

Keywords: aging, chronic kidney disease, hypertension, obesity, senescence
Abstract
Patients with hypertension or obesity can develop glomerular dysfunction characterized by injury and depletion of podocytes. To better understand the molecular processes involved, young mice were treated with either deoxycorticosterone acetate (DOCA) or fed a high-fat diet (HFD) to induce hypertension or obesity, respectively. The transcriptional changes associated with these phenotypes were measured by unbiased bulk mRNA sequencing of isolated podocytes from experimental models and their respective controls. Key findings were validated by immunostaining. In addition to a decrease in canonical proteins and reduced podocyte number, podocytes from both hypertensive and obese mice exhibited a sterile inflammatory phenotype characterized by increases in NLR family pyrin domain containing 3 (NLRP3) inflammasome, protein cell death-1, and Toll-like receptor pathways. Finally, although the mice were young, podocytes in both models exhibited increased expression of senescence and aging genes, including genes consistent with a senescence-associated secretory phenotype. However, there were differences between the hypertension- and obesity-associated senescence phenotypes. Both show stress-induced podocyte senescence characterized by increased p21 and p53. Moreover, in hypertensive mice, this is superimposed upon age-associated podocyte senescence characterized by increased p16 and p19. These results suggest that senescence, aging, and inflammation are critical aspects of the podocyte phenotype in experimental hypertension and obesity in mice.
NEW & NOTEWORTHY Hypertension and obesity can lead to glomerular dysfunction in patients, causing podocyte injury and depletion. Here, young mice given deoxycorticosterone acetate or a high-fat diet to induce hypertension or obesity, respectively. mRNA sequencing of isolated podocytes showed transcriptional changes consistent with senescence, a senescent-associated secretory phenotype, and aging, which was confirmed by immunostaining. Ongoing studies are determining the mechanistic roles of the accelerated aging podocyte phenotype in experimental hypertension and obesity.
INTRODUCTION
The incidence and prevalence of hypertension (1) and obesity (2) are increasing globally. Both are independent risk factors for cardiovascular disease and have major negative impacts on the kidney. Hypertension is considered the second leading cause of chronic kidney disease (3). The underlying mechanism(s) are likely multifactorial but include impairment of the glomerular autoregulatory mechanisms that normally protect the glomerulus from pathological alterations in systemic blood pressure (4–6). Several clinical studies show that hypertension contributes to podocyte injury. Wang et al. (7) showed that the number of podocytes per glomerulus was lower in patients with biopsy-proven hypertensive nephrosclerosis and that the patient’s urines exhibited higher levels of podocyte markers than those of healthy controls, suggesting active podocyte shedding. Naik et al. (8) demonstrated that urinary podocyte markers correlate with systemic blood pressure and that podocyte loss precedes apparent kidney disease in subjects with hypertension. The latter was confirmed in patients with hypertensive nondiabetes (9). Finally, examining autopsy samples and living kidney donors, lower podocyte density was also observed in subjects with hypertension without apparent kidney disease (10, 11). Animal models have validated these clinical reports. For example, podocyte injury in the deoxycorticosterone acetate (DOCA) hypertensive model in rats and mice has shown roles for TRPC5 (12) and claudin-5 (13), whereas deacetylation of Septin4 mitigated podocyte damage in hypertensive mice (14).
A similar detrimental effect has been observed for obesity, which is associated with low-grade albuminuria and a decline in kidney function (reviewed in Ref. 15). Like hypertension, changes to podocytes occur in obesity and obesity-related glomerulopathy. These include podocyte effacement, enlargement of foot processes, detachment, and reduced density (15). In addition, the podocyte transcriptome changes are characterized by the nascent expression of mesenchymal markers. Finally, patients with obesity-related glomerulopathy can lose up to 45% of their podocytes (16). Obesity leads to progressive increases in the glomerular filtration rate, renal plasma flow, and filtration fraction, synonymous with glomerular hyperfiltration. The relationship between obesity and kidney impairment was further reinforced in patients who lost weight following bariatric surgery with subsequent reduced glomerular hemodynamics (17). Mechanistically, reduced levels of adiponectin and leptin, together with the development of insulin resistance and hypercholesterolemia are some of the pathways that contribute to the decline in kidney function due to obesity (18) (reviewed in Ref. 19). A subset of patients with obesity develop more severe kidney damage due to obesity-related glomerulopathy, which is a distinct form of peri-hilar focal segmental glomerulosclerosis (FSGS) with higher levels of proteinuria but are typically not in the nephrotic range (15, 19). Obesity-related podocyte injury has been reproduced in experimental rodent studies using high-fat diet (HFD) (20–23). For example, obese mice develop podocyte damage, including increased foot process diameter, swollen and fused foot processes, and mitochondrial vacuolar degeneration (21, 22). These aberrant phenotypic changes were reduced upon treatment with the glucagon-like peptide-1 receptor agonist liraglutide (21).
