

Abstract
Transition metal multimetallic complexes have seen intense study due to their unique bonding and potential for cooperative reactivity, but actinide–transition metal (An–TM) species are far less understood. We have synthesized uranium– and thorium–osmium heterometallic polyhydride complexes in order to study An–Os bonding and investigate the reactivity of An–Os interactions. Computational studies suggest the presence of a significant bonding interaction between the actinide center and the four coordinated osmium centers supported by bridging hydrides. Upon photolysis, these complexes undergo intramolecular C–H activation with the formation of an Os–Os bond, while the thorium complex may activate an additional C–H bond of the benzene solvent, resulting in a μ-η1,η1 phenyl ligand across one Th–Os interaction.
We report the synthesis of actinide–osmium polyhydrides and demonstrate their ability to promote photo-induced intra- and intermolecular C–H activation.
Introduction
Metal hydrides have seen extensive interest due to their importance in processes such as catalytic hydrofunctionalization of olefins,1 reduction of organic substrates and small molecules,2,3 and electrocatalytic redox reactions.4 While d-block metal hydrides have been the focus of the majority of these investigations, it was soon discovered that f-block hydrides exhibit unique reactivity that, in some cases (particularly the hydrogenation and polymerization of unsaturated organic substrates), render them even better catalysts than their transition metal (TM) counterparts.5 Furthermore, heterometallic hydride complexes—derived from the combination of f-block and transition metals—have exciting potential for cooperative reactivity utilizing the unique properties of each metal center; they also offer the possibility of providing fundamental insight into f-block-TM bonding. Bridging hydrides are well suited to support these interactions, as their minimal steric profile allows for close metal–metal contact. In addition, hydride elimination to form metal–metal bonds is well-precedented in the transition metal literature, offering a potential route to unsupported metal–metal bonds.6 Lastly, heterometallic actinide hydrides are of wider interest due to their possible superconductive properties at high pressures, and intriguingly, for their potential as hydrogen storage materials.7
Significant strides have been made in the synthesis of rare earth/transition metal polyhydrides since the first example, a trimetallic Y2Zr tetrahydride, was synthesized by Evans in 1984.8 In subsequent years, several groups have reported the successful syntheses of such complexes using alkane elimination, H2 elimination, and salt metathesis.9–15 Hou has reported extensive studies on Y– and Lu–TM complexes, synthesizing polyhydride clusters with all of the 2nd and 3rd row transition metals between groups 6 and 9 (excluding Tc), several of which display hydrogen addition and release properties.16–22
In contrast, while actinide (An) borohydride and aluminohydride chemistry is well-established,5,23–27 multimetallic hydride chemistry of the actinides is underdeveloped. Ephritikhine reported a series of U/Re complexes supported by three bridging hydrides, utilizing potassium rhenate salts to install the rhenium center on uranium pentamethylcyclopentadienyl (Cp*) halide complexes.28–30 The U–Re distance in [K(18-crown-6)][(Cp*)2(Cl)U(μ-H)3Re(H)3(PPh3)2] was long (3.255(8) Å), leading the authors to conclude there was no direct U–Re interaction;29 limited reactivity was observed with all U–Re species described.
Following our report of hydride-supported An–Al bonds (An = Th, U),26 we have recently focused on applying this strategy to stabilize new An–TM interactions. Our efforts have resulted in the syntheses of several multimetallic actinide tetrairidate dodecahydride complexes, Th{[(μ-H)2(H)IrCp*]2[(μ-H)3IrCp*]2} (A) and U{(μ-H)3IrCp*}4 (B), from the iridium polyhydride starting material K[Cp*IrH3] and actinide halides (Scheme 1). These complexes featured the first reported computational evidence for An–Ir interaction.31 Given the efficacy of eliminating a bridging ligand to form An–Co bonds,32 we sought to engender An–TM bonds via dihydrogen elimination, but both A and B proved resistant to this process, either by thermolysis up to 80 °C, or via photolysis with 254 nm light.
Scheme 1. Previously reported syntheses of An–Ir multimetallic complexes A and B.

We turned to the osmium counterpart, Cp*OsH5, reported by Girolami, to pursue the synthesis of An–Os multimetallic complexes.33 This species is known to be photoactive, losing multiple hydrides to form the [Cp*OsH2]2 dimer. We reasoned that synthesis of actinide tetraosmate complexes analogous to tetrairidate complexes A and B might lead to more productive photolysis, ideally with the loss of hydrides and formation of An–Os bonds. Here, we report the syntheses, calculated bonding character, and photolytic chemistry of these uranium and thorium tetraosmate species.
Results and discussion
In order to install the osmium centers around the actinide center, Cp*OsH5 was converted in situ to the potassium osmate species K[Cp*OsH4] (1) with the addition of benzylpotassium in THF, forming a pale-yellow solution. 1H NMR confirms the stoichiometric deprotonation of Cp*OsH5, resulting in formation of 1 and toluene (Fig. S7†). Compared to Cp*OsH5, the hydride resonance of 1 shifts upfield from −11.00 ppm to −17.15 ppm, with a concomitant reduction in integration from five protons to four (relative to the Cp* resonance). The 1H NMR spectrum closely resembles that of the Li[Cp*OsH4] species reported by Suzuki, which contains a hydride resonance at −17.52 ppm.34
Reaction of four equivalents of 1 with ThCl4(DME)2 or UCl4 in THF led to the formation of Th{(μ-H)4OsCp*}4 (2-Th) and U{(μ-H)4OsCp*}4 (2-U), which can be isolated as colorless and yellow crystals from n-hexane in 66% and 68% yield, respectively (Scheme 2).
Scheme 2. Synthetic route to compounds 1 and 2-An.

The 1H NMR spectrum of diamagnetic 2-Th features two resonances for the Cp* methyl protons and hydrides at 2.13 ppm and −11.01 ppm, respectively. These integrate in a 15 : 4 ratio, indicating that all hydrides in the potassium metalate starting material are preserved. The 1H NMR spectrum of 2-U contains a sharp resonance at 3.65 ppm for the Cp* methyl protons, as well as a hydride resonance at 89.8 ppm, shifted significantly downfield due to the paramagnetic influence of the formally uranium(iv) center. Once again, the two peaks integrate in a 15 : 4 ratio.
