

DCAF1 degrades the ribosome assembly factor PWP1 to control ribosome biogenesis, cell proliferation, and development.
Abstract
Evolutionarily conserved DCAF1 is a major substrate receptor for the DDB1-CUL4-ROC1 E3 ubiquitin ligase (CRL4) and controls cell proliferation and development. The molecular basis for these functions is unclear. We show here that DCAF1 loss in multiple tissues and organs selectively eliminates proliferating cells and causes perinatal lethality, thymic atrophy, and bone marrow defect. Inducible DCAF1 loss eliminates proliferating, but not quiescent, T cells and MEFs. We identify the ribosome assembly factor PWP1 as a substrate of the CRL4DCAF1 ligase. DCAF1 loss results in PWP1 accumulation, impairing rRNA processing and ribosome biogenesis. Knockdown or overexpression of PWP1 can rescue defects or cause similar defects as DCAF1 loss, respectively, in ribosome biogenesis. DCAF1 loss increases free RPL11, resulting in L11-MDM2 association and p53 activation. Cumulatively, these results reveal a critical function for DCAF1 in ribosome biogenesis and define a molecular basis of DCAF1 function in cell proliferation and development.
INTRODUCTION
As the molecular factory for protein synthesis, the ribosome underlies the cell’s capacity to grow. It is estimated that in a rapidly growing yeast cell, 60% of total transcription is devoted to ribosomal RNA (rRNA), and 50% of RNA polymerase II transcription and 90% of mRNA splicing are devoted to ribosomal proteins (RPs) (1). This extraordinary use of resources makes it critical for the economy of the cell to tightly regulate ribosome biogenesis, the process of making ribosomes. Defects in the molecular components involved in ribosome biogenesis cause a range of pathologies in different organisms. In Drosophila, mutations in as many as 64 ribosomal protein genes are linked to Minute syndrome, a collection of haploinsufficient phenotypes characterized by prolonged development, short and thin bristles, and poor fertility and viability (2, 3). In humans, an assortment of inherited or sporadic disorders known as ribosomopathies are linked to mutations in either ribosomal protein genes or ribosome biogenesis factors (4–6). A number of studies have also established causal associations between inherited mutations affecting ribosome biogenesis and elevated cancer risk (7, 8). It is therefore expected that cells have developed a surveillance system to monitor the status of the translational machinery. For example, ribosomopathies lead to “ribosomal stress” signals that converge on the p53 signaling pathway in affected cells and tissues (9).
The eukaryotic ribosome consists of two subunits formed by the association of 79 ribosomal proteins with 4 distinct rRNAs. The small subunit (40S) comprises the 18S rRNA assembled to 33 ribosomal proteins, whereas the large subunit (60S) contains the 5S, 5.8S, and 28S (25S in yeast) rRNAs associated with 46 ribosomal proteins. Ribosome biogenesis is an ordered process that begins in the nucleoli, where RNA polymerase I (Pol I) transcribes rRNA gene repeats into long precursor rRNAs (pre-rRNAs). Each pre-rRNA contains three rRNA segments in the order of 18S, 5.8S, and 28S that are surrounded by external transcribed sequences at 5′ and 3′ ends (5′- and 3′-ETS) and separated by two internal transcribed spacers (ITS1 and ITS2). ETSs and ITSs are removed through a series of highly coordinated but poorly understood endonucleolytic cleavage reactions. Ribosomal proteins are deposited onto pre-rRNA during transcription and rRNA maturation to form nascent pre-ribosomes that are then cleaved into the precursors of 40S and 60S subunits in the nucleolus. Pre-60S and pre-40S are then exported through the nucleoplasm into the cytoplasm, where they join the pool of translationally competent 80S ribosomal subunits (10). This assembly process is very complex, involving an estimated 200 ribosome assembly factors in yeast and more than 300 in mammalian cells (11, 12). Most assembly factors were identified by genetic analyses or protein complex purification. Their biochemical function and regulation, and by extension the regulation of ribosome biogenesis, remain poorly understood.
The ubiquitin pathway plays a critical role in the regulation of most cellular processes via an enzymatic cascade, where E1 and E2 enzymes catalyze the activation and conjugation of ubiquitin, while E3s confer reaction specificity through substrate recruitment (13, 14). Comprising the largest family of E3 ligases is the cullin RING E3 ligase (CRL) complexes in which cullin serves as the scaffold to bind small RING finger protein ROC1 (RBX1/HRT1) through a C-terminal domain and a linker-substrate receptor dimer or a substrate receptor directly via an N-terminal domain. Mammalian cells express nine distinct cullins, including two cullin 4 (CUL4) proteins, CUL4A and CUL4B, that use DNA damage–binding protein 1 (DDB1) as the linker. DDB1 bridges the interaction between CUL4 and a subset of DDB1 binding WD40 repeat proteins (DWD or DCAFs for DDB1 cullin-associated factors) that function as substrate receptors to target specific substrates to the CRL4 E3 complexes (15). One of the most abundant DWD/DCAF proteins is DCAF1/VprBP. DCAF1 is evolutionarily conserved in mammals, Drosophila, Xenopus, Caenorhabditis elegans, and Arabidopsis, but has no apparent homolog in yeast (16, 17). It is ubiquitously expressed in all tissues and organs that have been examined (18). Genetic analyses revealed an essential function of Dcaf1 during embryonic development in plants, flies, and mammals, resulting in developmental arrest at the globular stage of Arabidopsis (19), late pupal stage in Drosophila (20), and early embryonic lethality before embryonic day 7.5 (E7.5) in mice (21), respectively. The substrate(s) and function of Dcaf1 critical for embryonic development have yet to be defined. In this study, we report that loss of the functions of Dcaf1 in embryonic mouse brain and adult thymus and bone marrow selectively eliminates proliferating cells and causes perinatal lethality and lymphoid cell defects, respectively. Our biochemical studies led to the discovery of a previously unknown substrate of the CRL4DCAF1 ligase, the ribosome assembly factor PWP1, and a critical function for DCAF1 in the regulation of rRNA processing and ribosome biogenesis. Loss of function of DCAF1 results in accumulation of free ribosomal protein L11 and increased L11-MDM2 association, leading to p53 activation. These results help to explain the critical function of DCAF1 in cell proliferation and tissue, organ, and animal development. They also provide in vivo evidence supporting a physiological relevant function of the L11-MDM2-p53 checkpoint pathway.
RESULTS
Conditional brain-specific Dcaf1 knockout in mice results in perinatal death and defects in brain and lens development
We previously generated two Dcaf1/VprBP mutant mouse strains and found that whole-body deletion of Dcaf1 resulted in embryonic lethality (21). Conditional deletion analyses identified a function of Dcaf1 in oocyte survival (22), B cell development and V(D)J recombination fidelity (23), and expansion of T cells (24). To determine the cellular mechanism of Dcaf1 in embryonic development, we established conditional brain-specific Dcaf1 knockout mice (CKO) by crossing the floxed Dcaf1 mice with Nestin-Cre mice that express Cre recombinase under the control of Nestin promoter in neuronal and glial cell precursors (fig. S1A). Dcaf1 gene deletion and protein depletion in neonatal mouse brains were confirmed by genomic polymerase chain reaction (PCR) and immunoblotting at day 0 (P0) (Fig. 1A). Although the conditional brain-specific Dcaf1 knockout mice (Dcaf1f/f;Nestin-Cre) are born alive, all CKO mice died within 40 hours after birth (Fig. 1B). The proportion of the Dcaf1 CKO mice in total neonates is 12.5%, less than 25% expected Mendelian ratio, suggesting that some embryos might have died from brain-specific deletion of Dcaf1 during embryogenesis. Hematoxylin and Eosin (H&E) staining revealed a thinner ventricular zone, smaller striatum, and hemorrhages from aberrantly grown capillary vessels in the brain cortex (Fig. 1C and fig. S1B). Furthermore, the lens in the eyes of Dcaf1 CKO mice were collapsed (Fig. 1D). These results suggested a critical function of Dcaf1 in brain development that is essential for perinatal survival.
Fig. 1. Conditional brain-specific Dcaf1 knockout in mice results in perinatal death and defects in brain and lens development.
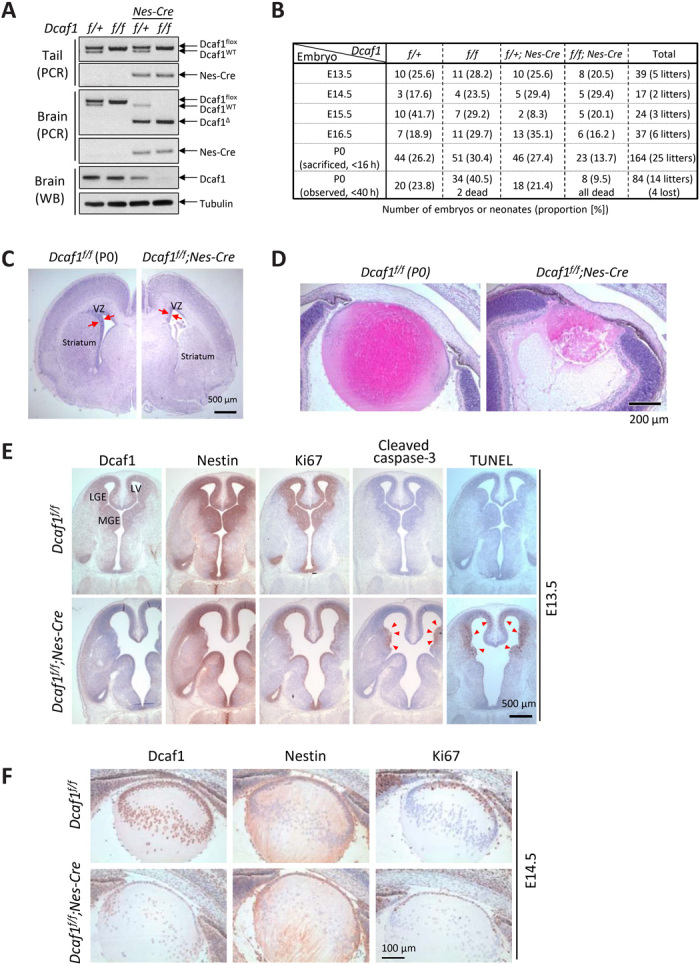
(A) Dcaf1 gene deletion and protein depletion in neonatal mouse brains were confirmed by genomic PCR and immunoblotting. (B) Brain-specific Dcaf1 knockout mice died perinatally within 40 hours of birth. Summary of the numbers of embryos and neonates and their proportion (numbers in the brackets) in each age from the mating of Dcaf1f/+;Nestin-Cre and Dcaf1f/f mice. Neonates were sacrificed soon after delivery (<16 hours after birth) for analysis or observed overnight (<40 hours). Four neonates were lost because of cannibalization. (C) H&E staining of the coronal brain sections from control and Dcaf1 knockout P0 mice. VZ, ventricular zone. (D) H&E staining of the horizontal lens sections. (E) IHC staining of the control and Dcaf1 knockout brains at E13.5. Arrowheads indicate the cleaved caspase-3 and TUNEL (terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling)–positive cells. (F) IHC staining of the control and Dcaf1 knockout lens at E14.5.