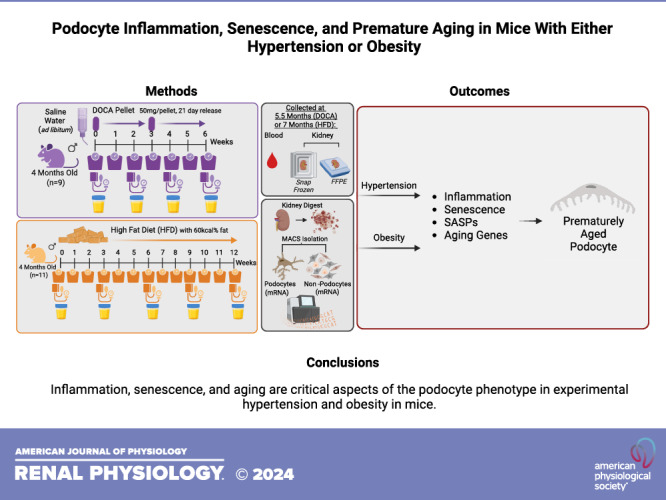
Despite advances in the understanding of podocyte damage in hypertension and obesity, the pathways underlying these changes are not fully elucidated. To close these knowledge gaps by identifying new pathways responsible for podocyte injury in hypertension and obesity, we undertook an unbiased approach, performing bulk mRNA sequencing of podocytes isolated from representative hypertensive (DOCA-treated) and obese [high-fat diet (HFD)-fed] mouse models. The data demonstrate that either hypertension or obesity in mice resulted in podocytes acquiring an inflammatory, senescent, and aged phenotype.
METHODS
Mice and Experimental Models
All mice were housed in the animal care facility at the University of Washington under specific pathogen-free (SPF) conditions. Mature 4-mo-old adult male C57BL/6J mice were purchased from The Jackson Laboratory (Stock No: 000664, Bar Harbor, ME) and allowed to acclimate for at least 1 wk in our facilities before starting experiments or being euthanized. All animals were co-housed in social groups of two to five animals, provided with environmental enrichment (nestlets), maintained on a 14:10-h light-dark cycle, at 70 ± 5°F and 50 ± 10% relative humidity, with ad libitum access to food and water. Mice were euthanized at specified time points by cervical dislocation according to American Veterinary Medical Association (AVMA) guidelines for the euthanasia of animals by certified personnel, and tissues were processed for RNA and histology. To address the Reduction aspect of the 3Rs (Animal Use Alternatives), we have a bank of aged male C57BL/6J mice, also obtained from The Jackson Laboratory, aged and euthanized at various time points for use as controls. Blood pressure was measured with the CODA 6 noninvasive tail-cuff system (Kent Scientific, Torrington, CT) as previously described (24–26). Pooled spot urines were collected on the specified collection days. Animal protocols were approved (2968-04) by the University of Washington Institutional Animal Care and Use Committee.
Deoxycorticosterone Acetate Salt Model
Four-month-old mice (n = 9) were given two sequential subcutaneous deoxycorticosterone acetate salt (DOCA) pellet implants (50 mg/pellet, 21-day release, Innovative Research of America, Sarasota, FL) 3 wk apart for a total of 6 wk of sustained DOCA release (Fig. 1A). For the 6-wk duration of the experiment, animals received supplemental ad libitum saline water for drinking and their regular house water. Animals were weighed weekly, and urine and blood pressure were collected every 2 wk. At the end of 6 wk, animals were euthanized and processed as aforementioned (Fig. 1, B and C). Kidneys from mice aged 4 (n = 5) and 5.5 (n = 5) mo of age served as baseline and normal controls, respectively.
