

Abstract
RhoGDI2 is a guanine nucleotide dissociation inhibitor (GDI) specific for the Rho family of small GTPases that plays dual opposite roles in tumor progression, being both a promoter in tissues such as breast and a metastasis suppressor in tissues such as the bladder. Despite a clear role for this protein in modulating the invasive and metastatic process, the mechanisms through which RhoGDI2 executes these functions remain unclear. This review will highlight the current state of our knowledge regarding how RhoGDI2 functions in metastasis with a focus on bladder cancer and will also seek to highlight other potential underappreciated avenues through which this protein may affect cancer cell behavior.
Keywords: RhoGDI2, GTPase, Metastasis, Bladder neoplasms
1. Introduction
The Rho family of GTPases is comprised of 20 members that can play regulatory roles in virtually all cellular processes including motility, cytoskeletal reorganization, cell growth, and differentiation. The actions and mechanisms of regulation for Rho family GTPases have been extensively reviewed elsewhere [1–3]. Briefly, Rho GTPases, the best characterized of which are RhoA, Rac1, and Cdc42, cycle between active GTP bound states and inactive GDP bound states. The activation and inactivation of GTPases is controlled by three classes of molecules: (1) guanine nucleotide exchange factors (GEFs) that enhance the rate of exchange of GDP for GTP; (2) GTPase activating proteins (GAPs) that accelerate the intrinsic hydrolysis of GTP to GDP; and (3) guanine nucleotide dissociation inhibitors (GDIs) that bind to GTPases and sequester them in the cytosol, effectively inactivating GTPases. Aberrant regulation and/or activation of Rho GTPases has been linked to tumorigenic phenotypes in a variety of human cancers [4–6], underscoring the importance of these proteins in normal cellular function, and delineating the need for a clear understanding of not only how and when they are regulated but also of how these proteins cross-talk to coordinate activity.
Rho GDIs consist of a family of three members: RhoGDI1, RhoGDI2 (also known as RhoGDIβ, D4-GDI, or Ly-GDI), and RhoGDI3. RhoGDI1 is ubiquitously expressed and is the best studied member of the family [7–9]. RhoGDI2 was initially believed to be expressed specifically in hematopoietic cells [10], but has subsequently been found to be highly expressed in a variety of other cell types as well [11]. RhoGDI3 is predominantly expressed in brain, lung, kidney, testis, and pancreas [12, 13], and is targeted to the Golgi via its unique N-terminal domain where it plays a role in vesicle transport [14, 15]. Despite a high degree of sequence similarity, RhoGDI1 and RhoGDI2 are highly divergent in their binding affinities for specific GTPases [16] and, more importantly, in their roles in tumorigenesis and metastasis [17]. For example, RhoGDI2 functions as a metastasis suppressor but not a tumor suppressor in bladder cancer cells, while RhoGDI1 is a ubiquitous suppressor of tumor growth in all sites so far examined in bladder cancer models [18], suggesting that their cellular functions must diverge to cause these differential effects.
While there are very clear links between the alteration of RhoGDI2 protein levels and disease progression and/or metastasis in several types of cancer, the mechanistic underpinnings of the mode of RhoGDI2 action under carcinogenic cellular conditions are poorly understood at best. This review will outline what is currently known about the actions of RhoGDI2. Particular emphasis will be given to its role as a bladder cancer metastasis suppressor, as RhoGDI2’s function is most widely characterized in this cancer model. However, we will also touch upon the evidence demonstrating that overexpression of RhoGDI2 promotes tumor progression. We will propose several hypotheses regarding how RhoGDI2 may regulate metastasis, and we will detail what we believe to be the challenges for the future study of RhoGDI2 function.
2. RhoGDI2 and cancer
Alterations in RhoGDI2 expression levels have been shown in a variety of human tumor types. Interestingly, the function of RhoGDI2 as a pro- or anti-tumorigenic and/or metastatic protein varies greatly among cancer tissue type, and even in some cases among histological subtype. For instance, RhoGDI2 expression is downregulated in bladder cancer [19], lung cancer [20], and Hodgkin lymphoma [21], but upregulated in ovarian cancer [22, 23] and stomach cancer [24]. In other tissues, the expression pattern of RhoGDI2 in normal versus cancer specimens is unclear. For example, in breast, RhoGDI2 expression has been reported to be upregulated in cancer [25] and to promote invasion of breast cancer cells [26], while another report found a biphasic expression pattern of RhoGDI2 in breast cancer with decreased expression correlating with lymph node metastasis [27]. Further, a recent report found RhoGDI2 expression was upregulated in human ovarian tumors, and this correlated with histological subtype and grade. Unexpectedly, stable suppression of RhoGDI2 protein expression in HeyA8 ovarian cancer cells increased anchorage-independent growth and Matrigel invasion in vitro and tail-vein lung colony metastatic growth in vivo [22]. Although it is unclear whether this is a universal phenomenon or merely limited to the cell line tested, this would suggest that RhoGDI2 functions as a metastasis suppressor in ovarian cancer despite enhanced expression in ovarian tumors. A model that would explain such findings is one in which the cell is overexpressing a suppressor gene such as RhoGDI2 in an attempt to unsuccessfully oppose or correct a malignant phenotype. In any event, it is clear that reports analyzing only expression levels in tumors must also be supplemented with experimental cellular data to verify any conclusions drawn. The following section will highlight our knowledge of the various roles RhoGDI2 plays in a variety of human tumor types.
2.1. RhoGDI2 is a bladder cancer metastasis suppressor
Studies to identify metastasis suppressor genes in bladder utilized the lineage related T24 and T24T bladder cancer cell lines. T24 cells are poorly tumorigenic and non-metastatic, while T24T cells are tumorigenic and highly metastatic, making this a useful model system to study bladder cancer progression [28]. Oligonucleotide arrays were utilized to compare gene expression levels between the T24 and T24T cell lines. As metastasis suppressor genes were being sought, analysis focused on genes that were downregulated in the metastatic T24T cell line as compared to T24 cells. These genes were further filtered based upon their correlation with tumor grade and stage in a panel of human tumors. Of the top 30 candidate metastasis suppressors, RhoGDI2 was the only one to also correlate with tumor grade and stage [19]. In addition, loss of RhoGDI2 expression in human bladder tumors correlates with shorter disease-free survival times in patients and is a prognostic indicator of bladder cancer metastasis [11]. In keeping with the definition of a metastasis suppressor [29], re-introduction of RhoGDI2 into T24T bladder cells had no effect on cell proliferation, anchorage-independent growth, or subcutaneous tumor growth in athymic mice, but drastically reduced the ability of these cells to colonize lung in experimental metastasis assays, strongly suggesting that RhoGDI2 functions as a metastasis suppressor gene in bladder cancer [19].
Despite a clear role for RhoDGI2 as a bladder cancer metastasis suppressor, the mechanisms through which RhoGDI2 functions as a metastasis suppressor are not yet clear. One such study to investigate these mechanisms found that mutations of RhoGDI2 that either enhance or decrease the binding affinity of RhoGDI2 for GTPases both led to a decrease in the metastasis suppressor function of RhoGDI2. Surprisingly, introduction of wild-type RhoGDI2 into bladder cancer cells led to enhanced Rac1 activation [18], suggesting that RhoGDI2 does not suppress bladder cancer through inhibition of Rac1 activity. It was postulated that perhaps RhoGDI2 enhances the association of Rac1 with a GEF that negatively regulates metastasis, although the potential partners or pathways through which this may occur have not been studied.