The solid-state IR spectra of 2-Th and 2-U each feature a single metal-hydride stretching signal at 1993 cm−1 and 1990 cm−1, respectively, shifted by about 100 cm−1 compared to the reported hydride stretch for Cp*OsH5 of 2083 cm−1.33 These values are significantly higher than the bridging hydride stretch of 1762 cm−1 found in [Cp*OsH2]2.35 Several other examples of bridging hydrides in Cp*-substituted osmium multimetallic complexes have been reported, but lack reported hydride stretches for comparison.34,36 However, our values are in good agreement with the bridging hydride stretching signals of 1962 and 1951 cm−1 for the iridate complexes A and B,31 which exhibit a similar decrease in frequency by approximately 100 cm−1 from the value for Cp*IrH4 of 2150 cm−1.37
Single crystals of 2-Th and 2-U suitable for X-ray diffraction (XRD) were grown from saturated solutions of cold n-pentane, and the solid-state structures of both were crystallographically characterized, confirming that four osmium centers are coordinated around the actinide centers. No hydrides could be located in the difference maps, most likely due to the close proximity of numerous heavy atoms, a phenomenon that we observed previously with the related actinide-iridium complexes.31 Complex 2-Th crystallizes in the space group P21/c with Th–Os distances ranging from 3.0183(6) Å to 3.0379(6) Å, well within the sum of covalent radii for Th and Os (3.50 Å).38 The Os centers are slightly distorted from an ideal tetrahedral geometry around Th, with a τ4 value39 of 0.93 (calculated with α = 115.80(2)°, β = 112.32(2)°), slightly less than that of A (0.97).31 The Os–Os distances range from 4.7446(7) Å to 5.1395(7) Å, which precludes any Os–Os interactions considering osmium's covalent radius of 1.44 Å.38 Unlike A, complex 2-Th has one consistent hydride binding mode across all osmate moieties. All osmate fragments feature fairly linear 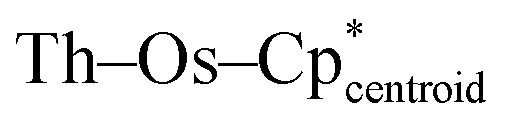 angles (171.10(15)° to 176.92(14)°). These values are sufficiently close to linearity to indicate that all four hydrides in each [Cp*OsH4]− fragment bridge the Th and Os centers. Therefore, 2-Th is formally 16-coordinate, with 16 bridging hydrides around the thorium center, matching the highest observed coordination number around an atom in an isolated compound;40,41 interestingly, higher coordination numbers have been proposed for early actinide-noble gas complexes.42 This is reminiscent of the 15-coordinate aminodiboranate thorium complex reported by Girolami, which also features thorium with an extraordinarily large number of bridging hydrides, in this case provided by borohydride-based ligands rather than transition metal polyhydrides.23
angles (171.10(15)° to 176.92(14)°). These values are sufficiently close to linearity to indicate that all four hydrides in each [Cp*OsH4]− fragment bridge the Th and Os centers. Therefore, 2-Th is formally 16-coordinate, with 16 bridging hydrides around the thorium center, matching the highest observed coordination number around an atom in an isolated compound;40,41 interestingly, higher coordination numbers have been proposed for early actinide-noble gas complexes.42 This is reminiscent of the 15-coordinate aminodiboranate thorium complex reported by Girolami, which also features thorium with an extraordinarily large number of bridging hydrides, in this case provided by borohydride-based ligands rather than transition metal polyhydrides.23
Complex 2-U crystallizes in the cubic space group Pa3̄, with a single osmate moiety centered on the 3-fold symmetry axis and another which generates the remaining three osmate fragments through symmetry. The on-axis U–Os distance is 2.9490(6) Å, while the off-axis U–Os distances are each 2.9501(4) Å, far shorter than the sum of covalent radii for U and Os (3.40 Å)38 and on average shorter than those of 2-Th by approximately 0.08 Å (3.027(6) Å vs. 2.950(6) Å). The Os centers are arranged in an essentially tetrahedral manner around uranium, with a τ4 value of 0.98 (calculated with α = 111.00(2)°, β = 111.00(2)°), significantly more tetrahedral than B, which has a τ4 value of 0.87.31 As in 2-Th, the Os–Os distances are long enough to rule out any possible interaction, varying from 4.7703(8) Å to 4.8615(7) Å. The 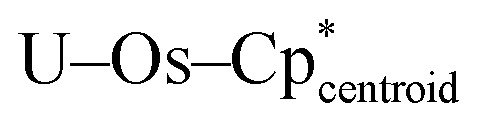 angles are quite linear, at 175.30(11)° for the on-axis fragment and 176.4(5)° for the off-axis fragments, indicating that as in 2-Th, all 16 hydrides of 2-U bridge the osmium centers and uranium. This is the first report of such a high coordination number for uranium, which previously had been limited to 14-coordinate complexes,43,44 and a second example of a formally 16-coordinate actinide complex.
angles are quite linear, at 175.30(11)° for the on-axis fragment and 176.4(5)° for the off-axis fragments, indicating that as in 2-Th, all 16 hydrides of 2-U bridge the osmium centers and uranium. This is the first report of such a high coordination number for uranium, which previously had been limited to 14-coordinate complexes,43,44 and a second example of a formally 16-coordinate actinide complex.
To further support our conclusions about the hydride binding modes in 2-Th and 2-U as well as investigate potential metal–metal interactions, we turned to density functional theory (DFT) calculations for further insight. All DFT computations employed the B3PW91 functional (full details in the ESI†). Computational modeling of 2-Th and 2-U predicts near-tetrahedral configurations for the osmium fragments around the actinide centers, as observed in the solid-state structures, as well as sixteen bridging hydrides for each species (Fig. 1). The calculated An–Os distances are consistently ∼0.04 Å shorter than the experimentally determined distances, but this and all other observed disparities between computational and experimental metrics are quite minor (Table 1). In addition, the two highest experimentally observed stretching frequencies for 2-Th and 2-U fall within the predicted ranges according to calculations, further evidence that these calculations are accurately modeling the hydrides within these compounds.
Fig. 1. Computed structures for 2-Th and 2-U (single-crystal XRD-derived ORTEP diagrams can be found in Fig. S32 and S33†). Color key: light blue (thorium), green (uranium), blue (osmium), white (hydrogen). Hydrogen atoms on the Cp* rings are omitted and Cp* ligands are wireframed for clarity.

