

Abstract
Rho GDP-Dissociation Inhibitors (RhoGDIs) are important regulators of the Rho family of small GTPases. The expression of RhoGDIs is altered in a variety of cancers and they have been shown to mediate several processes during tumorigenesis and cancer progression. Using examples of RhoGDI-mediated signaling and expression patterns in endothelial cells as well as pancreatic, breast, and bladder cancer, the multitude of potential cancer therapeutic targets presented by a better understanding of their function is illustrated. Several novel therapeutic strategies are proposed for intervening in RhoGDI signaling, and potential complications arising from their implementation are discussed.
Keywords: Guanine Nucleotide Dissociation, Inhibitors, RhoGDIs, RhoGDI1, RhoGDI2, RhoGTPase, Cancer therapy, Angiogenesis, Pancreatic cancer, Breast cancer, Bladder cancer
1. Introduction
Involvement of the Rho family of small GTP binding proteins in cancer is well established. A great deal of work has been done on the proteins that regulate their GTPase activity and the downstream effectors that are involved in cancer phenotypes. Much of this literature concerns the regulation of Rho activity by the GEFs (Guanine Nucleotide Exchange Factors) and GAPs (GTPase Activating Proteins).1,2 But a third type of protein, the Rho family GDP Dissociation Inhibitors (RhoGDIs), are just now being appreciated as critical components of the Rho regulating machinery, and an emerging collection of evidence suggests that their activity is altered in carcinogenesis and tumor progression. As more evidence of their multi-faceted influence on the cancer phenotype via Rho family members accumulates, many intriguing and promising therapeutic targets are emerging from the elucidation of how these proteins work. However, an examination of what we now know about RhoGDIs also indicates that their functions may result in tissue-specific effects and thus more extensive understanding of the pathways they regulate will be necessary for the complete realization of novel cancer treatments based on their function.
2. The RhoGDI family and their activities
Although RhoGDI proteins have been reviewed extensively elsewhere,3–5 a brief summary is necessary for understanding the potential opportunities and pitfalls for RhoGDI-centric intervention. Three RhoGDI proteins have been indentified in humans. They are referred to in the literature by various nomenclatures, but here they will be called RhoGDI1, 2, and 3. The prototype of the family, RhoGDI1, is known as RhoGDIα, arhgdi alpha or arhgdia. RhoGDI2 is variously called RhoGDIβ, arhgdi beta, arhgdib, ly-gdi, and D4-gdi. RhoGDI3 is also known as RhoGDIγ, arhgdi gamma or arhgdig. RhoGDI3 is the most divergent of the three and is associated with the golgi and vesicular membranes.6 Very little is known about RhoGDI3 in cancer; therefore, it will not be discussed further here. RhoGDI1 is the most ubiquitously expressed of the family. While the highest expression of RhoGDI2 is in cells of hematopoietic origin, it has recently been found in a variety of other tissues and cancers.3
RhoGDIs regulate a multitude of phenotypes including cell division, morphology, migration, vesicular trafficking and gene expression. They probably affect these diverse phenotypes principally by controlling the location and activity of members of the Rho family of small GTPases. Therefore, a full appreciation of the function of RhoGDIs is dependent on knowledge of the role of RhoGTPases, which have been reviewed extensively elsewhere.1,2,7,8 Rho proteins comprise a family within the larger superfamily of Ras-related proteins. They are signal transducers that alternate between an activated, GTP-bound state, and the inactive, GDP-bound form. The cycle between the GTP or GDP bound state is mediated by GTPase activator proteins (GAPs) and guanine nucleotide exchange factors (GEFs), which facilitate the exchange of GDP for GTP. When bound to GTP, active Rho proteins bind to variety of downstream effector molecules. Currently, the Rho family is thought to contain 20 members,8 and over 60 Rho effectors have been identified.1 This accounts for the number of signaling pathways regulated by RhoGTPases, and the complexity of the potential impact of RhoGDIs on signal transduction.
RhoGDIs are thought to affect Rho proteins in several ways. In general, RhoGDI and RhoGDI2 have been thought of as multi-modal inhibitors of the RhoGTPases. Methods of Rho inhibition by RhoGDIs include inhibition of Rho catalytic activity by inhibiting the GTPase function as well as inhibiting dissociation of GDP from the Rho protein after hydrolysis. RhoGDIs also interact with prenylated Rho proteins in a manner that removes them from membranes, causing a re-localization to the cytoplasm. In a related function, they are thought to shuttle Rho proteins between membrane domains. Therefore, they cannot be viewed simply as inhibitors of RhoGTPases, but probably also direct activated Rho proteins to certain sub-cellular compartments.9,10 By virtue of binding Rhos, they are also believed to restrict interactions of RhoGEFs and RhoGAPs with Rho proteins, and the binding of other modulators of Rho activity.
Many of the common Rho family of proteins have been shown or are suspected to interact with one or more of the RhoGDIs (Table 1 in3), and it seems likely that additional members of the Rho family will be found to be regulated by GDIs. Differences in the RhoGDIs are reflected in differing affinities for Rho family members. For example, RhoGDI2 has a much lower affinity for Cdc42 than for the Rac proteins, whereas RhoGDI1 has similar affinities for RhoA, Rac, and Cdc42.11,12 In addition to their innately different affinities for RhoGTPases, RhoGDI1 and RhoGDI2 also exhibit quite different effects on Rho family protein activation in some circumstances, which will be discussed further below.
