

Isotope labeling and electron paramagnetic resonance spectroscopy reveal a long-postulated bis-ethene chromium(I) motif.
Abstract
A bis-ethene chromium(I) species, which is the postulated key intermediate in the widely accepted metallacyclic mechanism for ethene oligomerization, is experimentally observed. This catalytic transformation is an important commercial route to linear α-olefins (primarily, 1-hexene and 1-octene), which act as comonomers for the production of polyethene. Here, electron paramagnetic resonance studies of a catalytic system based on [Cr(CO)4(PNP)][Al(OC(CF3)3)4] [PNP = Ph2PN(iPr)PPh2] activated with Et6Al2 provide the first unequivocal evidence for a chromium(I) bis-ethene complex. The concentration of this species is enhanced under ethene and isotope labeling studies that confirm its composition as containing [Cr(C2H4)2(CO)2(PNP)]+. These observations open a new route to mechanistic studies of selective ethene oligomerization.
INTRODUCTION
We report the first bis-ethene chromium(I) complex. These complexes have been postulated for over 40 years as the key intermediate in the metallacyclic mechanism of commercialized selective ethene oligomerization reactions (1), but none has ever been identified experimentally.
Linear α-olefins (LAOs) are valuable chemical building blocks for the production of surfactants, lubricants, and detergents, but they are most importantly used as comonomers for high-density and linear low-density polyethene (2, 3). Polyethene is one of the most widely used plastics in consumer products such as food packaging, coatings, bags, toys, power cable coatings, gas pipes, etc., and continues to replace other materials (4, 5). A total of 3.5 million tons of LAOs was produced in 2012, rising with a growth rate of about 3.3% per year (2012–2018) projected to reach a $11.5 billion market by 2025 (6). Even numbered LAOs are commonly produced by ethene oligomerization (Ziegler, modified Ziegler, Shell higher olefins, and Fischer-Tropsch processes) (2, 7, 8) with statistical product distributions (i.e., Schulz-Flory and Poisson distributions) including the formation of higher polymers hampering continuous flow operation (9, 10). However, increasing demand for specific alkenes (1-hexene and 1-octene) makes selective production highly desirable. Chromium catalysts give rise to selective 1-hexene formation, and different systems have been commercialized on the basis of their high activity and selectivity (1, 11, 12). A metallacyclic trimerization mechanism (Fig. 1) has initially been postulated from kinetic data (1). Deuterium isotope labeling studies (13) support this mechanism, the expanded mechanism for formation of 1-octene, and abundant by-products (14). The postulated mechanisms (Fig. 1) involve the following: (i) η2 coordination of two ethene molecules, (ii) oxidative coupling to give a metallacyclopentane, (iii) coordination and migratory insertion of further ethene irreversibly forming larger metallacycles (13), and (iv) either an H-shift (14) or a β-hydride elimination and reductive elimination to afford the LAO and regenerate the catalyst. Trimerization and tetramerization stem from a single catalyst precursor (15) with the selectivity of tetramerization catalysts guided by accessibility for further ethene and the relative stabilities of seven- and nine-membered metallacycles (14, 16). Computational (17) and experimental (18, 19) data back a CrI/III (20–22) redox couple. Chromium(I) ethene complexes and five- and seven-membered chromium(III) metallacycles have been observed (23), but the key bis-ethene complex has remained elusive until this work.
Fig. 1. Proposed metallacyclic and extended metallacyclic mechanisms.
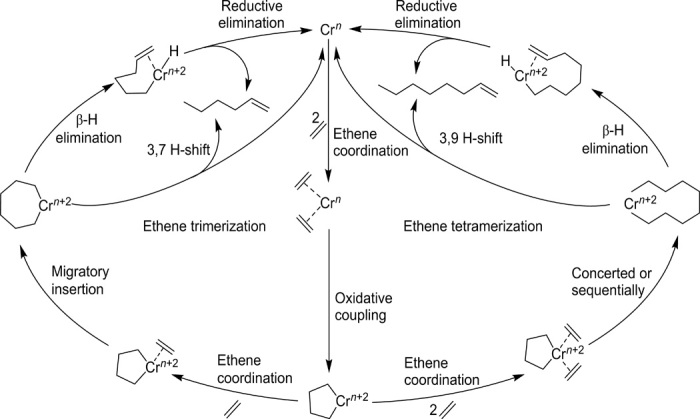
The formation of 1-hexene and 1-octene are shown here for ethene trimerization and tetramerization, respectively.
