, 1 NaH2PO4, 2 CaCl2, 25 NaHCO3, and 25 glucose (pH 7.4). The brain slices were then transferred to a custom-made perfusion chamber and imaged using an FV1000MPE two-photon microscope (Olympus) or Bruker two-photon microscope. CRF1.0, CCK1.0, and EGFP-CAAX were excited using a 920-nm two-photon laser, and dual-color imaging used a 950-nm two-photon laser for excitation, and electrode tips were placed near the CeA or CA1 region expressing CRF1.0, CCK1.0, or EGFP-CAAX. Electrical stimuli were applied using an S88 stimulator (Grass Instruments), with a stimulation voltage of 5 to 8 V and pulse duration of 1 ms. For CRF1.0 imaging experiments in acute brain slices, the electrode was placed on the surface of the brain slice, and the averaged depth of imaging regions was 52 ± 5 μm below the surface, which is calculated by aligning imaging regions to z-axis scanning images.
The gRNA sequences for Crh knockout or control scramble gRNA are as follows: sal: CCTCAGCCGGTTCTGATCCGC; sa2: GAAGAA-TACTTCCTCCGCCTG; sa3: GAGCCGGCCGAAC-GCGGCGCC; sa4: CCCAACTCCACGCCCCTCACC; sa5: CGCGACCCGCGGTGAGGGGCG; and saCtrl: GTGTAGTTCGACCATTCGTG.
Two-photon in vivo imaging in mice
Female C57BL/6N mice (6 to 8 weeks of age) were anesthetized with Avertin, and AAV9-hSyn-CRF1.0, AAV9-hSyn-CRFmut, or AAV9-hSyn-EGFP-CAAX (200 nl, full titer, WZ Biosciences) was injected into the motor cortex (AP: 1.0 mm relative to Bregma; ML: 1.5 mm relative to Bregma; DV: −0.5 mm from the dura) and PFC (AP: 2.8 mm relative to Bregma; ML: 0.5 mm; DV: −0.5 mm from the dura). A high-speed drill was then used to open a 4 mm by 4 mm square in the skull. After virus injection, craniotomies were installed with a glass coverslip affixed to the skull surface. A stainless-steel head holder was attached to the animal’s skull using dental cement to help restrain the animal’s head and reduce motion-induced artifacts during imaging. The imaging experiments were performed ~3 weeks after surgery. An awake mouse with head mounts was habituated for 10 min in the treadmill-adapted imaging apparatus to minimize the stress associated with head restraint and imaging. The motor cortex or PFC was imaged 100 to 200 μm below the pial surface to measure sensor fluorescence.
A Bruker Ultima Investigator two-photon microscope equipped with Spectra-Physics Insight X3 was used for in vivo imaging. A 920-nm laser was used for excitation, and a 490- to 560-nm filter was used to measure green fluorescence. All experiments were performed using a 16×/0.8 NA objective immersed in saline, and images were acquired at a frame rate of 1.5 Hz. For the forced running model, the running speed was set at ~15 cm/s; for the tail shock model, a 0.7-mA shock was delivered for a duration of 3 s.
After imaging, any motion-related artifacts were corrected using the Non-Rigid Motion Correction (NoRMscorre) algorithm. The fluorescence time course was measured using ImageJ software by averaging all pixels within the regions of interest (ROIs). ΔF/F0 was calculated using the following equation: ΔF/F0 = [(F - F0)/F0], in which F0 is the baseline fluorescence signal averaged over a 10-s period before theonset of theforced running or tail shock.
Fiber photometry recording of CRF1.0 with in vivo drug application
Male C57BL/6N mice bred at the NYULMC animal facility (10 to 12 weeks of age) were anesthetized with isoflurane and placed in a stereotaxic frame. AAV expressing hSyn-CRF1.0 or hSyn-CRFmut (Vigene Biosciences) was injected (160 nl per animal) into the PVN (AP: −0.75 mm relative to Bregma; ML: +0.22 mm relative to Bregma; DV: −4.7 mm from the dura). An optical fiber (400-μm diameter) was implanted 150 μm above the virus injection site (either at the time of virus injection or 2 weeks later). At the same time that the optical fiber was implanted, a bilateral cannula (Plastics One) for drug infusion was also implanted in the dorsal third ventricle or the left lateral ventricle. At least 4 weeks after virus injection, fiber photometry recording was performed in the PVN.
Before fiber photometry recording, a ferrule sleeve (ADAL1–5, Thorlabs) was used to connect a matching optic fiber to the implanted fiber, and recordings were performed on the head-fixed wheel. For recording, a 390-Hz sinusoidal 488-nm blue LED light (35 mW; M470F1; Thorlabs) driven by a LEDD1B driver (Thorlabs) was bandpass-filtered (passing band: 472 ± 15 nm, Semrock, FF02–472/30–25) and delivered to the brain to excite CRF1.0 or CRFmut. The emission light passed through the same optic fiber, through a bandpass filter (passing band: 534 ± 25 nm, Semrock, FF01–535/50), and into a Femtowatt Silicon Photoreceiver, which recorded the CRF1.0 or CRFmut emission using an RZ5 real-time processor (Tucker-Davis Technologies). The 390-Hz signals from the photoreceiver were extracted in real-time using a custom-written program (Tucker-Davis Technologies) and used to determine the intensity of theCRF1.0orCRFmutfluorescencesignal.