Experimentally and computationally derived bond distances, angles, and Os–H stretches for 2-Th and 2-U.
| 2-Th | 2-U | |||
|---|---|---|---|---|
| Expt. | Comp. | Expt. | Comp. | |
| An–Os dist. (Å) | 3.0183(6)–3.0379(6) | 2.977–2.978 | 2.9490(6)–2.9501(4) | 2.911–2.913 |
| Os–CCp* dist. (Å) | 2.173(11)–2.320(10) | 2.187–2.306 | 2.18(3)–2.29(3) | 2.187–2.300 |
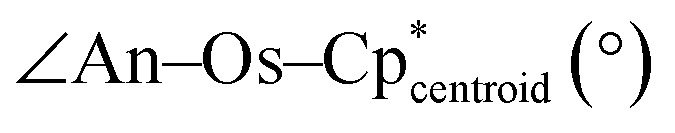
|
171.10(15)–176.92(14) | 178.9–179.3 | 175.30(11)–176.4(5) | 177.1–179.2 |
| Os–H (cm−1) | 1990 | 2051–1963 | 1993 | 2056–1947 |
| 862 | 947–838 | 863 | 939–818 | |
Natural Bond Order (NBO) analyses were carried out on 2-Th and 2-U as well, to analyze the degree of metal–metal interaction between the actinide and osmium centers. Wiberg Bond Indices (WBIs) of 0.72–0.73 were calculated for the Th–Os interactions in 2-Th, while values of 0.81–0.83 were found for the U–Os interactions in 2-U. These values suggest significant bonding interactions between the actinide and osmium centers, and are comparable to previously reported An–TM bonds as well as our An–Ir systems.31,45,46 Complexes 2-Th and 2-U are, to the best of our knowledge, the first reported compounds to evidence An–Os covalency. Analysis of the M–H bonds reveals a more covalent hydride interaction with Os than U or Th, with Os–H WBIs of 0.57–0.60 compared to An–H WBIs of 0.18–0.24. Similar to A and B, the identity of the actinide does not appear to affect hydride covalency in 2-An.
With these actinide-osmium polyhydrides in hand, we investigated the potential for thermolytic and photolytic H2 elimination in 2-U and 2-Th. Both complexes were stable in C6D6 when heated at 60 °C for 8 hours, as monitored by 1H NMR. However, upon irradiation by 254 nm light in C6H6, complex 2-U undergoes a color change from yellow to dark brown, forming complex 3-U (Scheme 3) via the C–H activation of a Cp* methyl group and the loss of hydrides as H2, as detected by 1H NMR spectroscopy at 4.47 ppm in C6D6 (Fig. S12†).47
Scheme 3. Photolysis of 2-U and 2-Th with 254 nm light in C6H6 to produce 3-U, 3-Th, and 4-Th.

Complex 3-U shows significantly reduced solubility compared to 2-U. It exhibits minimal solubility in n-hexane, requiring several drops of benzene to facilitate dissolution of the crude material. Upon workup in this manner, 3-U crystallizes at −40 °C as tiny brown crystals in 43% yield. Larger, X-ray quality crystals were grown from dilute solutions in n-hexane without benzene, from which the solid-state structure was determined (Fig. 2, left). As a result of photolysis, two of the Cp*Os moieties in 3-U feature formally Os(ii) centers, with a significantly reduced Os–Os distance of 2.4639(4) Å and the formation of a U–C bond between uranium and the activated Cp* methylene. The U–Os distances of the remaining two formally Os(iv) fragments are slightly lengthened compared to 2-U, ranging from 2.9571(7) Å to 3.0264(7) Å. The U(iv) center is disordered across two positions, asymmetrically bridging the diosmium fragment towards either osmium atom with near-50% occupancy. There is some variation in the U–C bond length depending on which direction the asymmetric U atom favors, at 2.603(7) Å and 2.657(9) Å. These values are similar to the U–C bond lengths found for other uranium “tuck-over” complexes.48,49 In addition, the two unreacted [Cp*OsH4]− fragments in complex 3-U splay further outward from the tetrahedral geometry of 2-U, with an Os–U–Os angle of 118.15(3)° or 120.75(3)° depending on the uranium position. As with the previous complexes, no hydrides could be resolved in the solid-state structure.
Fig. 2. Single-crystal XRD-derived ORTEP diagrams for 3-U (left), 3-Th (middle), and 4-Th (right), with ellipsoids drawn at the 50% probability level. Non-methylene hydrogen atoms have been omitted and non-cyclometallated Cp* ligands have been wireframed for clarity. Only the major component of the disordered actinide center is shown in 3-U and 3-Th. Hydrides were not resolved in the crystal structures due to the numerous heavy metal centers in proximity. Selected bond distances (Å) and angles (°) for 3-U: U1–Os1 2.9870(12), U1–Os2 3.0267(11), U1–Os3 3.2205(8), U1–Os4 2.7974(8), U1–C6 2.657(9), Os3–Os4, 2.4640(5), Os1–U1–Os2 118.15(3),  ,
,  . Selected bond distances (Å) and angles (°) for 3-Th: Th1–Os3 3.0260(3), Th1–Os2 3.2567(5), Th1–Os1 2.9539(4), Th1–C1 2.534(10), Os1–Os2, 2.4689(4), Os3–Th1–Os3′ 121.133(14),
. Selected bond distances (Å) and angles (°) for 3-Th: Th1–Os3 3.0260(3), Th1–Os2 3.2567(5), Th1–Os1 2.9539(4), Th1–C1 2.534(10), Os1–Os2, 2.4689(4), Os3–Th1–Os3′ 121.133(14),  ,
,  . Selected bond distances (Å) and angles (°) for 4-Th: Th1–Os1 3.0593(9), Th1–Os2′ 3.2965(11), Th1–Os2 2.9403(12), Th1–C12 2.703(12), Os2–Os2′, 2.4536(9), Os1–Th1–Os1′ 135.06(4),
. Selected bond distances (Å) and angles (°) for 4-Th: Th1–Os1 3.0593(9), Th1–Os2′ 3.2965(11), Th1–Os2 2.9403(12), Th1–C12 2.703(12), Os2–Os2′, 2.4536(9), Os1–Th1–Os1′ 135.06(4),  .
.

As discussed previously, photolytic loss of hydrides has precedence with Cp*OsH5, which is known to photolyze with the loss of 3 equivalents of H2 to form the dimer [Cp*OsH2]2. This species has an essentially identical Os–Os distance of 2.4568(6) Å to that of 3-U.35 The newly formed [(CH2)Me4C5Os(μ-H)3OsCp*]2− fragment closely resembles the doubly deprotonated dimer, with one deprotonation occurring at a Cp* methyl group and the other occurring from the bridging hydrides, coordinated side-on to the actinide center. Hou previously reported the reaction and side-on coordination of [Cp*OsH2]2 with rare earth dialkyl complexes of the form (C5Me4SiMe3)Ln(CH2SiMe3)2(THF) (Ln = Y, Lu) resulting in trinuclear, C–H activated complexes.17 However, in these species, the geometry of the [Cp*OsH2]2 fragment was not preserved due to the migration of several hydrides to bridge the lanthanide and osmium atoms, and reactivity was promoted via thermolysis rather than photolysis.