The most selective catalyst system for ethene tetramerization is generated in situ by activating a mixture of CrIII precursor and diphosphinoamine [Ph2PN(iPr)PPh2 = PNP] ligand with an excess of modified methylaluminoxane (12). The formation of a cationic chromium(I)/PNP species (20) stabilized by a weakly coordinating anion (MAO-Me)¯ is a crucial step. Cationic chromium(I)/PNP complexes with weakly coordinating anions show efficient tetramerization catalysis (20), making them attractive for mechanistic investigations. Electron paramagnetic resonance (EPR) spectroscopy is exclusively and exquisitely sensitive to paramagnetic centers and ideally suited for monitoring a CrI/III redox couple. EPR studies revealed the formation of deactivating sandwich complexes of chromium(I) in toluene solvent strongly correlating with diminished catalyst performance (24, 25).
A chromium(I) precursor was selected, as it enables highly resolved EPR spectra while it has been shown to lead to the same catalytical activity as the common and economically more viable chromium(III) precursors (20). In this work, chromium(I)-precatalyst 1 (1 = [Cr(CO)4(PNP)][Al(OC(CF3)3)4]) was activated in the absence and presence of ethene gas and the Cr speciation was monitored by EPR. Selective isotope labeling of carbonyl ligands and ethene gas gives strong evidence for the composition of a novel intermediate.
RESULTS
1 was activated by adding 5 equivalents of Et6Al2 at 273 K under an Ar atmosphere (see the “Optimized reaction conditions” section in Materials and Methods), and the formation of different intermediates was monitored in situ by EPR. On adding Et6Al2, the dark blue sample instantly changed color to orange brown. EPR spectra were taken at defined times after activation at 295 K (movie S1) and changed within minutes, showing a superposition of three different signals (Fig. 2). The corresponding powder spectra (i.e., frozen solutions) were recorded at 100 K for all three species. The spectra were broadened because of anisotropy in the g and A tensors, and therefore, the small hyperfine couplings were unresolved (fig. S1, left). Initial experiments at 10 K did not alter the spectra but slowed acquisition because of slower relaxation. Both frozen and liquid solution spectra (fig. S1) were simulated by a mixture of the three species using the spin Hamiltonian parameters given in table S1. Two of the room temperature spectra were in agreement with the previously reported species A (labeled A) with two magnetically equivalent 31P nuclei (nuclear spin, I = ½ and isotropic hyperfine coupling, PAiso = 52.3 MHz) resulting in a 1:2:1 triplet (indicated by dashed lines in green) and CrI-bisarene with 10 near-equivalent protons (1H; I = ½; HAiso = 9.5 MHz) resulting in an undecet with 11 superhyperfine lines (with a binomial intensity distribution of 1:10:45:120:210:252:210:120:45:20:1), with the exception of the two outer lines being too weak to be observed. 53Cr (I = 3/2; natural abundance, 9.5%; CrAiso = 50.5 MHz) causes quartet satellites where the undecet is further split into a 1:1:1:1 quartet. The inner two lines of the quartet overlap with the main multiplet and are not resolved. CrI-bisarene displays the lowest g value (Fig. 2) (26). The signal at intermediate g value has not been reported and was labeled X (shown by dashed lines in blue). Although its g factor is similar to the one assigned to species D (Fig. 2) (26), here, the 31P coupling constant is smaller (two 31P nuclei; I = ½; PAiso = 76.0 MHz) and a further splitting into seven observable lines for each line in the triplet by an additional, even smaller, hyperfine coupling (Aiso = 8.2 MHz) is present (the presence of further outer lines cannot be excluded; Fig. 2, 30 min). This coupling arises from an even number of six or more magnetically equivalent spin ½ nuclei. It has been simulated using eight equivalent I = ½ nuclei. Species D would not be expected to exhibit this coupling pattern. Thus, the hypothesis of in situ formation of ethene complexes (Fig. 3) was tested by performing the identical experiment under ethene gas (1 bar).