For generating dose-response curves, CRF (C3042, Sigma or AS-24254, Eurogentec) or AHCRF 9–41 (1184, Tocris) was infused into one of the ventricles through the implanted cannula using a syringe (65457–02, Hamilton). For the data shown in Fig. 6, B and C, 250 nl of CRF diluted to indicated concentrations (4.8, 1.6, 0.5, or 0.05 mg/ml) or 250 nl saline was infused; for the data shown in Fig. 6D, 100 nl of 1.6 mg/ml CRF and/or 300 nl of 0.25 mg/ml AHCRF 9–41 was infused. CRF was diluted in distilled water, and AHCRF 9–41 was diluted in distilled water containing 0.1 M NH4OH.
For the data shown in Fig. 6, B and D, Friedman’s test was performed, followed by Horm correction. For the data shown in Fig. 6C, the two-sided paired Wilcoxon signed rank test was performed. The peak values obtained after applying 1.6 mg/ml CRF in Fig. 6, B and D, were the average of all trials from each animal.
Fiber photometry recording of CRF1.0 during behavioral testing
Male C57BL/6N mice (10 to 12 weeks of age, from River Vital Laboratory) were anesthetized with an intraperitoneal injection of sodium pentobarbital (80 mg/kg body weight) and AAV9-hSyn-CRF1.0 or AAV9-hSyn-EGFP-CAAX (300 nl, 3 × 1013 to 5 × 1013 vg/ml, WZ Biosciences) was injected into the PVN (AP: −0.80 mm relative to Bregma; ML: −0.25 mm relative to Bregma; DV: −4.60 mm from the dura) at a rate of 40 nl/min. The optic fiber (200 μm inner core diameter, 0.37 fiber numerical aperture, Thinkerbiotech) was implanted 0.20 mm above the injection site and sealed with dental cement. After 4 to 5 weeks (to allow the mice to recover and to allow for virus expression), a Multi-Channel Fiber Photometry Device (Inper, 0PT-FPS-410/470/561) was used for recording. Signals were acquired at a frame rate of 50 Hz, with an exposure time of 9 ms, with gain 0, using 470-nm light at 30 to 40% power.
For the tail lift experiments, the mouse was suspended by the tail 50 cm above the floor for 30 s per trial. Three 30-s tail lift trials were performed at an interval of ~220 s; the signal recorded 150 s before the first lift was used as the baseline, and the average of the three responses recorded during the 30-s lifts was used as the lift signal.
For LiCl or saline injection, the signal recorded 500 s before injection was recorded as the baseline. The mice were then briefly anesthetized with isoflurane and given an intraperitoneal injection of saline (0.1 ml/10 g body weight) or LiCl (125 mg/kg body weight) dissolved in saline. The signals were recorded for 2400 s after intraperitoneal injection, and the average response measured during the first 1500 s was used as the LiCl or saline signal.
Fiber photometry recording of SST1.0 during olfactory Pavlovian learning
Male adult C57BL/6J mice (8 to 13 weeks of age, from River Vital Laboratory) were anesthetized under ketamine and xylazine (100 and 10 mg/kg intraperitoneally, respectively) and AAV9-hSyn-SST1.0 (300 nl, 3 × 1013 to 5 × 1013 vg/ml, WZ Biosciences) was injected into the BLA (AP: −1.5 mm relative to Bregma; ML: −3.25 mm relative to Bregma; DV: −4.6 mm from the dura) at a rate of 100 nl/min. The optic fiber (200 μm, 0.39 NA, Thorlabs) was implanted 0.10 mm above the injection site and sealed with dental cement. Mice were allowed at least 3 weeks to recover and to express the virus before behavioral training. Signals were recorded using FiberOptoMeter (F0M-02M, C-Light, SooChow, China), using a beam from a 470-nm LED reflected with a dichroic mirror, focused with a lens coupled to a photomultiplier tube. The LED power at the tip of the patch cord was between 25 and 30 μW.
Transcriptome-wide RNA-seq analysis
Cultured rat cortical neurons at DIV 5–7 were transfected with AAV virus (at a titer of 3 × 1013 to5 × 1013 vg/ml) expressing CRF1.0, SST1.0, or EGFP-CAAX, and the neurons were lysed by Trizol after 7-day expression for RNA extraction. Male C57BL/6N mice (6 to 8 weeks of age) were anesthetized with an intraperitoneal injection of tribromoethanol (Avertin; 500 mg/kg body weight), and the AAV9-hSyn-CRF1.0, AAV9-hSyn-SST1.0, AAV9-hSyn-mApple-CAAX, or AAV9-hSyn-EGFP-CAAX virus (300 nl, 3 × 1013 to 5 × 1013 vg/ml, WZ Biosciences) was bilaterally injected into the motor cortex (AP: 1.0 mm relative to Bregma; ML: ±1.5 mm relative to Bregma; DV: −0.5 mm from the dura). After 2-week expression, motor cortex regions were dissected and frozen by liquid nitrogen for RNA extraction. The mRNA library constructing and sequencing were conducted by AZENTA Life Sciences, performed in Illumina HiSeq/Illumina Novaseq/MGI2000 instrument. Sequencing qualities were filtered by Cutadapt (V1.9.1, phred cutoff: 20, error rate: 0.1, adapter overlap: 1bp, min. length: 75, proportion of N: 0.1). Data were aligned to reference genome by the software Hisat2 (v2.0.1) and gene expression levels were estimated from the pair-end clean data by the software HTSeq (v0.6.1). The FPKM (fragments per kilobase of transcript per million mapped reads) heatmaps of selected CREB responsive genes were plotted by R (v 4.2.2).
