

Termini restraining enables structural determination of small membrane proteins, greatly expanding their structural database.
Abstract
Small membrane proteins are difficult targets for structural characterization. Here, we stabilize their folding by restraining their amino and carboxyl termini with associable protein entities, exemplified by the two halves of a superfolder GFP. The termini-restrained proteins are functional and show improved stability during overexpression and purification. The reassembled GFP provides a versatile scaffold for membrane protein crystallization, enables diffraction to atomic resolution, and facilitates crystal identification, phase determination, and density modification. This strategy gives rise to 14 new structures of five vertebrate proteins from distinct functional families, bringing a substantial expansion to the structural database of small membrane proteins. Moreover, a high-resolution structure of bacterial DsbB reveals that this thiol oxidoreductase is activated through a catalytic triad, similar to cysteine proteases. Overall, termini restraining proves exceptionally effective for stabilization and structure determination of small membrane proteins.
INTRODUCTION
Integral membrane proteins constitute approximately 30% of proteomes in prokaryotes and eukaryotes, and they represent more than half of the drug targets. Elucidating their molecular functions and facilitating the development of new drugs requires knowledge of their three-dimensional structures. Structural studies generally start from protein overexpression in cells, followed by protein purification from the native membranes with selected detergents. These operations frequently disrupt the function and folding of membrane proteins, resulting in their denaturation and aggregation (Fig. 1). To overcome this difficulty, structural biologists often perform large screens of homologous proteins to identify a few that show sufficient stability during overexpression and purification (1). Most membrane proteins, however, need to have their instability and flexibility reduced before they can be structurally characterized. One approach to enhance protein thermostability is systematic mutagenesis of the entire protein to identify multiple synergistically stabilizing mutations, a trial-and-error method that requires tedious effort (2–4). Another time-consuming and costly approach is the search of suitable antibodies that can stabilize a flexible region of membrane proteins and provide a scaffold for cocrystallization (5) or cryo–electron microscopy (cryo-EM) analyses (6). Structure determination of heterotrimeric GTP-binding protein (G protein)–coupled receptors (GPCRs) can be accomplished by replacing an intracellular loop in GPCRs with a highly crystallizable soluble protein (7, 8). However, there are limited reports of applying this engineering method to obtain structures of membrane proteins from other families, because it is unknown which flexible loops should be replaced, and the replacement is restricted by the size of the soluble protein. Moreover, lessons learned from GPCRs are that their G protein coupling function is compromised by the replacement of the intracellular loop (7), by an antibody bound to this functionally essential loop (9), and even by heterologous overexpression in insect cells (10). Together, maintaining the native structure and function of membrane proteins in vitro remains the biggest hurdle to overcome for structural biologists (11).
Fig. 1. Termini restraining of small membrane proteins facilitates their entire structure determination process.
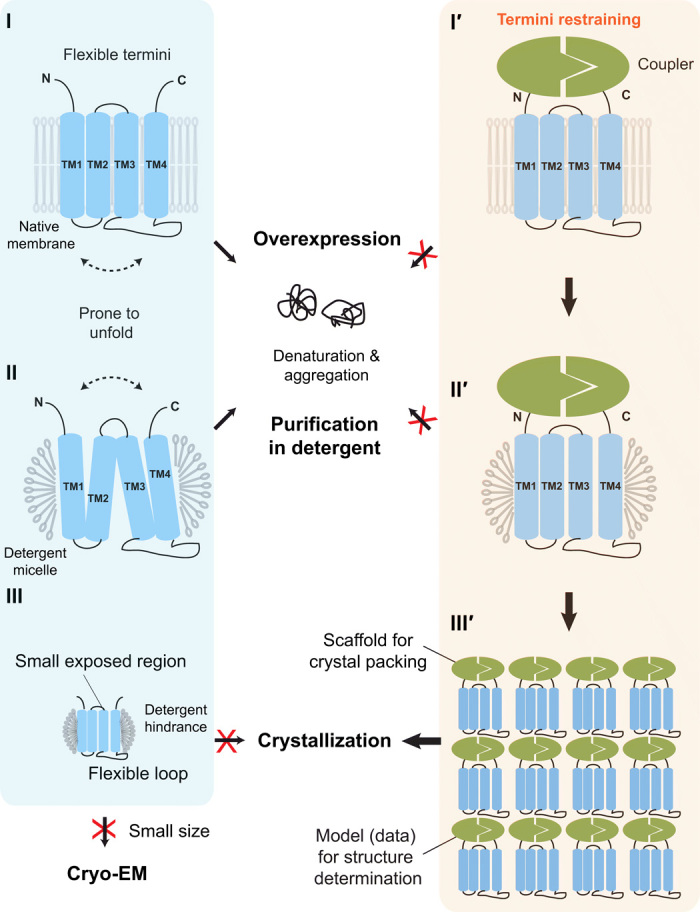
(Left) Difficulties with traditional structural biology approaches (blue box). (I) Overexpression of membrane proteins increases their tendency to unfold and aggregate in cells. The unfolding involves large relative motions between TMs (dashed arrow). (II) Protein purification in detergents is often disruptive to the native folded state of membrane proteins. (III) Small membrane proteins in detergent micelles often contain either small exposed regions or large flexible loops, both of which increase the difficulty of making crystal contact. These proteins are also too small for structural determination by cryo-EM. (Right) Termini-restraining strategy (orange box). (I′) Two associable protein entities (coupler; green) are fused to the flexible N and C termini of a membrane protein (blue), providing a loose restraint to stabilize its folded state during protein overexpression in cells (I′) and protein purification in detergents (II′). (III′) Introducing the coupler protein also provides a large surface for crystal packing and facilitates structure determination.
Structure determination of small membrane proteins poses additional challenges. More than 60% of membrane proteins have fewer than the seven transmembrane helices (TMs) that GPCRs have (12). These small proteins are difficult targets for crystallography. With their membrane portion buried in the detergent micelles, these proteins are often left with either small exposed regions that can barely provide crystal contacts or large flexible loops that may destabilize crystal contacts (Fig. 1). Because many of these proteins are not subunits of a large and stable assembly, their small size generally places them beyond the reach of cryo-EM techniques, which are currently striving toward a minimum size of ~100 kDa for membrane proteins. As a result, small membrane proteins are much underexplored by structural biology. Taking the example of four-TM proteins, structures have been reported so far for only six individual proteins—CD81 (13), CD9 (14), Cldn4 (15), Cldn9 (16), DHHC20 (17), and SCD1 (18)—from hundreds of these in the human genome (19). To meet this critical challenge, here, we develop a termini-restraining approach to facilitate the entire structure determination process of small membrane proteins. This new approach gives rise to 14 structures of five proteins from humans and other vertebrates, whose structures were unknown from traditional approaches, and a high-resolution structure of bacterial disulfide bond formation protein B (DsbB) that reveals its catalytic chemistry, thereby bringing a substantial expansion to the current structural database of small membrane proteins.
RESULTS
Design principles of termini restraining
Our strategy is to stabilize small membrane proteins by restraining their two termini with a coupler formed of two associable protein entities (Fig. 1). As a proof of concept, we fused the N and C termini of the membrane proteins to the N- and C-terminal halves of a superfolder green fluorescent protein (sfGFP), respectively; split sfGFP has been optimized for efficient self-association in previous works (20, 21). Tethering this soluble coupler protein to the two flexible termini of the membrane proteins provides a mild restraint, hampering the drastic TM motions that lead to unfolding and thus favoring their folded state during protein overexpression and purification. The flexible portions of the two termini loosely connect the coupler to the first and last TMs of membrane proteins, permitting moderate conformational changes that are usually sufficient for the normal protein function. This approach requires that the two termini of the target proteins are located on the same side of the membrane. Given that most membrane proteins are predicted to have even numbers of TMs and to have both termini in the cytosol (22, 23), this engineering strategy can be applied to numerous membrane proteins with such folding topology.
Selection of small membrane proteins
To test the generality of our approach, we selected seven model proteins from humans or other vertebrates (Table 1) that were considered difficult structural targets. These small membrane proteins (10 to 40 kDa) contain even numbers of TMs (2, 4, 6), but their three-dimensional structures were unknown. These proteins represent distinct functional families, including membrane enzymes, adhesion molecules, and transporters. Vitamin K epoxide reductase (VKOR) and VKOR-like (VKORL) are thiol oxidoreductases. Signal peptidase complex subunit 1 (SPCS1) is a serine protease subunit required for the processing of flaviviral proteins (24). CD53 is a member of the tetraspanins (25). Claudin 4 (Cldn4) forms the backbone of tight junction (15). Jagunal homolog 1 (JAGN1) is involved in vesicle-mediated transport (26). Mitoferrin-1 (Mfrn1) belongs to the SLC25 carrier family that transports solutes across the mitochondria inner membrane (27). We have also included DsbB, a bacterial oxidoreductase that has been previously cocrystallized with its partner protein (DsbA) or in the presence of an antibody (28, 29), but these tremendous measures produced structures of only moderate resolution (3.4 to 3.7 Ǻ) that lack protein side-chain information (30). Thus, DsbB serves as a benchmark that allows us to compare the result of structure determination by traditional methods and by our termini-restraining approach.