2.2. Effectors of RhoGDI2 metastasis suppression in bladder
To identify genes negatively regulated by RhoGDI2 that may function to mediate metastasis upon RhoGDI2 loss, microarray analysis was performed to compare gene expression in T24T parental cells versus T24T cells with forced re-expression of RhoGDI2. A subset of genes was identified with downregulated expression in the cells expressing RhoGDI2 and was further analyzed for expression levels in a panel of bladder tumors of various stages. One gene that was identified that was downregulated upon RhoGDI2 re-expression but had increased expression as a function of increasing tumor grade in human tumor specimens was endothelin 1 (EDN1) [30]. Endothelin 1 is a 21 amino acid secreted peptide that, along with its cognate receptors, plays important roles in vasoconstriction, tissue differentiation, and repair. Overexpression of endothelin 1 and/or its receptors is linked to the progression of several types of cancers and promotes proliferation, cell survival, angiogenesis, and metastasis [31]. In human bladder cancer specimens, EDN1 expression increases according to stage, is higher in muscle invasive bladder cancer versus non-muscle invasive cancer, and high EDN1 expression correlates with lower disease-specific survival time [30, 32]. Endothelin 1 signaling enhances migration, invasion, and proteolytic activity in bladder cancer cells in vitro, and is necessary for the lung colonization of bladder cancer cells in experimental metastasis assays in a mechanism involving recruitment of macrophages to lung and enhanced inflammation [32]. Pharmacological inhibition of the endothelin signaling pathway significantly reduces the incidence of lung metastasis of bladder cancer cells [30, 32], further demonstrating the importance of this pathway during metastasis.
The importance of RhoGDI2 in modulation of the tumor microenvironment was further established when a separate genetic screen, this time utilizing UM-UC-3 metastatic bladder cancer cells with reconstituted RhoGDI2 expression, identified versican (VCAN) as a gene with highly downregulated expression following RhoGDI2 expression. Versican is a proteoglycan that can bind to a variety of molecules such as extracellular matrix proteins and chemokines. In cancer, it is often correlated with enhanced motility and metastasis of cancer cells, and its overexpression has been found in a variety of human tumors and is often associated with poor prognosis [33]. This paradigm also holds true in bladder where VCAN mRNA levels are positively correlated with tumor stage [34], are strongly upregulated in muscle invasive bladder cancer versus non-muscle invasive cancer, and correlate with poor disease-specific survival [35]. Further, versican levels also correlate with tumor cell migration in vitro [34]. Expression of RhoGDI2 in metastatic bladder cells reduced the expression level of versican at the protein level and reduced the metastatic capability of the cells. Interestingly, reconstitution of versican in RhoGDI2 expressing bladder cancer cells reversed the metastasis suppressor function of RhoGDI2 in lung colonization assays, and increased levels of inflammatory cytokines and macrophages at the tumor site, altogether indicating that reduction of versican levels is important for the RhoGDI2 metastasis suppressor effect. Further, versican seems to mediate its prometastatic effect through enhanced CCL2 chemokine levels and macrophage recruitment [35]. The ET-1 and versican data strongly argue that RhoGDI2 may exert its metastasis suppressor effect, at least in part, by negatively regulating inflammation in the tumor microenvironment at sites of metastatic colonization (Fig. 1). Of interest would be to determine the mechanistic pathways through which RhoGDI2 downregulates ET-1 and versican.
Fig. 1.
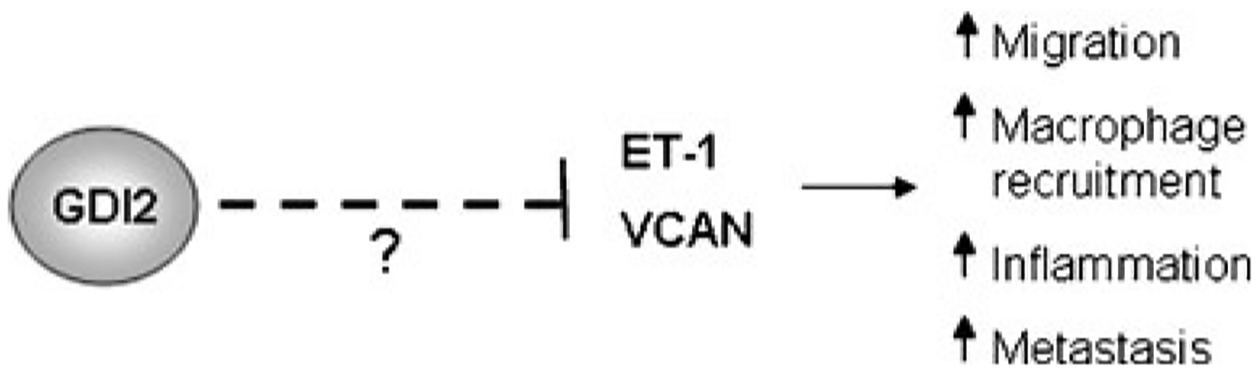
RhoGDI2 modulates the tumor microenvironment. Loss of RhoGDI2 expression in bladder cancer cells causes an increase in the expression levels of endothelin 1 and versican. Expression of these molecules leads to enhanced migration of cells, enhanced inflammatory response and macrophage recruitment to sites of metastatic colonization, and overall enhanced metastatic capability of cells. The mechanisms through which RhoGDI2 modulates endothelin 1 and versican expression are unknown
2.3. Post-translational mechanisms of RhoGDI2 regulation and bladder cancer metastasis
Using a proteomics-based approach, Src tyrosine kinase was found in complex with RhoGDI2. Expression of Src, at both the message and protein level, and activation of Src decreases with increasing stage in human bladder tumors [36, 37]. Interestingly, Src expression is inversely correlated with RhoGDI2 expression in bladder cancer, indicating that the two proteins are generally not both downregulated in the same tumors, and suggesting that they may be involved in the same signaling pathway [37]. Src phosphorylated RhoGDI2 at Tyr 153 both in vitro and in cells. A phospho-mimetic mutant of RhoGDI2, Y153E-RhoGDI2, exhibited reduced binding to Rac1 and a corresponding decrease in the ability of RhoGDI2 to extract Rac1 from membranes (Fig. 2). On the other hand, the Y153F-RhoGDI2 mutant that cannot be phosphorylated exhibited enhanced binding to Rac1 and increased ability to extract Rac1 from the membrane. Interestingly, Src phosphorylation of Tyr 153 on RhoGDI2 also caused enhanced localization of RhoGDI2 to membranes. When the Y153E-RhoGDI2 mutant was introduced into UMUC3 bladder cells and experimental metastasis assays were performed, Y153E-RhoGDI2 had enhanced metastasis suppressor capabilities as compared to WT-RhoGDI2 as was evidenced by a reduced tumor cell burden in mouse lung [37]. It was postulated that the membrane targeting of Y153E-RhoGDI2 could be a means through which RhoGDI2 functions as a metastasis suppressor, although the mechanism through which this occurs is unknown. Interestingly, RhoGDI1 is also phosphorylated at the analogous Tyr site, Tyr 156, in a Src-dependent manner. Like RhoGDI2, phosphorylation at Tyr 156 of RhoGDI1 decreased Rac binding, enhanced GTPase activity in cells as measured by cell ruffling, and led to relocalization of RhoGDI1 to the membrane [38]. This phosphorylation event was postulated to serve two purposes. First, to prolong GTPase activity by preventing binding to RhoGDI1, and second, to enhance the rate of GTPase recycling by targeting RhoGDI1 to the membrane at sites of Rac activation.
Fig. 2.
