

Abstract
Background:
Helios (encoded by IKZF2), a member of the Ikaros family of transcription factors, is a zinc finger protein involved in embryogenesis and immune function. Although predominantly recognized for its role in the development and function of T lymphocytes, particularly the CD4+ regulatory T cells (Tregs), the expression and function of Helios extends beyond the immune system. During embryogenesis, Helios is expressed in a wide range of tissues, making genetic variants that disrupt the function of Helios strong candidates for causing widespread immune-related and developmental abnormalities in humans.
Methods:
We performed detailed phenotypic, genomic, and functional investigations on two unrelated individuals with a phenotype of immune dysregulation combined with syndromic features including craniofacial differences, sensorineural hearing loss, and congenital abnormalities.
Results:
Genome sequencing revealed de novo heterozygous variants that alter the critical DNA-binding zinc fingers (ZF) of Helios. Proband 1 had a tandem duplication of ZFs 2 and 3 in the DNA-binding domain of Helios (p.Gly136_Ser191dup) and Proband 2 had a missense variant impacting one of the key residues for specific base recognition and DNA interaction in ZF2 of Helios (p.Gly153Arg). Functional studies confirmed that both these variant proteins are expressed, and that they interfere with the ability of the wild-type Helios protein to perform its canonical function—repressing IL2 transcription activity—in a dominant negative manner.
Conclusion:
This study is the first to describe dominant negative IKZF2 variants. These variants cause a novel genetic syndrome characterized by Immunodysregulation, Craniofacial anomalies, Hearing impairment, Athelia, and Developmental delay (ICHAD syndrome).
Keywords: IKZF2, Helios, bilateral sensorineural hearing loss, immune dysregulation, craniofacial differences, syndrome, exome sequencing, genome sequencing
INTRODUCTION
The Ikaros Zinc Finger (IKZF) family comprises five transcription factors – Ikaros (encoded by IKZF1), Helios (IKZF2), Aiolos (IKZF3), Eos (IKZF4), and Pegasus (IKZF5) – which play critical roles in both neurodevelopment1–6 and the development, maintenance, and regulation of hematopoietic cells7–9. Their role has been best characterized in lymphocyte development and function. Ikaros, Helios, Aiolos and Eos regulate the differentiation and function of CD4 T helper cell subsets by modulating cytokine pathways in a cell-specific manner8. In particular, Helios helps maintain the phenotype, stability, and function of regulatory T cells (Tregs), a T cell population critical for immune tolerance and autoimmunity prevention10 11. Helios stabilizes Treg function by suppressing IL2 transcription10. Interestingly, loss of Helios results in the de-repression of the IL2 locus, allowing the production of IL2 and other effector cytokines by Tregs10 11.
Expression of Helios extends beyond the immune system. During embryogenesis, Helios is expressed at varying levels in many tissues, including brain, eye, muscle, respiratory and gastrointestinal systems, kidney, skin, and all hematopoietic centers. In contrast, in adults, there is biased Helios expression in T cell lineages, the spleen, and the thymus12 13. During neurodevelopment, Helios is expressed in several brain structures including the lateral ganglionic eminence, the cingulate, insular and retrosplenial cortex, the hippocampus and the accessory olfactory bulb in mice6 14. Helios-deficient mice develop a host of neurodevelopmental features, notably deficits in motor skill acquisition resulting from impaired striatal neurogenesis14, and neuropsychiatric abnormalities resulting from dysfunction in the striatum and hippocampus4–6 14. In addition, Ikzf2 is postnatally expressed in outer hair cells in the cochlea and regulates outer hair cell maturation. Mice carrying a homozygous missense variant in IKZF2 (Ikzf2cello/cello) have sensorineural hearing loss2.
The dynamic, broad expression of Helios during development, and its role in Tregs, makes it a strong candidate for causing disease. Indeed, three recent studies described monoallelic and biallelic variants in IKZF2 causing immunodeficiency and dysregulation in nine individuals15–17. These variants largely fall outside known functional domains of the protein. Helios contains an N-terminal DNA-binding domain that directly interacts with gene regulatory elements, and a C-terminal protein-binding domain, which mediates homo- and heterodimerization with other Ikaros members and interaction with several transcriptional regulators18 19. The previously reported IKZF2 variants mainly affect the ability of Helios to interact with other transcriptional regulators and proteins, resulting in immune dysregulation and enhanced susceptibility to infections in the absence of syndromic features.
Here, we describe a novel syndrome in two unrelated children caused by heterozygous de novo IKZF2 variants that exclusively affect the highly conserved zinc fingers (ZF) in the DNA binding domain of Helios. The core phenotype in these two individuals comprises T cell lymphopenia, recurrent respiratory infections, craniofacial differences, cleft palate, sensorineural hearing impairment, athelia, and developmental delay. Our data suggest a dominant negative mechanism underlying the syndrome, resulting in a more profound phenotype than that associated with loss-of-function IKZF2 variants.
SUBJECTS AND METHODS
Sequencing, Genomic Data Analysis, and Variant Validation
Proband 1
Whole-genome libraries were prepared from peripheral blood DNA from Proband 1 and parents at The Centre for Applied Genomics (Toronto), using Illumina TruSeq PCR-Free library prep kit. The libraries were sequenced to a targeted depth of 40X using the Illumina HiSeq. Reads were mapped to human reference genome GRCh37 using BWA-MEM-v.0.7.1720. Single nucleotide variants (SNVs) and insertions/deletions (Indels) discovery was performed using Genome Analysis Toolkit (GATK HaplotypeCaller-v.3.8)21. SNVs and Indels were filtered out if QD < 2.0. Variants were annotated using VarSeq™ v2.2.1 (Bozeman, MT: Golden Helix). We filtered for rare variants (gnomAD v2.1.1 minor allele frequencies ≤ 1%) (https://gnomad.broadinstitute.org/). We further filtered for coding non-synonymous variants, and non-coding variants that had a SpliceAI score or had pathogenic or variant of uncertain significance records in ClinVar 22. Among these, de novo, compound heterozygous, homozygous, hemizygous, or parentally inherited in genes with evidence of involvement in the immune system were prioritized according to allele counts in gnomAD, damaging in silico predictions, inheritance, relevant phenotypes or functions, and gene constraint metrics (pLI ≥ 0.9, Missense Z-score > 3 for missense variants). Bioinformatic tools such as SIFT, PolyPhen223, CADD24, dbscSNV25, and SpliceAI assessed possible impacts on protein.
Structural variants (SVs) were called using CNVnator-v.0.326, LUMPY-v.0.2.1327, ERDS-v.1.128, and Manta-v.1.1.129, and were annotated using Annovar30. Variants were filtered out if were present in inhouse control databases (allele count >5) or were called by only one tool. SVs were prioritized based on inheritance, zygosity, presence in the reference population databases and other variant databases (gnomAD-SV-v2.131, ClinVar32, dbVar33, Decipher34, DGV35, LOVD-3.036), gene constraint metric (pLI ≥ 0.9), and relevant gene function or phenotypes.
Proband 2
Exome sequencing was performed by a clinical commercial lab on genomic DNA extracted from whole blood for Proband 2 and his parents. Enrichment was carried out using NimbleGen SeqCap EZ Exome Library SR protocol (Roche NimbleGen) and captured libraries were sequenced on the Illumina HiSeq. Sequencing reads were mapped to the human reference assembly (UCSC hg19) using Edico DRAGEN BioIT. Variants were called using Edico DRAGEN haplotype-based caller. CNV analysis of exome sequencing data was performed to identify exonic copy number changes. Variants were annotated using Emedgene. Synonymous variants, intronic variants not affecting splicing sites, and common benign variants were excluded from interpretation unless previously reported in the literature as possibly pathogenic variants. The candidate variants were re-annotated using VarSeq™ v2.2.1 (Bozeman, MT: Golden Helix) to keep the annotations consistent with those generated for Proband 1.
RT-PCR and Sanger sequencing
The mRNA was isolated from lymphoblastoid cell lines (LCLs) derived from Proband 1 and a healthy control, and cDNA was synthesized using Superscript III First-strand synthesis SuperMix (Thermo Fisher Scientific). Reverse transcription PCR (RT-PCR) was carried out with 2x FroggaMix Plus (FroggaBio) using a forward primer (catgacaagcacaaattcag) that spans exon 3-4 junction of the two main transcripts of IKZF2, and a reverse primer (ttgtggcagcgttccttgtg) that maps to the exon 6 region. The resulting PCR products were loaded on an agarose gel, and subsequently Sanger sequenced.
Protein 3D structure
Protein 3D structure for the wild-type (WT) protein was predicted by Alphafold237. Protein structure for the HeliosGly136_Ser191dup was modeled with AlphaFold237, and executed on Google Colab using the default parameters38. We utilized PyMOL (http://www.pymol.org/) to generate HeliosGly153Arg protein structure image by mutagenizing Gly153 on the WT protein structure.
Protein Expression Analysis
Helios expression was assessed by immunoblotting. Briefly, 8x105 HEK293 cells were seeded in 6-well culture plates in 2 mL of Dulbecco modified Eagle medium (DMEM) with 10% FBS (Gibco, Life Technologies), 1% Penicillin-Streptomycin (Gibco, Life Technologies), 2mM L-glutamine (HyClone, Thermo Fisher Scientific), and 1mM sodium pyruvate (Gibco, Life Technologies) and incubated for 24 hours at 37°C prior to transfection. Each well was transfected with 3 μg of relevant plasmids using Lipofectamine™ 3000 Transfection Reagent (Thermo Fisher Scientific) according to the manufacturer’s recommendations. Whole cell lysates were prepared 24 hrs post transfection by lysing cells in RIPA Lysis and Extraction Buffer (Thermo Fisher Scientific) supplemented with the Halt protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). The protein concentrations were measured using Pierce™ Coomassie Plus (Bradford) Assay Reagent. Lysates were separated by 10% SDS-PAGE and transferred onto polyvinylidene difluoride membranes (MilliporeSigma). Membranes were blocked using 5% BSA in Tris-buffered saline supplemented with Tween-20, incubated with anti-Helios (Cell Signalling Technology, cat# 89270, 1:1000), anti-FLAG (Sigma Aldrich, cat# F1804, 1:1000), and anti-β-Actin (Cell Signalling Technology, cat# 3700, 1:20,000) primary antibodies in blocking buffer overnight at 4°C, and lastly incubated with anti-rabbit (IgG DyLight 800, Rockland Immunochemicals) and anti-mouse (IgG IRDye 680RD, LI-COR) secondary antibodies at a concentration of 1:20,000 for 1hr at room temperature. The membranes were imaged using a LI-COR Odyssey infrared scanner (LI-COR Biosciences). The bands were quantified using Image Studio™ Lite (LI-COR) and normalized to their corresponding β-Actin expression levels. For total protein staining, Revert™ 700 Total Protein Stain Kit was used (LI-COR Biosciences).
