

A deep learning–based method to infer transcription factor binding in single cells has been developed by learning from bulk data.
Abstract
Characterizing genome-wide binding profiles of transcription factors (TFs) is essential for understanding biological processes. Although techniques have been developed to assess binding profiles within a population of cells, determining them at a single-cell level remains elusive. Here, we report scFAN (single-cell factor analysis network), a deep learning model that predicts genome-wide TF binding profiles in individual cells. scFAN is pretrained on genome-wide bulk assay for transposase-accessible chromatin sequencing (ATAC-seq), DNA sequence, and chromatin immunoprecipitation sequencing (ChIP-seq) data and uses single-cell ATAC-seq to predict TF binding in individual cells. We demonstrate the efficacy of scFAN by both studying sequence motifs enriched within predicted binding peaks and using predicted TFs for discovering cell types. We develop a new metric “TF activity score” to characterize each cell and show that activity scores can reliably capture cell identities. scFAN allows us to discover and study cellular identities and heterogeneity based on chromatin accessibility profiles.
INTRODUCTION
Transcription factors (TFs) bind to accessible or “open” promoter and enhancer regions, which play a pivotal role in regulating gene expression by aiding or inhibiting binding of RNA polymerase (1–3). Different binding events lead to heterogeneity of gene expression across a population of cells, which may result in distinct cellular identities. Therefore, characterizing TF binding profiles is critical for understanding gene regulatory mechanisms and differentiation of cells into distinct subpopulations.
Chromatin accessibility assays such as deoxyribonuclease hypersensitive sites sequencing (DNase-seq) (4), formaldehyde-assisted isolation of regulatory elements sequencing (FAIRE-seq) (5), and assay for transposase-accessible chromatin sequencing (ATAC-seq) (6) provide a way to study TF binding activity across the whole genome (7, 8). Of these methods, ATAC-seq is gaining popularity because of its low cost, efficiency, and simplicity. ATAC-seq profiles are generally designed to identify open chromatin regions, which can be used to infer TF binding events if these regions overlap with protein-binding sites.
A previously published model, HINT-ATAC, was designed to predict TF binding at a cell population level [based on either bulk ATAC-seq data or a combination of single-cell ATAC-seq (scATAC–seq) data as bulk data] (8). In recent years, deep learning techniques, such as convolutional neural networks (CNNs), have become a powerful tool for discovering TF binding patterns (9). Methods such as FactorNet (10) and deepATAC (11) leverage deep learning–based approaches to identify open chromatin regions and infer TF binding locations using bulk chromatin accessibility data. However, all these methods make population-level TF binding predictions and therefore do not take into account heterogeneity within cellular populations.
Recent advances in single-cell epigenomic sequencing permit characterization of chromatin accessibility at a single-cell level (12). For example, probing chromatin accessibility within single cells by scATAC-seq has become possible (13, 14), enabling the identification of cis- and trans-regulators and the study of how these regulators coordinate in different cells to influence cell fate (15–17). As in all single-cell sequencing technologies, using only scATAC-seq data is challenging because they are sparse and noisy due to not only technical constraints such as shallow sequencing (13) but also biological realities such as cellular heterogeneity (18).
To address these challenges, we present a deep learning–based framework called single-cell factor analysis network (scFAN). scFAN’s pipeline consists of a “pretrained model” trained on bulk data, which is then used to predict TF binding at a cellular level using a combination of DNA sequence data, aggregated similar scATAC-seq data, and mapability data (19). This approach alleviates the intrinsic sparsity and noise constraints of scATAC-seq. scFAN provides an effective tool to predict different TF profiles across individual cells and can be used for analyzing single-cell epigenomics and predicting cell types.
RESULTS
scFAN overview
We start with a brief overview of scFAN (Fig. 1A and fig. S1). scFAN is a deep learning model that predicts the probability of a TF binding at a given genomic region, with inputs of ATAC-seq, DNA sequence, and DNA mapability data from that region. scFAN is trained using publicly available “bulk” datasets, which contain genome-wide ATAC-seq and chromatin immunoprecipitation sequencing (ChIP-seq) profiles collected from multiple cell types measured at a population level. The data inputs (i.e., feature vectors) are 1000–base pair (bp) bins composed of bulk ATAC-seq data, DNA sequence, and mapability data for that bin. The feature vectors are fed into a three-layer CNN to extract high-level features. The CNN is then linked to two fully connected layers and a final sigmoid layer to make predictions. The ground-truth outputs are multiple binary labels indicating whether a particular TF binds to that genomic region, annotated on the basis of ChIP-seq peaks.
Fig. 1. scFAN pipeline and classification performance on bulk data.
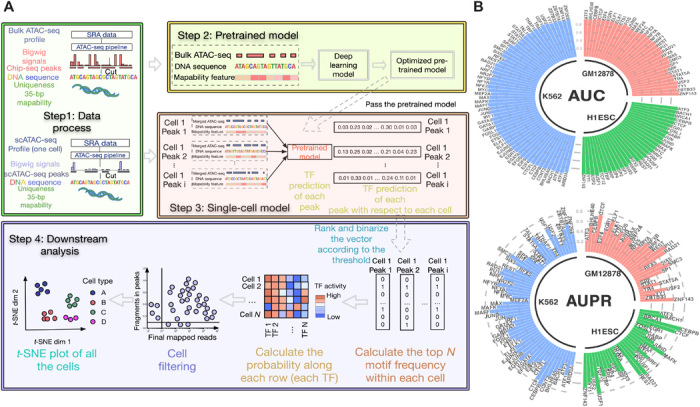
(A) scFAN pipeline. Bulk ATAC-seq, mapability data, and regions of DNA identified by ChIP-seq data are passed to the deep learning “pretrained model.” The trained model is then used to predict TF binding profiles based on regions of DNA called by scATAC-seq, mapability data, and a combination of scATAC-seq and bulk ATAC-seq. TF “activity scores” are calculated from the predictions by summing the number of times the top 2 most frequent TFs appear per cell. scFAN cluster cells from these activity scores. (B) Circular barplots showing AUC and auPR values of all the TFs from the pretrained model, from three different cell lines.