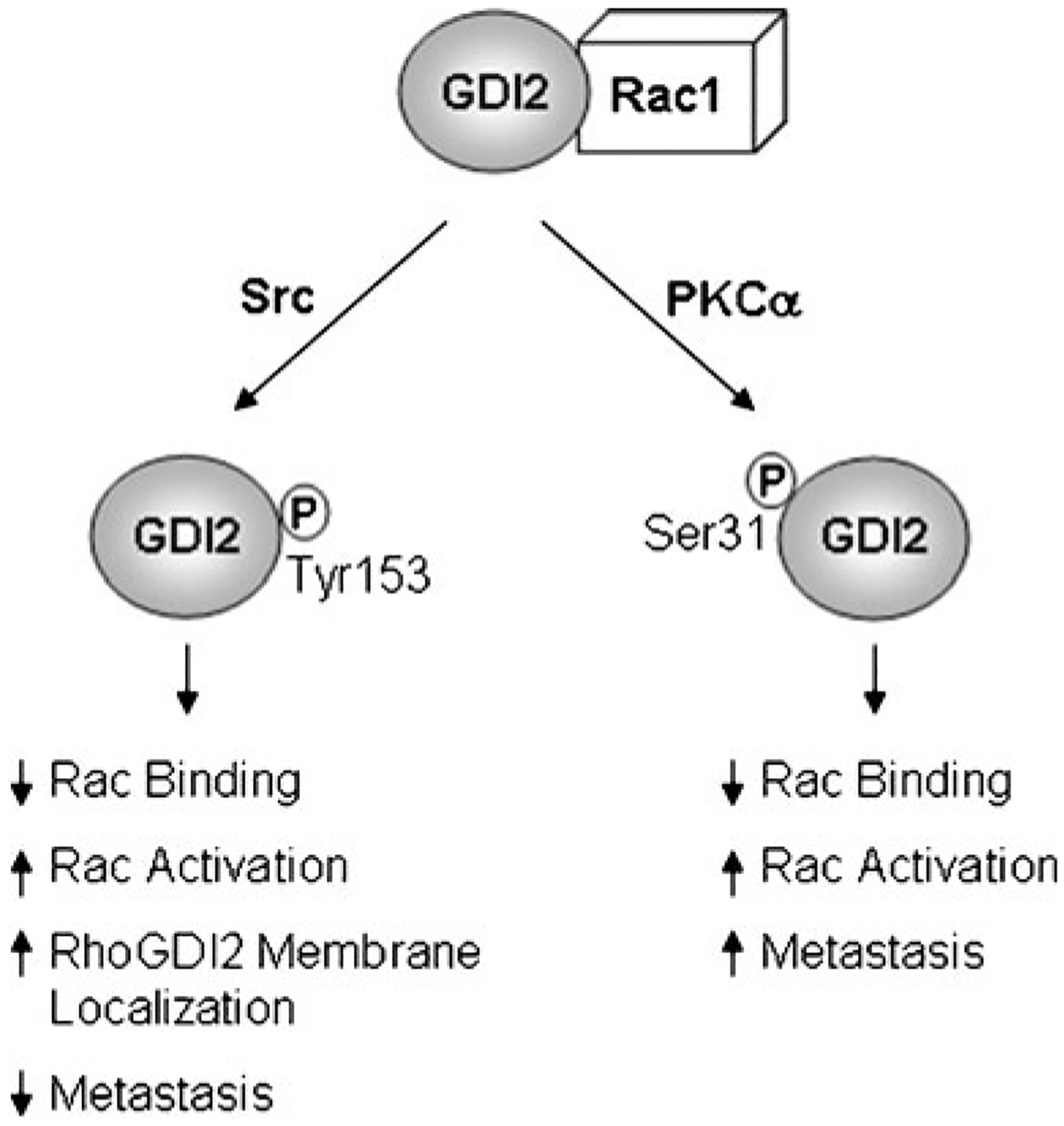
RhoGDI2 metastasis suppressor activity is modulated by post-translational mechanisms. RhoGDI2 can be phosphorylated on Tyr 156 by Src, and this phosphorylation causes dissociation of Rac1 and RhoGDI2, re-localization of both Rac1 and RhoGDI2 to membranes, a corresponding increase in Rac1 activation, and enhanced metastasis suppressor activity. Alternately, RhoGDI2 can be phosphorylated on Ser 31 by PKCα which causes dissociation of Rac1 and RhoGDI2, activation of Rac1, and enhanced metastasis
Recently, our laboratory has shown that Src suppresses metastasis via negative regulation of p190Rho GAP and subsequent inhibition of the RhoA and RhoC mediated Rho-associated protein kinase (ROCK) pathway that is important for cell migration [39]. Perhaps Src and RhoGDI2 are synergizing to affect the ROCK signaling pathway and suppress migration, possibly through modulation of Rho activity. This would be in keeping with our observation that Src-mediated phosphorylation of RhoGDI2 enhances its metastasis suppressor activity [37].
While loss of RhoGDI2 in human bladder cancer tumors often correlates with metastasis, approximately 35% of patients who develop metastatic bladder cancer retain RhoGDI2 protein expression. While other factors may be contributing to metastasis in these patients, it is also conceivable that RhoGDI2 may be inactivated through a post-translational mechanism in these patients. Recently, a PKCα-mediated phosphorylation site at Ser 31 on RhoGDI2 was identified (Fig. 2). Phosphorylation at this site reduces RhoGDI2’s association with Rac1 and causes a concomitant increase in Rac1 localization to membranes and Rac1 activation, indicating that Ser 31 phosphorylation decreases the GDI activity of RhoGDI2 [40]. Structural analysis of the region surrounding Ser 31 of RhoGDI2 indicated that Ser 31 caps the helical hairpin structure of the N-terminal region of RhoGDI2 that contacts the switch regions of GTPases and is essential for GTPase binding [41, 42]. Phosphorylation at Ser 31 is predicted to destabilize the structure of the N-terminal helical hairpin, likely accounting for the decrease in RhoGDI2 binding to Rac1 that was observed. Using UM-UC-3 metastatic bladder cancer cells that stably express either wild-type or S31E-RhoGDI2, we have found that while wild-type RhoGDI2 nearly completely blocks tumor formation in experimental metastasis assays in mice, S31E-RhoGDI2 does not function as a metastasis suppressor and lung metastases are formed similarly to control cells (E. Griner and D. Theodorescu, unpublished results). The mechanism through which this occurs is unclear at present, but could likely be due to altered GTPase binding through disruption of the structure of the N-terminal domain of RhoGDI2 that likely occurs upon phosphorylation.
The finding that S31E-RhoGDI2 does not function as a metastasis suppressor in mouse models of experimental metastasis is important in that it suggests that altered PKCα activation may also be linked to bladder cancer metastasis in tumor cells that still express RhoGDI2. Interestingly, several studies have shown via immunohistochemistry that PKCα protein levels are either elevated or have enhanced activation as a function of increasing grade in bladder tumors [43, 44]. While the sample sizes of these studies were relatively small and a more comprehensive analysis should be performed before definitive conclusions can be drawn, this information is suggestive that PKCα activation and subsequent phosphorylation on Ser 31 of RhoGDI2 in metastatic bladder tumors could be an alternative mechanism of inactivation of the metastasis suppressor function of RhoGDI2 in bladder. A more detailed analysis of the metastatic bladder tumors that retain RhoGDI2 expression needs to be performed to determine whether Ser 31 on RhoGDI2 is phosphorylated in these tumors and whether or not PKCα is concomitantly activated in these tumors. If PKCα-mediated phosphorylation is a mechanism through which RhoGDI2 becomes inactivated in bladder cancer tumors, this would potentially open up new avenues of PKCα targeted therapy for the treatment of patients with RhoGDI2 positive metastatic bladder cancer.