Luciferase Reporter Assay
The −580 to +57 region of the human IL2 promoter was cloned from genomic DNA obtained from HEK293 cells and the fragment inserted into PGL4.14 (Promega) firefly luciferase reporter plasmid using XhoI and HindIII restriction sites. Briefly, 1.5x105 HEK293 cells were seeded in 24-well culture plates in 0.5 mL of DMEM with 10% FBS, 1% Penicillin-Streptomycin, 2mM L-glutamine, and 1mM sodium pyruvate and incubated at 37°C for 24 hours prior to transfection. Cells were transfected with 250 ng of IL2 promoter plasmid, 250 ng of a plasmid encoding either empty vector (EV), WT and/or variants, and 10ng of PGL4.74 Renilla luciferase (Promega) control plasmid using Lipofectamine™ 3000 Transfection Reagent (Thermo Fisher Scientific) according to the manufacturer’s recommendations. Specifically, cells received either EV, WT, or variant plasmids (250 ng) along with the promoter and renilla plasmid or were co-transfected with increasing ratios of variant to WT plasmid (respectively 0.5:1, 1:1, 2:1, 4:1 and 8:1) adding up to a total amount of 250 ng of expression plasmid. Cell lysates were prepared using 1x Glo Lysis Buffer (Promega) and transferred to white flat-bottom 96-well plates in technical triplicates. Dual-Glo Luciferase Assay Kit (Promega) was used according to manufacturer’s recommendations and luciferase activity was measured using the Infinite M200 plate reader (Tecan) by integrating luminescence over 10 seconds per well.
Statistical Analysis
All data are presented as mean ± standard error of the mean (SEM). Statistical significance was evaluated by paired one-way ANOVA with the Dunnett’s test to account for multiple comparisons, paired two-tailed T-test, and Pearson correlation analysis. The following annotations were used to represent significance: p-val < 0.05 (*), p-val < 0.01 (**), p-val < 0.001 (***).
RESULTS
Identification of two unrelated individuals with syndromic developmental and immune abnormalities
Proband 1
Proband 1 is a female child born at 38 weeks of gestation after an unremarkable pregnancy. Details of her medical history are provided in Table 1 and summarized here. Her birth weight was within the lower normal range (10th centile). Shortly after birth, she experienced respiratory distress due to excessive mucus, which resolved without intervention. She continues to experience increased lower respiratory secretions and chronic rhinorrhea. Investigations have shown normal ciliary ultrastructure and mild bronchiectasis.
Table 1.
Clinical summary and variant description of Probands 1 and 2.
| Individual | Proband 1 | Proband 2 |
|---|---|---|
| Variant Information | ||
| Genomic change (GRCh37) | NC_000002.11:g.213900960_213921017dup | NC_000002.11:g.213914554C>G |
| cDNA change | NC_000002.11(NM_016260.3):c.406+540_574+13477dup | NM_016260.3:c.457G>C |
| Protein change | NP_057344.2:p.Gly136_Ser191dup | NP_057344.2:p.Gly153Arg |
| Exon(s) Affected | Exon 5 | Exon 5 |
| Variant Description | Exon 5 is duplicated in tandem, resulting in the inframe duplication of ZFs 2 and 3 and two linker sequences in the DNA binding domain | Missense variant is located in ZF2 in the DNA binding domain |
| Variant detection method | Trio genome sequencing– not detected by exome sequencing | Trio exome sequencing |
| Inheritance | De novo | De novo |
| General Information | ||
| Ancestry | European | European (Italian, Dutch) |
| Consanguinity | No | No |
| Sex | Female | Male |
| Pregnancy and Birth | ||
| Antenatal complications | • Unremarkable pregnancy • Normal growth and anatomy on ultrasounds at 18-20 weeks of gestation |
• Concern for gestational diabetes, treated with diet |
| Gestational age | • Spontaneous vaginal delivery at 38 weeks | • 35 weeks – caesarean section for breech presentation with premature rupture of membranes |
| Neonatal findings | • Abnormal results on Auditory Brainstem Response (ABR) test • Respiratory congestion due to excessive mucus – ongoing issue since birth • Excessive weight loss due to feeding difficulties related to cleft palate |
• Abnormal results on Auditory Brainstem Response (ABR) test • Respiratory distress after birth and was intubated/on supplemental oxygen for 8-weeks in the NICU |
| Birth weight (kg) | 2.86 (10th percentile) | 2.24 (30th percentile) |
| Most Recent Examination | ||
| Age | 4-6 years | 6-8 years |
| Weight (kg) | 15.5 (10th percentile) | 21.2 (10th percentile) |
| Height (cm) | 106.5 (25th percentile) | 119.6 (10th percentile) |
| Neurobiological Features | ||
| Developmental delay/ intellectual disability | • Global developmental delay affecting language, fine motor skills, and processing speed and emotional regulation | • Autism (CARS score 43 at 2.5y) – repetitive behaviors, head banging, poor eye contact, sensory-seeking/sensory-adverse behaviors, emotional outbursts |
| Motor development | • Delayed fine motor skills | • Borderline-mild delay in gross motor skills |
| Language and speech delay | • Receptive language is at the low end of normal for age • Expressive language is one year delayed • Phonological speech disorder |
• Significant language/communication delays • Nonverbal |
| Seizures | No | No |
| MRI scan | • Normal inner ear structures | • Cystic lesion within sella • Normal inner ear structures |
| Other Findings | ||
| Urological | No | • Hypospadias |
| Hearing/ENT | • Bilateral severe-to-profound sensorineural hearing loss • Chronic clear rhinorrhea and nasal congestion |
• Bilateral moderate sensorineural hearing loss • Mild asymmetric right bony choanal stenosis |
| Vision | • Blepharophimosis • Severe photophobia • Yellow crusting on eyelashes due to increased mucous production • Variable esotropia |
• Fusion of the right eyelid • Lagophthalmos • The cornea of both eyes had stellate epithelial and anterior stromal central opacity |
| Feeding | • Cleft palate causing feeding difficulties early in life – corrected by surgery • Feeding through NG tube starting 4 months of age until receiving a G-tube at 13 months of age • Mainly on oral diet starting 3 years of age |
• Cleft palate • Persistent feeding difficulties, received a G-tube at 3 months of age |
| Craniofacial differences | • Bitemporal narrowing • Ankyloglossia • Upturned nose • High forehead • Macrotia • Depressed nasal bridge |
• Brachycephaly • Midface hypoplasia • Narrow and long palpebral fissures • Short philtrum • Simplified ears • Prominent chin |
| Teeth | • Poor enamel • Thickened gums • Malaligned teeth |
• Dental caries |
| Skeletal | • No anomalies on examination and x-ray | • Juvenile osteochondrosis of the head of the left femur |
| Additional findings | • Athelia (absent nipples) • Mild bronchiectasis with increased lower airway secretions |
• Athelia (absent nipples) • Chronic lung disease - discoid atelectasis • Sleep problems |
| Immune Phenotype | ||
| Newborn screening for immunodeficiency | • Abnormal TREC assay – T cell lymphopenia | • Abnormal TREC assay – T cell lymphopenia |
| Susceptibility to infections | • Recurrent respiratory infections | • Recurrent pneumonia • Recurrent ear infections |
| Atopic disease | • Atopic dermatitis diagnosed at 10 months of age | • None noted |
| Autoimmune disease | • Autoimmune hemolytic anemia diagnosed at 2 months of age | • None noted |
| Additional findings | • Lymphopenia: low total CD4 and CD8 T cells • Normal IgA and IgM levels • Unable to accurately assess endogenous IgG production – patient on IVIG • Normal tetanus and diphtheria vaccine titers documented off IVIG |
• Lymphopenia: low total CD4, CD8 and B cells • Mildly low IgG with normal IgM and IgA • Poor Hib, tetanus, and S. pneumoniae vaccine titers |
Perinatally, she experienced failure to thrive and feeding difficulties. She was diagnosed with cleft palate at 2 months of age. She failed her newborn hearing screen and was diagnosed with sensorineural hearing loss at 6 weeks of age by auditory brainstem response (ABR) testing. Her visual reinforcement audiometry test at 17 months of age indicated moderately severe to profound hearing loss in the right ear, and moderately severe to severe hearing loss in the left ear (Figure 1A), which has since progressed to severe-to-profound hearing loss in both ears. Brain MRI and CT scan at 9 months of age was unremarkable and indicated no inner ear anomalies. She received bilateral cochlear implants between ages 2.5-3.5 years old that have aided her hearing and language development. Proband 1 continues to experience developmental delays across multiple domains, including language, fine motor skills, processing speed, and emotional regulation. Notable craniofacial features include microcephaly, deep-set eyes, blepharophimosis, blepharitis, photophobia, bitemporal narrowing, ankyloglossia, poor enamel, thickened gums, upturned nose, depressed nasal bridge, high forehead, and macrotia. She also has athelia (absent nipples) (Figure 1C–G).
Figure 1. Clinical phenotypes of Proband 1 and 2.
