

A balance of dynamic hydrophobic and hydrophilic contacts enables client specificity in mitochondrial membrane-protein chaperones.
Abstract
Chaperones are essential for assisting protein folding and for transferring poorly soluble proteins to their functional locations within cells. Hydrophobic interactions drive promiscuous chaperone-client binding, but our understanding of how additional interactions enable client specificity is sparse. Here, we decipher what determines binding of two chaperones (TIM8·13 and TIM9·10) to different integral membrane proteins, the all-transmembrane mitochondrial carrier Ggc1 and Tim23, which has an additional disordered hydrophilic domain. Combining NMR, SAXS, and molecular dynamics simulations, we determine the structures of Tim23/TIM8·13 and Tim23/TIM9·10 complexes. TIM8·13 uses transient salt bridges to interact with the hydrophilic part of its client, but its interactions to the transmembrane part are weaker than in TIM9·10. Consequently, TIM9·10 outcompetes TIM8·13 in binding hydrophobic clients, while TIM8·13 is tuned to few clients with both hydrophilic and hydrophobic parts. Our study exemplifies how chaperones fine-tune the balance of promiscuity versus specificity.
INTRODUCTION
Cellular survival and function fundamentally rely on an intact proteome. Proteins within cells need to be correctly folded to their functional conformation and be present at the cellular location where they function. Chaperones play a central role in maintaining this cellular protein homeostasis (1), either by helping other proteins to reach their functional three-dimensional (3D) structure after synthesis, by transporting them across the cytosol or organelles, or by sustaining their native fold along their lifetime. More than 20,000 different proteins are required to fulfill the functions of human cells, and it is believed that the majority rely on chaperones to reach and maintain their native fold (2). Given the diversity of the client proteins, many chaperones promiscuously interact with tens of different “client” proteins that may differ widely in size, structure, and physicochemical properties. However, the need for efficient binding and refolding of their clients also calls for some degree of specificity. Chaperones operate at this delicate balance of promiscuity and specificity to their clients. The interactions that determine the chaperone-client specificity are only partly understood.
Hydrophobic interactions play a crucial role for chaperone interactions, as most chaperones bind to hydrophobic patches on their clients and shield them from aggregation. Electrostatic charges also play a role in some chaperone complexes (3). The interaction motifs recognized by different chaperones differ by their physicochemical properties (4). For example, for interacting with the Hsp70 chaperone family, Ile, Phe, Leu, and Val residues are particularly important (5, 6); the SecB chaperone recognizes nine-residue-long stretches enriched in aromatic and basic residues (7); the chaperone Spy uses longer-range charge interactions for the formation of an initial encounter complex, followed by more tight binding mediated by hydrophobic interactions (8), whereby structurally frustrated sites on the client protein are particularly prone to binding (9).
Our understanding of the underlying principles of chaperone-client interactions is hampered by the lack of atomic-level views onto the structure and dynamics of these complexes. Their inherently dynamic and often transient nature represents a substantial experimental challenge toward structural characterization. Only a very limited number of chaperone complex structures have been reported [reviewed in (10)]. The modes of interactions that they revealed range from rather well-defined binding poses of client polypeptides in the chaperone’s binding pockets, reminiscent of complexes formed by globular proteins, to highly flexible ensembles of at least partly disordered conformations (“fuzzy complexes”). In the latter, a multitude of local chaperone-client interactions may result in a high overall affinity despite the low affinity and short lifetime of each individual intermolecular contact.
Multiple molecular chaperones are present in the cell with mutually overlapping functions and “clientomes” (2, 11, 12). It is poorly understood, however, whether a given client protein adopts a different conformation (or ensemble of conformations) when it is bound to different chaperones, and if different clients, when bound to a given chaperone, all show similar conformational properties. α-Synuclein appears to have similar interaction patterns with six different chaperones (13); outer membrane proteins (OmpA, OmpX, and FhuA) have similar properties—essentially fully unfolded—when bound to SurA and Skp chaperones (14, 15), at least as judged by their nuclear magnetic resonance (NMR) fingerprint spectra. Phosphatase A displays an extended dynamic conformation, but well-defined binding poses of its interacting parts, when bound to trigger factor (16), Hsp40 (17), or SecB (18). Thus, while these reports suggest that a given protein adopts similar properties on different chaperones, the scarcity of data and the absence of a direct comparison of complex structures leave open which interactions may confer specificity.
A pair of “holdase” chaperone complexes of the mitochondrial intermembrane space (IMS), TIM8·13 and TIM9·10, are structurally highly similar but have different substrate binding preferences. These chaperones transport precursors of membrane proteins with internal targeting sequence (henceforth denoted as “precursors”) to the membrane-insertase machineries in the inner membrane (TIM22) and outer mitochondrial membranes (SAM) (19). The TIM chaperones form hetero-hexameric structures of ca. 70 kDa, composed of an alternating arrangement of Tim9 and Tim10 or Tim8 and Tim13. TIM9·10 is essential to cellular viability (20–22); even single-point mutations in Tim9 or Tim10 that keep the chaperone structure intact but affect precursor protein binding can impair yeast growth and cause lethality (23). Although TIM8·13 is not essential in yeast (24), yeast cells depleted of Tim8 and Tim13 show conditional lethality (25). In addition, mutations in the human Tim8a protein have been identified as the cause of a neurodegenerative disorder known as Mohr-Tranebjærg syndrome or deafness-dystonia-optic neuropathy syndrome (26, 27).
In vivo experiments, predominantly in yeast, have identified mitochondrial membrane proteins whose biogenesis depends on small TIM chaperones. TIM9·10 is believed to interact with all members of the mitochondrial carrier (SLC25) family, which comprises more than 50 members in humans, such as the adenosine diphosphate (ADP)/adenosine triphosphate (ATP) carrier (Aac in yeast); furthermore, TIM9·10 transports the central components of the TIM22 and TIM23 insertion machineries (Tim23, Tim17, and Tim22) as well as outer membrane β barrel proteins (28). TIM8·13 has a narrower clientome and was shown to bind the precursors of the inner membrane proteins Tim23 (25, 29, 30) and Ca2+-binding aspartate-glutamate carriers (31), as well as the outer membrane β barrel proteins VDAC/Porin, Tom40 (32), and Tob55/Sam50 (33). There is evidence that TIM8·13 does bind neither the inner membrane protein ADP/ATP carrier (Aac) nor Tim17 (25). The inner membrane proteins that have been reported to interact with TIM8·13 have a hydrophilic domain in addition to transmembrane (TM) domains (fig. S1), but this does not hold true for the outer membrane β barrels. Thus, the mechanisms by which TIM8·13 binds its clients remain unclear.
Recently, we obtained the first structure of a complex of a small TIM chaperone, TIM9·10, with the mitochondrial guanosine diphosphate (GDP)/guanosine triphosphate (GTP) carrier (Ggc1) (23). The structure, composed of two chaperone complexes holding one precursor protein, revealed a highly dynamic ensemble of Ggc1 conformers that form multiple short-lived and rapidly interconverting (<1 ms) interactions with a hydrophobic binding cleft of the chaperone (fig. S2). The TIM9·10-Ggc1 complex can be described as a “fuzzy complex,” in which the high overall affinity is driven by a multitude of individually weak interactions with the hydrophobic TM parts of its clients.
To understand what confers specificity in the mitochondrial IMS chaperone system, we studied chaperone complexes of TIM9·10 and TIM8·13 with two precursor proteins, the Ggc1 and the insertase component Tim23. In their native state, Ggc1 comprises six TM helices without soluble domains, and Tim23 comprises four TM helices and a ca. 100-residue-long soluble IMS domain (Fig. 1A). By solving the complex structures of the two chaperone complexes holding Tim23, we reveal that the differential specificity of the two chaperones is based on an interplay of hydrophobic and hydrophilic interactions, which leads to different conformational properties of the precursor protein bound to these chaperones.
Fig. 1. Biochemical characterization of TIM chaperone-membrane protein complexes.

(A) Native topology of the two precursor proteins used in this study. (B) Schematic view of the pull-down experiment used to prepare chaperone-precursor complexes. (C) Formation of Tim23-chaperone complexes, monitored by SDS-PAGE. Either TIM8·13, TIM9·10, or a 1:1 mixture was added to NiNTA-bound Tim23. The lanes correspond to flow-through after applying chaperone (FT), additional wash (W), and imidazole elution (E). Protein bands corresponding to Tim10 and Tim8 overlap. Mw, molecular weight. (D) Relative amounts of complexes with Ggc1 and Tim23, obtained from three different experiments: (i) a pull-down assay where both chaperones were applied to bound precursor protein (black); (ii) preparation of a TIM9·10-precursor protein complex and addition of TIM8·13, and SDS-PAGE and MS analysis after 1 and 3 hours (red); (iii) preparation of TIM8·13-Tim23 followed by TIM9·10 addition and SDS-PAGE (blue) as in (ii). The protein amounts were determined from LC–ESI-TOF–MS (fig. S4); error estimates from two or more experiments. (E) Sequence alignment of the small Tims, numbered from the N- and C-terminal conserved Cys residues (“0”). Red, conserved hydrophobic residues; blue, hydrophilic Tim8 residues, K30 and S36L. See fig. S5 for comprehensive alignment. (F) Location of the residues in the hydrophobic cleft. (G) Comparison of Kyte-Doolittle hydrophobicity of the residues in the binding cleft of wild-type (WT) native Tim proteins and Tim8K30F,S36L. (H) Pull-down experiment of Ggc1 with TIM9·10, TIM8·13, and TIM8·13 (Tim8K30F,S36L). Lane descriptions are as in (C); in addition, the fraction obtained after final wash with urea and imidazole, to control the Ggc1 initially loaded onto the column, is shown [control (C)]. (I) Amount of complex obtained from pull-down experiments of WT and mutant chaperones; the same amount of Ggc1 was applied in all three experiments, and the total amount of eluted complex was determined spectroscopically.
RESULTS
TIM8·13 and TIM9·10 interact differently with membrane precursor proteins
We have developed an experimental protocol (23) to prepare complexes of the inherently insoluble membrane-protein precursors and chaperones (Fig. 1, B and C). Briefly, the approach involves the recombinant production of the His-tagged precursor protein, binding it to a His-affinity column (NiNTA) in denaturing conditions, followed by removal of the denaturant and simultaneous addition of a chaperone. The chaperone-precursor complex is then eluted for further biochemical, biophysical, and structural investigations.