Table 1. Summary of small membrane proteins, enhancement of protein stability after termini restraining, and improvement of structural resolution after termini shortening.
ER, endoplasmic reticulum; MW, molecular weight; Tm, melting temperature; TR, termini restraining; N/A, not analyzed or not available; War, warfarin; n.p., autobuilding not performed because of poor data quality; PDB, Protein Data Bank.
| Protein | SPCS1 | VKOR* | VKORL* | DsbB | JAGN1 | CD53 (43) | Cldn4 | Mfrn1 |
| Origin | Gallus gallus | Homo sapiens | Takifugu rubripes | Escherichia coli | Homo sapiens |
Homo
sapiens |
Homo
sapiens |
Oreochromis
niloticus |
| Protein function or family |
Signal peptidase subunit |
Thiol oxidoreductase |
Thiol oxidoreductase |
Thiol oxidoreductase |
Vesicle transport |
Tetraspanin | Claudin | Mitochondrial transporter |
| Subcellular location |
ER membrane |
ER membrane | ER membrane | Cytoplasmic membrane |
ER membrane | Cytoplasmic membrane |
Cytoplasmic membrane |
Mitochondria inner membrane |
| TM no. | 2 | 4 | 4 | 4 | 4 | 4 | 4 | 6 |
| MW (kDa) | 11.6 | 18.2 | 19.6 | 20.1 | 21.1 | 24.3 | 22.1 | 36.7 |
| Length (amino acids) |
101 | 163 | 175 | 176 | 183 | 219 | 209 | 338 |
| Protein stability | ||||||||
| Folded protein† | + | ++ | +++ | +/− | +++ | − | +/− | ++ |
| Tm before TR (°C) | 64.6 | 49.2 | 53.3 | 49.5 | 57.8 | 79.7 | 85.2 | 58.9 |
| Tm after TR (°C) | 64.4 | 52.1 | 62.1 | >90 | 66.3 | 80.2 | 73.0 | 60.2 |
| Activity or function |
N/A | Improved | Improved | Maintained | N/A | N/A | Maintained | Maintained |
| Crystallization | ||||||||
| Initial (amino acids) |
1–80 | Full length (War) | Full length | 6–170‡ | Full length | Full length | 1–184 | N/A |
| Resolution (Å) | ~5 | 5.7 | 4.3 | 3.7 | 2.25 | 2.9 | N/A | N/A |
| Space group | C222 | P222 | P43212 | P2 | C2 | P2 | ||
| Cell dimensions | ||||||||
| a, b, c (Å) | 69.8, 366.7, 44.4 |
65.2, 89.2, 96.2 | 88.5, 88.5, 335.6 | 83.5, 56.5, 120.3 | 90.8, 42.7, 111.8 |
49.4, 210.5, 73.4 |
||
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 | 90, 96.5, 90 | 90, 103.8, 90 | 90, 100.2, 90 | ||
| Solvent content | 69.2% | 59.3% | 65.5% | 62.3% | 67.0% | 45.6% | ||
| Phase improvement |
Solvent flattening |
Solvent flattening |
Cross-crystal | Solvent flattening |
Solvent flattening |
Twofold averaging |
||
| Auto building§ |
n.p. | n.p. | 57.9% | n.p. | 93.4% | 96.4% | ||
| Shortened (amino acids) |
1–75 | 3–155 (War) | 6–175 | 6–167 | N/A | N/A | N/A | N/A |
| Resolution (Å) | 2.45 | 2.2 | 2.4 | 2.9 | ||||
| Space group | C2 | P21212 | C2 | P3221 | ||||
| Cell dimensions | ||||||||
| a, b, c (Å) | 105.0, 55.3, 92.7 |
47.4, 70.9, 153.1 | 153.6, 49.1, 84.3 | 53.4, 53.4, 280.7 | ||||
| α, β, γ (°) | 90, 110.9, 90 | 90, 90, 90 | 90, 112.7, 90 | 90, 90, 120 | ||||
| Solvent content | 65.2% | 57.8% | 60.8% | 52.2% | ||||
| Phase improvement |
Solvent flattening |
Solvent flattening |
Solvent flattening |
Cross-crystal | ||||
| Auto building§ |
90.5% | 84.3% | 97.8% | 83.1% | ||||
| Rwork/Rfree (%) | 20.4/23.2 | 20.1/22.3 | 20.5/23.7 | 26.0/28.6 | 19.5/22.7 | 22.3/26.5 | N/A | N/A |
| PDB code | 6WVE | 6WV3, 6WV6, 6WVH, 6WV4, 6WV5, 6WV7 |
6WVI, 6WVB, 6WV8, 6WV9, 6WVA |
6WVF | 6WVD | 6WVG | N/A | N/A |
*Biological interpretations of the VKOR and VKORL structures are being reported in an accompanying manuscript (42).
†Changes in the fraction of folded membrane proteins after TR are roughly estimated (+ or − signs) from the whole-cell extraction of overexpressed proteins with SDS, SDS/urea, and DDM.
‡This construct was used according to Inaba et al. (28).
§Autobuilding was performed with BUCCANEER using the density modification maps generated from PARROT (see Materials and Methods).
Retained protein functions after termini restraining
Our first priority was to confirm that termini restraining does not compromise protein functions. The restrained VKOR and VKORL retain their cellular reductase activity and became slightly more sensitive to the inhibition by warfarin, an anticoagulant targeting these enzymes (Fig. 2A). A historically challenging example of protein instability, human VKOR rapidly loses enzymatic activity during purification; this prevented the biochemical identification of this protein before the genomic era (31). With the two termini restrained, purified human VKOR shows a specific activity 20 times higher than the unrestrained protein does (Fig. 2B), suggesting that the restraining markedly increases protein stability. Similarly, the specific activity of the restrained VKORL is five times higher than that of the wild-type VKORL (Fig. 2B). The restrained DsbB, like the wild-type DsbB, can promote disulfide bond formation in Escherichia coli; both constructs complement the motility defect of a ∆dsbB strain (32) and inactivate a disulfide-sensitive β-galactosidase fusion protein (Fig. 2C) (33). Cldn4, another protein that we could assay for function, retains interactions of forming high-order complexes at the cell junction after the sfGFP restraining (Fig. 2D). In particular, the restrained Mfrn1, which needs to transport iron via large rocking motions (34), exhibits high activity in liposomes as the unrestrained Mfrn1 does (Fig. 2E). Together, these examples indicate that termini restraining maintains the transport activity, enzymatic activity, and molecular interaction of these representative membrane proteins, even when large conformational changes are required for their functions.
Fig. 2. Termini restraining maintains the functions of small membrane proteins and enhances their stability during overexpression and purification.

(A) Cellular activities of termini-restrained (TR) and unrestrained constructs of VKOR and VKORL in response to warfarin inhibition. (B) Activities of purified VKOR and VKORL constructs at the same protein concentration. (C) DsbB-catalyzed disulfide bond formation. Left: Transformations restore the motility (measured by colony size) of ∆dsbB E. coli. EV, empty vector. Right: Disulfide-mediated inactivation (white colonies) of MalF-fused β-galactosidase. (D) Cldn4-mediated cell junction. Fluorescence images are of live human embryonic kidney (HEK) 293 cells transfected with Cldn4 constructs. (E) Fe2+ uptake activity of purified Mfrn1 in liposomes (50). Cal, calcimycin. See Materials and Methods for explanation of assays in (A) to (E). (F) Fluorescence images of live Pichia cells overexpressing ER membrane proteins with C-GFP tag or sfGFP restraining. The merged images combine GFP fluorescence with ER marker. Red arrows indicate potential protein accumulation in the ER and/or vacuoles. DIC, differential interference contrast. (G) Comparison of extractable folded membrane protein fraction with or without the termini restraining. The immunoblots are shown for target proteins extracted from whole cells with SDS, SDS/urea, or DDM. (H) FSEC-based thermostability assay of purified proteins showing change of Tm (ΔTm) with termini restraining. The unrestrained (U) and sfGFP-restrained constructs both carry a C-terminal mCherry tag for fluorescence detection.