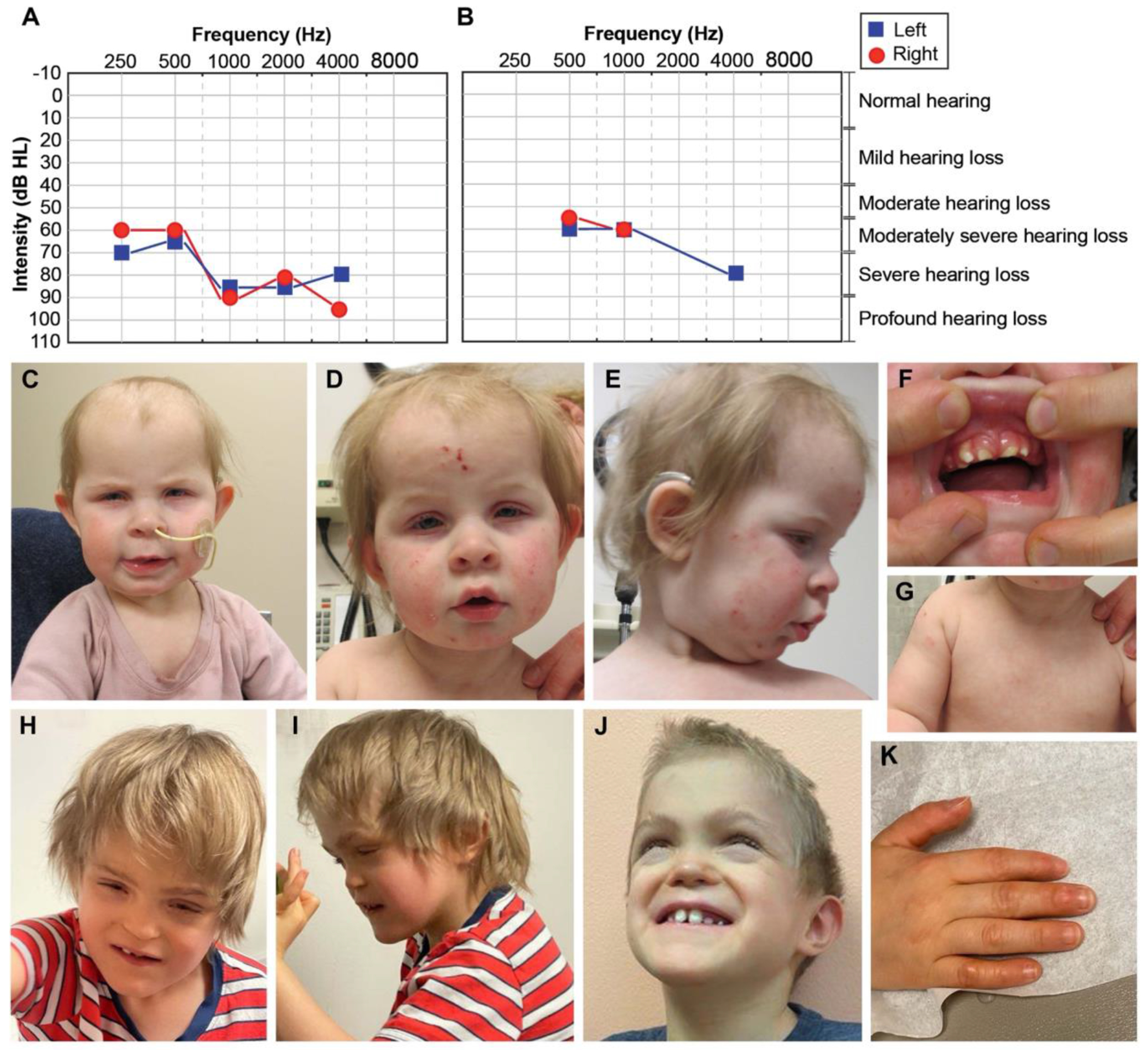
(A) Proband 1’s visual reinforcement audiometry (VRA) test completed at 17 months of age using insert headphones and warble tones. Air conduction thresholds from this test revealed moderately severe to profound hearing loss in the right ear, and moderately severe to severe hearing loss in the left ear. (B) Air conduction thresholds from Proband 2’s auditory brainstem response (ABR) test at 7 years of age indicated moderate to moderately severe hearing loss for the right ear and moderately severe to severe hearing loss for the left ear. (C-D) Face of Proband 1 at 10 months-1.5 years. In C and D note high forehead, macrotia, deep-set eyes, blepharophimosis, blepharitis (yellow crust on lashes), and bitemporal narrowing. In D and E note inflammation and excoriation marks from atopic dermatitis involving the whole face. In E note upturned nose and depressed nasal bridge. (F) Mouth of Proband 1. Note misaligned teeth and thickened gums. (G) Chest of Proband 1. Note athelia (absent nipples). (H-J) Face of Proband 2 at 6-7.5 years. Note brachycephaly, midface hypoplasia, narrow and long palpebral fissures, lagophthalmos (incomplete closure of eyelids), depressed nasal bridge, short philtrum, simplified ears, prominent chin, abnormal dentition, and misaligned teeth. (K) Hand of Proband 2. Note callused digits.
At two months of age, she was diagnosed with an unusual presentation of autoimmune hemolytic anemia. Her direct antiglobulin test (DAT) was strongly positive with mild red cell agglutination and a pan-reactive antibody screen. Bone marrow failure was ruled out by bone marrow biopsy. Proband 1 was also diagnosed with T cell lymphopenia from birth, detected by both an abnormal T cell receptor excision circle (TREC) assay and abnormal flow cytometry documenting low T cell numbers (total CD3, CD8, CD4) (Supplemental Table 1). Serum immunoglobulins showed normal levels of IgG, IgA, and IgM, but elevated IgE levels (Supplemental Table 1). At 10 months of age, she developed severe atopic dermatitis, affecting her face, limbs, and trunk, which responded to topical corticosteroids and emollients. Proband 1 experiences frequent respiratory infections. To support her immune function, she is treated with monthly intravenous immunoglobulin therapy (IVIG) and daily oral antibiotic prophylaxis with good clinical benefit as documented by fewer respiratory infections requiring health service utilization.
Proband 2
Proband 2 is a male child who was linked to this research project through GeneMatcher.org 39. He was born at 35 weeks of gestation. He experienced respiratory distress shortly after birth, and he was mechanically ventilated for the first 36 hours of life. He required supplemental oxygen for most of his 8-week stay in the neonatal intensive care unit. CT scan identified mucosal thickening and inflammation within the nasal cavity, in addition to mild asymmetric right bony choanal stenosis.
His birth weight was within the normal range (30th percentile). Similar to Proband 1, he was diagnosed with cleft palate and had feeding difficulties. He required gastrostomy tube placement at 3 months of age. He failed newborn hearing screening and later his ABR test indicated moderate to severe hearing loss affecting both ears (Figure 1B). His brain MRI demonstrated normal inner ear structures.
Proband 2 has a history of recurrent pneumonia with chronic right upper lobe atelectasis, chronic nasal congestion, and frequent wheezing. He has a history of recurrent sinus infections and otitis media. He was diagnosed with T cell lymphopenia, based on abnormal newborn TREC assay and abnormal flow cytometry revealing low T (total CD3 and CD4) and B cell numbers. Assessment of humoral immune function revealed mildly low serum IgG with normal IgM and IgA levels, and additionally poor responses to routine vaccines (Table 1).
Physical features of Proband 2 include brachycephaly, midface hypoplasia, narrow and long palpebral fissures, fusion of the right eyelid, lagophthalmos, corneal opacity, short philtrum, simplified ears, prominent chin, abnormal dentition (dental caries), athelia, and hypospadias (Figure 1H–K). Proband 2 has also been diagnosed with autism (Childhood Autism Rating Scale score = 43) with repetitive behaviors, head banging, reduced eye contact, sensory-seeking/sensory-adverse behaviors, and emotional outbursts. He is currently nonverbal with a mild delay in gross motor skills (Table 1).
Identification of two monoallelic de novo IKZF2 variants
Genome sequencing for Proband 1 and her unaffected parents revealed a 20 kb de novo heterozygous intragenic tandem duplication (2:213900960-213921017) in IKZF2 spanning exon 5 and parts of the flanking introns 4 and 5 of the Refseq transcript NM_016260.3 (NC_000002.11(NM_016260.3): c.406+540_574+13477dup) (Table 1; Figure 2A and 2C). Two bases were inserted at the join-point which was confirmed by Sanger sequencing (Figure 2C). This duplication is not present in reference population databases (DGV, gnomAD) or other variant databases (ClinVar, LOVD, Decipher, dbVar). It was detected by four of four in silico tools (Manta, Lumpy, CNVnator, ERDS), and was supported by evidence from split reads crossing the breakpoint junction, discordantly mapped read pairs, and increased read depth (Figure 2C). Comparison of common SNP allelic frequencies located in this region in Proband 1, and her parents indicated that the duplication arose on the maternal allele. Loading the RT-PCR products on the gel revealed four bands, two of which corresponded to the expected wild-type products for the two main transcripts of IKZF2, while the other two bands were approximately 168 bps larger than the corresponding wild-type products, indicating the presence of the duplication of exon 5 that encodes 56 amino acids in both transcripts (Figure 2D). Subsequent Sanger sequencing of the RT-PCR products revealed a complete duplication of exon 5 on the coding sequence, and confirmed that the duplication occurs without any alteration in the reading frame or splicing (Figure 2D and Supplemental Figure 1). These findings indicate that this variant results in an in-frame duplication of residues Gly136 to Ser191 (NP_057344.2:p.Gly136_Ser191dup) (Table 1; Figure 2E and 2F).
Figure 2. Family pedigrees, RT-PCR, Sanger results, and schematic of the IKZF2 variants on the gene and protein.