Figure 1.

Transcriptomic changes in podocytes measured by RNA-sequencing from deoxycorticosterone acetate (DOCA)-treated mice. A–C: schematic of the experimental design. Following 6 wk of DOCA treatment (A), podocytes were isolated by magnetic-activated cell sorting (MACS) and underwent bulk RNA-sequencing (B), and kidneys were also collected and fixed for immunostaining (C). D: principal component analysis (PCA). PCA shows clear separation of the DOCA-treated mice (blue color) from the control mice (red color). E: volcano plot shows separation of individual genes that are increased (red), decreased (blue), or unchanged (gray) in hypertensive mice compared with controls. F–H: Hallmark pathways. F: Hallmark gene sets of the highest pathways enriched by Gene Set Enrichment Analysis (GSEA) show a predominance of inflammatory pathways in DOCA-treated mice. G: the top 20 genes in the inflammatory response pathway are shown. H: GSEA Hallmark pathways downregulated in podocytes upon DOCA treatment shows that oxidative phosphorylation was the most dysregulated Hallmark pathway.
High-Fat Diet-Induced Obesity Model
Four-month-old mice (n = 11) were provided ad libitum high-fat diet (HFD) with 60 kcal% fat (D12492, Research Diets Inc., New Brunswick, NJ) for 12 wk, which changed weekly. Weights were taken weekly for the duration of the study, whereas blood pressure and urine were taken every 3 wk. At the end of 12 wk, animals were euthanized and processed as aforementioned (Fig. 4, A–C). Kidneys were taken from mice at 4 (n = 5) and 7 (n = 5) mo of age that were maintained on house chow with 13 kcal% fat (LabDiet 5053, Purina Mills, Gray Summit, MO) and served as baseline and normal controls.
Figure 4.

Inflammatory pathways are increased in podocytes in mice treated with a high-fat diet (HFD). A–C: schema of experimental protocol for HFD in mice. Following a HFD for 12 wk (A), podocytes isolated by magnetic-activated cell sorting (MACS) underwent bulk RNA-sequencing (B) and kidneys were collected and fixed for immunostaining (C). D: principal component analysis (PCA). There is no overlap between HFD mice (purple) and their controls (blue). Shown too are the PCA for mice on deoxycorticosterone acetate (DOCA, green) and their controls (red). The latter are a different age to controls for HFD. The PCA shows no overlap between DOCA and HFD mice. E: volcano plot shows genes positively and negatively enriched by HFD. F–H: Hallmark pathway analyses. F: inflammatory pathways are the predominant pathways upregulated by HFD, with examples of the top 20 genes shown for the Hallmark inflammatory response pathway (H). G: downregulated Hallmark pathways include several metabolic processes. I: genes in the NLR family pyrin domain containing 3 (NLRP3) and PD1 signaling pathways are increased in podocytes of HFD mice.