Deletion of Dcaf1 selectively eliminates proliferating brain and lens cells in vivo
To determine the cellular basis of the perinatal lethality and severe brain development defects caused by conditional Dcaf1 deletion, we examined cell proliferation and death in the brains of mouse embryos. Consistent with the intense Nestin expression pattern, Dcaf1 protein began to decrease at E12.5 and was depleted after E14.5 (fig. S1C) in brain tissues from Dcaf1f/f;Nestin-Cre mice. H&E staining of E13.5 Dcaf1 CKO brain revealed noticeable phenotypes, including enlarged ventricles in the cortex and collapse of ganglionic eminences (fig. S1D). Immunohistochemical (IHC) staining was performed to determine the Dcaf1 expression pattern and the phenotypes in Dcaf1 CKO brain. In the control mouse brain, the area of Dcaf1 expression was very similar to Nestin-positive and Ki67 (a proliferation marker)–positive area (Fig. 1E, upper panel). Dcaf1 depletion in Nestin-positive cells in Dcaf1 CKO mouse brain was confirmed (lower panel). Notably, Ki67-positive cells were markedly decreased in Dcaf1f/f;Nestin-Cre brain when compared with the control Dcaf1f/f brain, indicating that deletion of Dcaf1 results in loss of proliferating cells. IHC using antibodies to cleaved caspase-3 and TUNEL (terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling) revealed apoptosis at the edge of collapsed ganglionic eminence and in the middle layer of brain cortex of Dcaf1 CKO brain (Fig. 1E). These results link the loss of Dcaf1 to increased apoptosis of active proliferating cells in the brain.
Nestin is also expressed in the lens epithelium, which are the parental cells responsible for growth and development of the entire ocular lens (25). In the lens of E14.5 mouse eyes, Dcaf1 depletion was detected in the lens epithelium, accompanied by a marked decrease of Ki67-positive cells (Fig. 1F, lower). These defects likely caused the collapse of the lens in Dcaf1 CKO neonates. Collectively, these results show that loss of Dcaf1 inhibits cell proliferation and increases cell apoptosis in the brain and lens, resulting in the damage of brain progenitor cells and leading to brain hemorrhages and atrophy, providing a plausible cellular mechanism for neonatal lethality after conditional Dcaf1 deletion in brain. Apoptotic markers were positive only in the Ki67-positive area, suggesting that deletion of Dcaf1 preferentially eliminated the proliferating, but not nondividing, cells in the brain and lens. Notably, both brain and lens defects caused by the conditional deletion of Dcaf1 phenocopy those reported in the brain and lens after conditional deletion of Ddb1 (26), including selective elimination of proliferating cells, increased apoptosis, neuronal and lens degeneration, and perinatal lethality. These phenotypical similarities are consistent with the fact that DCAF1 is the major binding partner of DDB1 (21).
Inducible disruption of Dcaf1 in adult mice results in marked thymic atrophy and bone marrow defect
To further explore the in vivo function of Dcaf1, we established inducible Dcaf1 knockout mice by crossing the floxed Dcaf1 mice with Cre-ERT2 mice that express tamoxifen-inducible Cre recombinase under the ubiquitin C promoter (fig. S2A). Dcaf1 protein depletion after tamoxifen administration was observed in multiple tissues (fig. S2B). In the course of analyzing these mice, we noted a marked thymic atrophy in tamoxifen-treated Dcaf1f/−;Cre-ERT2 mice (Fig. 2A). Dcaf1 protein depletion after tamoxifen administration was confirmed in thymus (fig. S2C). In addition to reduced thymic size, total thymocyte cell counts were significantly reduced in thymus from tamoxifen-treated Dcaf1f/−;Cre-ERT2 mice (1.36 ± 0.67 × 107 in knockout versus 15.52 ± 1.37 × 107 in control animals, P = 0.0005; Fig. 2B). Histological analysis of thymus from tamoxifen-treated Dcaf1f/−;Cre-ERT2 mice revealed a clear lack of typical corticomedullary architecture (Fig. 2C). These results suggested a critical function of Dcaf1 in thymus and T cell development.
Fig. 2. Inducible disruption of Dcaf1 in adult mice results in marked thymic atrophy and bone marrow defect.
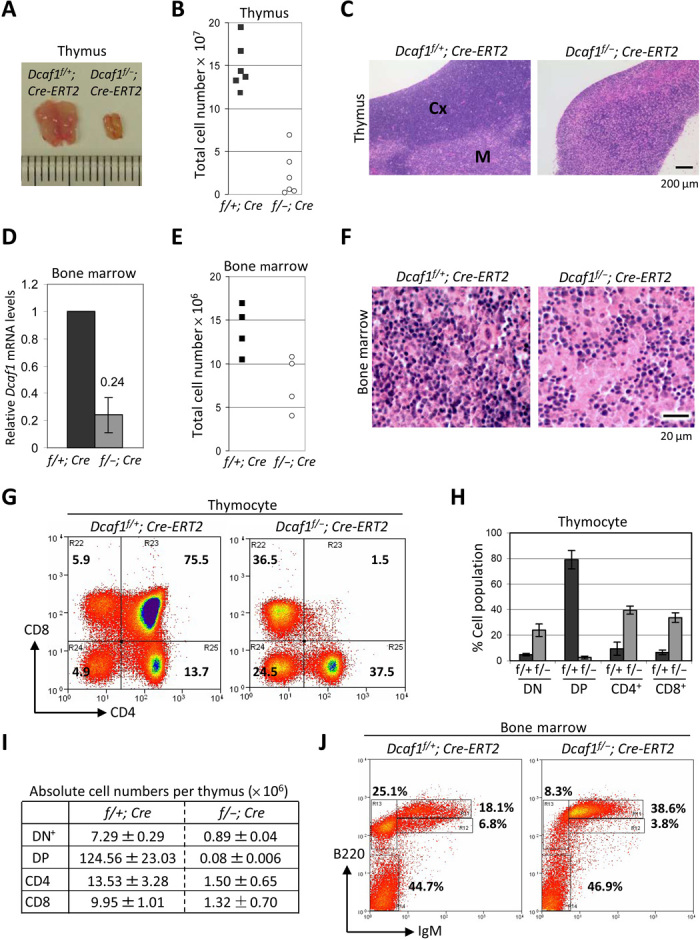
(A) Thymi were isolated from Dcaf1f/+;Cre-ERT2 and Dcaf1f/−;Cre-ERT2 mice 6 days after tamoxifen injection (Photo credit: Sarah C. Jackson, taken at University of North Carolina at Chapel Hill). (B) Total thymic cellularity was quantified using a hemocytometer (n = 6). (C) H&E staining of formalin-fixed, paraffin-embedded thymus sections. Cortex (Cx) and medulla (M) are indicated in control mice, but are not well defined in Dcaf1 knockout mice. (D) Bone marrow cells from femur were isolated 6 days after tamoxifen injection, and Dcaf1 mRNA levels were determined by RT-qPCR in triplicate. (E) Total bone marrow cellularity was quantified using a hemocytometer (n = 4). (F) H&E staining of decalcified, paraffin-embedded bone marrow sections. (G) Flow cytometric analysis of thymocytes from tamoxifen-treated control and Dcaf1 knockout mice, stained with CD8 and CD4 antibodies. (H) Bar diagram showing average percentages for CD4−CD8− (DN), CD4+CD8+ (DP), CD4+ SP, and CD8+ SP subpopulations. Error bars represent ±SEM for triplicate experiments. (I) Absolute cell numbers for thymocyte subsets in (H) were calculated by multiplying percentages by total cell numbers. (J) Flow cytometric analysis of bone marrow cells from tamoxifen-treated control and Dcaf1 knockout mice, stained with B220 antibody and immunoglobulin M (IgM) antibody.
Having observed a clear requirement for Dcaf1 in T cell development, we sought to determine whether Dcaf1 also played a role in myeloid cell development by examining the effect of Dcaf1 disruption in bone marrow. The efficiency of Dcaf1 disruption was assessed by quantitative reverse transcription PCR (RT-qPCR) of isolated bone marrow cells (Fig. 2D). The total number of bone marrow cells from femur was reduced (7.76 ± 1.60 × 106 in knockout versus 13.94 ± 1.41 × 106 in control animals; Fig. 2E), although this difference was not statistically significant. Histological analysis of bone marrow tissue supported the notion that bone marrow cellularity decreased following tamoxifen administration in Dcaf1f/−;Cre-ERT2 mice (Fig. 2F), suggesting that Dcaf1 also plays a critical function for bone marrow tissue development.
Deletion of Dcaf1 selectively eliminates proliferating T and B cells in vivo
We determined the cellular basis of severe immune cell development defects caused by Dcaf1 deletion. T cell precursors migrate to the thymus as double negative (CD4−CD8−, DN), mature into double-positive (CD4+CD8+, DP) cells, and subsequently commit to become single-positive (CD4+ or CD8+, SP) cells. Active cell division occurs during T cell development, especially as DN cells progress during their development toward DP cells. In the Dcaf1 knockout thymus, CD4/CD8 staining notably revealed a near-complete loss of the DP cells, with a corresponding relative increase in the percentage of DN and SP cells (Fig. 2, G and H). While this could suggest a block in the DN to DP transition (with residual SP cells developed before tamoxifen treatment), both the absolute numbers of DN and SP cells were also greatly reduced (Fig. 2I). One explanation is that Dcaf1 deletion results in the loss of proliferating DN T cells, leading to the loss of DP and SP cells. Furthermore, we also noted a marked increase of apoptotic and dead cells in Dcaf1 knockout DP population compared with control cells (fig. S2D). We conclude that the marked decrease of thymocytes is due to a combination of impaired DN proliferation and increased apoptosis of DP population.
Development of pre- and pro-B cells to immature B cells in bone marrow is associated with active cell proliferation. The B220+IgM− cells, which represent pre- and pro-B cell populations, were reduced in bone marrow of tamoxifen-treated Dcaf1f/−;Cre-ERT2 mice (8.3% in knockout versus 25.1% in control, Fig. 2J), as were the B220loIgM+ cells, which represent immature B cells (3.8% in knockout versus 6.8% in control). Concomitantly, a relative increase in the percentage of recirculating, mature B cells marked as B220hiIgM+ was found in bone marrow of tamoxifen-treated Dcaf1f/−;Cre-ERT2 mice (38.6% in knockout versus 18.1% in control). This suggests that B cells that matured before tamoxifen injection in Dcaf1f/−;Cre-ERT2 mice were largely unaffected by Dcaf1 disruption, but naïve B cell development in bone marrow was inhibited. Collectively, we conclude that deletion of Dcaf1 selectively eliminates proliferating T cells and B cells in thymus and bone marrow.