The measurement of dissociation constants of chaperones and membrane precursor proteins, using methods such as isothermal titration calorimetry (ITC) or surface plasmon resonance, is not possible, because the complexes cannot be formed in solution [e.g., flash-dilution methods, which work for other chaperones (14), failed]. Thus, to characterize the relative affinities of the precursor proteins to the two chaperones, we performed different types of competition experiments. In a first experiment, the precursor protein was bound to the affinity resin, and both chaperones were simultaneously added, before washing excess chaperone and eluting the chaperone-precursor complexes (Fig. 1C). NMR spectroscopy shows that the two chaperones do not form mixed hetero-hexameric complexes, implying that TIM9·10 and TIM8·13 stay intact in such competition experiments (fig. S3). In a second class of experiments, we prepared one type of complex (e.g., TIM9·10-Tim23) and added the other chaperone (e.g., TIM8·13) in its apo state, allowing the precursor protein to be transferred. These experiments also demonstrate that membrane precursor proteins can be transferred between these two chaperones on the time scale that we investigated (minutes to hours). We used SDS–polyacrylamide gel electrophoresis (PAGE) analyses and electrospray ionization mass spectrometry (ESI-MS) to systematically quantify the amount of obtained complexes (Fig. 1D and fig. S4). Consistently, we find that Ggc1 has a strong preference for TIM9·10 (ca. 5- to 10-fold), while Tim23 shows a slight preference for TIM8·13 (ca. 1.5-fold).
Together, we established that the two chaperones bind with different affinities to two inner membrane precursor proteins, whereby TIM8·13 is barely able to hold Ggc1, in contrast to TIM9·10, while it can hold Tim23 slightly better than TIM9·10.
The small TIM chaperones use a conserved hydrophobic cleft for membrane precursor protein binding
To understand the different binding properties, we performed a sequence alignment of the small TIMs, which reveals a well conserved set of hydrophobic residues that point toward the binding cleft formed between the inner (N terminus) and outer tentacles (Fig. 1, E and F) (23). The overall hydrophobicity of these residues is lower in Tim8 and Tim13 than in Tim9 and Tim10 (Fig. 1G). In particular, Tim8 has a charged residue in position −14 (Lys30). (The sequences are numbered starting with negative numbering at the twin CX3C motif toward the N terminus and positive numbering from the last Cys to the C terminus, and the hydrophobic motif residues are at positions −20, −14, −11, −8, −7, +5, +11, and +15.) This positive charge at position −14, either Lys or Arg, is conserved among eukaryotes (fig. S5). In yeast, position −8 of the hydrophobic motif is polar (Ser36), although this position is not strictly conserved. Overall, these residues make the hydrophobic binding cleft of Tim8 less hydrophobic than in the other small Tims.
We speculated that the less hydrophobic nature of TIM8·13’s binding cleft reduces its affinity to TM parts of membrane precursor proteins. To test this hypothesis, we generated a mutant TIM8·13 with increased hydrophobicity (Tim8K30F,S36L; Fig. 1G). This more hydrophobic TIM8·13(Tim8K30F,S36L) chaperone allowed us to obtain significantly larger amounts of complex with Ggc1 than native TIM8·13, under otherwise identical conditions (Fig. 1, H and I). This observation establishes the importance of the hydrophobic cleft for binding hydrophobic TM parts of precursor proteins. Equivalent experiments with the full-length (FL) Tim23, shown in fig. S6, reveal that the additional hydrophobic residues in the binding cleft of Tim8K30F,S36L do not improve its capacity to bind Tim23. This observation suggests that the binding mechanisms in place for binding these two different precursor proteins differ.
To better understand the client-binding properties of the two chaperones, we turned to structural studies. Solution-NMR spectra of apo TIM8·13 (Fig. 2A) and residue-wise resonance assignments allowed the identification of the residues forming secondary structure and estimating their local flexibility. In agreement with the crystal structure, the core of rather rigid tentacles comprises the top part of the chaperone between the CX3C motifs and ca. 15 to 25 residues before and after these motifs. About 10 to 20 residues on each of the N and C termini are flexible (fig. S7).
Fig. 2. Solution-NMR and binding of a VDAC fragment to TIM8·13.

(A) 1H-15N NMR spectrum of TIM8·13 at 35°C. (B) CSP in TIM8·13 upon addition of five molar equivalents of cyclic VDAC257–279. (C) CSP effects of VDAC257–279 binding. The data for TIM9·10 are from (23). (D) Plot of CSP data on the TIM8·13 structure. (E) Photo-induced cross-linking of the linear (left) and cyclic (right) VDAC257–279 peptides to TIM8·13. While hardly any adducts are observed for the linear one, the cyclic peptide forms cross-linking photo-adducts (PA), including of higher molecular weight, resulting from multiple cross-links, as reported earlier (23, 34, 35). (F) Schematic structure of the two last strands of VDAC, as found in the NMR structure (61) of the full β barrel, showing that the hydrophobic and hydrophilic side chains cluster on the two opposite faces of the β-turn.
To probe the binding of a TM segment of a membrane precursor protein, we performed NMR-detected titration experiments of TIM8·13 with a cyclic peptide corresponding to the two C-terminal strands of the β barrel voltage-dependent anion channel (VDAC257–279) that has a propensity to form a β-turn (34). Addition of this cyclic VDAC257–279 induces chemical shift perturbations (CSPs) (Fig. 2B) that are primarily located in the hydrophobic cleft formed between the inner and outer rings of helices (Fig. 2, C and D). This binding site matches very closely the site on TIM9·10 to which VDAC257–279 binds (Fig. 2C) (23). The VDAC257–279–induced CSP effects in TIM8·13 are overall only about half of the magnitude of CSPs found in TIM9·10, pointing to a higher population of the TIM9·10–VDAC257–279 complex compared to TIM8·13–VDAC257–279 at comparable conditions (Fig. 2C). This finding suggests a lower affinity of TIM8·13 to VDAC257–279, as expected from its lower hydrophobicity.
Photo-induced cross-linking experiments of a Bpa-modified VDAC257–279 peptide to TIM8·13 show that only the cyclic peptide forms cross-linking adducts, while the linear, mostly disordered (34) form does not (Fig. 2E). The same behavior was also found for TIM9·10 (23) and yeast cytosolic chaperones Ssa1, Ydj1, Djp1, and Hsp104 (35). A rationale for this finding is the fact that in a β-turn the side chains of consecutive residues point to the two opposing faces, thus creating one hydrophobic and one more hydrophilic face (Fig. 2F). In contrast, because of its disorder, the linear VDAC257–279 peptide does not have a stable hydrophobic face, reducing its affinity to the hydrophobic binding cleft on the chaperone. In line with these findings, NMR titration data with the linear peptide show small CSPs that are spread across the protein, thus pointing to unspecific interaction (fig. S8).
Collectively, the experiments with the client fragment VDAC257–279 provide a first evidence that both chaperones use the same conserved binding cleft to interact with hydrophobic membrane precursor protein sequences, and that TIM9·10 interacts more efficiently with TM parts, and thus with Ggc1 and the VDAC fragment. We propose that the more hydrophobic nature of the binding cleft in TIM9·10 allows it to interact more strongly with TM parts of its clients. In light of this observation, how does TIM8·13 achieve a binding affinity to Tim23, which is slightly higher than the one of TIM9·10 (Fig. 1D)?
Hydrophilic fragments interact differently with TIM8·13 and TIM9·10
Tim23 has a hydrophilic N-terminal segment in addition to four TM helices (Fig. 3A), and we investigated whether this part interacts with the chaperones. NMR spectra of the soluble Tim23IMS fragment (residues 1 to 98) in isolation show the hallmark features of a highly flexible intrinsically disordered protein with low spectral dispersion of 1H-15N NMR signals (Fig. 3, B and C, orange spectrum), as previously reported (36). Upon addition of TIM9·10, the Tim23IMS 1H-15N spectrum (Fig. 3B, left) shows only small changes: All cross-peaks are still detectable, and small CSPs are only observed for a few residues at the N terminus, which has higher hydrophobicity (Fig. 3D). This finding suggests only very weak, possibly nonspecific interactions between the very N terminus of Tim23IMS and TIM9·10. In line with this finding, the interaction is not detectable by ITC measurements (Fig. 3E).
Fig. 3. Tim23 has markedly different properties when binding to TIM8·13 and to TIM9·10.

(A) Hydrophobicity of Tim23 (Kyte-Doolittle). (B) NMR spectra of the 15N-labeled soluble Tim23IMS fragment in the presence of TIM9·10 (left, black) and of FL Tim23 bound to TIM9·10 (right, black) are compared to the Tim23IMS fragment in isolation (orange), under identical buffer conditions and NMR parameters. (C) As in (B) but with TIM8·13 instead of TIM9·10. (D) CSP of residues in Tim23IMS upon addition of one (light orange) or five (dark orange) molar equivalents of TIM9·10. (E) Calorimetric titrations for the interaction of TIM9·10 or TIM8·13 (54 μM in the calorimetric cell) with Tim23IMS (1.15 mM in the injecting syringe). Thermograms are displayed in the upper plots, and binding isotherms (ligand-normalized heat effects per injection as a function of the molar ratio, [Tim23IMS]/[chaperone]) are displayed in the lower plots. Control experiments, injecting into a buffer, are shown in blue. (F) Intensity ratio of residues in Tim23IMS in the presence of four molar equivalents of TIM8·13 compared to Tim23IMS alone. (G) CSP of the detectable residues in FL Tim23 attached to TIM9·10 (brown), compared to the soluble Tim23IMS fragment. (H) Intensity ratio of detectable residues in Tim23FL attached to TIM8·13. Note that the ratio was not corrected for differences in sample concentration, and the scale cannot be compared to the one in (G).
The interaction of the hydrophilic Tim23IMS fragment with TIM8·13 is significantly stronger, with pronounced binding effects detected by ITC, and a dissociation constant of Kd = 66 ± 8 μM (Fig. 3E, right; see table S1). The 1H-15N NMR spectrum of Tim23IMS in the presence of TIM8·13 shows strongly reduced peak intensities for most of the residues (Fig. 3C, left). Such a peak broadening is expected when a highly flexible polypeptide binds to a relatively large object such as TIM8·13, thereby inducing faster nuclear spin relaxation and thus broader signals of lower intensity. Analysis of the peak intensity reduction reveals two regions of Tim23 that are particularly involved in the binding: (i) the N-terminal hydrophobic residues, which are also involved in interacting with TIM9·10, and (ii) a long sequence stretch comprising residues from ca. 30 to 80 (Fig. 3F).
To investigate whether TIM8·13 may interact with another soluble protein from the IMS, we performed ITC experiments with the globular protein cytochrome c. No interaction could be detected (fig. S9), suggesting that the TIM8·13-Tim23IMS interaction may be related to the unfolded, flexible character of the latter.
To characterize the conformation of FL Tim23 bound to TIM8·13 and TIM9·10, we prepared Tim23FL-labeled Tim23-chaperone complexes using the method outlined in Fig. 1B. Very similar to the experiments with the Tim23IMS fragment, the signals corresponding to the N-terminal half of Tim23 are still intense in the Tim23FL-TIM9·10 complex (Fig. 3, B and G), revealing that the N-terminal half of Tim23FL does not interact strongly with TIM9·10. The small observed CSPs are localized primarily at the hydrophobic N terminus. In contrast, when Tim23FL is bound to TIM8·13, the signals corresponding to its N-terminal half are severely reduced in intensity, revealing tight contact of the flexible N-terminal half of Tim23 to TIM8·13 (Fig. 3, C and H).