2.4. RhoGDI2 and its pro-tumorigenic role in other tissues
In contrast to its anti-metastatic role in bladder, RhoGDI2 appears to be pro-tumorigenic in other tissue types such as breast and stomach, although the mechanisms through which this effect is mediated are relatively unknown. Several groups have demonstrated that depletion of RhoGDI2 in highly invasive MDA-MB-231 breast cancer cells decreased migration and invasion of these cells [24, 26, 45]. This effect may be due to the loss of β1-integrin expression that was observed upon RhoGDI2 depletion [26], although the mechanisms through which RhoGDI2 may control β1-integrin expression are unknown. Knockdown of RhoGDI2 in MDA-MB-231 cells also caused cells to adopt a more epithelial morphology in 2-D culture, and imparted the ability to form acinar structures in 3-D culture [26]. RhoGDI2 expression was also found to positively regulate Cox-2 expression in MDA-MB-231 cells [45], which is important as Cox-2 has been positively linked to invasion and metastasis in breast and other tumor types [46, 47]. It is worthy to note that the role of RhoGDI2 in breast cancer tumorigenesis has only been studied in one cell line, and it would be interesting to determine whether this phenomenon occurs in other breast cancer cell lines. In gastric cancer cells, depletion of RhoGDI2 also reduced invasion in vitro, while overexpression of RhoGDI2 in a gastric cancer cells line with low RhoGDI2 expression enhanced invasion in vitro, and increased subcutaneous tumor growth, angiogenesis, and metastasis in vivo [24]. While these studies clearly demonstrate that RhoGDI2 can function as a promoter of tumor progression, the mechanisms through which it acts are as yet only hinted at, and a full understanding of how RhoGDI2 acts to promote tumor progression would greatly enhance our understanding of RhoGDI2’s function.
3. The role of RhoGDIs in GTPase activity networks
Emerging evidence suggests that RhoGDI proteins can no longer be thought of as merely passive regulators of Rho GTPase function, but as essential mediators of RhoGTPase cross-talk and activity within the cell. Several published reports have identified Rho family GTPase cross talk pathways in which expression and activity levels of one GTPase can be regulated by another, some suggesting that RhoGDIs may play a role in this cross-regulation [48–53]. To understand this phenomenon, Boulter and colleagues recently described how knockdown of RhoGDI1 led to destabilization and degradation of unbound RhoGTPases resulting in a reduction of GTPase protein levels, but enhanced activation of the remaining GTPases. Paradoxically, while GTPase activity was enhanced, functional assays, such as the assessment of migration, indicated that GTPase activity was actually reduced in the cells, perhaps owing to the mis-localization of active GTPases to the endoplasmic reticulum instead of the plasma membrane [48]. This speaks to the important role that RhoGDIs play in ensuring the proper localization of GTPases so they may optimally interact with effectors. Further, Boulter et al. showed that GTPases cross-talk by competing for binding to RhoGDI1. Exogenously expressed GTPases cause dissociation from RhoGDI1 and degradation of endogenous GTPases [48], arguing that fluctuations in the relative amount of one GTPase within the cell would also impact the amount of other GTPases that also rely on binding to RhoGDIs for stabilization.
While it is well accepted that altered GTPase function is involved in most aspects of tumorigenesis and metastasis, the importance of RhoGDIs as mediators of GTPase cross-talk within a cancer setting has largely been unappreciated. Depletion of RhoC in prostate cancer cells decreased migration, anchorage independent growth, and tumor growth in mice. This effect was due to enhanced RhoA expression, and was dependent on RhoGDI1 expression, likely because RhoC silencing decreased competition for RhoGDI1 binding and stabilization of RhoA [50]. It is interesting to note that in the prostate cancer cell lines used, RhoGDI1 expression preferentially modulated the expression levels of RhoA and RhoC, and only modulated Rac1 or Cdc42 expression levels when both RhoA and RhoC were depleted. This would very much argue to the importance of the relative abundance of the various Rho family GTPases within the cell in determining the specific effect that modulation of RhoGDI levels would cause to GTPase expression. Further, it has been shown that the amount of the predominant RhoGDI in the cell is roughly equivalent to the amount of Rho family GTPases present in the cell [54], and all GTPases must compete for binding to the limited amount of RhoGDI available. This would indicate that not only would cells be exquisitely sensitive to slight perturbations of GTPase levels within the cell but also that the type and relative abundance of both GTPase and RhoGDI isoforms would largely dictate how these perturbations would alter cellular function. Indeed, differential RhoGDI and/or Rho family GTPase expression levels may largely be responsible for the vastly different effects that RhoGDIs have as either pro- or anti-tumorigenic/metastatic proteins depending on the cell type studied.
Given the important role RhoGDI1 plays in regulating RhoGTPase stability and homeostasis in both a normal and cancer setting, it is intriguing to speculate that loss of RhoGDI2 during bladder cancer progression could also alter the relative levels of RhoGTPase proteins to promote a more metastatic phenotype (Fig. 3). In this case, we would predict that Rac1 levels would be most acutely affected, as this is the major small GTPase binding partner of RhoGDI2 [37]. In keeping with the model set forth by Boulter et al., we would predict that loss of RhoGDI2 would alter the stability of Rac1, thus decreasing the pool of Rac1 in the cell. Reduced Rac1 activity within a cell could have many possible consequences in regulating the metastatic phenotype, and it is unknown at this time what role Rac1 plays in bladder cancer and whether or not it functions in a pro- or anti-metastatic manner in bladder, if at all. Rac1 has been shown to downregulate RhoA activity and suppress the migratory phenotype of fibroblasts [51], suggesting that decreased Rac1 expression in bladder cells could lead to enhanced RhoA activation and a more migratory phenotype. Loss of Rac1 in bladder cells could also enhance stability and/or activation of other GTPases that function in a prometastatic manner by decreasing the level of competition for binding to the remaining RhoGDIs in the cell. It is also important not to overlook the potential role that other lesser studied Rho family GTPases could be playing during bladder cancer metastasis.
Fig. 3.