Protein stability during overexpression and purification
Because overexpression often causes misfolding or aggregation of membrane proteins (10, 35), we compared the sfGFP-restrained proteins with those tagged with a C-terminal GFP (C-GFP). Fluorescence imaging showed that termini restraining does not change the subcellular localization of these proteins (Fig. 2F and fig. S1). Moreover, the reconstituted fluorescence of sfGFP indicates that proteins with even numbers of TMs fold properly in their predicted topologies (36). Some of the C-GFP–tagged proteins, such as VKORL, appear to accumulate in the endoplasmic reticulum (ER) and/or vacuoles, whereas the sfGFP-restrained proteins are distributed evenly at the ER membrane boundary. To understand this difference, we compared the whole-cell extraction of the restrained and unrestrained proteins in the presence of SDS, SDS/urea, and n-dodecyl-β-D-maltoside (DDM) detergents, the latter of which preferably solubilizes correctly folded membrane proteins (10). For C-GFP–tagged VKOR, VKORL, JAGN1, SPCS1, and Mfrn1, the amount of DDM-extractable protein was much less than that extractable by SDS and/or SDS/urea, indicating that a large portion of these recombinant proteins are expressed in aggregated or insoluble form (Fig. 2G). In contrast, termini restraining substantially increases the DDM-extractable fraction of these ER and mitochondrial proteins, indicating that the restraining can prevent protein aggregation during overexpression. The relative level of DDM-extractable protein, however, is similar for the restrained and unrestrained DsbB from E. coli cytoplasmic membrane. For the human proteins located on the cytoplasmic membrane, we found that the DDM-extractable fraction remains nearly unchanged for restrained Cldn4 and becomes less for restrained CD53, probably because these cell surface proteins are intrinsically stable and therefore their stability is not improved by termini restraining. Overall, although we did not observe improvement for every protein, the restraining affords a readily testable approach to increase the effective production of membrane proteins, presumably through stabilizing their folded state in cells.
The ability of proteins to maintain their folded state usually correlates with their thermostability (37). Therefore, we measured the melting temperature (Tm) of proteins purified in detergent solution by using fluorescence-detected size exclusion chromatography (FSEC), a well-established assay for membrane protein stability (38). To allow precise comparison, we tagged the sfGFP-restrained and unrestrained constructs with a C-terminal mCherry for FSEC detection. This design avoids the Tm comparison between split and integral GFPs; instead, the sfGFP serves only as the coupler, and the Tm is measured through the change of mCherry signal. The restraining raised the Tm by 8.8°C for VKORL and 8.5°C for JAGN1 (Fig. 2H). The Tm change of DsbB after restraining is even more substantial, from 50°C to more than 90°C. The comparison of VKOR proteins required further stabilization with bound warfarin, under which condition the restraining increased the Tm by 2.9°C. The restraining slightly changes the Tm of SPCS1, CD53, and Mfrn1, and lowers that of Cldn4, probably because the unstrained Cldn4 has an inherently high Tm (85°C). This restraining approach thus appears to be more effective at improving the detergent stability of proteins with relatively low Tm.
Crystallization and structure determination
Seven restrained membrane proteins—VKOR, VKORL, CD53, SPCS1, Cldn4, JAGN1, and DsbB—could be readily crystallized in lipidic-cubic phase (LCP) (Table 1). We optimized all the crystals except Cldn4, whose structure was reported during our study (15). The effort of optimization was immediately justified, because the sfGFP coupler can be detected under fluorescence microscope. Even tiny protein crystals can be distinguished from other particles made of salt, detergent, or lipids (Fig. 3A). Traditionally, it requires months of effort to optimize the initial hits and allow crystals to grow large enough to confirm the presence of the target protein by crystal diffraction pattern or by silver staining of dissolved crystals. The sfGFP coupler bypasses this technical bottleneck.
Fig. 3. The sfGFP coupler enables crystallization of small membrane proteins.
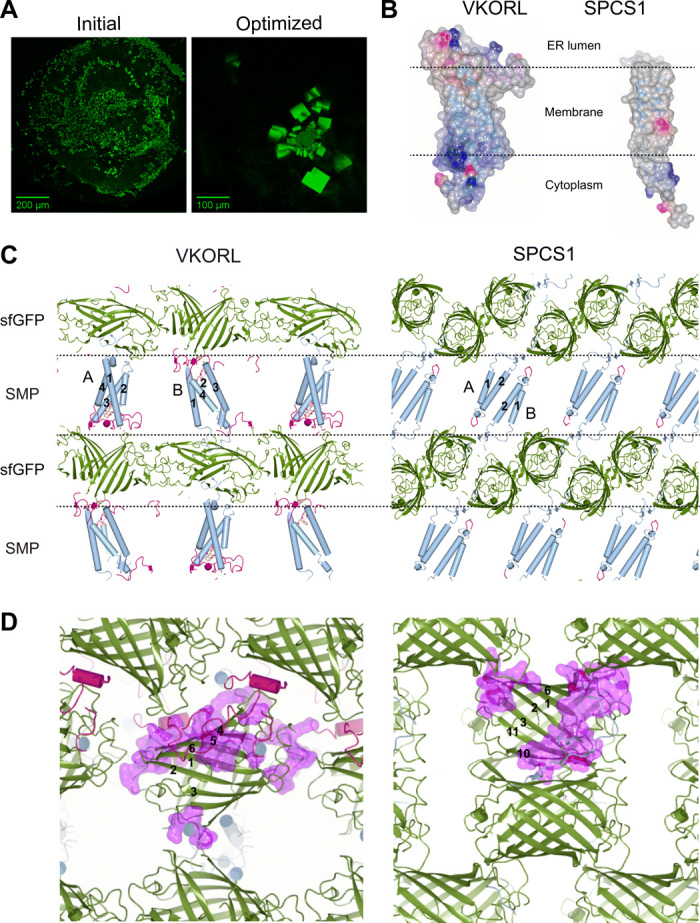
VKORL and SPCS1 are shown as examples here; other proteins are shown in fig. S2. (A) Identification of initial crystallization hits (left) of sfGFP-restrained VKORL through fluorescence imaging. The optimized crystals (right) are shown for comparison. (B) Electrostatic surfaces of VKORL and SPCS1 showing their different shapes and surface charges (blue, positive charge; red, negative charge). (C) sfGFP serves as a crystal packing scaffold that is highly adaptable to accommodate various small membrane proteins. Each crystal is packed with alternative layers (dashed lines) of sfGFP (green) and small membrane protein (SMP) molecules. The TMs (numbered) are shown in blue, and extramembrane regions are shown in red. Different membrane protein molecules in the crystals are indicated (A and B). (D) Zoom-in view of versatile crystal packing interactions formed among sfGFP molecules. The large polar surface of sfGFP can accommodate a multitude of crystal contacts (purple surface). The β strands in sfGFP are numbered to illustrate location of the contacts.
The sfGFP coupler affords a highly versatile scaffold for the crystallization of small membrane proteins, which have different sizes, shapes, and surface properties (Fig. 3B and fig. S2A). Each of the sfGFP-restrained proteins crystallized in a different space group or with different unit cell dimensions (Table 1, table S1, Fig. 3C, and fig. S2B). Even for the same protein, VKOR or VKORL, alternative reactive states or different bound ligands generated five and three different crystal forms, respectively (table S2 and fig. S2C). Despite these differences, the crystal lattices are always packed into two alternative layers, one made by sfGFP molecules and another by the small membrane proteins (Fig. 3C and fig. S2, B and C). Crystal contacts can also be formed between the TM regions of the membrane protein molecules, likely induced by the LCP crystallization condition. To accommodate the different membrane proteins, sfGFP molecules form various lattice packing interactions using nearly every part of its large polar surface (Fig. 3D). None of the 12 crystal forms exhibits the same packing pattern in the layer of sfGFP molecules (fig. S2, B and C). This remarkable adaptability shows that termini restraining by sfGFP is broadly applicable to crystallizing small membrane proteins.
The quality of crystal diffraction is markedly improved by shortening the flexible termini regions of membrane proteins tethered to sfGFP. This shortening induced alternative crystal packing. For instance, crystals of the restrained full-length VKORL only diffracted to 4.3 Å (Fig. 4A). Shortening the N terminus of VKORL by a mere five amino acids changed the space group from P43212 to C2 (Table 1) with completely different crystal packing interactions (Fig. 4B), probably by restricting the relative motion between sfGFP and VKORL molecules. As a result, the remaining part of the VKORL N terminus becomes ordered in the C2 crystal form, and the orientation of VKORL with respect to sfGFP becomes different (Fig. 4C). The solvent content decreases by 5%, and the crystal diffraction improves to 2.4 Å (Fig. 4A). Similar termini optimizations changed the crystal packing of VKOR, DsbB, and SPCS1 (Table 1) and markedly improved their crystal diffractions to near-atomic resolution (up to 2.0 Ǻ).
Fig. 4. Restraining-based methods for crystal diffraction and phase improvements.