Fig. 2. EPR data showing formation of three different species over time.

1 dissolved in a 1:1 solvent mixture of dichloromethane (DCM) and methylcyclohexane (MCH) by volume was activated in situ at 273 K by adding 5 equivalents of Et6Al2. The continuous-wave (CW) EPR spectra were recorded every minute for 16 hours at 295 K. Individual time slices are shown with field positions marked by dashed lines showing the following: (i) A (0 min, green), (ii) X (30 min, blue), and (iii) CrI-bisarene (253 min). The structures of 1 and previously reported species (26) A and D and CrI-bisarene are presented.
Fig. 3. Proposed mechanism showing intermediates formed upon activation.

The transformation of CrI-precatalyst 1 upon activation with 5 equivalents of Et6Al2 in a solvent mixture of 1:1 ratio of DCM and MCH by volume.
An identical but more intense signal of X formed under ethene within 2 to 3 min [Fig. 4A (a and b) and fig. S2]. The experimental spectra can be simulated by a mixture of CrI-bisarene and X (13CO)0 (Fig. 4B, a and b). However, activation under deuterated ethene (1 bar) saw the smaller hyperfine pattern collapse into the linewidth (Fig. 4A, c). This is reproduced by simulations (Fig. 4B, a and e) replacing hydrogen with deuterium (eight equivalent 2H nuclei, I = 1) nuclei and scaling the hyperfine coupling by the gyromagnetic ratios to 1.3 MHz (γH/γD ≈ 6.5, γH/γD ≈ 6.51, i.e., DAiso = HAiso/6.5 = 1.3 MHz). This result indicates that X is an ethene bound Cr/PNP complex. One could speculate that the coupling to eight protons, which are replaced by deuterons upon introduction of d4-ethene, might originate from a d8-metallacyclopentane formed in situ. However, in this molecule, α and β protons would no longer be equivalent and would give rise to different hyperfine couplings. Furthermore, after oxidative cyclization, the chromium is in oxidation state III and would not be expected to have a doublet ground state. The resolution that we retrieve here has not been achieved with chromium(III) complexes in solution EPR where the additional zero-field splitting interaction broadens the lines substantially. Last, one could hypothesize a metallacyclopentane that has subsequently been reduced to an anionic chromium(I) complex. We have computed the expected proton hyperfine couplings (see the Supplementary Materials), which give much worse agreement with the experiment. On this basis, we rule out that species X is the metallacyclopentane and conclude that it has to be the bis-ethene complex. The hyperfine pattern resulting from eight equivalent 1H nuclei reproduces the experimental spectra and is consistent with two η2-ethene ligands (fig. S3). Gas chromatography–mass spectrometry (GC-MS) showed that the formation of 1-hexene and 1-octene is occurring under 1-bar ethene, demonstrating catalytic competence under the conditions stabilizing the bis-ethene intermediate (fig. S4). Moreover, GC-MS showed formation of ethene under in situ EPR conditions (fig. S5), explaining the formation of X in the absence of added ethene gas.
Fig. 4. CW EPR data for X.

(A) The CW EPR spectrum recorded for X at 295 K under identical conditions (a) Ar atmosphere, (b) ethene (1 bar), (c) deuterated ethene (1 bar), and subsequently, the activation of 60% 13CO enriched 1 was performed under (d) ethene (1 bar) and (e) deuterated ethene (1 bar). (B) The simulated EPR spectra for individual components are shown in blue (a) CrI-bisarene (giso = 1.9875; 10 1H nuclei: I = ½, HAiso = 9.5 MHz; one 53Cr nucleus: I = 3/2; natural abundance, 9.5%; CrAiso = 50.5 MHz), (b) X (13CO)0 (two 31P and eight 1H nuclei), (c) X (13CO)1 (two 31P, eight 1H, and one 13C nuclei), (d) X (13CO)2 (two 31P, eight 1H, and two 13C nuclei), (e) d8-X (13CO)0 (two 31P and eight 2H nuclei), (f) d8-X (13CO)1 (two 31P, eight 2H, and one 13C nuclei), and (g) d8-X (13CO)2 (two 31P, eight 2H, and two 13C nuclei). Additional spin Hamiltonian parameters: giso = 1.9970; 31P nuclei: I = ½, PAiso = 76.0 MHz; 1H nuclei: I = ½, HAiso = 8.2 MHz; DH nuclei: I = 1, DAiso = 1.3 MHz; and 13C nuclei: I = ½, CAiso = 25 MHz. The corresponding simulations are shown in red using the relative weightings for the individual components in blue (a) 0.25:0.75:0:0:0:0:0, (b) 0.3:0.7:0:0:0:0:0, (c) 0.02:0:0:0:0.98:0:0, (d) 0.3:0.112:0.336:0.252:0:0:0, and (e) 0.15:0:0:0:0.136:0.408:0.306, respectively.