3. Regulation of RhoGDI effector interactions
Post-translational modifications such as phosphorylation of RhoGDIs are thought to regulate their interactions with their target effectors. Binding and displacement of RhoGDIs from Rho proteins are coordinated with phosphorylation events to carry out complex cellular signaling. An example of these complex signaling cascades in angiogenesis was recently reported by Elfenbein et al.13 Polarized Rac1 activation in endothelial cells causes migration and is necessary for angiogenesis, and Rac1 activation is preceded by activation of RhoG. FGF2 is a potent endothelial cell motility factor that is a ligand for syndecan 4 (S4). It was shown that RhoGDI1 binds in a complex with S4 and its adaptor synectin, and that this binding increases the affinity of RhoGDI1 for RhoG. Upon ligand binding, and S4 clustering, protein kinase Cα (PKCα) is activated which phosphorylates RhoGDI1 at Ser96. Phosphorylation of RhoGDI1 inhibits its binding to RhoG, resulting in activation of RhoG and the subsequent activation of Rac1. Surprisingly, phosphorylation of RhoGDI1 at Ser96 did not change the affinity of RhoGDI1 for Rac1. Knezevic et al.14 also found that phospho-Ser96 did not alter the activation of Rac1 in endothelial cells. However, the same group has shown that RhoGDI1 is phosphorylated at Ser96 by PKCα in response to thrombin engagement of endothelial cell surface Protease-Activated Receptor 1 (PAR-1). This phosphorylation results in the release of RhoA by RhoGDI1 and activation of RhoA, which is required for thrombin-induced vascular permeability.14,15 In these examples, RhoGDI1 directly and indirectly regulates three different RhoGTPases, in two signaling pathways, in one cell type.
Such involvement of RhoGDI1 in angiogenesis and vascular permeability, two hallmarks of the neo-vasculature induced by tumors, illustrates the target-rich therapeutic environment created by a better understanding of RhoGDI function. The ligand/receptor interactions, PKCα, and the RhoGDI1/Rho protein interactions all become viable candidates for inhibiting tumor-induced angiogenesis. This understanding could allow rational and selective drug development. For example, PKC inhibitors could be screened for ability to inhibit the Ser96 phosphorylation on RhoGDI1, while minimizing inhibition of PKC activity on other substrates. Hence, knowledge of RhoGDI signaling could lead to more effective, better tolerated angiogenesis inhibitors.
Other kinases, including P-21 Activated Kinase 1 (PAK1) and Src, phosphorylate RhoGDIs are likely to regulate RhoGDI signaling pathways as complex as those regulated by PKCα. PAK1 phosphorylates RhoGDI1 at two sites, which causes the selective release of Rac1, but not RhoA.16 Interestingly, PAK1 is also a major downstream effector of Rac1, so this likely represents a positive feed-forward mechanism for signal amplification. Src phosphorylation of RhoGDIs will be discussed further below in the context of bladder cancer.
4. RhoGDI expression and function in human cancer
The pattern of RhoGDI1 and RhoGDI2 expression within a single type of cancer, expression of a single RhoGDI between cancer types, and their activity toward particular Rho proteins, can be strikingly divergent. This is highly suggestive that the functions of RhoGDI1 and RhoGDI2 are not entirely redundant, and that there are additional layers of RhoGDI regulation that are context-dependent. This lack of redundancy is supported by work in transgenic mice where deletion of the RhoGDI1 and RhoGDI2 genes has profoundly different consequences. RhoGDI1 knock-out mice die from renal failure at about one year of age and the males are infertile.17 In contrast, the defects noted in RhoGDI2 knock-out mice are relatively mild, with some diminution of superoxide production by phagocytes18 and slightly decreased lymphocyte survival and responsiveness19 being noted as the major phenotypes. Therefore, the menu of therapeutic intervention designed to target RhoGDI-mediated events in cancer cells will very likely be dependent on the cancer type or even sub-type (i.e., estrogen receptor-positive versus -negative breast cancer). This discussion will focus on RhoGDI expression and function in three cancers where it is best understood and thus most ready to be translated therapeutically. Speculative examples of such translation will be provided.
5. Pancreatic Cancer
Studies found an association of elevated RhoGDI2 expression and ability of pancreatic cancer cells to invade along nerve tracts (perineural invasion). Koide et al.20 developed an ex vivo model for isolating perineural invasive pancreatic cancer cells and then used microarrays to identify genes with differential expression. Perineural invasive cells had 41-fold higher RhoGDI2 expression than poorly invasive cells, a result confirmed by others.21 They also demonstrated that knockdown of RhoGDI2 expression in these cells impaired invasion.
Vav1 is a RacGEF normally expressed in T cells where it is necessary for the stimulation of the transcription factor Nuclear Factor of Activated T cells, or NFAT. Induction of NFAT by Vav1 proceeds through a Rac-mediated pathway.22 It was recently discovered that Vav1 binds both RhoGDI2 and Rac in a complex and surprisingly, RhoGDI2 increases Vav1’s ability to activate NFAT.23,24 This is significant because ectopic expression of Vav1 has been detected in numerous cancers and especially pancreatic cancer.25–27 How much of Vav1’s transforming ability is attributable to NFAT remains to be seen, but the immunosuppressive drugs cyclosporine and FK506 inhibit NFAT. Therefore, such drugs that are safely used in transplantation today could be used to target downstream RhoGDI2 effectors in pancreatic cancer invasion. While immunosuppression is not a desired side effect of therapy in a cancer patient, at least conceptually this signaling pathway demonstrates a clinically tractable target.
RhoGDI1 activity appears to be quite different from that of RhoGDI2 in pancreatic cancer. Very recently, it was shown that a member of the trk neurotrophin receptor family, TrkBT1, a variant of tropomyosin-related kinase B, is over-expressed in the cytoplasm of pancreatic cancers metastatic to the liver. Moreover, it was shown that TrkBT1 promotes cell proliferation and enhanced metastatic potential by virtue of its sequestration of RhoGDI1 and the subsequent activation of RhoA signaling.28 Since the trk variant involved lacks a kinase domain and is mainly cytoplasmic, it would appear that ligand-based analogs or kinase inhibitors do not present a viable means of affecting its interaction with RhoGDI1. However, Ohira et al.29 found that Brain-Derived Neurotrophic Factor (BDNF) could induce the release of RhoGDI1 from TrkBT1 in astrocytes, suggesting that ligand blockade via small molecules could be efficacious in some scenarios if such agents could be developed.
