

Malfunctions in the formaldehyde clearance processes induce DNA damage and overload the DNA repair system and cause AMeD syndrome.
Abstract
Rs671 in the aldehyde dehydrogenase 2 gene (ALDH2) is the cause of Asian alcohol flushing response after drinking. ALDH2 detoxifies endogenous aldehydes, which are the major source of DNA damage repaired by the Fanconi anemia pathway. Here, we show that the rs671 defective allele in combination with mutations in the alcohol dehydrogenase 5 gene, which encodes formaldehyde dehydrogenase (ADH5FDH), causes a previously unidentified disorder, AMeD (aplastic anemia, mental retardation, and dwarfism) syndrome. Cellular studies revealed that a decrease in the formaldehyde tolerance underlies a loss of differentiation and proliferation capacity of hematopoietic stem cells. Moreover, Adh5−/−Aldh2E506K/E506K double-deficient mice recapitulated key clinical features of AMeDS, showing short life span, dwarfism, and hematopoietic failure. Collectively, our results suggest that the combined deficiency of formaldehyde clearance mechanisms leads to the complex clinical features due to overload of formaldehyde-induced DNA damage, thereby saturation of DNA repair processes.
INTRODUCTION
Reactive aldehydes, such as acetaldehyde and formaldehyde, are cytotoxic and carcinogenic because they damage DNA and interfere with transcription and replication. Whereas acetaldehyde is mostly produced by oxidative degradation of ingested alcohol, formaldehyde is an ordinary one-carbon (1C) metabolite that is generated from various in vivo biochemical reactions, including enzymatic demethylation of histones and nucleic acids (1, 2). These free aldehydes are swiftly oxidized to innocuous carboxylic acids by cellular dehydrogenases. Aldehyde dehydrogenase 2 (ALDH2) detoxifies acetaldehyde into acetate, but this enzyme is inactivated in ~50% of the population in East Asian countries because of a functional single-nucleotide polymorphism, rs671 [ALDH2*2, c.1510G>A, p.E504K; MAF (minor allele frequency) = 0.27 in Japanese]. Rs671 is known to cause “alcohol flushing response” after drinking (3–6). Apparently, alcohol flushing is not a disease; however, the rs671 defective (A) allele is protective against alcoholism and is also associated with an increased risk of various clinical conditions, including cardiovascular disorders (7, 8) and certain types of cancer (4, 9–12). In particular, the incidence of gastrointestinal cancers, represented by esophageal squamous cell carcinoma, is significantly higher in individuals with the rs671 defective allele when they regularly drink alcohol (10–12). Despite a multitude of known genetic associations, no disease with a true digenic/oligogenic inheritance (13–15), under complete penetrance, due to rs671 has been reported. With regard to the formaldehyde elimination, alcohol dehydrogenase 5 (also known as formaldehyde dehydrogenase or S-nitrosoglutathione reductase, ADH5/FDH/GSNOR) is the principal enzyme converting formaldehyde to formic acid in a glutathione-dependent manner (16). Because GSNOR is also a key enzyme for the modulation of cellular nitric oxide signaling, thereby regulating circulatory functions, ADH5 polymorphisms are known to be associated with an increased risk of cardiovascular disorders (16). Nevertheless, to date, no congenital disorders due to the loss of ADH5 function has been reported.
When the metabolic processes of aldehyde clearance become uncapable or the capacity overflows, various types of endogenous DNA damage increase. Aldehydes primarily produce DNA interstrand cross-links (ICL) and nonenzymatic DNA-protein cross-links (DPC) (17). These DNA lesions prevent replication fork progression; therefore, they are thought to be largely repaired by the following replication-coupled DNA repair mechanisms: (i) ICL repair pathway involves “FANC” proteins that are mutated in Fanconi anemia (FA), a rare inherited bone marrow failure syndrome (IBMFS) (18, 19). This pathway (otherwise known as FA pathway) is activated in S phase and eliminates ICL, by unhooking of covalently bridged Watson/Crick strands with structure-specific endonucleases, followed by sequential actions involving translesion synthesis (TLS), homologous recombination, and nucleotide excision repair (NER). (ii) DPC repair is similarly initiated by the stalling of DNA polymerases at replication forks, where metalloproteases, such as SPRTN, degrade DNA-bound proteins to remnant peptides before TLS and further excision of remaining lesions by NER. Mutations in the SPRTN gene also cause a rare disorder, Ruijs-Aalfs syndrome (RJALS), characterized by segmental progeria and early-onset hepatocellular carcinoma (20).
In this study, we report a number of families with a new form of IBMFS cases. On the basis of genome analysis of the patients, we identified disease-causing digenic mutations in the ALDH2 and ADH5 genes. Cellular and animal studies demonstrate that the simultaneous loss of ALDH2 and ADH5 activities leads to an increase of cellular formaldehyde sensitivity and multisystem abnormalities including hematopoietic failure. Our results suggest that the formaldehyde clearance is as important as the DNA repair system for normal development of both humans and mice.
RESULTS
Case reports: Families with a new form of IBMFS with mental retardation and dwarfism
We report 10 cases in eight unrelated families, presenting a previously unclassified trait, characterized by aplastic anemia (AA), mental retardation, and short stature and microcephaly (dwarfism), termed AMeD syndrome (AMeDS). Pedigrees of the families (Fig. 1A), summaries of clinical manifestations (Table 1), and hematological complications (Table 2) of the affected individuals are shown (see also detailed clinical episodes below). Each of the cases was initially diagnosed as AA, FA, refractory anemia (RA), Bloom syndrome (BS) or Dubowitz syndrome (DS), largely based on their facial appearance and hematological manifestations (18, 21, 22). All cases developed myelodysplasia during infancy to childhood, and of seven cases with detailed clinical records, four patients received bone marrow transplants (BMTs). The possibility of FA is often considered in the differential diagnosis of IBMFS. Notably, severe dwarfism (height and head circumference < −4.0 SD) and intellectual disabilities both typical in AMeDS cases are uncommon in FA; these additional symptoms rather resemble those of patients with transcription-coupled NER deficiency, Cockayne syndrome (CS) (23), and its related disorders [cerebro-oculo-facio-skeletal syndrome (COFS) and XFE progeroid syndrome (XFEPS)] (24, 25). Also in contrast to typical FA cases, neither polydactyly nor chromosome fragility was observed in any of the AMeDS cases. Dyskeratosis congenita (DC) was also excluded by normal telomere length tested in some of the cases. These families and cases were extracted from the Genome Instability Syndrome Diagnosis Project, a part of the Rare/Intractable Disease (nanbyo) Project of Japan, as well as from a collection of undiagnosed IBMFS children analyzed by the central review system of the Japanese Society of Pediatric Hematology and Oncology and the targeted sequencing system for IBMFS at Nagoya University Pediatrics Department.
Fig. 1. Identification of digenic variants in ADH5 and ALDH2 in patients with AMeDS.
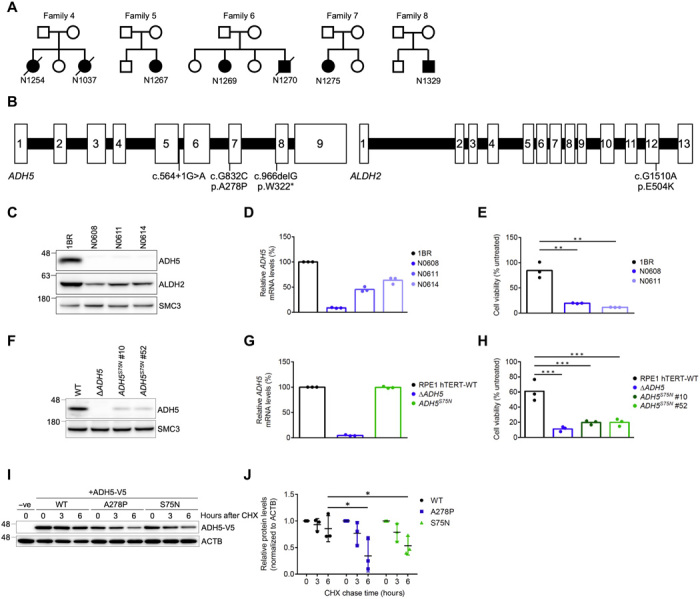
(A) Pedigrees of AMeDS families 4 to 8. (B) Pathogenic variants identified in ADH5 and ALDH2. (C) Immunoblots of ADH5 and ALDH2 in primary fibroblasts from normal (1BR) and patients with AMeDS (N0608, N0611, and N0614). SMC3 is a loading control. (D) ADH5 transcript of normal (1BR) and AMeDS (N0608, N0611, and N0614) cells. The relative transcript levels analyzed by the ∆∆CT method are shown for triplicate experiments. (E) Cell viability after continuous 30 μM formaldehyde treatment. Results from triplicate experiments (means ± SD) are shown. **P < 0.01, two-tailed unpaired t test. (F) Immunoblots showing a reduced stability of ADH5 p.S75N identified in a healthy individual, NAG16714. Gene-edited hTERT-immortalized RPE1 (RPE1 hTERT) cells expressing the homozygous ADH5 p.S75N alleles (clones no. 10 and no. 52), and ∆ADH5 cells are examined. (G) Stable expression of the p.S75N mRNA. (H) Cell viability after continuous 40 μM formaldehyde treatment. Results from triplicate experiments (means ± SD) are shown. ***P < 0.001, two-tailed unpaired t test. (I) ADH5 p.S75N is unstable as with p.A278P. U2OS cells transfected with V5-tagged ADH5 WT (wild type), p.A278P, or p.S75N were harvested at the indicated times following cycloheximide (CHX) treatment. Cell lysates were immunoblotted with V5 and ACTB antibodies. (J) Quantification of ADH5-V5 levels in (I) by image analysis, normalized to ACTB levels. Means (± SD) from triplicate experiments are shown. *P < 0.05; one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test.