To inform on the fate of the CO ligands and identify the remainder of the chromium ligand sphere, 13C labeling of the carbonyl groups in 1 was performed and 60% 13C enrichment was confirmed by mass spectrometry and infrared spectroscopy (figs. S6 to S8). The activation under identical conditions as for natural abundance 1 resulted in additional lines for both C2H4 and C2D4, confirming the presence of CO as ligand (Fig. 4A, d and e). Spectral simulations (Fig. 4A) were performed as the weighted sum of different species and isotopologs expected for 60% 13CO enrichment (Fig. 4B). The isotopologs with 0, 1, and 2 13CO ligands formed upon activation under deuterated ethene result in a singlet, doublet, and triplet splitting of each of the 31P triplet lines, respectively (Fig. 4B, e to g), producing a triplet of apparent quintets (Fig. 4A, e). The EPR spectrum upon activation under protonated ethene (Fig. 4A, d) was also found in agreement with the weighted sum of isotopologs with 0, 1, and 2 13CO ligands (Fig 4B, b to d) and CrI-bisarene. This confirms that two CO fragments remain bound while two are replaced by ethene ligands. The deconvolution of each spectral simulation is described in the Supplementary Materials (figs. S9 to S13), and the spin Hamiltonian parameters are listed in table S2. While alkylation of the carbonyls to form acyl ligands is possible in theory, these can be excluded because of a much larger density functional theory (DFT)–computed 13C hyperfine coupling not supported by the experiment (figs. S14 to S16 and tables S3 and S4).
DISCUSSION
The combination of in situ EPR spectroscopy with selective isotope labeling of catalyst precursor and substrate allowed dissecting the composition of the transient species in the catalytically active mixture. All evidence agrees with X being the PNP bound cationic CrI-bis(ethene)bis(CO) species ([Cr(C2H4)2(CO)2(PNP)]+). On the basis of the DFT studies, the structures of cis- and trans-CO isomers differed in energy by 84 kJ/mol, with the trans complex being more stable. In addition, this isomer shows better agreement between the DFT-calculated and experimental spin Hamiltonian parameters. In conclusion, the magnetic equivalence of both carbonyls and ethene protons hints at the symmetrical and most stable trans-CO isomer with fast rotation of ethene. This structural motif has been proposed early on as the crucial intermediate in the widely accepted catalytic mechanism. Nevertheless, for over four decades, no bis-ethene species of chromium has ever previously been detected. Here, this species was sufficiently stabilized to be observed at room temperature for tens of minutes, allowing EPR detection with high resolution. We attribute this to the two residual CO ligands enforcing a doublet ground state, as the low ratio of activator did not remove all CO ligands. The active catalyst most likely, as suggested in literature, would be without the carbonyl ligands, and the absence of experimental evidence of such a species is expected. The loss of carbonyl ligands on addition of excess Et6Al2 would lead to available vacant sites, resulting in higher activity and selectivity toward LAOs. In the future, activation of commercially used CrIII precursors using a low amount of Et6Al2 in the presence of CO can be monitored to reproduce the bis-ethene intermediates. Investigation of bis-ethene intermediates might well develop to a platform for further improving catalyst design. Here, the low amounts of ethene either being generated in situ or added as nonpressurized gas resulted in “starved catalysis,” and this might well have contributed to the long lifetime of the bis-ethene species, allowing characterization by EPR. GC-MS showed that oligomerization is occurring under 1-bar ethene, demonstrating catalytic competence under the conditions stabilizing the bis-ethene intermediate.