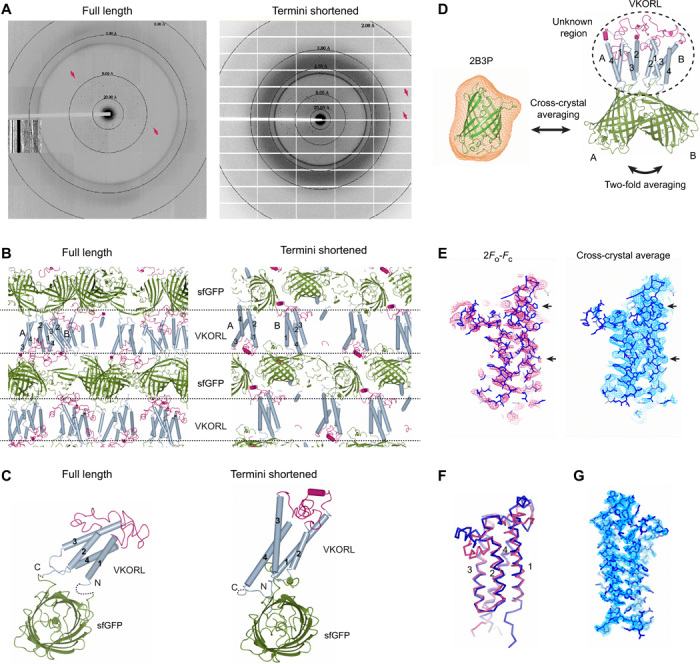
(A) Shortening the restrained termini improves the crystal diffraction of VKORL. Diffraction patterns are shown for full-length (left) and termini-shortened (right) VKORL restrained by sfGFP. Arrows indicate visible diffraction spots near the diffraction limit. (B) Change of crystal packing interactions. The coloring and annotations are the same as in Fig. 3C. (C) Change of relative orientation between the membrane protein and sfGFP after termini shortening. The dashed lines indicate disordered termini. (D) Improvement of electron density maps through cross-crystal averaging with the coupler protein. The cross-crystal averaging is performed between the sfGFP (2B3P) crystal (averaging mask shown in orange) and the sfGFP-restrained VKORL crystal. In addition, the twofold NCS (molecules A and B) is used. (E) Electron density maps of the VKORL region before and after cross-crystal averaging of the low-resolution data. Arrows indicate regions with improved electron density. (F) Superimposition of the low-resolution (4.3 Å; blue) and high-resolution models (2.4 Å; red) showing that the cross-crystal averaged map allows building of a relatively reliable model at low resolution. (G) Electron density maps of the high-resolution VKORL structure after solvent flattening. All the maps are contoured at 1σ.
The crystallographic phase problem was solved using the structural model and diffraction data of the sfGFP coupler. Molecular replacement (MR) with the sfGFP model [Protein Data Bank (PDB) code 2B3P] readily generated solutions for all datasets, because the membrane proteins are small and sfGFP constitutes a relatively large portion of the diffraction mass. For the high-resolution datasets, after rigid body refinement of the MR model and solvent flattening with automatically generated mask, the electron density maps (figs. S3 and S4), which are unbiased at the membrane protein region, allowed automatic model building for a major part (often above 90%) of the target proteins (Table 1). Even at low resolution, the initial MR phases could be markedly improved by cross-crystal averaging (39) with the diffraction data of sfGFP alone (Fig. 4D). For instance, averaging the sfGFP data with the 4.3-Å VKORL data [twofold noncrystallographic symmetry (NCS)] generates a well-featured electron density map (Fig. 4E) that allowed correct building of nearly all TMs (Fig. 4F). The same cross-crystal averaging approach is also useful for data at higher resolution and without NCS, such as the 2.9-Å data of DsbB (fig. S5). Overall, model phasing with the sfGFP coupler avoids experimental phasing, a tedious and sometimes unsuccessful process.
The termini-restraining approach yields the first structures of five small membrane proteins from humans and other vertebrates (Fig. 5A). The structures of VKOR and VKORL, unlike their bacterial homolog structures (fig. S6) (40, 41), elucidate almost their entire catalytic cycle and their mechanism of inhibition by warfarin and other antagonists, which are oral anticoagulants commonly used to treat and prevent thromboembolic diseases such as stroke and heart attack (42). The crystal structure of CD53 is the first structure ever captured of the partner-interacting state for the entire tetraspanin family (43), which mediates molecular interactions for myriad cellular processes (25). SPCS1 is a major therapeutic target of Zika virus (24), and its structure suggests a regulatory role for this signal peptidase subunit during the maturation process of Zika and other flaviviruses. Last, the JAGN1 structure reveals a unique structural fold that may allow specific interaction with proteins involved in vesicle-mediated transport. Thorough analyses of these new structures are being reported elsewhere (42, 43); here, we focus on the benchmark example, DsbB, a disulfide bond formation enzyme.
Fig. 5. High-resolution crystal structures of small membrane proteins provide new mechanistic insights into their functions.
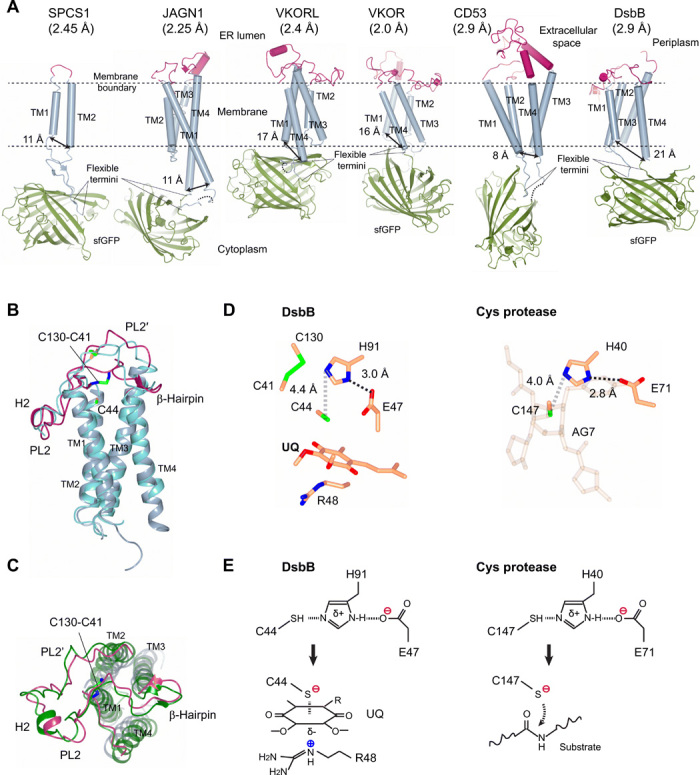
(A) New membrane protein structures (resolution in parenthesis) determined by the termini-restraining strategy. The membrane boundary and flexible ends of membrane proteins fused to the sfGFP are indicated. The double arrows indicate the distance (Cα-Cα atoms) between the ends of N- and C-terminal TMs. (B) Side view of the restrained DsbB Cys104Ser structure [same colors as in (A)] superimposed with an independently published crystal structure (PDB code 2ZUQ; blue). PL, periplasmic loop; H, helix. The Fab bound to DsbB in 2ZUQ is omitted for clarity. (C) Top view of the restrained DsbB structure, superimposed with an NMR structure (2K74; green) that captures a similar electron-transfer state (fig. S7). (D) Structural comparison of the catalytic triad in E. coli DsbB and hepatitis A virus 3C protease (1CQQ), a representative cysteine protease (45, 46). (E) Similarity of catalytic triads in DsbB and cysteine proteases. Histidine deprotonation of the catalytic cysteine (top) generates the reactive thiolate (bottom). The thiolate in DsbB forms a charge-transfer complex with the quinone (UQ), and the thiolate in cysteine protease attacks the scissile bond carbonyl carbon in a peptide substrate.
Maintained DsbB structure after termini restraining
The overall structure of termini-restrained DsbB (Fig. 5A) closely matches previously determined structures of DsbB, indicating that the restraining does not disrupt the protein conformation. The transmembrane region of restrained DsbB has a root mean square deviation (RMSD) of 1.1 Å to that of 2ZUQ (Fig. 5B) (29). The short β-hairpin that folds on top of TM4 is also maintained. The conformation of a long loop connecting TM3 and TM4 is similar to the nuclear magnetic resonance (NMR) structure of a Cys44Ser/Cys104Ser mutant (44) because of formation of the same interloop disulfide, Cys130-Cys41 (Fig. 5C). This interloop disulfide is induced by a Cys104Ser mutation, which also allows a charge-transfer complex to form between Cys44 and the quinone cofactor of DsbB in our structure (fig. S7).
DsbB activation by a catalytic triad
The high-resolution structure reveals that thiolate activation in E. coli DsbB uses a catalytic triad that is reminiscent of the classical mechanisms used by cysteine or serine proteases (Fig. 5D). The electron density maps clearly show the protein side chains and the quinone ring at the active site (fig. S5C). The quinone ring is stacked between Arg48 at the bottom and Cys44 and Met142 on the top. Cys44, His91, and Glu47 form the catalytic triad, whose spatial arrangement is essentially the same as that observed in the cysteine proteases (Fig. 5D) (45, 46). Thus, a similar mechanism may be used by the thiol oxidoreductase DsbB to activate Cys44 (Fig. 5E). This process starts from the deprotonation of His91, probably with assistance from Glu47. Next, His91 abstracts the proton from the sulfhydryl group of Cys44, generating a nucleophile to attack the quinone. The negative charge of Cys44 thiolate is not stabilized by Arg48 (29) because it is on the other side of the quinone ring. Instead, Arg48 may stabilize the entire charge-transfer complex and also facilitate its formation through inducing polarization of the quinone ring. Consistent with this mechanism, Arg48 and His91 are two of the most conserved residues in DsbB homologs (47). Moreover, mutations of Arg48, His91, and Glu47 show accumulation of DsbB-DsbA complex and/or reduced quinone binding (47, 48), both of which may result from impaired Cys44 reactivity. The quinone reduction activity of His91Ala and Glu47Ala mutants is also drastically reduced (fig. S8). Together, these new insights gained from our termini-restraining approach offer a much clearer picture of the redox chemistry of E. coli DsbB.