In neither of the two Tim23FL complexes, any additional signals, which may correspond to Tim23’s TM helices, are visible. We ascribe this lack of detectable signals of residues in the TM part to extensive line broadening. The origin of this line broadening may be ascribed to the large size of the complex and likely to additional millisecond time scale dynamics of Tim23’s TM parts in the hydrophobic binding cleft of the chaperones. Such millisecond motions have been found in the TIM9·10-Ggc1 complex (23).
TIM8·13 uses an additional hydrophilic face for protein binding
We probed the binding sites that the chaperones use to interact with Tim23IMS or Tim23FL using NMR spectroscopy on samples, in which only the chaperone was isotope-labeled. The CSPs in the two chaperones upon addition of Tim23IMS reveal distinct binding patterns (Fig. 4A): In TIM8·13, the largest effects involve residues in the hydrophilic top part of the chaperone, between the CX3C motifs, as well as a few residues toward the C-terminal outer ring of helices; in contrast, the corresponding top part of TIM9·10 does not show any significant effects, but CSPs are observed at residues in the hydrophobic binding cleft and, in particular, the N-terminal helix (Fig. 4, B and C). These data, together with the Tim23IMS-detected data in Fig. 3, establish that TIM8·13 uses its hydrophilic top part to bind Tim23’s N-terminal half, while only a short stretch of hydrophobic residues at the very N terminus of Tim23 interacts with the hydrophobic cleft of TIM9·10, which is also the binding site of TM parts (Figs. 1 and 2).
Fig. 4. Tim23IMS and FL Tim23 differ in their interactions with TIM9·10 and TIM8·13 chaperones.
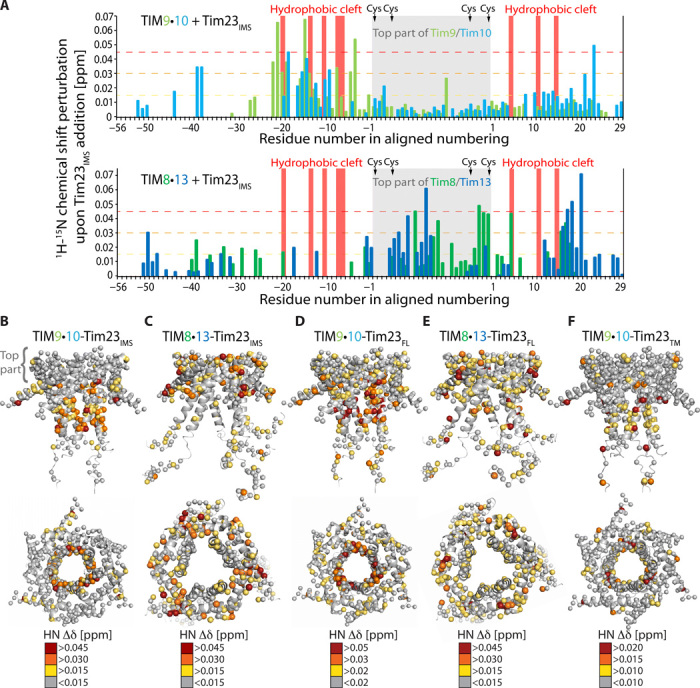
(A) CSPs observed upon addition of the Tim23IMS fragment to TIM9·10 (top) and TIM8·13 (bottom). The chaperone:Tim23IMS ratios were 1:1 (TIM8·13) and 1:3 (TIM9·10). Mapping of Tim23IMS-induced CSPs on TIM9·10 (B) and TIM8·13 (C), showing that while the top part of TIM9·10 does not show any significant CSPs, the corresponding part is the main interacting region of TIM8·13. CSP in complexes of TIM9·10 (D) and TIM8·13 (E) bound to FL Tim23. Tim23TM-induced CSP mapped on TIM9·10 (F), showing similar binding as the FL Tim23.
Chaperone-labeled complexes with Tim23FL confirm these findings and point to the additional effects induced by the bound TM part: In TIM9·10-Tim23FL, large CSP effects are located primarily in the binding cleft, in line with the view that the top part of TIM9·10 is not involved in binding Tim23. In contrast, Tim23FL-induced CSPs are found across the whole TIM8·13, including the hydrophilic top and the hydrophobic cleft (Fig. 4, D and E, and fig. S10).
We furthermore prepared complexes of a truncated Tim23 fragment (Tim23TM, residues 92 to 222), which allows to selectively detect the interaction of the TM part with the chaperones. The TIM9·10-Tim23TM complex features the largest CSPs in the hydrophobic binding cleft, qualitatively similar to the binding site detected with Tim23FL (Fig. 4F and fig. S11). The complex of Tim23TM with TIM8·13 appears to be much less stable than TIM9·10-Tim23TM: In the pull-down experiment, only a very small amount of complex could be obtained, and the complex rapidly precipitated (not shown), excluding NMR analyses. This observation reflects that the hydrophobic cleft of TIM8·13 is less capable of holding a hydrophobic polypeptide than the one of TIM9·10.
Collectively, NMR, ITC, and mutagenesis have revealed that the hydrophobic cleft of both TIM8·13 and TIM9·10 is essential to hold the hydrophobic parts of the clients and that TIM8·13, but not TIM9·10, additionally interacts with the hydrophilic part of Tim23 to increase its affinity. This interaction, which is mediated by the hydrophilic top part of TIM8·13, reduces the conformational flexibility of Tim23’s N-terminal half. The observation that the interaction is driven by hydrophilic contacts supports previous findings of the protein import (25): TIM8·13 was found to interact with hydrophobic membrane precursor only when they were fused to the hydrophilic Tim23IMS part.
We have also investigated whether a given FL Tim23 chain may interact simultaneously with TIM9·10 and TIM8·13, using hydrophobic and hydrophilic interactions, respectively, to form a ternary complex. However, samples containing all three components do not contain detectable amounts of such complexes, and we conclude that the affinity is too low to simultaneously bind two chaperones (fig. S12).
Structural ensembles of chaperone-Tim23 complexes
We integrated the NMR data with further biophysical, structural, and numerical techniques to obtain a full structural and dynamical description of the complexes. We first investigated the complex stoichiometry using size exclusion chromatography coupled to multiangle light scattering (SEC-MALS), NMR-detected diffusion coefficient measurements, and analytical ultracentrifugation (AUC). These methods, which provide estimates of molecular mass (and shape) from orthogonal physical properties (gel filtration and light scattering; translational diffusion), reveal properties best compatible with a 1:1 (chaperone:precursor) stoichiometry (Fig. 5A and fig. S13). Small-angle x-ray scattering (SAXS) data of both TIM9·10-Tim23 and TIM8·13-Tim23 also point to a molecular weight corresponding to a 1:1 complex (SAXS; Fig. 5B). This stoichiometry contrasts the 2:1 (chaperone:precursor) stoichiometry for TIM9·10 holding the 35-kDa large carrier Ggc1 (fig. S2) (23).
Fig. 5. Architecture of the TIM8·13 and TIM9·10 holdases in complex with FL Tim23.
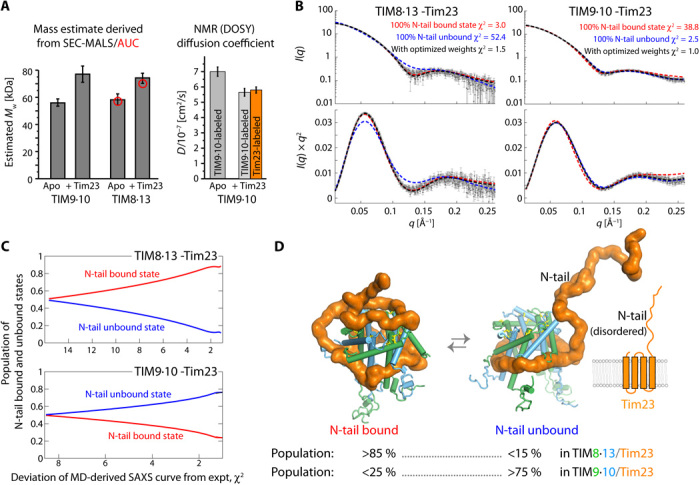
(A) Left: Apparent molecular weights of apo and holo chaperone complexes from SEC-MALS and AUC (red) circles. Right: Translational diffusion coefficients of TIM9·10 (apo) and TIM9·10-Tim23FL from NMR DOSY measurements. Two independent samples were used for the complex, in which either the chaperone or the precursor protein was labeled, as indicated. See also fig. S13. (B) Small-angle x-ray scattering (SAXS) curves (top) and Kratky plot representations thereof for the two chaperone-precursor complexes. The lines are SAXS curves calculated from structural ensembles obtained over 4.25-μs-long MD trajectories, in which the N-terminal half of Tim23 was either in a conformation bound to the top part of the chaperone (red) or in a loose unbound conformation (blue), or from an ensemble in which these two classes of states were present with optimized weights. (C) Goodness of fit of the back-calculated SAXS curves to the experimental SAXS data as a function of the relative weights of the two classes of conformations (bound/unbound). (D) Snapshots of conformations in which Tim23N-tail is either bound or unbound and the best-fit relative weights of the two classes of states as derived from SAXS/MD. More SAXS/MD data and ensemble views are provided in fig. S14 and in movies S1 and S2.
SAXS provides significantly more information, namely, the overall shape of the ensemble of conformations present in solution. Given the flexibility of the complex, these SAXS data are best analyzed by considering explicitly a dynamic ensemble. We used molecular dynamics (MD) simulations to account for the breadth of possible conformations that, collectively, result in the observed scattering. To effectively sample the conformational space of the chaperone-Tim23 complex, we constructed two distinct structural models, in which the N-terminal half of Tim23 is either modeled as a floppy unstructured tail or bound to the hydrophilic upper part of chaperone, denoted as “N-tail unbound” and “N-tail bound” conformations, respectively. In both models, the hydrophobic C-terminal TM domain of Tim23 is bound to the hydrophobic cleft of the chaperone, as identified by NMR (Fig. 4 and fig. S10, C and D) (23). Initiating from both conformations, explicit-solvent atomistic MD simulations (∼4.25 μs in total) were performed to collect the structures for the N-tail unbound and N-tail bound ensembles. In the case of TIM8·13, the N-tail bound ensemble recapitulates the experimentally observed pattern better than the N-tail unbound ensemble (Fig. 3, C and E). We then constructed a mixed ensemble consisting of a mixture of N-tail bound and N-tail unbound states. We used this pool of conformations for further ensemble refinement, with the relative populations of these two ensembles of states as free parameter, using the Bayesian maximum entropy (BME) method guided by experimental SAXS data (23, 37, 38). We found that the experimental SAXS data of TIM8·13-Tim23 are very well reproduced when the mixed ensemble has >85% of the N-tail bound state (Fig. 5, C and D). In contrast, the experimental data of TIM9·10-Tim23 are only well reproduced when the TIM9·10-Tim23 ensemble comprises predominantly the N-tail unbound state. These refined ensembles guided by experimental SAXS data are in excellent agreement with the NMR data, which showed that (i) in the TIM9·10-Tim23 complex, the N-terminal part of Tim23 is predominantly free and flexible, and Tim23 makes contacts only to the hydrophobic cleft of the chaperone, while (ii) in TIM8·13-Tim23, the Tim23IMS part is largely bound to the upper part of the chaperone (Figs. 3 and 4).