We also examined mature lymphocytes in the spleen. Flow cytometric analysis of B and T cell populations in spleen showed no change in the relative percentage of lymphocytes following Dcaf1 disruption (fig. S2, E and F). This suggests that developing B and T cells are more severely affected by Dcaf1 disruption than mature lymphocytes, possibly reflecting the G0 state of naïve, mature T and B cells.
Deletion of Dcaf1 selectively eliminates proliferating cells in vitro
We hypothesized that the developmental defect present in T cells was likely due to a failure of thymocytes to proliferate, rather than a requirement for Dcaf1 in pathways specific to T cell development [e.g., V(D)J recombination, or positive or negative selection]. To test for a requirement of Dcaf1 in cell proliferation independent of its role in T cell development, we stimulated mature T cell proliferation in vitro and subsequently monitored cell divisions. We isolated mature lymphocytes from tamoxifen-treated Dcaf1f/−;Cre-ERT2 and control Dcaf1f/+;Cre-ERT2 mice and confirmed a decrease in Dcaf1 by immunoblotting (Fig. 3A). To follow cell divisions, lymphocytes were labeled with CFSE (carboxyfluorescein diacetate succinimidyl ester), a fluorescent dye that forms stable adducts on intracellular proteins and provides a quantitative measurement of cell division by using flow cytometry as it is diluted by half following each cell division. T cell proliferation was activated by anti-CD3 and anti-CD28 costimulation followed by in vitro culture. We collected cells at 42, 54, and 66 hours after stimulation or mock treatment and monitored the proliferation of T cells by flow cytometry. Whereas control cells showed continued, robust proliferation in response to activation as seen by the discrete CFSE peak resulting from replication dilution, we found that very few knockout T cells proliferated in response to activation at 42 and 54 hours and less than half had undergone any proliferation at 66 hours (Fig. 3B). The total cell number following T cell stimulation was decreased in Dcaf1-disrupted primary lymphocytes compared to control primary lymphocytes (Fig. 3C). Notably, the ratio of costimulated versus mock-treated cell number is less than 1 in Dcaf1-disrupted lymphocytes at 2 days after stimulation, suggesting that Dcaf1 disruption results in cell death after T cell stimulation (Fig. 3C). Therefore, we conclude that Dcaf1 is required for the proliferation of T cells during costimulation and suggest that loss of proliferative capacity may account for the developmental defects observed in T cells.
Fig. 3. Deletion of Dcaf1 selectively eliminates proliferating cells in vitro.
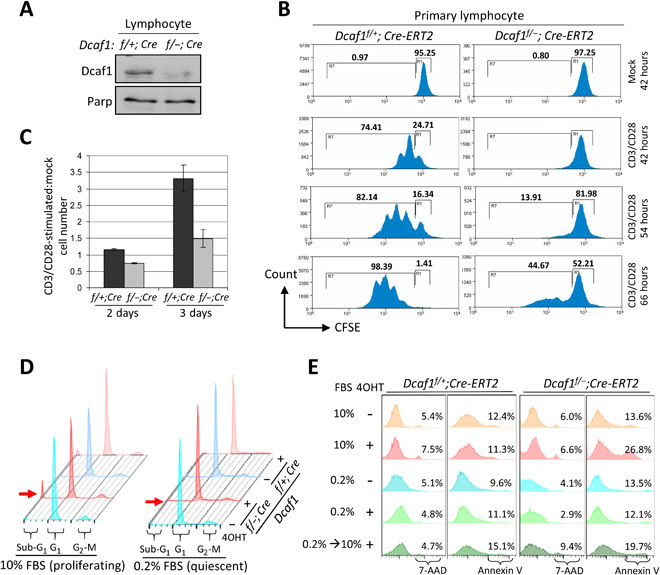
(A) Immunoblotting of primary lymphocyte lysates derived from Dcaf1f/+;Cre-ERT2 and Dcaf1f/−;Cre-ERT2 mice 6 days after tamoxifen injection. (B) Primary lymphocytes were labeled with CFSE and followed by CD3/CD28 stimulation or mock. Cells were cultured in vitro for 42, 54, or 66 hours before collection. Flow cytometric analysis for CFSE was used to determine the proliferation, gated by the 7-AAD–negative T cells. (C) Primary T cells were costimulated with CD3/CD28 or mock-treated and then plated at equal cell numbers. The total cell numbers following activation, expressed relative to the numbers of mock-treated cells, are reported at 2 and 3 days after stimulation. (D) MEFs were cultured in 10 or 0.2% FBS 1 day before 4OHT treatment for 3 days. DNA content was analyzed by PI staining followed by flow cytometry. Red arrow indicates sub-G1 population, representing apoptotic cells. (E) MEFs were cultured in 10 or 0.2% FBS 1 day before 4OHT treatment for 4 days. One group of MEFs was restimulated by 10% FBS after 2-day serum starvation. Cell death and apoptosis analysis was carried out by staining with 7-AAD and annexin V, respectively.
To further demonstrate that Dcaf1 functions in proliferating cells, we generated tamoxifen-inducible Dcaf1f/+;Cre-ERT2 and Dcaf1f/−;Cre-ERT2 conditional mouse embryonic fibroblast (MEF) lines. We arrested both Dcaf1f/+;Cre-ERT2 and Dcaf1f/−;Cre-ERT2 MEFs in G0-G1 quiescence by serum deprivation and then treated with 4OHT (4-Hydroxytamoxifen) to induce Dcaf1 deletion, followed by fluorescence-activated cell sorting (FACS) analysis of cell cycle phase distribution (Fig. 3D). This experiment showed that compared to the marked (10%; Fig. 3D, left panels) sub-G1 population detected in proliferating cells, deletion of Dcaf1 caused very little death of quiescent cells (1.9%; Fig. 3D, right panels). We further confirmed this finding by annexin V (apoptotic) and 7-AAD (7-aminoactinomycin D, necrotic) staining and FACS analyses (Fig. 3E). This assay showed that apoptotic cells in control Dcaf1f/+;Cre-ERT2 MEFs, as measured by positive staining to annexin V, ranged between 9.6 and 15.1% and was not markedly affected by either 4OHT treatment or whether cells were proliferating [cultured in the presence of 10% fetal bovine serum (FBS)] or in a quiescent state (cultured in 0.2% FBS). The Dcaf1f/−;Cre-ERT2 MEFs showed similar apoptosis as the control Dcaf1f/+;Cre-ERT2 MEFs when cultured in 10% FBS, and untreated by 4OHT (13.6%) or treated with 4OHT, but cultured in 0.2% FBS (12.1%). In contrast to the control MEFs, however, 4OHT treatment of Dcaf1f/−;Cre-ERT2 MEFs cultured in 10% FBS resulted in a marked increase of apoptotic cells (26.8%), indicating that loss of Dcaf1 preferentially eliminates proliferating MEF cells. Furthermore, switching of 4OHT-treated Dcaf1f/−;Cre-ERT2 MEFs from 0.2% FBS (quiescent) to 10% FBS (proliferating) for 2 days increased annexin V–positive cells to 19.7%, indicating that loss of Dcaf1 potently killed cells once they started proliferating but had little effect on quiescent nondividing cells. Together, these results demonstrate that Dcaf1 plays a critical role in controlling cell cycle and, when it is deleted, selectively eliminates the proliferating cells both in vivo during embryonic brain development and T cell and B cell development and in vitro in anti-CD3/CD28–stimulated T cells and cultured MEFs.
Ribosome assembly factor PWP1 is a substrate of CRL4DCAF1 E3 ubiquitin ligase
Several CRL4DCAF1 substrates have been reported (16), but none of them can explain the essential function of DCAF1 during early development or cell proliferation. To identify additional CRL4DCAF1 substrates, we performed two immunoprecipitations of endogenous DCAF1 complexes and mass spectrometry analyses (IP-mass spec) to search for DCAF1 interacting proteins. One used the anti-DCAF1 antibody to immunopurify DCAF1 complex from HeLa cells. The other was to knock-in three tandem FLAG epitopes (3xFLAG) to the N terminus of the endogenous DCAF1 gene in HeLa cells, followed by immunopurification of 3xFLAG-DCAF1 complex using the anti-FLAG antibody. Successful in-frame knock-in of 3xFLAG tag into endogenous DCAF1 was verified by DNA sequencing (fig. S3A), immunoblotting (Fig. 4A), and immunofluorescence (IF) staining (fig. S3, B and C). Both immunopurifications were stained with SYPRO Ruby (Fig. 4B) and subjected to mass spectrometry analyses (Fig. 4C).
Fig. 4. Ribosome assembly factor PWP1 is a substrate of CRL4DCAF1 E3 ubiquitin ligase.
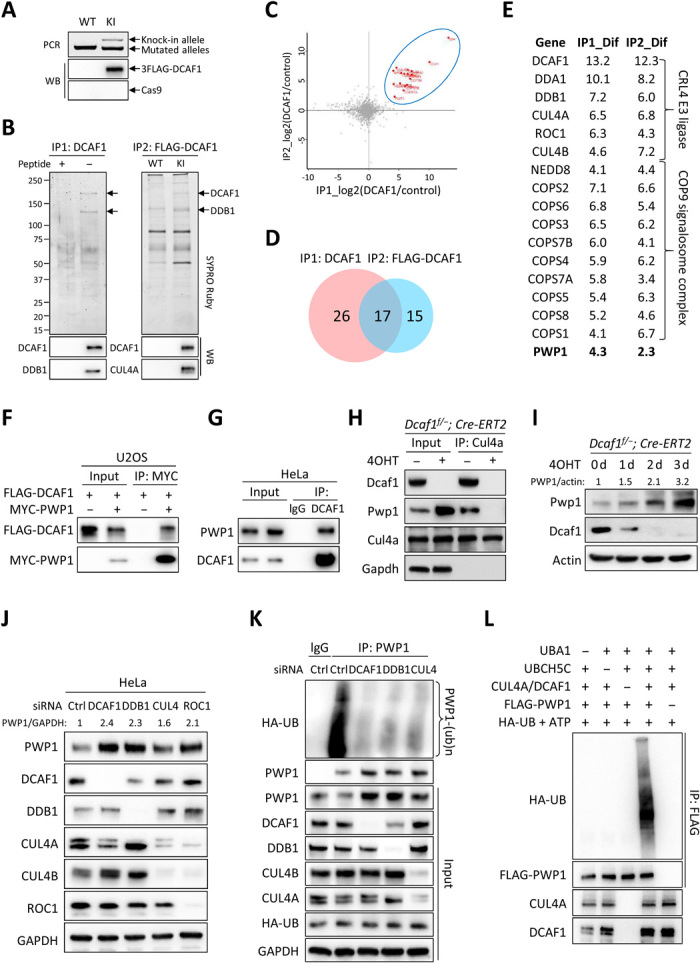
(A) 3xFLAG knock-in (KI) to endogenous DCAF1 in HeLa cells verified by genomic PCR and immunoblotting. WT, wild type; WB, Western blot. (B) Immunopurification of two endogenous DCAF1 complexes from HeLa cells. Molar excess competing antigen peptides added in control immunocomplexes. (C) Scatter plot of log2 ratios of 626 proteins identified by mass spectrometry analyses of two DCAF1 immunocomplexes. (D) Venn diagram of proteins with greater than fourfold abundance change. (E) List of 17 DCAF1-interacting proteins identified in both DCAF1 immunocomplexes. (F) Interaction between ectopically expressed PWP1 and DCAF1 in U2OS cells by coimmunoprecipitation (co-IP). (G) Endogenous interaction between DCAF1 and PWP1 in HeLa cells treated with 20 μM MG132 for 6 hours. (H) Dcaf1f/−;Cre-ERT2 MEFs with 200 nM 4OHT or dimethyl sulfoxide (DMSO) for 2 days. Protein-protein interactions by co-IP. (I) MEFs with 200 nM 4OHT for indicated days. (J) HeLa cells transfected with small interfering RNAs (siRNAs) targeting indicated genes individually. (K) HeLa cells stably expressing HA-tagged ubiquitin transfected with siRNAs targeting indicated genes and treated with MG132. Endogenous PWP1 was immunoprecipitated and immunoblotted. (L) Immunopurified PWP1 protein incubated with CRL4DCAF1 E3 immune complex in the presence or absence of UBA1 (E1), UBCH5C (E2), adenosine triphosphate (ATP), and ubiquitin in vitro for 1 hour. Ubiquitylated PWP1 was immunoprecipitated and immunoblotted.