Yamashita and Tohyama30 have reported that another member of the neurotrophin receptor family, p75NTR, can bind RhoGDI1, displacing it from RhoA and thereby activating RhoA in neurons. They developed a peptide, TAT-Pep5, which can disrupt the RhoGDI1/p75NTR interaction that maintains RhoA in an inactive state, bound to RhoGDI1. This protein is internalized by cells by virtue of fusion with the human immunodeficiency virus TAT domain protein and therefore has therapeutic potential in cells expressing p75NTR. It also provides a paradigm for how other interactions that displace RhoGDIs from Rho proteins could be inhibited.
6. Breast Cancer
Although several groups have looked at RhoGDI expression in breast tumors and cell lines, a clear picture of the role of RhoGDIs in breast cancer has not emerged. Fritz et al.31 first reported an increase in RhoGDI1 in breast tumors vs. matching normal samples from a small cohort of patients.15 Jiang et al.32 examined RhoGDI1 and RhoGDI2 in breast cancers from a larger cohort of patients undergoing radical mastectomy (120 cancers and 32 normal tissues). They found a significant reduction of RhoGDI1 expression in tumor versus normal breast by both quantitative RT-PCR and immunohistochemistry (IHC) staining. Furthermore, they showed that the reduction of RhoGDI1 had a significant, bad prognostic correlation when tumors were stratified by node status or by recurrence and disease-specific death. Differences in RhoGDI2 expression, measured by qRT-PCR only, showed no significant pattern between tumor and normal, node-negative and node-positive tumors, or in relation to tumor grade. Hu et al.33 revisited the question of RhoGDI2 expression by IHC in breast cancers from a cohort of 71 patients. When looking at various regions of tumor within the same slide, it appeared that RhoGDI2 expression increased in the progression of normal breast epithelia to hyperplastic lesions, which had the highest expression, but then remarkably decreased from carcinoma in situ to invasive cancer. Importantly, they also found a statistically significant negative correlation between RhoGDI2 expression and nodal involvement. Hu et al. hypothesized that RhoGDI2 expression is biphasic in breast cancer progression, with an increase in the earliest stages of tumor development followed by loss of expression in metastasis.
These differences in expression patterns and correlations for RhoGDI1 and RhoGDI2 in breast cancer are likely due to methodology, size of the cohorts, cohort type (preneoplastic, invasive, etc.) and even unappreciated differences in coincident, underlying abnormalities represented in some cohorts. For example, some breast cancers express a splice variant of Rac1, called Rac1b,34 which contains a 19-amino-acid insertion near the switch region that is important for binding of Rac1 regulators. It has been found that Rac1b does not interact with RhoGDI135,36 and is capable of growth transformation in NIH3T3 cells.36 Since Rac1b may evade regulation, RhoGDI expression patterns may not be meaningful in those tumors.
It is also possible that RhoGDIs can mediate both protumorigenic and anti-tumorigenic signaling pathways. Schunke et al.37 found that RhoGDI2 decreased migration by MDA-MB-231 breast cancer cells but also increased expression of COX-2, a gene associated with progression in breast and other cancers. As in the previous studies in T cells cited above, RhoGDI2 and vav1 cooperated to increase COX-2 expression, probably through NFAT activation, in breast cancer cells. In order to assess the net effect of these antagonistic phenotypes, they assessed RhoGDI2 status by mRNA levels in 263 samples, and by protein levels in 117 samples, of breast cancer and found no association with disease-free survival or overall survival. They hypothesized that on balance, these two opposing effects cancel each other out. However, that does not rule out the possibility that for any one individual tumor, this balance could be tipped one way or the other depending on numerous factors, including host response and the occurrence of other mitigating alterations in the cancer cells.
An example of the possible dependency of RhoGDI function on underlying tumor characteristics is the finding that RhoGDI2 is part of the prognostic signature of ER-positive, but not ER-negative breast cancers. Wang et al.38 developed a 76-gene signature that predicts metastasis-free survival for five years, using Affymetrix microarrays to measure gene expression in tumor samples from patients that were lymphnode-negative and had not received systemic adjuvant therapy. While their 76-gene signature applies to both ER-positive and ER-negative tumors, RhoGDI2 was incorporated into the signature because a decrease in RhoGDI2 expression was indicative of poor prognosis in ER-positive tumors. Significantly, this same predictive signature has been validated by the same group39 and by an independent lab40 in different patient cohorts.
Notwithstanding the unresolved question of RhoGDI expression and prognostic significance in breast cancer tissues, in vitro studies have found interesting functional consequences of RhoGDI expression that may be relevant to breast cancer phenotypes and treatment. Su et al.41,42 have reported that RhoGDI1 can increase transcriptional activity of Estrogen Receptor α (ERα) via a RhoGTPase-dependent pathway that acts on estrogen receptor co-activators GRIP1 and CBP/p300. Furthermore, RhoGDI1 could increase both ligand-dependent and -independent ERα activity. Likewise, El Marzouk et al.43 found that RhoGDI1 could enhance ERα-regulated transcription, but that this activity was due in part to direct binding of RhoGDI1 and ERα in the nucleus. While the therapeutic implications of two possible forms of ERα regulation by RhoGDI1 are not understood, the significance of RhoGDI expression with regard to ER status and effectiveness of antiestrogen therapy warrants investigation.