The amount of N-tail bound relative to N-tail unbound states is expected to depend on the affinity of the N-tail of Tim23 to the chaperone. The ITC-derived TIM8·13-Tim23IMS affinity (Kd = 66 μM; Fig. 3H) predicts that the population of N-tail bound states is of the order of 75 to 98% (see Methods for details), in excellent agreement with the MD/SAXS-derived value (>85%). This good match of data from the Tim23IMS fragment and Tim23FL suggests that the binding of Tim23’s hydrophilic N-tail does not strongly depend on the presence of the TM part. The low affinity of the N-tail to TIM9·10, reflected by the inability to detect TIM9·10-Tim23IMS binding by ITC, is mirrored by the small population of the N-tail bound states in the FL complex.
To identify the molecular mechanisms underlying the observed differences in N-tail binding, we studied the interactions formed between Tim23 and the chaperones along the MD simulation. An interesting pattern emerges from the analysis of the electrostatic interactions. The top part of TIM8·13 has predominantly polar and negatively charged residues, which are in transient contact with the positive charges of Tim23 N-tail, within a dynamic ensemble of conformations (Fig. 6). For example, three key aspartate or glutamate residues in TIM8·13 appear to be involved in binding of lysine or arginine residues of Tim23IMS (Fig. 6A). In Tim9, a lysine (K51) is present in the top part and contributes a positive charge (the equivalent position in TIM8·13 is a noncharged, polar residue) (Fig. 6B). We hypothesized that the less complementary electrostatic properties of TIM9·10’s top part and Tim23’s N-tail, as compared to TIM8·13, may diminish the affinity of the N-tail to TIM9·10. We attempted to investigate the importance of these charged residues experimentally and prepared single and double mutants that invert the pattern of charged residues. In TIM8·13, we introduced lysine or arginine instead of negatively charged residues, expecting to thereby reduce the affinity to Tim23IMS; conversely, in TIM9·10, we introduced negative charges to promote the Tim23IMS interaction. However, ITC experiments show that most of these mutants do not significantly differ in their binding affinity to Tim23IMS (fig. S9 and table S1). In one of the TIM8·13 mutants, the binding affinity even increases, despite the additional positive charge in the chaperone. These findings suggest that due to the disordered nature of Tim23’s N-tail, its binding with the chaperones might not be dominated by a few strong interactions but instead be contributed by a complex interaction network with many weak and widely distributed interactions, which are tolerant to introduction of the single point mutants that we explored. The MD ensemble (Fig. 6C), due to its limited time scale and force field imperfections (39), may only be able to identify a rather small number of key interaction sites. Unlike the case of this hydrophilic interaction, we were able to identify several single-point mutations in the TIM9·10 hydrophobic motif that abrogate the binding, with a strong phenotype (23).
Fig. 6. Tentative identification of electrostatic interactions from the MD ensemble.
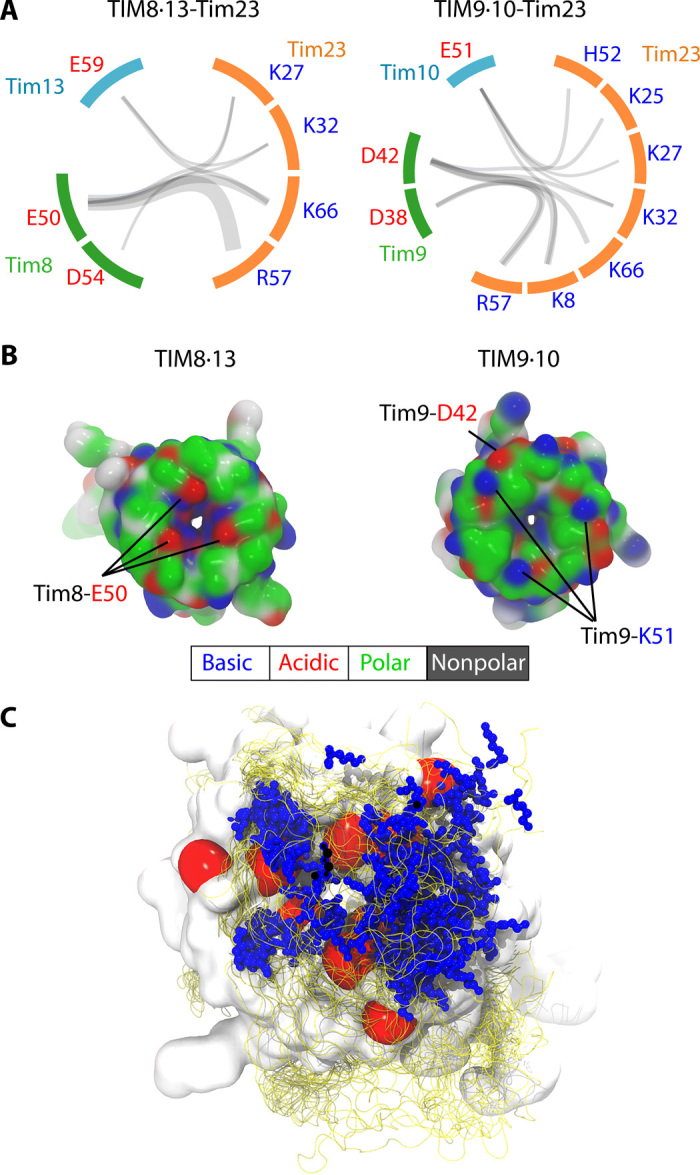
(A) The charged residue pairs forming salt bridges are connected by gray semitransparent lines whose thickness linearly scales with the frequency of the corresponding salt bridge observed in MD simulations. Although more diverse salt bridges were observed in TIM9·10-Tim23 (10 in TIM9·10-Tim23 and 7 in TIM8·13-Tim23), these salt bridges were, on average, less stable than the ones in TIM8·13-Tim23, likely resulting in overall weaker interactions. (B) Snapshots of top views of the two chaperones along MD simulations of their holo forms in complex with TIM23. The top views of the chaperones in the apo forms are shown in fig. S14 (E and F). Residues are color-coded according to the scheme reflected below the figure. (C) Ensemble view of the N-tail bound state of TIM8·13-Tim23. The red surface represents the negatively charged E59 of Tim13 and E50 and D54 of Tim8. Blue stick and ball represents the side chain of positively charged residues (K8, K25, K27, K32, R57, and K66) of Tim23, which is shown as an ensemble of 25 structures. Ensemble view of the N-tail bound state of TIM9·10-Tim23 is shown in fig. S14G.
DISCUSSION
Transfer chaperones (holdases) need to fulfill two contradicting requirements, holding their clients very tightly to avoid their premature release and aggregation while, at the same time, allowing release at the downstream factor. This apparent contradiction is solved by a subtle balance of multiple individually weak interactions and a resulting dynamic complex, wherein the precursor protein samples a wide range of different conformations. This ensemble of conformations results in a high overall affinity; yet, a downstream foldase/insertase can detach the precursor protein from the chaperone without significant energy barrier (40). Balancing the interaction strengths is, thus, crucial to chaperone function. Here, we have revealed a fine-tuning of chaperone-client specificity that involves hydrophobic interactions with the chaperone’s binding cleft and additional hydrophilic interactions, mostly mediated by charged residues, with the chaperone’s top part. Lower hydrophobicity within the binding cleft of TIM8·13 compared to TIM9·10 arises by overall less hydrophobic residues and a positively charged residue (Lys/Arg) that is highly conserved in Tim8. As a consequence, TIM8·13 is less able to hold the TM parts of its clients than TIM9·10 by ca. one order of magnitude. As we showed, replacement of two charged/polar side chains in TIM8·13’s cleft brings TIM8·13 to a similar level as TIM9·10 for holding an all-TM client.
For binding of its native client Tim23, TIM8·13 uses additional hydrophilic interactions to its client’s IMS segment, which is ineffective in the TIM9·10-Tim23 interaction. The additional interaction effectively compensates for the lower affinity of TIM8·13 to the client’s TM part. In the case of Tim23, this additional interaction involves a sequence stretch of at least 35 to 40 residues (Fig. 3, E and G). TIM8·13 has also been shown to be involved in the transport of a Ca2+-regulated mitochondrial carrier, the Asp/Glu carrier (31), which has an additional soluble calmodulin-like domain. Whether this soluble domain is folded or disordered while the TM domain is attached to the hydrophobic chaperone cleft remains to be investigated. It is tempting to speculate that interactions between TIM8·13’s top part and the calmodulin-like hydrophilic part of these carriers are important for this binding, similarly as for the case of Tim23 (fig. S1). Membrane precursor proteins that have been shown not to interact with TIM8·13, such as mitochondrial carriers (Ggc and Aac) and Tim17, lack extended hydrophilic stretches, underlining the importance of those parts in binding (fig. S1). From the sequences of known clients and known “nonclients” of TIM8·13, we propose that a minimum sequence length of about 20 to 25 residues is required for binding.
The nature of these additional hydrophilic interactions appears to involve primarily charged residues that form a complex and wide-connected interaction network that could be hard to suppress by mutating individual sites.
This study provides a rationale why mitochondria contain two very similar IMS chaperone complexes, the essential TIM9·10 and the nonessential TIM8·13 complex. The observation that this dual system is conserved even in humans suggests that the presence of the TIM8·13 system is not just the result of gene duplication, which appears rather often in yeast. The current results propose that for some substrates (like Tim23, or Asp-Glu carrier; see fig. S1), TIM8·13 can contribute stabilizing interactions with the hydrophilic soluble parts. Our competition experiments have also revealed that mitochondrial membrane precursor proteins may be transferred from one TIM chaperone to the other, opening the possibility that these two chaperones truly cooperate in precursor protein transfer to downstream insertases.
Together, our study reveals how a subtle balance of hydrophobic and hydrophilic interactions is used to tune promiscuity versus specificity in molecular chaperones. We propose that a similar balance of interactions determines the clientome of the cellular chaperones.