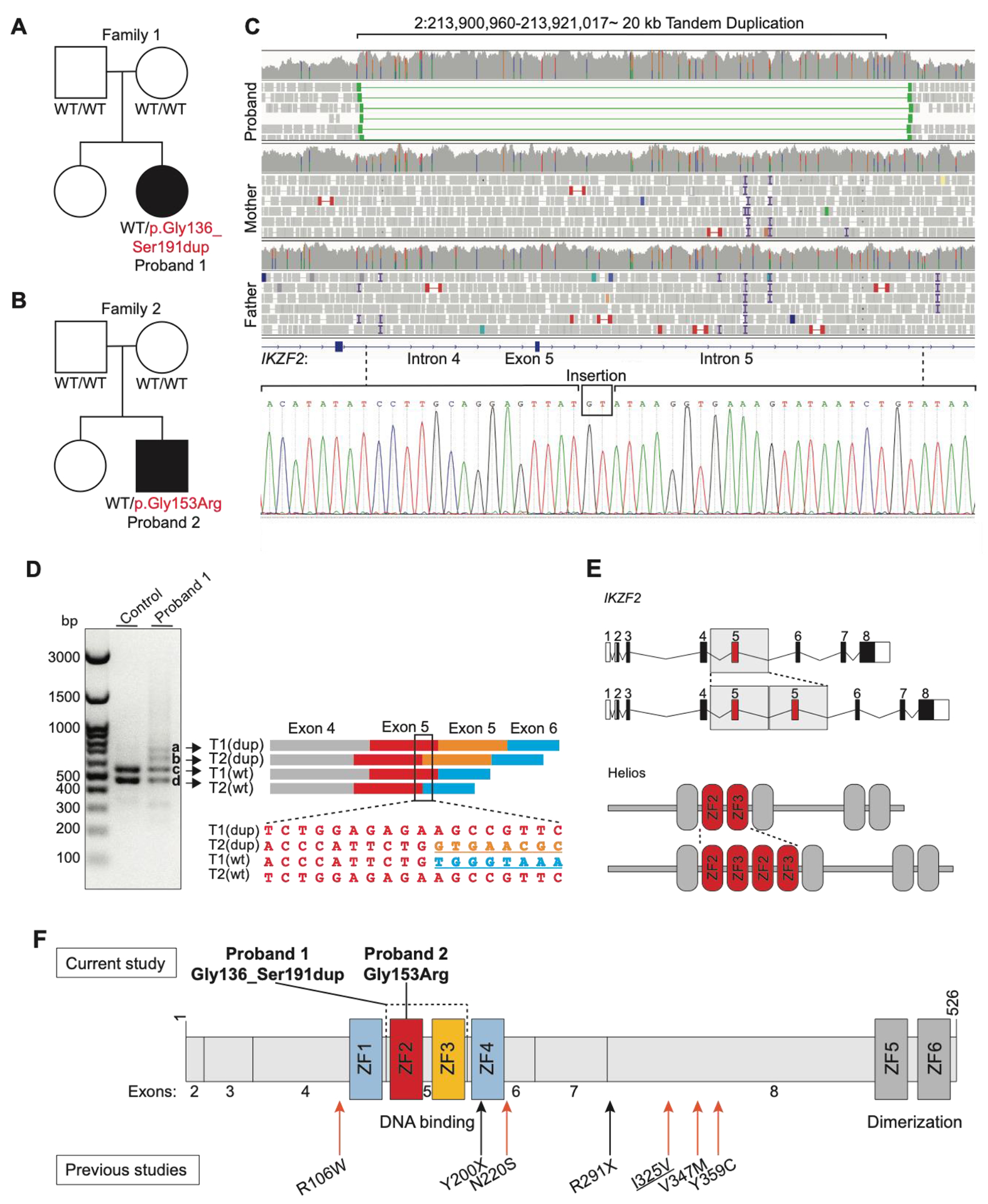
(A and B) The pedigrees of Proband 1 and 2. For individuals tested by genome and/or Sanger sequencing, the genotypes are noted. WT indicates a wild-type allele. Black boxes show individuals with ICHAD. Proband 1 harbors a de novo intragenic tandem duplication and Proband 2 harbors a de novo missense variant (C) The Integrative Genomics Viewer visualization of the duplicated region, with discordant read pairs and increased read depth providing evidence of the genomic duplication in Proband 1 (on top). Sanger sequencing chromatogram of the tandem duplication breakpoint junction on DNA and a GT insertion (at the bottom) (D) The gel image on the left shows the results of the RT-PCR. The two bottom bands (c and d) in the first and second lanes represent the amplicons from the two wild-type transcripts (T1, NM_016260.3; T2, NM_001079526.2; wt, wild-type), while the two top bands that are only visible in the second lane (a and b) correspond to the products from the two transcripts with exon 5 duplication (dup, exon 5 duplication). On the right side of the figure, a schematic of the RT-PCR products is shown alongside the Sanger trace from the region marked on the four RT-PCR products. The second sequence maps to the exon 5-exon 5 junction on altered T2. (E) Schematic of the wild-type and altered gene (top) and protein (bottom). (F) Helios protein and its functional domains (ZFs 1-6). The de novo tandem duplication (p.Gly136_Ser191dup) in Proband 1 and the de novo missense variant (p.Gly153Arg) in Proband 2 are shown on top of the protein, and the previously reported variants in the literature are shown at the bottom of the protein. Both variants presented in this study are located in the DNA binding domain of Helios. ZF, zinc finger; Refseq NM_016260.3; Underlined variants are homozygous variant; Orange arrow indicates missense variant; Black arrow indicates stop gain variant.
Exon 5 of IKZF2 encodes N-terminal Cys2His2 (C2H2) zinc fingers (ZF) 2 and 3 and two linker sequences. ZFs 2 and 3 are highly conserved and are critical for DNA binding activity of Helios (Figure 2E, 2F, and 3A)40 41. Figure 3B illustrates a predicted structure of the wild-type and the altered protein. Notably, somatic tandem duplications in exon 5 of IKZF2 have been reported previously in lymphoma patients (Supplemental Table 2)42.
Figure 3. Multiple alignments of ZFs 2 and 3 and the linker sequences in orthologs and paralogs of IKZF2.
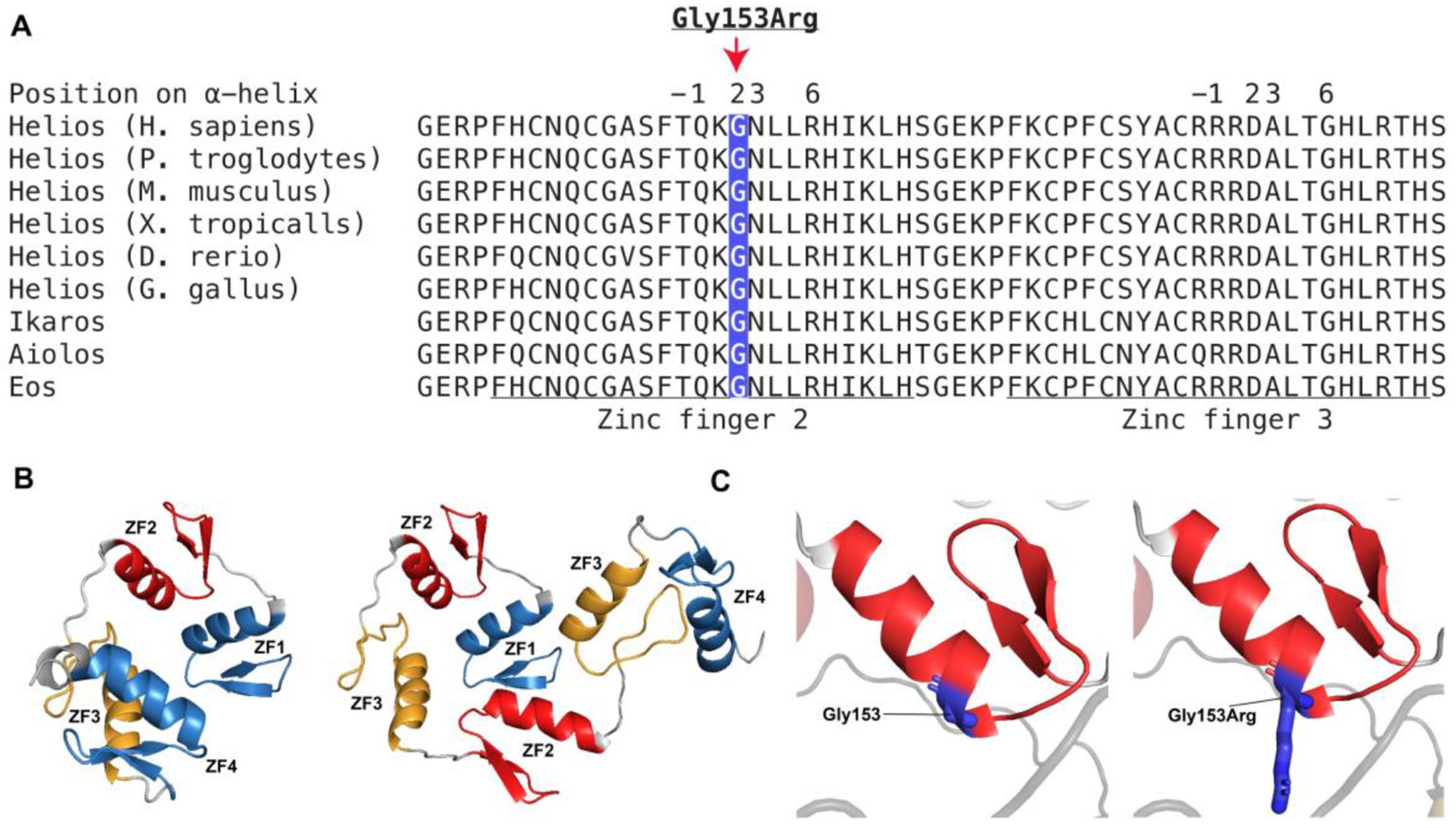
(A) The tandem duplication (p.Gly136_Ser191dup) affects ZFs 2 and 3 and linker sequences which are highly conserved across vertebrates and the paralogs, IKZF1, IKZF3, and IKZF4. The underlined sequence shows ZFs 2 and 3, and the remaining sequence represents the linkers. The missense variant (p.Gly153Arg) affects an amino acid which is situated at position 2 in reference to the start of α helix of ZF2 and has a critical role in DNA recognition and binding. (B) The Alphafold predicted structure of ZFs 1-4. The left structure shows four ZFs (ZFs 1-4), and the right structure shows six ZFs including the duplication (ZFs 1, 2, 3, 2, 3, 4). ZF, zinc finger. (C) The Alphafold predicted structure of human ZF2. ZF2 is shown in red and the amino acid Gly153 shown in dark blue. The left structure shows the wild-type residue, Gly153, and the right structure shows the altered residue, Gly153Arg.