Magnetic-Activated Cell Sorting of Podocytes
Kidney tissue (w/o the kidney capsule and surrounding fat) was placed into ice-cold RPMI 1640 medium (w/o l-glutamine and phenol red, GE Healthcare Bio-Sciences, Pittsburgh, PA) (Figs. 1B and 4B). After removal of the medulla, the remaining cortex was minced into fine pieces and digested in 0.2 mg/mL Liberase TL (Sigma Aldrich, St. Louis, MO), 100 U/mL DNAse I (Sigma Aldrich, St. Louis, MO) in RPMI 1640 medium (w/o l-glutamine and phenol red) by shaking at 37°C for 30 min. The digest was passed through an 18-G needle (Becton Dickenson, Franklin Lakes, NJ) 10 times. Enzymes were inactivated by adding 5 mL of RPMI 1640 medium (w/o l-glutamine and phenol red) supplemented with 1 mM sodium pyruvate (Thermo Fisher Scientific, Waltham, MA), 9% Nu-Serum IV Growth Medium Supplement (Corning Incorporated—Life Sciences, Durham, NC), and 100 U/mL penicillin-streptomycin (Thermo Fisher Scientific, Waltham, MA). The cell suspension was passed through a 100-µm and a 40-µm cell strainer (BD Biosciences, San Jose, CA) and pelleted by centrifugation at 200 g at 4°C for 5 min. Cells were resuspended in media containing two rabbit anti-nephrin antibodies (1:100, Abcam, Cambridge, MA). After 1 h at 4°C, cells were pelleted, washed in media, and incubated with anti-rabbit microbeads (Miltenyi Biotec, Auburn, CA) along with Alexa Fluor 594-conjugated AffiniPure donkey anti-rabbit IgG 1:200 (to visualize binding of the nephrin antibodies to the podocytes) for 30 min at 4°C. Cells were pelleted and washed in PBS with 0.5% BSA and 2 mM EDTA and applied to magnetic-activated cell sorting (MACS) LS columns (Miltenyi Biotec, Auburn, CA) to separate microbead-bound podocytes from the other kidney cells gently. Cells eluted through the magnetic field were collected, pelleted, and designated nonpodocyte (NP) cell fractions. LS columns were removed from the magnetic field and then washed with PBS with 0.5% BSA and 2 mM EDTA, and podocytes were collected. A small aliquot of each fraction was imaged using an EVOS FL Cell Imaging System to verify podocyte isolation based on the presence of a nephrin antibody. In addition, qRT-PCR for a panel of podocyte genes was performed in both podocyte and nonpodocyte fractions to confirm cell type identity (27).
RNA Isolation, qRT-PCR, and Library Preparation and Sequencing
Messenger RNA was isolated using the RNeasy Mini Kit (Qiagen, Germantown, MD) as per the manufacturer’s instructions and used for bulk mRNA sequencing or converted to cDNA by reverse transcription with the High-Capacity RNA-to-cDNA Kit (Thermo Fisher, Waltham, MA) and used for quantitative real-time PCR analysis. qRT-PCR was performed using iTaq SYBR Green Supermix (Bio-Rad, Hercules, CA), QuantiTect Primer Assay (Qiagen, Germantown, MD) for Cdkn2a (p16 INK4A variant 2 QT01164898, p19ARF variant 1 QT01164891) and Ckn1a (p21 QT01752562) on the QuantStudio 6 Flex real-time PCR System (Applied Biosystems) as we have previously described (27). Relative mRNA expression levels were normalized to glyceraldehyde-3-phosphate dehydrogenase (Gapdh). Psomagen, Inc. (Rockville, MD) performed library generation and bulk next-generation mRNA sequencing using TruSeq RNA Library Prep Kits (Illumina, San Diego, CA) and the Illumina platform. The transcriptomic data were submitted to the NCBI’s Gene Expression Omnibus (GEO) sequence repository and are available under GEO Series Accession No. GSE255723.
Data Analysis: Alignment and Quantification
Transcript abundance was estimated without aligning reads using Salmon (28) against an index of coding sequences from the Ensembl GRCm38 assembly. Transcript-level abundance was imported and count and offset matrices generated using the tximport R/Bioconductor package (29). Differential expression analysis was performed using the DESeq2 R/Bioconductor package (30). Principal component analysis (PCA) was performed using pcaExplorer (v2.26.1), considering the top 500 genes (31). Note that samples with low read counts did not cluster with the other samples and were flagged as outliers and removed. Gene Set Enrichment Analysis (GSEA) was performed using the Molecular Signature Database (MSigDB) as described (32). GSEA bar graphs were created in R (v.4.3.1) using GGplot2 (v.3.4.2), Dplyr (v.1.1.2), and Tidyverse (v.2.0.0).