Table 1. Digenic inheritance of the ALDH2 rs671 defective allele and ADH5 biallelic mutations identified in AMeDS individuals.
|
Family
ID |
Individual
ID |
Sex |
Initial
diagnosis* |
ADH5 mutations | ALDH2 mutations |
Hematological
abnormalities** |
Other
complications*** |
Remarks**** | ||
| Allele1 | Allele2 | Allele1 | Allele2 | |||||||
| 1 | N0608 | F | FA | p.W322* | p.W322* | p.E504K | WT | NA | NA | UPD (ADH5 locus) |
| 2 | N0611 | F | AA |
Splicing
c.564+1G>A |
p.A278P | p.E504K | WT | NA | NA | |
| Father2 | Normal | c.564+1G>A | WT | p.E504K | WT | |||||
| Sibling2-1 | F | Normal | WT | WT | WT | WT | ||||
| 3 | N0614 | M | AA | p.W322* | p.A278P | p.E504K | WT | NA | NA | |
| 4 | N1037 | F | BS | p.W322* | p.W322* | p.E504K | p.E504K | MDS (2) |
SS (n.d.), MC, MD,
NR, TC, HP, ID |
Died from
interstitial pneumonia (9 years) |
| N1254 | F | RA | p.W322* | p.W322* | p.E504K | p.E504K | AML (2) |
SS (−5.2), MC,
MD, NR, TC |
Died after BMT (3
years) |
|
| Father4 | Normal | p.W322* | WT | p.E504K | p.E504K | |||||
| Mother4 | Normal | p.W322* | WT | p.E504K | WT | |||||
| Sibling4-1 | F | Normal | p.W322* | WT | p.E504K | p.E504K | ||||
| 5 | N1267 | F | DS | p.W322k | p.A278P | p.E504K | WT | MDS (7) |
SS (−5.4), MC,
MG, PLSVC, ID |
Alive after second
BMT (14 years) |
| Sibling5-1 | M | Normal | WT | WT | p.E504K | WT | ||||
| 6 | N1269 | F | FA | p.W322* | p.W322* | p.E504K | WT | MDS (3) |
SS (−4.0), MD,
ASD, ADHD, ID |
Alive after BMT
(8 years) |
| N1270 | M | FA | p.W322* | p.W322* | p.E504K | p.E504K | MDS (0) |
SS (−4.7), MC
(−5.8), MD, TC, AH, GA, HP, ID |
Died from an
infection (2 years) |
|
| Father6 | Normal | p.W322* | WT | p.E504K | WT | |||||
| Mother6 | Normal | p.W322* | WT | p.E504K | WT | |||||
| Sibling6-1 | F | Normal | WT | WT | p.E504K | WT | ||||
| Sibling6-2 | F | Normal | p.W322* | WT | p.E504K | WT | ||||
| 7 | N1275 | F | FA | p.W322* | p.A278P | p.E504K | WT | MDS (8) |
MC, SS (−4.1), HP,
UH, DC, HD, LD, ID |
Alive after BMT
(12 years) |
| Father7 | Normal | p.W322* | WT | WT | WT | |||||
| Mother7 | Normal | p.A278P | WT | p.E504K | WT | |||||
| Sibling7-1 | F | Normal | WT | WT | WT | WT | ||||
| 8 | N1329 | M | FA | p.W322* | p.A278P | p.E504K | WT | MDS (12) |
MC, SS (−3.2),
HP, ID, PP |
Alive (16 years) |
| Father8 | Normal | p.W322* | WT | p.E504K | p.E504K | |||||
| Sibling8-1 | Normal | WT | WT | p.E504K | WT | |||||
| 9 | NAG16714 | F | Normal | p.S75N | p.S75N | WT | WT |
No previous
disease (55 years) |
||
Affected individuals and Nagahama case are shown in bold.
*FA, Fanconi anemia; AA, aplastic anemia; RA, refractory anemia; BS, Bloom syndrome; DS, Dubowitz syndrome.
**Ages at onset are in parentheses. MDS, myelodysplastic syndromes; AML, acute myeloid leukemia. NA, not analyzed.
***SD in parentheses. n.d., not determined; SS, short stature; MC, microcephaly; MD, motor deterioration; NR, nasal ridge; TC, telecanthus; HP, hyperpigmentation; ID, intellectual disability; MG, micrognathia; PLSVC, persistent left superior vena cava; ASD, autistic spectrum disorder; ADHD, attention-deficit hyperactivity disorder; AH, adrenal hypoplasia; GA, gonadal abnormality; UH, uterus hypoplasia; DC, dolichocephaly; HD, hyperdactyly (long thumb); LD, learning disability; PP, Precocious puberty.
****UPD, uniparental isodisomy; BMT, bone marrow transplant.
Table 2. Detailed hematological findings in AMeDS individuals.
NA, not analyzed. WBC, white blood cells; Neut, neutrophils; Mon, monocytes; Hb, hemoglobin; MCV, mean corpuscular volume; Ret, reticulocytes; Plt, platelets; HbF, fetal hemoglobin; PB, peripheral blood; BM, bone marrow; RCMD, refractory cytopenia with multilineage dysplasia; RAEB-1, refractory anemia with excess blasts-1.
|
Individual
ID |
WBC
(×109/liter) |
Neut
(×109/liter) |
Mon
(×109/liter) |
Hb
(g/dl) |
MCV
(fl) |
Ret
(‰) |
Plt
(×109/liter) |
HbF
(%) |
PB blast (%) | BM blast (%) |
Morphological
diagnosis |
Karyotype |
| N1037* | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA |
| N1254* | 1.4 | 0.4 | 0.4 | 9.3 | NA | NA | 47 | NA | NA | NA | NA | 45,XX,-7[8/10] 46,XX,der(5;17) (p10;q10),+8[4/20] |
| N1267 | 2.9 | 0.5 | 0.9 | 9.7 | 113 | 24 | 61 | 37 | 0 | 3 | RCMD (MDS-MLD) |
45,XX,+1,der(1;21) (q10;q10),-7[12/20] 46,idem,+7,add(18) (p11.2)[8/20] |
| N1269† | 9.8 | 3.9 | 1.3 | 7.7 | 75 | 10.9 | 56 | 4 | 0 | 3 | RAEB-1 (MDS-EB-1) |
45,idem,add(7) (p11.2),-8,add(19)(p13) [14/20] 46,XX,ins(1;?)(q12;?) [2/20] |
| N1270† | 3.4 | 1 | 0.9 | 7.4 | 101 | 36 | 17 | NA | 0 | 0 | RCMD (MDS-MLD) |
46,XY,add(7)(p15)[1/20] |
| N1275 | 1.9 | 0.1 | 0.04 | 10.7 | 98 | 5 | 24 | NA | 0 | 1 | RAEB-1 (MSD-EB-1) |
46,XX,+1,der(1;22) (q10;q10)[2/20] 47,idem,del(7) (q?),add(17) (p11.2),+mar1[9/20] 47,idem,add(17),del(20) (q1?),+mar1[5/20] |
| N1329 | 7.5 | 4.8 | 0.6 | 8.3 | 86.3 | NA | 37 | NA | 4 | 5 | RAEB-1 (MSD-EB-1) |
46,XY,+1,der(1;15) (q10;q10),add(17)(p11.2) 46,idem,add(17) |
*Affected siblings in Family4.
†Affected siblings in Family6.
N1254 (Family4), the first daughter of nonconsanguineous Japanese parents, was born at 39 weeks and 3 days with a birth weight of 2880 g (−0.12 SD), after an uneventful antenatal period. She had neither obvious malformations nor abnormalities at birth. At 3 weeks of age, she had poor weight gain (Kaup index of 13) and presented with telecanthus and broad nasal tip. At 1 year of age, she was repeatedly admitted to the hospital because of prolonged fever and pancytopenia. Skin hyperpigmentation and displacement of the left third toe were pointed out. She was initially diagnosed with RA. At 2 years of age, she had severe growth retardation (height: 71 cm, −2.3 SD; weight: 6850 g, −2.7 SD), delayed motor and language development (spoke only two or three meaningful words), and was diagnosed with acute myeloid leukemia (AML; monosomy 7). She received a cord blood stem cell transplant from an unrelated donor. She had successful engraftment of neutrophils, but the thrombocyte levels did not return to normal. She had leukoencephalopathy and cerebral abscess and died at 2 years and 10 months of age. She had no episode of sunburn.
N1037 (Family4), a younger sister of N1254, was born at 41 weeks and 3 days with a birth weight of 2870 g (−1.27 SD), after an uneventful antenatal period. She had neither obvious malformations nor abnormalities at birth. At 1 year of age, she was repeatedly admitted to the hospital because of prolonged fever and thrombocytopenia. At 5 years of age, she was initially diagnosed with possible BS based on her facial characteristics and hematological abnormalities. She presented with short stature, microcephaly, delayed motor and language developments (spoke only two or three meaningful words), analgia, hypohidrosis, hypothyroidism, and displacement of the left third toe. She died of interstitial pneumonia at 9 years and 8 months of age. She had no episode of sunburn.
N1267 (Family5), the second child of nonconsanguineous Japanese parents, was born at 40 weeks and 5 days with a birth weight of 2606 g (−1.81 SD), after an uneventful antenatal period. No physical abnormalities were noted at birth. She was undergoing medical follow-up care because of low birth weight, failure to thrive, and short stature. She presented with pancytopenia at 7 years of age. She was initially diagnosed with DS. Bone marrow examination showed trilineage dysplasia with an abnormal karyotype: 46,XX,+1,der(1:21)(q10;q10) [7/20]; 46,idem,add(18)(p11.2) [13/20]. Regular bone marrow examination performed 1 year after the diagnosis revealed monosomy 7 clonal evolution. BMT from a human leukocyte antigen (HLA)–matched sibling donor (sibling5-1) appeared to be successful; however, the disease relapsed 3 months after the transplant. She underwent her second BMT from an HLA-matched unrelated donor. She is alive and disease-free 5 years after the transplant.
N1269 (Family6), the second daughter of nonconsanguineous Japanese parents, was born at 39 weeks and 2 days with a birth weight of 2442 g (−1.72 SD), after an uneventful antenatal period. No physical abnormalities were noted at birth. At 2 years of age, she presented with thrombocytopenia, short stature, and developmental delay. Following a transient elevation in platelet count, her thrombocytopenia and anemia subsequently progressed and she was diagnosed with myelodysplastic syndrome (MDS) with trilineage dysplasia at 3 years of age. Cytogenetic analysis revealed a complex karyotype with trisomy 8: 46,XX,der(5;17)(p10;q10),+8 [4/20]; 45,idem,add(7)(p11.2),-8,add(19)(p13) [14/20]; 46,XX,ins(1;?)(q12;?) [2/20]. BMT from an HLA-matched sibling donor was successful, and she is alive 6 years after the transplant.
N1270 (Family6), a younger brother of N1269, was born at 39 weeks and 0 days with a birth weight of 2366 g (−2.14 SD), after an uneventful antenatal period. No physical abnormalities were noted at birth. At 3 months of age, he was admitted to the hospital because of poor weight gain and possible developmental abnormalities. He had bicytopenia (thrombocytopenia and anemia), hypothyroidism, skin hyperpigmentation, agenesis of corpus callosal, and recurrent epileptic seizures. Bone marrow examination showed MDS with a normal karyotype. His motor and intellectual disabilities were severe. He needed a gastrostomy tube for feeding at 1 year and 8 months of age. He died of infection at 2 years of age: height, 76.7 cm (−4.5 SD); weight, 8.85 kg (−3.0 SD); and head circumference, 40 cm (−5.8 SD). Telecanthus, displacement of the right fourth toe, and low-set ears were pointed out. He had hypertrophic cardiomyopathy and frontal lobe atrophy.
N1275 (Family7), the first daughter of nonconsanguineous Japanese parents, was born at 41 weeks and 5 days with a birth weight of 2984 g (−0.65 SD). She had initially presented with pancytopenia at 8 years of age. Bone marrow examination revealed hypoplastic MDS with the following abnormal karyotype: 46,XX,+1,der(1;22) (q10;q10) [2/20]; 47,idem,del(7)(q?),add(17)(p11.2),+mar1 [9/20]; 47,idem,add(17),del(20)(q1?),+mar1 [5/20]. She presented with FA-like physical anomalies, such as short stature, skin hyperpigmentation, and developmental delay. However, her chromosomal breakage test and FANCD2 ubiquitination were normal. BMT from a mismatched unrelated donor with reduced intensity conditioning was successful. She is alive 5 years after transplant.