Exome sequencing for Proband 2 and his unaffected parents (Family 2) revealed a de novo heterozygous p.Gly153Arg missense variant in IKZF2 (NM_016260.3:c.457G>C) (Figure 2B and 2F). This variant is absent in reference population databases including gnomAD, TOPMed, Decipher, ClinVar, and LOVD. All five in silico tools (SIFT=”Damaging”, Polyphen-2=”Probably damaging”, REVEL=”0.5”, FATHMM-XF=”1.0”, and CADD=”32”) predict that this missense variant has a damaging impact on the protein. Gly153 is a highly conserved residue across vertebrates and the paralogs of IKZF2 (Figure 3A). The missense variant is in ZF2 on the +2 residue which is one of four residues (+6, +3, −1 and +2 in reference to the beginning of α helix) critical for DNA recognition and binding (Figure 3A)43. Figure 3C shows the predicted structure of ZF2 and the Gly153 and Gly153Arg. Pathogenic variants affecting the four residues of the α helix have been reported in Ikaros and Aiolos44. Specifically, residues in the +2 position in the ZF domains have an important function in DNA binding by recognizing and making contact with the complementary strand of DNA43. A similar amino acid substitution, p.Gly159Arg, affecting the same homologous amino acid in IKZF3 (Aiolos) has been functionally shown to be pathogenic, as it impairs binding of Aiolos homodimers and Aiolos-Ikaros heterodimers to their corresponding canonical DNA sequences in a dominant negative manner (Supplemental Figure 2)45.
HeliosGly136_Ser191dup protein expression is reduced while HeliosGly153Arg protein expression is comparable to WT Helios
To understand the impact of p.Gly136_Ser191dup on protein expression, we utilized Human Embryonic Kidney (HEK293) cells which lack basal expression of Helios and subsequently transfected them with pCMV6-XL4 plasmids containing either FLAG-tagged wild-type IKZF2 (WT), p.Gly136_Ser191dup (Dup) IKZF2, or an empty vector (EV). As expected, EV-transfected cells lacked Helios expression as assessed by both FLAG and Helios antibody probes, confirming absence of detectable endogenous expression of Helios protein in HEK293 cells. In contrast, in WT IKZF2 transfected cells, a prominent band at ~70 kDa was detected by FLAG and Helios antibodies, coinciding with the expected size of the major isoform of the Helios protein (Figure 4A and 4B). HeliosGly136_Ser191dup was expressed at ~75 kDa, reflecting the 56 amino acid duplication of ZFs 2 and 3. However the relative expression of the variant was significantly reduced to ~50% of the WT Helios protein, as quantified by densitometry (Figure 4A–C). To model the heterozygous state of Proband 1, we co-transfected cells with both WT and Dup IKZF2 plasmids. The expression of HeliosGly136_Ser191dup remained significantly lower than the WT protein (Figure 4D). We also show that the expression of the WT protein is independent of Heliosp.Gly136_Ser191dup, as the amount of the WT protein co-expressed with the variant remains unaffected compared to when it is co-expressed with the EV (Figure 4E). However, we show that the co-expression of Heliosp.Gly136_Ser191dup with the WT protein leads to significantly higher expression levels of Heliosp.Gly136_Ser191dup compared to its co-expression with EV. This suggests that the presence of the WT protein may affect the stability of the variant protein (Figure 4F).
Figure 4. Assessment of variant protein expression in HEK293 cells.
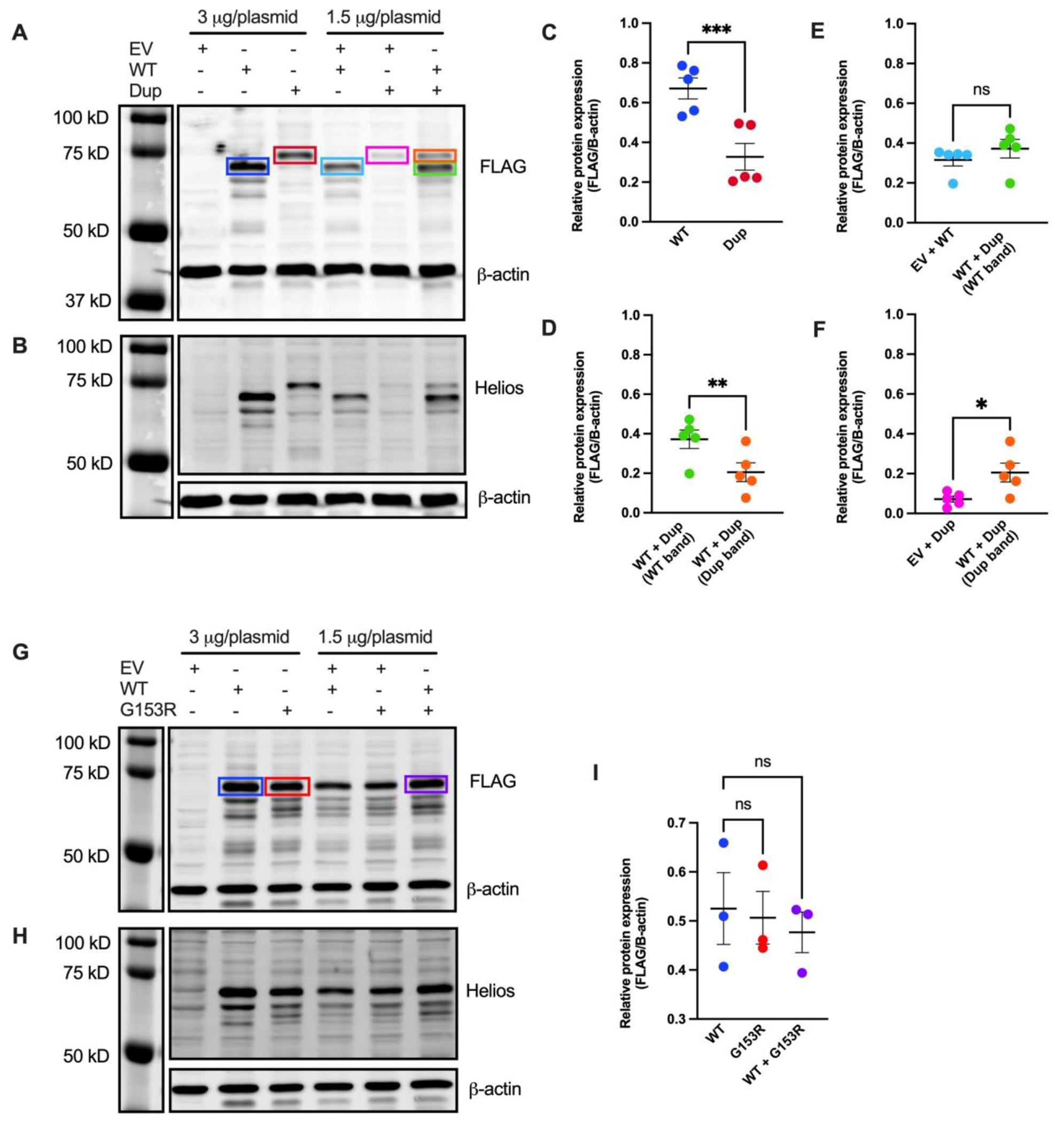
(A and B) Immunoblot of lysates obtained from HEK293 cells transfected with with pCMV6-XL4 plasmids expressing FLAG-tagged wild-type IKZF2 (WT), p.Gly136_Ser191dup variant (Dup), and/or empty vector control (EV). Protein was detected using anti-FLAG antibody in A and anti-Helios antibody in B. An anti-β actin antibody was used as loading control. A, B are representative blots from five independent experiments. (C, D, E and F) Band intensities of FLAG and β-actin fluorescence signals were quantified using Image Studio™ Lite Quantification Software (Licor). Data presented are mean ± SEM from five independent experiments. Paired two-tailed t-test was used for statistical analysis: *p≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001. (G and H) Immunoblot of lysates obtained from HEK293 cells transfected with pCMV6-XL4 plasmids expressing FLAG-tagged WT IKZF2, p.Gly153Arg (G153R), and/or EV. Protein was detected using anti-FLAG antibody in G and anti-Helios antibody in H. An anti-β-actin antibody was used as loading control. G and H are representative blots from three independent experiments. (I) Quantification of band intensities of FLAG and β-actin fluorescence signals using Image Studio™ Lite. Data presented are mean ± SEM from three independent experiments. Paired two-tailed t-test was used for statistical analysis; ns, not significant.
The expression of HeliosGly153Arg was indistinguishable from WT protein, as quantified by densitometry (Figure 4G–I). To recapitulate the heterozygous state of Proband 2, we co-transfected cells with both WT and p.G153R (G153R) plasmids. Total Helios protein expression remained unchanged and statistically comparable to that of the WT Helios and HeliosGly153Arg alone (Figure 4I). This suggests the expression of WT and HeliosGly153Arg are independent of each other. In addition, to confidently interpret changes to protein expression levels for both variants, total protein staining used to ensure even loading of the gel (Supplemental Figure 3).
HeliosGly136_Ser191dup and HeliosGly153Arg cannot effectively repress IL2 promoter activity and interfere with WT protein function in a dominant negative manner
Exon 5 encodes ZFs 2 and 3, which are critical domains for DNA recognition and binding capability of Helios 8. Helios binds to and epigenetically modifies the interleukin 2 (IL2) promoter, supressing its expression 10. We employed a luciferase reporter assay to assess the ability of both variant proteins to suppress IL2 expression. We co-transfected HEK293 cells with a firefly luciferase reporter plasmid containing the −580 to +57 region of the human IL2 promoter, EV, or WT/variant plasmids, and Renilla luciferase plasmid (Figure 5A). We measured firefly luciferase luminescence as an indicator of IL2 promoter activity and normalized this value to Renilla luciferase luminescence to account for differences in transfection efficiency. HeliosWT (represented by 0% variant to WT plasmid ratio) was able to repress IL2 promoter activity significantly more effectively than HeliosGly136_Ser191dup and HeliosGly153Arg expressed alone (100% variant to WT plasmid ratio). When the WT plasmid was co-transfected with increasing ratios of p.Gly136_Ser191dup or p.Gly153Arg plasmids (respectively 0.5:1, 1:1, 2:1, 4:1 and 8:1 ratios of variant to WT plasmids), we observed a significant increase in the promoter activity of IL2, which indicated a decrease in the ability of the variant proteins to repress the IL2 promoter in a dominant negative manner (Figure 5B and 5C).
Figure 5. Luciferase reporter assay to assess variants’ ability to repress IL2 promoter activity.