RhoGDI1 expression could also influence the choice of cancer therapy since it confers a protective effect from some chemotherapeutics in in vitro studies. RhoGDI1 protected MDA-MB-231 breast cancer cells and JLP-119 lymphoma cells from etoposide and doxorubicin treatment44 and HEK293 cells from doxorubicin, taxol or busulfan-induced cytotoxicity.45
7. Bladder Cancer
The discovery and function of RhoGDI2 as a metastasis suppressor in bladder cancer was recently reviewed by us.46 Briefly, RhoGDI2 expression was found to be decreased in bladder cancer cells with greater metastatic potential.47 Reintroduction of RhoGDI2 expression in a metastatic bladder cell line (T24T) did not alter in vitro growth, but did inhibit cell motility and invasion in a bladder organotypic invasion assay. When introduced into immunocompromised murine hosts, RhoGDI2 did not suppress subcutaneous growth, but did inhibit lung metastasis after tail vein injection. In clinical tissue microarrays from radical cystectomies, low RhoGDI2 expression was a negative prognostic indicator for disease-free survival.48 Our conclusion from these data is that RhoGDI2 acts as a metastasis suppressor gene in bladder cancer. Interestingly, while RhoGDI1 is expressed in bladder cancer cell lines and tissues, its expression is not altered or correlated with prognosis.
While patterns of RhoGDI2 expression in bladder cancer correlate well with the in vitro and in vivo phenotypes, as well as clinical profiles, the mechanism underlying suppression of bladder cancer metastasis by RhoGDI2 revealed several surprises. Src kinase decreases in bladder cancer progression in the same fashion as that seen with RhoGDI2. A decrease in Src and RhoGDI2 is rarely observed in the same individual tumor, suggesting that they share a common pathway. It was found that Src phosphorylates RhoGDI2 in bladder cancer cells,49 just as Src was previously shown to phosphorylate RhoGDI1.50 Moreover, phosphorylation of RhoGDI2 by Src was necessary for its metastasis suppressor function in bladder cancer cells. This is suggestive that Src kinase inhibitors may be contraindicated in the treatment of bladder cancer and suggests that Src may suppress metastasis in bladder cancer and this may be entirely or partly mediated via RhoGDI2. Further experiments will likely clarify this issue.
Equally surprising is the finding that RhoGDI2 does not inhibit Rac activation in bladder cancer cells and that mutations that either increase or decrease RhoGDI2’s affinity for RhoGTPases, abolish its ability to suppress lung metastasis. The authors suggest that RhoGDI2 may target RhoGTPases (probably Rac1) to novel effector pathways that are necessary for metastasis suppression.51 This result diminishes the prospect of therapeutic inhibition of bladder cancer metastasis by somehow increasing RhoGDI2 binding to Rac1.
A promising method of unraveling the metastasis suppressing pathway of RhoGDI2 is to look at downstream effectors. We have identified two target genes that are repressed upon expression of RhoGDI2 in bladder cancer cells and whose expression increases as a function of stage in bladder cancers. These genes encode endothelin-1 (ET1)52 and neuromedin U (NmU).53 They are both small peptide agonists of G-protein-coupled receptors.54,55 Two NmU receptors, NmU-R1 and NmU-R2, have been cloned.54 NmU was shown by us to increase the number of lung metastases by metastatically competent bladder cancer cells in a murine xenograft model.53 Although NmU has had little attention as a therapeutic target in cancer, an inhibitor of one of its receptors has been developed.56 This drug would be an excellent candidate for further investigation.
Endothelin and its connection to cancer have been reviewed recently and extensively.55,57,58 Endothelin signaling pathways, collectively termed the “endothelin axis”, have been implicated in a multitude of tumor types and are thought to promote a host of cancer phenotypes, including proliferation, resistance to apoptosis, invasion, angiogenesis, immune suppression, osteogenesis in bone metastases, and pain. ET1 belongs to a family of small related peptides that includes ET2 and ET3. Endothelins have two known receptors, endothelin receptor A (ETA) and endothelin receptor B (ETB). The ligand/receptor interaction most often associated with the aforementioned cancer phenotypes is the activation of ETA by ET1.58 For this reason, several ETA inhibitors have been developed, and some have entered clinical trials.
Atrasentan, a selective ETA inhibitor was used in two recent phase 3 trials for treatment of prostate cancer. In the first, atrasentan was used to treat patients with metastatic, hormone-refractory prostate cancer.59 While atrasentan did not delay disease progression, the increase in bone alkaline phosphates (BAP), a measure of the extent of bone metastasis, and prostate-specific antigen (PSA), a measure of tumor burden, were both delayed. The second study with atrasentan focused on men with nonmetastatic, hormone-refractory prostate cancer.60 A delay in the time to progression (TTP) was noted in this study, but because of the large inter-institutional variation in TTP, the delay was not significant. Once again, there was a decrease in the rise of PSA and BAP. ZD4054 is another ETA-selective inhibitor that has been used in a clinical trial for hormone-resistant, bone metastatic prostate cancer.61 In this study, ZD4054 did not delay disease progression, but overall time of survival was increased in the treatment group. Taken together, these trials indicate that selective ETA inhibitors are well tolerated and demonstrate some clinically meaningful effects.
In our own pre-clinical studies, atrasentan52 and ZD4054 (Said and Theodorescu, submitted) significantly inhibited the formation of lung metastases by a human bladder cancer cell line after tail vein injection of murine hosts. This result recapitulated the phenotype obtained after re-introducing RhoGDI2 expression in the same cell line. Based on these promising results, and the favorable tolerability of atrasentan and ZD4054 in previous human trials, we have currently designed a clinical trial with ZD4054 as an adjuvant therapy for lung metastasis in patients with bladder cancer.