Western blot
Primary antibodies to SSTR2 (Abcam, ab134152, 1:2000), primary antibodies to CB1R (Abcam, ab23703,1:500) GFP antibody (Abcam, ab6556, 1:1000), β-actin antibody (CWBIO, CW0096M, 1:2000), Goat anti Rabbit IgG-HRP conjugated (CWBIO, CW0103S, 1:3000), and Goat anti Mouse IgG-HRP conjugated (CWBIO, CW0102S, 1:3000) were used in this study for Western blots. Standard Western blot protocols were applied. Male C57BL/6N mice (10 to 12 weeks of age, from River Vital Laboratory) expressing SST1.0 or CRF1.0 sensors and EGFP-CAAX for 2 weeks were used. In brief, mice motor cortex were lysed, protein concentration was quantified by bicinchoninic acid assay, and an equal amount of each lysate was loaded for SDS-polyacrylamide gel electrophoresis. Then, sample proteins were transferred onto hybridization nitrocellulose filter (Millipore, HATF00010) and immunoblotted with primary antibodies, followed by secondary antibody incubation and exposure using cECL Western Blot Kit (CWBIO, CW0048M) by ChemiDoc XRS System (BIO-RAD).
Immunohistochemistry
Mice were anaesthetized (using Avertin) and intracardially perfused with saline followed by 4% paraformaldehyde (PFA) in 0.1 M PBS buffer, and brains were dissected and fixed at 4 °C overnight by 4% PFA in 0.1 M PBS buffer. Brains were sectioned at 40 μm thickness using a VT1200 vibratome (Leica). Sections were placed in blocking solution containing 5% normal goat serum (NGS) and 0.1% Triton X-100 and 2 mM MgCl2 in 1× PBS for 1 hour, then incubated with primary antibodies to CRF (Peninsula Laboratories International, T-4037, 1:1000), primary antibodies to SSTR2 (Abcam, ab134152, 1:2000), and diluted AGT solution (0.5% NGS, 0.1% Triton, and 2 mM MgCl2 in 1× PBS) overnight at 4 °C. Sections were rinsed three times in AGT and incubated for 1 hour at room temperature with secondary antibodies Alexa-555-conjugated goat-anti-rabbit IgG (H+L) (AAT Bioquest, 16690,1:500) and Nissl Stained using NeuroTrace 640/660 (Invitrogen, N21483,1:300). Sections were rinsed three times in AGT and mounted on slides using DAPI Fluoromount-G (SouthernBiotech, 0100–20) mounting medium. Sections were imaged on Olympus VS120 slide scanner.
Slice electrophysiology
The procedures for preparing acute brain slices were similar to the parts of fluorescence imaging of peptide sensors in acute brain slices. Briefly, male C57BL/6N mice (6 to 8 weeks of age) were anesthetized with Avertin, and the AAV9-hSyn-SST1.0, AAV9-hSyn-CRF1.0, or AAV9-hSyn-EYFP virus (300 nl, 3 × 1013 to 5 × 1013 vg/ml, WZ Biosciences) was injected into the mPFC (AP: 2.0 mm relative to Bregma; ML: 0.5 mm; DV: −2.0 mm from the dura) at a rate of 30 nl/min. After 3 weeks of virus expression, coronal medial PFC slices were prepared in a solution containing (in millimolar concentrations): 228 sucrose, 26 NaHCO3,11 glucose, 2.5 KCI, 1 NaH2PO4, 7 MgSO4, and 0.5 CaCI2 and recovered at 35°C in ACSF containing (in millimolar concentrations): 119 NaCI, 26 NaHCO3,11 glucose, 2.5 KCI, 1 NaH2PO4,1.3 MgSO4, and 2.5 CaCI2. After 1 hour incubation, the slices were transferred to a recording chamber bathed with oxygenated ACSF at 35.5°C. Layer 5 pyramidal neurons were visualized with an upright infrared differential interference contrast (IR-DIC) microscope (BX51WI; Olympus). Whole-cell recordings were performed with a MultiClamp 700B amplifier and Digidata 1550B4 (Molecular Devices, USA). The resistance of patch pipette was 4 to6 MΩ. Signals were filtered at 10 kHz and then sampled at 20 kHz using Clampex v10.4 (Molecular Devices).