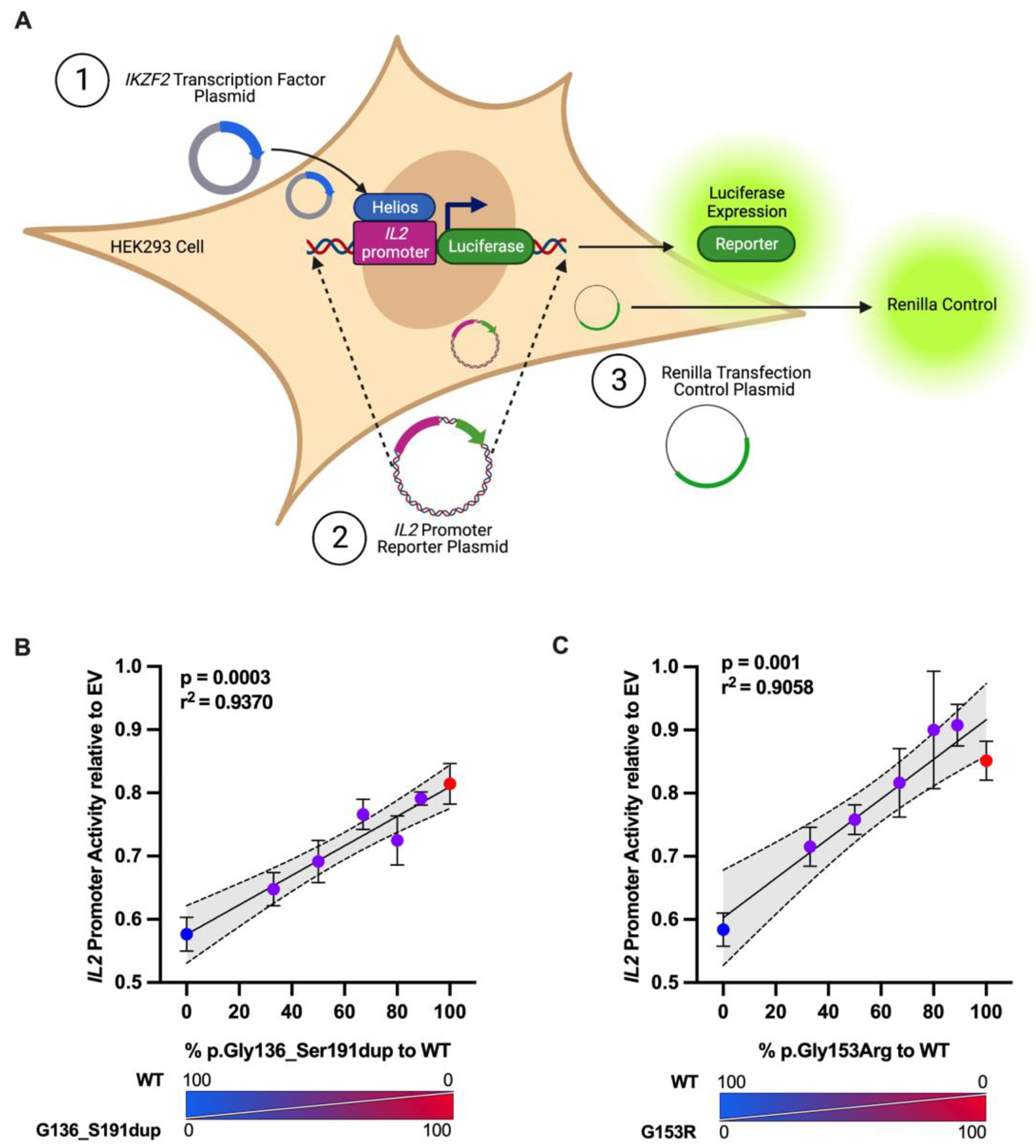
(A) Schematic representing the experiment. HEK293 cells were transfected with an empty pCMV6-XL4 (EV) plasmid or a plasmid encoding the wild-type (WT) and/or the indicated ratios of IKZF2 variants, a pGL4.14 firefly luciferase reporter plasmid carrying a region of the human IL2 promoter, and pGL4.74 renilla luciferase plasmid as transfection control. (B and C) After 24 hours, cells were lysed and measured for firefly luciferase and renilla luciferase activity. Data presented are mean ± SEM from three independent experiments, and each independent experiment is an average of three technical replicates. Dotted lines indicate 95% confidence intervals for Pearson’s Correlation test.
DISCUSSION
In this study we describe a novel multisystem disorder in two unrelated patients caused by heterozygous pathogenic de novo variants affecting the DNA binding domain of Helios. Both variants result in syndromic features including craniofacial differences, athelia, early-onset bilateral sensorineural hearing loss, cleft palate, developmental delays, combined with immune system abnormalities including T cell lymphopenia and recurrent respiratory infections. We propose the acronym ICHAD, short for Immunodysregulation, Craniofacial anomalies, Hearing impairment, Athelia, and Developmental delay.
Although some functional roles of Helios remain poorly understood, its broad expression during embryogenesis4 13 14 likely underlies the developmental phenotypes observed in both probands. There are phenotypic similarities between our patients and various murine models of Helios deficiency. For example, both probands have developmental abnormalities of their eyelids. Proband 1 has blepharophimosis and blepharitis (Figure 1C and 1D), and Proband 2 has fusion of his right eyelid, lagophthalmos, and corneal opacity (Figure 1H and 1J). Replicating this human phenotype, Ikzf2−/− mice have smaller eye openings46, and incomplete palpebral closure due to inflammation of the sebaceous glands in the eyelids6, while Ikzf2cello/cello mice exhibit microphthalmia (small eyes) and a ‘cloudy eye’ phenotype2. In addition, replicating the early-onset bilateral sensorineural hearing loss seen in both patients (Figure 1A and 1B), Ikzf2cello/cello mice show elevated ABR thresholds and postnatal hearing impairment, as explained by the role of Helios in the maturation of outer hair cells of the cochlea2.
Consistent with the role of Helios in the development of T lymphocytes and regulation of the immune system, both probands have T cell lymphopenia and enhanced susceptibility to infections, while Proband 1 is also affected by immune dysregulation in the form of atopy and autoimmunity. Similar immune features are seen in global and selective murine models of Helios deficiency. Mice with Helios-deficient Tregs develop progressive systemic immune activation over time47, and Ikzf2−/− mice develop autoimmune disease by 6–8 months of age11.
Helios deficiency in mice has also been linked to neurodevelopmental abnormalities. Martín-Ibáñez et al. have shown that Helios has a regulatory role in development of the striatum, cortex, and hippocampus in mice, and Ikzf2−/− mice show a decrease in striatopallidal neurons of the dorsomedial striatum that control motor skill learning during embryonic development 4 14. Ikzf2−/− mice also exhibit schizophrenic-like behaviors related to dysfunction in the striatum and hippocampus5. When considered in aggregate, the murine models support a role of Helios impairment in causing developmental delays.
Since both HeliosGly136_Ser191dup and HeliosGly153Arg proteins are expressed (Figure 4), haploinsufficiency is unlikely to be the mechanism of disease. Both patients are heterozygous for variants that affect critical and highly conserved zinc fingers (ZF) in the DNA binding domain of Helios that recognize unique DNA motifs and thus determine the overall sequence specificity and affinity of Helios48. As such, we hypothesized that both variants would alter the function of WT Helios in a dominant negative manner. The pathogenicity and dominant negative impact of p.Gly136_Ser191dup and p.Gly153Arg are supported by our luciferase reporter assay using the IL2 promoter which showed that co-transfecting WT plasmid with increasing amounts of variant plasmids resulted in significant increase in the transcriptional activity of the IL2 locus, thereby demonstrating that the variant proteins fail to suppress IL2 transcription in a dominant negative manner (Figure 5). We anticipate this assay will be useful in assessing the functional impact of additional novel IKZF2 variants likely to emerge.
Other evidence supporting a dominant negative mechanism comes from similar tandem duplications involving ZFs 2 and 3 of Helios that have been reported in cases of peripheral T-cell lymphoma and a dominant negative function has been hypothesized for these variants (Supplemental Table 2)42. Somatic mutations in Helios with dominant negative function have been detected in hematopoietic malignancies49. Furthermore, the discordance of the number of ZFs between HeliosGly136_Ser191dup and the WT protein may affect the size and structure of the altered Helios homodimers and heterodimers, consequently impacting their ability to bind to canonical DNA sequences. The dominant negative effect of HeliosGly136_Ser191dup may result from variant protein forming dysfunctional dimers with WT protein. Finally, based on the role of the gene and zinc finger duplications as an evolutionary process in the functional diversification of the poly-zinc-finger gene family50, we speculate that the duplication of zinc fingers in our study may have a neomorphic effect by recognizing novel DNA binding sites. However, we did not experimentally investigate this possibility, and further studies would be necessary to confirm or refute this hypothesis.
Evidence for pathogenicity of HeliosGly153Arg can be drawn from study of human diseases related to missense variants in other Ikaros family members. Gly153 is a highly conserved residue across paralogs (Figure 3A) and is homologous to Gly159 in Aiolos (Supplemental Figure 2). Recently, Yamashita et al. reported a pathogenic missense variant (p.Gly159Arg) in Aiolos, resulting in T and B cell defects in the affected individuals due to dominant negative and neomorphic effects45. AiolosGly159Arg was expressed comparably to WT and formed homodimers with WT Aiolos, but failed to recognize the canonical Aiolos DNA sequences. Furthermore, AiolosGly159Arg interfered with Ikaros function by forming defective heterodimers that were unable to bind to canonical Aiolos-Ikaros DNA binding sequences. AiolosGly159Arg also showed a neomorphic effect by binding to novel sequences. Based on the homology between Gly153 in Helios and the pathogenic Gly159 in Aiolos, we propose that the Gly153Arg substitution not only exerts a dominant negative effect, but may also result in a neomorphic effect as does AiolosGly159Arg. Nevertheless, the focus of our study was on the dominant negative effect, and further investigations are needed to confirm this proposition.
In conclusion, our data demonstrate that dominant negative-acting variants in Helios, a key transcription factor involved in hematopoietic development and embryogenesis, are associated with a syndromic disorder, ICHAD, involving developmental and immune abnormalities.