N1329 (Family8), the second son of nonconsanguineous Japanese parents, was born at around 40 weeks with a birth weight of 3480 g (0.72 SD), after an uneventful antenatal period. He presented with short stature (−2.52 SD) and had a delayed bone age at 6 years of age. He had Tanner stage 4 and an advanced bone age and was diagnosed with precocious puberty at 10 years of age. He had bicytopenia (anemia and leukopenia), and his bone marrow examination revealed MDS with an abnormal karyotype: 46,XY,+1,der(1;15)(q10;q10),add(17)(p11.2)x2 at 12 years of age. He was initially diagnosed with FA. At 15 years of age, he had short stature (height: 149.7 cm, −3.2 SD; weight: 31.35 kg, −2.7 SD), microcephaly (head circumference: 51.5 cm, −4.7 SD), and intellectual disability. He has no episode of sunburn.
Digenic mutations in the ADH5 and ALDH2 genes cause AMeDS
We have implemented whole-exome sequencing (WES) for genetic screening of undiagnosed cases (standard WES procedure, see Materials and Methods). From the WES and follow-up studies, we identified biallelic mutations in the ADH5 gene in all of the AMeDS cases. By the WES analyses, we did not find any other potential causative genes shared among more than two of the cases under an autosomal recessive model of inheritance; we were not aware of any reported pathogenic variants of known disorders in any of the identified potential causative genes; no biallelic variants were detected in known FA-associated genes (table S1) (18). The patients were homozygous or compound heterozygous for the following ADH5 variant alleles: c.966delG, p.W322*; c.G832C, p.A278P; c.564+1G>A, 5′ splice site (Fig. 1B and Table 1). Immunoblot analysis of AMeDS fibroblasts demonstrated a significant reduction of the ADH5 protein levels, indicating that the identified variants led to loss-of-function (LOF) changes causing a lack of gene expression or involving a severe protein destabilization (Fig. 1, C and D).
Previous animal studies demonstrated that combined inactivation of the endogenous aldehydes detoxification and the FA pathway leads to very severe attrition of hematopoietic stem and progenitor cells (HSPCs) and abnormal fetal development (26–30). In these processes, ADH5 is the key enzyme in the protection against DNA damage induction, by eliminating endogenous formaldehyde (30). Consistent with this notion, ADH5-deficient AMeDS cells exhibited increased sensitivity to formaldehyde treatment (Fig. 1E), although the cells displayed normal ubiquitination of FANCD2 and resistance to ICL-inducing mitomycin C, indicating that the FA pathway is proficient in the AMeDS cases (fig. S1, A to C). From these results, we anticipated that the ADH5 deficiency was the primary cause of AMeDS.
In contrary, Adh5 null mice did not show any devastating phenotype that causes a survival disadvantage (31). To further evaluate the pathogenicity of the ADH5 deficiency in humans, we first searched for individuals with biallelic ADH5 rare variants in genotype-available databases. No homozygous ADH5 LOF variants were detected within ~140,000 individuals in gnomAD (v.2.1.1). Because all the AMeDS cases were of Japanese origin, we conducted an additional search of ~5600 Japanese individuals within ToMMo (Tohoku Medical Megabank; 2036 individuals), HGVD (Human Genetic Variation Database, Kyoto University; 300 individuals), BBJ (BioBank Japan, RIKEN Institute; 1006 individuals), and Nagahama Study (Nagahama Prospective Cohort for Comprehensive Human Bioscience, Kyoto University; 1321 individuals) datasets, as well as in-house genome databases. Furthermore, we genotyped the ADH5 pathogenic variant alleles (p.W322*, p.A278P, and c.564+1G>A) in ~26,000 Japanese individuals (Hospital-Based Epidemiologic Research Program at Aichi Cancer Center, Aichi Cancer Center). From these screenings, we identified a healthy individual (female, age 55, no preexisting conditions) with a homozygous mutation, c.G224A (p.S75N) in the ADH5 gene (NAG16714 in Nagahama Study; Table 1). As we could not obtain cellular materials from this individual, we generated hTERT-immortalized RPE1 (RPE1 hTERT) and U2OS cells with ∆ADH5 and with the site-specific ADH5 p.S75N homozygous mutation using the CRISPR-Cas9–based gene editing technique (without silent mutations, see Materials and Methods; table S2). Immunoblot analysis revealed severely decreased levels of the ADH5-p.S75N protein in the mutant cells (Fig. 1F and fig. S1D), although the ADH5 mRNA expression was unaffected (Fig. 1G). The ADH5-p.S75N mutant cells were as sensitive to formaldehyde as ∆ADH5 cells (Fig. 1H and fig. S1E) due to destabilization of the ADH5 protein (Fig. 1, I and J). These initial results indicate that the loss of ADH5 expression or deficiency in formaldehyde detoxification is not associated with any obvious disease phenotype. These data suggest that the ADH5 monogenic deficiency is not sufficient to cause AMeDS.
We therefore considered a possibility of digenic/oligogenic inheritance. We focused on the ALDH2 gene and rs671 because ALDH2 retains a weak catabolic activity for formaldehyde (32) and for various endogenous active aldehyde species, such as 4-hydroxy-2-nonenal (4-HNE) that arises from membrane lipid peroxidation products (33), in addition to its primary function of detoxifying acetaldehyde. These active aldehydes generate ICL-DNA damage (17); consequently, they can put loads on the FA pathway and other DNA repair pathways, such as base excision repair and DPC repair. Therefore, the phenotypes of patients with AMeDS may result from the lack of enzymatic activity of ALDH2 in combination with the loss of ADH5 function for endogenous aldehydes.
Individuals harboring either one or two copies of the ALDH2 rs671 defective allele display a severe deficiency in acetaldehyde catabolic activity because the active enzyme complex requires the wild-type ALDH2 homotetramer (5, 34). By examining the ALDH2 rs671 genotype, we indeed found that all 10 patients with AMeDS carry at least one copy of the defective allele (G/A or A/A) (Table 1). Despite the high allele frequency in Japanese population, appearance of the rs671 defective allele in the AMeDS cases deviates substantially from the Hardy-Weinberg equilibrium (Pearson’s χ2 test; P = 0.0007), suggesting that this locus is strongly associated with the disease development. The healthy individual, NAG16714 with the homozygous ADH5 p.S75N defective variant, harbors the homozygous rs671 wild-type (G/G) alleles (Table 1). Furthermore, all three cases homozygous for the rs671 defective alleles (N1037, N1254, and N1270) manifested more severe phenotypes, including neurological abnormalities, prominent motor deterioration (confined to a wheelchair or bed), and early death (Tables 1 and 2). This suggests that the aldehyde detoxification activity determined by the rs671 genotype underlies the severity of AMeDS clinical features. Collectively, we conclude that the ALDH2 rs671 defective allele in combination with biallelic LOF mutations in the ADH5 gene is necessary and sufficient to cause a true digenic disorder, AMeDS, classified as IBMFS.
ALDH2 and ADH5 may cope with the formaldehyde clearance and prevent induction of DNA damage
To determine potential substrates of the ADH5 and ALDH2 enzymes, we have measured growth inhibition profiles of ADH5- and/or ALDH2-deficient U2OS cells after treatments with various active aldehydes (fig. S2, A to C). Nine major endogenous aldehydes—including α,β-unsaturated aldehydes [4-hydroxyhexenal, (4-HHE), 4-HNE, 4-oxononenal, acrolein, and crotonaldehyde], the simplest aldehyde (formaldehyde), dialdehydes (glyoxal and methylglyoxal), and a saturated aldehyde (heptanal)—whose chemical properties have been widely studied, were chosen for the proliferation assay.
While the treatments with α,β-unsaturated aldehydes (4-HHE, 4-HNE, and acrolein) inhibited cell proliferation of ALDH2E504K U2OS cells, ADH5−/− cells were not affected by the same treatment (fig. S2D); this may imply that these aldehydes are preferentially metabolized and detoxified by the ALDH2 enzyme. We found that formaldehyde and methylglyoxal treatments suppressed cell proliferation of ADH5−/−ALDH2E504K double-deficient U2OS cells compared to single-deficient ADH5−/− or ALDH2E504K cells, indicating that these aldehydes are possible substrates of both ADH5 and ALDH2 enzymes (fig. S2D). The concentration of formaldehyde in human plasma is estimated to be ~100 μM (35, 36), while that of methylglyoxal is much less than 1 μM (37), which implies that near physiological levels of formaldehyde, but not methylglyoxal, can perturb cell proliferation. To further investigate the effects of formaldehyde treatment in ADH5−/−ALDH2E504K double-deficient U2OS cells more precisely, we analyzed replication inhibitory profiles by flow cytometry (Fig. 2A). While either LOF of ADH5 or ALDH2 modestly attenuated the progression of cell cycle compared to wild-type cells, digenic loss of ADH5 and ALDH2 led to the significant inhibition of DNA replication after formaldehyde treatment (Fig. 2, A and B).
Fig. 2. Formaldehyde is a possible endogenous aldehyde metabolized by both ADH5 and ALDH2.
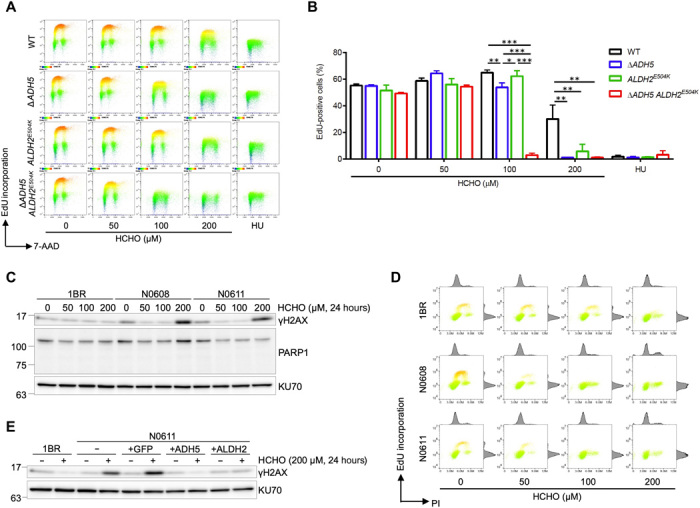
(A) Formaldehyde treatment inhibits DNA replication in ADH5 and ALDH2 double-deficient cells. 5-ethynyl-2′-deoxyuridine (EdU) incorporation in WT, ∆ADH5, ALDH2E504K, or ∆ADH5 ALDH2E504K double-mutant U2OS cells after formaldehyde treatment is measured by fluorescence-activated cell sorting (FACS) analysis. Cells were incubated with indicated concentration of formaldehyde or 10 mM hydroxyurea (HU) as a positive control for 8 hours followed by EdU incorporation for 1 hour. Then, cells were fixed with 70% ethanol and analyzed by FACS for Alexa Fluor 488–labeled EdU and DNA stained with 7-aminoactinomycin D (7-AAD). Representative FACS images are shown. (B) Quantification of data in (A). Graph shows the percentage of EdU-positive cells. Means (± SD) from three independent experiments are shown. *P < 0.05, **P < 0.01, and ***P < 0.001, one-way ANOVA with Tukey’s multiple comparisons test. (C) Formaldehyde treatment induces DNA damage in cells from AMeDS-affected individuals. Immunoblots showing phospho-Ser139 histone H2AX (γH2AX), a DNA damage marker, and PARP1, an apoptosis marker in normal (1BR) and AMeDS (N0608 and N0611) cells. KU70 is a loading control. (D) EdU incorporation in normal and AMeDS cells after formaldehyde treatment measured by FACS analysis. Cells were incubated with indicated concentration of formaldehyde for 22 hours followed by EdU incorporation for 2 hours. Then, cells were fixed with 70% ethanol and analyzed by FACS for Alexa Fluor 488–labeled EdU and DNA stained with propidium iodide (PI). (E) Formaldehyde-induced DNA damage in AMeDS cells (N0611) is ameliorated with expression of either the wild-type ADH5 or ALDH2 cDNA. Green fluorescent protein as a mock control.