These two IP-mass spec analyses identified combined 626 putative DCAF1-interacting proteins with at least two unique peptides for each individual protein (table S1). The statistically significant label-free quantification (LFQ) ratios that are proportional to the relative DCAF1 binding of individual proteins were averaged over three technical replicates. A stringent analysis was performed to identify the high-confidence DCAF1-interacting proteins with greater than fourfold abundance change compared to control immune complexes. This led to the identification of 43 and 32 proteins in DCAF1 or FLAG-DCAF1 immune complexes, respectively, including 17 proteins that were identified in both immune complexes (Fig. 4, C and D, and table S1). Of these 17 proteins, DCAF1 and 15 known components of CRL4 E3 complex, including both CUL4A and CUL4B, DDB1, DDA1, ROC1, NEDD8, and 9 subunits of COP9 signalosome complex (Fig. 4E), were identified. The only previously unknown protein identified in both DCAF1 and FLAG-DCAF1 immune complexes was the ribosome assembly factor PWP1.
We confirmed the binding between DCAF1 and PWP1 by the coimmunoprecipitation assays and demonstrated binding between ectopically (Fig. 4F) and endogenously (Fig. 4G) expressed DCAF1 and PWP1 proteins. Furthermore, we found that deletion of Dcaf1 in MEFs prevented Pwp1 from binding with Cul4a (Fig. 4H), demonstrating that Dcaf1 bridges Pwp1 to Cul4a. To determine whether PWP1 is a substrate of the CRL4DCAF1 E3 ligase, we performed knockout and knockdown of DCAF1 and individual components in CRL4DCAF1 E3 complex in MEF and HeLa cells. Deletion of Dcaf1 resulted in marked accumulation of Pwp1 in MEFs in a time-dependent manner (Fig. 4I). Moreover, depletion of DCAF1, DDB1, and ROC1 individually and in combination with both CUL4A and CUL4B all resulted in accumulation of PWP1 in HeLa cells (Fig. 4J). To demonstrate the ubiquitylation of PWP1 by the CRL4DCAF1 E3 ligase, we performed both in vivo and in vitro ubiquitylation assays. We found that endogenously expressed PWP1 is actively ubiquitylated in HeLa cells and that knockdown of either DDB1 or DCAF1 individually or in combination with CUL4A and CUL4B resulted in substantial reduction of PWP1 ubiquitylation (Fig. 4K). An in vitro ubiquitylation assay demonstrated that the CRL4DCAF1 E3 immunocomplexes caused robust ubiquitylation of PWP1, which is dependent on the addition of E1, E2, and E3 complex and substrate PWP1 (Fig. 4L). Together, these results demonstrate that the ribosome assembly factor PWP1 is a substrate of the CRL4DCAF1 E3 ligase.
Loss of function of DCAF1 results in defects in ribosome biogenesis
PWP1 is a highly conserved protein in eukaryotes and is widely expressed across different tissues in mice and humans (27, 28). It was initially identified in budding yeast as a protein that contains multiple periodic tryptophan (W) residues defining a WD40 repeat and, when deleted, results in severe growth retardation and marked reduction of protein synthesis (29). In yeast, Pwp1 is a component of a subcomplex that associates with the ITS2 regions of the rRNA gene, and deletion of Pwp1 results in accumulation of 35S rRNA precursor and reduction in other rRNA precursors, as well as mature 25S and 18S rRNA (27, 30, 31). In Drosophila, Pwp1 (dPwp1) mutants result in substantial reduction in 5.8S, 18S, and 28S rRNA and develop the Minute phenotype linked to mutations in many individual ribosomal protein genes (2, 32). These studies suggest a conserved function of PWP1 for a critical step during ribosome biogenesis such as rRNA processing.
A mosaic analysis in Drosophila wing discs revealed that Dcaf1/Mahjong−/− cells developed defects similar to the Minute mutants (20). Furthermore, a recent genome-wide RNA interference (RNAi) screen in human HeLa cells identified several components of the CRL4 E3 ligase, including CUL4A, CUL4B, DDB1, ROC1, and CSN subunits, that are required for the early steps of nucleolar ribosome synthesis (33). The substrate receptor for the CRL4 E3 ligase involved in the ribosome biogenesis was not identified. These findings prompted us to determine whether the CRL4DCAF1 E3 ligase, by ubiquitylating PWP1, is necessary for rRNA processing and ribosome biogenesis. We adapted procedures developed by Badertscher et al. (33) that used dynamic translocation of BYSTIN/ENP1, a 40S trans-acting factor, to monitor biogenesis in HeLa cells. Under steady-state conditions, BYSTIN predominantly localizes to nucleoli and shuttles into the cytoplasm along with newly synthesized 40S subunits. Blocking pre-40S nuclear export with leptomycin B (LMB) caused BYSTIN to accumulate in the nucleoplasm in unperturbed cells, but trapped in the nucleoli if an early step of ribosome biogenesis was blocked (Fig. 5, A to C). We found that either overexpression of PWP1 or knockdown of DCAF1, like knockdown of either DDB1, in combination with CUL4A and CUL4B or ribosomal assembly factor rRNA 2′-O-methyltransferase fibrillarin (FBL), resulted in a failure of BYSTIN translocating out the nucleoli, indicative of an early step defect in nucleolar ribosome biogenesis before the transport of ribosomal precursor subunits into the nucleoplasm (Fig. 5, A and B; overexpression and knockdown efficiency were shown in fig. S4, A and B). To demonstrate that depletion of DCAF1 impairs ribosome biogenesis by accumulating PWP1, we performed a double knockdown of DCAF1 and PWP1 in HeLa cells. We found that BYSTIN was mostly accumulated in the nucleoplasm of double knockdown cells after LMB treatment, with some portion remaining in the nucleoli, similarly as control cells (Fig. 5C; knockdown efficiency was shown in fig. S4C). This result indicates that depletion of DCAF1-induced nucleolar ribosome biogenesis defect is largely rescued by knockdown of PWP1.
Fig. 5. Loss of function of DCAF1 results in defects in ribosome biogenesis.
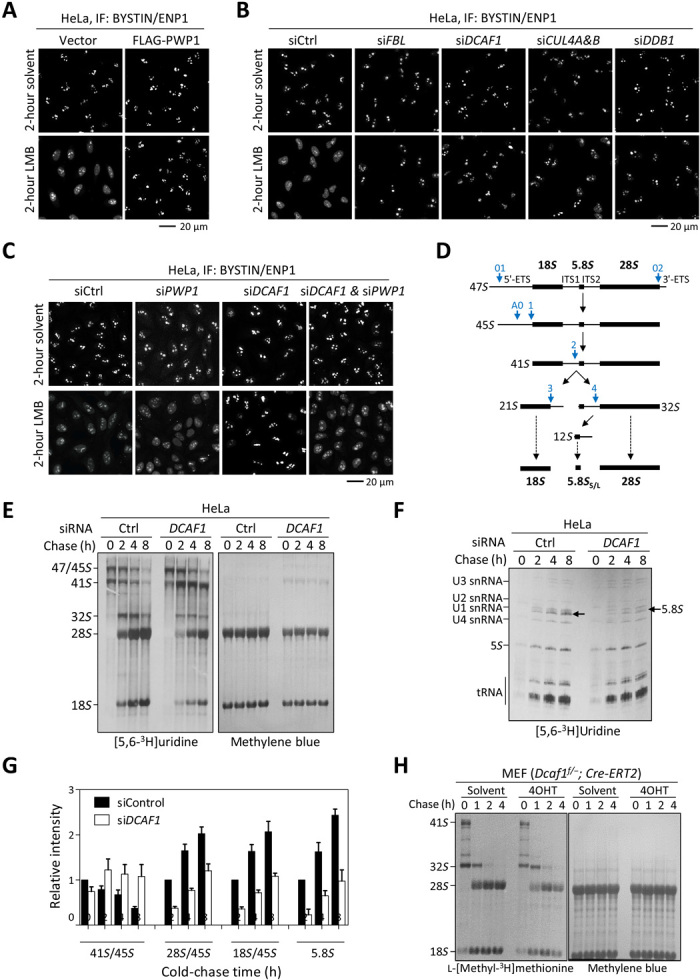
(A) HeLa cells stably expressing PWP1 or empty vector were treated with 20 nM LMB or solvent for 2 hours. The subcellular distribution of BYSTIN/ENP1 was determined by IF. (B and C) HeLa cells were transfected with siRNAs targeting indicated genes individually or in combination and then treated with 20 nM LMB or solvent for 2 hours. (D) Diagram illustrating the major steps of mammalian rRNA processing. Blue arrows indicate the major cleavage sites in human cells. (E and F) HeLa cells were transfected with siRNAs targeting DCAF1 or control siRNA and then followed by pulse-chase analysis of the [3H]uridine-labeled rRNAs. The newly synthesized and total rRNAs were determined by autoradiography and methylene blue staining, respectively. (G) The relative intensities of newly synthesized 41S, 28S, and 18S rRNAs compared to 45S precursors at 0 hours in (E) were quantified. The intensities of newly synthesized 5.8S rRNAs in (F) were quantified and normalized against 5S rRNAs. Error bars represent ±SD for duplicate experiments. (H) Dcaf1f/−;Cre-ERT2 MEFs were treated with 200 nM 4OHT or solvent for 3 days, followed by pulse-chase analysis of the l-[methyl-3H]methionine–labeled rRNAs.