Supplementary Material
KEY MESSAGES.
What is already known on this topic?
Helios (encoded by IKZF2) is known to be essential for regulating the development and function of T cells, regulatory T cells, and natural killer cells, with recent studies linking IKZF2 variants to immunodeficiency and immune dysregulation.
What this study adds?
Our study reveals that dominant negative variants in the DNA binding domain of Helios are associated with a more complex syndrome in humans, characterized by immunodysregulation, craniofacial anomalies, hearing impairment, athelia, and developmental delay (designated as ICHAD). This highlights the broader role of Helios in human physiology, extending beyond the immune system to include fetal development and neurodevelopment, which is consistent with murine models of Helios deficiency.
How might this study affect research, practice, or policy?
This study expands the spectrum of Helios-related diseases in humans, broadening our understanding of Helios’ role in human physiology, prompting further investigation into the molecular mechanisms underlying ICHAD, informing clinical management of affected individuals, and potentially influencing policy development for screening and treatment strategies in patients with syndromic disorders involving immunodysregulation and developmental abnormalities.
Acknowledgements:
We would like to acknowledge the patients and their families for their continued support and trust. We thank The Centre for Applied Genomics (TCAG) for providing sequencing services.
Funding:
MV-S is funded by the Vanier Canada Graduate Scholarship (Vanier CGS) and the University of British Columbia Four Year Doctoral Fellowship (4YF). CMB holds a Health Professional-Investigator award from Michael Smith Health Research BC and is supported by the Providence Healthcare Research Institute Early Career Investigator award. SET holds a Translational Research Grant Award from the Jeffrey Modell Foundation, a Tier 1 Canada Research Chair in Pediatric Precision Health, and the Aubrey J. Tingle Professor of Pediatric Immunology. SET and CMB hold a Project Grant from the Canadian Institutes of Health Research (PJT 178054). Part of this work was funded by a joint award held by AL, CvK, and SM from Genome Canada, Genome BC, and BC Children’s Hospital Research Institute (F17-04161). Additionally, research reported in this manuscript was supported by the NIH Common Fund, through the Office of Strategic Coordination/Office of the NIH Director under Award Number U01HG007709. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
List of abbreviations
- ABR
Auditory Brainstem Response
- C2H2
Cys2His2
- EV
Empty vector
- FOXP3
Forkhead box P3
- HEK293
Human Embryonic Kidney 293
- IEI
Inborn errors of immunity
- IKZF
IKAROS Family Zinc Finger
- IL2
Interleukin 2
- Indels
Insertions/deletions
- LoF
Loss-of-function
- SLE
Systemic lupus erythematosus
- SNV
Single nucleotide variants
- Th
T helper
- Treg
Regulatory T cells
- WT
Wild-type
- ZF
Zinc finger
Footnotes
Competing interests: The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing completed at Baylor Genetics Laboratories.
Ethics approval: Written informed consent was obtained from participants for specimen collection, genetic analyses, clinical and research data generation, and the publication of relevant findings and photographs. The research study protocols were approved by The University of British Columbia Clinical Research Ethics Board (H18-02912) and the National Institutes of Health Intramural Institutional Review Board (15HG0130).
Collaborators: Members of the Undiagnosed Diseases Network include: Maria T. Acosta, Margaret Adam, David R. Adams, Raquel L. Alvarez, Justin Alvey, Laura Amendola, Ashley Andrews, Euan A. Ashley, Carlos A. Bacino,Guney Bademci, Ashok Balasubramanyam, Dustin Baldridge, Jim Bale, Michael Bamshad, Deborah Barbouth, Pinar Bayrak-Toydemir, Anita Beck, Alan H. Beggs, Edward Behrens, Gill Bejerano, Hugo J. Bellen, Jimmy Bennett, Beverly Berg-Rood, Jonathan A. Bernstein, Gerard T. Berry, Anna Bican, Stephanie Bivona, Elizabeth Blue, John Bohnsack, Devon Bonner, Lorenzo Botto, Brenna Boyd, Lauren C. Briere, Elly Brokamp, Gabrielle Brown, Elizabeth A. Burke, Lindsay C. Burrage, Manish J. Butte, Peter Byers, William E. Byrd, John Carey, Olveen Carrasquillo, Thomas Cassini, Ta Chen Peter Chang, Sirisak Chanprasert, Hsiao-Tuan Chao, Gary D. Clark, Terra R. Coakley, Laurel A. Cobban, Joy D. Cogan, Matthew Coggins, F. Sessions Cole, Heather A. Colley, Cynthia M. Cooper, Heidi Cope, Rosario Corona, William J. Craigen, Andrew B. Crouse, Michael Cunningham, Precilla D’Souza, Hongzheng Dai, Surendra Dasari, Joie Davis, Jyoti G. Dayal, Esteban C. Dell’Angelica, Katrina Dipple, Daniel Doherty, Naghmeh Dorrani, Argenia L. Doss, Emilie D. Douine, Laura Duncan, Dawn Earl, David J. Eckstein, Lisa T. Emrick, Christine M. Eng, Marni Falk, Elizabeth L. Fieg, Paul G. Fisher, Brent L. Fogel, Irman Forghani, William A. Gahl, Ian Glass, Bernadette Gochuico, Page C. Goddard, Rena A. Godfrey, Katie Golden-Grant, Alana Grajewski, Don Hadley, Sihoun Hahn, Meghan C. Halley, Rizwan Hamid, Kelly Hassey, Nichole Hayes, Frances High, Anne Hing, Fuki M. Hisama, Ingrid A. Holm, Jason Hom, Martha Horike-Pyne, Alden Huang, Sarah Hutchison, Wendy Introne, Rosario Isasi, Kosuke Izumi, Fariha Jamal, Gail P. Jarvik, Jeffrey Jarvik, Suman Jayadev, Orpa Jean-Marie, Vaidehi Jobanputra, Lefkothea Karaviti, Jennifer Kennedy, Shamika Ketkar, Dana Kiley, Gonench Kilich, Shilpa N. Kobren, Isaac S. Kohane, Jennefer N. Kohler, Susan Korrick, Mary Kozuira, Deborah Krakow, Donna M. Krasnewich, Elijah Kravets, Seema R. Lalani, Byron Lam, Christina Lam, Brendan C. Lanpher, Ian R. Lanza, Kimberly LeBlanc, Brendan H. Lee, Roy Levitt, Richard A. Lewis, Pengfei Liu, Xue Zhong Liu, Nicola Longo, Sandra K. Loo, Joseph Loscalzo, Richard L. Maas, Ellen F. Macnamara, Calum A. MacRae, Valerie V. Maduro, Rachel Mahoney, May Christine V. Malicdan, Laura A. Mamounas, Teri A. Manolio, Rong Mao, Kenneth Maravilla, Ronit Marom, Gabor Marth, Beth A. Martin, Martin G. Martin, Julian A. Martínez-Agosto, Shruti Marwaha, Jacob McCauley, Allyn McConkie-Rosell, Alexa T. McCray, Elisabeth McGee, Heather Mefford, J. Lawrence Merritt, Matthew Might, Ghayda Mirzaa, Eva Morava, Paolo Moretti, John Mulvihill, Mariko Nakano-Okuno, Stanley F. Nelson, John H. Newman, Sarah K. Nicholas, Deborah Nickerson, Shirley Nieves-Rodriguez, Donna Novacic, Devin Oglesbee, James P. Orengo, Laura Pace, Stephen Pak, J. Carl Pallais, Christina G.S. Palmer, Jeanette C. Papp, Neil H. Parker, John A. Phillips III, Jennifer E. Posey, Lorraine Potocki, Barbara N. Pusey Swerdzewski, Aaron Quinlan, Deepak A. Rao, Anna Raper, Wendy Raskind, Genecee Renteria, Chloe M. Reuter, Lynette Rives, Amy K. Robertson, Lance H. Rodan, Jill A. Rosenfeld, Natalie Rosenwasser, Francis Rossignol, Maura Ruzhnikov, Ralph Sacco, Jacinda B. Sampson, Mario Saporta, Judy Schaechter, Timothy Schedl, Kelly Schoch, Daryl A. Scott, C. Ron Scott, Vandana Shashi, Jimann Shin, Edwin K. Silverman, Janet S. Sinsheimer, Kathy Sisco, Edward C. Smith, Kevin S. Smith, Emily Solem, Lilianna Solnica-Krezel, Ben Solomon, Rebecca C. Spillmann, Joan M. Stoler, Kathleen Sullivan, Jennifer A. Sullivan, Angela Sun, Shirley Sutton, David A. Sweetser, Virginia Sybert, Holly K. Tabor, Queenie K.-G. Tan, Amelia L. M. Tan, Mustafa Tekin, Fred Telischi, Willa Thorson, Cynthia J. Tifft, Camilo Toro, Alyssa A. Tran, Rachel A. Ungar, Tiina K. Urv, Adeline Vanderver, Matt Velinder , Dave Viskochil, Tiphanie P. Vogel, Colleen E. Wahl, Melissa Walker, Stephanie Wallace, Nicole M. Walley, Jennifer Wambach, Jijun Wan, Lee-kai Wang, Michael F. Wangler, Patricia A. Ward, Daniel Wegner, Monika Weisz Hubshman, Mark Wener, Tara Wenger, Monte Westerfield, Matthew T. Wheeler, Jordan Whitlock, Lynne A. Wolfe, Kim Worley, Changrui Xiao, Shinya Yamamoto, John Yang, Zhe Zhang, Stephan Zuchner
References
- 1.Alsiö JM, Tarchini B, Cayouette M, et al. Ikaros promotes early-born neuronal fates in the cerebral cortex. PNAS 2013;110(8):E716–E25. doi: 10.1073/pnas.1215707110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chessum L, Matern MS, Kelly MC, et al. Helios is a key transcriptional regulator of outer hair cell maturation. Nature 2018;563(7733):696–700. doi: 10.1038/s41586-018-0728-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Javed A, Mattar P, Cui A, et al. Ikaros family proteins regulate developmental windows in the mouse retina through convergent and divergent transcriptional programs: Cold Spring Harbor Laboratory, 2021. [Google Scholar]
- 4.Martín-Ibáñez R, Pardo M, Giralt A, et al. Helios expression coordinates the development of a subset of striatopallidal medium spiny neurons. Development 2017;144(8):1566–77. doi: 10.1242/dev.138248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sancho-Balsells A, Brito V, Fernandez B, et al. Lack of Helios During Neural Development Induces Adult Schizophrenia-Like Behaviors Associated With Aberrant Levels of the TRIF-Recruiter Protein WDFY1. Front Cell Neurosci 2020;14:93. doi: 10.3389/fncel.2020.00093 [published Online First: 2020/06/02] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Giralt A, Brito V, Pardo M, et al. Helios modulates the maturation of a CA1 neuronal subpopulation required for spatial memory formation. Exp Neurol 2020;323:113095. doi: 10.1016/j.expneurol.2019.113095 [published Online First: 2019/11/13] [DOI] [PubMed] [Google Scholar]