We next studied the cooperative actions of ADH5 and ALDH2 on the prevention of DNA damage induction. We assessed increased cellular DNA damage levels as a consequence of diminished formaldehyde processing activity in patients with AMeDS cells. We measured formaldehyde-induced DNA damage by immunoblotting of histone H2AX Ser139 phosphorylation (γH2AX) as a DNA damage marker. AMeDS cells (N0608 and N0611) showed significant increase of γH2AX levels after 200 μM formaldehyde treatment, although normal cells were resistant to even such a high dose, indicating that unrepairable DNA damage are indeed increased in the AMeDS cells supposedly because of the lack of formaldehyde processing capacity (Fig. 2C). Similar to ADH5−/−ALDH2E504K double-deficient U2OS cells, significant inhibition of DNA replication after formaldehyde treatment was also confirmed in patient with AMeDS cells (Fig. 2D). Ectopic expression of either of the wild-type ADH5 or ALDH2 complementary DNA (cDNA) in the AMeDS cells completely eliminated the induction of formaldehyde-induced DNA damage, suggesting that both ADH5 and ALDH2 deficiencies underlie the decrease of formaldehyde detoxification capacity (Fig. 2E and fig. S2, E and F). Together, formaldehyde is metabolized by both ADH5 and ALDH2, and even naturally occurring concentration of formaldehyde may have a negative effect on cell proliferation and genome integrity in ADH5 and ALDH2 double-deficient cells.
ADH5 and ALDH2 enzymatic activities are crucial for the normal differentiation and proliferation of HSPCs in humans
We next investigated the effects of ADH5 and ALDH2 digenic deficiency on the progenitor cell capacity of HSPCs in humans. We performed colony-forming unit (CFU) assays of CD34+ umbilical cord blood–derived HSPCs, which were prepared from healthy Japanese donors (RIKEN BRC). The ALDH2 rs671 genotype was confirmed in each HSPC pool, and the ADH5 expression was eliminated by the CRISPR-Cas9–based gene editing with specific single-guide RNAs (sgRNAs) (Fig. 3A and fig. S3, A and B). The loss of ADH5 did not induce unexpected DNA damage (determined by γH2AX induction shown in fig. S3B), and it did not affect the proliferation of HSPCs regardless of the rs671 genotype during culture in hematopoietic maintenance medium (fig. S3C), suggesting that decrease in the formaldehyde detoxification capacity does not involve any growth disadvantage of HSPCs in ex vivo conditions. However, the differentiation and proliferation potential of HSPCs was severely compromised when ADH5 was deleted in HSPCs with the ALDH2 rs671 defective (G/A) but not with the wild-type (G/G) alleles (Fig. 3, B and C). In addition, these ADH5 and ALDH2 rs671 double-deficient HSPCs had reduced capacity to differentiate into common progenitor cells and/or their progeny cells (fig. S3D), suggesting that formaldehyde detoxification deficiency causes a wide range of hematopoietic abnormalities in humans.
Fig. 3. Deletion of ADH5 attenuates differentiation and proliferation capacity of CD34+ HSPCs harboring the ALDH2 rs671 allele.
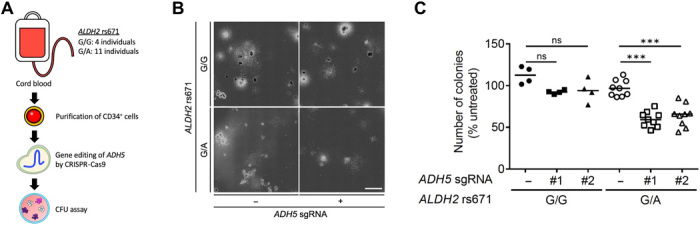
(A) Schematic representation of CFU assay. CD34+ HSPCs are derived from umbilical cord blood of Japanese healthy donors. The numbers of HSPC pools with the designated ALDH2 rs671 alleles are shown. ADH5 was deleted in each HSPC pool by CRISPR-Cas9–based gene editing. (B) CFU assay of gene-edited CD34+ HSPCs was performed using a methylcellulose medium. Representative images are shown. Scale bar, 3 mm. (C) Total number of colonies after 14-day CFU assay of gene-edited CD34+ HSPCs. The number of colonies was normalized to untreated control. Statistical analysis was performed using one-way ANOVA with Tukey’s multiple comparisons test (***P < 0.001; ns, not significant). Lines represent median.
Mice with the Adh5−/− and Aldh2-E506K digenic mutations recapitulate key clinical features of AMeDS
To investigate the consequences of the ADH5 and ALDH2 digenic deficiency for the development of multisystem abnormalities in AMeDS, we generated gene-edited mice with Adh5−/− and Aldh2-E506K (equivalent to the human ALDH2-E504K and hereafter called Aldh2-KI) double mutation using the CRISPR-Cas9 technique. Adh5+/−Aldh2+/KI female mice were interbred with Adh5+/−Aldh2KI/KI male mice, and the offspring genotypes were measured. Adh5−/−Aldh2KI/KI mice were born at near Mendelian ratios (Fig. 4A). The weight and size of the Adh5−/−Aldh2KI/KI double-deficient neonates were indistinguishable from those of their littermates, indicating of no prenatal growth retardation, which is similar to human AMeDS cases (Fig. 4, B and C). Severe growth failure with poor weight gain was prominent in all of the Adh5−/−Aldh2KI/KI mice at 1 to 2 weeks after birth (Fig. 4, B and C); computed tomography (CT) and dissection analyses also revealed multisystem abnormalities, including small body size, extremely shrunken organs, diminished muscle and subcutaneous fat volumes, and kyphosis at 3 weeks after birth (fig. S4A), although all the animals received breast-feeding from their mothers without problem. Intriguingly, all the Adh5−/−Aldh2KI/KI mice displayed anemia and severe debility and eventually died, possibly due to cachexia or overall weakness, within 4 weeks after birth before weaning, although Adh5−/−Aldh2+/KI mice or mice with other genotypes did not show any survival disadvantage during this period (Fig. 4D). Notably, Adh5−/− or Aldh2KI/KI animals did not show any obvious developmental defects, which is consistent with previous reports (31, 38).
Fig. 4. Loss of Adh5 function in combination with reduced Aldh2 activity recapitulates the phenotype of AMeDS in mice.

(A) Observed and expected frequencies of mice at 2 weeks of age from intercrossed of Adh5+/−Aldh2+/KI female mice with Adh5+/−Aldh2KI/KI male mice. Chi-square test shows no significant difference between observed and expected (P = 0.17). (B) Postnatal growth defects of Adh5−/−Aldh2KI/KI mice. Representative pictures are shown. Blue arrows indicate Adh5−/−Aldh2KI/KI mice. Photo credit: Yasuyoshi Oka, Nagoya University. (C) Body weights of individual mice at 0 days, 2 weeks, or 6 weeks of age. **P < 0.01 and ***P < 0.001, one-way ANOVA with Tukey’s multiple comparisons test. (D) Kaplan-Meier curves with log-rank (Mantel-Cox) test show a significant decrease in survival of Adh5−/−Aldh2KI/KI compared to the mice with other genotypes (P < 0.0001). (E) Quantification of nucleated bone marrow cells in bilateral femurs and tibias from 3-week-old mice is shown (means ± SD; n = at least 5 animals). *P < 0.05 and ***P < 0.001; one-way ANOVA with Tukey’s multiple comparisons test. (F and G) Quantification of hematopoietic subset: LKS (Lin−c-Kit+Sca-1+), HSC (Lin−c-Kit+Sca-1+CD150+CD48−), MPP (Lin−c-Kit+Sca-1+CD150−CD48−), HPC1 (Lin−c-Kit+Sca-1+CD150−CD48+), HPC2 (Lin−c-Kit+Sca-1+CD150+CD48+), CLP (Lin−c-KitlowSca-1lowCD127+CD135+), CMP (Lin−c-Kit+Sca-1−CD34+CD16/32−), MEP (Lin−c-Kit+Sca-1−CD34−CD16/32−), and GMP (Lin−c-Kit+Sca-1−CD34+CD16/32+) in individual mice at 3 weeks of age in (F) and at 8 to 9 months of age in (G). Means ± SD; n = at least 5 animals. *P < 0.05, **P < 0.01, and ***P < 0.001, one-way ANOVA with Tukey’s multiple comparisons test.
Adh5−/−Aldh2+/KI mice also displayed smaller body weight compared to that of Adh5−/− or Aldh2KI/KI animals from 2 to 6 weeks after birth (Fig. 4C). Similar but much milder manifestations compared to the Adh5−/−Aldh2KI/KI mice were also detected in the Adh5−/−Aldh2+/KI animals at 6 months (fig. S4B). Furthermore, we noticed that all middle-aged Adh5−/−Aldh2+/KI animals (8 to 9 months) displayed skin hyperpigmentation on the tails, indicative of FA-like features (fig. S4C). A previous report described a skin hyperpigmentation induced in Aldh2−/− mice continuously administrated with ethanol (acetaldehyde precursor) (39). The Adh5−/−Aldh2+/KI male and female animals were fertile.
To study hematopoietic functions of the Adh5 and Aldh2 double-deficient mice in detail, we examined peripheral blood hematological parameters. Adh5−/−Aldh2KI/KI mice at the moribund stage (3 weeks of age) exhibited decreased red blood cells (RBCs), hemoglobin (HGB) levels, hematocrit (HCT) values, and increased levels of mean corpuscular volume (MCV), indicating macrocytic anemia, although age-matched Adh5−/−Aldh2+/KI mice did not present apparent hematopoietic defects at this point (fig. S4D). The Adh5−/−Aldh2+/KI mice eventually displayed similar abnormalities of erythrocytes at 8 to 9 months after birth (fig. S4E).