The 1H NMR spectrum of Cs symmetric 3-U contains six observable resonances, allowing all protons to be assigned besides the hydrides of the diosmium fragment. The mirror plane passes through the Os–Os and U–CH2 bonds, resulting in the observation of two Cp* resonances (in a 2 : 1 ratio) and two methyl resonances from the activated Cp* (in a 1 : 1 ratio). The activated methylene resonance is shifted far upfield to −89.1 ppm. The hydrides of 3-U are observed at 55.5 ppm, significantly more shielded than the hydrides of 2-U, which are shifted to 89.8 ppm. This downfield signal integrates to about eight protons, and is therefore assigned to the hydrides of the two unactivated [Cp*OsH4]− fragments. However, the hydrides associated with the diosmium fragment were not observed between −100 to 600 ppm in the 1H NMR spectrum, perhaps due to the paramagnetic nature of 3-U. We therefore hypothesized that a diamagnetic Th(iv) photolysis product could serve as a useful analogue to 3-U.
Complex 2-Th displays slightly divergent reactivity from 2-U, photolyzing over four hours with a color change from colorless to orange to produce both 3-Th and 4-Th (Scheme 3). As with the photolysis of 2-U, H2 formation can be detected by 1H NMR spectroscopy (Fig. S15†). However, complex 3-Th, the analogous species to 3-U, is the minor product in this reaction. The major product, 4-Th, results from the additional activation of one equivalent of the benzene solvent, replacing one bridging hydride from one [Cp*OsH4]− fragment with an asymmetrical bridging μ-η1,η1-phenyl ligand. This benzene activation appears to lend stability to the photolysis product, as reactions in n-hexane instead result in dark brown intractable mixtures and yield no crystalline product. In contrast, the photolysis product 3-U can be prepared and isolated from n-hexane in satisfactory yields.
Compounds 3-Th and 4-Th consistently co-crystallize from the crude photolysis mixture, typically in a 22 : 78 3-Th to 4-Th ratio (mol/mol%) as measured by 1H NMR. Complex 4-Th could be isolated in sparing (<20%) yield with photolysis times in excess of two days. Tracking of the reaction by 1H NMR shows that 4-Th forms in significant quantities with 3-Th upon photolysis of 2-Th and gradually increases in concentration over time, such that 3-Th cannot be isolated with short reaction times. Addition of H2 gas to a sample of 4-Th did not lead to hydrogenolysis of the bridging phenyl ligand to form 3-Th. Separation of 3-Th from 4-Th was achieved a single time by recrystallization of a particularly high-percentage 3-Th-containing photolysis crop (47% 3-Th by 1H NMR) from a 1 : 1 hexane/benzene solution at −40 °C, yielding crystals containing 88% 3-Th. As a result, the 1H NMR signals of each compound can be distinctly identified, although quantities sufficient for high-quality 2D NMR spectra were not obtained. Spectroscopic analyses were instead carried out on the crystalline mixture of 3-Th and 4-Th. As confirmation that two species are present in this material, Diffusion Ordered Spectroscopy (DOSY) experiments on a nearly 1 : 1 solution of 3-Th/4-Th demonstrate that the 1H NMR peaks associated with 3-Th and 4-Th have slightly different diffusion coefficients (Fig. S31†). This slight difference in diffusion rate is unsurprising given the relatively small difference in molecular weight and steric bulk between the two complexes, and is well outside the margin of error in these experiments.
The 1H NMR spectrum of 3-Th contains one more resonance than the spectrum of 3-U. Analogous to 3-U, there are four resonances in the methyl region, in addition to a singlet methylene resonance at 1.34 ppm. In contrast to the uranium species, there are two hydride signals, one integrating to eight hydrides and the other integrating to three hydrides, found at −9.74 ppm and −11.06 ppm, respectively. These signals are in turn assigned to the hydrides of the two [Cp*OsH4]− fragments and the hydrides of the diosmium fragment, [(CH2)Me4C5Os(μ-H)3OsCp*]2−. Therefore, we assign three hydrides to the diosmium fragment of 3-U as well, resulting in 11 total hydrides for both 3-U and 3-Th.
The 1H NMR spectrum of 4-Th is significantly more complex, as the bridging phenyl ligand renders each osmate fragment distinct. The phenyl signals of 4-Th are observed between 7.07 and 7.98 ppm. The three unactivated Cp* moieties and four methyl groups of the activated Cp* each appear as separate signals, and the methylene resonances arising from the Cp* cyclometallation are split as an AX system centered at 1.08 and 1.37 ppm.1H,13C HSQC experiments confirm that the protons giving rise to these signals are bonded to the same carbon, which appears at 61.07 ppm in the 13C NMR spectrum. In the hydride region, the three hydrides of the [Cp*OsH3(C6H5)]− fragment are distinct in solution, and splitting can be observed between the three signals. The hydride distal to the activated benzene appears furthest downfield, at −8.16 ppm, and is split into a triplet by the two proximal hydrides, which appear as doublets at −10.54 and −10.84 ppm. The bridging hydrides of the [(CH2)Me4C5Os(μ-H)3OsCp*]2− and [Cp*OsH4]− fragments are observed as two singlets at −9.80 and −11.59 ppm, respectively.
Both 3-Th and 4-Th are highly soluble in benzene and toluene while only sparingly so in n-hexane. Recrystallization of the aforementioned singular 3-Th sample from n-hexane and minimal benzene afforded single crystals of 3-Th suitable for diffraction experiments. X-ray quality crystals of 4-Th were grown from n-hexane by recrystallizing the primarily 4-Th crystalline material isolated from long photolysis times (>1 day).
Complex 3-Th crystallizes with one equivalent of n-hexane in the space group Pmn21, with half of the molecule generated by a mirror plane (Fig. 2, middle). It is isostructural to 3-U, but unlike 3-U, the disorder of the central actinide atom strongly favors one orientation, at over 90% occupancy, indicating a preference for the molecules to align in the same orientation. Further discussion of the structure of 3-Th will primarily refer to this major component. The diosmium fragment in 3-Th is nearly identical to that of 3-U, with a similar Os–Os distance of 2.4689(4) Å, but a shorter Th–C bond measuring 2.534(10) Å (2.729(10) Å in the minor component). The monoosmate fragments feature slightly longer Th–Os bond lengths than the U–Os distances in 3-U at 3.0260(3) Å, with a similar Os–Th–Os angle of 121.133(14)°.
In comparison, the primary notable feature in the structure of 4-Th is the presence of an asymmetric bridging phenyl ligand across one Th–Os interaction (Fig. 2, right). The structure is completely asymmetric, but due to disorder 4-Th crystallizes in the space group P42/ncm, with the thorium center and the bridging phenyl ligand disordered equivalently across two sites, resulting in four possible conformations for the molecule. The diosmium fragment is isostructural to that of 3-Th, with an Os–Os distance of 2.4536(9) Å and a Th–C bond length of 2.703(12) Å between Th and the activated Cp* methylene.