METHODS
Plasmids
Genes coding for Saccharomyces cerevisiae Tim8 and Tim13 were cloned in the coexpression plasmid pETDuet1. The expressed protein sequences were MSSLSTSDLASLDDTSKKEIATFLEGENSKQKVQMSIHQFTNICFKKCVESVNDSNLSSQEEQCLSNCVNRFLDTNIRIVNGLQNTR (Tim8) and MGSSHHHHHHSQDPSQDPENLYFQGGLSSIFGGGAPSQQKEAATTAKTTPNPIAKELKNQIAQELAVANATELVNKISENCFEKCLTSPYATRNDACIDQCLAKYMRSWNVISKAYISRIQNASASGEI (Tim13). A tobacco etch virus (TEV) cleavage site on Tim13 allows the generation of the final construct starting with GGLSS (the native Tim13 sequence starts with MGLSS). The same approach was used for preparing TIM9·10, including coexpression of the two proteins, with a cleavable His6-tag on one of the proteins (Tim10), as described elsewhere (23). The gene coding for FL S. cerevisiae Tim23 (C98S, C209S, and C213A) with a C-terminal His6-tag was cloned in the expression plasmid pET21b(+). The plasmid for expression of the intrinsically disordered N-terminal domain of S. cerevisiae Tim23IMS (residues 1 to 98) with an N-terminal glutathione S-transferase (GST) tag is described in (36), and the pET10N plasmid encoding Tim23TM is described in (41). The S. cerevisiae Ggc1(C222S) construct was designed with a C-terminal His6-tag in pET21a expression plasmid, reported earlier (23).
Protein expression and purification
We found that TIM9·10 and TIM8·13 chaperone complexes can be obtained by overexpression in either SHuffle T7 or BL21(DE3) Escherichia coli cells. Expression in the former results in soluble protein with correctly formed disulfide bonds, while the latter requires refolding from inclusion bodies. The proteins obtained with either method have indistinguishable properties (SEC, NMR). For TIM9·10, SHuffle expression results in better yield, while we obtain higher TIM8·13 yields with refolding from BL21(DE3). Accordingly, TIM9·10 and unlabeled TIM8·13 were overexpressed in the SHuffle T7 E. coli cells and purified as described previously (23). Overexpression of the isotope-labeled TIM8·13 chaperone complex from the BL21(DE3) E. coli cells was induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG), and the cells were incubated for 4 hours at 37°C. Cell pellets were sonicated, and the inclusion body fraction was resuspended sequentially, first in buffer A [50 mM tris(tris(hydroxymethyl)aminomethane), 150 mM NaCl (pH 7.4)] supplemented with 1% lauryldimethylamine oxide (LDAO) and 1% Triton X-100, then in buffer A supplemented with 1 M NaCl and 1 M urea, and, lastly, in buffer B [50 mM tris, 250 mM NaCl (pH 8.5)]. The last pellet fraction was solubilized in buffer B supplemented with 50 mM dithiothreitol and 3 M guanidine-HCl at 4°C overnight. The TIM8·13 complex was refolded by rapid dilution in buffer B containing 5 mM glutathione and 0.5 mM glutathione disulfide. The complex was purified on a NiNTA affinity column, and the affinity tag was removed with TEV protease and an additional NiNTA purification step. FL precursor proteins, Tim23 and Ggc1, were expressed as inclusion bodies from BL21(DE3) cells, at 37°C during 1.5 and 3 hours, respectively, after adding 1 mM IPTG. Precursor proteins were solubilized in buffer A supplemented with 4 M guanidine-HCl for Tim23 and 6 M guanidine-HCl for Ggc1 at 4°C overnight. Precursor proteins were purified by affinity chromatography in the same denaturating conditions used for solubilization. Imidazole was removed from the precursor protein sample with dialysis in buffer A supplemented with 4 M guanidine-HCl.
GST-tagged Tim23IMS was expressed in the soluble protein fraction from BL21(DE3)Ril+ cells during 4 hours at 25°C, after adding 1 mM IPTG. After sonication of the cell pellets, the soluble protein fraction was incubated with glutathione-agarose resin for 2 hours at 4°C. After washing the unspecifically bound proteins with 10 column volumes (CVs) of buffer A, the GST-tag was cleaved from the Tim23IMS by incubating the resin with 1 mg of TEV protease per 50 mg of the precursor protein, at 4°C overnight. Cleaved Tim23IMS and the protease were collected in the flow-through, and an additional NiNTA purification step was applied to remove the TEV protease from the protein sample. Soluble Tim23IMS was subjected to gel filtration on a Superdex 75 10/300 column and stored in buffer A.
Tim23TM, comprising residues 92 to 222 (41), was produced in E. coli BL21(DE3)Ril+ during 3 hours at 37°C and purified in denaturing conditions as described for the FL Tim23. Chaperone proteins used for detection by NMR experiments were expressed in D2O M9 minimal medium and either labeled with 15NH4Cl (1 g/liter) and D-[2H,13C]glucose (2 g/liter) or specifically labeled on isoleucine, alanine, leucine, and valine side chains using a QLAM-A βIδ1LproRVproR kit from NMR-Bio (www.nmr-bio.com) according to the manufacturer’s instructions. The proteins not detected by NMR in complex samples (i.e., the precursor proteins in complexes directed toward chaperone detection or the chaperone in preprotein-detected experiments) were unlabeled and produced in LB medium. Chaperone-bound Tim23FL was deuterated (produced in D2O M9 medium), while the Tim23IMS fragment was prepared in H2O M9 medium.
The fragments of human VDAC1 peptide (cyclic or linear VDAC257–279) were prepared by solid-phase synthesis as described elsewhere (34), lyophilized, and resolubilized first in dimethyl sulfoxide (DMSO) and then stepwise diluted into buffer, as described elsewhere (23). The peptide used for photo-induced cross-linking differed from the one used for NMR by the substitution of L263 by a Bpa side chain, as used earlier (23, 34).
Preparation of chaperone-precursor protein complexes
Purified precursor protein, i.e., either FL Tim23, the TM Tim23TM fragment, or Ggc1, was bound to NiNTA resin in 4 M guanidine-HCl. The column was washed with five CVs of buffer A supplemented with 4 M guanidine-HCl and with five CVs of buffer A. A twofold excess of the chaperone complex was passed through the column twice. The column was washed with 10 CVs of buffer A, and the precursor-chaperone complex was eluted in 5 CVs of buffer A supplemented with 300 mM imidazole. The precursor-chaperone complex was immediately subjected to dialysis against buffer A before concentrating on Amicon 30-kDa molecular weight cut-off (MWCO) centrifugal filters (1000g). Immediate removal of imidazole was particularly important for the preparation of the less stable Tim23FL-TIM9·10 and Tim23TM-TIM8·13 complex. Complexes of Tim23IMS with TIM8·13 or TIM9·10 were prepared by mixing two purified protein samples and dialysis against buffer A. Formation of the precursor-chaperone complex was verified by SEC on a Superdex 200 column. The resulting complex was further characterized by SEC-MALS. TIM8·13 and TIM8·13-Tim23 were analyzed by AUC. Both experiments were performed at 10°C in buffer A. The amount of eluted complex was estimated from the protein concentration, measured absorbance of the sample at 280 nm, and the sum of the molecular weights and extinction coefficients of the chaperone and precursor protein.
Competition assays
The first competition assay was performed by adding an equimolar mixture of TIM8·13 and TIM9·10 chaperones to the NiNTA-bound precursor protein, Tim23FL or Ggc1. After washing the column, precursor-chaperone complex was eluted in buffer A supplemented with 300 mM imidazole. In the time-dependent competition assay, the complex of a precursor protein and one of the chaperones (TIM8·13 or TIM9·10) was prepared, and then an equimolar amount of the other chaperone was added (time point 0). The reaction mixture was incubated at 30°C. After 0.5, 1, and 3 hours, an aliquot of the reaction mixture was taken and the (newly formed) precursor-chaperone complex was isolated on a NiNTA affinity column. The difference in the amount of specific chaperone, TIM8·13 or TIM9·10, bound to the precursor protein was analyzed by SDS-PAGE and liquid chromatography coupled with mass spectrometry (LC–ESI-TOF–MS, 6210, Agilent Technologies, at the MS platform, IBS Grenoble). Samples for analysis by MS were heat-shocked for 15 min at 90°C, resulting in the dissociation and precipitation of the precursor protein, while the apo-chaperones were recovered in the supernatant after cooling the sample and centrifugation for 10 min at 39 kg. As a reference, samples of precursor proteins, Tim23FL and Ggc1, bound to individual chaperone, TIM8·13 or TIM9·10, were prepared and analyzed in parallel. To be noted, preparation of the TIM8·13-Ggc1 complex, in quantity sufficient for the analysis, was unsuccessful. To calculate the difference in the amount of specific chaperone bound to the precursor protein, normalized areas under the chromatography peaks corresponding to each Tim monomer were used.
ITC experiments
Calorimetric binding experiments of Tim23IMS and TIM chaperones were performed using a MicroCal ITC200 instrument (GE Healthcare). Sixteen successive 2.5-μl aliquots of 1.15 mM Tim23IMS were injected into a sample cell containing 55 μM TIM9·10 or TIM8·13. All ITC data were acquired in buffer A at 20°C. Control experiments included titrating Tim23IMS into buffer A. The enthalpy accompanying each injection was calculated by integrating the resultant exotherm, which corresponds to the released heat as a function of ligand concentration added at each titration point. ITC data were analyzed via the MicroCal Origin software using a single-site binding model and nonlinear least squares fit of thermodynamic binding parameters (∆H, K, and n). An identical procedure was performed for TIM8·13–cytochrome c and TIM9·10–cytochrome c ITC experiments. Cytochrome c was from horse heart (Merck/Sigma-Aldrich). We also performed ITC experiments with the VDAC peptides; no effects could be detected, in line with a millimolar affinity, as already reported for the TIM9·10–cyclic VDAC257–279 peptide (23).
Cross-linking of VDAC257–279
In vitro cross-linking of VDAC257–279 used precisely the protocol described in (23) for TIM9·10. Briefly, 5 μM TIM8·13 was mixed with VDAC257–279 at 0, 25, or 50 μM; incubated for 10 min on ice; and ultraviolet (UV)–illuminated (30 min, 4°C).
We detected several cross-linking adducts of the β-hairpin peptide and the TIM components. Such multiband behavior is similar to the pattern of cross-linking products of this peptide with either TOM (translocase of the outer membrane) subunits or cytosolic chaperones (34, 35). We suppose that this variability can result from a variable number of peptides bound to one molecule of protein and from different cross-linking sites on the protein, which, in turn, can cause different migration behavior in the SDS-PAGE.
SEC-MALS experiments
SEC-MALS experiments were performed at the Biophysical platform (AUC-PAOL) in Grenoble. The experimental setup comprised a high-performance liquid chromatography (HPLC) system (Shimadzu, Kyoto, Japan) consisting of a DGU-20 AD degasser, an LC-20 AD pump, a SIL20-ACHT autosampler, an XL-Therm column oven (WynSep, Sainte Foy d’Aigrefeuille, France), a CBM-20A communication interface, an SPD-M20A UV-visible detector, a miniDAWN TREOS static light scattering detector (Wyatt, Santa Barbara, USA), a DynaPro NanoStar dynamic light scattering detector, and an Optilab rEX refractive index detector. The samples were stored at 4°C, and a volume of 20, 40, 50, or 90 μl was injected on a Superdex 200, equilibrated at 4°C; the buffer was 50 mM tris and 150 mM NaCl filtered at 0.1 μm, at a flow rate of 0.5 ml/min. Bovine serum albumin was used for calibration. Two independent sets of experiments conducted with two different batches of protein samples were highly similar.