We further investigated the maintenance of HSPCs in the Adh5 and Aldh2 double-deficient mice. In Adh5−/−Aldh2KI/KI mice at the moribund stage (3 weeks of age), total number of nucleated bone marrow cells from tibiae and femora significantly decreased compared with that of other animals (Fig. 4E). Consistently, the number of multipotent self-renewing HSCs defined by Lin−c-Kit+Sca-1+ CD150+CD48− (CD150+ long-term HSCs) was significantly reduced in Adh5−/−Aldh2KI/KI mice (Fig. 4F and fig. S4I); similar trends were observed for immature hematopoietic progenitors, including Lin−c-Kit+Sca-1+CD150−CD48− [CD150− multipotent progenitors (MPPs)] and Lin−c-Kit+Sca-1+CD150−CD48+ [CD48+ restricted progenitors (HPC1)] cells. We found that the number of further differentiated progenitor cells—including Lin−c-KitlowSca-1low CD127+CD135+ [common lymphoid progenitors (CLPs)], Lin−c-Kit+ Sca-1−CD34+CD16/32− [CD34+ common myeloid progenitors (CMPs)], and Lin−c-Kit+Sca-1−CD34−CD16/32− [bipotent megakaryocyte/erythrocyte lineage-restricted progenitors (MEPs)]—was also significantly diminished in Adh5−/−Aldh2KI/KI mice. In connection with the decrease of CLPs in Adh5−/−Aldh2KI/KI mice, the number of lymphocytes in peripheral blood and the weights of thymus and spleen were reduced without any significant alteration of lymphocyte distribution in these organs (fig. S4, F to H). Although Adh5−/−Aldh2+/KI mice did not show any anomaly of bone marrow cells at 3 weeks of age (Fig. 4F), the number of MPPs and CLPs was significantly decreased compared with that of Adh5 or Aldh2 single-deficient mice at 8 to 9 months of age (Fig. 4G). Collectively, hematopoietic abnormalities in the Adh5 and Aldh2 double-deficient animals are due to exhaustion of HSPCs. These findings demonstrate that the combined LOFs in the Adh5 and Aldh2 genes cause multisystem abnormalities potentially due to the lack of formaldehyde clearance capacity in the double-deficient animals, and the Aldh2-E506K allele defines the severities of manifestations, which clearly mimic the major clinical features of AMeDS in humans.
DISCUSSION
Digenic inheritance (DI) is the simplest genetic trait describing complex oligogenic disorders caused by the malfunctions of two or more genes (13–15). In the past human genetics studies, thousands of monogenic disorders have been identified. However, to date, only tens of diseases with solid evidence for DI have been reported (13–15). Moreover, even in well-documented DI disorders, such as retinitis pigmentosa and Bardet-Biedl syndrome, affected individuals often display heterogeneous clinical features because of incomplete penetrance, and unexplained pedigrees are frequently observed (40, 41). Our AMeDS cases all develop generally uniform clinical symptoms (AA, mental retardation, and dwarfism), and they are genetically characterized by true DI, i.e., mutations in two distinct genes are necessary and sufficient to cause a disease, with no exception. The ALDH2 rs671 defective allele is also involved in the severity of AMeDS clinical features; however, unlike other coinheriting genetic modifiers, it is essential for the disease development in addition with the ADH5 deficiency.
The accumulation of unrepaired endogenous DNA damage ultimately triggers cancer and aging through a failure in the essential functions of various cellular processes (42–44). DNA lesions may induce mutations and chromosomal aberrations that cause genome instability and an increased risk of cancer. In parallel, major DNA lesions can also interfere with transcription and replication, resulting in the loss of accurate gene expression profiles, delay of cell cycle progression, and induction of cell death, which contribute to aging. The cellular defense against DNA damage involves serial mechanisms (two-tier protection): (tier 1) enzymatic detoxification processes of highly reactive genotoxic chemicals, such as reactive oxygen species (ROS) and active aldehydes, and (tier 2) DNA repair processes to eliminate various types of DNA damage and restore genetic information. In this regard, dysfunctions in either of these mechanisms may result in carcinogenesis and aging-related phenotypes. In particular, cancer predisposition and progeroid symptoms are naturally observed in a variety of human genetic disorders due to mutations in DNA repair genes (tier 2) (45). In contrast, there is only a small number of congenital diseases that are caused by abnormalities in the detoxification systems of chemical compounds that induce DNA damage (tier 1). Recent clinical reports have shown that a complete absence of the superoxide dismutase 1 (SOD1) enzyme, which is involved in the removal of ROS, causes an extreme oxygen sensitivity in patient’s cells and is associated with autosomal recessive progressive spastic tetraplegia and axial hypotonia (STAHP), characterized by severe and progressive psychomotor retardation in humans (46, 47). Note that mutations in the SOD1 gene usually cause autosomal dominant amyotrophic lateral sclerosis because of the toxic effects of protein aggregation rather than by the loss of enzymatic activity (48). The STAHP phenotype with the lack of SOD1 may be due to an overload of oxidative DNA damage in the single-strand break (SSB) repair pathway; this postulation is corroborated by a report that SSB repair deficiency by the XRCC1 gene mutation causes spinocerebellar ataxia (SCA) (49). Our AMeDS cases and the animal model further support the idea that malfunctions in the detoxification systems of active genotoxic compounds cause symptoms of cancer predisposition and accelerated aging.
Patients with AMeDS display many characteristic clinical features that overlap with other DNA repair deficiency disorders (table S3). Here, we propose that FA-like hematopoietic abnormalities observed in AMeDS may result from the overload of the FA pathway (ICL repair pathway) due to limitations of cellular detoxification properties against endogenous formaldehyde. During differentiation including hematopoiesis, various histone demethylases erase methyl marks on lysine residues of histones associated with gene regulation, leading to the release of active formaldehyde (50). Under a limited capacity of the FA pathway, the rs671 defective allele would significantly contribute to the increase of unrepaired formaldehyde-induced DNA lesions during hematopoiesis. This idea is consistent with previous reports that rs671 is a genetic modifier of the severity of BMF in Japanese FA cases (51), as well as in children with sporadic AA (52); this is also true for the FA pathway–proficient AMeDS cases, as rs671 genotype defines the severity of AMeDS clinical features. AMeDS cases display premalignant MDS or leukemia, indicative of cancer predisposition, although no solid tumor is present at the moment. Adulthood patients with FA commonly develop solid tumors including head and neck squamous cell carcinoma, in addition to MDS and leukemia (53). Because the ages of AMeDS cases with clinical records range from 2 to 16 years, follow-up studies are necessary to investigate the etiology of cancers. On the other hand, in canonical FA, severe dwarfism and neurological abnormalities, as well as psychomotor retardation, are uncommon. In this respect, AMeDS clinical features have substantial similarities to those of segmental progeroid disorders, RJALS and CS (and its severe forms, COFS and XFEPS). Since RJALS cells with mutations in the SPRTN gene display hypersensitivity to DPC-inducing chemicals, including formaldehyde (54), the phenotypes of AMeDS that overlap with RJALS could be explained from the overload of DPC repair pathway. From these perspectives, the severe phenotypes of AMeDS may be due to a combined failure of multiple DNA repair processes as represented by BRCA1 (FANCS)– or XPF (FANCQ)–deficient atypical FA cases, as well as by patients with ERCC1/XPF-deficient COFS/XFEPS (25, 55–57), because BRCA1 and the ERCC1/XPF complex, respectively, contribute to DNA double-strand break repair and NER, together with the FA pathway. ADH5 and ALDH2 double deficiency would also induce various types of DNA damage apart from ICL and DPC, such as simple aldehyde base adducts, which can be repaired independently of the ICL and DPC repair pathways (58–60). Consequently, in AMeDS cases, these synergistic effects may trigger the severe clinical features. Likewise, CS-like clinical features observed in AMeDS rather suggest an additional failure in TCR; further analyses will address this possibility. In conclusion, our data propose that the combined deficiency in formaldehyde metabolic processes, by harboring the prevalent polymorphism rs671 in ALDH2 together with biallelic mutations in ADH5, overburdens the multiple DNA repair pathways and leads to a true digenic disorder, AMeDS, which is similar both in the clinical features and molecular pathogenesis but distinct from other DNA repair deficiency disorders.
MATERIALS AND METHODS
Human studies
Affected individuals and normal control samples were obtained with local ethical approvals (the Ethics Committee for Human Genome Studies in Research Institute of Environmental Medicine, Nagoya University; the ethics committee of the Nagoya University Graduate School of Medicine). Written informed consent was obtained from the patients.
Exome sequencing
Next-generation sequencing (NGS) was performed in-house or by macrogen. Genomic DNA of the individuals was enriched by using the Agilent SureSelect Human All Exon Kit version 5/6 (Agilent, Santa Clara, CA, USA). The captured genomic fragments were sequenced on the Illumina HiSeq 2500 sequencer (Illumina, San Diego CA, USA) using paired-end (PE) flow cells to obtain 100– to 150–base pair PE reads of 100× to 200× coverage.
Bioinformatic analysis of the exome data
The NGS data were analyzed by our standard exome pipeline. Briefly, low-quality reads were trimmed out by Trimmomatic (version 3.36) (61). The reads were then aligned to the human reference genome (GRC h37/hg19) with the Burrows-Wheeler Aligner (version 0.7.12-r1039) (https://arxiv.org/abs/1303.3997). Duplicate reads were removed using Biobambam2 (version 2.0.72) (doi: 10.1186/1751-0473-9-13). The aligned reads were locally realigned, and base quality scores were recalibrated using the IndelRealigner and BaseRecalibrator programs in Genome Analysis Toolkit (GATK; version 3.5) (62). Single-nucleotide variants were identified by the HaplotypeCaller program in GATK. All the variants were annotated with ANNOVAR (63) based on the GENCODE release 19 (GRCh37.p13). To further determine potentially pathogenic changes, commonly observed variants (MAF > 0.01) were excluded using public databases and functionally significant changes were extracted. According to an autosomal recessive inheritance model, genes that carried homozygous or compound heterozygous changes were determined. We considered 2 to 18 potential causative genes in each of the affected individuals (table S1), and we identified ADH5 as only pathogenic candidate gene shared among any subset of the affected individuals.
Cell lines and culture
The following cell lines were used in this study: U2OS; RPE1 hTERT; HEK293 (human embryonic kidney–293), immortalized normal human embryonic kidney cells; 1BR, normal human primary fibroblast; and FA20P, primary fibroblast from an FA-A individual. N0608, N0611, and N0614 were obtained from JCRB Cell Bank. All cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Wako) supplemented with 10% fetal bovine serum (FBS; Invitrogen) and antibiotics, unless otherwise noted. Mycoplasma testing was performed routinely.
Genome editing using CRISPR-Cas9 technology
For plasmid-based genome editing experiments, a guide RNA (gRNA) coding sequence was cloned into the pX459 vector. The designated plasmid was transfected into U2OS cells using X-tremeGENE HP DNA Transfection Reagent (Merck). Cells were selected for 48 hours with puromycin (1 μg/ml) in DMEM with 10% FBS. Single clones were isolated by limiting dilution. For ribonucleoprotein-based genome editing experiments, HiFi Cas9 Nuclease V3 (Integrated DNA Technologies) was mixed with crRNA (CRISPR RNA):tracrRNA (trans-activating CRISPR RNA) complex and single-stranded oligodeoxynucleotide (ssODN). The mixture was electroporated into U2OS or RPE1 hTERT cells using 4D-Nucleofector (Lonza). Cells were recovered by DMEM with 10% FBS and cultured on a 35-mm dish for 24 hours. Single-cell clones were isolated using a limiting dilution in 96-well plates. All gRNA and ssODN sequence information are listed in table S2.