In the remaining portion of 4-Th, the two monoosmate fragments splay further outward to accommodate the activated phenyl ring, with an expanded Os–Th–Os angle of 135.06(4)°, compared to 121.133(14)° in 3-Th. Within this expanded pocket, one equivalent of activated benzene is present as a bridging μ-η1,η1-phenyl across one Th–Os interaction, replacing one of the bridging hydrides. This phenyl ring is coordinated asymmetrically, angled at 67.0(14)° relative to the Th–Os bond. It is heavily skewed toward the osmium center, with an Os–C distance of 2.12(5) Å compared to a Th–C distance of 2.96(4) Å. Examples of bridging μ-η1,η1-aryl ligands involving osmium are limited to triosmium carbonyl clusters and feature symmetric coordination modes.50–52 Several late transition metal multimetallic species feature similar μ-η1,η1-aryls, including two related Au–Ag and Au–Cu polymeric chains,53 as well as Pt–Ag and Pt–Cu complexes.54,55 The polymeric complex [Au2Ag2(C6F5)4(N CCH3)2]n most closely resembles the highly asymmetric phenyl coordination mode of 4-Th, with Ag–Cphenyl distances of 2.508(6) and 2.687(6) Å, Au–Cphenyl distances of 2.055(6) and 2.088(6) Å, and a Cphenyl–Au–Ag angle of 66.00(15)°. In all these cases, however, the bridging aryl ligand is coordinated to a metal center in the starting material, whereas in 4-Th it arises from the C–H activation of benzene. In the rare earths, a symmetric η1-phenyl bridging two scandium centers has been proposed as an intermediate in the activation of benzene by (1,1′-fc(NSitBuMe2)2)ScI(THF)2 and KC8, though no bridging aryl species were isolated.56
Photolysis reactions of 2-Th in substituted aryl solvents such as toluene, mesitylene, and fluorobenzene were attempted as well. Photolysis in toluene led to a color change to orange, similar to the reaction in benzene. 1H NMR analysis reveals a mixture of numerous products, presumably from the activation of the aryl ring at the ortho, meta, and para positions, as well as the formation of 3-Th, further evidence that 3-Th is formed without the activation of benzene (or any solvent). There does not appear to be a strong preference for activation at any position, making isolation of a single product untenable. Photolysis was less fruitful in mesitylene, with detection of 3-Th as the major product, but less evidence of aryl activation products in the 1H NMR spectrum. In contrast, reactions in fluorobenzene rapidly became brown, but the major identifiable products were Cp*OsH5 and [Cp*OsH2]2, with no isolable C–H or C–F activation products of fluorobenzene.
In order to investigate potential metal–metal bonding interactions in compounds 3-U, 3-Th, and 4-Th, better elucidate the positions of the hydrides in these photolysis products, and gain insight into the benzene activation by 4-Th, we undertook a similar computational investigation as for complexes 2-U and 2-Th, at the same level of theory. Complexes with different numbers of hydrides were computed, considering different spin states for each (see Fig. S34 and Table S5 in ESI†). For 3-U and 3-Th, the most stable structures are found with eleven hydrides in a triplet and singlet spin state, respectively, which is in line with the presence of a U(iv) and Th(iv) actinide center (Fig. 3, top and S34†). In 4-Th, a singlet spin state is once again most stable, indicating a Th(iv) center, while only ten hydrides are found due to the substitution of one hydride with the bridging phenyl ligand (Fig. 3, bottom). It is worth noting that the three optimized structures are the only structures with linear 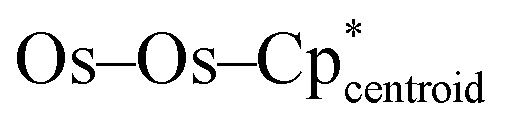 angles in line with the experimental structures; simulations with greater or fewer hydrides converged to structures with bent
angles in line with the experimental structures; simulations with greater or fewer hydrides converged to structures with bent 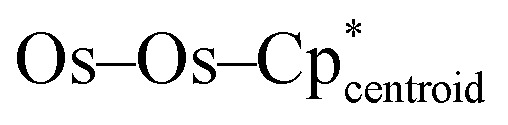 geometries. As with complexes 2-An, the optimized geometries compare well with the experimental data (Table S6†). Among others, the Os–Os distance is well reproduced computationally at 2.47 Å, versus about 2.46 Å experimentally.
geometries. As with complexes 2-An, the optimized geometries compare well with the experimental data (Table S6†). Among others, the Os–Os distance is well reproduced computationally at 2.47 Å, versus about 2.46 Å experimentally.
Fig. 3. Calculated structures for 3-U (top) and 4-Th (bottom), with WBIs labeled for select bonds. Color key: green (uranium), light blue (thorium), blue (osmium), gray (carbon), white (hydrogen). Methyl hydrogen atoms have been omitted and non-cyclometallated Cp* ligands have been wireframed for clarity.

NBO analyses were carried out on the most stable optimized structures for the three complexes. WBIs of 0.53–0.79 were found for the U–Os interactions in 3-U, while WBIs of 0.55–0.70 were found for the Th–Os interactions in 3-Th, with comparable values of 0.55–0.71 in 4-Th. These values are lower than that found in the parent complexes 2-An but indicates that a substantial covalent bonding An–Os interaction remains in these complexes. As in complexes 2-U and 2-Th, the M–H bonds are more covalent with Os than U or Th, with Os–H WBIs of 0.30–0.60 compared to An–H WBIs of 0.02–0.21 for 3-An and 4-An. NBO analysis of the M–Cphenyl bonding in 4-Th shows a covalent Os–C bond (45% Os/55% C) which appears to be delocalized toward Th (second-order perturbation analysis, see Table S9†). It is thus a three-center-two-electron bond, as with the bridging hydrides (Fig. S36†).
Interestingly, in the three complexes an Os–Os WBI of 0.73–0.74 is found in the diosmium fragment. This result is in line with a substantial bonding interaction between the two Os centers. The interaction is further corroborated at the NBO level where a single covalent Os–Os bond is found in 3-U and 3-Th (only a second order perturbation was found for 4-Th). This bond implies a 5d–5d overlap between the two Os centers in an almost non-polarized covalent bond (Table S8†); the hydride-mediated metal–metal interaction can also be observed in the HOMO-3 orbital of 3-Th and 4-Th (Fig. 4, left). As previously mentioned, the diosmium fragment in complexes 3-An and 4-Th is similar to [Cp*OsH2]2, which Girolami and coworkers concluded to contain no Os–Os bond, citing the results of Morokuma on the analogous diruthenium complex.34,35,57,58 There are several key differences between the compounds and computational methods used which may contribute to the discrepancy. First, the calculations in the 1993 paper were done using the ab initio, restricted Hartree-Fock method, in contrast to the semi-empirical method employed in this study. The geometry of the antibonding osmium, 5dz2 orbitals is different between the thorium compounds and [Cp*RuH2]2 as well. In the HOMO-4 orbital of 3-Th and 4-Th, the osmium 5dz2 orbitals are oriented such that they overlap with a thorium 6d orbital (Fig. 4, right), resulting in δ-antibonding interactions between the osmium orbitals rather than the σ-antibonding overlap predicted for [Cp*RuH2]2. This should decrease the repulsive interaction experienced between the osmium centers, and may account for the difference in the calculated Os–Os bonding.