Immunostaining
Immunoperoxidase staining was performed on 4-µm thick formalin-fixed paraffin-embedded (FFPE) mouse and human kidney sections as previously described (33) (Figs. 1C and 4C). Briefly, paraffin-embedded, formalin-fixed sections were incubated in Histoclear (National Diagnostics, Atlanta, GA) and graded a series of ethanol for rehydration. Sections were boiled in antigen retrieval buffer (citric acid buffer pH 6.0 or EDTA buffer pH 6.0, pH 8.0). Background Buster (Accurate Chemical & Scientific, Westbury, NY), goat anti-rabbit Fab fragment, and rabbit Fab fragment (Jackson ImmunoResearch Laboratories, West Grove, PA) were used to avoid nonspecific protein binding. Endogenous biotin activity was suppressed using an Avidin/Biotin Blocking Kit (Vector Laboratories, Burlingame, CA). Initial primary antibodies were incubated overnight at 4°C. In the case of double immunostaining, subsequent primary antibodies were incubated overnight at 4°C or for 3 h at room temperature. Secondary antibodies and streptavidin conjugates were incubated for 1 h at room temperature. The primary antibodies used in the study are summarized in Supplemental Table S1. Diaminobenzidine (Sigma Aldrich, St. Louis, MO), with or without 8% nickel chloride, was precipitated to visualize immunoperoxidase staining.
Microscopy and Imaging
Kidney sections were analyzed under an EVOS FL Cell Imaging System (Life Technologies) using the ×20 objective. Quantification of p57, podocin, Wilms’ tumor 1 (WT-1), vascular endothelial growth factor A (VEGFa), synaptopodin, p16, p21, p19, and senescence-associated-β-galactosidase (SA-β-gal) staining was performed using ImageJ 1.46r software (National Institutes of Health). A minimum of 40 glomeruli were assessed for quantification.
Podocyte density was determined through p57 nuclear staining. The calculation involved counting the number of p57-positive cells in a single glomerulus and dividing it by the measured glomerular tuft area. The values were then adjusted by referring to Venkatareddy et al.’s (34) methods. The final podocyte density was expressed as podocytes per total tuft volume (pods/×106 μm3). Once the podocyte densities were calculated, the values were averaged (34).
SA-β-Galactosidase Activity Staining
Frozen sections (Figs. 1C and 4C, 10 μm thick) were warmed from −80°C to room temperature and allowed to air dry. Sections were then treated in accordance with the manufacturer’s protocol for the SA-β-gal staining kit (Cell Signaling Technology, 9860) as previously described (33).
In Situ Hybridization
Cdkn2a (p16 INK4A, variant 2, and p19 ARF, variant 1) mRNA expression was determined by RNAscope 2.0 following Advanced Cell Diagnostics (ACD, Newark, CA) kit instructions. In brief, freshly cut 4-µm formalin-fixed paraffin-embedded (FFPE) (Figs. 1C and 4C) slides were baked for 1 h at 60°C. Samples were deparaffinized and pretreated. Target probes for Cdkn2a (p16 INK4A, variant 2, and p19 ARF, variant 1) were hybridized. The signal was amplified and developed. Sections were dehydrated through graded ethanol (50%, 70%, and 100%) and xylene and then mounted with Cytoseal XYL (Thermo Fisher Scientific Inc., Waltham, MA). Staining data were recorded according to the presence of punctate nuclear and cytoplasmic staining and signal intensity.
Statistics
Statistical analysis was performed using GraphPad Prism 10.0 (GraphPad Software, Inc., San Diego, CA). t tests were performed when comparing only two groups of data, whereas repeated-measures one-way ANOVAs were performed when making multiple comparisons. A P value threshold of 0.05 was used to determine statistical significance and data are shown as the means ± SD. For the RNA-sequencing results, a P adjusted value threshold of 0.05 was used to determine statistical significance.