- 7.Heizmann B, Kastner P, Chan S. The Ikaros family in lymphocyte development. Curr Opin Immunol 2018;51:14–23. doi: 10.1016/j.coi.2017.11.005 [DOI] [PubMed] [Google Scholar]
- 8.Powell MD, Read KA, Sreekumar BK, et al. Ikaros Zinc Finger Transcription Factors: Regulators of Cytokine Signaling Pathways and CD4+ T Helper Cell Differentiation. Front Immunol 2019;10 doi: 10.3389/fimmu.2019.01299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoshida T, Georgopoulos K. Ikaros fingers on lymphocyte differentiation. Int J Hematol 2014;100(3):220–29. doi: 10.1007/s12185-014-1644-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baine I, Basu S, Ames R, et al. Helios Induces Epigenetic Silencing of Il2 Gene Expression in Regulatory T Cells. J Immunol 2013;190(3):1008–16. doi: 10.4049/jimmunol.1200792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim H-J, Barnitz RA, Kreslavsky T, et al. Stable inhibitory activity of regulatory T cells requires the transcription factor Helios. Science 2015;350(6258):334–39. doi: 10.1126/science.aad0616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hosokawa Y, Maeda Y, Seto M. Human Helios, an Ikaros -related zinc finger DNA binding protein: cDNA cloning and tissue expression pattern. Immunogenetics 1999;50(1-2):106–08. doi: 10.1007/s002510050696 [DOI] [PubMed] [Google Scholar]
- 13.Kelley CM, Ikeda T, Koipally J, et al. Helios, a novel dimerization partner of Ikaros expressed in the earliest hematopoietic progenitors. Curr Biol 1998;8(9):508–S1. doi: 10.1016/s0960-9822(98)70202-7 [DOI] [PubMed] [Google Scholar]
- 14.Martín-Ibáñez R, Crespo E, Esgleas M, et al. Helios Transcription Factor Expression Depends on Gsx2 and Dlx1&2 Function in Developing Striatal Matrix Neurons. Stem Cells Dev 2012;21(12):2239–51. doi: 10.1089/scd.2011.0607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hetemaki I, Kaustio M, Kinnunen M, et al. Loss-of-function mutation in IKZF2 leads to immunodeficiency with dysregulated germinal center reactions and reduction of MAIT cells. Sci Immunol 2021;6(65):eabe3454. doi: 10.1126/sciimmunol.abe3454 [published Online First: 2021/11/27] [DOI] [PubMed] [Google Scholar]
- 16.Shahin T, Kuehn HS, Shoeb MR, et al. Germline biallelic mutation affecting the transcription factor Helios causes pleiotropic defects of immunity. Sci Immunol 2021;6(65):eabe3981. doi: 10.1126/sciimmunol.abe3981 [published Online First: 2021/11/27] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shahin T, Mayr D, Shoeb MR, et al. Identification of germline monoallelic mutations in IKZF2 in patients with immune dysregulation. Blood Adv 2022;6(7):2444–51. doi: 10.1182/bloodadvances.2021006367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.John LB, Ward AC. The Ikaros gene family: transcriptional regulators of hematopoiesis and immunity. Mol Immunol 2011;48(9-10):1272–8. doi: 10.1016/j.molimm.2011.03.006 [published Online First: 2011/04/12] [DOI] [PubMed] [Google Scholar]
- 19.Payne MA. Zinc finger structure-function in Ikaros Marvin A Payne. World J Biol Chem 2011;2(6):161. doi: 10.4331/wjbc.v2.i6.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25(14):1754–60. doi: 10.1093/bioinformatics/btp324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20(9):1297–303. doi: 10.1101/gr.107524.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019;176(3):535–48.e24. doi: 10.1016/j.cell.2018.12.015 [DOI] [PubMed] [Google Scholar]
- 23.Adzhubei I, Jordan DM, Sunyaev SR. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr Protoc Hum Genet 2013;76(1):7.20.1–7.20.41. doi: 10.1002/0471142905.hg0720s76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rentzsch P, Witten D, Cooper GM, et al. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 2019;47(D1):D886–D94. doi: 10.1093/nar/gky1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jian X, Boerwinkle E, Liu X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res 2014;42(22):13534–44. doi: 10.1093/nar/gku1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abyzov A, Urban AE, Snyder M, et al. CNVnator: An approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res 2011;21(6):974–84. doi: 10.1101/gr.114876.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Layer RM, Chiang C, Quinlan AR, et al. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol 2014;15(6):R84. doi: 10.1186/gb-2014-15-6-r84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu M, Anna Han Y, et al. Using ERDS to Infer Copy-Number Variants in High-Coverage Genomes. Am J Hum Genet 2012;91(3):408–21. doi: 10.1016/j.ajhg.2012.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen X, Schulz-Trieglaff O, Shaw R, et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 2016;32(8):1220–22. doi: 10.1093/bioinformatics/btv710 [DOI] [PubMed] [Google Scholar]
- 30.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 2010;38(16):e164–e64. doi: 10.1093/nar/gkq603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Collins RL, Brand H, Karczewski KJ, et al. A structural variation reference for medical and population genetics. Nature 2020;581(7809):444–51. doi: 10.1038/s41586-020-2287-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 2018;46(D1):D1062–D67. doi: 10.1093/nar/gkx1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lappalainen I, Lopez J, Skipper L, et al. dbVar and DGVa: public archives for genomic structural variation. Nucleic Acids Res 2012;41(D1):D936–D41. doi: 10.1093/nar/gks1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bragin E, Chatzimichali EA, Wright CF, et al. DECIPHER: database for the interpretation of phenotype-linked plausibly pathogenic sequence and copy-number variation. Nucleic Acids Res 2014;42(D1):D993–D1000. doi: 10.1093/nar/gkt937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Macdonald JR, Ziman R, Yuen RKC, et al. The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res 2014;42(D1):D986–D92. doi: 10.1093/nar/gkt958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fokkema IFAC, Taschner PEM, Schaafsma GCP, et al. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat 2011;32(5):557–63. doi: 10.1002/humu.21438 [DOI] [PubMed] [Google Scholar]
- 37.Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021;596(7873):583–89. doi: 10.1038/s41586-021-03819-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mirdita M, Schütze K, Moriwaki Y, et al. ColabFold: making protein folding accessible to all. Nat Methods 2022;19(6):679–82. doi: 10.1038/s41592-022-01488-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sobreira N, Schiettecatte F, Valle D, et al. GeneMatcher: A Matching Tool for Connecting Investigators with an Interest in the Same Gene. Hum Mutat 2015;36(10):928–30. doi: 10.1002/humu.22844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hahm K, Cobb BS, McCarty AS, et al. Helios, a T cell-restricted Ikaros family member that quantitatively associates with Ikaros at centromeric heterochromatin. Genes Dev 1998;12(6):782–96. doi: 10.1101/gad.12.6.782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schjerven H, McLaughlin J, Arenzana TL, et al. Selective regulation of lymphopoiesis and leukemogenesis by individual zinc fingers of Ikaros. Nat Immunol 2013;14(10):1073–83. doi: 10.1038/ni.2707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Watatani Y, Sato Y, Miyoshi H, et al. Molecular heterogeneity in peripheral T-cell lymphoma, not otherwise specified revealed by comprehensive genetic profiling. Leukemia 2019;33(12):2867–83. doi: 10.1038/s41375-019-0473-1 [DOI] [PubMed] [Google Scholar]
- 43.Wolfe SA, Nekludova L, Pabo CO. DNA Recognition by Cys2His2 Zinc Finger Proteins. Annu Rev Bioph Biom 2000;29(1):183–212. doi: 10.1146/annurev.biophys.29.1.183 [DOI] [PubMed] [Google Scholar]
- 44.Yamashita M, Morio T. AIOLOS Variants Causing Immunodeficiency in Human and Mice. Front Immunol 2022;13:866582. doi: 10.3389/fimmu.2022.866582 [published Online First: 2022/04/22] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamashita M, Kuehn HS, Okuyama K, et al. A variant in human AIOLOS impairs adaptive immunity by interfering with IKAROS. Nat Immunol 2021;22(7):893–903. doi: 10.1038/s41590-021-00951-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cai Q, Dierich A, Oulad-Abdelghani M, et al. Helios Deficiency Has Minimal Impact on T Cell Development and Function. J Immunol 2009;183(4):2303–11. doi: 10.4049/jimmunol.0901407 [DOI] [PubMed] [Google Scholar]
- 47.Sebastian M, Lopez-Ocasio M, Metidji A, et al. Helios Controls a Limited Subset of Regulatory T Cell Functions. J Immunol 2016;196(1):144–55. doi: 10.4049/jimmunol.1501704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Molnar A, Georgopoulos K. The Ikaros gene encodes a family of functionally diverse zinc finger DNA-binding proteins. Mol Cell Biol 1994;14(12):8292–303. doi: 10.1128/mcb.14.12.8292-8303.1994 [published Online First: 1994/12/01] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tabayashi T, Ishimaru F, Takata M, et al. Characterization of the short isoform of Helios overexpressed in patients with T-cell malignancies. Cancer Sci 2007;98(2):182–8. doi: 10.1111/j.1349-7006.2006.00372.x [published Online First: 2007/02/14] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Emerson RO, Thomas JH. Adaptive Evolution in Zinc Finger Transcription Factors. PLoS Genet 2009;5(1):e1000325. doi: 10.1371/journal.pgen.1000325 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.