The standard treatment for patients with high-grade invasive disease is radical cystectomy. However, up to 50% of all patients who underwent surgery later developed recurrent disease that was mostly metastatic and fatal. Based on neoadjuvant and adjuvant clinical trial data, many oncologists believe that there are sufficient data at this time to support the use of either pre- or post-surgical chemotherapy in patients with locally advanced bladder cancer. In particular, treatment seems justified for those considered to be at high risk of recurrence; this would include patients who have undergone cystectomy and were found to have tumors that penetrate the bladder wall (T3 or T4) or that have spread to regional lymph nodes (N1–3). However, since the expected benefit would be limited with an absolute increase in disease-free and overall survival within the range of 5–15%, there is a great need to develop new agents and treatment strategies to further reduce the rate of recurrence.
Given our preclinical animal studies, we hypothesize that ET axis blockade will suppress or delay the onset of metastatic disease in these patients and have proposed a randomized phase II study to test this hypothesis (Fig. 1). Patients who have had a radical cystectomy for locally advanced TCC of the bladder (stage T3, T4, and/or N1–3 disease) will be randomized to daily ZD4054 or placebo for two years following four cycles of cisplatin and gemcitabine chemotherapy. The two groups of patients will then be followed to determine the relapse-free survival (RFS) rate at two years. The study will have 80% power to detect a hazard ratio of 1.76, which corresponds to an absolute improvement in the two-year relapse rate from 60% to 75%. Secondary goals of the randomized phase II study are to establish the safety of prolonged administration of ZD4054 and estimate the relative frequency of local relapse versus failure with metastatic disease in the two groups. The latter may provide evidence that ZD4054 selectively inhibits or delays onset of metastatic disease.
Fig. 1 –
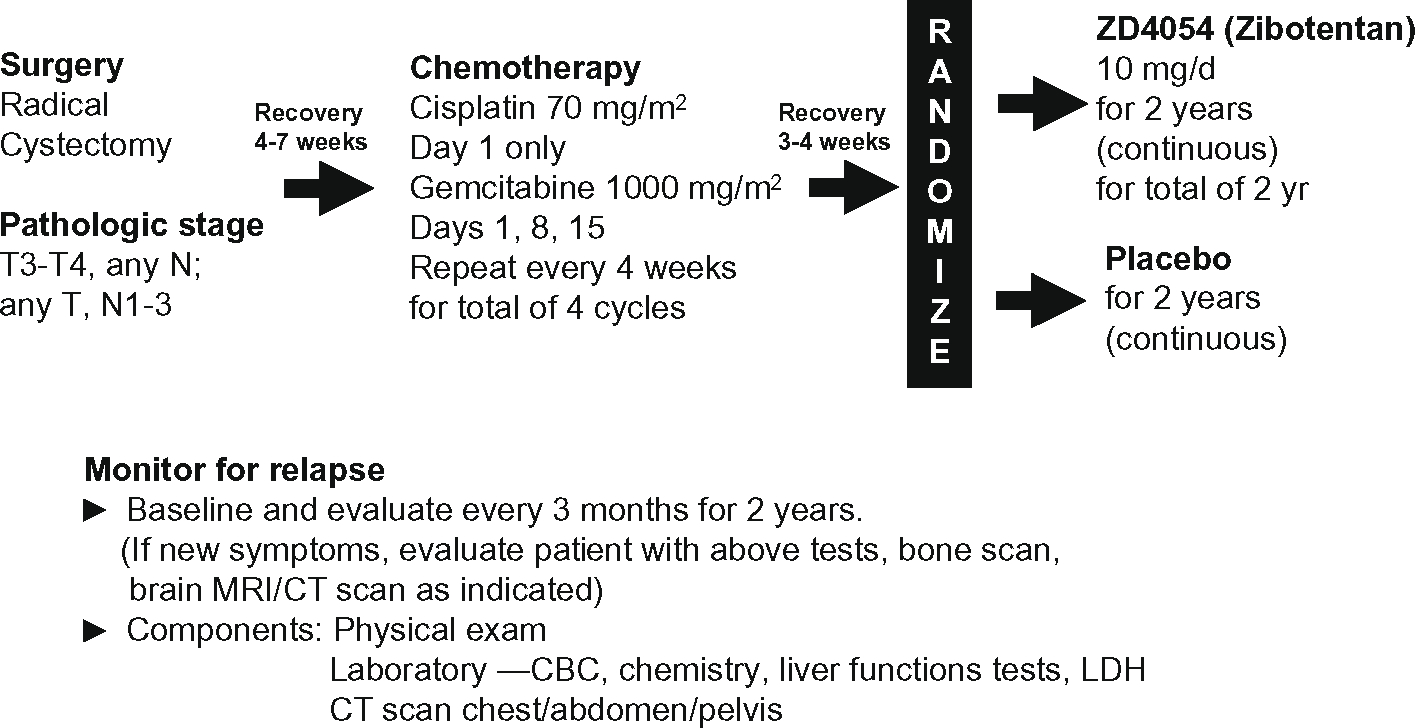
We hypothesize that ET axis blockade will suppress or delay the onset of metastatic disease in these patients and have proposed a randomized phase II study to test this hypothesis.
8. Conclusions
It is clear from the multitude of processes regulated by Rho family GTPases, the complex regulation of association between RhoGDIs with Rho proteins, and the diverse consequences of RhoGTPase interaction with RhoGDIs, that there are multiple points to attack in these pathways. However, this complexity also poses problems and risks. Indeed, the pattern of expression and activities of RhoGDIs in human cancers clearly demonstrate that a universal, simplistic strategy targeting RhoGDIs will not work for all types of cancer. It seems possible that a successful therapeutic in one type of cancer could be very deleterious in another. However, armed with a new found appreciation of the role of RhoGDIs in tumor formation and progression, and a growing arsenal of tools with which to target RhoGDI-mediated signaling, there is the future promise of meaningful therapeutic intervention.