Once the model is fully trained, scFAN predicts TF bindings in each individual cell based on its scATAC-seq profile. Because of the intrinsic sparsity of current scATAC-seq technology, we smoothed the scATAC-seq signal from the individual bases of each cell by aggregating scATAC-seq data from similar cells. For each single cell, we calculated similarity scores between it and other cells, then aggregated chromatin accessibility signals of its near neighbors to boost chromatin accessibility coverage, and used the aggregated data as inputs to our model. This approach allows us to increase the chromatin accessibility coverage while retaining cellular specificity. The input vectors in the prediction step are the aforementioned aggregated scATAC-seq data, DNA regions called by scATAC-seq, and mapability data.
Validation of scFAN accuracy on bulk data
We trained scFAN on three bulk ATAC-seq datasets, GM12878, H1-ESC, and K562, in which ChIP-seq data for a number of TFs were also available from the ENCODE consortium (with 33, 31, and 60 TFs in each dataset, respectively), and generated three pretrained scFAN models—one for each dataset. We then validated the accuracy of the trained models on test datasets (hold-out chromosome regions were not used during training). Similar to the TF binding annotations in the training data, the ground-truth labels of the TF binding in the testing data are also based on ChIP-seq peaks. Because our dataset has more negative samples than positive samples, we measured the prediction accuracy using the area under the ROC (receiver operating characteristic) curve (AUC), the area under the precision-recall curve (AUPR), the recall value, and the F1 score corresponding to each TF (Fig. 1B, fig. S2, and table S1) to comprehensively evaluate the performance of our model. Our trained model captured most of the TF binding information correctly: All the TF prediction AUC values are more than 0.80, and nearly half of the TF AUPR values are more than 0.8 (table S1). Moreover, we and others have reported that CNNs could capture TF binding motif information (10, 20). We used the same method from FactorNet and visualized TF kernels of SPI1, CREB1, JUND, and MAFK from the trained model based on cell line GM12878. These kernels were first converted to position weight matrices and then aligned with motifs from JASPAR (21) using TOMTOM (22). All these kernels successfully matched the TFs that were identified by known database like JASPER with matched E-values all less than 10−3, e.g., 9.02 × 10−4 for TF SPI1 (Fig. 2A).
Fig. 2. Validation of TF predictions.
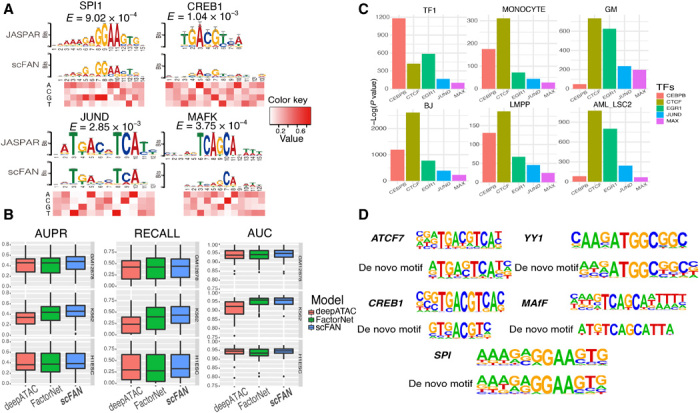
scFAN can predict both bulk and single-cell TF binding. (A) Four convolutional kernels that matched with four known motifs derived from JASPAR database. The heatmap denotes the value of each nucleotide corresponding to the above position. (B) Box plot of the performance of the pretrained model and two other models predicting bulk cell TF binding on the same dataset. (C) Enrichment analysis of the five predicted most active TFs from six randomly chosen cells. scFAN predicts the most likely TF per bin and adds up the number of times each TF is the highest predicted TF. Homer takes all the candidate peaks that need to be predicted and generates the enrichment analysis. All these TFs were significantly enriched in all these peaks. (D) Several example regions were used for enrichment analysis. scFAN was used to predict these regions’ most active TFs, which are ATCF7, YY1, CREB1, MAFF, and SPI. De novo matched motifs were compared to known motifs from Homer.
We then compared scFAN with two other state-of-the-art bulk TF binding profile prediction methods, FactorNet (10), and deepATAC (11). Similar to FactorNet and deepATAC, scFAN uses convolutional neural nets as its basic building structure but simplifies the model structure to include fewer convolution layers with fewer parameters. A key difference between the input of scFAN and the input of the previous two models is the continuous ATAC-seq signal used by scFAN, as opposed to the binarized signal used by deepATAC and the DNase-seq data adopted by FactorNet. Binarizing the data may result in loss or change of ATAC-seq signal coverage across the genome. All three models were trained and tested on the same datasets. Encouragingly, scFAN more accurately predicted bulk TF binding than either FactorNet or deepATAC, based on mean values of AUC, AUPR, and recall in three cell lines (Fig. 2B). Per the GM12878, K562, and H1ESC cell lines, 85% (61%), 90% (55%), and 81% (71%) of TF predictions have better recall values compared to deepATAC (FactorNet). The improvements are statistically significant for two comparisons (two-tailed t test, P < 0.05).
In addition, we tested the transferability of the model by focusing on the 17 shared TFs that have ChIP-seq data in all three cell lines. For each of these TFs, we trained a TF model on one cell line and then evaluated its performance (in terms of AUC) on the other two cell lines. Of the 17 tested TFs, the majority (75%) showed robust model transferability across cell lines (fig. S3A). There are still four to five TFs showing reduced performance across cell lines; however, these TFs exhibit clear cell type specificity.
Single-cell TF predictions are consistent with enrichment analysis
Next, we evaluated scFAN’s predictive performance at a single-cell level. We ran scFAN TF binding prediction on two scATAC-seq datasets. The first one consists of 2210 cells with multiple cell types: chronic myelogenous leukemia cell line K562 (both treated and untreated with drug), lymphoblastoid cell lines (GM12878) (including replicates), human embryonic stem cells (H1ESC), fibroblasts (BJ), erythroblasts (TF-1), promyeloblast (HL60), patients with acute myeloid leukemia (AML), lymphoid-primed multipotent progenitors (LMPPs), and monocyte cells from Buenrostro et al. (13) and Corces et al. (23). For simplicity, we denote this dataset as “Corces.” The second dataset is the peripheral blood mononuclear cell (PBMC) dataset from Buenrostro et al. (24, 25), which consists of 10 fluorescence-activated cell sorting (FACS)–sorted cell populations from CD34+ human bone marrow, namely, hematopoietic stem cells (HSCs), multipotent progenitors (MPPs), LMPPs, common myeloid progenitors, granulocyte-macrophage progenitors (GMPs), megakaryocyte-erythrocyte progenitors, common lymphoid progenitors, plasmacytoid dendritic cells, monocytes, and other uncharacterized cells (26). We ran TF binding predictions on each individual cell using each of the three pretrained scFAN models and then concatenated these predictions (see Materials and Methods) to generate the binding profiles of 124 TFs in each of the 2210/2034 cells.