To provide additional evidence supporting the function of DCAF1 during ribosome biogenesis, we performed pulse-labeling analysis using [3H]uridine in HeLa cells after knocking down DCAF1. This experiment showed that depletion of DCAF1 resulted in substantial accumulation of 41S rRNA precursor with a concomitant decrease of newly synthesized 28S, 18S, and 5.8S rRNA (Fig. 5, D to G; knockdown efficiency was shown in fig. S4D). Analyzing low–molecular weight RNA by 7 M urea polyacrylamide gel electrophoresis (PAGE) confirmed the decrease of newly synthesized 5.8S rRNA after DCAF1 knockdown (Fig. 5, F and G). To further confirm the role of DCAF1 in rRNA processing, we treated Dcaf1f/−;Cre-ERT2 MEF cells with 4OHT for 3 days to induce Dcaf1 deletion and examined rRNA synthesis and processing by pulse-chase analysis. Because the uridine salvage pathway is not very active in MEF cells, we replaced [3H]uridine with l-[methyl-3H]methionine, which can be more rapidly incorporated because of the rapid turnover of the cellular methionine pool. 47S transcript (pre-rRNA) is extensively modified by methylation once it is transcribed. l-[Methyl-3H]methionine is converted readily to S-adenosylmethionine, which donates the 3H-methyl in the methylation of rRNA precursors (34). Depletion of Dcaf1 did not substantially affect the total 28S and 18S rRNA, as measured by methylene blue staining, but decreased both newly synthesized 28S and 18S rRNA (Fig. 5H; knockout efficiency was shown in fig. S4E). Together, these results demonstrate that the CRL4DCAF1 E3 ligase is important for rRNA processing and ribosome biogenesis, and this function of CRL4DCAF1 is achieved, at least in part, by regulating PWP1.
Loss of Dcaf1 activates RP-Mdm2-p53 checkpoint pathway
We previously reported that impairment of ribosome biogenesis caused by the inhibition of RNA Pol I–dependent ribosomal DNA (rDNA) transcription via a low dose (5 nM) of actinomycin D accumulates free, unassembled ribosomal protein L11 that binds to MDM2 and inhibits MDM2-mediated p53 ubiquitylation, leading to an increase of p53 (35). To determine whether impairment of rRNA processing and ribosome biogenesis by loss of DCAF1, like the inhibition of Pol I–dependent rDNA transcription by actinomycin D, also activates the p53 pathway, we examined the histology of thymus sections from moderately affected Dcaf1f/+;Cre-ERT2 or Dcaf1f/−;Cre-ERT2 animals 6 days after tamoxifen administration, at which time there was robust deletion of the Dcaf1. Incorporation of the thymidine analog 5-bromo-2′-deoxyuridine (BrdU) was almost undetectable in thymus from knockout mice, indicating a strong inhibition of DNA replication (Fig. 6A). Concurrently, cleaved caspase-3 and p53 were more abundantly detected in knockout mice, suggesting a strong increase of apoptosis induced by p53 accumulation. Consistent with these in vivo observations, deletion of Dcaf1 in MEF cells resulted in substantial decrease of Ki67 signals and accumulation of p53 and PUMA and BAX, two proapoptotic p53 target genes (Fig. 6B). To explore the mechanism of p53 activation in the Dcaf1 loss, we separated assembled ribosomes from ribosome-free proteins by ultracentrifugation and analyzed for free unassembled ribosomal proteins in the S100 fraction of Dcaf1f/+;Cre-ERT2 or Dcaf1f/−;Cre-ERT2 MEFs. Immunoblot analysis revealed that depletion of Dcaf1 results in the accumulation of free ribosomal protein RPL11, while the total amount of RPL11 protein was largely not affected (Fig. 6C). Knockdown of DCAF1 in U2OS cells increased the MDM2-L11 association, the levels of p53 and CDK inhibitor p21, a p53 transcriptional target (Fig. 6D). To provide genetic evidence linking the loss Dcaf1 to increased MDM2-L11 association and p53 activation, we knocked down Dcaf1 in wild-type or Mdm2C305F/C305F primary MEF cells. The C305F mutation was first identified in human osteosarcoma (36) and disrupts MDM2’s binding with L11 (37, 38). We found that compared with the wild-type MEFs, knockdown of Dcaf1 in Mdm2C305F/C305F MEFs similarly resulted in an increase in Pwp1 protein but failed to accumulate p53 and p21 (Fig. 6E), consistent with L11 not being able to bind Mdm2. Together, these results provide a molecular mechanism—increased free L11 and L11-MDM2 binding leading to activation of p53—for the G1 cell cycle arrest and apoptosis in cells with impaired ribosome biogenesis after loss of DCAF1 (Fig. 6F).
Fig. 6. Loss of Dcaf1 impairs ribosome biogenesis and activates RP-Mdm2-p53 checkpoint pathway.
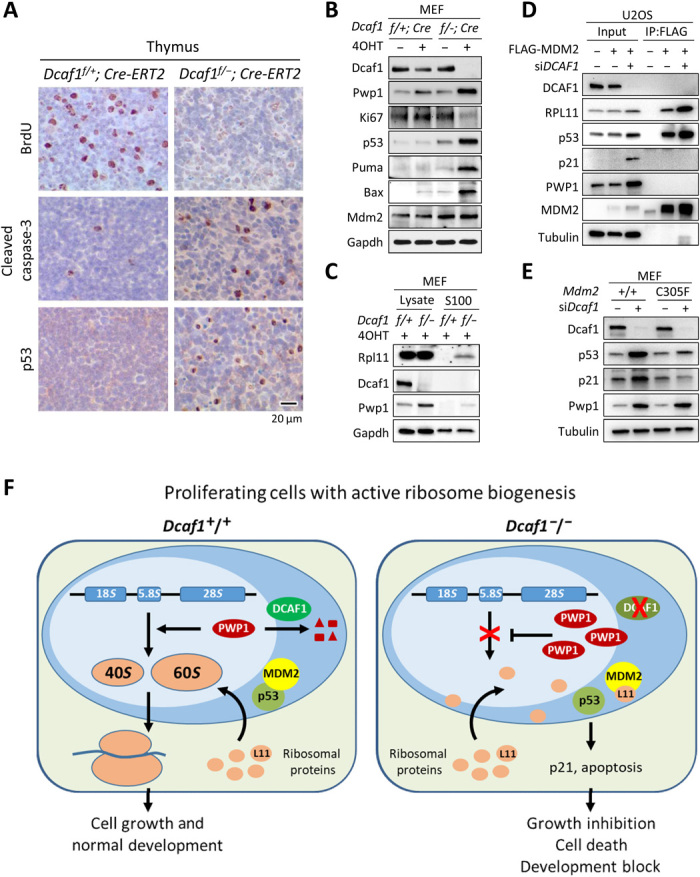
(A) IHC staining of BrdU, cleaved caspase-3, and p53 from sections of formalin-fixed, paraffin-embedded thymus tissue following tamoxifen treatment. Positive staining is indicated by brown color; sections were counterstained with hematoxylin. (B) Dcaf1f/+;Cre-ERT2 and Dcaf1f/−;Cre-ERT2 MEFs were treated with 200 nM 4OHT for 3 days. (C) 4OHT-treated Dcaf1f/+;Cre-ERT2 and Dcaf1f/−;Cre-ERT2 MEFs were homogenized in hypotonic buffer (designated as total cell lysates). Ribosomes were pelleted from lysates by ultracentrifugation at 100,000g for 3 hours at 4°C, and the ribosome-free supernatants were collected (designated as the S100 fractions). (D) U2OS cells stably expressing FLAG-tagged MDM2 or empty vector were transfected with siRNAs targeting DCAF1 or control siRNAs. The interactions between MDM2 and RPL11 proteins were determined by co-IP analysis. (E) Activation of p53 by loss of DCAF1 depends on intact RP-MDM2-p53 pathway. Primary Mdm2+/+ or Mdm2C305F/C305F MEFs were transfected with siRNAs targeting Dcaf1 or control siRNAs. (F) Schematic model for the function of DCAF1-based E3 ubiquitin ligase in the regulation of PWP1 protein, ribosome biogenesis, cell proliferation, and development.
DISCUSSION
Much of our knowledge on the control of ribosome biogenesis were gained from extensive studies in yeast. In contrast, very little is known about this process in higher eukaryotes that are much more complex because of the elaborate signaling networks that control ribosome biogenesis and different nutrient responses. Previous studies using inhibitors of the proteasome and neddylation have implicated the ubiquitin-proteasome system and cullin E3 ligases in ribosome biogenesis in human cells (39, 40). Recent genome-wide screening directly linked a CUL4-DDB1–based E3 ligase in the regulation of ribosome biogenesis (33). Our study supports these previous findings and identifies the first ribosome assembly factor that is regulated by degradation by a CRL E3 ligase in mammalian cells (Fig. 4). DCAF1 binds directly with PWP1, is required for PWP1 ubiquitylation both in vivo and in vitro, and, when deleted or depleted, results in accumulation of PWP1 (Figs. 4 and 6). Functionally, overexpression of PWP1 causes a similar defect in nucleolar ribosome biogenesis as DCAF1 loss (Fig. 5, A and B). Simultaneous knockdown of PWP1 and DCAF1 prevented the defects of nucleolar ribosome biogenesis caused by DCAF1 loss (Fig. 5C). Together, these results demonstrate that PWP1 is a direct and major substrate of the CRL4DCAF1 E3 ligase that can regulate ribosome biogenesis (Fig. 6F).
In yeast, PWP1 is a component of a subcomplex that associates with ITS2 and plays an important role in pre-rRNA processing and creating stable pre-ribosomes (27, 30, 31). In Drosophila, Pwp1 (dPwp1) mutants develop a Minute phenotype that was originally identified as similar to haploinsufficient defects in ribosomal proteins in development and later linked to mutations in many individual ribosomal protein genes (2, 32). These studies suggest a conserved function of PWP1 in rRNA processing and early steps of ribosome biogenesis. The biochemical mechanism of PWP1 in rRNA processing in human cells is currently unknown. Loss of function of DCAF1, like knockdown of DDB1, ROC1, or combination of CUL4A and CUL4B, blocked nucleolar ribosome biogenesis (Fig. 5). DCAF1 loss resulted in PWP1 accumulation and defects in the 41S cleavage, suggesting that PWP1 may function in 41S cleavage in the nucleolus. This notion is supported by the finding that overexpression of PWP1 similarly blocked nucleolar ribosome biogenesis (Fig. 5A). These findings indicate that the level of PWP1 protein needs to be properly regulated and, when deleted or abnormally accumulated, impairs ribosome biogenesis, implying a checkpoint-like function for PWP1. The function of PWP1 has recently been extended beyond rRNA processing to Pol I–mediated rRNA transcription (32) and telomere maintenance (41). Whether these two processes are also regulated by DCAF1 and collectively contribute to defects in cell proliferation and development, and p53 activation is an interesting possibility that will require further study.