Layer 5 pyramidal neurons expressing SST1.0, CRF1.0, or EYFP in mPFC were recorded using an internal solution containing (in millimolar concentrations): 140 K-gluconate, 10 HEPES, 0.25 EGTA, 2 MgATP, 0.3 Na3GTP, 7 phosphocreatine (pH 7.25 to 7.3; osmolarity 294 to 298). To isolate voltage-dependent potassium currents, 0.5 μM TTX and 300 μM Cd2+ were included in the bath. We initially held the recorded neuron at −60 mV and applied a series of 200-ms test potentials (−140 to0 mV) to activate voltage-dependent potassium currents. The current recorded under pretreatment conditions was defined as the constitutive potassium current. Perfusion of 20 μM baclofen was used to activate the GIRK channel currents. The baclofen-activated currents (IBac) were defined by subtracting the constitutive current from those recorded during baclofen perfusion. Liquid junction potential (~16 mV) was not corrected for the membrane potential values in the text and figures.
Behavioral assays
Male C57BL/6J mice [8 weeks of age, from SPF (Beijing) Biotechnology Co., LTD.] were anesthetized, and AAV9-hSyn-CRF1.0 or AAV9-hSyn-EGFP-CAAX was injected into the PVN, similar to the procedure for the PVN fiber photometry recording experiment. The AAV9-hSyn-SST1.0 or AAV9-hSyn-EYFP was injected into the BLA, similar to the procedure for the BLA fiber photometry recording experiment. The same cohort of mice was subjected to various behavioral tests at least 26 days after surgery in the following order: open-field test, elevated plus maze test, tail suspension test, forced swimming test, and sucrose preference test. Mice were handled for 3 days for 3 min each day before experiments. During behavioral procedures, all of the mice were singly housed. The behavior assays were performed as previously described (60, 80).
Olfactory Pavlovian learning
The olfactory Pavlovian conditioning assays were performed using protocols described in previous research (81). In brief, mice were trained to associate odors with reinforcing outcomes in head-fixed configuration using a custom-made apparatus. The odors used in this study were ethyl acetate, 2-pentanone, and (R)-(+)-limonene, which have neutral value to mice. Mice were water-deprived and habituated to the head-fixed recording configuration. In the reward trials, water was delivered. In the neutral trials, nothing happened after cue delivery. In the punishment trials, an air puff was delivered to the eye of mice. Mice were trained for 4 days, and each day’s session contained 180 trials.
Metabolism
Four or five mice were co-housed, and body weights were measured every 4 days after the virus injection. Twenty-nine days after virus injection, mice were singly housed, and water and food consumption was recorded from day 29 12:00 to day 30 11:00.
Open-field test
The open-field test was performed in a non-transparent square box (50 cm by 50 cm by 40 cm), with smooth interior walls. The center area of the open field was defined as a 25 cm by 25 cm zone centered in the arena. At the start of the test, mice were placed in one of four corners of the arena and were allowed to freely explore the environment for 5 min. Locomotion traces were recorded by video camera for 10 min for each mouse. Time and entries in the center area and total distance were analyzed by EthoVision XT 8.5 software.
Elevated plus maze test
The elevated plus maze has two opposite open arms without walls (30 cm by 6 cm), two opposite closed arms (30 cm by 6 cm by 15 cm) and a central platform (5 cm by 5 cm). The maze was elevated 80 cm above the floor. At the start of the session, animals were first placed in the center zone facing one of the open arms. Mice were allowed to explore the maze for 6 min. Locomotion traces were recorded by video camera. The time and entries were quantified and analyzed. Data were analyzed using EthoVision XT 5.1 software.
Tail suspension test
The mouse was suspended by the tail 50 cm above the floor, ensuring that the mouse could not make any other contact or climb during the assay. The immobility behavior in a 6-min session was recorded, and immobility time for 2 to 6 min was analyzed.
Forced swimming test
The mouse was placed in a transparent cylinder (25 cm high, 25 cm in diameter) filled with water to a depth of 18 cm and maintained at between 24° and 25°C for each 6-min session. After each session, the mouse was dried with a towel and returned to its home cage. The total immobility time for 2 to 6 min was recorded and analyzed. Mice were considered immobile when they did not make any struggling movements.
Sucrose preference test
The mice were singly housed with two bottles of 1% sucrose water and adapted for 1 day before the test. The mice were singly housed with one bottle of 1% sucrose water and one bottle of water. The consumption from day 35 20:00 to day 36 12:00 was recorded, and the sucrose preference [sucrose water consumption/(sucrose water consumption + water consumption)] was analyzed.
Quantification and statistical analysis
Summary data with error bars are presented as the mean ± SEM. Except where indicated otherwise, groups were compared using Student’s t test or a one-way analysis of variance with post hoc test, and differences were considered significant at P ≤ 0.05. Where applicable, *P ≤ 0.05. **P ≤ 0.01, ***P ≤ 0.001, and n.s. means not significant.
Supplementary Material
ACKNOWLEDGMENTS
We thank Y. Rao for sharing the two-photon microscope. We thank X. Lei at PKU-CLS, the National Center for Protein Sciences at Peking University, and State Key Laboratory of Membrane Biology at Tsinghua University for providing support for the Opera Phenix high-content screening system. Cartoons in figures were created with BioRender.com. We thank members of the Li lab for helpful suggestions and comments on the manuscript.