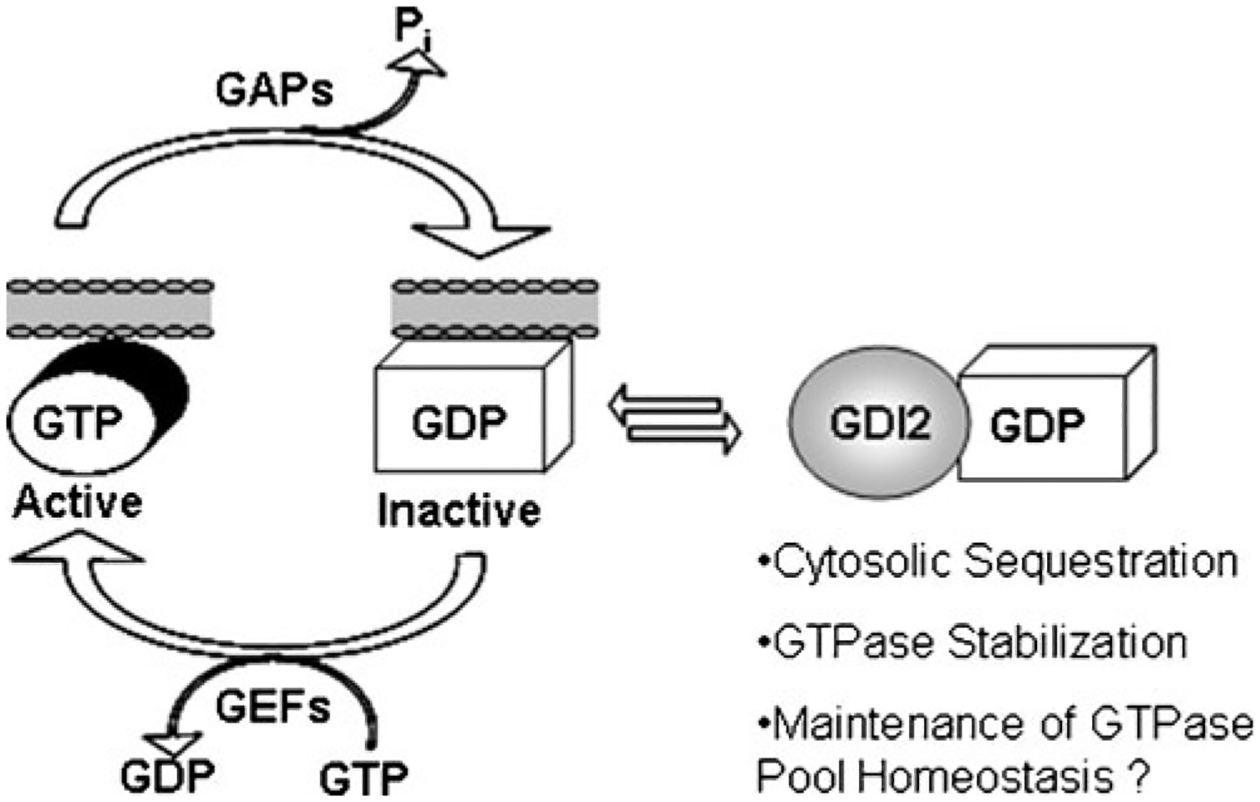
RhoGDI2 regulates localization and activation of Rho family GTPases. GTPases cycle between active GTP bound states and inactive GDP bound states, and activation of GTPases occurs at membranes. RhoGDI2 binds to GTPases and extracts them from membranes, essentially sequestering them in an inactive state in the cytosol. Binding of GDIs to GTPases also stabilizes GTPases and prevents their degradation, maintaining the normal homeostasis of the cellular GTPase pool. Loss of RhoGDI2 in a cancer cell is predicted to not only affect the activation levels of GTPases but could also alter the relative levels of GTPases in the cells in such a way as to favor the metastatic phenotype
It is clear that Rho family GTPases are intimately linked in their regulation and activation, and several key questions remain to be answered to understand how loss of RhoGDI2 during bladder cancer progression may affect the Rho family GTPase repertoire. Does RhoGDI2 function in a similar manner as RhoGDI1 to protect Rho family GTPases from degradation? If so, how does loss of RhoGDI2 affect the expression or activation levels of Rho family GTPases within the cell? How does alteration of GTPase expression mechanistically modulate metastasis in bladder or other tissues? These questions will need to be answered in a comprehensive manner, and consideration will need to be given to all GTPases within the cell.
4. Apoptosis: a divergent role for RhoGDI2
While RhoGDI1 and RhoGDI2 have a high degree of sequence similarity and the two proteins are generally believed to function similarly, the N-terminal amino acid residues of RhoGDI2 are highly divergent from that of RhoGDI1, suggesting that this area of the protein may confer differential functions to RhoGDI2. Interestingly, the N-terminal portion of the RhoGDI2 sequence has two consensus sites for caspase cleavage that are not conserved in RhoGDI1, indicating that RhoGDI1 and 2 may have differential roles in caspase-mediated apoptosis.
Caspase 3-mediated cleavage occurs after aspartic acid 19 at the consensus site DELD19. Cleavage at this site has been shown in response to such stimuli as Fas ligand [55] or anti-Fas antibody [56], staurosporine [55–57], TNF-α [58], ionizing radiation [59], proteosome inhibition [57], and chemotherapeutic drugs such as etoposide [56], Taxol [60], epirubicin [60], or daunorubicin [61]. A caspase 1 cleavage site is located following aspartic acid 55 at the sequence LLGD55. Cleavage at this site has been shown in vitro and in cells following LPS/ATP stimulation. Truncation of the first 55 amino acids of RhoGDI2 disallows Rac1 binding in vitro and fails to regulate Rac1 as measured by GDP dissociation from Rac1 [62].
It is unclear whether cleavage of RhoGDI2 is necessary for apoptosis or merely a by-product of the apoptotic process, and few studies have rigorously tried to determine the role of RhoGDI2 in apoptosis. Interestingly, the cleaved form of RhoGDI2 translocates to the nucleus of cells following treatment with apoptotic stimuli [56, 57, 59]. There is conflicting evidence regarding whether or not nuclear translocation of cleaved RhoGDI2 mediates apoptosis. One study found that when a D19A-RhoGDI2 mutant that cannot be cleaved was transfected into CHO cells, staurosporine still induced apoptosis of cells [56], suggesting that cleavage of RhoGDI2 is dispensable for apoptosis to occur. On the other hand, another study found that expression of the D19A-RhoGDI2 mutant in K562 leukemia cells led to reduced apoptosis in response to proteosome inhibitor treatment, while a truncation mutant of RhoGDI2 with the first 19 amino acids removed conferred enhanced sensitivity to proteosome inhibitor-mediated apoptosis. The addition of a nuclear export sequence to the truncation mutant abolished the effect, leading them to conclude that nuclear localization of cleaved RhoGDI2 was required for the enhanced sensitivity to apoptotic stimuli observed [57]. From these studies, it is likely that the role of RhoGDI2 in apoptosis may vary depending on the nature of the cell type and apoptotic stimuli used.
If caspase-mediated cleavage of RhoGDI2 in response to apoptotic stimuli does indeed play a role in the regulation of apoptosis, it is intriguing to speculate that perhaps loss of RhoGDI2 in tumors may enable tumor cells to evade apoptosis and modulate cell survival during tumorigenesis and metastasis. This may be especially important during metastatic colonization when cells must be resistant to anoikis and mechanical stresses in the vasculature, and survive at the secondary site following extravasation [63]. Indeed, one study found that a RhoGDI2 truncation mutant lacking the first 55 amino acid residues inhibited lung colonization of v-src-transformed fibroblasts in experimental metastasis assays by promoting anoikis of the cells [64], suggesting that cleavage of RhoGDI2 negatively regulates cell survival. A second study found that expression of RhoGDI2 in a Hodgkin lymphoma cell line enhanced apoptosis of these cells in a small but significant manner [21]. Conversely, studies in colon and gastric cancer cells showed that RhoGDI2 expression conferred resistance to various chemotherapeutic drugs, possibly via upregulation of the anti-apoptotic Bcl-2 protein [65, 66]. These studies would suggest that RhoGDI2 is an anti-apoptotic protein, although neither study determined whether RhoGDI2 was cleaved during the apoptotic process or determined the mechanism through which RhoGDI2 was causing drug resistance. It is also interesting to note that RhoGDI2 conferred resistance to apoptotic drugs in colon and gastric cells [65, 66], two cell types that correlate increased expression of RhoGDI2 with tumorigenesis, perhaps suggesting that RhoGDI2 and its role in apoptosis may be a mechanism through which RhoGDI2 differentially affects tumorigenesis or metastasis. To this end, further studies must be performed to answer a variety of questions: Is RhoGDI2 necessary for apoptosis? If so, is it pro-apoptotic or anti-apoptotic? What are the mechanisms/downstream effectors through which it regulates apoptosis? Does RhoGDI2’s role in apoptosis modulate the tumorigenicity or metastatic capability of cancer cells? All these questions must also be asked in a tissue and stimulus specific manner.