Confirmation of accurate gene editing and indel frequencies
Genomic DNA was extracted from gene-edited cells using the MightyAmp Genotyping Kit (Takara) according to the manufacturer’s instruction. Mutations and indel frequencies of gene-edited cells (CD34+ HSPCs, U2OS cells, RPE1 hTERT cells, and mice) were confirmed by Sanger sequencing and TIDE (tracking of indels by decomposition) analysis (64). Untreated cells were always used as a negative control for calculating indel frequencies with TIDE. All primer sequence information are listed in table S2.
Quantitative reverse-transcription polymerase chain reaction
Total RNA was isolated using an RNeasy mini kit (Qiagen), according to the manufacturer’s instructions, and cDNA was generated with SuperScript IV (Thermo Fisher Scientific), according to the manufacturer’s instructions. The quantitative reverse transcription polymerase chain reaction (RT-qPCR) was performed using LightCycler 96 System (Roche). For the detection of target genes, SYBR green (Qiagen) was used according to the manufacturer’s instructions. Expression of mRNAs was quantitated using the following set of primers: ADH5 (forward, 5′-CCAGCACATTTTCTGAATACAC-3′; reverse, 5′-ACCAAAGACGGCACAAAC-3′) and ACTB (forward, 5′-TCACCCACACTGTGCCCATCTACGA-3′; reverse, 5′-CAGCGGAACCGCTCATTGCCAATGG-3′). The LightCycler was programmed to run an initial heat-denaturing step at 95°C for 15 min, 45 cycles at 94°C for 15 s, an annealing step for 20 s at 58°C, and an extension step for 10 s at 72°C coupled with fluorescence measurements. Following amplification, melting curves of the PCR products were monitored from 65° to 97°C to determine the specificity of amplification. Each sample was run in triplicate, and expression of target genes was normalized against ACTB.
Immunochemical methods
Cells were lysed in EBC buffer [50 mM tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 0.5% NP-40, and 1 mM dithiothreitol (DTT)] or denaturing buffer [20 mM tris (pH 7.5), 50 mM NaCl, 1 mM EDTA, 0.5% NP-40, 0.5% SDS, 0.5% sodium deoxycholate, and 1 mM DTT] supplemented with protease inhibitor cocktail (Roche) and phosphatase inhibitor cocktail (Nacalai Tesque) and incubated on ice, cleared by centrifugation. Purified proteins were resolved by 6, 12.5, or 5 to 20% gradient SDS–polyacrylamide gel electrophoresis. Resolved protein samples were transferred to polyvinylidene difluoride membrane for immunodetection. Antibodies used for immunochemical experiments were as follows: rabbit monoclonal anti-ADH5 (ab174283, Abcam), rabbit polyclonal anti-ADH5 (HPA044578, Atlas Antibodies), mouse monoclonal anti-ALDH2 (MA5-17029, Invitrogen), rabbit polyclonal anti-SMC3 (A300-060A, Bethyl Laboratories), rabbit polyclonal anti-γH2AX (no. 2577, Cell Signaling Technology), rabbit monoclonal anti-KU70 (no. 4588, Cell Signaling Technology), rabbit monoclonal anti-FANCD2 (ab108928, Abcam), rabbit polyclonal anti-FANCA (A301-980A, Bethyl Laboratories), mouse monoclonal anti–ACTB (sc-47778, Santa Cruz Biotechnology), rabbit polyclonal anti–V5-tag (PM003, MBL), and mouse monoclonal anti-PARP1 (sc-8007, Santa Cruz Biotechnology).
Aldehyde sensitivity screening
U2OS cells were seeded in 96-well plates (10,000 cells per well) and treated with the following aldehydes for 8 hours: 4-HHE (Cayman Chemical), 4-hydroxynonenal (Cayman Chemical), 4-oxononenal (Cayman Chemical), acrolein (Wako), crotonaldehyde (Tokyo Chemical Industry), formaldehyde (Nacalai Tesque), glyoxal (Tokyo Chemical Industry), heptanal (Tokyo Chemical Industry), or methylglyoxal (Sigma-Aldrich). After incorporation of 5 μM 5-ethynyl-2′-deoxyuridine (EdU) for 1 hour, cells were fixed and permeabilized for 20 min in phosphate-buffered saline (PBS) containing 2% formaldehyde and 0.5% Triton X-100. After washing with PBS, cells were then incubated with coupling buffer with 10 μM Alexa Fluor 488 azide (Invitrogen), 50 mM tris-HCl (pH 7.3), 4 mM CuSO4, 10 mM sodium ascorbate, and 4′,6-diamidino-2-phenylindole (DAPI) for 60 min, followed by washing with PBST (PBS + 0.05% Tween 20). Fluorescent image acquisition and data processing were automated using CellInsight NXT (Thermo Fisher Scientific).
Measurement of EdU incorporation by flow cytometry
Cells were labeled with 5 μM EdU for 1 hour (U2OS cells) or 2 hours (primary fibroblasts) followed by fixing in 70% ethanol overnight at −30°C. Cells were incubated with coupling buffer with 10 μM Alexa Fluor 488 azide (Invitrogen), 50 mM tris-HCl (pH 7.3), 4 mM CuSO4, and 10 mM sodium ascorbate for 60 min. DNA was stained with 7-aminoactinomycin D (7-AAD) or propidium iodide. Data were acquired on a Cytomics FC500 FACS analyzer (Beckman Coulter) or CytoFLEX S FACS analyzer (Beckman Coulter) and analyzed with FlowJo version 10.6.2.
Lentivirus infection
For gene expression, HEK293 cells were transfected with the pLenti6.3 construct encoding gene of interest together with ViraPower Packaging Mix (Invitrogen) using Lipofectamine 2000 (Invitrogen). Viral particles were collected 48 hours after transfection and concentrated using PEG-it Virus Precipitation Solution (System Biosciences). For virus complementation experiments, viral particles produced by transfection of pLenti6.3 were used to infect cells. Selectin under blasticidin (5 μg/ml) was carried out.
Cell viability assay
Cells were seeded in 96-well plates (500 to 1000 cells per well) and fixed and stained with 2% formaldehyde and DAPI at 4 days (U2OS and RPE1 hTERT cells) or 7 days (primary fibroblasts) after formaldehyde treatment. Cells were identified and quantified on the basis of DAPI signal using CellInsight NXT (Thermo Fisher Scientific).
Measurement of ALDH2 Activity
The ALDH2 activity was analyzed using the colorimetric ALDH2 Activity Assay Kit (Abcam) according to the manufacturer’s instruction.
Electroporation of CD34+ HSPCs
CD34+ HSPCs from normal cord blood were procured from RIKEN BioResource Center (RIKEN BRC). Frozen CD34+ HSPCs were thawed and cultured in StemSpan SFEM II medium supplemented with StemSpan CC110 cocktail (STEMCELL Technologies) for 48 hours before electroporation. CD34+ HSPCs were electroporated using 4D-Nucleofector (Lonza). The following conditions were used: 50,000 cells were pelleted and resuspended in Lonza P3 solution containing TrueCut Cas9 protein v2 (Thermo Fisher Scientific) complexed with synthetic chemically modified sgRNA (ADH5#1, 5′-UCAGGGUAUAGGCAUCGGUG-3′; ADH5#2, 5′-CUGAUAGAUCAUUGCCACUG-3′; Synthego) at a 1:3 molar ratio. This mixture was electroporated using the Lonza 4D-Nucleofector (program EH-100). Electroporated cells were recovered and transferred to culture in StemSpan SFEM II medium supplemented with StemSpan CC110 cocktail.
Methylcellulose CFU assay
CD34+ HSPCs at 2 days after electroporation were resuspended in Iscove’s MDM (modified Dulbecco’s medium) and plated on methylcellulose-based media (MethoCult Optimum, STEMCELL Technologies) according to the manufacturer’s instruction. Cells were plated onto 35-mm petri dishes, in duplicate, and incubated for 14 days at 37°C with 5% CO2 and ≥95% humidity. CFU-erythroid; burst-forming unit–erythroid; CFU granulocyte and macrophage; and CFU granulocyte, erythroid, macrophage and megakaryocyte were classified and counted according to standard morphological criteria under microscopy in a blind fashion.
Animal studies
All the animal studies were conducted in compliance with the ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines. The experiments using genetically modified mice were approved by the Animal Care and Use Committee and the recombinant DNA experiment committee of Nagoya University and Osaka University.
Animals
C57BL/6JJcl mice were purchased from CLEA Japan. The animals were kept under conditions of 50% humidity and a 12-hour light/12-hour dark cycle. They were fed a standard pellet diet (MF, Oriental Yeast) and tap water ad libitum, unless otherwise noted.
Genome editing of mouse embryos
The following reagents were purchased: HiFi Cas9 Nuclease V3, tracrRNA, crRNA, and ssODN (Integrated DNA Technologies). To design gRNA sequence, software tools (http://crispor.tefor.net/ and https://crispr.dbcls.jp/) predicting unique target sites throughout the mouse genome were used. Pronuclear-stage mouse embryos were prepared by thawing frozen embryos (CLEA Japan) and cultured in a KSOM (potassium simplex optimization medium) (ARK Resource). For electroporation, 100 to 150 embryos at 1 hour after thawing were placed into a chamber with 40 μl of serum-free media (Opti-MEM, Thermo Fisher Scientific) containing HiFi Cas9 Nuclease V3 (100 ng/μl), Adh5 gRNA (100 ng/μl), Aldh2 gRNA (100 ng/μl), and ssODN (300 ng/μl). They were electroporated with a 5-mm gap electrode (CUY505P5, Nepa Gene) in a NEPA21 super electroporator (Nepa Gene). The poring pulses for the electroporation were voltage of 225 V, pulse width of 1 ms for mouse embryos, pulse interval of 50 ms, and number of pulses of 4. The first and second transfer pulses were voltage of 20 V, pulse width of 50 ms, pulse interval of 50 ms, and number of pulses of 5. Mouse embryos that developed to the two-cell stage after the electroporation were transferred into the oviducts of female surrogates anesthetized with sevoflurane or isoflurane (Mylan). All gRNA and ssODN sequence information are listed in table S2.
CT imaging
CT analysis was performed on anesthetized mice using a CosmoScan FX system (RIGAKU), with the following parameters: x-ray tube (90 kV), current (88 μA), FOV (field of view) (60 mm), and voxel size (240 μm). Data were analyzed and visualized by 3D Slicer version 4.10.2.
Mice hematological analysis
Peripheral blood from the animals was subjected to complete blood cell count analysis. RBC, platelet (Plt), white blood cell (WBC), HGB, HCT, MCV, mean corpuscular hemoglobin concentration, and lymphocyte were measured using an IDEXX ProCyte Dx (IDEXX Laboratories).