Funding:
This research was supported by grants from the National Natural Science Foundation of China (31925017), the National Key R&D Program of China (2019YFE011781), the Beijing Municipal Science and Technology Commission (Z220009), and the NIH BRAIN Initiative (1U01NS120824) to Y.L. Support was also provided by the Feng Foundation of Biomedical Research, the Clement and Xinxin Foundation, the New Cornerstone Science Foundation through the New Cornerstone Investigator Program and the XPLORER PRIZE (to Y.L.) and by grants from the Peking-Tsinghua Center for Life Sciences and the State Key Laboratory of Membrane Biology at Peking University School of Life Sciences to Y.L., the NIH BRAIN Initiative (1U01NS113358) to Y.L. and D.L., grants from the National Natural Science Foundation of China (82171492) to Y.Z., the Osamu Hayaishi Memorial Scholarship to T.O., and the Levy Leon Postdoctoral Fellowship to M.L.
Footnotes
Competing interests: All authors declare no competing interests.
Data and materials availability:
DNA plasmids of GRAB peptide sensors used in this study have been deposited to Addgene (Addgene ID 208654–208685). All other data needed to evaluate the conclusions in the paper are available in the main text or the supplementary materials.
REFERENCES AND NOTES
- 1.De Wied D, De Kloet ER, Pro-opiomelanocortin (POMC) as homeostatic control system. Ann. N. Y. Acad. Sci. 512, 328–337 (1987). doi: 10.1111/j.1749-6632.1987.tb24971.x; [DOI] [PubMed] [Google Scholar]
- 2.Spiess J, Rivier J, Rivier C, Vale W, Primary structure of corticotropin-releasing factor from ovine hypothalamus. Proc. Natl. Acad. Sci. U.S.A. 78, 6517–6521 (1981). doi: 10.1073/pnas.78.10.6517; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schally AV, Arimura A, Kastin AJ, Hypothalamic regulatory hormones. Science 179, 341–350 (1973). doi: 10.1126/science.179.4071.341; [DOI] [PubMed] [Google Scholar]
- 4.Du Vigneaud V, Hormones of the posterior pituitary gland: Oxytocin and vasopressin. Harvey Lect. 50, 1–26 (1954-1955). [PubMed] [Google Scholar]
- 5.Alhadeff AL et al. , A neural circuit for the suppression of pain by a competing need state. Cell 173, 140–152.e15 (2018). doi: 10.1016/j.cell.2018.02.057; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Lecea L et al. , The hypocretins: Hypothalamus-specific peptides with neuroexcitatory activity. Proc. Natl. Acad. Sci. U.S.A. 95, 322–327 (1998). doi: 10.1073/pnas.95.1.322; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sakurai T et al. , Orexins and orexin receptors: A family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell 92, 573–585 (1998). doi: 10.1016/S0092-8674(00)80949-6; [DOI] [PubMed] [Google Scholar]
- 8.Vale W, Spiess J, Rivier C, Rivier J, Characterization of a 41-residue ovine hypothalamic peptide that stimulates secretion of corticotropin and β-endorphin. Science 213, 1394–1397 (1981). doi: 10.1126/science.6267699; [DOI] [PubMed] [Google Scholar]
- 9.Hauser AS, Attwood MM, Rask-Andersen M, Schiöth HB, Gloriam DE, Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 16, 829–842 (2017). doi: 10.1038/nrd.2017.178; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davenport AP, Scully CCG, de Graaf C, Brown AJH, Maguire JJ, Advances in therapeutic peptides targeting G protein-coupled receptors. Nat. Rev. Drug Discov. 19, 389–413 (2020). doi: 10.1038/s41573-020-0062-z; [DOI] [PubMed] [Google Scholar]
- 11.Hökfelt T, Bartfai T, Bloom F, Neuropeptides: Opportunities for drug discovery. Lancet Neurol. 2, 463–472 (2003). doi: 10.1016/S1474-4422(03)00482-4; [DOI] [PubMed] [Google Scholar]
- 12.Marvin JS et al. , An optimized fluorescent probe for visualizing glutamate neurotransmission. Nat. Methods 10, 162–170 (2013). doi: 10.1038/nmeth.2333; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Unger EK et al. , Directed evolution of a selective and sensitive serotonin sensor via machine learning. Cell 183, 1986–2002.e26 (2020). doi: 10.1016/j.cell.2020.11.040; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Borden PM et al. , A fast genetically encoded fluorescent sensor for faithful in vivo acetylcholine detection in mice, fish, worms and flies. bioRxiv 2020.02.07.939504 [Preprint] (2020); 10.1101/2020.02.07.939504. [DOI] [Google Scholar]