In conclusion, the detection of a PNP bound cationic CrI-bis(ethene) species is the first ever experimental detection of any bis-ethene complex of chromium. This crucial motif in the selective ethene oligomerization catalysis has been commonly accepted in the literature for over four decades but never directly identified. Its detection further points toward a CrI/III redox couple being active for ethene oligomerization. Thus, our results not only go beyond merely confirming the prior postulations but also move the science onto a much stronger and surer footing. Now that conditions for generating this species are known, it can be subjected to systematic investigation. The identification of conditions making it sufficiently persistent will aid the development of further improved catalytic systems for ethene oligomerization.
MATERIALS AND METHODS
Experimental design
Materials
Ag[Al(OC(CF3)3)4] was obtained from IoLiTec. PNP (27), Cr(CO)4(PNP), and 1 (20, 28) were synthesized following the standard procedures as reported. The L-CAA (L-CAA = [(C18H37)2N(CH3)H][Al(OC(CF3)3)4]) salt was synthesized using the same procedure as for L-CAB (L-CAB = [(C18H37)2N(CH3)H][B(C6F5)4]) (29), except that (Et2O)2.5LiB(C6F5)4 was replaced by Li[Al(OC(CF3)3)4]. C2H4, C2D4 and 13CO were obtained from BOC. Et6Al2 was obtained from AkzoNobel, and all solvents were obtained from Sigma-Aldrich. Quartz EPR tubes (4 mm) from Wilmad, syringes from Hamilton, precision seal rubber septa from Sigma-Aldrich, and GC vials from Agilent Technologies were obtained.
Optimized reaction conditions
Our reaction design was optimized on the basis of several considerations: (i) The activator was directly injected into a vial containing the CrI-precatalyst 1 for homogeneous mixing. (ii) The activation and the sample transfer to an EPR tube was performed at 273 K to slow down the reaction kinetics. (iii) Various solvent systems were tested, and a mixture of dichloromethane (DCM) and methylcyclohexane (MCH) (1:1 by volume) was selected. This mixture gave good resolution of the continuous-wave EPR signal at room temperature because addition of the alkane reduces the dielectric constant of the solvent mixture (solvents with large dielectric constant lead to microwave absorption and signal broadening). Furthermore, this solvent mixture forms a glass on freezing rather than crystallizing, thus giving higher sensitivity and resolution for EPR measurements when frozen. (iv) Activator (2.5 to 5 equivalents) is crucial for reproducibility of intermediates. (v) The reaction was performed under an argon atmosphere.
Sample preparation
All manipulations for chromium complexes were carried out in a glove box (MBraun LABmaster DP) under an argon atmosphere with oxygen and water below 0.1 parts per million using solvents purified and dried via standard procedures by passing through alumina and deoxygenated by freeze-pump-thaw cycles. A 13 mM stock solution of 1 (7.8 mg, 5 μmol) was prepared separately in a 2:1 mixture by volume of DCM and MCH referred to as CrI-precatalyst solution. Aliquots (75 μl) of the CrI-precatalyst solution were transferred into separate GC vials. A 0.4 M stock solution of Et6Al2 (0.11 ml, 0.4 mmol) was prepared separately in 2 ml of MCH. Aliquots (100 μl) of the Et6Al2 stock solution were transferred into separate GC vials. The Et6Al2 solution must be handled under an inert atmosphere, and for in situ activation, the concentration was kept below its pyrophoric limit (<0.7 M) for safe handling. The activation was performed in situ outside the glove box by injecting 25 μl of Et6Al2 solution (5 equivalents with respect to CrI) into a GC vial containing CrI-precatalyst solution at 273 K using an argon-flushed Hamilton syringe. The activated mixture was quickly transferred to a 4-mm quartz EPR tube (presealed with a rubber septum in the glove box) and immediately frozen at 77 K by transferring into liquid N2. A control sample for CrI-precatalyst was also prepared in the glove box by replacing 25 μl of Et6Al2 stock solution with pure MCH, thereby keeping the same CrI concentration in all the samples. The activations were performed under identical conditions as above, except that a presealed tube with argon was flushed with 1 bar of ethene (C2H4 or C2D4) using a gas/vacuum manifold before the activation.