Unlike the bulk data, acquiring TF information via simultaneous ChIP-seq and ATAC-seq measurements in the same single cell is still technologically challenging. Hence, we could not evaluate the accuracy of our single-cell TF binding predictions by directly comparing to a ground-truth label as in the case of the bulk data. To assess the quality of our predictions, we instead used two indirect approaches.
First, we verified whether there are sequence motifs enriched in the predicted TF regions and whether these motifs matched known binding profiles of the TFs. For this purpose, we used the software Homer (27) to discover and evaluate the enrichment of motifs with scFAN-predicted peaks from the Corces dataset. The result showed that five of the active TFs predicted by scFAN in six cells were all significantly enriched in Homer (P < 10−10; Fig. 2C). TFs critical to monocyte differentiation such as SPI1 (a.k.a. PU.1), EGR, CREB, and YY1 were highly enriched in monocyte cells (P < 10−5) (28, 29). To further explore whether each of these TF binding predictions matched the known motifs, we implemented TF predictions in all the candidate peaks in the monocyte cell using scFAN, selected those peaks that were predicted to bind with each one of these TFs, and performed de novo enrichment analysis using Homer. For each TF result, we used one of the most enriched de novo assembled motifs to match with corresponding TF motifs. We found that the de novo assembled motifs from scFAN closely matched the known motifs from Homer (Fig. 2D).
Using single-cell TF prediction to cluster cell types
Next, we studied whether the predicted TF binding profiles can be used to differentiate cell types. We reasoned that if the TF binding predictions are accurate, they should be sufficient to cluster cells into different groups that share similar cell identities. Fortunately, the cell types of individual cells in the scATAC-seq datasets are known. We can therefore assess the quality of the cell clusters derived from TF binding profiles by comparing them to their true cell type labels.
To explore the ability of scFAN to cluster cell types based on the TF binding predictions, we developed a metric called “TF activity scores” to characterize the state of single cells. The TF’s activity score of a cell summarizes the intensity of its predicted occurrences across the genome in the cell—the higher the score, the more active the TF is (see Materials and Methods). Overall, the state of each cell is characterized by a TF activity vector of dimension 124, one component for each TF (all three pretrained models’ predictions were used to generate TF activity scores; see Materials and Methods). Both datasets were clustered using hierarchical clustering based on Euclidean distances between the TF activity vectors, shown in t-distributed stochastic neighbor embedding (t-SNE) plots (Figs. 3A and 4A). To comprehensively show the clusters of cells without ground-truth labels, we marked each of those “unknown” clusters with a numerical label. The number of clusters was computed via community detection using open-source python packages “networkx” and “community.” The predicted clusters did not entirely overlap with the clusters defined by these labels, even if they showed an overall consistency to external cell type labels (fig. S3B). It is possible that some of these clusters discovered by the model potentially correspond to previously unidentified cell types not recognized in the original annotations.
Fig. 3. Comparison of scFAN to seven other count matrix–based methods or open chromatin accessibility–based methods applied to the Corces dataset.
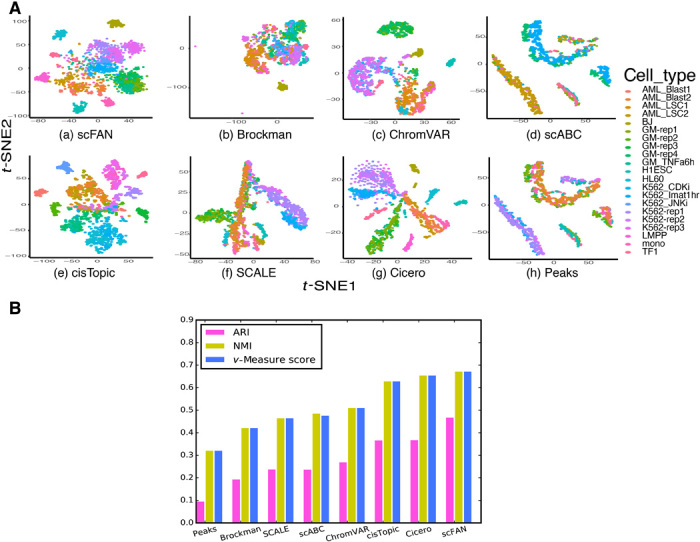
(A) t-SNEs of all seven different open chromatin–based or count matrix–based clustering methods. (B) Comparison of seven different clustering metrics of each method. ARI, NMI, and v-measure score were used to measure each method. The higher the score, the better the clustering performance.
Fig. 4. Comparison of scFAN to seven other count matrix–based methods or open chromatin accessibility–based methods applied to the PBMC dataset.
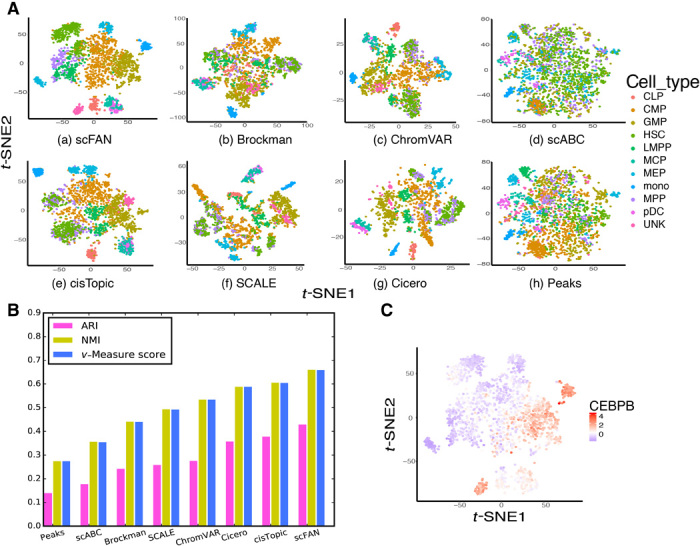
(A) t-SNEs of all seven different open chromatin–based or peak-cell count matrix–based clustering methods. (B) Comparison of seven different clustering metrics of each method. ARI, NMI, and v-measure score were used to measure each method. The higher the score, the better the clustering performance. (C) t-SNE plot on the PBMC dataset, colored by CEBPB activity score; CEBPB is more active in GMP and monocyte cells as their colors are more red than other cells.