DCAF1 is an evolutionary conserved gene. Deletion of Dcaf1 resulted in an early development block in plant, fly, and mouse and inhibition of cell proliferation (19–21). Mutation of Drosophila Dcaf1/Mahjong gene results in a “loser cell” phenotype that is shared, notably, with Drosophila cells harboring mutations in ribosomal protein genes collectively known as Minute phenotype (2, 20). A number of potential substrates of the CRL4DCAF1 ligase have been reported (16), but none of these reported substrates can explain an evolutionarily conserved and essential function of DCAF1 in development or ribosome biogenesis. The studies presented here provide a cellular and molecular mechanism explaining the essential function of DCAF1 in cell proliferation and development. We found that Dcaf1 deficiency in brain and lens caused increased apoptosis, neuronal and lens degeneration, brain hemorrhages, and neonatal death (Fig. 1). We also found that systemic inducible deletion of Dcaf1 in adult mice resulted in marked thymic atrophy and bone marrow associated with the decrease of cellularity (Fig. 2). The brain and lens defects phenocopy those previously reported for the conditional deletion of Ddb1 after crossing with the same Nestin-Cre (26). One notable cellular defect shared by the loss of either Dcaf1 or Ddb1 is the selective elimination of proliferating cells, as seen in vivo in brain and lens (Fig. 1) (26), in skin (42), in liver hepatocytes (43), and in T and B cells undergoing development (Fig. 2). In vitro, we found that Dcaf1 deletion eliminates proliferating but not quiescent T cells and MEFs (Fig. 3). These marked phenotypic similarities, at both tissue and cellular levels, are consistent with the finding that DCAF1 is the major binding partner of DDB1 [e.g., (21)]. Furthermore, our studies provide a plausible molecular mechanism for the elimination of proliferating cells caused by DCAF1 loss. We showed that loss of DCAF1 increases free ribosomal protein L11 and L11-MDM2 association and results in p53 activation, leading to increased p21 and cleaved caspase (Fig. 6, A to D). p53 and p21 accumulation in cells with DCAF1 knockdown was blocked by a knock-in tumor-derived mutation in MDM2, C305F, that disrupts MDM2’s binding to L11 (Fig. 6E). Together, these results support a model that the CRL4DCAF1 E3 ligase controls PWP1 ubiquitylation and ribosome biogenesis that, when impaired, activates the L11-MDM2-p53 checkpoint pathway to prevent and protect cells from entering cell cycle with impaired ribosome (Fig. 6F).
MATERIALS AND METHODS
Plasmids and reagents
Expression constructs for DCAF1 were previously described (44). Full-length human PWP1 was inserted into pcDNA3-3myc or p3FLAG-CMV vectors for transient expression, and PWP1, MDM2, and UBC were constructed into lentiviral vectors for stable transduction. Detailed information for antibodies, chemicals, oligonucleotides, and other key resources used in this study are reported in table S2.
Cell culture and cell transfection
HeLa and U2OS cells were cultured at 37°C with 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) or RPMI 1640 medium, respectively, supplemented with 10% FBS (Gibco). 293F cells were adapted to suspension culture in FreeStyle 293 Expression Medium (Thermo Fisher Scientific). Cells were authenticated using short tandem repeat assays. Mycoplasma test results were negative. Cell transfection was performed using Lipofectamine 2000 (Invitrogen) for plasmid DNAs and small interfering RNAs (siRNAs) following the manufacturer’s instructions. Stable cell lines were established by lentivirus transduction, selected, and maintained in medium containing puromycin (1 μg/ml; Amresco).
MEF cell lines were established from the tamoxifen-inducible conditional Dcaf1 knockout mouse embryos. E13.5 embryos were isolated by removing head, thoracic, and abdominal organs. The body tissue was minced by aspiration-ejection cycle three times using an 18-gauge needle with syringe in DMEM supplemented with 15% FBS and 1% penicillin-streptomycin (Corning), followed by incubation overnight at 37°C with 5% CO2. Mdm2C305F/C305F primary MEF was a gift from Y. Zhang (University of North Carolina at Chapel Hill).
Primary lymphocytes from mouse lymph nodes and spleen were obtained from 6-week-old tamoxifen-treated mice 6 days following the first injection. After lysis of red blood cells, cells were cultured in RPMI 1640 medium supplemented with 10% FBS, penicillin-streptomycin, 2 mM l-glutamine, 50 μM β-mercaptoethanol, and 100 nM 4OHT. Cells were cultured for 8 hours before CFSE labeling and T cell activation.
RNAi and CRISPR-Cas9
All siRNA oligonucleotides were synthesized with 3′ dTdT overhangs by Sigma-Aldrich in a purified and annealed duplex form. The sense sequences targeting human DCAF1, CUL4A, CUL4B, DDB1, ROC1, or FBL are reported in table S2 (33, 45). RNAi-mediated down-regulation was performed by transfecting siRNAs in accordance with the manufacturer’s instructions. A nontargeting control siRNA duplex (sense 5′-UUCUCCGAACGUGUCACGUTT-3′) was included as a negative control. The knockdown efficiency was assessed 48 to 72 hours after transfection by immunoblotting.
The procedures for CRISPR-Cas9–mediated knock-in followed the protocols previously published (46). The single guide RNA (sgRNA) targeting the start codon of human DCAF1 (exon 3) was designed by using the online CRISPR Design Tool (http://crispr.mit.edu/) and constructed into CRISPR-Cas9 vector, pSpCas9(BB)-2A-GFP (PX458) (Addgene #48138). The single-stranded oligodeoxynucleotide (ssODN) template containing 3xFLAG sequences was synthesized by Integrated DNA Technologies. The sequence of sgRNA and ssODN was provided in table S2. HeLa cells were transfected with PX458-sgRNA plasmid and ssODN, and the green fluorescent protein (GFP)–positive clonal cell populations were isolated by FACS 2 days after transfection. The nonhomologous end joining (NHEJ) inhibitor SCR7 (10 μM) was used to increase the knock-in efficiency. The recovered clones were validated by genomic PCR sequencing and immunoblotting.
Mouse procedures
Mice were bred and maintained strictly according to protocol approved by the University of North Carolina at Chapel Hill Institutional Animal Care and Use Committee (IACUC). The generation of conditional Dcaf1 mice was previously described (21). All mice were backcrossed to C57BL/6 for at least six generations. Germline transmission of Dcaf1 knockout allele was obtained by crossing conditional mice with EIIa-cre mice (The Jackson Laboratory, stock 003724). Nestin-Cre mice were previously obtained from The Jackson Laboratory [B6.Cg-Tg(Nes-cre)1Kln/J, stock no. 003771].
All control experiments were performed using littermate-matched mice. Dcaf1-F1 and Dcaf1-R primers were used for genotyping Dcaf1 knockout allele by detecting 280–base pair (bp) PCR products. Dcaf1-F2 and Dcaf1-R primers were used for genotyping the wild-type and floxed Dcaf1 alleles by detecting 434- and 468-bp PCR products, respectively. Cre-F and Cre-R were used for genotyping the Nestin-Cre allele by detecting 281-bp PCR products.
Immunoblotting and immunoprecipitation
To prepare total cell lysates, mouse tissues were minced in cold radioimmunoprecipitation assay buffer supplemented with protease inhibitors, phosphatase inhibitors, micrococcal nuclease (2 U/ml), and 1 mM CaCl2. Culture cells were washed with cold phosphate-buffered saline (PBS) once and lysed in 1× Laemmli loading buffer for immunoblotting or 0.5% NP-40 lysis buffer supplemented with protease inhibitors and phosphatase inhibitors for immunoprecipitation. For immunoprecipitation, cell lysates were incubated with specific antibody for 3 hours at 4°C, followed by addition of protein A–agarose for 1 hour at 4°C. Immunoprecipitates were washed three times with lysis buffer, and proteins were eluted from beads with 1× Laemmli loading buffer. For immunoblotting, cell lysates were mixed with Laemmli loading buffer to 1× and heated at 99°C for 5 min. Proteins were resolved on SDS-PAGE and visualized by chemiluminescence. Images were taken by a ChemiDoc MP Imaging system (Bio-Rad). Protein bands were quantitated using Bio-Rad Image Lab 6.0.
Mass spectrometry analysis
The DCAF1 immune complexes were derived using either anti-DCAF1 or anti-FLAG antibodies as described in the text and validated by SYPRO Ruby staining (Thermo Fisher Scientific) and immunoblotting. The immune complexes were then subjected to in-solution tryptic digestion using a filter-aided sample preparation approach (47). Briefly, the immune complexes were eluted by incubating with the molar excess competing antigen peptides and transferred to Vivacon 500 ultrafiltration devices (10,000 MWCO, Hydrosart membrane, Sartorius). Protein samples were washed twice with a urea buffer (8 M urea, 0.1 M tris-HCl, pH 8.0) supplemented with 100 mM dithiothreitol (DTT) followed by centrifugation at 10,000g for 10 min at room temperature. After denaturation and reduction, proteins were then alkylated in urea buffer containing 50 mM iodoacetamide at room temperature for 30 min, shielded from light. Samples were washed twice with 50 mM ABC buffer (ammonium bicarbonate in ddH2O). Two hundred microliters of 50 mM ABC containing trypsin (5 μg/ml) was added to the samples and incubated at 37°C overnight. The digested peptides were collected by centrifugation with ABC buffer and desalted over homemade C18 tips. The clean peptide mixtures were separated on nano-C18 reversed-phase columns connected to an EASY-nLC 1000 system (Thermo Fisher Scientific) and analyzed with a Q Exactive mass spectrometer. Three technical liquid chromatography–mass spectrometry replicates were obtained for each sample.
Mass spectra were processed, and peptide identification was performed using the Andromeda search engine found in MaxQuant software version 1.5.7.4 (Max Planck Institute, Germany). All protein database searches were performed against the UniProt human protein sequence database (UP000005640). Peptide inference was made with a false discovery rate (FDR) of 1%, and peptides were assigned to proteins with a protein FDR of 5%. Peptide identifications are reported by filtering of reverse and contaminant entries and assigning to their leading razor protein. LFQ was performed on the basis of peak area. The measured area under the curve of mass/charge ratio and the retention time-aligned extracted ion chromatogram of a peptide were performed via the LFQ module. Data processing and statistical analysis were performed on Perseus (version 1.6.0.7). Protein quantitation was performed on three technical replicates, and two-sample t test was used with a P value of 5% to report statistically significant protein abundance fold changes.
Histology
Mouse tissues were fixed in PBS-buffered 4% paraformaldehyde (PFA) (VWR) overnight at 4°C, embedded in paraffin, cut into 4-μm sections, and attached on Superfrost Plus Micro Slides (VWR). The slides were deparaffinized with SafeClear II Xylene Substitutes (Thermo Fisher Scientific) and hydrophilized following standard protocol and stained with H&E dye. For IHC, the hydrophilized slides were boiled in 10 mM citrate buffer (pH 6.0) for 30 min for antigen retrieval and washed thoroughly with PBS. Sections were incubated with specific antibodies overnight at 4°C. After PBS wash, the primary antibodies were detected by diaminobenzidine (DAB) with EnVision+ kit [Dual Link System-HRP (DAB+) kit, DAKO] according to the manufacturer’s instruction, followed by hematoxylin counterstaining. Both H&E and IHC slides were dehydrated and mounted with Permount Mounting Medium (Thermo Fisher Scientific).