DISCUSSION
With termini restraining, structural determination of small membrane proteins at near- atomic resolution becomes relatively routine. We have obtained a total of 15 crystal structures of six different proteins (42, 43), three of which are of human origin and two from other vertebrates (Table 1 and Fig. 5A). Obtaining the structure of an individual 2-TM protein (SPCS1) by crystallography is, to our knowledge, unprecedented; the 2-TM proteins alone constitute ~10% of the membrane proteome (12). Without the sfGFP coupler, SPCS1 and JAGN1 only contain small exposed loops, whereas VKOR, VKORL, DsbB, and CD53 have large and flexible extramembrane regions, making them difficult targets for traditional membrane crystallography. Unlike any other approach, termini restraining now affords an effective way to crystallize these proteins.
The underlying principle of termini restraining is fundamentally different from conventional protein engineering methods used in structural biology. The termini restraining affords a global stabilization by inserting the entire membrane protein into a highly stable coupler protein, thereby enhancing the overall stability of the restrained constructs. Unlike systematic mutagenesis (2–4), the restraining is a rational way of stabilizing overall membrane protein fold (Fig. 1) and requires minimal construct screening. An effective mutation (2) generally increases the Tm by 1° to 3°C, whereas the restraining increases the Tm of DsbB by more than 40°C and the Tm of two other proteins by 8° to 9°C. Increased protein thermostability should limit the frequency of transiently unfolded states, thereby lowering the probability of protein aggregation in the cellular environment and in vitro (37). Several restrained proteins show much less aggregation during overexpression (Fig. 2G). Consequently, we obtained much higher yield of functional proteins after purification, some with increased specific activity (Fig. 2B).
This restraining strategy requires only that the two protein termini are located at the same side of the membrane, and therefore, it is nearly universally useful for proteins with even numbers of TMs from any protein family (Table 1). The varying distances between the ends of N- and C-terminal TMs of different proteins do not affect the ability of termini restraining to stabilize and crystallize these proteins (Fig. 5A). Furthermore, termini restraining does not change the integrity of the target membrane protein by replacing an essential structural component. As a result, the biological functions are retained for the various membrane proteins that we have analyzed (Fig. 2, A to E). In particular, the example of Mfrn1 suggests that the termini restraining is applicable to many classes of transporter proteins by permitting the large rocking motions required for their activities. Furthermore, formation of high-order complex, such as the cell junction (extracellular side) mediated by Cldn4 (Fig. 2D), may not be interfered by termini restraining at the other side of the membrane (intracellular side). Cautions should be taken, however, in that the fused coupler protein may interfere with protein complex formation or other protein functions at the same side of the membrane where the coupler protein is introduced.
The termini restraining affords a combined strategy to obtain near-atomic-resolution structures, which are often essential to mechanistic understandings of protein function, as with the redox chemistry of DsbB that we report here. These well-diffracting crystals only form in the presence of the coupler protein, sfGFP, which affords a large and versatile polar interface for stable packing interactions (Fig. 3, B to D, and fig. S2, B and C) and also increases the relative diffraction mass. The restrained constructs are compatible with the LCP crystallization method (49) that further improves the structural resolution. Linking both termini of the membrane protein to sfGFP reduces their relative motions, and optimizing the termini length effectively improves crystal diffraction (Fig. 4 and Table 1). The sfGFP also enables crystal identification, phase determination, and density modification (Figs. 3A and 4D). Together, introducing the sfGFP coupler facilitates the entire pipeline of obtaining high-resolution crystal structures of small membrane proteins.
Our method is currently limited to small membrane proteins with even numbers of TMs but is potentially applicable to those with odd numbers of TMs by fusing one component of the associable protein entities to an internal loop. The restraining can be extended to use other self-associable entities, such as protein domains, protein oligomers, or complexes, or to use intein-mediated polypeptide joining. This strategy is not limited to structural studies, as the idea of termini restraining may inspire future developments of protein engineering and protein design. Moreover, functional membrane proteins with improved stability and increased yield enable their biochemical and biophysical studies and fulfil many drug discovery and therapeutic needs.
MATERIALS AND METHODS
Constructs and cloning
Termini restraining was performed by fusing sfGFP residues 1 to 144 and 146 to end (20) to the N and C termini of target membrane proteins (Table 1), respectively. The constructs used for functional assays, fluorescence imaging, whole-cell extraction, and crystallization have a 10× His tag or FLAG tag added to the C terminus of truncated sfGFP (1 to 230). For Tm measurements, the constructs are fused into a full-length sfGFP, followed by a PreScission protease cleavage site, mCherry, and 10× His tag. The unrestrained constructs were generated by tagging the membrane proteins with the PreScission site, enhanced GFP (EGFP; or mCherry), and 10× His tag or FLAG tag. The unrestrained VKOR and Mfrn1 required the addition of an N-terminal small ubiquitin-like modifier protein (SUMO) tag to improve their expression level in Pichia pastoris (50). To be consistent, the restrained VKOR and Mfrn1 used for the Tm measurements also contain the N-SUMO tag. The coding sequences of these constructs were cloned into a modified pPICZ-A vector for expression in P. pastoris and a pBudCE4.1 vector for expression in HEK293 cells. DsbB constructs were cloned into pET28b for FSEC-Tm analysis and crystallization and cloned into pET25b for activity assays. Termini truncations and mutations of the crystallization constructs were performed by one-step site-directed mutagenesis (51).
Protein expression
For protein expression in P. pastoris, plasmids were linearized and transformed into the Pichia cells by electroporation. Clones with the highest expression level were selected by FSEC (38). The cells were grown at 30°C for 20 hours in the buffered minimal glycerol (BMG) media [1.2% glycerol, 0.34% yeast nitrogen base, 1% ammonium sulfate, biotin (0.4 μg/ml), and 100 mM potassium phosphate (pH 6.0)]. After exchanging to the buffered minimal methanol (BMM) media (0.34% yeast nitrogen base, 1% ammonium sulfate, biotin (0.4 μg/ml), and 200 mM potassium phosphate (pH 6.0)], the cells were induced with 0.7% methanol and grown for 1 to 3 days at 25°C. The harvested cells were flash-frozen in liquid nitrogen.
The DsbB-containing plasmids were transformed to E. coli C41 cells. The cells were grown in LB medium at 37°C to 0.6 optical density (OD). Subsequently, the cells were induced with 0.4 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and grown at 25°C overnight. The harvested cells were used immediately for protein purification and other assays.
Expression of CD53, Cldn4, and Mfrn1 in HEK293 cells was through transient transfection of 0.6 μg of plasmids with Lipofectamine 3000. The cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum at 37°C for 2 days.
Cell imaging
Live Pichia and HEK293 cells were stained with different dyes according to the expected subcellular location of different membrane proteins. ER-Tracker Blue-White DPX (5 μM) was used for the ER membrane, FM 4-64 (10 μg/ml) was used for the plasma membrane, and MitoTracker Red (10 μg/ml) was used for the mitochondria. The excitation/emission wavelengths were 405/430 nm for ER-Tracker, 561/575 nm for FM4-64 and MitoTracker, and 488/561 nm for sfGFP or EGFP fused to membrane proteins. The live cells were viewed under a Zeiss LSM 880 II Airyscan FAST confocal microscope with 63× oil immersion objective for Pichia cells and 40× oil objective for HEK293 cells.
Whole-cell fractionation
Frozen Pichia cells were disrupted with a mixer mill (Retsch) to break the cell wall. The cells were resuspended in tris-buffered saline (TBS) buffer [0.15 M NaCl and 20 mM tris-HCl (pH 8.0)], and cell membranes were lysed via microfluidization. The same amount of the whole-cell suspension was extracted in 2% DDM, 2% SDS, or a mixture of 2% SDS and 6 M urea (adjusted to the same final volume using TBS buffer with protease inhibitors) at room temperature for 30 min. After ultracentrifugation at 88,760g, the supernatant was heated in the SDS sample buffer [with 40 mM dithiothreitol (DTT)] at 90°C for 10 min and loaded onto SDS–polyacrylamide gel electrophoresis. The Western blots used the His tag antibody (GenScript, A00186) for most proteins. For human VKOR, anti-VKORC1 antibody (Thermo Fisher Scientific, PA560093) was used.