Analytical ultracentrifugation
AUC experiments of TIM8·13 and TIM8·13-Tim23 were performed at 50,000 rpm and 10°C, on an XLI analytical ultracentrifuge, with An-60 Ti and An-50 Ti rotors (Beckman Coulter, Palo Alto, USA) and double-sector cells of optical path length 12 and 3 mm equipped with Sapphire windows (Nanolytics, Potsdam, DE). Acquisitions were made using absorbance at 250- and 280-nm wavelength and interference optics. The reference is the buffer 50 mM tris and 150 mM NaCl. The data were processed by Redate software version 1.0.1. The c(s) and Non Interacting Species (NIS) analysis was done with SEDFIT software version 15.01b and Gussi 1.2.0, and the multiwavelength analysis was done with SEDPHAT software version 12.1b.
NMR spectroscopy
All NMR experiments were performed on Bruker Avance III spectrometers operating at 600-, 700-, 850-, or 950-MHz 1H Larmor frequency. The samples were in the NMR buffer [50 mM NaCl, 50 mM tris (pH 7.4)] with 10% (v/v) D2O, unless stated differently. All multidimensional NMR data were analyzed with CCPN (version 2) (42). Diffusion ordered spectroscopy (DOSY) data were analyzed with in-house written python scripts. For calculating CSP data, the contribution of each different nuclei was weighted by the gyromagnetic ratios of the respective nucleus: e.g., the combined 1H-15N CSP was calculated as , where γ are the gyromagnetic ratios.
TIM8·13 and Tim23IMS resonance assignments
For the resonance assignment of TIM8·13, the following experiments were performed: 2D 15N-1H-BEST-TROSY heteronuclear single-quantum coherence (HSQC), 3D BEST-TROSY HNCO, 3D BEST-TROSY HNcaCO, 3D BEST-TROSY HNCA, 3D BEST-TROSY HNcoCA, 3D BEST-TROSY HNcocaCB, and 3D BEST-TROSY HNcaCB (43, 44) and a 3D 15N-NOESY HSQC. The experiments were performed with a 0.236 mM [2H,15N,13C]-labeled TIM8·13 at 308 and 333 K. The NMR resonance assignment of TIM9·10 was reported earlier (23). We collected BEST-TROSY HNCA, HNCO, and HNcoCA experiments to assign Tim23IMS, aided by the previously reported assignment (36).
VDAC titration experiments
Cyclic hVDAC1257–279 peptide was synthesized and lyophilized as described elsewhere (34). The peptide was dissolved in DMSO, and the DMSO concentration was reduced to 10% by stepwise addition of NMR buffer (1:1 in each step). Chaperone, TIM9·10 or TIM8·13, in buffer A was added to yield a final DMSO concentration of 6% and a chaperone concentration of 0.15 mM (TIM9·10) or 0.1 mM (TIM8·13). Combined 15N-1H CSP was calculated from the chemical shifts obtained from the 15N-1H HSQC spectra of the complex samples with a molar ratio of 1:4 for TIM9·10:VDAC and a molar ratio of 1:5 for TIM8·13:VDAC, in comparison to the chemical shifts from the apo-chaperone spectrum. The NMR experiments were performed at 308 K.
Tim23IMS titration experiments
For each titration point, individual samples were prepared by mixing two soluble protein samples and monitored using 15N-1H-BEST-TROSY HSQC experiments at 283 K (for Tim23 observed experiment) or at 308 K (for chaperone observed experiments). Titration samples with 100 μM [15N]-labeled Tim23IMS with molar ratios for Tim23IMS:TIM8·13 from 1:0 to 1:4 and molar ratios for Tim23IMS:TIM9·10 from 1:0 to 1:5 were used. For the chaperone observed experiments, used samples contained 200 μM [2H,13C,15N]-labeled TIM8·13 with 1:0 and 1:1 molar ratios of Tim23IMS and 350 μM [2H,13C,15N]-labeled TIM9·10 with 1:0 and 1:3 molar ratios of Tim23IMS.
NMR experiments with the Tim23FL
Complexes of the chaperones with the FL Tim23 were prepared as indicated above (preparation of chaperone-precursor protein complexes). Peak positions (chemical shifts) of the amide backbone sites of TIM8·13, apo and in complex with Tim23FL, were obtained from the 1H-15N HSQC experiments at 308 K, with 120 μM [13CH3-ILV]-TIM8·13-Tim23FL sample. Similarly, to calculate combined 15N-1H and 13C-1H CSPs, chemical shifts of the amide backbone and ILVA-13CH3 groups of TIM9·10, apo and in complex with Tim23FL, were obtained from the 1H-15N HSQC and 1H-13C heteronuclear multiple-quantum coherence (HMQC) experiments at 288 K. Sample of [13CH3-ILVA]-TIM9·10 with Tim23FL was at 140 μM concentration. For the CSP calculations with the complexes of [2H-15N]-labeled Tim23FL and the chaperones (190 μM complex with TIM8·13 and 61 μM complex with TIM9·10), chemical shifts from 1H-15N HSQC experiments at 288 K were used in comparison to the chemical shifts of apo-Tim23IMS.
Diffusion ordered spectroscopy
Diffusion-ordered NMR spectroscopy (DOSY) experiments were performed at 288 K and 600-MHz 1H Larmor frequency. Diffusion constants were derived from a series of 1D 1H spectra either over the methyl region (methyl-selective DOSY experiments, for 13CH3-ILVA–labeled apo and Tim23FL-bound TIM9·10) or over the amide region (for [15N]Tim23FL-TIM9·10). Diffusion coefficients were obtained from fitting integrated 1D intensities as a function of the gradient strength at constant diffusion delay.
SAXS data collection and analysis
SAXS data were collected at ESRF (European Synchrotron Radiation Facility) BM29 beamline (45) with a Pilatus 1M detector (Dectris) at a distance of 2.872 m from the 1.8-mm-diameter flow-through capillary. Data on TIM8·13 were collected in a batch mode. The x-ray energy was 12.5 keV, and the accessible q range was 0.032 to 4.9 nm−1. The incoming flux at the sample position was in the order of 1012 photons/s in 700 mm × 700 mm. All images were automatically azimuthally averaged with pyFAI (46). SAXS data of pure TIM8·13 were collected at 1, 2.5, and 5 mg/ml using the BioSAXS sample changer (47). Ten frames of 1 s were collected for each concentration. Exposures with radiation damage were discarded, the remaining frames were averaged, and the background was subtracted by an online processing pipeline (48). Data from the three concentrations were merged following standard procedures to create an idealized scattering curve, using PRIMUS from the ATSAS package (49). The pair distribution function p(r) was calculated using GNOM (50).
Online purification of the TIM8·13-Tim23FL and TIM9·10-Tim23FL complexes using gel-filtration column (HiLoad 16/600 Superdex 200 PG) was performed with an HPLC system (Shimadzu, France), as described in (51). The HPLC system was directly coupled to the flow-through capillary of SAXS exposure unit. The flow rate for all online experiments was 0.2 ml/min. Data collection was performed continuously throughout the chromatography run at a frame rate of 1 Hz. All SAXS data have been deposited on SASBDB (Small Angle Scattering Biological Data Bank).
MD simulations and fitting of SAXS data
The initial model of Tim23 was built using I-TASSER (Iterative Threading ASSEmbly Refinement) (52) and QUARK web servers (53), which predicted a long unstructured N-terminal tail and four/five helical structures in the TM domain. The structure of TIM9·10 hexamer built in our previous work (23) was used as the initial model of TIM9·10 chaperone and as the template to build the model of TIM8·13 chaperone based on the sequence of yeast Tim8 and Tim13 (UniProt IDs: P57744 and P53299) by homology modeling with MODELLER (54). [Note that in the crystal structure of TIM8·13 (PDB-ID 3CJH), more than 75 residues are missing in each Tim8-Tim13 pair, thus requiring model building.] The disulfide bonds related to the twin CX3C motif were also kept in these models. The structures of the TIM8·13 hexamer and Tim23 were subsequently used to build the full structure of the TIM8·13-Tim23 complex by manually wrapping the helical structures of the TM domain of Tim23 around the hydrophobic cleft of TIM8·13, which has been identified by NMR, and leaving the unstructured N terminus of Tim23 as a floppy tail. The complex structure was further optimized by energy minimization and relaxation in 100-ns MD simulations using the simulation protocol as described in the following section. This model was used to generate the so-called N-tail unbound ensemble, in which the N-terminal half of Tim23 is free in solution. On the basis of the N-tail unbound model of the TIM8·13-Tim23 complex, we further constructed the N-tail bound model, in which the N-terminal half of Tim23 is in contact with the upper part of TIM8·13. This was achieved by adding a restraint term in the force field using PLUMED plugin (55), Vrestraints, which is a half-harmonic potential of the form of k(R − R0)2 when R is larger than R0, and zero when R is less than R0. Here, R is the distance between the center of mass of Tim23Nter and the top part of the chaperone. We used R0 = 1 nm and k = 400 kJ mol−1. The N-tail bound and unbound models for the TIM9·10-Tim23 complex were constructed by replacing TIM8·13 with TIM9·10 based on the corresponding TIM8·13-Tim23 models.
The TIM8·13-Tim23 complex in the N-tail unbound conformation was placed into a periodic cubic box with sides of 17.5 nm solvated with TIP3P water molecules containing Na+ and Cl− ions at 0.10 M, resulting in ∼700,000 atoms in total. To reduce the computational cost, the complex in N-tail bound conformation was placed in a smaller cubic box with sides of 12.9 nm, resulting in ∼300,000 atoms in total. The systems of the TIM9·10-Tim23 complex have similar size as the TIM8·13-Tim23 systems in the corresponding states. The apo TIM8·13 chaperone was placed into a periodic cubic box with sides of 12.0 nm, containing ∼230,000 atoms.
The Amber ff99SB-disp force field (56) was used for all simulations. The temperature and pressure were kept constant at 300 K using the v-rescale thermostat and at 1.0 bar using the Parrinello-Rahman barostat with a 2-ps time coupling constant, respectively. Neighbor searching was performed every 10 steps. The particle-meshed-Ewald (PME) algorithm was used for electrostatic interactions. A single cutoff of 1.0 nm was used for both the PME algorithm and van der Waals interactions. A reciprocal grid of 96 × 96 × 96 cells was used with fourth-order B-spline interpolation. The hydrogen mass repartitioning technique (57) was used with a single linear constraint solver (LINCS) iteration (expansion order 6) (58), allowing simulations to be performed with an integration time step of 4 fs. MD simulations were performed using GROMACS 2018 or 2019 (59).