5. Challenges for the future
The challenges for the future of RhoGDI2 study are many. As stated previously, RhoGDI2 appears to be either anti-metastatic, as in the case of bladder cancer, or protumorigenic, as illustrated in breast or gastric cancer, indicating that RhoGDI2 must regulate distinct pathways in each cell type. What then, may be the key processes or effectors through which RhoGDI2 may be exerting its disparate effects on tumor progression? One can envision several means through which RhoGDI2 may have differential effects on this process depending on the cell type involved. For example, each tissue type likely has a distinct repertoire of GTPases and their positive and negative regulators. Perhaps the relative levels or activation of GTPases and the interplay among different GTPases within the cellular pool dictates whether or not RhoGDI2 functions as a metastasis suppressor or tumor promoter. RhoGDI2 has also been linked to apoptosis, although its role in this process is as yet unclear. Perhaps a differential ability to affect apoptosis in different tissue types may be another means through which differential RhoGDI2 functions are manifested in cancer cells. In bladder cells, RhoGDI2 may function as a metastasis suppressor by negatively regulating inflammation in the tumor microenvironment through regulation of ET-1 and versican expression, but it is unknown whether or not RhoGDI2 expression similarly affects the tumor microenvironment in other cell types. Differential modulation of signaling pathways involved in this process may be yet another means through which RhoGDI2 functions differently depending on cell type.
Given the frequency with which RhoGDI2 expression is modulated in human cancers, it is clear that RhoGDI2 is playing a role in the carcinogenic process. This review has presented several hypotheses regarding how RhoGDI2 may function, and has identified several key questions that need to be answered in order to better understand RhoGDI2 function and how this function is altered in cancer. First, RhoGDI2 function must be better understood under normal conditions so that we can also understand how alteration of RhoGDI2 expression or regulation can play a role in cancer. Particular attention should be paid to how RhoGDI2 differs from RhoGDI1, as these key differences may be, in part, what defines whether or not one is pro-tumorigenic or a metastasis suppressor. Secondly, we must study RhoGDI2 function in a broader context, and not in isolation with a single GTPase. It is clear that GTPases are intimately linked in their expression and activation, and by extension, RhoGDI2 must also be integrated into the analysis of how Rho GTPases interplay, and how alterations of one protein may influence levels or activation of other proteins. Third, we must rigorously attempt to uncover the mechanisms through which RhoGDI2 promotes tumor progression in order to compare this information to what we already know regarding RhoGDI2’s progression suppressor function. Perhaps through this comparison, we may identify common pathways that may allow us to more clearly define how RhoGDI2 functions. Finally, we must also consider that RhoGDI2 may be functioning through an as yet unidentified pathway that may not relate to GTPase signaling.
Acknowledgments
This study was supported by National Institutes of Health grants CA143971 to D.T. and CA910935 to E.M.G.
Contributor Information
Erin M. Griner, Center for Cell Signaling and Department of Microbiology, Immunology and Cancer Biology, University of Virginia, Charlottesville, VA 22908, USA
Dan Theodorescu, Department of Pharmacology, University of Colorado, Aurora, CO 80045, USA; Department of Surgery, University of Colorado Comprehensive Cancer Center, Aurora, CO 80045, USA.
References
- 1.Heasman SJ, & Ridley AJ (2008). Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nature Reviews Molecular Cell Biology, 9(9), 690–701. doi: 10.1038/nrm2476. [DOI] [PubMed] [Google Scholar]
- 2.Jaffe AB, & Hall A (2005). Rho GTPases: biochemistry and biology. Annual Review of Cell and Developmental Biology, 21, 247–269. [DOI] [PubMed] [Google Scholar]
- 3.VanAelst L, & DsouzaSchorey C (1997). Rho GTPases and signaling networks. Genes & Development, 11(18), 2295–2322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- 4.Kamai T, Tsujii T, Arai K, Takagi K, Asami H, Ito Y, et al. (2003). Significant association of Rho/ROCK pathway with invasion and metastasis of bladder cancer. Clinical Cancer Research, 9 (7), 2632–2641. [PubMed] [Google Scholar]
- 5.Pervaiz S, Cao J, Chao OSP, Chin YY, & Clement MV (2001). Activation of the RacGTPase inhibits apoptosis in human tumor cells. Oncogene, 20(43), 6263–6268. doi: 10.1038/sj.onc.1204840. [DOI] [PubMed] [Google Scholar]
- 6.Vega FM, & Ridley AJ (2008). Rho GTPases in cancer cell biology. Febs Letters, 582(14), 2093–2101. doi: 10.1016/j.febslet.2008.04.039. [DOI] [PubMed] [Google Scholar]
- 7.DerMardirossian C, & Bokoch GM (2005). GDIs: central regulatory molecules in Rho GTPase activation. Trends in Cell Biology, 15(7), 356–363. doi: 10.1016/j.tcb.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 8.Dovas A, & Couchman JR (2005). RhoGDI: multiple functions in the regulation of Rho family GTPase activities. Biochemical Journal, 390, 1–9. doi: 10.1042/bj20050104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garcia-Mata R, Boulter E, & Burridge K (2011). The ‘invisible hand’: regulation of RHO GTPases by RHOGDIs. Nature Reviews Molecular Cell Biology, 12(8), 493–504. doi: 10.1038/nrm3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scherle P, Behrens T, & Staudt LM (1993). LY-GDI, a GDP-dissociation inhibitor of the RhoA GTP-binding protein, is expressed preferentially in lymphocytes. Proceedings of the National Academy of Sciences of the United States of America, 90(16), 7568–7572. doi: 10.1073/pnas.90.16.7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Theodorescu D, Sapinoso LM, Conaway MR, Oxford G, Hampton GM, & Frierson HF (2004). Reduced expression of metastasis suppressor RhoGD12 is associated with decreased survival for patients with bladder cancer. Clinical Cancer Research, 10(11), 3800–3806. doi: 10.1158/1078-0432.ccr-03-0653. [DOI] [PubMed] [Google Scholar]
- 12.Adra CN, Manor D, Ko JL, Zhu SC, Horiuchi T, VanAelst L, et al. (1997). RhoGDI gamma: a GDP-dissociation inhibitor for Rho proteins with preferential expression in brain and pancreas. Proceedings of the National Academy of Sciences of the United States of America, 94(9), 4279–4284. doi: 10.1073/pnas.94.9.4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zalcman G, Closson V, Camonis J, Honore N, RousseauMerck MF, Tavitian A, et al. (1996). RhoGDI-3 is a new GDP dissociation inhibitor (GDI)—identification of a non-cytosolic GDI protein interacting with the small GTP-binding proteins rhoB and rhoG. Journal of Biological Chemistry, 271 (48), 30366–30374. [DOI] [PubMed] [Google Scholar]