CD53, Cldn4, and Mfrn1 were expressed in HEK293 cells because the Pichia expression of these proteins results in incorrect subcellular locations even for the C-GFP–tagged constructs. The cells were resuspended in TBS buffer and extracted in 2% DDM, 0.1% SDS/1% Triton X-100, or a mixture of 0.1% SDS/1% Triton X-100 and 6 M urea at room temperature for 30 min. After ultracentrifugation, the fractionated samples were subjected to Western blot using anti-FLAG antibody (Sigma-Aldrich, F2555).
Protein purification
For the purification of most proteins, frozen Pichia cells were broken by mixer mill (Retsch) and resuspended in a buffer containing 150 mM NaCl, 20 mM tris-HCl (pH 8.0), deoxyribonuclease I (10 μg/ml), and protease inhibitor cocktail. Subsequently, DDM was added (2% final concentration) to solubilize the cell membranes for 3 hours at 4°C. After ultracentrifugation at 130,000g for 20 min, the supernatant was incubated with TALON metal affinity resin (Clontech) for 3 hours at 4°C. The resin was washed with 10 mM imidazole and eluted with 250 mM imidazole in a buffer containing 150 mM NaCl, 0.2% DDM, and 20 mM tris (pH 8.0). The eluted proteins were concentrated and further purified by size exclusion chromatography (Superdex 200 or Superose 6) in a buffer containing 150 mM NaCl, 20 mM tris (pH 8.0), 0.05% DDM, or 0.05% lauryl maltose neopentyl glycol (LMNG); DDM was used for Tm measurement and activity assay, and LMNG was used for crystallization.
Purification of human VKOR requires the addition of warfarin, other vitamin K antagonists, or vitamin K epoxide before the DDM solubilization (42). The same ligand concentration was maintained during the entire purification process.
The purification of Mfrn1 proteins (50) has the following modifications (50). The broken cells were resuspended in 150 mM KCl, 1 mM tris(2-carboxyethyl)phosphine (TCEP), and 50 mM Hepes-KOH (pH 8.0). For metal affinity chromatography, the washing (10 mM imidazole) and elution (250 mM imidazole) buffers contained 150 mM KCl, 1 mM TCEP, cardiolipin (0.05 mg/ml), 0.2% DDM, and 20 mM Mops-KOH (pH 7.5). The buffer used for size exclusion chromatography contained 140 mM KCl, 1 mM TCEP, cardiolipin (0.05 mg/ml), 0.2% DDM, and 20 mM Mops-KOH (pH 7.5). For functional assays, C-EGFP and N-SUMO tags were removed from the proteins by digestion with PreScission protease (20:1, w/w) before the size exclusion chromatography.
For the purification of DsbB proteins, E. coli cells were resuspended in 150 mM NaCl and 20 mM tris (pH 8.0) and then broken by sonication. After centrifugation at 4700g for 15 min, the cell membranes were collected by ultracentrifugation at 130,000g for 35 min. The membrane fraction was solubilized in a buffer containing 2% DDM, 150 mM NaCl, and 20 mM tris (pH 8.0) for 2 hours at 4°C. The membrane debris was removed by ultracentrifugation, and the supernatant was incubated with 3 ml of TALON metal affinity resin for 2 hours. The resin was washed with 5 mM imidazole and eluted with 250 mM imidazole in a buffer containing 0.05% DDM, 150 mM NaCl, and 20 mM tris (pH 8.0). The proteins were further purified through a Superdex 200 column in a buffer containing either 0.05% DDM, 150 mM NaCl, and 20 mM tris (pH 8.0) for crystallization or 0.05% DDM, 50 mM NaCl, and 20 mM Hepes (pH 7.5) for the activity assay.
Thermostability FSEC
The peak fractions from size exclusion chromatography were pooled, and the protein concentrations of restrained and unrestrained proteins were adjusted to a similar level (usually 1 mg/ml). The restrained and unrestrained pairs were simultaneously incubated at different temperatures (in a thermocycler) for 10 min. After centrifugation at 20,000g for 1 hour, the supernatant was analyzed by FSEC (Shimadzu) using a Superdex 200 or Superose 6 column in a buffer containing 0.05% DDM, 150 mM NaCl, and 20 mM tris (pH 8.0). The restrained and unrestrained proteins that had been heated at the same temperature were loaded to FSEC consecutively for precise Tm comparison. Fluorescence of mCherry was monitored with the excitation wavelength at 587 nm and emission at 610 nm. The peak heights were normalized to that of 4°C samples, and the Tm was obtained by fitting the thermostability curves with a sigmoidal dose-response equation (52).
Functional assays
The cellular carboxylation assay of VKOR and VKORL activities was performed as previously described (53, 54). Briefly, VKORL and VKORL clones, along with a luciferase gene, in a pBudCE4.1 vector were transfected into a cell line that contains a chimeric factor IX Gla and protein C (FIXgla-PC) gene and has endogenous VKORC1 and VKORL1 genes knocked out. The carboxylation level of secreted FIXgla-PC was measured by a sandwich enzyme-linked immunosorbent assay (ELISA) by using the cell culture medium, with luciferase activity serving as the control for transfection efficiency. The transfected cells were treated with warfarin at 11 different concentrations for inhibition analysis.
In vitro analysis of the epoxide reductase activity of VKOR and VKORL used a fluorometric-based assay (31, 40). The same concentration of restrained and unrestrained protein was added to a buffer containing 20 μM vitamin K epoxide, 6 mM DTT, 0.1% DDM, 0.1 M NaCl, and 20 mM tris (pH 7.6). Fluorescence of the reduced product, vitamin K hydroquinone, was detected with excitation at 248 nm and emission at 430 nm.
For the E. coli motility assay of DsbB activity, the restrained DsbB was transformed to a ΔdsbB HK320 strain (32, 33). Empty pET25b vector and pET25b-DsbB-EGFP were used as negative and positive controls, respectively. To analyze motility associated with each construct, a single colony was grown on a M63 minimal plate with 0.3% agar, 0.2% glucose, 0.5 mM IPTG, and ampicillin (100 μg/ml) at 30°C for 2 days.
The DsbB activity was also assessed through a disulfide-sensitive β-galactosidase assay. The assay was carried out by transforming the constructs into a ΔdsbB E. coli strain with a malF-lacZ fusion gene (HK325) (33). The transformants were streaked onto M63 minimal medium plates [X-galactosidase (100 μg/ml), 0.5 mM IPTG, 1.5% agar, 0.2% glucose, 0.2% maltose, and ampicillin (100 μg/ml)] and grown at 30°C for 2 days. The maltose/maltodextrin transport system permease protein (MALF) fusion to the β-galactosidase (malF-lacZ) brings part of the protein to the E. coli periplasm and inactivates the enzyme though disulfide bond formation. Thus, the β-galactosidase is active in the ΔdsbB strain (blue colonies) and becomes inactive after the complementation of functional DsbB (white colonies).
To analyze the cell junction formed by Cldn4, the restrained and unrestrained proteins were transiently expressed in HEK293 cells. The cells were viewed under a Zeiss Airyscan FAST confocal microscope.
To analyze the iron transport activity of restrained and unrestrained Mfrn1, the purified proteins were reconstituted into liposomes. A dried mixture of 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), and cardiolipin in a 55:40:5 ratio (w/w) was resuspended (with sonication) to 10 mg/ml in 10 mM Mops (pH 7.5), 140 mM KCl, 1 mM sorbitol, and 0.6% sodium cholate. The Mfrn1 proteins were added at a 1:50 ratio (w/w) to total lipids and incubated at 4°C for 30 min. Subsequently, the mixture was incubated with pretreated Bio-Beads at 4°C overnight. Calcein was incorporated into the proteoliposomes by three freeze-thaw cycles, and excess calcein was removed by passing the mixture through a G50 column. The Fe2+ uptake assay was carried out in 96-well plates with calcein fluorescence measured (excitation, 494 nm; emission, 516 nm) using a plate reader (Molecular Dimensions). After 5 min of equilibration, 20 μM FeSO4 was added to initiate the Mfrn1-mediated transport. After the transport was saturated, 1 μM calcimycin was added to allow metal influx through this ionophore. The traces of calcein fluorescence were plotted as a percentage of maximal quenching after the addition of calcimycin.
The quinone reduction activity of wild-type and mutant DsbB constructs was assayed as previously described (55). Briefly, the 200-μl reaction mixture contained 2 nM DsbB, 20 μM reduced DsbA, and 25 μM 2,3-dimethoxy-5-methyl-6-decyl-1,4-benzoquinone (Q0C10) in 0.05% DDM, 50 mM NaCl, and 20 mM Hepes (pH 7.5). The decrease of absorption at 275 nm due to quinone reduction was recorded in a ultraviolet-transparent 96-well plate with a plate reader (Molecular Dimensions).