Acknowledgments
This work was supported by the National Institute of Health grant R01CA075115 to DT.
Footnotes
Conflict of interest statement
D.T. has received research funding from Astra Zeneca for preclinical work with ZD4054.
REFERENCES
- 1.Lu Q, Longo FM, Zhou H, Massa SM, Chen YH. Signaling through Rho GTPase pathway as viable drug target. Curr Med Chem 2009;16(11):1355–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oxford G, Theodorescu D. Ras superfamily monomeric G proteins in carcinoma cell motility. Cancer Lett 2003;189(2):117–28. [DOI] [PubMed] [Google Scholar]
- 3.DerMardirossian C, Bokoch GM. GDIs: central regulatory molecules in Rho GTPase activation. Trends Cell Biol 2005;15(7):356–63. [DOI] [PubMed] [Google Scholar]
- 4.Dovas A, Couchman JR. RhoGDI: multiple functions in the regulation of Rho family GTPase activities. Biochem J 2005;390(Pt 1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dransart E, Olofsson B, Cherfils J. RhoGDIs revisited: novel roles in Rho regulation. Traffic 2005;6(11):957–66. [DOI] [PubMed] [Google Scholar]
- 6.Dransart E, Morin A, Cherfils J, Olofsson B. RhoGDI-3, a promising system to investigate the regulatory function of rhoGDIs: uncoupling of inhibitory and shuttling functions of rhoGDIs. Biochem Soc Trans 2005;33(Pt 4):623–6. [DOI] [PubMed] [Google Scholar]
- 7.Karlsson R, Pedersen ED, Wang Z, Brakebusch C. Rho GTPase function in tumorigenesis. Biochim Biophys Acta 2009;1796(2):91–8. [DOI] [PubMed] [Google Scholar]
- 8.Vega FM, Ridley AJ. Rho GTPases in cancer cell biology. FEBS Lett 2008;582(14):2093–101. [DOI] [PubMed] [Google Scholar]
- 9.Johnson JL, Erickson JW, Cerione RA. New insights into how the Rho guanine nucleotide dissociation inhibitor regulates the interaction of Cdc42 with membranes. J Biol Chem 2009;284(35):23860–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lin Q, Fuji RN, Yang W, Cerione RA. RhoGDI is required for Cdc42-mediated cellular transformation. Curr Biol 2003;13(17):1469–79. [DOI] [PubMed] [Google Scholar]
- 11.Gorvel JP, Chang TC, Boretto J, Azuma T, Chavrier P. Differential properties of D4/LyGDI versus RhoGDI: phosphorylation and rho GTPase selectivity. FEBS Lett 1998;422(2):269–73. [DOI] [PubMed] [Google Scholar]
- 12.Lelias JM, Adra CN, Wulf GM, Guillemot JC, Khagad M, Caput D, et al. CDNA cloning of a human mRNA preferentially expressed in hematopoietic cells and with homology to a GDP-dissociation inhibitor for the rho GTP-binding proteins. Proc Natl Acad Sci USA 1993;90(4):1479–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elfenbein A, Rhodes JM, Meller J, Schwartz MA, Matsuda M, Simons M. Suppression of RhoG activity is mediated by a syndecan 4-synectin-RhoGDI1 complex and is reversed by PKCalpha in a Rac1 activation pathway. J Cell Biol 2009;186(1):75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Knezevic N, Roy A, Timblin B, Konstantoulaki M, Sharma T, Malik AB, et al. GDI-1 phosphorylation switch at serine 96 induces RhoA activation and increased endothelial permeability. Mol Cell Biol 2007;27(18):6323–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mehta D, Rahman A, Malik AB. Protein kinase C-alpha signals rho-guanine nucleotide dissociation inhibitor phosphorylation and rho activation and regulates the endothelial cell barrier function. J Biol Chem 2001;276(25):22614–20. [DOI] [PubMed] [Google Scholar]
- 16.DerMardirossian C, Schnelzer A, Bokoch GM. Phosphorylation of RhoGDI by Pak1 mediates dissociation of Rac GTPase. Mol Cell 2004;15(1):117–27. [DOI] [PubMed] [Google Scholar]
- 17.Togawa A, Miyoshi J, Ishizaki H, Tanaka M, Takakura A, Nishioka H, et al. Progressive impairment of kidneys and reproductive organs in mice lacking Rho GDIalpha. Oncogene 1999;18(39):5373–80. [DOI] [PubMed] [Google Scholar]
- 18.Guillemot JC, Kruskal BA, Adra CN, Zhu S, Ko JL, Burch P, et al. Targeted disruption of guanosine diphosphate-dissociation inhibitor for Rho-related proteins, GDID4: normal hematopoietic differentiation but subtle defect in superoxide production by macrophages derived from in vitro embryonal stem cell differentiation. Blood 1996;88(7):2722–31. [PubMed] [Google Scholar]
- 19.Yin L, Schwartzberg P, Scharton-Kersten TM, Staudt L, Lenardo M. Immune responses in mice deficient in Ly-GDI, a lymphoid-specific regulator of Rho GTPases. Mol Immunol 1997;34(6):481–91. [DOI] [PubMed] [Google Scholar]
- 20.Koide N, Yamada T, Shibata R, Mori T, Fukuma M, Yamazaki K, et al. Establishment of perineural invasion models and analysis of gene expression revealed an invariant chain (CD74) as a possible molecule involved in perineural invasion in pancreatic cancer. Clin Cancer Res 2006;12(8):2419–26. [DOI] [PubMed] [Google Scholar]