- 14.Brunet N, Morin A, & Olofsson B (2002). RhoGDl-3 regulates RhoG and targets this protein to the Golgi complex through its unique N-terminal domain. Traffic, 3(5), 342–357. doi: 10.1034/j.1600-0854.2002.30504.x. [DOI] [PubMed] [Google Scholar]
- 15.Dransart E, Morin A, Cherfils J, & Olofsson B (2005). Uncoupling of inhibitory and shuttling functions of Rho GDP dissociation inhibitors. Journal of Biological Chemistry, 280(6), 4674–4683. doi: 10.1074/jbc.M409741200. [DOI] [PubMed] [Google Scholar]
- 16.Gorvel JP, Chang TC, Boretto J, Azuma T, & Chavrier P (1998). Differential properties of D4/LyGDI versus RhoGDI: phosphorylation and rho GTPase selectivity. Febs Letters, 422 (2), 269–273. doi: 10.1016/s0014-5793(98)00020-9. [DOI] [PubMed] [Google Scholar]
- 17.Harding MA, & Theodorescu D (2010). RhoGDI signaling provides targets for cancer therapy. European Journal of Cancer, 46(7), 1252–1259. doi: 10.1016/j.ejca.2010.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moissoglu K, McRoberts KS, Meier JA, Theodorescu D, & Schwartz MA (2009). Rho GDP dissociation inhibitor 2 suppresses metastasis via unconventional regulation of RhoGTPases. Cancer Research, 69(7), 2838–2844. doi: 10.1158/0008-5472.can-08-1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gildea JJ, Seraj MJ, Oxford G, Harding MA, Hampton GM, Moskaluk CA, et al. (2002). RhoGD12 is an invasion and metastasis suppressor gene in human cancer. Cancer Research, 62 (22), 6418–6423. [PubMed] [Google Scholar]
- 20.Niu H, Li H, Xu C, & He P (2010). Expression profile of RhoGDI2 in lung cancers and role of RhoGDI2 in lung cancer metastasis. Oncology Reports, 24(2), 465–471. doi: 10.3892/or_00000880. [DOI] [PubMed] [Google Scholar]
- 21.Ma L, Xu G, Sotnikova A, Szczepanowski M, Giefing M, Krause K, et al. (2007). Loss of expression of LyGDI (ARHGDIB), a rho GDP-dissociation inhibitor, in Hodgkin lymphoma. British Journal of Haematology, 139(2), 217–223. doi: 10.1111/j.1365-2141.2007.06782.x. [DOI] [PubMed] [Google Scholar]
- 22.Stevens EV, Banet N, Onesto C, Plachco A, Alan JK, Nikolaishvili-Feinberg N, et al. (2011). RhoGDI2 antagonizes ovarian carcinoma growth, invasion and metastasis. Small GTPases, 2(4), 202–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhen H, Yang S, Wu H, Wang S, Lv J, Ma L, et al. (2010). LyGDI is a promising biomarker for ovarian cancer. International Journal of Gynecological Cancer, 20(3), 316–322. doi: 10.1111/IGC.0b013e3181d0b02d. [DOI] [PubMed] [Google Scholar]
- 24.Cho HJ, Baek KE, Park S-M, Kim I-K, Choi Y-L, Cho H-J, et al. (2009). RhoGDI2 expression is associated with tumor growth and malignant progression of gastric cancer. Clinical Cancer Research, 15(8), 2612–2619. doi: 10.1158/1078-0432.ccr-08-2192. [DOI] [PubMed] [Google Scholar]
- 25.Moon H-G, Jeong S-H, Ju Y-T, Jeong C-Y, Lee JS, Lee Y-J, et al. (2010). Up-regulation of RhoGDI2 in human breast cancer and its prognostic implications. Cancer Research and Treatment: Official Journal of Korean Cancer Association, 42(3), 151–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang YQ, & Zhang BL (2006). D4-GDI, a Rho GTPase regulator, promotes breast cancer cell invasiveness. Cancer Research, 66(11), 5592–5598. doi: 10.1158/0008-5472.can-05-4004. [DOI] [PubMed] [Google Scholar]
- 27.Hu LD, Zou HF, Zhan SX, & Cao KM (2007). Biphasic expression of RhoGDI2 in the progression of breast cancer and its negative relation with lymph node metastasis. Oncology Reports, 17(6), 1383–1389. [PubMed] [Google Scholar]
- 28.Gildea JJ, Golden WL, Harding MA, & Theodorescu D (2000). Genetic and phenotypic changes associated with the acquisition of tumorigenicity in human bladder cancer. [Article]. Genes Chromosomes & Cancer, 27(3), 252–263, doi:. [DOI] [PubMed] [Google Scholar]
- 29.Steeg PS (2003). Metastasis suppressors alter the signal transduction of cancer cells. Nature Reviews Cancer, 3(1), 55–63. doi: 10.1038/nrc967. [DOI] [PubMed] [Google Scholar]
- 30.Titus B, Frierson HF, Conaway M, Ching K, Guise T, Chirgwin J, et al. (2005). Endothelin axis is a target of the lung metastasis suppressor gene RhoGD12. Cancer Research, 65(16), 7320–7327. doi: 10.1158/0008-5472.can-05-1403. [DOI] [PubMed] [Google Scholar]
- 31.Nelson J, Bagnato A, Battistini B, & Nisen P (2003). The endothelin axis: emerging role in cancer. Nature Reviews Cancer, 3 (2), 110–116. doi: 10.1038/nrc990. [DOI] [PubMed] [Google Scholar]
- 32.Said N, Smith S, Sanchez-Carbayo M, & Theodorescu D (2011). Tumor endothelin-1 enhances metastatic colonization of the lung in mouse xenograft models of bladder cancer. Journal of Clinical Investigation, 121(1), 132–147. doi: 10.1172/jci42912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ricciardelli C, Sakko AJ, Ween MP, Russell DL, & Horsfall DJ (2009). The biological role and regulation of versican levels in cancer. Cancer and Metastasis Reviews, 28(1–2), 233–245. doi: 10.1007/s10555-009-9182-y. [DOI] [PubMed] [Google Scholar]
- 34.Wu Y, Siadaty MS, Berens ME, Hampton GM, & Theodorescu D (2008). Overlapping gene expression profiles of cell migration and tumor invasion in human bladder cancer identify metallothionein 1E and nicotinamide N-methyltransferase as novel regulators of cell migration. Oncogene, 27(52), 6679–6689. doi: 10.1038/onc.2008.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Said N, Sanchez-Carbayo M, Smith SC & Theodorescu D (2012). RhoGDI2 suppresses lung metastasis in mice by reducing tumor versican expression and macrophage infiltration. Journal of Clinical Investigation 122(4), 1503–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fanning P, Bulovas K, Saini KS, Libertino JA, Joyce AD, & Summerhayes IC (1992). Elevated expression of PP60C-SRC in low-grade human bladder carcinomas. Cancer Research, 52(6), 1457–1462. [PubMed] [Google Scholar]
- 37.Wu Y, Moissogiu K, Wang H, Wang X, Frierson HF, Schwartz MA, et al. (2009). Src phosphorylation of RhoGDI2 regulates its metastasis suppressor function. Proceedings of the National Academy of Sciences of the USA, 106(14), 5807–5812. doi: 10.1073/pnas.0810094106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DerMardirossian C, Rocklin G, Seo J-Y, & Bokoch GM (2006). Phosphorylation of RhoGDI by Src regulates Rho GTPase binding and cytosol-membrane cycling. Molecular Biology of the Cell, 17(11), 4760–4768. doi: 10.1091/mbc.E06-06-0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thomas S, Overdevest JB, Nitz MD, Williams PD, Owens CR, Sanchez-Carbayo M, et al. (2011). Src and caveolin-1 reciprocally regulate metastasis via a common downstream signaling pathway in bladder cancer. Cancer Research, 71 (3), 832–841. doi: 10.1158/0008-5472.can-10-0730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Griner EM, Churchill MEA, Brautigan DL, & Theodorescu D (2012). PKCalpha phosphorylation of RhoGDI2 at Ser31 disrupts interactions with Rac1 and decreases GDI activity. Oncogene. doi: 10.1038/onc.2012.124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Golovanov AP, Chuang TH, DerMardirossian C, Barsukov I, Hawkins D, Badii R, et al. (2001). Structure–activity relationships in flexible protein domains: regulation of rho GTPases by RhoGDI and D4 GDI. Journal of Molecular Biology, 305(1), 121–135. doi: 10.1006/jmbi.2000.4262. [DOI] [PubMed] [Google Scholar]