Fig. 4. Kohn–Sham Os–Os bonding orbitals for 3-Th derived from DFT (isovalue = 0.03). The orbitals for 4-Th are nearly identical and may be found in Fig. S35.†.

Conclusions
Complexes 3-U, 3-Th, and 4-Th are notable as products of photolysis-driven C–H activation by both actinide and transition metal centers. All feature the intramolecular activation of sp3 C–H bonds, while 4-Th also features the uncommon intermolecular activation of a benzene sp2 C–H bond by an actinide complex, resulting in a bridging Th–C–Os bond. Actinide-promoted benzene activation has been documented in uranium inverse sandwich complexes, resulting in the direct borylation of benzene and naphthalene, but in this instance did not result in the formation of a new An–C bond following activation.59 Despite the report of thermolytic benzene C–H activation by thorium neopentyl species in 1981, subsequent examples of benzene activation by f-block metal centers remain few and far between. These reports have been limited to the rare-earth elements and involve thermolysis at high temperatures60,61 or strong reductants such as a potassium mirror or KC8.56,62 In contrast, the formation of 4-Th is photolytically driven, occurring at ambient conditions. The osmium polyhydride OsH6(PiPr3)2 has been known to activate a variety of polycyclic, N-substituted aromatic species upon thermolysis,63–67 but no reactivity with smaller, less substituted aromatics such as toluene or benzene has been reported. In addition, the photolytic reactivity of 2-U and 2-Th is not solely dependent on the osmium centers, as both Cp*OsH5 and [Cp*OsH2]2 do not form stable Os–C bonds upon photolysis in benzene, although H/D exchange is observed when Cp*OsH5 is photolyzed to form [Cp*OsH2]2 in C6D6 (Fig. S30†). This is particularly exciting, as heterobimetallic cooperative C–H activation is a field of growing interest, yet systems featuring An–TM cooperativity are rare.68
In the context of actinide photolysis reactivity, the reactions of 2-U and 2-Th are also unique. In contrast to previously reported photolytic actinide C–H activations, which were driven by irradiation of stable or transient terminal uranium nitrides69,70 or Th–C bond cleavage,71 here the chemistry is driven by the loss of hydrides as H2. This contributes to the minimal record of photolytic elimination of hydrides in the actinides,28 as well as photolysis-driven actinide–metal bond formation.32 It is, to the best of our knowledge, the first documented example of the formation of new actinide bonds via photolytic hydride elimination. Future work will focus on the synthesis of heterobimetallic hydride-supported An–TM systems, hydride elimination to drive unsupported metal–metal bonding in those systems, and potential cooperative reactivity with small molecules.
Data availability
The experimental and computational data associated with this article are provided in the ESI.†
Author contributions
C. Z. Ye performed the experimental work. S. N. Kelly and I. J. Brackbill recorded and interpreted the XRD data. I. Del Rosal and L. Maron performed and interpreted the computational studies. C. Z. Ye wrote the original draft of the manuscript. J. Arnold and C. Camp conceptualized the research, found the funds, administrated the project, and supervised the work. I. Del Rosal, L. Maron, J. Arnold, and C. Camp revised and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was supported by the Director, Office of Science, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences Heavy Element Chemistry Program of the U.S. Department of Energy (DOE) at LBNL under Contract DE-AC02-05CH11231 and the CNRS International Emerging Action – IEA 2022 project AnTM. The authors acknowledge the HPCs CALcul en Midi-Pyrénées (CALMIP-EOS grant 1415). C. Z. Y. and S. N. K. acknowledge the U.S. DOE Integrated University Program for graduate research fellowships. The authors thank the DOE National Nuclear Security Administration through the Nuclear Science and Security Consortium for a fellowship (to I. J. B.) under Award No. DE-NA0003996. We thank Dr Hasan Celik and UC Berkeley's NMR facility in the College of Chemistry (CoC-NMR) for spectroscopic assistance. Instruments in the CoC-NMR are supported in part by NIH S10OD024998, and by the National Science Foundation under Grant No. 2018784. The Advanced Light Source (ALS) is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. DOE under Contract No. DE-AC02-05CH11231. Dr Simon J. Teat is thanked for his assistance during crystallography experiments at the ALS. We also acknowledge Dr Nick Settineri and UC Berkeley's CheXray facility for crystallographic assistance. David Hales is thanked for helpful discussions.