- 21.Abiatari I, DeOliveira T, Kerkadze V, Schwager C, Esposito I, Giese NA, et al. Consensus transcriptome signature of perineural invasion in pancreatic carcinoma. Mol Cancer Ther 2009;8(6):1494–504. [DOI] [PubMed] [Google Scholar]
- 22.Katzav S Flesh and blood: the story of Vav1, a gene that signals in hematopoietic cells but can be transforming in human malignancies. Cancer Lett 2007;255(2):241–54. [DOI] [PubMed] [Google Scholar]
- 23.Groysman M, Hornstein I, Alcover A, Katzav S. Vav1 and Ly-GDI two regulators of Rho GTPases, function cooperatively as signal transducers in T cell antigen receptor-induced pathways. J Biol Chem 2002;277(51):50121–30. [DOI] [PubMed] [Google Scholar]
- 24.Groysman M, Russek CS, Katzav S. Vav, a GDP/GTP nucleotide exchange factor, interacts with GDIs, proteins that inhibit GDP/GTP dissociation. FEBS Lett 2000;467(1):75–80. [DOI] [PubMed] [Google Scholar]
- 25.Denicola G, Tuveson DA. VAV1: a new target in pancreatic cancer? Cancer Biol Ther 2005;4(5):509–11. [DOI] [PubMed] [Google Scholar]
- 26.Fernandez-Zapico ME, Gonzalez-Paz NC, Weiss E, Savoy DN, Molina JR, Fonseca R, et al. Ectopic expression of VAV1 reveals an unexpected role in pancreatic cancer tumorigenesis. Cancer Cell 2005;7(1):39–49. [DOI] [PubMed] [Google Scholar]
- 27.Katzav S Vav1: a hematopoietic signal transduction molecule involved in human malignancies. Int J Biochem Cell Biol 2009;41(6):1245–8. [DOI] [PubMed] [Google Scholar]
- 28.Li Z, Chang Z, Chiao LJ, Kang Y, Xia Q, Zhu C, et al. TrkBT1 induces liver metastasis of pancreatic cancer cells by sequestering Rho GDP dissociation inhibitor and promoting RhoA activation. Cancer Res 2009;69(19):7851–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohira K, Kumanogoh H, Sahara Y, Homma KJ, Hirai H, Nakamura S, et al. A truncated tropomyosin-related kinase B receptor, T1, regulates glial cell morphology via Rho GDP dissociation inhibitor 1. J Neurosci 2005;25(6):1343–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamashita T, Tohyama M. The p75 receptor acts as a displacement factor that releases Rho from Rho-GDI. Nat Neurosci 2003;6(5):461–7. [DOI] [PubMed] [Google Scholar]
- 31.Fritz G, Brachetti C, Bahlmann F, Schmidt M, Kaina B. RhoGTPases in human breast tumours: expression and mutation analyses and correlation with clinical parameters. Br J Cancer 2002;87(6):635–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jiang WG, Watkins G, Lane J, Cunnick GH, Douglas-Jones A, Mokbel K, et al. Prognostic value of rho GTPases and rho guanine nucleotide dissociation inhibitors in human breast cancers. Clin Cancer Res 2003;9(17):6432–40. [PubMed] [Google Scholar]
- 33.Hu LD, Zou HF, Zhan SX, Cao KM. Biphasic expression of RhoGDI2 in the progression of breast cancer and its negative relation with lymph node metastasis. Oncol Rep 2007;17(6):1383–9. [PubMed] [Google Scholar]
- 34.Schnelzer A, Prechtel D, Knaus U, Dehne K, Gerhard M, Graeff H, et al. Rac1 in human breast cancer: overexpression, mutation analysis, and characterization of a new isoform, Rac1b. Oncogene 2000;19(26):3013–20. [DOI] [PubMed] [Google Scholar]
- 35.Matos P, Collard JG, Jordan P. Tumor-related alternatively spliced Rac1b is not regulated by Rho-GDP dissociation inhibitors and exhibits selective downstream signaling. J Biol Chem 2003;278(50):50442–8. [DOI] [PubMed] [Google Scholar]
- 36.Singh A, Karnoub AE, Palmby TR, Lengyel E, Sondek J, Der CJ. Rac1b, a tumor associated, constitutively active Rac1 splice variant, promotes cellular transformation. Oncogene 2004;23(58):9369–80. [DOI] [PubMed] [Google Scholar]
- 37.Schunke D, Span P, Ronneburg H, Dittmer A, Vetter M, Holzhausen HJ, et al. Cyclooxygenase-2 is a target gene of rho GDP dissociation inhibitor beta in breast cancer cells. Cancer Res 2007;67(22):10694–702. [DOI] [PubMed] [Google Scholar]
- 38.Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet 2005;365(9460):671–9. [DOI] [PubMed] [Google Scholar]
- 39.Foekens JA, Atkins D, Zhang Y, Sweep FC, Harbeck N, Paradiso A, et al. Multicenter validation of a gene expression-based prognostic signature in lymph node-negative primary breast cancer. J Clin Oncol 2006;24(11):1665–71. [DOI] [PubMed] [Google Scholar]
- 40.Desmedt C, Piette F, Loi S, Wang Y, Lallemand F, Haibe-Kains B, et al. Strong time dependence of the 76-gene prognostic signature for node-negative breast cancer patients in the TRANSBIG multicenter independent validation series. Clin Cancer Res 2007;13(11):3207–14. [DOI] [PubMed] [Google Scholar]
- 41.Su LF, Knoblauch R, Garabedian MJ. Rho GTPases as modulators of the estrogen receptor transcriptional response. J Biol Chem 2001;276(5):3231–7. [DOI] [PubMed] [Google Scholar]