13C isotope enrichment of the carbonyl groups was performed by heating a Cr(CO)4(PNP) solution to 150°C in 13CO (2 bars). This complex was oxidized to the 13C-enriched 1 with Ag[Al(OC(CF3)3)4] following standard procedures, and activation was performed under identical conditions as above for the natural abundance 1.
EPR spectroscopy
The EPR spectra were acquired using an X-band (~9.34 GHz) Bruker EMX 10/12 spectrometer equipped with an ELEXSYS super-high-sensitivity probehead (Bruker ER4122SHQE) and an ESR900 He cryostat. The sample temperature was stabilized to 100 K with a variable temperature helium flow cryostat (Oxford Instruments) using liquid N2. The spectra were obtained using 10-mW microwave power, 0.1-mT modulation amplitude, 40-ms sampling time, and 100-kHz modulation frequency. The solution state spectra were obtained at 295 K. For monitoring the spectral changes over time, incremental scans were performed at 295 K with a delay of 60 s. All simulations were made using EasySpin 5.0.7 (30) with the functions “pepper” for frozen sate and “garlic” for isotropic spectra measured in solution state.
Acknowledgments
Dedicated to the memory of Paul C. J. Kamer, a fine chemist, colleague and friend. We thank C. Logan Mackay and F. Cruickshank, School of Chemistry, University of Edinburgh for recording the mass spectrometry data. Funding: This work was supported by Sasol Ltd. S.C. is grateful for a St Leonard’s postgraduate fellowship by the University of St Andrews. Author contributions: S.C. performed all EPR sample preparations, EPR measurements, data analysis, with support and advice from B.E.B., and DFT calculations jointly with M.B. D.M.S. provided the materials required for the EPR sample preparations and performed infrared spectroscopy measurements and GC-MS analysis on the EPR samples. D.M.S. performed isotope enrichment with contributions from N.L.B., A.J.B.W., and S.C. D.J.C.-H. interpreted the data and contributed to the design of isotope labeling experiments. S.C. and B.E.B. wrote the manuscript with contributions from all the authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The research data supporting this publication can be accessed at https://doi.org/10.17630/8565cf75-30d0-419f-881a-6d3c86dd0710 (44). Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/51/eabd7057/DC1
REFERENCES AND NOTES
- 1.Manyik R. M., Walker W. E., Wilson T. P., A soluble chromium-based catalyst for ethylene trimerization and polymerization. J. Catal. 47, 197–209 (1977). [Google Scholar]
- 2.Breuil P.-A. R., Magna L., Olivier-Bourbigou H., Role of homogeneous catalysis in oligomerization of olefins: Focus on selected examples based on group 4 to group 10 transition metal complexes. Catal. Lett. 145, 173–192 (2015). [Google Scholar]
- 3.Peitz S., Aluri B. R., Peulecke N., Müller B. H., Wöhl A., Müller W., Al-Hazmi M. H., Mosa F. M., Rosenthal U., An alternative mechanistic concept for homogeneous selective ethylene oligomerization of chromium-based catalysts: Binuclear metallacycles as a reason for 1-octene selectivity? Chem. Eur. J. 16, 7670–7676 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Younkin T. R., Connor E. F., Henderson J. I., Friedrich S. K., Grubbs R. H., Bansleben D. A., Neutral, single-component nickel (II) polyolefin catalysts that tolerate heteroatoms. Science 287, 460–462 (2000). [DOI] [PubMed] [Google Scholar]
- 5.A. H. Charles, Modern Plastics Handbook (McGraw-Hill Education, 2000). [Google Scholar]
- 6.Alpha olefin—Worldwide market size, share & trends analysis & forecast (2019–2025): 1-Hexene is anticipated to exhibit a revenue-based CAGR of 5.8% from 2019 to 2025—ResearchAndMarkets.com; www.businesswire.com/news/home/20190410005555/en/Alpha-Olefin---Worldwide-Market-Size-Share [accessed 28 April 2020].