- 42.Scheffzek K, Stephan I, Jensen ON, Illenberger D, & Gierschik P (2000). The Rac–RhoGDI complex and the structural basis for the regulation of Rho proteins by RhoGDI. Nature Structural Biology, 7(2), 122–126. doi: 10.1038/72392. [DOI] [PubMed] [Google Scholar]
- 43.Aaltonen V, Koivunen J, Laato M, & Peltonen J (2006). Heterogeneity of cellular proliferation within transitional cell carcinoma: correlation of protein kinase C alpha/betal expression and activity. Journal of Histochemistry & Cytochemistry, 54(7), 795–806. doi: 10.1369/jhc.5A6839.2006. [DOI] [PubMed] [Google Scholar]
- 44.Koren R, Langzam L, Paz A, Livne PM, Gal R, & Sampson SR (2000). Protein kinase C (PKC) isoenzymes immunohistochemistry in lymph node revealing solution-fixed, paraffin-embedded bladder tumors. Applied Immunohistochemistry & Molecular Morphology, 8(2), 166–171. doi: 10.1097/00022744-200006000-00013. [DOI] [PubMed] [Google Scholar]
- 45.Schunke D, Span P, Ronneburg H, Dittmer A, Vetter M, Holzhausen H-J, et al. (2007). Cyclooxygenase-2 is a target gene of rho GDP dissociation inhibitor beta in breast cancer cells. Cancer Research, 67(22), 10694–10702. doi: 10.1158/0008-5472.can-07-1621. [DOI] [PubMed] [Google Scholar]
- 46.Harris RE (2007). Cyclooxygenase-2 (cox-2) and the inflammogenesis of cancer. Sub-cellular Biochemistry, 42, 93–126. [DOI] [PubMed] [Google Scholar]
- 47.Larkins TL, Nowell M, Singh S, & Sanford GL (2006). Inhibition of cyclooxygenase-2 decreases breast cancer cell motility, invasion and matrix metalloproteinase expression. Bmc Cancer, 6. doi:181 10.1186/1471-2407-6-181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boulter E, Garcia-Mata R, Guilluy C, Dubash A, Rossi G, Brennwald PJ, et al. (2010). Regulation of Rho GTPase cross-talk, degradation and activity by RhoGDI1. Nature Cell Biology, 12(5), 477–U136. doi: 10.1038/ncb2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ho TTG, Merajver SD, Lapiere CM, Nusgens BV, & Deroanne CF (2008). RhoA-GDP regulates RhoB protein stability—potential involvement of RhoGDI alpha. Journal of Biological Chemistry, 283(31), 21588–21598. doi: 10.1074/jbc.M710033200. [DOI] [PubMed] [Google Scholar]
- 50.Ho TTG, Stultiens A, Dubail J, Lapiere CM, Nusgens BV, Colige AC, et al. (2011). RhoGDI alpha-dependent balance between RhoA and RhoC is a key regulator of cancer cell tumorigenesis. Molecular Biology of the Cell, 22(17), 3263–3275. doi: 10.1091/mbc.E11-01-0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sander EE, ten Klooster JP, van Delft S, van der Kammen RA, & Collard JG (1999). Rac downregulates Rho activity: reciprocal balance between both GTPases determines cellular morphology and migratory behavior. Journal of Cell Biology, 147(5), 1009–1021. doi: 10.1083/jcb.147.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Simpson KJ, Dugan AS, & Mercurio AM (2004). Functional analysis of the contribution of RhoA and RhoC GTPases to invasive breast carcinoma. Cancer Research, 64(23), 8694–8701. doi: 10.1158/0008-5472.can-04-2247. [DOI] [PubMed] [Google Scholar]
- 53.Huang C-Y, Yang L-C, Liu K-Y, Chang IC, Liao P-H, Chou JI-Y, et al. (2009). ZAK negatively regulates RhoGDI beta-induced Rac1-mediated hypertrophic growth and cell migration. Journal of Biomedical Science, 16. doi:56 10.1186/1423-0127-16-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Michaelson D, Silletti J, Murphy G, D’Eustachio P, Rush M, & Philips MR (2001). Differential localization of Rho GTPases in live cells: regulation by hypervariable regions and RhoGDI binding. Journal of Cell Biology, 152(1), 111–126. doi: 10.1083/jcb.152.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Na SQ, Chuang TH, Cunningham A, Turi TG, Hanke JH, Bokoch GM, et al. (1996). D4-GDI, a substrate of CPP32, is proteolyzed during Fas-induced apoptosis. Journal of Biological Chemistry, 271(19), 11209–11213. [DOI] [PubMed] [Google Scholar]
- 56.Krieser RJ, & Eastman A (1999). Cleavage and nuclear translocation of the caspase 3 substrate Rho GDP-dissociation inhibitor, D4-GDI, during apoptosis. Cell Death and Differentiation, 6(5), 412–419. doi: 10.1038/sj.cdd.4400515. [DOI] [PubMed] [Google Scholar]
- 57.Choi MR, Groot M, & Drexler HCA (2007). Functional implications of caspase-mediated RhoGDI2 processing during apoptosis of HL60 and K562 leukemia cells. Apoptosis, 12(11), 2025–2035. doi: 10.1007/s10495-007-0121-5. [DOI] [PubMed] [Google Scholar]
- 58.Kettritz R, Xu YX, Faass B, Klein JB, Muller EC, Otto A, et al. (2000). TNF-alpha-mediated neutrophil apoptosis involves Ly-GDI, a Rho GTPase regulator. Journal of Leukocyte Biology, 68(2), 277–283. [PubMed] [Google Scholar]
- 59.Zhou XW, Suto S, Ota T, & Tatsuka M (2004). Nuclear translocation of cleaved LyGDI dissociated from Rho and Rac during Trp53-dependent ionizing radiation-induced apoptosis of thymus cells in vitro. Radiation Research, 162(3), 287–295. doi: 10.1667/rr3220. [DOI] [PubMed] [Google Scholar]
- 60.Essmann F, Wieder T, Otto A, Muller EC, Dorken B, & Daniel PT (2000). GDP dissociation inhibitor D4-GDI (Rho-GDI 2), but not the homologous Rho-GDI 1, is cleaved by caspase-3 during drug-induced apoptosis. Biochemical Journal, 346, 777–783. doi: 10.1042/0264-6021:3460777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kwon KB, Park EK, Ryu DG, & Park BH (2002). D4-GDI is cleaved by caspase-3 during daunorubicin-induced apoptosis in HL-60 cells. Experimental and Molecular Medicine, 34(1), 32–37. [DOI] [PubMed] [Google Scholar]
- 62.Danley DE, Chuang TH, & Bokoch GM (1996). Defective Rho GTPase regulation by IL-1 beta-converting enzyme-mediated cleavage of D4 GDP dissociation inhibitor. Journal of Immunology, 157(2), 500–503. [PubMed] [Google Scholar]
- 63.Mehlen P, & Puisieux A (2006). Metastasis: a question of life or death. Nature Reviews Cancer, 6(6), 449–458. doi: 10.1038/nrc1886. [DOI] [PubMed] [Google Scholar]
- 64.Ota T, Maeda M, Suto S, & Tatsuka M (2004). LyGDl functions in cancer metastasis by anchoring Rho proteins to the cell membrane. Molecular Carcinogenesis, 39(4), 206–220. doi: 10.1002/mc.20006. [DOI] [PubMed] [Google Scholar]
- 65.Cho HJ, Baek KE, Park S-M, Kim I-K, Nam I-K, Choi Y-L, et al. (2011). RhoGDI2 confers gastric cancer cells resistance against cisplatin-induced apoptosis by upregulation of Bcl-2 expression. Cancer Letters, 311(1), 48–56. doi: 10.1016/j.canlet.2011.06.024. [DOI] [PubMed] [Google Scholar]
- 66.Zheng Z, Li J, He X, Chen X, Yu B, Ji J, et al. (2010). Involvement of RhoGDI2 in the resistance of colon cancer cells to 5-fluorouracil. Hepato-Gastroenterology, 57(102–03), 1106–1112. [PubMed] [Google Scholar]