Flow cytometry
All assays were performed on the Attune NxT Acoustic Focusing Cytometer (Thermo Fisher Scientific), and data were analyzed using FlowJo v10.0.7 software. To analyze DNA content by propidium iodide (PI) staining, cells were fixed in 70% cold ethanol overnight at 4°C, washed two times with PBS containing 1% bovine serum albumin (BSA), and then incubated for 30 min at 37°C with ribonuclease A (RNase A) (0.1 mg/ml) in PBS. Cells were then stained with PI (50 μg/ml) in PBS for 20 min at room temperature, shielded from light. For cell death and apoptosis analysis, cells were harvested by trypsinization, and staining was carried out using the annexin V Apoptosis Detection Kit (BD Biosciences). Briefly, cells were resuspended in 1× binding buffer and incubated with fluorochrome-conjugated annexin V and 7-AAD for 15 min in darkness at room temperature.
For thymocyte, splenocyte, and bone marrow cells, single-cell suspensions were prepared in PBS containing 2% FBS (FACS buffer) after lysis of red blood cells in four parts 0.8% NH4Cl, 0.1 mM EDTA (pH 7.45), and 1 part FACS buffer. Live cells (106) were stained in FACS buffer containing Fc block (BioLegend, TruStain FcX; 1:200) with the antibodies indicated [Pacific Blue–conjugated CD8a, allophycocyanin-AF750–conjugated CD4, phycoerythrin-conjugated B220, or fluorescein isothiocyanate–conjugated immunoglobulin M (IgM)] and immediately analyzed by flow cytometry.
For in vitro T cell proliferation experiments, primary lymphocytes were labeled with 5 μM CFSE (Molecular Probes) in PBS at room temperature for 10 min, and labeling was quenched with the addition of an equal volume of FBS followed by two washes in RPMI. T cells were then stimulated by anti-CD3 and CD28 antibodies (Caltag) and cultured in RPMI for the indicated time before flow cytometry analysis.
Immunofluorescence
Cells were cultured on 35-mm glass bottom dishes, fixed in 4% PFA for 10 min, and permeabilized in 0.1% Triton X-100 in PBS for 5 min. After blocking with 5% goat serum in 2% BSA/PBS for 30 min, cells were incubated with primary antibodies diluted in blocking solution overnight at 4°C. Cells were washed three times in 2% BSA/PBS and incubated with secondary antibody (Alexa Fluor dye–labeled goat anti-rabbit or anti-mouse antibody, Thermo Fisher Scientific) diluted in blocking solution for 1 hour at room temperature in the dark. After washing with PBS, cells were stained with 4′,6-diamidino-2-phenylindole (DAPI) (1.0 μg/ml) at room temperature and then washed with PBS. All images were taken under a confocal microscope.
In vivo and in vitro ubiquitination assay
The procedures for both assays were performed as described previously (48, 49). Briefly, for the in vivo ubiquitylation assay, HeLa cells stably expressing HA-tagged ubiquitin were transfected with indicated siRNA oligos for 72 hours and treated with 20 μM MG132 for 6 hours before collecting the cells. Cells were lysed under denaturing conditions in 1% SDS buffer [50 mM tris-HCl (pH 7.5), 0.5 mM EDTA, 1 mM DTT] by boiling for 10 min. Lysates were clarified by centrifugation at 13,000 rpm and diluted 10-fold with 0.5% NP-40 lysis buffer and then subjected to immunoprecipitation by anti-PWP1 antibody along with protein A–agarose and subsequent SDS-PAGE. Ubiquitylated PWP1 was determined by immunoblotting with anti-HA antibody.
For the in vitro ubiquitylation assay, Myc-tagged CUL4A-DCAF1 immune complexes and FLAG-tagged PWP1 were ectopically expressed in 293F cells, extracted in 0.5% NP-40 lysis buffer, immunoprecipitated using anti–c-MYC or anti-FLAG agarose, and eluted with MYC or FLAG antigen peptides, respectively. Ubiquitylation reactions were performed in a 50-μl reaction volume, containing 100 nM E1 (UBA1), 1 μM E2 (UBCH5C/UBE2D3), 20 μM human recombinant HA-ubiquitin (Boston Biochem), 20 mM Hepes (pH 7.0), 20 mM NaCl, 2 mM MgCl2, 2 mM adenosine triphosphate (ATP), 100 ng of eluted CUL4A-DCAF1 complexes as the source of E3, and 100 ng of FLAG-PWP1 as substrate. The reactions were incubated at 37°C for 1 hour, then terminated by adding 1% final concentration of SDS and boiling for 10 min, followed by 10-fold dilution with a 0.5% NP-40 buffer, and subjected to immunoprecipitation by anti-FLAG agarose and subsequent SDS-PAGE. Ubiquitylated PWP1 was determined by immunoblotting with anti-HA antibody.
Pulse-chase analysis of rRNA synthesis
For monitoring rRNA processing, pulse-chase experiments using [5,6-3H]uridine and l-[methyl-3H]methionine were carried out in HeLa and MEF cells, respectively. HeLa cells were transfected with siRNA targeting DCAF1 for 48 hours, pulsed with [5,6-3H]uridine (10 μCi/ml) for 30 min, and then chased with unlabeled uridine (0.5 mM)–containing medium for the indicated time points. For the pulse-chase analysis in MEFs, 4OHT or solvent-treated cells were cultured in methionine-cysteine–free medium supplemented with dialyzed serum for 30 min and labeled with l-[methyl-3H]-methionine (50 μCi/ml) for additional 30 min. Cold methionine (100 μM) was added to chase the label for various lengths of times. Total RNA was extracted by TRIzol Reagent (Thermo Fisher Scientific) following the manufacturer’s instructions. To assay rRNA precursors and mature 18S and 28S rRNA, 5 μg of 3H-labeled RNA was separated on a 1% formaldehyde agarose gel at 35 V for 24 hours in 1× Mops buffer. To assay the 5.8S rRNA, 10 μg of 3H-labeled RNA was separated on a 10% urea polyacrylamide gel at 400 V for 2.5 hours in 1× Tris-borate-EDTA buffer. Denatured RNA was transferred onto a nylon Hybond-N+ membrane (Amersham). The membrane was dried, ultraviolet cross-linked, and stained with methylene blue dye in 300 mM NaAc (pH 5.2) to visualize the total RNAs. The dried membrane was sprayed with EN3HANCE (PerkinElmer) and exposed to Hyperfilm MP (Amersham Pharmacia) at −80°C with an intensifying screen for 3 to 10 days. The film was subjected to autofluorography. Newly synthesized RNAs in the scanned films were quantitated using the software provided by Amersham Typhoon Imaging System.
Ribosome purification and isolation of ribosome-free ribosomal proteins
Procedures for the preparation of ribosome-free ribosomal protein were modified as previously described (50). Briefly, MEF cells of different genotypes (Dcaf1f/+;Cre-ERT2 and Dcaf1f/−;Cre-ERT2) were grown on 10-cm plate and then treated with 200 nM 4OHT for 48 hours to induce Dcaf1 knockout. Cells were harvested using trypsin, rinsed with PBS, and then resuspended in hypotonic buffer [5 mM tris-HCl (pH 7.5), 2.5 mM MgCl2, and 1.5 mM KCl] supplemented with protease inhibitors. Cells were lysed by homogenization using a Dounce homogenizer. Ribosomes and polysomes were pelleted by ultracentrifugation at 100,000g for 3 hours at 4°C. Ribosome-free supernatants were carefully collected (S100 sample), proteins were precipitated by trichloroacetic acid (15%), and pellets were dissolved in SDS sample buffer. Free ribosomal protein L11 and other proteins were analyzed by immunoblotting.
Statistical analysis
Statistical analyses were performed with a paired, two-tailed Student’s t test. The values of P < 0.05 were considered statistically significant. Data presented were means ± SD, unless otherwise indicated.