- 7.Sydora O. L., Selective ethylene oligomerization. Organometallics 38, 997–1010 (2019). [Google Scholar]
- 8.McGuinness D. S., Olefin oligomerization via metallacycles: Dimerization, trimerization, tetramerization, and beyond. Chem. Rev. 111, 2321–2341 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Wass D. F., Chromium-catalysed ethene trimerisation and tetramerisation—breaking the rules in olefin oligomerisation. Dalton Trans. , 816–819 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Agapie T., Selective ethylene oligomerization: Recent advances in chromium catalysis and mechanistic investigations. Coord. Chem. Rev. 255, 861–880 (2011). [Google Scholar]
- 11.Briggs J. R., The selective trimerization of ethylene to hex-1-ene. J. Chem. Soc. Chem. Commun. , 674–675 (1989). [Google Scholar]
- 12.Bollmann A., Blann K., Dixon J. T., Hess F. M., Killian E., Maumela H., McGuinness D. S., Morgan D. H., Neveling A., Otto S., Overett M., Slawin A. M. Z., Wasserscheid P., Kuhlmann S., Ethylene tetramerization: A new route to produce 1-octene in exceptionally high selectivities. J. Am. Chem. Soc. 126, 14712–14713 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Agapie T., Schofer S. J., Labinger J. A., Bercaw J. E., Mechanistic studies of the ethylene trimerization reaction with chromium−diphosphine catalysts: Experimental evidence for a mechanism involving metallacyclic intermediates. J. Am. Chem. Soc. 126, 1304–1305 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Overett M. J., Blann K., Bollmann A., Dixon J. T., Haasbroek D., Killian E., Maumela H., McGuinness D. S., Morgan D. H., Mechanistic investigations of the ethylene tetramerisation reaction. J. Am. Chem. Soc. 127, 10723–10730 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Hirscher N. A., Labinger J. A., Agapie T., Isotopic labelling in ethylene oligomerization: Addressing the issue of 1-octene vs. 1-hexene selectivity. Dalton Trans. 48, 40–44 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Agapie T., Labinger J. A., Bercaw J. E., Mechanistic studies of olefin and alkyne trimerization with chromium catalysts: Deuterium labeling and studies of regiochemistry using a model chromacyclopentane complex. J. Am. Chem. Soc. 129, 14281–14295 (2007). [DOI] [PubMed] [Google Scholar]
- 17.McGuinness D. S., Chan B., Britovsek G. J. P., Yates B. F., Ethylene trimerisation with Cr-PNP catalysts: A theoretical benchmarking study and assessment of catalyst oxidation state. Aust. J. Chem. 67, 1481–1490 (2014). [Google Scholar]
- 18.Skobelev I. Y., Panchenko V. N., Lyakin O. Y., Bryliakov K. P., Zakharov V. A., Talsi E. P., In situ EPR monitoring of chromium species formed during Cr−pyrrolyl ethylene trimerization catalyst formation. Organometallics 29, 2943–2950 (2010). [Google Scholar]
- 19.Do L. H., Labinger J. A., Bercaw J. E., Spectral studies of a Cr(PNP)–MAO system for selective ethylene trimerization catalysis: Searching for the active species. ACS Catal. 3, 2582–2585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rucklidge A. J., McGuinness D. S., Tooze R. P., Slawin A. M. Z., Pelletier J. D. A., Hanton M. J., Webb P. B., Ethylene tetramerization with cationic chromium(I) complexes. Organometallics 26, 2782–2787 (2007). [Google Scholar]
- 21.Hirscher N. A., Perez Sierra D., Agapie T., Robust chromium precursors for catalysis: Isolation and structure of a single-component ethylene tetramerization precatalyst. J. Am. Chem. Soc. 141, 6022–6029 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chabbra S., Smith D. M., Bode B. E., Isolation of EPR spectra and estimation of spin-states in two-component mixtures of paramagnets. Dalton Trans. 47, 10473–10479 (2018). [DOI] [PubMed] [Google Scholar]
- 23.Emrich R., Heinemann O., Jolly P. W., Krüger C., Verhovnik G. P. J., The role of metallacycles in the chromium-catalyzed trimerization of ethylene. Organometallics 16, 1511–1513 (1997). [Google Scholar]
- 24.Brückner A., Jabor J. K., McConnell A. E. C., Webb P. B., Monitoring structure and valence state of chromium sites during catalyst formation and ethylene oligomerization by in situ EPR spectroscopy. Organometallics 27, 3849–3856 (2008). [Google Scholar]