Crystallization and data collection
The restrained proteins were concentrated to approximately 40 mg/ml and reconstituted into LCP by thorough mixing with monoolein at 2:3 ratio (v/v) using a restrained syringe. With a Gryphon LCP robot (Art Robbins), 100-nl drops of the reconstituted protein were dispensed onto a glass plate and 800 nl of precipitant solutions was overlaid (table S4). The LCP plates were incubated at 22°C. Crystals generally appeared overnight and grew to an optimal size after 1 week. The crystals were harvested from the mesophase bolus and flash-frozen in liquid nitrogen. Diffraction data were collected with a microfocus beam and a PILATUS detector at the ID-24 beamlines of Advanced Photon Source.
Structure determination
MR was conducted with PHASER (56) using the fused sfGFP as the search model (PDB code 2B3P) (57). This MR model was rigid body–refined with REFMAC (58). The partial model phases were improved by solvent flattening and NCS averaging using the programs PARROT (59) and DM (60).
For VKORL (the low-resolution data) and DsbB, cross-crystal averaging (39) was performed between the existing model and data of sfGFP (2B3P) and those of the sfGFP-restrained membrane proteins using the program DMMULTI (60). Molecular masks were generated and modified using programs NCSMASK and MAPMASK, installed in the CCP4 suite (61), and MAPMAN and MAMA, installed in the UPPSALA software factory (62). Rotation and translation matrices that match the reference molecule to the target molecule were calculated using program PDBSET in the CCP4 suite.
The density-modified maps were used for automatic model building by the BUCCANEER software (59). For most datasets, the phases after rigid body refinement of the sfGFP model and solvent flattening with PARROT with automatically generated mask were sufficient for the automatic building of most parts of the membrane protein models (Table 1). The models were manually built to completeness using program COOT (63). The model refinement was by programs installed in the PHENIX suites (64).
Acknowledgments
We thank A. Krezel, J. Robertson, and D. Fremont for critical reading of the manuscript; C. Landeta for providing the ΔdsbB strains; and R. Zhang and M. Diamond for providing the SPCS1 sequence. Funding: W.L. is supported by the W. M. Keck Foundation (Forefront of Science Award), NHLBI (R01 HL121718), Children’s Discovery Institute (MCII 2020-854), NEI (R21 EY028705), and NIGMS (R01 GM131008). This work used NE-CAT beamlines (GM124165), a Pilatus detector (RR029205), an Eiger detector (OD021527) at the APS (DE-AC02-06CH11357). Author contributions: W.L. and S. Liu designed the study. S. Liu purified and crystallized VKOR, VKORL, DsbB, and SPCS1. Y.Y. purified and crystallized CD53 and JAGN1. W.L. solved the structures. S. Liu and S. Li performed functional assays and protein stability analyses. W.L. oversaw the studies, analyzed the results, and wrote the manuscript with inputs from other authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Atomic coordinates and structure factors for the reported crystal structures have been deposited in the PDB under accession codes 6WVD (JAGN1), 6WVE (SPCS1) and 6WVF (DsbB). Those of CD53 (43), VKOR, and VKORL have also been deposited (42).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/51/eabe3717/DC1
REFERENCES AND NOTES
- 1.Kang H. J., Lee C., Drew D., Breaking the barriers in membrane protein crystallography. Int. J. Biochem. Cell Biol. 45, 636–644 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Serrano-Vega M. J., Magnani F., Shibata Y., Tate C. G., Conformational thermostabilization of the β1-adrenergic receptor in a detergent-resistant form. Proc. Natl. Acad. Sci. U.S.A. 105, 877–882 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tate C. G., A crystal clear solution for determining G-protein-coupled receptor structures. Trends Biochem. Sci. 37, 343–352 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Magnani F., Serrano-Vega M. J., Shibata Y., Abdul-Hussein S., Lebon G., Miller-Gallacher J., Singhal A., Strege A., Thomas J. A., Tate C. G., A mutagenesis and screening strategy to generate optimally thermostabilized membrane proteins for structural studies. Nat. Protoc. 11, 1554–1571 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou Y., Morais-Cabral J. H., Kaufman A., MacKinnon R., Chemistry of ion coordination and hydration revealed by a K+ channel–Fab complex at 2.0 Å resolution. Nature 414, 43–48 (2001). [DOI] [PubMed] [Google Scholar]
- 6.Kim J. M., Wu S., Tomasiak T. M., Mergel C., Winter M. B., Stiller S. B., Robles-Colmanares Y., Stroud R. M., Tampé R., Craik C. S., Cheng Y., Subnanometre-resolution electron cryomicroscopy structure of a heterodimeric ABC exporter. Nature 517, 396–400 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosenbaum D. M., Cherezov V., Hanson M. A., Rasmussen S. G., Thian F. S., Kobilka T. S., Choi H.-J., Yao X.-J., Weis W. I., Stevens R. C., Kobilka B. K., GPCR engineering yields high-resolution structural insights into β2-adrenergic receptor function. Science 318, 1266–1273 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Chun E., Thompson A. A., Liu W., Roth C. B., Griffith M. T., Katritch V., Kunken J., Xu F., Cherezov V., Hanson M. A., Stevens R. C., Fusion partner toolchest for the stabilization and crystallization of G protein-coupled receptors. Structure 20, 967–976 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rasmussen S. G., Choi H.-J., Rosenbaum D. M., Kobilka T. S., Thian F. S., Edwards P. C., Burghammer M., Ratnala V. R., Sanishvili R., Fischetti R. F., Schertler G. F., Weis W. I., Kobilka B. K., Crystal structure of the human β2 adrenergic G-protein-coupled receptor. Nature 450, 383–387 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Thomas J. A., Tate C. G., Quality control in eukaryotic membrane protein overproduction. J. Mol. Biol. 426, 4139–4154 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alexandrov A. I., Mileni M., Chien E. Y., Hanson M. A., Stevens R. C., Microscale fluorescent thermal stability assay for membrane proteins. Structure 16, 351–359 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Wallin E., von Heijne G., Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 7, 1029–1038 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zimmerman B., Kelly B., McMillan B. J., Seegar T. C. M., Dror R. O., Kruse A. C., Blacklow S. C., Crystal structure of a full-length human tetraspanin reveals a cholesterol-binding pocket. Cell 167, 1041–1051.e11 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Umeda R., Satouh Y., Takemoto M., Nakada-Nakura Y., Liu K., Yokoyama T., Shirouzu M., Iwata S., Nomura N., Sato K., Ikawa M., Nishizawa T., Nureki O., Structural insights into tetraspanin CD9 function. Nat. Commun. 11, 1606 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shinoda T., Shinya N., Ito K., Ohsawa N., Terada T., Hirata K., Kawano Y., Yamamoto M., Kimura-Someya T., Yokoyama S., Shirouzu M., Structural basis for disruption of claudin assembly in tight junctions by an enterotoxin. Sci. Rep. 6, 33632 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vecchio A. J., Stroud R. M., Claudin-9 structures reveal mechanism for toxin-induced gut barrier breakdown. Proc. Natl. Acad. Sci. U.S.A. 116, 17817–17824 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rana M. S., Kumar P., Lee C.-J., Verardi R., Rajashankar K. R., Banerjee A., Fatty acyl recognition and transfer by an integral membrane S-acyltransferase. Science 359, eaao6326 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang H., Klein M. G., Zou H., Lane W., Snell G., Levin I., Li K., Sang B.-C., Crystal structure of human stearoyl-coenzyme A desaturase in complex with substrate. Nat. Struct. Mol. Biol. 22, 581–585 (2015). [DOI] [PubMed] [Google Scholar]
- 19.S. White, laboratory at UC, Irvine; https://blanco.uci.edu/mpstruc/.