To further verify the effectiveness of the TF activity score, we included two bulk expression datasets from ENCODE for an additional analysis—experiment ENCSR000AEE corresponding to cell type GM12878 and experiment ENCSR109IQO corresponding to cell type K562. For each cell type, we randomly selected 10 cells and calculated their mean TF activity scores. Then, we extracted the TF expression based on the fragments per kilobase million (FPKM) and calculated their Pearson correlations with the TF activity scores. Most of the TF expression values are well correlated with these TF activity scores, with correlation coefficient R > 0.7 and P < 0.01 (fig. S3C).
To validate the clustering result, we evaluated the clustering performance of scFAN by comparing the predicted clusters to ground-truth cell type labels. The performance of scFAN was benchmarked against several other popular methods that cluster cells on chromatin accessibility data—scABC (30), cisTopic (31), SCALE (32), Cicero (33), Brockman (34), and ChromVAR (15). For the Corces dataset, we performed the same filtering procedure for all the cells and used the same parameter settings (fig. S4A). We retained all cells of the PBMC dataset because those cells were filtered in the original study. We used three common metrics to quantitatively measure the clustering performance of scFAN and other compared methods: adjusted Rand index (ARI), normalized mutual information (NMI), and v-measure score (V-score). ARI correlates with clustering accuracy, so the higher the ARI is, the more accurately the model clustered the cells. NMI/v-measure score measures the mutual information of different clusters: If the two clusters have similar boundaries, they might show similar NMI/v-measure score and vice versa. Our model had the highest metric scores of these methods on the Corces(PBMCs) datasets, with ARI, NMI, and v-measure score equaling 0.470(0.432), 0.674(0.663), and 0.674(0.662), respectively (Figs. 3B and 4B). These results indicate that clustering cell types based on TF activity scores is consistently better than previous methods based on peak-cell matrix or chromatin accessibility. In addition, it further shows the transferability of our model because the PBMC dataset of 2034 cells is totally independent from the GM12878, K562, and H1-ESC cells and was not used for training the model.
Having demonstrated that TF activity scores are effective in differentiating cell types, we explored the contribution of individual TFs in defining cell identities. For this purpose, we plotted the activity scores of three TFs (EGR1, CEBPB, and SPI1) across the Corces dataset of 2210 cells on top of the cluster t-SNE plots (Fig. 5A). A couple of observations are notable from these plots. First, individual TFs show considerable amount of variation in their activity scores across different cell types. For instance, LMPP cells have the highest EGR1 activity score, with a mean activity score of 1.879, suggesting EGR1’s prominent role in the transcriptional regulation of LMPP cells. CEBPB, on the other hand, has the highest mean activity score in fibroblast cells (3.136). Second, there is also large heterogeneity among different TFs in their involvement in different cell types. SPI1 is more active than EGR1 in monocyte cells, with EGR1 mean activity score value higher in AML cells than monocyte cells. These observations seemed to be consistent with previously published studies, which not only indicate that EGR1 is highly enriched in LMPP cells (35) but also show that CEBPB is involved in fibroblast cell development and so is SPI1 in LMPP cells (36). We also found that in both datasets, the activity scores of CEBPB are relatively high in GMP and monocyte cells compared with others (Figs. 4C and 5A). Similar findings were shown in the original study (24). Overall, the computed TF activity score exhibits useful biological meaning to delineate the differentiation process of those cells.
Fig. 5. TF activity score varies across cell types, and H1 cells are well separated by TF activity scores.
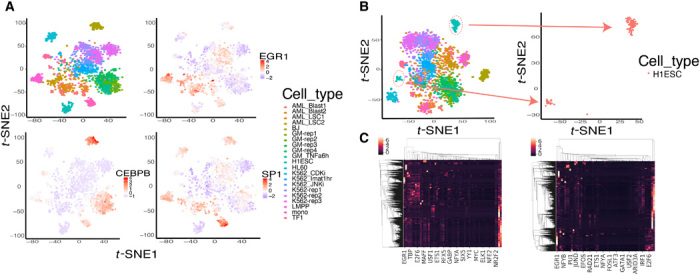
(A) TFs have varying activity scores across cell types. EGR1 is most active in the LMPP cells, CEBPB is most active in fibroblasts (BJ) cells, and SPI1 is most active in monocyte cells. (B) Separation of H1 embryonic stem cells (ESCs) colored red when using scATAC-seq as model input. The H1ESC cells clearly separated into two distinct groups (subclusters 1 and 2). (C) Heatmap plot of all the TFs and across the whole chromosome from one H1ESC cell. The left heatmap was generated by aggregated scATAC-seq data as input, and the right heatmap was generated by raw scATAC-seq data as input. The left heatmap contains more TF prediction information than the right plot.
The use of scFAN and TF activity score–based clustering can potentially help alleviate single-cell sparsity and further improve clustering performance. When only raw scATAC-seq data without aggregation were used to predict TF and cluster cells on the Corces dataset, scFAN subclustered nominally genetically identical H1ESCs (Fig. 5B). However, when we adopted the aggregated scATAC-seq data as our input, scFAN grouped the subclusters back into one cluster. The aggregation of the scATAC-seq signals probably helped recover chromatin accessibility signals of H1ESC cells, which made the model prediction more accurate. The heatmap plots of TF prediction on one H1ESC cell across all the peaks using raw scATAC-seq data and the aggregated scATAC-seq data showed that the TF prediction results of scFAN contain higher probability on some TFs compared with the heatmap without scATAC-seq aggregation (indicated by the brighter colors) (Fig. 5C). Furthermore, we randomly selected the regions in chromosome 1 to visualize the chromatin accessibility signals (fig. S5). We found that the signal coverage in some regions became dense after borrowing information from neighboring cells, which might be helpful for further TF prediction in those separated H1ESC cells. From the improvement of ARI and NMI metric values compared to previous models, we could indirectly infer that scFAN potentially has the ability to help alleviate the data sparsity and find missing signals of scATAC-seq data in low-coverage cells and thus provide a better performance on TF prediction across the genome.