- 25.McDyre L., Carter E., Cavell K. J., Murphy D. M., Platts J. A., Sampford K., Ward B. D., Gabrielli W. F., Hanton M. J., Smith D. M., Intramolecular formation of a CrI(bis-arene) species via TEA activation of [Cr(CO)4(Ph2P(C3H6)PPh2)]+: An EPR and DFT investigation. Organometallics 30, 4505–4508 (2011). [Google Scholar]
- 26.Carter E., Cavell K. J., Gabrielli W. F., Hanton M. J., Hallett A. J., McDyre L., Platts J. A., Smith D. M., Murphy D. M., Formation of [Cr(CO)x(Ph2PN(iPr)PPh2)]+ structural isomers by reaction of triethylaluminum with a chromium N,N-bis(diarylphosphino)amine complex [Cr(CO)4(Ph2PN(iPr)PPh2)]+: An EPR and DFT investigation. Organometallics 32, 1924–1931 (2013). [Google Scholar]
- 27.Blann K., Bollmann A., de Bod H., Dixon J. T., Killian E., Nongodlwana P., Maumela M. C., Maumela H., McConnell A. E., Morgan D. H., Overett M. J., Prétorius M., Kuhlmann S., Wasserscheid P., Ethylene tetramerisation: Subtle effects exhibited by N-substituted diphosphinoamine ligands. J. Catal. 249, 244–249 (2007). [Google Scholar]
- 28.McGuinness D. S., Brown D. B., Tooze R. P., Hess F. M., Dixon J. T., Slawin A. M. Z., Ethylene trimerization with Cr−PNP and Cr−SNS complexes: Effect of ligand structure, metal oxidation state, and role of activator on catalysis. Organometallics 25, 3605–3610 (2006). [Google Scholar]
- 29.R. K. Rosen, D. D. VanderLende, Highly soluble olefin polymerization catalyst activator, U.S. Patent 5,919,983A (1999).
- 30.Stoll S., Schweiger A., EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 178, 42–55 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Bond A. M., McGarvey B. R., Rieger A. L., Rieger P. H., On the failure to observe isotropic electron paramagnetic resonance spectra for certain chromium(I) carbonyl complexes. Inorg. Chem. 39, 3428–3429 (2000). [DOI] [PubMed] [Google Scholar]
- 32.Perdew J. P., Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B Condens. Matter 33, 8822–8824 (1986). [DOI] [PubMed] [Google Scholar]
- 33.Becke A. D., Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 38, 3098–3100 (1988). [DOI] [PubMed] [Google Scholar]
- 34.Perdew J. P., Erratum: Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B Condens. Matter 34, 7406 (1986). [PubMed] [Google Scholar]
- 35.Bühl M., Kabrede H., Geometries of transition-metal complexes from density-functional theory. J. Chem. Theory Comput. 2, 1282–1290 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Grimme S., Antony J., Ehrlich S., Krieg H., A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Becke A. D., Johnson E. R., A density-functional model of the dispersion interaction. J. Chem. Phys. 123, 154101 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Weigend F., Ahlrichs R., Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297–3305 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Adamo C., Barone V., Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 110, 6158–6170 (1999). [Google Scholar]
- 40.Munzarová M., Kaupp M., A critical validation of density functional and coupled-cluster approaches for the calculation of EPR hyperfine coupling constants in transition metal complexes. J. Phys. Chem. A 103, 9966–9983 (1999). [Google Scholar]
- 41.W. Kutzelnigg, U. Fleischer, M. Schindler, in NMR Basic Princ. Progress Vol. 23, P. Diehl, E. Fluck, H. Günther, R. Kodfeld, J. Seelig, Eds. (Springer, 1991), pp. 165–262. [Google Scholar]
- 42.M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian 09 (Gaussian Inc., 2013).
- 43.Neese F., The ORCA program system. WIREs Comput. Mol. Sci. 2, 73–78 (2012). [Google Scholar]
- 44.S. Chabbra, D. M. Smith, N. L. Bell, A. J. B. Watson, M. Bühl, D. J. Cole-Hamilton, B. E. Bode, First experimental evidence for a bis-ethene chromium(I) complex forming from an activated ethene oligomerization catalyst (dataset). Dataset. University of St Andrews Research Portal (2020); 10.17630/8565cf75-30d0-419f-881a-6d3c86dd0710. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/51/eabd7057/DC1