- 15.S. R. Foster et al. , Discovery of human signaling systems: pairing peptides to G protein-coupled receptors. Cell 179, 895–908.e21 (2019). doi: 10.1016/j.cell.2019.10.010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Isberg V et al. , GPCRdb: An information system for G protein-coupled receptors. Nucleic Acids Res. 44, D356–D364 (2016). doi: 10.1093/nar/gkv1178; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jing M et al. , A genetically encoded fluorescent acetylcholine indicator for in vitro and in vivo studies. Nat. Biotechnol. 36, 726–737 (2018). doi: 10.1038/nbt.4184; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun F et al. , A genetically encoded fluorescent sensor enables rapid and specific detection of dopamine in flies, fish, and mice. Cell 174, 481–496.e19 (2018). doi: 10.1016/j.cell.2018.06.042; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feng J et al. , A genetically encoded fluorescent sensor for rapid and specific in vivo detection of norepinephrine. Neuron 102, 745–761.e8 (2019). doi: 10.1016/j.neuron.2019.02.037; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jing M et al. , An optimized acetylcholine sensor for monitoring in vivo cholinergic activity. Nat. Methods 17, 1139–1146 (2020). doi: 10.1038/s41592-020-0953-2; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patriarchi T et al. , Ultrafast neuronal imaging of dopamine dynamics with designed genetically encoded sensors. Science 360, eaat4422 (2018). doi: 10.1126/science.aat4422; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Qian T et al. , A genetically encoded sensor measures temporal oxytocin release from different neuronal compartments. Nat. Biotechnol. 41, 944–957 (2023). doi: 10.1038/s41587-022-01561-2; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ino D, Tanaka Y, Hibino H, Nishiyama M, A fluorescent sensor for real-time measurement of extracellular oxytocin dynamics in the brain. Nat. Methods 19, 1286–1294 (2022). doi: 10.1038/s41592-022-01597-x; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duffet L et al. , A genetically encoded sensor for in vivo imaging of orexin neuropeptides. Nat. Methods 19, 231–241 (2022). doi: 10.1038/s41592-021-01390-2; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Otaki JM, Firestein S, Length analyses of mammalian G-protein-coupled receptors. J. Theor. Biol. 211, 77–100 (2001). doi: 10.1006/jtbi.2001.2272; [DOI] [PubMed] [Google Scholar]
- 26.Unal H, Karnik SS, Domain coupling in GPCRs: The engine for induced conformational changes. Trends Pharmacol. Sci. 33, 79–88 (2012). doi: 10.1016/j.tips.2011.09.007; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hollenstein K et al. , Structure of class B GPCR corticotropin-releasing factor receptor 1. Nature 499, 438–443 (2013). doi: 10.1038/nature12357; [DOI] [PubMed] [Google Scholar]
- 28.Ma S et al. , Molecular basis for hormone recognition and activation of corticotropin-releasing factor receptors. Mol. Cell 77, 669–680. e4 (2020). doi: 10.1016/j.molcel.2020.01.013; [DOI] [PubMed] [Google Scholar]
- 29.Zhao W et al. , Structural insights into ligand recognition and selectivity of somatostatin receptors. Cell Res. 32, 761–772 (2022). doi: 10.1038/s41422-022-00679-x; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paul A et al. , Transcriptional architecture of synaptic communication delineates GABAergic neuron identity. Cell 171, 522–539.e20 (2017). doi: 10.1016/j.cell.2017.08.032; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith SJ et al. , Single-cell transcriptomic evidence for dense intracortical neuropeptide networks. eLife 8, e47889 (2019). doi: 10.7554/eLife.47889; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhong W et al. , The neuropeptide landscape of human prefrontal cortex. Proc. Natl. Acad. Sci. U.S.A. 119, e2123146119 (2022). doi: 10.1073/pnas.2123146119; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun F et al. , Next-generation GRAB sensors for monitoring dopaminergic activity in vivo. Nat. Methods 17, 1156–1166 (2020). doi: 10.1038/s41592-020-00981-9; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wan J et al. , A genetically encoded sensor for measuring serotonin dynamics. Nat. Neurosci. 24, 746–752 (2021). doi: 10.1038/s41593-021-00823-7; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dautzenberg FM et al. , Cell-type specific calcium signaling by corticotropin-releasing factor type 1 (CRF1) and 2a (CRF2(a)) receptors: phospholipase C-mediated responses in human embryonic kidney 293 but not SK-N-MC neuroblastoma cells. Biochem. Pharmacol. 68, 1833–1844 (2004). doi: 10.1016/j.bcp.2004.07.013; [DOI] [PubMed] [Google Scholar]
- 36.Wamsley B, Fishell G, Genetic and activity-dependent mechanisms underlying interneuron diversity. Nat. Rev. Neurosci. 18, 299–309 (2017). doi: 10.1038/nrn.2017.30; [DOI] [PubMed] [Google Scholar]
- 37.Persoon CM et al. , Pool size estimations for dense-core vesicles in mammalian CNS neurons. EMBO J. 37, e99672 (2018). doi: 10.15252/embj.201899672; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arora S et al. , SNAP-25 gene family members differentially support secretory vesicle fusion. J. Cell Sci. 130, 1877–1889 (2017). doi: 10.1242/jcs.201889; [DOI] [PubMed] [Google Scholar]