Alleviating batch effects
scFAN could potentially reduce batch effects compared with other models. Identical cell types derived from different batches (or samples) may group into multiple subclusters due to batch effects (Fig. 4A and fig. S4B). For example, in the PBMC dataset, 160 LMPP cells are from two different batches, and certain peaks are more likely to appear in one batch than the other. scFAN therefore reduced those peaks to help alleviate batch effects from different batches (details in Materials and Methods). We performed batch effect correction on the PBMC dataset because we identified multiple batches within some cell types (such as LMPP cells, MPP cells, and HSCs) in this dataset. Our model “dragged” LMPP and MPP cells together while keeping other cells well clustered compared with other models [Fig. 4A (a, e, and g)]. After batch effect correction, some cell types such as HSCs still partitioned into two subclusters [fig. S6A (c)]. We then performed gene enrichment pathway analysis and found differential expression of genes between the two subclusters, suggesting that these two subclusters likely represent different cell identities (fig. S6, B and C).
As for the Corces dataset consisting of the three K562 replicate cells and four GM12878 replicate cells, we compared the clustering results between the raw data (i.e., without batch correction) and the batch-corrected data by computing ARI, NMI, and v-measure score (v-score). We found that the metric changes between the raw data and the batch-corrected data were quite small, suggesting that these data do not suffer from meaningful batch effects (fig. S6D). On the other hand, we reevaluated the clustering scores of scFAN under a new setting in which all the replicates in GM12878 and K562 are considered as one cell type. We compared scFAN to two other state-of-the-art models—cisTopic and Cicero. We saw that scFAN still outperforms these models, while the ARI scores on GM12878 and K562 replicates are similar or lower than other two models, meaning no batch effect overfitting on these cells (fig. S7A); we therefore did not perform additional batch effect corrections on these replicates.
In addition, we also evaluated the performance of the compared methods with the addition of batch effect correction on the PBMC dataset. We found that some batch effects might have been alleviated because LMPP cells were “dragged” back together, but some other cells remained mixed (fig. S7, B and C); however, the low ARI/NMI/V-score might indicate that these methods would have difficulty handling single-cell data with complex batch effects.
Performance and sensitivity
Because scFAN can cluster cells accurately, we wanted to characterize how sensitive the clustering is to different parameters. We used the Corces dataset and started by varying the number of top predicted TFs per cell the clustering algorithm takes into account. scFAN by default uses the top 2 predicted TFs per cell. We compared the original clustering result to the clustering based on the activity scores of only the most active TF and the top 5 most active TFs. There is a slight improvement in the NMI and v-measure when choosing the top 5 TFs, but top 2 yields the highest ARI score. Overall, the clustering is robust to the chosen number of most active TFs (Fig. 6A).
Fig. 6. Clustering performance comparison when different thresholds and parameters are changed on the Corces dataset.
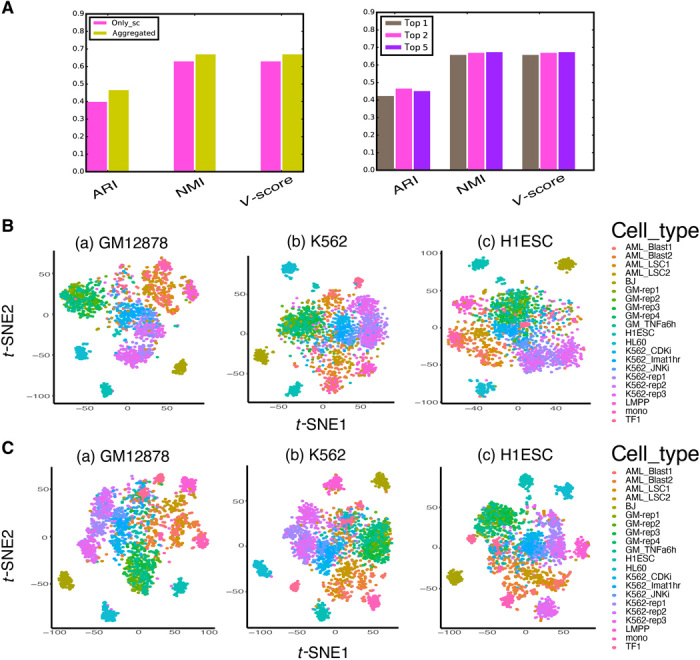
(A) Sensitivity of clustering to different ATAC-seq data that were used in the model and sensitivity of clustering to number of top TFs used. scFAN by default cluster cells on the aggregated ATAC-seq data and on the activity score of the top 2 most active TFs. (B) Clustering performance using three different pretrained models adopting scATAC-seq data as input. (C) Clustering performance using three different pretrained models adopting aggregated ATAC-seq data as input.
Next, we combined different models for predictions and tested the clustering performance on the Corces dataset (fig. S7D). While the overall ARI/NMI/V-measure scores decreased a little when choosing one or two models, they are still comparable with the default combined result and are better than most compared methods (Fig. 3B), suggesting the robustness of scFAN with respect to the choice of models. To alleviate the bias a single model may bring to the result, scFAN could combine the three models to enhance the overall performance.
We also verified the performance of TF binding prediction and clustering with the raw scATAC-seq data, for which the pretrained model was the same but the scATAC-seq signal was not aggregated. Using the same six cells shown in Fig. 2C, we found that those peaks, which were enriched with specific TFs in the aggregated scATAC-seq data model, remain mostly enriched in those cells (fig. S4C). Similar findings were obtained for the PBMC dataset (fig. S4D), demonstrating the consistency between the raw data approach and the aggregation approach and indicating the capability of scFAN to preserve the cell heterogeneity. We found that including similar scATAC-seq data to alleviate the data sparsity actually improves clustering performance over only using unaggregated scATAC-seq data (Fig. 6 and fig. S6A), probably due to the aforementioned sparsity and noisiness of scATAC-seq. Overall, the clustering performance of scFAN on both datasets using only scATAC-seq data still outperformed most of the compared methods, including cisTopic and Cicero (the other two best clustering models), further confirming the robustness of our model (fig. S6A).
DISCUSSION
Here, we developed a pipeline to predict TF binding not only at a cellular level but also in a specific genomic region within a single cell. scFAN is a deep learning–based single-cell analysis pipeline that mitigates the fundamental difficulties in analyzing scATAC-seq by leveraging bulk ATAC-seq data. At the bulk level, we found that scFAN can predict TF binding motifs more accurately than other deep learning models. At the single-cell level, scFAN robustly identifies cellular identities, even in cells that are genetically similar. Detecting cellular identities at a chromatin accessibility level may enable more faithful identification of distinct cell types. scFAN is also effective in dealing with batch effects across multiple samples.