Acknowledgments
We thank members of the Xiong and Marzluff laboratories for discussions and support throughout this study, in particular X. Yang, R. Meganck, and C. Holmquist for aid with pulse-labeling analysis and Northern blotting. Funding: This study was supported by NIH grants R01 CA212407 to Y.Z., GM29832 to W.F.M., GM067113 to Y.X., GM133107 to X.C., and UNC UCRF to X.C. Author contributions: Y.X. conceived the project and discussed with W.F.M. X.-R.H., N.S., S.C.J., P.W., and Z.L. performed the experiments and analyzed the data. M.D.S. assisted with VprBP/Dcaf1 mouse genetic analyses, L.X. and X.C. carried out the mass spectrometry analysis, and Y.Z. planned the experiment involving Mdm2C305F/C305F MEFs with Y.X. Y.X., X.-R.H., N.S., S.C.J. W.F.M. wrote the manuscript, and all authors reviewed and/or edited the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/51/eabd6078/DC1
REFERENCES AND NOTES
- 1.Warner J. R., The economics of ribosome biosynthesis in yeast. Trends Biochem. Sci. 24, 437–440 (1999). [DOI] [PubMed] [Google Scholar]
- 2.Marygold S. J., Roote J., Reuter G., Lambertsson A., Ashburner M., Millburn G. H., Harrison P. M., Yu Z., Kenmochi N., Kaufman T. C., Leevers S. J., Cook K. R., The ribosomal protein genes and Minute loci of Drosophila melanogaster. Genome Biol. 8, R216 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lambertsson A., The Minute genes in Drosophila and their molecular functions. Adv. Genet. 38, 69–134 (1998). [DOI] [PubMed] [Google Scholar]
- 4.Freed E. F., Bleichert F., Dutca L. M., Baserga S. J., When ribosomes go bad: Diseases of ribosome biogenesis. Mol. Biosyst. 6, 481–493 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Teng T., Thomas G., Mercer C. A., Growth control and ribosomopathies. Curr. Opin. Genet. Dev. 23, 63–71 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Mills E. W., Green R., Ribosomopathies: There’s strength in numbers. Science 358, eaan2755 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Pelletier J., Thomas G., Volarević S., Ribosome biogenesis in cancer: New players and therapeutic avenues. Nat. Rev. Cancer 18, 51–63 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Sulima S. O., Hofman I. J. F., De Keersmaecker K., Dinman J. D., How ribosomes translate cancer. Cancer Discov. 7, 1069–1087 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Y., Deisenroth C., Zhang Y., RP-MDM2-p53 pathway: Linking ribosomal biogenesis and tumor surveillance. Trends Cancer 2, 191–204 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henras A. K., Plisson-Chastang C., O’Donohue M. F., Chakraborty A., Gleizes P.-E., An overview of pre-ribosomal RNA processing in eukaryotes. Wiley Interdiscip. Rev. RNA 6, 225–242 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klinge S., Woolford J. L. Jr., Ribosome assembly coming into focus. Nat. Rev. Mol. Cell Biol. 20, 116–131 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bohnsack K. E., Bohnsack M. T., Uncovering the assembly pathway of human ribosomes and its emerging links to disease. EMBO J. 38, e100278 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pickart C. M., Back to the future with ubiquitin. Cell 116, 181–190 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Hershko A., Ciechanover A., The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 (1998). [DOI] [PubMed] [Google Scholar]
- 15.Jackson S., Xiong Y., Targeting protein ubiquitylation: DDB1 takes its RING off. Nat. Cell Biol. 11, 379–381 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakagawa T., Mondal K., Swanson P. C., VprBP (DCAF1): A promiscuous substrate recognition subunit that incorporates into both RING-family CRL4 and HECT-family EDD/UBR5 E3 ubiquitin ligases. BMC Mol. Biol. 14, 22 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schabla N. M., Mondal K., Swanson P. C., DCAF1 (VprBP): Emerging physiological roles for a unique dual-service E3 ubiquitin ligase substrate receptor. J. Mol. Cell Biol. 11, 725–735 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang S., Feng Y., Narayan O., Zhao L.-J., Cytoplasmic retention of HIV-1 regulatory protein Vpr by protein-protein interaction with a novel human cytoplasmic protein VprBP. Gene 263, 131–140 (2001). [DOI] [PubMed] [Google Scholar]
- 19.Zhang Y., Feng S., Chen F., Chen H., Wang J., McCall C., Xiong Y., Deng X. W., Arabidopsis DDB1-CUL4 associated factor1 forms a nuclear E3 ubiquitin ligase with DDB1 and CUL4 that is involved in multiple plant developmental processes. Plant Cell 20, 1437–1455 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tamori Y., Bialucha C. U., Tian A.-G., Kajita M., Huang Y.-C., Norman M., Harrison N., Poulton J., Ivanovitch K., Disch L., Liu T., Deng W.-M., Fujita Y., Involvement of Lgl and Mahjong/VprBP in cell competition. PLOS Biol. 8, e1000422 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McCall C. M., Miliani de Marval P. L., Chastain P. D. II, Jackson S. C., He Y. J., Kotake Y., Cook J. G., Xiong Y., Human immunodeficiency virus type 1 Vpr-binding protein VprBP, a WD40 protein associated with the DDB1-CUL4 E3 ubiquitin ligase, is essential for DNA replication and embryonic development. Mol. Cell. Biol. 28, 5621–5633 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yu C., Zhang Y.-L., Pan W.-W., Li X.-M., Wang Z.-W., Ge Z.-J., Zhou J.-J., Cang Y., Tong C., Sun Q.-Y., Fan H.-Y., CRL4 complex regulates mammalian oocyte survival and reprogramming by activation of TET proteins. Science 342, 1518–1521 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Kassmeier M. D., Mondal K., Palmer V. L., Raval P., Kumar S., Perry G. A., Anderson D. K., Ciborowski P., Jackson S., Xiong Y., Swanson P. C., VprBP binds full-length RAG1 and is required for B-cell development and V(D)J recombination fidelity. EMBO J. 31, 945–958 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guo Z., Kong Q., Liu C., Zhang S., Zou L., Yan F., Whitmire J. K., Xiong Y., Chen X., Wan Y. Y., DCAF1 controls T-cell function via p53-dependent and -independent mechanisms. Nat. Commun. 7, 10307 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang J., Bian W., Gao X., Chen L., Jing N., Nestin expression during mouse eye and lens development. Mech. Dev. 94, 287–291 (2000). [DOI] [PubMed] [Google Scholar]
- 26.Cang Y., Zhang J., Nicholas S. A., Bastien J., Li B., Zhou P., Goff S. P., Deletion of DDB1 in mouse brain and lens leads to p53-dependent elimination of proliferating cells. Cell 127, 929–940 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Zhang W., Morris Q. D., Chang R., Shai O., Bakowski M. A., Mitsakakis N., Mohammad N., Robinson M. D., Zirngibl R., Somogyi E., Laurin N., Eftekharpour E., Sat E., Grigull J., Pan Q., Peng W.-T., Krogan N., Greenblatt J., Fehlings M., van der Kooy D., Aubin J., Bruneau B. G., Rossant J., Blencowe B. J., Frey B. J., Hughes T. R., The functional landscape of mouse gene expression. J. Biol. 3, 21 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Honoré B., Baandrup U., Nielsen S., Vorum H., Endonuclein is a cell cycle regulated WD-repeat protein that is up-regulated in adenocarcinoma of the pancreas. Oncogene 21, 1123–1129 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Duronio R. J., Gordon J. I., Boguski M. S., Comparative analysis of the β transducin family with identification of several new members including PWP1, a nonessential gene of Saccharomyces cerevisiae that is divergently transcribed from NMT1. Proteins 13, 41–56 (1992). [DOI] [PubMed] [Google Scholar]
- 30.Krogan N. J., Peng W.-T., Cagney G., Robinson M. D., Haw R., Zhong G., Guo X., Zhang X., Canadien V., Richards D. P., Beattie B. K., Lalev A., Zhang W., Davierwala A. P., Mnaimneh S., Starostine A., Tikuisis A. P., Grigull J., Datta N., Bray J. E., Hughes T. R., Emili A., Greenblatt J. F., High-definition macromolecular composition of yeast RNA-processing complexes. Mol. Cell 13, 225–239 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Talkish J., Campbell I. W., Sahasranaman A., Jakovljevic J., Woolford J. L. Jr., Ribosome assembly factors Pwp1 and Nop12 are important for folding of 5.8S rRNA during ribosome biogenesis in Saccharomyces cerevisiae. Mol. Cell. Biol. 34, 1863–1877 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu Y., Mattila J., Ventelä S., Yadav L., Zhang W., Lamichane N., Sundström J., Kauko O., Grénman R., Varjosalo M., Westermarck J., Hietakangas V., PWP1 mediates nutrient-dependent growth control through nucleolar regulation of ribosomal gene expression. Dev. Cell 43, 240–252.e5 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Badertscher L., Wild T., Montellese C., Alexander L. T., Bammert L., Sarazova M., Stebler M., Csucs G., Mayer T. U., Zamboni N., Zemp I., Horvath P., Kutay U., Genome-wide RNAi screening identifies protein modules required for 40S subunit synthesis in human cells. Cell Rep. 13, 2879–2891 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Warner J. R., [30] Labeling of RNA and phosphoproteins in Saccharomyces cerevisiae. Methods Enzymol. 194, 423–428 (1991). [DOI] [PubMed] [Google Scholar]
- 35.Zhang Y., Wolf G. W., Bhat K., Jin A., Allio T., Burkhart W. A., Xiong Y., Ribosomal protein L11 negatively regulates oncoprotein MDM2 and mediates a p53-dependent ribosomal-stress checkpoint pathway. Mol. Cell. Biol. 23, 8902–8912 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schlott T., Reimer S., Jahns A., Ohlenbusch A., Ruschenburg I., Nagel H., Droese M., Point mutations and nucleotide insertions in the MDM2 zinc finger structure of human tumours. J. Pathol. 182, 54–61 (1997). [DOI] [PubMed] [Google Scholar]
- 37.Lindström M. S., Jin A., Deisenroth C., White Wolf G., Zhang Y., Cancer-associated mutations in the MDM2 zinc finger domain disrupt ribosomal protein interaction and attenuate MDM2-induced p53 degradation. Mol. Cell. Biol. 27, 1056–1068 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zheng J., Lang Y., Zhang Q., Cui D., Sun H., Jiang L., Chen Z., Zhang R., Gao Y., Tian W., Wu W., Tang J., Chen Z., Structure of human MDM2 complexed with RPL11 reveals the molecular basis of p53 activation. Genes Dev. 29, 1524–1534 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stavreva D. A., Kawasaki M., Dundr M., Koberna K., Muller W. G., Tsujimura-Takahashi T., Komatsu W., Hayano T., Isobe T., Raska I., Misteli T., Takahashi N., McNally J. G., Potential roles for ubiquitin and the proteasome during ribosome biogenesis. Mol. Cell. Biol. 26, 5131–5145 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bailly A., Perrin A., Bou Malhab L. J., Pion E., Larance M., Nagala M., Smith P., O’Donohue M.-F., Gleizes P.-E., Zomerdijk J., Lamond A. I., Xirodimas D. P., The NEDD8 inhibitor MLN4924 increases the size of the nucleolus and activates p53 through the ribosomal-Mdm2 pathway. Oncogene 35, 415–426 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Yu Y., Jia W., Lyu Y., Su D., Bai M., Shen J., Qiao J., Han T., Liu W., Chen J., Chen W., Ye D., Guo X., Zhu S., Xi J., Zhu R., Wan X., Gao S., Zhu J., Kang J., Pwp1 regulates telomere length by stabilizing shelterin complex and maintaining histone H4K20 trimethylation. Cell Discov. 5, 47 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cang Y., Zhang J., Nicholas S. A., Kim A. L., Zhou P., Goff S. P., DDB1 is essential for genomic stability in developing epidermis. Proc. Natl. Acad. Sci. U.S.A. 104, 2733–2737 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yamaji S., Zhang M., Zhang J., Endo Y., Bibikova E., Goff S. P., Cang Y., Hepatocyte-specific deletion of DDB1 induces liver regeneration and tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 107, 22237–22242 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakagawa T., Lv L., Nakagawa M., Yu Y., Yu C., D’Alessio A. C., Nakayama K., Fan H.-Y., Chen X., Xiong Y., CRL4VprBP E3 ligase promotes monoubiquitylation and chromatin binding of TET dioxygenases. Mol. Cell 57, 247–260 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hu J., McCall C. M., Ohta T., Xiong Y., Targeted ubiquitination of CDT1 by the DDB1-CUL4A-ROC1 ligase in response to DNA damage. Nat. Cell Biol. 6, 1003–1009 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Ran F. A., Hsu P. D., Wright J., Agarwala V., Scott D. A., Zhang F., Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wiśniewski J. R., Zougman A., Nagaraj N., Mann M., Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 (2009). [DOI] [PubMed] [Google Scholar]
- 48.Furukawa M., Andrews P. S., Xiong Y., Assays for RING family ubiquitin ligases. Methods Mol. Biol. 301, 37–46 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Han X.-R., Zha Z., Yuan H.-X., Feng X., Xia Y.-K., Lei Q.-Y., Guan K.-L., Xiong Y., KDM2B/FBXL10 targets c-Fos for ubiquitylation and degradation in response to mitogenic stimulation. Oncogene 35, 4179–4190 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morita M., Alain T., Topisirovic I., Sonenberg N., Polysome profiling analysis. Bio-protocol 3, e833 (2013). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/51/eabd6078/DC1