RESULTS
Acute Hypertension in Mice Is Accompanied by Senescence and a Senescent-Associated Secretory Phenotype in Podocytes
In control mice, the mean arterial blood pressure (MAP) increased by 3% from 4 to 5.5 mo of age (94 ± 12 vs. 97 ± 15 mmHg, P = 0.7162) (Supplemental Fig. S1A). To investigate the glomerular phenotype due to hypertension, a separate group of 4-mo-old mice were implanted with deoxycorticosterone acetate (DOCA) pellets and provided ad libitum access to saline water. Following the initiation of this regime, the MAP was assessed every other week, and the glomerular state was assessed at week 6 by bulk mRNA sequencing and immunofluorescence/histology analyses (Fig. 1, A–C). The average MAP was 95 ± 20 mmHg at 4 mo of age in this group of mice pre-DOCA implantation. Thereafter, the MAP increased by 24% at 4.5 mo (118 ± 23 mmHg, P = 0.009), by 44% at 5.0 mo (137 ± 42 mmHg, P = 0.0001), and by 22% at 5.5 mo (116 ± 21 mmHg, P = 0.09) (Supplemental Fig. S1B). As expected, the changes in MAP were paralleled by a decrease in renin-positive cells in the DOCA-treated animals (Supplemental Fig. S1, C–E).
Podocytes were isolated and processed for bulk mRNA sequencing to obtain an unbiased view of the transcriptional changes caused by hypertension. PCA analysis and a volcano plot comparing the two conditions (control vs. DOCA) identified dramatic differences (Fig. 1, D and E). Gene Set Enrichment Analysis (GSEA) examining the Hallmark gene sets showed that the highest pathways enriched in podocytes from DOCA-treated mice were related to inflammation, such as those characterizing the inflammatory response, allograft rejection, IL6-JAK-Stat3, interferon α/γ IL2-Stat5, and TNFa signaling (Fig. 1F). A heatmap depicting expression of the top 20 genes in the inflammatory response Hallmark pathway is shown in Fig. 1G. We recently reported that specific inflammatory pathways, i.e., programmed cell death protein 1 (PD-1) and NLR family pyrin domain containing 3 (NLRP3) inflammasome signaling, contribute to injury in middle-aged and aged podocytes (33, 35). In fact, the expression of the transcripts for programmed death ligand 1 (Cd274) and programmed death ligand 2 (Pdc1lg2) were increased 2.1- and 1.8-fold, respectively, in podocytes from DOCA-treated mice compared with control mice. Similarly, NLRP3 inflammasome pathway components, including Nlrp3 and Casp1, were increased by RNA and protein levels (Supplemental Fig. 2, A–G). Finally, upstream regulators of the NLRP3 inflammasome, Toll-like receptors (TLRs) such as Tlr4 (36), were increased in podocytes from DOCA-treated mice (Supplemental Fig. S2H). Together, these data demonstrate that podocytes from hypertensive mice acquire an extensive inflammatory phenotype.
Reduced Hallmark Pathways in DOCA-Treated Mice
Next, we investigated the GSEA pathways downregulated in podocytes upon DOCA treatment (Fig. 1H). Among these, oxidative phosphorylation was the most dysregulated Hallmark pathway with Uqcrh (ubiquinol-cytochrome c reductase hinge protein, 1.53-fold downregulated) and Ndufb3 (NADH: ubiquinone oxidoreductase subunit B3, 1.32-fold downregulated). Other notably downregulated pathways were Kirsten rat sarcoma virus (KRAS) signaling and apical surface signaling. The latter includes genes such as EPH receptor B4 (Ephb4, 2.0-fold downregulated), a signaling pathway reported to allow podocytes to survive transient capillary collapse during glomerular disease (37), actin filament-associated protein 1-like 2 (Afap1l2, 1.6-fold downregulated), ephrin A5 (Efna5, 2.3-fold downregulated), and GATA binding protein 3 (Gata3, 2.7-fold downregulated). Finally, the Wnt/β-catenin signaling Hallmark pathway, which includes genes like Jag1, Notch4, Hey2, Jag2, Fzd8, Hdac11, Dll1, and Gnai1, which are known to regulate several key downstream events required for normal podocyte function (38, 39), was downregulated. Together, these data show that several pathways and individual genes important for podocyte homeostasis were lowered in podocytes from hypertensive mice due to DOCA administration.