- 39.Kumar U, Singh S, Role of somatostatin in the regulation of central and peripheral factors of satiety and obesity. Int. J. Mol. Sci. 21, 2568 (2020). doi: 10.3390/ijms21072568; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hauge-Evans AC et al. , Somatostatin secreted by islet δ-cells fulfills multiple roles as a paracrine regulator of islet function. Diabetes 58, 403–411 (2009). doi: 10.2337/db08-0792; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rorsman P, Huising MO, The somatostatin-secreting pancreatic d-cell in health and disease. Nat. Rev. Endocrinol. 14, 404–414 (2018). doi: 10.1038/s41574-018-0020-6; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Salehi A, Qader SS, Grapengiesser E, Hellman B, Pulses of somatostatin release are slightly delayed compared with insulin and antisynchronous to glucagon. Regul. Pept. 144, 43–49 (2007). doi: 10.1016/j.regpep.2007.06.003; [DOI] [PubMed] [Google Scholar]
- 43.Hellman B, Salehi A, Grapengiesser E, Gylfe E, Isolated mouse islets respond to glucose with an initial peak of glucagon release followed by pulses of insulin and somatostatin in antisynchrony with glucagon. Biochem. Biophys. Res. Commun. 417, 1219–1223 (2012). doi: 10.1016/j.bbrc.2011.12.113; [DOI] [PubMed] [Google Scholar]
- 44.Reisine T, Bell GI, Molecular biology of somatostatin receptors. Endocr. Rev. 16, 427–442 (1995). [DOI] [PubMed] [Google Scholar]
- 45.Nakagawasai O et al. , Immunohistochemical fluorescence intensity reduction of brain somatostatin in the impairment of learning and memory-related behaviour induced by olfactory bulbectomy. Behav. Brain Res. 142, 63–67 (2003). doi: 10.1016/S0166-4328(02)00383-2; [DOI] [PubMed] [Google Scholar]
- 46.Davies P, Katzman R, Terry RD, Reduced somatostatin-like immunoreactivity in cerebral cortex from cases of Alzheimer disease and Alzheimer senile dementia. Nature 288, 279–280 (1980). doi: 10.1038/288279a0; [DOI] [PubMed] [Google Scholar]
- 47.Kluge C, Stoppel C, Szinyei C, Stork O, Pape HC, Role of the somatostatin system in contextual fear memory and hippocampal synaptic plasticity. Learn. Mem. 15, 252–260 (2008). doi: 10.1101/lm.793008; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stujenske JM et al. , Prelimbic cortex drives discrimination of non-aversion via amygdala somatostatin interneurons. Neuron 110, 2258–2267.e11 (2022). doi: 10.1016/j.neuron.2022.03.020; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gründemann J, Lüthi A, Ensemble coding in amygdala circuits for associative learning. Curr. Opin. Neurobiol. 35, 200–206 (2015). doi: 10.1016/j.conb.2015.10.005; [DOI] [PubMed] [Google Scholar]
- 50.Janak PH, Tye KM, From circuits to behaviour in the amygdala. Nature 517, 284–292 (2015). doi: 10.1038/nature14188; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jo YS, Namboodiri VMK, Stuber GD, Zweifel LS, Persistent activation of central amygdala CRF neurons helps drive the immediate fear extinction deficit. Nat. Commun. 11, 422 (2020). doi: 10.1038/s41467-020-14393-y; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sanford CA et al. , A central amygdala CRF circuit facilitates learning about weak threats. Neuron 93, 164–178 (2017). doi: 10.1016/j.neuron.2016.11.034; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pomrenze MB et al. , A corticotropin releasing factor network in the extended amygdala for anxiety. J. Neurosci. 39, 1030–1043 (2019). doi: 10.1523/JNEUROSCI.2143-18.2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Guglielmo G et al. , Inactivation of a CRF-dependent amygdalofugal pathway reverses addiction-like behaviors in alcohol-dependent rats. Nat. Commun. 10, 1238 (2019). doi: 10.1038/s41467-019-09183-0; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim J et al. , Rapid, biphasic CRF neuronal responses encode positive and negative valence. Nat. Neurosci. 22, 576–585 (2019). doi: 10.1038/s41593-019-0342-2; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yuan Y et al. , Reward inhibits paraventricular CRH neurons to relieve stress. Curr. Biol. 29, 1243–1251.e4 (2019). doi: 10.1016/j.cub.2019.02.048; [DOI] [PubMed] [Google Scholar]
- 57.Daviu N et al. , Paraventricular nucleus CRH neurons encode stress controllability and regulate defensive behavior selection. Nat. Neurosci. 23, 398–410 (2020). doi: 10.1038/s41593-020-0591-0; [DOI] [PubMed] [Google Scholar]
- 58.Gallopin T, Geoffroy H, Rossier J, Lambolez B, Cortical sources of CRF, NKB, and CCK and their effects on pyramidal cells in the neocortex. Cereb. Cortex 16, 1440–1452 (2006). doi: 10.1093/cercor/bhj081; [DOI] [PubMed] [Google Scholar]
- 59.Deussing JM, Chen A, The corticotropin-releasing factor family: physiology of the stress response. Physiol. Rev. 98, 2225–2286 (2018). doi: 10.1152/physrev.00042.2017; [DOI] [PubMed] [Google Scholar]
- 60.Chen P et al. , Prefrontal cortex corticotropin-releasing factor neurons control behavioral style selection under challenging situations. Neuron 106, 301–315.e7 (2020). doi: 10.1016/j.neuron.2020.01.033; [DOI] [PubMed] [Google Scholar]