Because of the limited availability of ChIP-seq TF binding data, we chose three standard cell lines in our model. Because the number of datasets used in this study lacks full coverage of all TFs in humans, some TF activities may be missing. With the increase in more TF-related data covering more TFs across multiple cell types, merging such TF-related single-cell information into one dataset could lead to a better prediction of TF binding and avoid calibration on prediction results, which is also a further expectation of implementing scFAN to more data. scFAN allows easy incorporation of new TF-related ChIP-seq data for other biological systems, and the users can choose their own dataset to retrain the model. While comparing predicted motifs to previously known ones or clustering cells based on TF activity can provide validation to some degree, experimental validations need to be carried out, for instance, using single-cell ChIP-seq (37) and scATAC-seq data together for further confirmation of scFAN’s. Last, scFAN’s downstream analyses, such as pseudotime analysis, can also be expanded and refined.
Overall, scFAN is a highly promising tool for single-cell analysis, not only for predicting TF binding and TF motifs but also for determining cellular identities. Being able to correlate open chromatin regions and binding activity of TFs in individual cells enables better understanding of cellular dynamics and regulations. This study shows that deep learning techniques can significantly improve our capability of using single-cell data to discern cell fate decisions.
MATERIALS AND METHODS
Data processing
DNA sequence
The sequence data were processed using the pipeline of Quang et al. (10). The genome was segmented into 200-bp bins, containing both the forward and reverse strands, with 50-bp intervals. Bins that overlapped with a known TF binding site were considered bound sites, and the rest of the bins excluding the blacklist regions (38) were considered as unbound sites. The bins were then expanded to 1000 bp, centered around the middle of each bin locus.
Chromatin accessibility
We processed the raw bulk ATAC-seq files by trimming with cutadapt (39), mapping to the human genome (hg19) using Bowtie2, and discarding the redundancy read pairs using Picard. We processed the scATAC-seq data with the ENCODE ATAC-seq pipeline protocol (https://github.com/ENCODE-DCC/ATAC-seq-pipeline) to obtain the filtered reads and called peaks using MACS2. The filtered bam files from both scATAC-seq and bulk ATAC-seq were converted into normalized bigwig files using deepTools2 (40). When we aggregated similar neighbor scATAC-seq signals, we adopted the bigWigMerge tool from the UCSC Genome Browser website and then converted the bedGraph file into bigwig files using a custom script. Bulk ATAC-seq and 35-bp uniqueness mapability signal values were also binned to 1000 bp with loci consistent with each ChIP-seq region.
Data preparation for machine learning
Bulk data
The bulk data were prepared as follows. Each bin was extracted from human genome DNA sequence, which was then one-hot encoded into a 4 × 1000 feature vector S. To fully use the sequencing signal data, the feature vector S was concatenated with both forward and reverse strands of ATAC-seq/mapability feature to form a 2 × 1000 ATAC-seq feature vector A and the 2 × 1000 mapability feature vector U, which were all concatenated to form the input feature vector SBulk. U refers to the uniqueness of a 35-bp subsequence on the positive strand starting at a particular base.
Single-cell data
The single-cell input feature vector SSC was prepared in a similar manner to the bulk input feature vector. The DNA sequence feature matrix S is now of the same length as SBulk but is extracted from peaks called from scATAC-seq data. We define the feature vector Asc, which is the aggregated scATAC-seq input data. Asc is a feature vector identical to A in the pretrained model. The mapability feature vector U remains the same. The feature vectors S, Asc, and U were concatenated into the single-cell input feature vector SSC.
Aggregated scATAC-seq data
The aggregated scATAC-seq data were computed by calculating the cell-cell similarity matrix using the scATAC-seq binarized cell-peak count matrix, which also helped alleviate batch effects (fig. S7E). We used cisTopic to calculate a low-dimensional cell-topic latent feature and used cosine similarity to calculate the similarity between one cell and other cells. For each cell, we considered its most 100 similar neighbor cells and aggregated their signals together to form its aggregated scATAC-seq data (fig. S8A).
Batch effect correction
First, for each cell type with multiple batches, we collected all the peaks from different batches separately. Second, for all the peaks from each single cell, we used the pyBedtools software to detect the peaks that are overlapped with all the peaks in other batches and retained those peaks. Third, we removed the peaks that did not overlap with any peaks in all the other batches, because those nonoverlapped peaks are potentially artifact sequencing regions that could eventually cause batch effects. The retained peaks were then used for further analysis.
Training and prediction
Calibration on the TF binding prediction
To train our pretrained models, we chose datasets from three different cell lines. For the TFs that are only present in the dataset from one cell line, those TF outputs were directly used to represent the final TF prediction. If the same TF appeared in multiple cell lines, we calculated the probability of intersecting peaks between called peaks in the single-cell dataset and called peaks of each bulk dataset separately. scFAN predicted these TFs on all the three models but only chose one model result whose corresponding cell line has the highest matched probability to the single cell and used its result to represent TF binding (see details in fig. S8B).
Deep learning calculations
Because deep learning models, such as CNNs (9), provide a natural and convenient way to make TF binding predictions, we either put the input feature vectors SBulk for the pretrained model or SSC for single-cell prediction in a three-layer two-dimensional CNN to extract the feature map. Two fully connected layers were connected to the output feature map, the output of which was passed to a sigmoid function to obtain the prediction of TF binding. Three different pretrained models were trained on bulk data SBulk from three different cell lines (GM12878, K562, and H1ESC). Each model was optimized using the Adam algorithm and then individually used to predict TF binding on the single-cell data SSC. The overall deep learning framework is shown in fig. S1. The TF binding predictions were then used to calculate the TF activity score to prepare for further clustering.
Our convolution calculation can be defined as follows
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
where SF is either SBulk or SSC, and F1, F2, F3, z1 denote the feature maps of each convolutional layer and the output of the first fully connected layer. W1, W2 refer to the weight matrix of the two fully connected layers. The final output of the network is the probability z of TF k binding to a peak n in each cell i, i.e., zi, n, k, where k ∈ 1⋯M, M being the total number of TFs of each cell from the pretrained model and n ∈ 1⋯N, N being the total number of peaks per cell. The parameters of the network are mostly in default settings, and other settings such as the number of CNN layers and the kernel sizes are adopted from FactorNet (10) and deepATAC (11). All the parameters of the network are shown in table S2.