- 42.Su LF, Wang Z, Garabedian MJ. Regulation of GRIP1 and CBP Coactivator activity by Rho GDI modulates estrogen receptor transcriptional enhancement. J Biol Chem 2002;277(40):37037–44. [DOI] [PubMed] [Google Scholar]
- 43.El Marzouk S, Schultz-Norton JR, Likhite VS, McLeod IX, Yates JR, Nardulli AM. Rho GDP dissociation inhibitor alpha interacts with estrogen receptor alpha and influences estrogen responsiveness. J Mol Endocrinol 2007;39(4):249–59. [DOI] [PubMed] [Google Scholar]
- 44.Zhang B, Zhang Y, Dagher MC, Shacter E. Rho GDP dissociation inhibitor protects cancer cells against drug-induced apoptosis. Cancer Res 2005;65(14):6054–62. [DOI] [PubMed] [Google Scholar]
- 45.Reimer J, Bien S, Sonnemann J, Beck JF, Wieland T, Kroemer HK, et al. Reduced expression of Rho guanine nucleotide dissociation inhibitor-alpha modulates the cytotoxic effect of busulfan in HEK293 cells. Anticancer Drugs 2007;18(3):333–40. [DOI] [PubMed] [Google Scholar]
- 46.Harding MA, Theodorescu D. RhoGDI2: a new metastasis suppressor gene: discovery and clinical translation. Urol Oncol 2007;25(5):401–6. [DOI] [PubMed] [Google Scholar]
- 47.Seraj MJ, Harding MA, Gildea JJ, Welch DR, Theodorescu D. The relationship of BRMS1 and RhoGDI2 gene expression to metastatic potential in lineage related human bladder cancer cell lines. Clin Exp Metastasis 2000;18(6):519–25. [DOI] [PubMed] [Google Scholar]
- 48.Theodorescu D, Sapinoso LM, Conaway MR, Oxford G, Hampton GM, Frierson HF Jr. Reduced expression of metastasis suppressor RhoGDI2 is associated with decreased survival for patients with bladder cancer. Clin Cancer Res 2004;10(11):3800–6. [DOI] [PubMed] [Google Scholar]
- 49.Wu Y, Moissoglu K, Wang H, Wang X, Frierson HF, Schwartz MA, et al. Src phosphorylation of RhoGDI2 regulates its metastasis suppressor function. Proc Natl Acad Sci U S A 2009;106(14):5807–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DerMardirossian C, Rocklin G, Seo JY, Bokoch GM. Phosphorylation of RhoGDI by Src regulates Rho GTPase binding and cytosol-membrane cycling. Mol Biol Cell 2006;17(11):4760–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moissoglu K, McRoberts KS, Meier JA, Theodorescu D, Schwartz MA. Rho GDP dissociation inhibitor 2 suppresses metastasis via unconventional regulation of RhoGTPases. Cancer Res 2009;69(7):2838–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Titus B, Frierson HF Jr, Conaway M, Ching K, Guise T, Chirgwin J, et al. Endothelin axis is a target of the lung metastasis suppressor gene RhoGDI2. Cancer Res 2005;65(16):7320–7. [DOI] [PubMed] [Google Scholar]
- 53.Wu Y, McRoberts K, Berr SS, Frierson HF Jr, Conaway M, Theodorescu D. Neuromedin U is regulated by the metastasis suppressor RhoGDI2 and is a novel promoter of tumor formation, lung metastasis and cancer cachexia. Oncogene 2007;26(5):765v 73. [DOI] [PubMed] [Google Scholar]
- 54.Brighton PJ, Szekeres PG, Willars GB. Neuromedin U and its receptors: structure, function, and physiological roles. Pharmacol Rev 2004;56(2):231–48. [DOI] [PubMed] [Google Scholar]
- 55.Nelson J, Bagnato A, Battistini B, Nisen P. The endothelin axis: emerging role in cancer. Nat Rev Cancer 2003;3(2):110–6. [DOI] [PubMed] [Google Scholar]
- 56.Liu JJ, Payza K, Huang J, Liu R, Chen T, Coupal M, et al. Discovery and pharmacological characterization of a small-molecule antagonist at neuromedin U receptor NMUR2. J Pharmacol Exp Ther 2009;330(1):268–75. [DOI] [PubMed] [Google Scholar]
- 57.Bagnato A, Rosano L. The endothelin axis in cancer. Int J Biochem Cell Biol 2008;40(8):1443–51. [DOI] [PubMed] [Google Scholar]
- 58.Lalich M, McNeel DG, Wilding G, Liu G. Endothelin receptor antagonists in cancer therapy. Cancer Invest 2007;25(8):785–94. [DOI] [PubMed] [Google Scholar]
- 59.Carducci MA, Saad F, Abrahamsson PA, Dearnaley DP, Schulman CC, North SA, et al. A phase 3 randomized controlled trial of the efficacy and safety of atrasentan in men with metastatic hormone-refractory prostate cancer. Cancer 2007;110(9):1959–66. [DOI] [PubMed] [Google Scholar]
- 60.Nelson JB, Love W, Chin JL, Saad F, Schulman CC, Sleep DJ, et al. Phase 3, randomized, controlled trial of atrasentan in patients with nonmetastatic, hormone-refractory prostate cancer. Cancer 2008;113(9):2478–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.James ND, Caty A, Borre M, Zonnenberg BA, Beuzeboc P, Morris T, et al. Safety and efficacy of the specific endothelin-A receptor antagonist ZD4054 in patients with hormone-resistant prostate cancer and bone metastases who were pain free or mildly symptomatic: a double-blind, placebo-controlled, randomised, phase 2 trial. Eur Urol 2009;55(5):1112–23. [DOI] [PubMed] [Google Scholar]