Electronic supplementary information (ESI) available. CCDC 2346841 (2-Th), 2346842 (2-U), 2346843 (3-U), 2346844 (3-Th) and 2346845 (4-Th). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4sc02380c
Notes and references
- Crossley S. W. M. Obradors C. Martinez R. M. Shenvi R. A. Chem. Rev. 2016;116:8912–9000. doi: 10.1021/acs.chemrev.6b00334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteruelas M. A. Oro L. A. Chem. Rev. 1998;98:577–588. doi: 10.1021/cr970322u. [DOI] [PubMed] [Google Scholar]
- Jordan A. J. Lalic G. Sadighi J. P. Chem. Rev. 2016;116:8318–8372. doi: 10.1021/acs.chemrev.6b00366. [DOI] [PubMed] [Google Scholar]
- Eberhardt N. A. Guan H. Chem. Rev. 2016;116:8373–8426. doi: 10.1021/acs.chemrev.6b00259. [DOI] [PubMed] [Google Scholar]
- Ephritikhine M. Chem. Rev. 1997;97:2193–2242. doi: 10.1021/cr960366n. [DOI] [PubMed] [Google Scholar]
- Perutz R. N. Procacci B. Chem. Rev. 2016;116:8506–8544. doi: 10.1021/acs.chemrev.6b00204. [DOI] [PubMed] [Google Scholar]
- Havela L. Legut D. Kolorenč J. Rep. Prog. Phys. 2023;86:056501. doi: 10.1088/1361-6633/acbe50. [DOI] [PubMed] [Google Scholar]
- Evans W. J. Meadows J. H. Hanusa T. P. J. Am. Chem. Soc. 1984;106:4454–4460. doi: 10.1021/ja00328a027. [DOI] [Google Scholar]
- Alvarez D. Jr. Caulton K. G. Evans W. J. Ziller J. W. J. Am. Chem. Soc. 1990;112:5674–5676. doi: 10.1021/ja00170a061. [DOI] [Google Scholar]
- Alvarez D. Jr. Caulton K. G. Evans W. J. Ziller J. W. Inorg. Chem. 1992;31:5500–5508. doi: 10.1021/ic00052a028. [DOI] [Google Scholar]
- Green M. L. H. Hughes A. K. Michaelidou D. M. Mountford P. J. Chem. Soc., Chem. Commun. 1993:591–593. doi: 10.1039/C39930000591. [DOI] [Google Scholar]
- Radu N. S. Gantzel P. K. Tilley T. D. J. Chem. Soc., Chem. Commun. 1994:1175–1176. doi: 10.1039/C39940001175. [DOI] [Google Scholar]
- Schwartz D. J. Ball G. E. Andersen R. A. J. Am. Chem. Soc. 1995;117:6027–6040. doi: 10.1021/ja00127a016. [DOI] [Google Scholar]
- Butovskii M. V. Tok O. L. Wagner F. R. Kempe R. Angew. Chem., Int. Ed. 2008;47:6469–6472. doi: 10.1002/anie.200800407. [DOI] [PubMed] [Google Scholar]
- Sobaczynski A. P. Bauer T. Kempe R. Organometallics. 2013;32:1363–1369. doi: 10.1021/om301039g. [DOI] [Google Scholar]
- Shima T. Hou Z. Chem. Lett. 2008;37:298–299. doi: 10.1246/cl.2008.298. [DOI] [Google Scholar]
- Shima T. Hou Z. Organometallics. 2009;28:2244–2252. doi: 10.1021/om900024q. [DOI] [Google Scholar]
- Takenaka Y. Shima T. Baldamus J. Hou Z. Angew. Chem., Int. Ed. 2009;48:7888–7891. doi: 10.1002/anie.200903660. [DOI] [PubMed] [Google Scholar]
- Takenaka Y. Hou Z. Organometallics. 2009;28:5196–5203. doi: 10.1021/om900453j. [DOI] [Google Scholar]
- Shima T. Luo Y. Stewart T. Bau R. McIntyre G. J. Mason S. A. Hou Z. Nat. Chem. 2011;3:814–820. doi: 10.1038/nchem.1147. [DOI] [PubMed] [Google Scholar]
- Shima T. Hou Z. Chem.–Eur. J. 2013;19:3458–3466. doi: 10.1002/chem.201203495. [DOI] [PubMed] [Google Scholar]
- O W. W. N. Kang X. Luo Y. Hou Z. Organometallics. 2014;33:1030–1043. doi: 10.1021/om401216v. [DOI] [Google Scholar]
- Daly S. R. Piccoli P. M. B. Schultz A. J. Todorova T. K. Gagliardi L. Girolami G. S. Angew. Chem., Int. Ed. 2010;49:3379–3381. doi: 10.1002/anie.200905797. [DOI] [PubMed] [Google Scholar]
- Dunbar A. C. Wright J. C. Grant D. J. Girolami G. S. Inorg. Chem. 2021;60:12489–12497. doi: 10.1021/acs.inorgchem.1c01710. [DOI] [PubMed] [Google Scholar]
- Lastowski R. J. Caroff C. M. Vogiatzis K. D. Girolami G. S. Organometallics. 2023;42:2839–2848. doi: 10.1021/acs.organomet.3c00306. [DOI] [Google Scholar]
- Altman A. B. Brown A. C. Rao G. Lohrey T. D. Britt R. D. Maron L. Minasian S. G. Shuh D. K. Arnold J. Chem. Sci. 2018;9:4317–4324. doi: 10.1039/C8SC01260A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward R. J. Rungthanaphatsophon P. Huang P. Kelley S. P. Walensky J. R. Chem. Sci. 2023;14:12255–12263. doi: 10.1039/D3SC04857H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudry D. Ephritikhine M. J. Organomet. Chem. 1986;311:189–192. doi: 10.1016/0022-328X(86)80231-5. [DOI] [Google Scholar]
- Cendrowski-Guillaume S. M. Lance M. Nierlich M. Vigner J. Ephritikhine M. J. Chem. Soc., Chem. Commun. 1994:1655–1656. doi: 10.1039/C39940001655. [DOI] [Google Scholar]
- Cendrowski-Guillaume S. M. Ephritikhine M. J. Chem. Soc., Dalton Trans. 1996:1487–1491. doi: 10.1039/DT9960001487. [DOI] [Google Scholar]
- Ye C. Z. Rosal I. D. Boreen M. A. Ouellette E. T. Russo D. R. Maron L. Arnold J. Camp C. Chem. Sci. 2023;14:861–868. doi: 10.1039/D2SC04903A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward A. L. Lukens W. W. Lu C. C. Arnold J. J. Am. Chem. Soc. 2014;136:3647–3654. doi: 10.1021/ja413192m. [DOI] [PubMed] [Google Scholar]
- Gross C. L. Wilson S. R. Girolami G. S. J. Am. Chem. Soc. 1994;116:10294–10295. doi: 10.1021/ja00101a057. [DOI] [Google Scholar]
- Shima T. Suzuki H. Organometallics. 2005;24:3939–3945. doi: 10.1021/om0503996. [DOI] [Google Scholar]
- Gross C. L. Girolami G. S. Organometallics. 2007;26:160–166. doi: 10.1021/om060920b. [DOI] [Google Scholar]
- Shima T. Ichikawa T. Suzuki H. Organometallics. 2007;26:6329–6337. doi: 10.1021/om700606q. [DOI] [Google Scholar]
- Gilbert T. M. Bergman R. G. Organometallics. 1983;2:1458–1460. doi: 10.1021/om50004a037. [DOI] [Google Scholar]