A total of 4.25-μs trajectories was collected to sample the conformational space of the chaperone-Tim23 complexes in both N-tail bound and N-tail unbound states. Four-microsecond trajectories were also collected to sample the ensemble of apo TIM8·13 chaperone. These sampled conformations were used for further ensemble refinement using the BME method (37, 38) guided by experimental SAXS data as described in our previous work (23). The distribution of both states, in principle, could be identified from the force field but needs substantial sampling. Therefore, instead of estimating the prior by large-scale MD simulations, we assigned equal weight (50%) for both states by inputting equal number of conformations (5000) into the mixed ensemble so without bias to either state. By tuning the regularization parameter in the BME reweighting algorithm, we adjusted the conformational weights in variant degrees to improve the fitting with experimental SAXS data.
The hydrogen bond and salt bridge formation between the N-terminal tail of Tim23 (residues 1 to 100) and the top surface of the chaperones was analyzed by GetContacts scripts (https://getcontacts.github.io/) and visualized using Flareplot (https://gpcrviz.github.io/flareplot/). Protein structures were visualized with PyMOL and VMD.
Calculations of affinities and populations
Estimation of the population of N-tail bound states from ITC-derived K d
We attempted to link the ITC-derived dissociation constant of the Tim23IMS fragment to the populations of bound and unbound states in the Tim23FL-chaperone complexes, using a rationale akin to the one outlined earlier for binding of disordered proteins to two subsites (60). Briefly, we treat the N-terminal tail of Tim23 as a ligand and the remaining bound complex as the target protein, and then the relationship between the population of the bound state (Pbound) and the binding affinity (Kd) can be written as Pbound/(1 − Pbound) = Ceff/Kd, where Ceff is the effective concentration of the disordered N-tail, which was estimated to be between 0.2 and 3 mM from the MD simulations, resulting in the estimation of Pbound to be between 75 and 98%.
Estimation of the Kd ratio from competition assays
Determining dissociation constants of TIM chaperones to its insoluble client proteins is hampered by the impossibility to form the complexes by solution methods such as titration, as it requires the pull-down method outlined in Fig. 1A. Nonetheless, the amount of TIM8·13-Tim23 and TIM9·10-Tim23 complexes obtained in the competition assays (Fig. 1) can provide an estimate of the relative affinities. The dissociation constants can be written from the concentrations as follows
where [TIM8·13 − Tim23] denotes the concentration of the formed chaperone-precursor complex, and [TIM8·13] and [Tim23] are the concentrations of free chaperone and precursor protein in solution. The latter is negligible, as no free precursor protein is eluted from the column (some aggregated precursor protein was removed from the equilibrium). Both chaperones have been applied at the same concentration c0 = [TIM8 · 13] + [TIM8 · 13 − Tim23] = [TIM9 · 10] + [TIM9 · 10 − Tim23] to the resin-bound precursor protein that was present at a concentration b0 = [Tim23] + [TIM9 · 10 − Tim23] + [TIM8 · 13 − Tim23].
Using the ratio of formed complex obtained in the competition assay, r = [TIM9 · 10 − Tim23]/[TIM8 · 13 − Tim23] leads to
The experimental protocol does not allow to determine with precision the concentrations of precursor protein (b0) and each chaperone (c0), as the former is bound to a resin. As the chaperone was added in excess, and some of the precursor protein precipitated on the column, we can safely assume c0 ≤ b0. With an experimentally found ratio of formed complexes of r = 5 and assuming that c0/b0 assumes the values of 1 to 5, the Kd ratio falls in the range of 1:25 to 1:6, i.e., ca. one order of magnitude.
Acknowledgments
We thank N. Wiedemann and C. Lindau for many insightful discussions and exploratory in vivo experiments. We thank A. Le Roy and C. Ebel (SEC-MALS; AUC), L. Signor (MS), and C. Mas (ITC) for excellent support. We thank H. Kalbacher (Universität Tübingen) for synthesis of the VDAC peptides, K. Giandoreggio-Barranco for preparing Tim23 samples, and U. Guillerm for support with protein production. We are grateful to B. Brutscher, A. Vallet, and A. Favier for excellent NMR platform operation and management. We are grateful to M. Zweckstetter and N. Pfanner for providing the Tim23IMS and Tim23TM plasmids, respectively. Funding: This study was supported by the European Research Council (StG-2012-311318-ProtDyn2Function) and the Agence Nationale de la Recherche (ANR-18-CE92-0032-MitoMemProtImp). This work used the platforms of the Grenoble Instruct-ERIC center (ISBG; UMS 3518 CNRS-CEA-UGA-EMBL) within the Grenoble Partnership for Structural Biology (PSB), supported by FRISBI (ANR-10-INSB-05-02 and ANR-10-INBS-0005) and GRAL (ANR-10-LABX-49–01), financed within the University Grenoble Alpes graduate school (Ecoles Universitaires de Recherche) CBH-EUR-GS (ANR-17-EURE-0003). Y.W. and K.L.-L. were supported by the BRAINSTRUC structural biology initiative from the Lundbeck Foundation. We acknowledge access to computational resources from the Danish National Supercomputer for Life Sciences (Computerome) and the ROBUST Resource for Biomolecular Simulations (supported by the Novo Nordisk Foundation). This work was furthermore supported by the Deutsche Forschungsgemeinschaft (RA 1028/8-1,2 to D.R.) Author contributions: K.W. and P.S. initiated this project. I.S., A.H., O.D., D.C., B.B., and K.W. prepared protein samples. I.S. performed the pull-down and competition assays. I.S., K.W., B.B., O.D., and P.S. performed and analyzed NMR data. K.W. and M.B. performed and analyzed SAXS experiments. Y.W. and K.L.-L. performed and analyzed MD simulations. T.J. and D.R. performed/analyzed cross-linking experiments. All authors contributed ideas and discussion. I.S., Y.W., and P.S. made the figures. P.S., I.S., and Y.W. wrote the manuscript with input from all authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The chemical shift assignments of TIM8·13 have been deposited in the BioMagResBank (www.bmrb.wisc.edu) under accession number 50213. All MD models and SAXS data have been deposited in the SASBDB (www.sasbdb.org) under accession numbers SASDH89 (TIM8·13-Tim23), SASDJP4 (TIM9·10-Tim23), SASDJQ4 (TIM8·13), and SASDEF2 [TIM9·10 (23)]. All CSP data have been deposited on Mendeley data (http://dx.doi.org/10.17632/8cr8rvtddm.1). Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/51/eabd0263/DC1
REFERENCES AND NOTES
- 1.Powers E. T., Morimoto R. I., Dillin A., Kelly J. W., Balch W. E., Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 78, 959–991 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Kim Y. E., Hipp M. S., Bracher A., Hayer-Hartl M., Hartl F. U., Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 82, 323–355 (2013). [DOI] [PubMed] [Google Scholar]
- 3.Kim D. H., Lee J.-E., Xu Z.-Y., Geem K. R., Kwon Y., Park J. W., Hwang I., Cytosolic targeting factor AKR2A captures chloroplast outer membrane-localized client proteins at the ribosome during translation. Nat. Commun. 6, 6843 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Bose D., Chakrabarti A., Substrate specificity in the context of molecular chaperones. IUBMB Life 69, 647–659 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Rüdiger S., Germeroth L., Schneider-Mergener J., Bukau B., Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. EMBO J. 16, 1501–1507 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blond-Elguindi S., Cwirla S. E., Dower W. J., Lipshutz R. J., Sprang S. R., Sambrook J. F., Gething M. J., Affinity panning of a library of peptides displayed on bacteriophages reveals the binding specificity of BiP. Cell 75, 717–728 (1993). [DOI] [PubMed] [Google Scholar]
- 7.Knoblauch N. T., Rüdiger S., Schönfeld H. J., Driessen A. J., Schneider-Mergener J., Bukau B., Substrate specificity of the SecB chaperone. J. Biol. Chem. 274, 34219–34225 (1999). [DOI] [PubMed] [Google Scholar]
- 8.Koldewey P., Stull F., Horowitz S., Martin R., Bardwell J. C. A., Forces driving chaperone action. Cell 166, 369–379 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He L., Sharpe T., Mazur A., Hiller S., A molecular mechanism of chaperone-client recognition. Sci. Adv. 2, e1601625 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hiller S., Burmann B. M., Chaperone–client complexes: A dynamic liaison. J. Magn. Reson. 289, 142–155 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Genevaux P., Keppel F., Schwager F., Langendijk-Genevaux P. S., Hartl F. U., Georgopoulos C., In vivo analysis of the overlapping functions of DnaK and trigger factor. EMBO Rep. 5, 195–200 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bukau B., Deuerling E., Pfund C., Craig E. A., Getting newly synthesized proteins into shape. Cell 101, 119–122 (2000). [DOI] [PubMed] [Google Scholar]
- 13.Burmann B. M., Gerez J. A., Matečko-Burmann I., Campioni S., Kumari P., Ghosh D., Mazur A., Aspholm E. E., Šulskis D., Wawrzyniuk M., Bock T., Schmidt A., Rüdiger S. G. D., Riek R., Hiller S., Regulation of α-synuclein by chaperones in mammalian cells. Nature 577, 127–132 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burmann B. M., Wang C., Hiller S., Conformation and dynamics of the periplasmic membrane-protein-chaperone complexes OmpX-Skp and tOmpA-Skp. Nat. Struct. Mol. Biol. 20, 1265–1272 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Thoma J., Burmann B. M., Hiller S., Müller D. J., Impact of holdase chaperones Skp and SurA on the folding of β-barrel outer-membrane proteins. Nat. Struct. Mol. Biol. 22, 795–802 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Saio T., Guan X., Rossi P., Economou A., Kalodimos C. G., Structural basis for protein antiaggregation activity of the trigger factor chaperone. Science 344, 1250494 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang Y., Rossi P., Kalodimos C. G., Structural basis for client recognition and activity of Hsp40 chaperones. Science 365, 1313–1319 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang C., Rossi P., Saio T., Kalodimos C. G., Structural basis for the antifolding activity of a molecular chaperone. Nature 537, 202–206 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wiedemann N., Pfanner N., Mitochondrial machineries for protein import and assembly. Annu. Rev. Biochem. 86, 685–714 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Sirrenberg C., Endres M., Fölsch H., Stuart R. A., Neupert W., Brunner M., Carrier protein import into mitochondria mediated by the intermembrane proteins Tim10/Mrs11 and Tim12/Mrs5. Nature 391, 912–915 (1998). [DOI] [PubMed] [Google Scholar]