Podocytes Express Aging Genes in DOCA-Treated Mice
Activating inflammatory pathways such as PD-1 and NLRP3 inflammasome signaling is part of healthy podocyte aging (33, 35). We thus analyzed genes considered to represent an aged phenotype, i.e., the RODWELL_AGING_KIDNEY_UP gene list (40) (Table 1). Among the genes listed, many were statistically significantly increased in the podocytes of DOCA-treated mice. Examples included Mmp7 (27-fold higher), Fn1 (10.7-fold higher), Timp1 (10.2-fold higher), Vcam1 (4.7-fold higher), Lmnb1 (3.0-fold higher), Ccl2 (4.4-fold higher), Serpine1 (2.2-fold higher), and Nfkb1 (1.9-fold higher). Importantly, validation of the increase in Timp1 (Supplemental Fig. S3, A–C), Lmnb1 (Supplemental Fig. S3, D–F), Nfkb1 (Supplemental Fig. S3, G–I), Vcam1 (Supplemental Fig. S3, J–L), Mmp2 (Supplemental Fig. S3, M–O), and Tgfb1 (Supplemental Fig. S3, P–R) were confirmed at the protein level by immunostaining.
Table 1.
Rodwell_Aging_Kidney_Up in podocytes of DOCA mice vs. control mice
| RODWELL_AGING_KIDNEY UP Genes |
RODWELL_AGING_KIDNEY DOWN Genes |
||
|---|---|---|---|
| Gene Name | *Fold Change Higher in DOCA vs. Control | Gene Name | *Fold Change Lower in DOCA vs. Control |
| Mmp7 | 27.4 | Adra2a | −3.4 |
| Havcr1 | 22.5 | Cdo1 | −3.0 |
| Gpnmb | 19.4 | Spon2 | −2.9 |
| Fn1 | 10.7 | Fzd3 | −2.9 |
| Alpk2 | 10.3 | Gabre | −2.5 |
| Timp1 | 10.2 | Nr2f1 | −2.5 |
| Col3a1 | 9.4 | Rarres1 | −2.4 |
| Adam8 | 7.2 | Tesc | −2.3 |
| Vcan | 7.1 | Lix1 | −2.3 |
| Inhba | 7.0 | Erich3 | −2.2 |
| Msr1 | 6.6 | Gfra1 | −2.2 |
| Col1a1 | 6.4 | Tspan18 | −2.2 |
| C7 | 5.6 | Gpx8 | −2.0 |
| Mrc1 | 5.2 | Vtcn1 | −2.0 |
| Ms4a7 | 5.1 | Tspan1 | −2.0 |
| Vcam1 | 4.8 | Slc1a3 | −2.0 |
| Ccl2 | 4.4 | Rbms3 | −2.0 |
| Col14a1 | 4.3 | Kcnd3 | −2.0 |
| Slc15a3 | 4.3 | Rflnb | −1.9 |
| Clec7a | 4.2 | Prickle1 | −1.9 |
| Ccr1 | 4.0 | Slc18a2 | −1.9 |
| Csf1r | 3.9 | Adgrg1 | −1.8 |
| C1qc | 3.9 | Aebp1 | −1.8 |
| Mpeg1 | 3.9 | Cdh6 | −1.8 |
| C1qa | 3.9 | Wfdc2 | −1.8 |
| Cd14 | 3.8 | Arvcf | −1.7 |
| Nnmt | 3.7 | Shank2 | −1.7 |
| Lcp2 | 3.7 | Itga1 | −1.7 |
| Rad54l | 3.6 | Pam | −1.6 |
| Myo1f | 3.6 | Asns | −1.6 |
| Tgfbi | 3.6 | Rab34 | −1.6 |