Cell isolation and FACS analysis
Bone marrow cells were flushed from femurs and tibias using a 26-gauge needle, and spleens and thymuses were dissociated by crushing followed by passing through a cell strainer in Ca2+- and Mg2+-free Hank’s buffered salt solution (Gibco) supplemented with 1% heat-inactivated bovine serum (Gibco). RBCs were lysed by resuspending the cells in RBC lysis buffer (eBioscience) for 5 min at room temperature. Cells were filtered through a 70-μm cell strainer to obtain a single-cell suspension. Number of cells was measured with a hemocytometer. Antibodies used for fluorescence-activated cell sorting (FACS) analysis were as follows: fluorescein isothiocyanate (FITC)–conjugated lineage cocktail (no. 133302, BioLegend), CD41 (FITC, no. 133903, BioLegend), FcεRIα (FITC, no. 134305, BioLegend), CD117 (APC, no. 105811, BioLegend), Sca-1 (PE, no. 108107, BioLegend), CD48 (Brilliant Violet 421, no. 103428, BioLegend), CD150 (APC/Fire 750, no. 115940, BioLegend), CD135 (Brilliant Violet 421, no. 135313, BioLegend), CD127 (PE/Cy7, no. 135014, BioLegend), CD16/32 (Brilliant Violet 421, no. 135313, BioLegend), CD34 (APC/Fire 750, no. 135014, BioLegend), CD3ε (Alexa Fluor 488, no. 100321, BioLegend), CD19 (APC, no. 152410, BioLegend), CD4 (PE, no. 130310, BioLegend), and CD8a (APC, no. 100712, BioLegend). Antibody staining was performed at 4°C for 20 min. Dead cells were excluded by staining with 7-AAD (BioLegend). Data were acquired on a CytoFLEX S FACS analyzer (Beckman Coulter) and analyzed with FlowJo version 10.6.2.
Acknowledgments
We would like to thank the families and clinicians for their involvement and participation. We are grateful to A. Lehmann for helpful comments and discussions on the manuscript. We thank M. Nakashima for comments on the animal analyses. We are grateful to S. Hashimoto, M. Isono, and K. Horiba, as well as M. Toyama, Y. He, and K. Katoh for the technical assistance. We thank JCRB Cell Bank (Osaka, Japan) for primary fibroblasts from patients, RIKEN BioResource Center (Tukuba, Japan) for fresh CD34+ umbilical cord blood cells and NIH for the use of dbGaP repositories (project no. 19720). Funding: This work was supported by the Special Coordination Funds for Rare and Intractable Diseases from the Japan Agency for Medical Research and Development (AMED) (JP19ek0109280, JP19dm0107090, JP19ek0109301, JP19ek0109348, and JP18kk020501 to N.Ma. and JP19ek0109281, JP19ek0109229, and JP19ek0109301 to T.O.); Grants in Aid for Scientific Research KAKENHI from the Japan Society for the Promotion of Science (JP16K21084 and JP18H03372 to Y. Oka, JP17K07255 and JP17KT0125 to K.Hi., JP17H01539 to N.Ma., 26253041, 15H02524, 16H06277, 18H03045, and 19K19425 to K.M., and JP15H02654 and JP17H00783 to T.O.); Grants in Aid for Scientific Research from the Ministry of Education, Science, Sports, Culture, and Technology of Japan, consisting of Priority Areas of Cancer (17015018), Innovative Areas (221S0001), and a Grant-in-Aid for the Third Term Comprehensive 10-year Strategy for Cancer Control from the Ministry of Health, Labour, and Welfare of Japan to K.M.; a medical research grant from Daiichi Sankyo Foundation of Life Science to Y. Oka; a grant from Daiko Foundation to T.O.; Science Research Grants from Uehara Memorial foundation to Y. Oka and T.O.; and medical research grants from Takeda Science Foundation to Y. Oka and T.O. Author contributions: Y. Oka and T.O. designed the study and the experiments. Y. Oka, Y.Oku., K.Hi., N.Mit., Y.H., N.Miy., Y.Kaw., K.T., M.N., N.Ma., F.M., K.M., and T.O. analyzed the genetics data. Y. Oka, Y.N., Y.Oku., K.Ha., H.T., M.S., Y.Kas., S.N., and T.O. performed molecular and cell biological experiments. Y. Oka, M.S., Y.Ko., M.Y., M.T., T.S., S.Ki., and T.M. performed animal studies. Y. Oka., M.H., H.M., Y.Oku., K.Hi., K.Ha., T.H., T.K., H.S., T.I., S.O., K.Y., Y.W., K.K., S.M., K.I., M.O., H.K., F.M., Y.T., S.K., and T.O. analyzed clinical manifestations of the affected individuals and healthy control cases. M.H., H.M., K.Ha., T.K., H.S., T.I., S.O., K.Y., Y.W., K.I., M.O., H.K., F.M., K.M., Y.T., and S.K. contributed the patients’ and control samples. Y. Oka and T.O. wrote the manuscript. M.H., Y.N., H.M., Y.Oku., and K.Hi. contributed equally to the study. All authors commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/51/eabd7197/DC1
REFERENCES AND NOTES
- 1.Burgos-Barragan G., Wit N., Meiser J., Dingler F. A., Pietzke M., Mulderrig L., Pontel L. B., Rosado I. V., Brewer T. F., Cordell R. L., Monks P. S., Chang C. J., Vazquez A., Patel K. J., Mammals divert endogenous genotoxic formaldehyde into one-carbon metabolism. Nature 548, 549–554 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Walport L. J., Hopkinson R. J., Schofield C. J., Mechanisms of human histone and nucleic acid demethylases. Curr. Opin. Chem. Biol. 16, 525–534 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Harada S., Agarwal D. P., Goedde H. W., Aldehyde dehydrogenase deficiency as cause of facial flushing reaction to alcohol in Japanese. Lancet 2, 982 (1981). [DOI] [PubMed] [Google Scholar]
- 4.Brooks P. J., Enoch M.-A., Goldman D., Li T.-K., Yokoyama A., The alcohol flushing response: An unrecognized risk factor for esophageal cancer from alcohol consumption. PLOS Med. 6, e1000050 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yoshida A., Huang I. Y., Ikawa M., Molecular abnormality of an inactive aldehyde dehydrogenase variant commonly found in Orientals. Proc. Natl. Acad. Sci. U.S.A. 81, 258–261 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li H., Borinskaya S., Yoshimura K., Kal’ina N., Marusin A., Stepanov V. A., Qin Z., Khaliq S., Lee M.-Y., Yang Y., Mohyuddin A., Gurwitz D., Mehdi S. Q., Rogaev E., Jin L., Yankovsky N. K., Kidd J. R., Kidd K. K., Refined geographic distribution of the oriental ALDH2*504Lys (nee 487Lys) variant. Ann. Hum. Genet. 73, 335–345 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takeuchi F., Yokota M., Yamamoto K., Nakashima E., Katsuya T., Asano H., Isono M., Nabika T., Sugiyama T., Fujioka A., Awata N., Ohnaka K., Nakatochi M., Kitajima H., Rakugi H., Nakamura J., Ohkubo T., Imai Y., Shimamoto K., Yamori Y., Yamaguchi S., Kobayashi S., Takayanagi R., Ogihara T., Kato N., Genome-wide association study of coronary artery disease in the Japanese. Eur. J. Hum. Genet. 20, 333–340 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hiura Y., Tabara Y., Kokubo Y., Okamura T., Miki T., Tomoike H., Iwai N., A genome-wide association study of hypertension-related phenotypes in a Japanese population. Circ. J. 74, 2353–2359 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Masaoka H., Ito H., Soga N., Hosono S., Oze I., Watanabe M., Tanaka H., Yokomizo A., Hayashi N., Eto M., Matsuo K., Aldehyde dehydrogenase 2 (ALDH2) and alcohol dehydrogenase 1B (ADH1B) polymorphisms exacerbate bladder cancer risk associated with alcohol drinking: Gene-environment interaction. Carcinogenesis 37, 583–588 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Matsuo K., Oze I., Hosono S., Ito H., Watanabe M., Ishioka K., Ito S., Tajika M., Yatabe Y., Niwa Y., Yamao K., Nakamura S., Tajima K., Tanaka H., The aldehyde dehydrogenase 2 (ALDH2) Glu504Lys polymorphism interacts with alcohol drinking in the risk of stomach cancer. Carcinogenesis 34, 1510–1515 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Baan R., Straif K., Grosse Y., Secretan B., El Ghissassi F., Bouvard V., Altieri A., Cogliano V.; WHO International Agency for Research on Cancer Monograph Working Group , Carcinogenicity of alcoholic beverages. Lancet Oncol. 8, 292–293 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Yokoyama A., Muramatsu T., Ohmori T., Higuchi S., Hayashida M., Ishii H., Esophageal cancer and aldehyde dehydrogenase-2 genotypes in Japanese males. Cancer Epidemiol. Biomarkers Prev. 5, 99–102 (1996). [PubMed] [Google Scholar]
- 13.Badano J. L., Katsanis N., Beyond Mendel: An evolving view of human genetic disease transmission. Nat. Rev. Genet. 3, 779–789 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Deltas C., Digenic inheritance and genetic modifiers. Clin. Genet. 93, 429–438 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Schäffer A. A., Digenic inheritance in medical genetics. J. Med. Genet. 50, 641–652 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barnett S. D., Buxton I. L. O., The role of S-nitrosoglutathione reductase (GSNOR) in human disease and therapy. Crit. Rev. Biochem. Mol. Biol. 52, 340–354 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Voulgaridou G.-P., Anestopoulos I., Franco R., Panayiotidis M. I., Pappa A., DNA damage induced by endogenous aldehydes: Current state of knowledge. Mutat. Res. 711, 13–27 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Che R., Zhang J., Nepal M., Han B., Fei P., Multifaceted Fanconi anemia signaling. Trends Genet. 34, 171–183 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bluteau O., Sebert M., Leblanc T., Peffault de Latour R., Quentin S., Lainey E., Hernandez L., Dalle J.-H., Sicre de Fontbrune F., Lengline E., Itzykson R., Clappier E., Boissel N., Vasquez N., Da Costa M., Masliah-Planchon J., Cuccuini W., Raimbault A., De Jaegere L., Ades L., Fenaux P., Maury S., Schmitt C., Muller M., Domenech C., Blin N., Bruno B., Pellier I., Hunault M., Blanche S., Petit A., Leverger G., Michel G., Bertrand Y., Baruchel A., Socié G., Soulier J., A landscape of germ line mutations in a cohort of inherited bone marrow failure patients. Blood 131, 717–732 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Lessel D., Vaz B., Halder S., Lockhart P. J., Marinovic-Terzic I., Lopez-Mosqueda J., Philipp M., Sim J. C. H., Smith K. R., Oehler J., Cabrera E., Freire R., Pope K., Nahid A., Norris F., Leventer R. J., Delatycki M. B., Barbi G., von Ameln S., Högel J., Degoricija M., Fertig R., Burkhalter M. D., Hofmann K., Thiele H., Altmüller J., Nürnberg G., Nürnberg P., Bahlo M., Martin G. M., Aalfs C. M., Oshima J., Terzic J., Amor D. J., Dikic I., Ramadan K., Kubisch C., Mutations in SPRTN cause early onset hepatocellular carcinoma, genomic instability and progeroid features. Nat. Genet. 46, 1239–1244 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bachrati C. Z., Hickson I. D., RecQ helicases: Suppressors of tumorigenesis and premature aging. Biochem. J. 374, 577–606 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huber R. S., Houlihan D., Filter K., Dubowitz syndrome: A review and implications for cognitive, behavioral, and psychological features. J. Clin. Med. Res. 3, 147–155 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Calmels N., Botta E., Jia N., Fawcett H., Nardo T., Nakazawa Y., Lanzafame M., Moriwaki S., Sugita K., Kubota M., Obringer C., Spitz M.-A., Stefanini M., Laugel V., Orioli D., Ogi T., Lehmann A. R., Functional and clinical relevance of novel mutations in a large cohort of patients with Cockayne syndrome. J. Med. Genet. 55, 329–343 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Laugel V., Cockayne syndrome: The expanding clinical and mutational spectrum. Mech. Ageing Dev. 134, 161–170 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Niedernhofer L. J., Garinis G. A., Raams A., Lalai A. S., Robinson A. R., Appeldoorn E., Odijk H., Oostendorp R., Ahmad A., van Leeuwen W., Theil A. F., Vermeulen W., van der Horst G. T. J., Meinecke P., Kleijer W. J., Vijg J., Jaspers N. G. J., Hoeijmakers J. H. J., A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature 444, 1038–1043 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Garaycoechea J. I., Crossan G. P., Langevin F., Mulderrig L., Louzada S., Yang F., Guilbaud G., Park N., Roerink S., Nik-Zainal S., Stratton M. R., Patel K. J., Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature 553, 171–177 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rosado I. V., Langevin F., Crossan G. P., Takata M., Patel K. J., Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA-repair pathway. Nat. Struct. Mol. Biol. 18, 1432–1434 (2011). [DOI] [PubMed] [Google Scholar]