Partition choice
Our pretrained model was restricted to the same dataset partition choice as in Quang et al. (10) for GM12878, H1-ESC, and K562: Chromosomes 1, 8, and 21 were used for testing, chromosome 11 was used for evaluation, and the remaining chromosomes were used for training (chromosome Y was excluded).
TF activity score
Here, we selected the top 2 potential predicted TF motifs of each peak and aggregated all the predicted TFs of all the peaks in each cell. We then normalized the value ci,k by calculating the probability across all peaks within a cell, which can be defined as the activity score pci, k for TF k in cell i, shown as follows
| (6) |
| (7) |
| (8) |
| (9) |
Cell clustering
We clustered cells on the TF activity scores. To use all the activity scores, we concatenated all the TF activity score results by column from all three models for all the cells without artificially cutting off any scores. We performed principal components analysis reduction and hierarchical clustering and drew the t-SNE plots using the concatenated feature. To filter the low-quality cells in the Corces dataset, we set the threshold of the fraction of total read counts per total number of peaks per cell to be 0.05 and also set the threshold of total read counts of each cell to be at least 1000 (fig. S2A). The scFAN pipeline is shown in Fig. 1A.
Method comparison
There are several recently published models, such as scABC (30), cisTopic (31), Cicero (33), SCALE (32), Brockman (34), and ChromVAR (15), that are also designed to cluster single cells based on scATAC-seq data. The first four methods all work with peak-by-cell binarized read count matrix. In particular, scABC uses the read count matrix to cluster cells via a weighted K-medoids clustering algorithm. cisTopic adopts latent Dirichlet allocation to convert the read count matrix into a topic-cell low-dimensional matrix, which is further used to clustering cells. Cicero applies latent semantic indexing to reduce the high-dimensional matrix into low-dimensional matrix similar to cisTopic. SCALE is a Variational AutoEncoder–based deep learning model that uses a Gaussian mixture model to initialize and model the cell clusters using binarized peak-cell matrix and then uses the latent features to cluster the cells. ChromVAR is based on scATAC-seq read counts and motifs in every peak: single-cell read count matrix and corrected peak-motif matched binary matrix are combined to calculate bias-corrected deviation and z-score matrix. The “corrected” z-score matrix is used to cluster each individual cell. Brockman uses adopted peaks to calculate k-mer frequency within each sample cell, generating more than 1000 kinds of k-mer frequency vectors of each cell and uses the combined matrix to cluster the cells. We also used the raw binarized matrix to directly cluster the cells as a benchmark. We used ARI, NMI, and V-measure score to quantitatively measure the clustering performance of these methods. We determined every cell label from each method using Euclidean distance and hierarchical clustering based on t-SNE projections of each method, which are the low-dimensional t-SNE embedding matrices from scABC, cisTopics, Cicero, and SCALE, k-mer t-SNE embedding matrix from Brockman, the motif correlation t-SNE embedding matrix from chromVAR, and the TF appearance probability t-SNE embedding matrix from our model.
Acknowledgments
We would like to acknowledge support from the NVIDIA. Funding: This work was supported by the National Natural Science Foundation of China (grant number 61872288) and the China Scholarship Council (to L.F.), NSF grants DMS1763272 and IIS1715017, a grant from the Simons Foundation (594598; to Q.N.), and NIH grants U01AR073159, P30AR075047, and R01GM123731. Author contributions: X.X. conceived the project. L.F. and L.Z. conducted the research. Q.N., Q.P., and X.X. supervised the research. L.F., E.D., Q.N., and X.X. contributed to the writing of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors (https://github.com/sperfu/scFAN/).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/51/eaba9031/DC1
REFERENCES AND NOTES
- 1.Jacob F., Monod J., Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 3, 318–356 (1961). [DOI] [PubMed] [Google Scholar]
- 2.Mercer T. R., Mattick J. S., Understanding the regulatory and transcriptional complexity of the genome through structure. Genome Res. 23, 1081–1088 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thomas M. C., Chiang C.-M., The general transcription machinery and general cofactors. Crit. Rev. Biochem. Mol. Biol. 41, 105–178 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Boyle A. P., Davis S., Shulha H. P., Meltzer P., Margulies E. H., Weng Z., Furey T. S., Crawford G. E., High-resolution mapping and characterization of open chromatin across the genome. Cell 132, 311–322 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giresi P. G., Kim J., McDaniell R. M., Iyer V. R., Lieb J. D., FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin. Genome Res. 17, 877–885 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buenrostro J. D., Giresi P. G., Zaba L. C., Chang H. Y., Greenleaf W. J., Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsompana M., Buck M. J., Chromatin accessibility: A window into the genome. Epigenetics Chromatin 7, 33 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Z., Schulz M. H., Look T., Begemann M., Zenke M., Costa I. G., Identification of transcription factor binding sites using ATAC-seq. Genome Biol. 20, 45 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Alipanahi B., Delong A., Weirauch M. T., Frey B. J., Predicting the sequence specificities of DNA- and RNA-binding proteins by deep learning. Nat. Biotechnol. 33, 831–838 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Quang D., Xie X., FactorNet: A deep learning framework for predicting cell type specific transcription factor binding from nucleotide-resolution sequential data. Methods 166, 40–47 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hiranuma N., Lundberg S., Lee S.-I., DeepATAC: A deep-learning method to predict regulatory factor binding activity from ATAC-seq signals. bioRxiv 2017, 172767 (2017). [Google Scholar]