- 20.Baird G. S., Zacharias D. A., Tsien R. Y., Circular permutation and receptor insertion within green fluorescent proteins. Proc. Natl. Acad. Sci. U.S.A. 96, 11241–11246 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cabantous S., Terwilliger T. C., Waldo G. S., Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol. 23, 102–107 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Daley D. O., Rapp M., Granseth E., Melén K., Drew D., von Heijne G., Global topology analysis of the Escherichia coli inner membrane proteome. Science 308, 1321–1323 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Kim H., Melén K., Osterberg M., von Heijne G., A global topology map of the Saccharomyces cerevisiae membrane proteome. Proc. Natl. Acad. Sci. U.S.A. 103, 11142–11147 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang R., Miner J. J., Gorman M. J., Rausch K., Ramage H., White J. P., Zuiani A., Zhang P., Fernandez E., Zhang Q., Dowd K. A., Pierson T. C., Cherry S., Diamond M. S., A CRISPR screen defines a signal peptide processing pathway required by flaviviruses. Nature 535, 164–168 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Charrin S., Jouannet S., Boucheix C., Rubinstein E., Tetraspanins at a glance. J. Cell Sci. 127, 3641–3648 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Boztug K., Jarvinen P. M., Salzer E., Racek T., Monch S., Garncarz W., Gertz E. M., Schaffer A. A., Antonopoulos A., Haslam S. M., Schieck L., Puchalka J., Diestelhorst J., Appaswamy G., Lescoeur B., Giambruno R., Bigenzahn J. W., Elling U., Pfeifer D., Conde C. D., Albert M. H., Welte K., Brandes G., Sherkat R., van der Werff Ten Bosch J., Rezaei N., Etzioni A., Bellanne-Chantelot C., Superti-Furga G., Penninger J. M., Bennett K. L., von Blume J., Dell A., Donadieu J., Klein C., JAGN1 deficiency causes aberrant myeloid cell homeostasis and congenital neutropenia. Nat. Genet. 46, 1021–1027 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shaw G. C., Cope J. J., Li L., Corson K., Hersey C., Ackermann G. E., Gwynn B., Lambert A. J., Wingert R. A., Traver D., Trede N. S., Barut B. A., Zhou Y., Minet E., Donovan A., Brownlie A., Balzan R., Weiss M. J., Peters L. L., Kaplan J., Zon L. I., Paw B. H., Mitoferrin is essential for erythroid iron assimilation. Nature 440, 96–100 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Inaba K., Murakami S., Suzuki M., Nakagawa A., Yamashita E., Okada K., Crystal structure of the DsbB-DsbA complex reveals a mechanism of disulfide bond generation. Cell 127, 789–801 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Inaba K., Murakami S., Nakagawa A., Iida H., Kinjo M., Ito K., Suzuki M., Dynamic nature of disulphide bond formation catalysts revealed by crystal structures of DsbB. EMBO J. 28, 779–791 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bushweller J. H., Protein disulfide exchange by the intramembrane enzymes DsbB, DsbD, and CcdA. J. Mol. Biol. 432, 5091–5103 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chu P.-H., Huang T.-Y., Williams J., Stafford D. W., Purified vitamin K epoxide reductase alone is sufficient for conversion of vitamin K epoxide to vitamin K and vitamin K to vitamin KH2. Proc. Natl. Acad. Sci. U.S.A. 103, 19308–19313 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dutton R. J., Wayman A., Wei J.-R., Rubin E. J., Beckwith J., Boyd D., Inhibition of bacterial disulfide bond formation by the anticoagulant warfarin. Proc. Natl. Acad. Sci. U.S.A. 107, 297–301 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Landeta C., Blazyk J. L., Hatahet F., Meehan B. M., Eser M., Myrick A., Bronstain L., Minami S., Arnold H., Ke N., Rubin E. J., Furie B. C., Furie B., Beckwith J., Dutton R., Boyd D., Compounds targeting disulfide bond forming enzyme DsbB of Gram-negative bacteria. Nat. Chem. Biol. 11, 292–298 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ruprecht J. J., King M. S., Zögg T., Aleksandrova A. A., Pardon E., Crichton P. G., Steyaert J., Kunji E. R. S., The molecular mechanism of transport by the mitochondrial ADP/ATP carrier. Cell 176, 435–447.e15 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Drew D. E., von Heijne G., Nordlund P., de Gier J.-W., Green fluorescent protein as an indicator to monitor membrane protein overexpression in Escherichia coli. FEBS Lett. 507, 220–224 (2001). [DOI] [PubMed] [Google Scholar]
- 36.Lee H., Kim H., Membrane topology of transmembrane proteins: Determinants and experimental tools. Biochem. Biophys. Res. Commun. 453, 268–276 (2014). [DOI] [PubMed] [Google Scholar]
- 37.Ellgaard L., Helenius A., Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 4, 181–191 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Kawate T., Gouaux E., Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins. Structure 14, 673–681 (2006). [DOI] [PubMed] [Google Scholar]
- 39.Li W., Li F., Cross-crystal averaging with search models to improve molecular replacement phases. Structure 19, 155–161 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li W., Schulman S., Dutton R. J., Boyd D., Beckwith J., Rapoport T. A., Structure of a bacterial homologue of vitamin K epoxide reductase. Nature 463, 507–512 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu S., Cheng W., Fowle Grider R., Shen G., Li W., Structures of an intramembrane vitamin K epoxide reductase homolog reveal control mechanisms for electron transfer. Nat. Commun. 5, –3110 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu S., Li S., Shen G., Sukumar N., Krezel A. M., Li W., Structural basis of antagonizing the vitamin K catalytic cycle for anticoagulation. Science 2020, eabc5667 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang Y., Liu X. R., Greenberg Z. J., Zhou F., He P., Fan L., Liu S., Shen G., Egawa T., Gross M. L., Schuettpelz L. G., Li W., Open conformation of tetraspanins shapes interaction partner networks on cell membranes. EMBO J. 39, e105246 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhou Y., Cierpicki T., Jimenez R. H. F., Lukasik S. M., Ellena J. F., Cafiso D. S., Kadokura H., Beckwith J., Bushweller J. H., NMR solution structure of the integral membrane enzyme DsbB: Functional insights into DsbB-catalyzed disulfide bond formation. Mol. Cell 31, 896–908 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Phan J., Zdanov A., Evdokimov A. G., Tropea J. E., Peters H. K. III, Kapust R. B., Li M., Wlodawer A., Waugh D. S., Structural basis for the substrate specificity of tobacco etch virus protease. J. Biol. Chem. 277, 50564–50572 (2002). [DOI] [PubMed] [Google Scholar]
- 46.Matthews D. A., Dragovich P. S., Webber S. E., Fuhrman S. A., Patick A. K., Zalman L. S., Hendrickson T. F., Love R. A., Prins T. J., Marakovits J. T., Zhou R., Tikhe J., Ford C. E., Meador J. W., Ferre R. A., Brown E. L., Binford S. L., Brothers M. A., DeLisle D. M., Worland S. T., Structure-assisted design of mechanism-based irreversible inhibitors of human rhinovirus 3C protease with potent antiviral activity against multiple rhinovirus serotypes. Proc. Natl. Acad. Sci. U.S.A. 96, 11000–11007 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sevier C. S., Kadokura H., Tam V. C., Beckwith J., Fass D., Kaiser C. A., The prokaryotic enzyme DsbB may share key structural features with eukaryotic disulfide bond forming oxidoreductases. Protein Sci. 14, 1630–1642 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kobayashi T., Takahashi Y., Ito K., Identification of a segment of DsbB essential for its respiration-coupled oxidation. Mol. Microbiol. 39, 158–165 (2001). [DOI] [PubMed] [Google Scholar]
- 49.Cherezov V., Lipidic cubic phase technologies for membrane protein structural studies. Curr. Opin. Struct. Biol. 21, 559–566 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Christenson E. T., Gallegos A. S., Banerjee A., In vitro reconstitution, functional dissection, and mutational analysis of metal ion transport by mitoferrin-1. J. Biol. Chem. 293, 3819–3828 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zheng L., Baumann U., Reymond J.-L., An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 32, e115 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hattori M., Hibbs R. E., Gouaux E., A fluorescence-detection size-exclusion chromatography-based thermostability assay for membrane protein precrystallization screening. Structure 20, 1293–1299 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shen G., Cui W., Zhang H., Zhou F., Huang W., Liu Q., Yang Y., Li S., Bowman G. R., Sadler J. E., Gross M. L., Li W., Warfarin traps human vitamin K epoxide reductase in an intermediate state during electron transfer. Nat. Struct. Mol. Biol. 24, 69–76 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tie J.-K., Jin D.-Y., Tie K., Stafford D. W., Evaluation of warfarin resistance using transcription activator-like effector nucleases-mediated vitamin K epoxide reductase knockout HEK293 cells. J. Thromb. Haemost. 11, 1556–1564 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bader M. W., Xie T., Yu C.-a., Bardwell J. C. A., Disulfide bonds are generated by quinone reduction. J. Biol. Chem. 275, 26082–26088 (2000). [DOI] [PubMed] [Google Scholar]
- 56.McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J., Phaser crystallographic software. J. Appl. Cryst. 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pédelacq J.-D., Cabantous S., Tran T., Terwilliger T. C., Waldo G. S., Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 24, 79–88 (2006). [DOI] [PubMed] [Google Scholar]
- 58.Murshudov G. N., Vagin A. A., Dodson E. J., Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 (1997). [DOI] [PubMed] [Google Scholar]
- 59.Cowtan K. D., Recent developments in classical density modification. Acta Crystallogr. D Biol. Crystallogr. 66, 470–478 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cowtan K. D., ‘Dm’: An automated procedure for phase improvement by density modification. Joint CCP4 ESF-EACBM Newslet. Protein Crystallogr. 31, 34–38 (1994). [Google Scholar]
- 61.Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S., Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kleywegt G. J., Jones T. A., Software for handling macromolecular envelopes. Acta Crystallogr. D Biol. Crystallogr. 55, 941–944 (1999). [DOI] [PubMed] [Google Scholar]
- 63.Emsley P., Lohkamp B., Scott W. G., Cowtan K. D., Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., Adams P. D., Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/51/eabe3717/DC1