- 28.Garaycoechea J. I., Crossan G. P., Langevin F., Daly M., Arends M. J., Patel K. J., Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature 489, 571–575 (2012). [DOI] [PubMed] [Google Scholar]
- 29.Langevin F., Crossan G. P., Rosado I. V., Arends M. J., Patel K. J., Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 475, 53–58 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Pontel L. B., Rosado I. V., Burgos-Barragan G., Garaycoechea J. I., Yu R., Arends M. J., Chandrasekaran G., Broecker V., Wei W., Liu L., Swenberg J. A., Crossan G. P., Patel K. J., Endogenous formaldehyde is a hematopoietic stem cell genotoxin and metabolic carcinogen. Mol. Cell 60, 177–188 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu L., Yan Y., Zeng M., Zhang J., Hanes M. A., Ahearn G., McMahon T. J., Dickfeld T., Marshall H. E., Que L. G., Stamler J. S., Essential roles of S-nitrosothiols in vascular homeostasis and endotoxic shock. Cell 116, 617–628 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Wang R. S., Nakajima T., Kawamoto T., Honma T., Effects of aldehyde dehydrogenase-2 genetic polymorphisms on metabolism of structurally different aldehydes in human liver. Drug Metab. Dispos. 30, 69–73 (2002). [DOI] [PubMed] [Google Scholar]
- 33.Yoval-Sánchez B., Rodríguez-Zavala J. S., Differences in susceptibility to inactivation of human aldehyde dehydrogenases by lipid peroxidation byproducts. Chem. Res. Toxicol. 25, 722–729 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Larson H. N., Weiner H., Hurley T. D., Disruption of the coenzyme binding site and dimer interface revealed in the crystal structure of mitochondrial aldehyde dehydrogenase "Asian" variant. J. Biol. Chem. 280, 30550–30556 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heck H. D., Casanova-Schmitz M., Dodd P. B., Schachter E. N., Witek T. J., Tosun T., Formaldehyde (CH2O) concentrations in the blood of humans and Fischer-344 rats exposed to CH2O under controlled conditions. Am. Ind. Hyg. Assoc. J. 46, 1–3 (1985). [DOI] [PubMed] [Google Scholar]
- 36.Luo W., Li H., Zhang Y., Ang C. Y. W., Determination of formaldehyde in blood plasma by high-performance liquid chromatography with fluorescence detection. J. Chromatogr. B Biomed. Sci. Appl. 753, 253–257 (2001). [DOI] [PubMed] [Google Scholar]
- 37.Dhananjayan K., Irrgang F., Raju R., Harman D. G., Moran C., Srikanth V., Münch G., Determination of glyoxal and methylglyoxal in serum by UHPLC coupled with fluorescence detection. Anal. Biochem. 573, 51–66 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Kitagawa K., Kawamoto T., Kunugita N., Tsukiyama T., Okamoto K., Yoshida A., Nakayama K., Nakayama K.-i., Aldehyde dehydrogenase (ALDH) 2 associates with oxidation of methoxyacetaldehyde; in vitro analysis with liver subcellular fraction derived from human and Aldh2 gene targeting mouse. FEBS Lett. 476, 306–311 (2000). [DOI] [PubMed] [Google Scholar]
- 39.Matsumoto A., Ito S., Wakamatsu K., Ichiba M., Vasiliou V., Akao C., Song B.-J., Fujita M., Ethanol induces skin hyperpigmentation in mice with aldehyde dehydrogenase 2 deficiency. Chem. Biol. Interact. 302, 61–66 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Daiger S. P., Sullivan L. S., Bowne S. J., Genes and mutations causing retinitis pigmentosa. Clin. Genet. 84, 132–141 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khan S. A., Muhammad N., Khan M. A., Kamal A., Rehman Z. U., Khan S., Genetics of human Bardet-Biedl syndrome, an updates. Clin. Genet. 90, 3–15 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Hoeijmakers J. H. J., DNA damage, aging, and cancer. N. Engl. J. Med. 361, 1475–1485 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Maynard S., Fang E. F., Scheibye-Knudsen M., Croteau D. L., Bohr V. A., DNA damage, DNA repair, aging, and neurodegeneration. Cold Spring Harb. Perspect. Med. 5, a025130 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marteijn J. A., Lans H., Vermeulen W., Hoeijmakers J. H. J., Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 15, 465–481 (2014). [DOI] [PubMed] [Google Scholar]
- 45.Schumacher B., Garinis G. A., Hoeijmakers J. H. J., Age to survive: DNA damage and aging. Trends Genet. 24, 77–85 (2008). [DOI] [PubMed] [Google Scholar]
- 46.Park J. H., Elpers C., Reunert J., McCormick M. L., Mohr J., Biskup S., Schwartz O., Rust S., Grüneberg M., Seelhöfer A., Schara U., Boltshauser E., Spitz D. R., Marquardt T., SOD1 deficiency: A novel syndrome distinct from amyotrophic lateral sclerosis. Brain 142, 2230–2237 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Andersen P. M., Nordström U., Tsiakas K., Johannsen J., Volk A. E., Bierhals T., Zetterström P., Marklund S. L., Hempel M., Santer R., Phenotype in an infant with SOD1 homozygous truncating mutation. N. Engl. J. Med. 381, 486–488 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Kim G., Gautier O., Tassoni-Tsuchida E., Ma X. R., Gitler A. D., ALS genetics: Gains, losses, and implications for future therapies. Neuron 10.1016/j.neuron.2020.08.022, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hoch N. C., Hanzlikova H., Rulten S. L., Tétreault M., Komulainen E., Ju L., Hornyak P., Zeng Z., Gittens W., Rey S. A., Staras K., Mancini G. M. S., McKinnon P. J., Wang Z.-Q., Wagner J. D.; Care4Rare Canada Consortium, Yoon G., Caldecott K. W., XRCC1 mutation is associated with PARP1 hyperactivation and cerebellar ataxia. Nature 541, 87–91 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cloos P. A. C., Christensen J., Agger K., Helin K., Erasing the methyl mark: Histone demethylases at the center of cellular differentiation and disease. Genes Dev. 22, 1115–1140 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hira A., Yabe H., Yoshida K., Okuno Y., Shiraishi Y., Chiba K., Tanaka H., Miyano S., Nakamura J., Kojima S., Ogawa S., Matsuo K., Takata M., Yabe M., Variant ALDH2 is associated with accelerated progression of bone marrow failure in Japanese Fanconi anemia patients. Blood 122, 3206–3209 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kawashima N., Narita A., Wang X., Xu Y., Sakaguchi H., Doisaki S., Muramatsu H., Hama A., Nakanishi K., Takahashi Y., Kojima S., Aldehyde dehydrogenase-2 polymorphism contributes to the progression of bone marrow failure in children with idiopathic aplastic anaemia. Br. J. Haematol. 168, 460–463 (2015). [DOI] [PubMed] [Google Scholar]
- 53.Alter B. P., Giri N., Savage S. A., Rosenberg P. S., Cancer in the National Cancer Institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica 103, 30–39 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vaz B., Popovic M., Newman J. A., Fielden J., Aitkenhead H., Halder S., Singh A. N., Vendrell I., Fischer R., Torrecilla I., Drobnitzky N., Freire R., Amor D. J., Lockhart P. J., Kessler B. M., McKenna G. W., Gileadi O., Ramadan K., Metalloprotease SPRTN/DVC1 orchestrates replication-coupled DNA-protein crosslink repair. Mol. Cell 64, 704–719 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sawyer S. L., Tian L., Kähkönen M., Schwartzentruber J., Kircher M.; University of Washington Centre for Mendelian Genomics; FORGE Canada Consortium, Majewski J., Dyment D. A., Innes A. M., Boycott K. M., Moreau L. A., Moilanen J. S., Greenberg R. A., Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov. 5, 135–142 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bogliolo M., Schuster B., Stoepker C., Derkunt B., Su Y., Raams A., Trujillo J. P., Minguillon J., Ramírez M. J., Pujol R., Casado J. A., Baños R., Rio P., Knies K., Zúñiga S., Benítez J., Bueren J. A., Jaspers N. G. J., Schärer O. D., de Winter J. P., Schindler D., Surrallés J., Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am. J. Hum. Genet. 92, 800–806 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kashiyama K., Nakazawa Y., Pilz D. T., Guo C., Shimada M., Sasaki K., Fawcett H., Wing J. F., Lewin S. O., Carr L., Li T.-S., Yoshiura K.-i., Utani A., Hirano A., Yamashita S., Greenblatt D., Nardo T., Stefanini M., McGibbon D., Sarkany R., Fassihi H., Takahashi Y., Nagayama Y., Mitsutake N., Lehmann A. R., Ogi T., Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes Cockayne syndrome, xeroderma pigmentosum, and Fanconi anemia. Am. J. Hum. Genet. 92, 807–819 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stingele J., Bellelli R., Boulton S. J., Mechanisms of DNA-protein crosslink repair. Nat. Rev. Mol. Cell Biol. 18, 563–573 (2017). [DOI] [PubMed] [Google Scholar]
- 59.Brooks P. J., Theruvathu J. A., DNA adducts from acetaldehyde: Implications for alcohol-related carcinogenesis. Alcohol 35, 187–193 (2005). [DOI] [PubMed] [Google Scholar]
- 60.De Bont R., van Larebeke N., Endogenous DNA damage in humans: A review of quantitative data. Mutagenesis 19, 169–185 (2004). [DOI] [PubMed] [Google Scholar]
- 61.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M. A., The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang K., Li M., Hakonarson H., ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 38, e164 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brinkman E. K., Chen T., Amendola M., van Steensel B., Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42, e168 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/51/eabd7197/DC1