- Cordero B. Gómez V. Platero-Prats A. E. Revés M. Echeverría J. Cremades E. Barragán F. Alvarez S. Dalton Trans. 2008:2832–2838. doi: 10.1039/B801115J. [DOI] [PubMed] [Google Scholar]
- Yang L. Powell D. R. Houser R. P. Dalton Trans. 2007:955–964. doi: 10.1039/B617136B. [DOI] [PubMed] [Google Scholar]
- Popov I. A. Jian T. Lopez G. V. Boldyrev A. I. Wang L.-S. Nat. Commun. 2015;6:8654. doi: 10.1038/ncomms9654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak D. Goddard R. Pörschke K.-R. J. Am. Chem. Soc. 2016;138:9444–9451. doi: 10.1021/jacs.6b02590. [DOI] [PubMed] [Google Scholar]
- Yang L. Cooper S. Kaltsoyannis N. Phys. Chem. Chem. Phys. 2021;23:4167–4177. doi: 10.1039/D0CP06175A. [DOI] [PubMed] [Google Scholar]
- Bernstein E. R. Hamilton W. C. Keiderling T. A. La Placa S. J. Lippard S. J. Mayerle J. J. Inorg. Chem. 1972;11:3009–3016. doi: 10.1021/ic50118a027. [DOI] [Google Scholar]
- Blake A. V. Fetrow T. V. Theiler Z. J. Vlaisavljevich B. Daly S. R. Chem. Commun. 2018;54:5602–5605. doi: 10.1039/C8CC02862A. [DOI] [PubMed] [Google Scholar]
- Hlina J. A. Pankhurst J. R. Kaltsoyannis N. Arnold P. L. J. Am. Chem. Soc. 2016;138:3333–3345. doi: 10.1021/jacs.5b10698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng G. McCabe K. N. Wang S. Maron L. Zhu C. Chem. Sci. 2020;11:7585–7592. doi: 10.1039/D0SC00389A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulmer G. R. Miller A. J. M. Sherden N. H. Gottlieb H. E. Nudelman A. Stoltz B. M. Bercaw J. E. Goldberg K. I. Organometallics. 2010;29:2176–2179. doi: 10.1021/om100106e. [DOI] [Google Scholar]
- Evans W. J. Miller K. A. DiPasquale A. G. Rheingold A. L. Stewart T. J. Bau R. Angew. Chem., Int. Ed. 2008;47:5075–5078. doi: 10.1002/anie.200801062. [DOI] [PubMed] [Google Scholar]
- Gardner B. M. McMaster J. Lewis W. Blake A. J. Liddle S. T. J. Am. Chem. Soc. 2009;131:10388–10389. doi: 10.1021/ja904459q. [DOI] [PubMed] [Google Scholar]
- Adams R. D. Rassolov V. Zhang Q. Organometallics. 2012;31:2961–2964. doi: 10.1021/om300235n. [DOI] [Google Scholar]
- Adams R. D. Rassolov V. Zhang Q. Organometallics. 2013;32:6368–6378. doi: 10.1021/om4007399. [DOI] [Google Scholar]
- Adams R. D. Luo Z. Wong Y. O. J. Organomet. Chem. 2015;784:46–51. doi: 10.1016/j.jorganchem.2014.08.009. [DOI] [Google Scholar]
- Fernández E. J. Laguna A. López-de-Luzuriaga J. M. Monge M. Montiel M. Olmos M. E. Rodríguez-Castillo M. Organometallics. 2006;25:3639–3646. doi: 10.1021/om060181z. [DOI] [Google Scholar]
- Moret M.-E. Chen P. Organometallics. 2008;27:4903–4916. doi: 10.1021/om800403y. [DOI] [Google Scholar]
- Moret M.-E. Chen P. J. Am. Chem. Soc. 2009;131:5675–5690. doi: 10.1021/ja900449y. [DOI] [PubMed] [Google Scholar]
- Huang W. Dulong F. Khan S. I. Cantat T. Diaconescu P. L. J. Am. Chem. Soc. 2014;136:17410–17413. doi: 10.1021/ja510761j. [DOI] [PubMed] [Google Scholar]
- Suzuki H. Omori H. Lee D. H. Yoshida Y. Morooka Y. Organometallics. 1988;7:2243–2245. doi: 10.1021/om00100a032. [DOI] [Google Scholar]
- Koga N. Morokuma K. J. Mol. Struct. 1993;300:181–189. doi: 10.1016/0022-2860(93)87016-3. [DOI] [Google Scholar]
- Arnold P. L. Mansell S. M. Maron L. McKay D. Nat. Chem. 2012;4:668–674. doi: 10.1038/nchem.1392. [DOI] [PubMed] [Google Scholar]
- Thompson M. E. Baxter S. M. Bulls A. R. Burger B. J. Nolan M. C. Santarsiero B. D. Schaefer W. P. Bercaw J. E. J. Am. Chem. Soc. 1987;109:203–219. doi: 10.1021/ja00235a031. [DOI] [Google Scholar]
- Maron L. Werkema E. L. Perrin L. Eisenstein O. Andersen R. A. J. Am. Chem. Soc. 2005;127:279–292. doi: 10.1021/ja0451012. [DOI] [PubMed] [Google Scholar]
- Coles M. P. Hitchcock P. B. Lappert M. F. Protchenko A. V. Organometallics. 2012;31:2682–2690. doi: 10.1021/om2009364. [DOI] [Google Scholar]
- Esteruelas M. A. Masamunt A. B. Oliván M. Oñate E. Valencia M. J. Am. Chem. Soc. 2008;130:11612–11613. doi: 10.1021/ja8048892. [DOI] [PubMed] [Google Scholar]
- Esteruelas M. A. Fernández I. Herrera A. Martín-Ortiz M. Martínez-Álvarez R. Oliván M. Oñate E. Sierra M. A. Valencia M. Organometallics. 2010;29:976–986. doi: 10.1021/om901030q. [DOI] [Google Scholar]
- Eguillor B. Esteruelas M. A. Fernández I. Gómez-Gallego M. Lledós A. Martín-Ortiz M. Oliván M. Oñate E. Sierra M. A. Organometallics. 2015;34:1898–1910. doi: 10.1021/acs.organomet.5b00174. [DOI] [Google Scholar]
- Esteruelas M. A. Larramona C. Oñate E. Organometallics. 2016;35:1597–1600. doi: 10.1021/acs.organomet.6b00264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancela L. Esteruelas M. A. López A. M. Oliván M. Oñate E. San-Torcuato A. Vélez A. Organometallics. 2020;39:2102–2115. doi: 10.1021/acs.organomet.0c00156. [DOI] [Google Scholar]
- Lachguar A. Pichugov A. V. Neumann T. Dubrawski Z. Camp C. Dalton Trans. 2024;53:1393–1409. doi: 10.1039/D3DT03571A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson R. K. Cantat T. Scott B. L. Morris D. E. Batista E. R. Kiplinger J. L. Nat. Chem. 2010;2:723–729. doi: 10.1038/nchem.705. [DOI] [PubMed] [Google Scholar]
- King D. M. Tuna F. McInnes E. J. L. McMaster J. Lewis W. Blake A. J. Liddle S. T. Nat. Chem. 2013;5:482–488. doi: 10.1038/nchem.1642. [DOI] [PubMed] [Google Scholar]
- Settineri N. S. Arnold J. Chem. Sci. 2018;9:2831–2841. doi: 10.1039/C7SC05328B. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The experimental and computational data associated with this article are provided in the ESI.†