- 21.Koehler C. M., Jarosch E., Tokatlidis K., Schmid K., Schweyen R. J., Schatz G., Import of mitochondrial carriers mediated by essential proteins of the intermembrane space. Science 279, 369–373 (1998). [DOI] [PubMed] [Google Scholar]
- 22.Lionaki E., de Marcos Lousa C., Baud C., Vougioukalaki M., Panayotou G., Tokatlidis K., The essential function of Tim12 in vivo is ensured by the assembly interactions of its C-terminal domain. J. Biol. Chem. 283, 15747–15753 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weinhäupl K., Lindau C., Hessel A., Wang Y., Schütze C., Jores T., Melchionda L., Schönfisch B., Kalbacher H., Bersch B., Rapaport D., Brennich M., Lindorff-Larsen K., Wiedemann N., Schanda P., Structural basis of membrane protein chaperoning through the mitochondrial intermembrane space. Cell 175, 1365–1379.e25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koehler C. M., Leuenberger D., Merchant S., Renold A., Junne T., Schatz G., Human deafness dystonia syndrome is a mitochondrial disease. Proc. Natl. Acad. Sci. U.S.A. 96, 2141–2146 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paschen S. A., Rothbauer U., Káldi K., Bauer M. F., Neupert W., Brunner M., The role of the TIM8-13 complex in the import of Tim23 into mitochondria. EMBO J. 19, 6392–6400 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roesch K., Curran S. P., Tranebjaerg L., Koehler C. M., Human deafness dystonia syndrome is caused by a defect in assembly of the DDP1/TIMM8a-TIMM13 complex. Hum. Mol. Genet. 11, 477–486 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Kang Y., Anderson A. J., Jackson T. D., Palmer C. S., De Souza D. P., Fujihara K. M., Stait T., Frazier A. E., Clemons N. J., Tull D., Thorburn D. R., Conville M. J. Mc, Ryan M. T., Stroud D. A., Stojanovski D., Function of hTim8a in complex IV assembly in neuronal cells provides insight into pathomechanism underlying Mohr-Tranebjærg syndrome. eLife 8, e48828 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morgenstern M., Stiller S. B., Lübbert P., Peikert C. D., Dannenmaier S., Drepper F., Weill U., Höß P., Feuerstein R., Gebert M., Bohnert M., van der Laan M., Schuldiner Mc, Schütze C., Oeljeklaus S., Pfanner N., Wiedemann N., Warscheid B., Definition of a high-confidence mitochondrial proteome at quantitative scale. Cell Rep. 19, 2836–2852 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leuenberger D., Bally N. A., Schatz G., Koehler C. M., Different import pathways through the mitochondrial intermembrane space for inner membrane proteins. EMBO J. 18, 4816–4822 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davis A. J., Sepuri N. B., Holder J., Johnson A. E., Jensen R. E., Two intermembrane space TIM complexes interact with different domains of Tim23p during its import into mitochondria. J. Cell Biol. 150, 1271–1282 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roesch K., Hynds P. J., Varga R., Tranebjaerg L., KoehlerKarin Roesch C. M., Hynds P. J., Varga R., Tranebjaerg L., Koehler C. M., The calcium-binding aspartate/glutamate carriers, citrin and aralar1, are new substrates for the DDP1/TIMM8a-TIMM13 complex. Hum. Mol. Genet. 13, 2101–2111 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Hoppins S. C., Nargang F. E., The Tim8-Tim13 complex of neurospora crassa functions in the assembly of proteins into both mitochondrial membranes. J. Biol. Chem. 279, 12396–12405 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Habib S. J., Waizenegger T., Lech M., Neupert W., Rapaport D., Assembly of the TOB complex of mitochondria. J. Biol. Chem. 280, 6434–6440 (2005). [DOI] [PubMed] [Google Scholar]
- 34.Jores T., Klinger A., Groß L. E., Kawano S., Flinner N., Duchardt-Ferner E., Wöhnert J., Kalbacher H., Endo T., Schleiff E., Rapaport D., Characterization of the targeting signal in mitochondrial β-barrel proteins. Nat. Commun. 7, 12036 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jores T., Lawatscheck J., Beke V., Franz-Wachtel M., Yunoki K., Fitzgerald J. C., Macek B., Endo T., Kalbacher H., Buchner J., Rapaport D., Cytosolic Hsp70 and Hsp40 chaperones enable the biogenesis of mitochondrial β-barrel proteins. J. Cell Biol. 217, 3091–3108 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.de la Cruz L., Bajaj R., Becker S., Zweckstetter M., The intermembrane space domain of Tim23 is intrinsically disordered with a distinct binding region for presequences. Protein Sci. 19, 2045–2054 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Orioli S., Larsen A. H., Bottaro S., Lindorff-Larsen K., How to learn from inconsistencies: Integrating molecular simulations with experimental data. Prog. Mol. Biol. Transl. Sci. 170, 123–176 (2020). [DOI] [PubMed] [Google Scholar]
- 38.Bottaro S., Bengtsen T., Lindorff-Larsen K., Integrating molecular simulation and experimental data: A bayesian/maximum entropy reweighting approach. Methods Mol. Biol. 2112, 219–240 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Ahmed M. C., Papaleo E., Lindorff-Larsen K., How well do force fields capture the strength of salt bridges in proteins? PeerJ 6, e4967 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hiller S., Chaperone-bound clients: The importance of being dynamic. Trends Biochem. Sci. 44, 517–527 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Truscott K. N., Kovermann P., Geissler A., Merlin A., Meijer M., Driessen A. J., Rassow J., Pfanner N., Wagner R., A presequence- and voltage-sensitive channel of the mitochondrial preprotein translocase formed by Tim23. Nat. Struct. Biol. 8, 1074–1082 (2001). [DOI] [PubMed] [Google Scholar]
- 42.Vranken W. F., Boucher W., Stevens T. J., Fogh R. H., Pajon A., Llinas M., Ulrich E. L., Markley J. L., Ionides J., Laue E. D., The CCPN data model for NMR spectroscopy: Development of a software pipeline. Proteins 59, 687–696 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Schanda P., Van Melckebeke H., Brutscher B., Speeding up three-dimensional protein NMR experiments to a few minutes. J. Am. Chem. Soc. 128, 9042–9043 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Favier A., Brutscher B., Recovering lost magnetization: Polarization enhancement in biomolecular NMR. J. Biomol. NMR 49, 9–15 (2011). [DOI] [PubMed] [Google Scholar]
- 45.Pernot P., Round A., Barrett R., De Maria Antolinos A., Gobbo A., Gordon E., Huet J., Kieffer J., Lentini M., Mattenet M., Morawe C., Mueller-Dieckmann C., Ohlsson S., Schmid W., Surr J., Theveneau P., Zerrad L., McSweeney S., Upgraded ESRF BM29 beamline for SAXS on macromolecules in solution. J. Synchrotron Radiat. 20, 660–664 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ashiotis G., Deschildre A., Nawaz Z., Wright J. P., Karkoulis D., Picca F. E., Kieffer J., The fast azimuthal integration Python library: PyFAI. J. Appl. Crystallogr. 48, 510–519 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Round A., Felisaz F., Fodinger L., Gobbo A., Huet J., Villard C., Blanchet C. E., Pernot P., Sweeney S. M., Roessle M., Svergun D. I., Cipriani F., BioSAXS sample changer: A robotic sample changer for rapid and reliable high-throughput X-ray solution scattering experiments. Acta Crystallogr. Sect. D Biol. Crystallogr. 71, 67–75 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brennich M. E., Kieffer J., Bonamis G., De Maria Antolinos A., Hutin S., Pernot P., Round A., Online data analysis at the ESRF bioSAXS beamline, BM29. J. Appl. Crystallogr. 49, 203–212 (2016). [Google Scholar]
- 49.Petoukhov M. V., Franke D., Shkumatov A. V., Tria G., Kikhney A. G., Gajda M., Gorba C., Mertens H. D. T., Konarev P. V., Svergun D. I., New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 45, 342–350 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Svergun D. I., Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 (1992). [Google Scholar]
- 51.Brennich M. E., Round A. R., Hutin S., Online size-exclusion and ion-exchange chromatography on a SAXS beamline. J. Vis. Exp. 2017, e54861 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yang J., Yan R., Roy A., Xu D., Poisson J., Zhang Y., The i-tasser suite: Protein structure and function prediction. Nat. Methods 12, 7–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu D., Zhang Y., Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins 80, 1715–1735 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Webb B., Sali A., Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics 47, 5–6 (2014). [DOI] [PubMed] [Google Scholar]
- 55.Bonomi M., Bussi G., Camilloni C., Tribello G. A., Banáš P., Barducci A., Bernetti M., Bolhuis P. G., Bottaro S., Branduardi D., Capelli R., Carloni P., Ceriotti M., Cesari A., Chen H., Chen W., Colizzi F., De S., De La Pierre M., Donadio D., Drobot V., Ensing B., Ferguson A. L., Filizola M., Fraser J. S., Fu H., Gasparotto P., Gervasio F. L., Giberti F., Gil-Ley A., Giorgino T., Heller G. T., Hocky G. M., Iannuzzi M., Invernizzi M., Jelfs K. E., Jussupow A., Kirilin E., Laio A., Limongelli V., Lindorff-Larsen K., Löhr T., Marinelli F., Martin-Samos L., Masetti M., Meyer R., Michaelides A., Molteni C., Morishita T., Nava M., Paissoni C., Papaleo E., Parrinello M., Pfaendtner J., Piaggi P., Piccini G. M., Pietropaolo A., Pietrucci F., Pipolo S., Provasi D., Quigley D., Raiteri P., Raniolo S., Rydzewski J., Salvalaglio M., Sosso G. C., Spiwok V., Šponer J., Swenson D. W. H., Tiwary P., Valsson O., Vendruscolo M., Voth G. A., White A., Promoting transparency and reproducibility in enhanced molecular simulations. Nat. Methods 16, 670–673 (2019). [DOI] [PubMed] [Google Scholar]
- 56.Robustelli P., Piana S., Shaw D. E., Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. U.S.A. 115, E4758–E4766 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hopkins C. W., Grand S. L., Walker R. C., Roitberg A. E., Long-time-step molecular dynamics through hydrogen mass repartitioning. J. Chem. Theory Comput. 11, 1864–1874 (2015). [DOI] [PubMed] [Google Scholar]
- 58.Feenstra K. A., Hess B., Berendsen H. J. C., Improving efficiency of large time-scale molecular dynamics simulations of hydrogen-rich systems. J. Comput. Chem. 20, 786–798 (1999). [DOI] [PubMed] [Google Scholar]
- 59.Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., Lindahl E., Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015). [Google Scholar]
- 60.Zhou H.-X., Pang X., Lu C., Rate constants and mechanisms of intrinsically disordered proteins binding to structured targets. Phys. Chem. Chem. Phys. 14, 10466–10476 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hiller S., Garces R. G., Malia T. J., Orekhov V. Y., Colombini M., Wagner G., Solution structure of the integral human membrane protein VDAC-1 in detergent micelles. Science 321, 1206–1210 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/51/eabd0263/DC1