- 12.Thurman R. E., Rynes E., Humbert R., Vierstra J., Maurano M. T., Haugen E., Sheffield N. C., Stergachis A. B., Wang H., Vernot B., Garg K., John S., Sandstrom R., Bates D., Boatman L., Canfield T. K., Diegel M., Dunn D., Ebersol A. K., Frum T., Giste E., Johnson A. K., Johnson E. M., Kutyavin T., Lajoie B., Lee B.-K., Lee K., London D., Lotakis D., Neph S., Neri F., Nguyen E. D., Qu H., Reynolds A. P., Roach V., Safi A., Sanchez M. E., Sanyal A., Shafer A., Simon J. M., Song L., Vong S., Weaver M., Yan Y., Zhang Z., Zhang Z., Lenhard B., Tewari M., Dorschner M. O., Hansen R. S., Navas P. A., Stamatoyannopoulos G., Iyer V. R., Lieb J. D., Sunyaev S. R., Akey J. M., Sabo P. J., Kaul R., Furey T. S., Dekker J., Crawford G. E., Stamatoyannopoulos J. A., The accessible chromatin landscape of the human genome. Nature 489, 75–82 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buenrostro J. D., Wu B., Litzenburger U. M., Ruff D., Gonzales M. L., Snyder M. P., Chang H. Y., Greenleaf W. J., Single-cell chromatin accessibility reveals principles of regulatory variation. Nature 523, 486–490 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cusanovich D. A., Daza R., Adey A., Pliner H. A., Christiansen L., Gunderson K. L., Steemers F. J., Trapnell C., Shendure J., Multiplex single-cell profiling of chromatin accessibility by combinatorial cellular indexing. Science 348, 910–914 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schep A. N., Wu B., Buenrostro J. D., Greenleaf W. J., chromVAR: Inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clark S. J., Lee H. J., Smallwood S. A., Kelsey G., Reik W., Single-cell epigenomics: Powerful new methods for understanding gene regulation and cell identity. Genome Biol. 17, 72 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin W., Tang Q., Wan M., Cui K., Zhang Y., Ren G., Ni B., Sklar J., Przytycka T. M., Childs R., Levens D., Zhao K., Genome-wide detection of DNase I hypersensitive sites in single cells and FFPE tissue samples. Nature 528, 142–146 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Altschuler S. J., Wu L. F., Cellular heterogeneity: Do differences make a difference? Cell 141, 559–563 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Derrien T., Estellé J., Marco Sola S., Knowles D. G., Raineri E., Guigó R., Ribeca P., Fast computation and applications of genome mappability. PLOS ONE 7, e30377 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park Y., Kellis M., Deep learning for regulatory genomics. Nat. Biotechnol. 33, 825–826 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Mathelier A., Fornes O., Arenillas D. J., Chen C.-Y., Denay G., Lee J., Shi W., Shyr C., Tan G., Worsley-Hunt R., Zhang A. W., Parcy F., Lenhard B., Sandelin A., Wasserman W. W., JASPAR 2016: A major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 44, D110–D115 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gupta S., Stamatoyannopoulos J. A., Bailey T. L., Noble W. S., Quantifying similarity between motifs. Genome Biol. 8, R24 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corces M. R., Buenrostro J. D., Wu B., Greenside P. G., Chan S. M., Koenig J. L., Snyder M. P., Pritchard J. K., Kundaje A., Greenleaf W. J., Majeti R., Chang H. Y., Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat. Genet. 48, 1193–1203 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buenrostro J. D., Corces M. R., Lareau C. A., Wu B., Schep A. N., Aryee M. J., Majeti R., Chang H. Y., Greenleaf W. J., Integrated single-cell analysis maps the continuous regulatory landscape of human hematopoietic differentiation. Cell 173, 1535–1548.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gnad F., Baucom A., Mukhyala K., Manning G., Zhang Z., Assessment of computational methods for predicting the effects of missense mutations in human cancers. BMC Genomics 14, S7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen H., Lareau C., Andreani T., Vinyard M. E., Pinello L., Assessment of computational methods for the analysis of single-cell ATAC-seq data. Genome Biol. 20, 241 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heinz S., Benner C., Spann N., Bertolino E., Lin Y. C., Laslo P., Cheng J. X., Murre C., Singh H., Glass C. K., Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Valledor A. F., Borràs F. E., Cullell-Young M., Celada A., Transcription factors that regulate monocyte/macrophage differentiation. J. Leukoc. Biol. 63, 405–417 (1998). [DOI] [PubMed] [Google Scholar]
- 29.Wen A. Y., Sakamoto K. M., Miller L. S., The role of the transcription factor CREB in immune function. J. Immunol. 185, 6413–6419 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zamanighomi M., Lin Z., Daley T., Chen X., Duren Z., Schep A., Greenleaf W. J., Wong W. H., Unsupervised clustering and epigenetic classification of single cells. Nat. Commun. 9, 2410 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.González-Blas C. B., Minnoye L., Papasokrati D., Aibar S., Hulselmans G., Christiaens V., Davie K., Wouters J., Aerts S., cisTopic: Cis-regulatory topic modeling on single-cell ATAC-seq data. Nat. Methods 16, 397–400 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xiong L., Xu K., Tian K., Shao Y., Tang L., Gao G., Zhang M., Jiang T., Zhang Q. C., SCALE method for single-cell ATAC-seq analysis via latent feature extraction. Nat. Commun. 10, 4579 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pliner H. A., Packer J. S., McFaline-Figueroa J. L., Cusanovich D. A., Daza R. M., Aghamirzaie D., Srivatsan S., Qiu X., Jackson D., Minkina A., Adey A. C., Steemers F. J., Shendure J., Trapnell C., Cicero predicts cis-regulatory DNA Interactions from single-cell chromatin accessibility data. Mol. Cell 71, 858–871.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Boer C. G., Regev A., BROCKMAN: Deciphering variance in epigenomic regulators by k-mer factorization. BMC Bioinform. 19, 253 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miyai T., Takano J., Endo T. A., Kawakami E., Agata Y., Motomura Y., Kubo M., Kashima Y., Suzuki Y., Kawamoto H., Ikawa T., Three-step transcriptional priming that drives the commitment of multipotent progenitors toward B cells. Genes Dev. 32, 112–126 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.D’Alò F., di Ruscio A., Guidi F., Fabiani E., Greco M., Rumi C., Hohaus S., Voso M. T., Leone G., PU.1 and CEBPA expression in acute myeloid leukemia. Leuk. Res. 32, 1448–1453 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Grosselin K., Durand A., Marsolier J., Poitou A., Marangoni E., Nemati F., Dahmani A., Lameiras S., Reyal F., Frenoy O., Pousse Y., Reichen M., Woolfe A., Brenan C., Griffiths A. D., Vallot C., Gérard A., High-throughput single-cell ChIP-seq identifies heterogeneity of chromatin states in breast cancer. Nat. Genet. 51, 1060–1066 (2019). [DOI] [PubMed] [Google Scholar]
- 38.ENCODE Project Consortium , An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin M., Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 17, 10–12 (2011). [Google Scholar]
- 40.Ramírez F., Ryan D. P., Grüning B., Bhardwaj V., Kilpert F., Richter A. S., Heyne S., Dündar F., Manke T., deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/51/eaba9031/DC1