- 61.Uribe-Mariño A et al. , Prefrontal cortex corticotropin-releasing factor receptor 1 conveys acute stress-induced executive dysfunction. Biol. Psychiatry 80, 743–753 (2016). doi: 10.1016/j.biopsych.2016.03.2106; [DOI] [PubMed] [Google Scholar]
- 62.Nielsen TA, DiGregorio DA, Silver RA, Modulation of glutamate mobility reveals the mechanism underlying slow-rising AMPAR EPSCs and the diffusion coefficient in the synaptic cleft. Neuron 42, 757–771 (2004). doi: 10.1016/j.neuron.2004.04.003; [DOI] [PubMed] [Google Scholar]
- 63.Rice ME, Gerhardt GA, Hierl PM, Nagy G, Adams RN, Diffusion coefficients of neurotransmitters and their metabolites in brain extracellular fluid space. Neuroscience 15, 891–902 (1985). doi: 10.1016/0306-4522(85)90087-9; [DOI] [PubMed] [Google Scholar]
- 64.Weiss AN, Bittner MA, Holz RW, Axelrod D, Protein mobility within secretory granules. Biophys. J. 107, 16–25 (2014). doi: 10.1016/j.bpj.2014.04.063; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.DiGruccio MR et al. , Comprehensive alpha, beta and delta cell transcriptomes reveal that ghrelin selectively activates delta cells and promotes somatostatin release from pancreatic islets. Mol. Metab. 5, 449–458 (2016). doi: 10.1016/j.molmet.2016.04.007; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Huising MO, van der Meulen T, Huang JL, Pourhosseinzadeh MS, Noguchi GM, The difference δ-cells make in glucose control. Physiology 33, 403–411 (2018). doi: 10.1152/physiol.00029.2018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Smith SJ, Hawrylycz M, Rossier J, Sümbül U, New light on cortical neuropeptides and synaptic network plasticity. Curr. Opin. Neurobiol. 63, 176–188 (2020). doi: 10.1016/j.conb.2020.04.002; [DOI] [PubMed] [Google Scholar]
- 68.Ludwig M, Leng G, Dendritic peptide release and peptide-dependent behaviours. Nat. Rev. Neurosci. 7, 126–136 (2006). doi: 10.1038/nrn1845; [DOI] [PubMed] [Google Scholar]
- 69.van den Pol AN, Neuropeptide transmission in brain circuits. Neuron 76, 98–115 (2012). doi: 10.1016/j.neuron.2012.09.014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Overington JP, Al-Lazikani B, Hopkins AL, How many drug targets are there? Nat. Rev. Drug Discov. 5, 993–996 (2006). doi: 10.1038/nrd2199; [DOI] [PubMed] [Google Scholar]
- 71.Nederpelt I, Bunnik J, IJzerman AP, Heitman LH, Kinetic profile of neuropeptide–receptor interactions. Trends Neurosci. 39, 830–839 (2016). doi: 10.1016/j.tins.2016.09.008; [DOI] [PubMed] [Google Scholar]
- 72.Kaiser A, Coin I, Capturing peptide-GPCR interactions and their dynamics. Molecules 25, 4724 (2020). doi: 10.3390/molecules25204724; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.De Souza EB, Corticotropin-releasing factor receptors in the rat central nervous system: Characterization and regional distribution. J. Neurosci. 7, 88–100 (1987). doi: 10.1523/JNEUROSCI.07-01-00088.1987; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Castro M, Nikolaev VO, Palm D, Lohse MJ, Vilardaga JP, Turn-on switch in parathyroid hormone receptor by a two-step parathyroid hormone binding mechanism. Proc. Natl. Acad. Sci. U.S.A. 102, 16084–16089 (2005). doi: 10.1073/pnas.0503942102; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nederpelt I et al. , Characterization of 12 GnRH peptide agonists – a kinetic perspective. Br. J. Pharmacol. 173, 128–141 (2016). doi: 10.1111/bph.13342; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yin W et al. , A complex structure of arrestin-2 bound to a G protein-coupled receptor. Cell Res. 29, 971–983 (2019). doi: 10.1038/s41422-019-0256-2; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yusa K, Zhou L, Li MA, Bradley A, Craig NL, A hyperactive piggyBac transposase for mammalian applications. Proc. Natl. Acad. Sci. U.S.A. 108, 1531–1536 (2011). doi: 10.1073/pnas.1008322108; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kroeze WK et al. , PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat. Struct. Mol. Biol. 22, 362–369 (2015). doi: 10.1038/nsmb.3014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wan Q et al. , Mini G protein probes for active G protein–coupled receptors (GPCRs) in live cells. J. Biol. Chem. 293, 7466–7473 (2018). doi: 10.1074/jbc.RA118.001975; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Qin XY et al. , PSD-93 up-regulates the synaptic activity of corticotropin-releasing hormone neurons in the paraventricular nucleus in depression. Acta Neuropathol. 142, 1045–1064 (2021). doi: 10.1007/s00401-021-02371-7; [DOI] [PubMed] [Google Scholar]
- 81.Zhu Y et al. , Dynamic salience processing in paraventricular thalamus gates associative learning. Science 362, 423–429 (2018). doi: 10.1126/science.aat0481; [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
DNA plasmids of GRAB peptide sensors used in this study have been deposited to Addgene (Addgene ID 208654–208685). All other data needed to evaluate the conclusions in the paper are available in the main text or the supplementary materials.