| Hgf | 3.6 | Nt5e | −1.5 |
| Jchain | 3.5 | Pxdn | −1.5 |
| Gbp5 | 3.5 | Prom1 | −1.5 |
| C1qb | 3.5 | Mcam | −1.5 |
| Cxcl14 | 3.5 | ||
| Cx3cr1 | 3.4 | ||
| Itgb2 | 3.4 | ||
| Rac2 | 3.3 | ||
| Lacc1 | 3.3 | ||
| Axl | 3.3 | ||
| Ncf1 | 3.2 | ||
| Fcer1g | 3.2 | ||
| Tbxas1 | 3.2 | ||
| Laptm5 | 3.2 | ||
| Lst1 | 3.2 | ||
| Grn | 3.2 | ||
| Stab1 | 3.2 | ||
| Tyrobp | 3.1 | ||
| Slamf8 | 3.1 | ||
| Rgs10 | 3.1 | ||
| Pik3cg | 3.1 | ||
| Runx3 | 3.1 | ||
| Apbb1ip | 3.1 | ||
| Dock2 | 3.0 | ||
| Gng2 | 3.0 | ||
| Ptpre | 3.0 | ||
| Rgs1 | 3.0 | ||
| Milr1 | 3.0 | ||
| Arhgap30 | 3.0 | ||
| Evi2a | 3.0 | ||
| Cxcl16 | 2.9 | ||
| Cxcl1 | 2.9 | ||
| Rnf213 | 2.9 | ||
| Parp14 | 2.9 | ||
| Celf2 | 2.9 | ||
| Arl4c | 2.9 | ||
| Ifitm1 | 2.8 | ||
| Hcst | 2.8 | ||
| Nlrc5 | 2.8 | ||
| Sting1 | 2.8 | ||
| Ptprc | 2.8 | ||
| Vsir | 2.8 | ||
| Loxl1 | 2.8 | ||
| Themis2 | 2.7 | ||
| Hcls1 | 2.7 | ||
| Zeb2 | 2.7 | ||
| Nfkbiz | 2.7 | ||
| Calhm6 | 2.7 | ||
| Rab8b | 2.7 | ||
| Pik3cd | 2.7 | ||
| Cd53 | 2.7 | ||
| Myof | 2.7 | ||
| Ikzf1 | 2.7 | ||
| Parvg | 2.6 | ||
| Antxr2 | 2.6 | ||
| Prex1 | 2.6 | ||
| Samhd1 | 2.6 | ||
| Birc3 | 2.6 | ||
| Man2b1 | 2.6 | ||
| Cxcr4 | 2.6 | ||
| Trafd1 | 2.6 | ||
| Ppp1r18 | 2.6 | ||
| Selplg | 2.6 | ||
| Fnbp1 | 2.6 | ||
| Igkc | 2.6 | ||
| Arpc1b | 2.5 | ||
| Cmtm3 | 2.5 | ||
| Arhgef6 | 2.5 | ||
| Coro7 | 2.5 | ||
| Il10ra | 2.5 | ||
| Lyn | 2.5 | ||
| Ehbp1l1 | 2.5 | ||
| Ptpn6 | 2.5 | ||
| Coro1a | 2.4 | ||
| Akna | 2.3 | ||
| Ifnar2 | 2.3 | ||
| Tap1 | 2.3 | ||
| Psmb8 | 2.3 | ||
| Casp1 | 2.3 | ||
| Jaml | 2.3 | ||
| Psmb9 | 2.3 | ||
| Dtx3l | 2.2 | ||
| Irf8 | 2.2 | ||
| Slc12a9 | 2.2 | ||
| Arhgap25 | 2.2 | ||
| Ccdc88a | 2.2 | ||
| Dok3 | 2.2 | ||
| Marchf1 | 2.1 | ||
| Fxyd5 | 2.1 | ||
| Clec10a | 2.1 | ||
| Calhm2 | 2.1 | ||
| Dok1 | 2.1 | ||
| Gmfg | 2.1 | ||
| Stk10 | 2.1 | ||
| Srgn | 2.1 | ||
| Vim | 2.1 | ||
| Arhgdib | 2.1 | ||
| Timp2 | 2.1 | ||
| Rps6ka1 | 2.1 | ||
| Rgs19 | 2.1 | ||
| Dock11 | 2.1 | ||
| Cybc1 | 2.0 | ||
| Pgm2l1 | 2.0 | ||
| Emp3 | 2.0 | ||
| Bhlhe41 | 2.0 | ||
| Mthfd1l | 2.0 | ||
| Ggta1 | 2.0 | ||
| Ifnar1 | 2.0 | ||
| Rtn1 | 2.0 | ||
| Acer3 | 2.0 | ||
| Glipr2 | 2.0 | ||
| Irak2 | 1.9 | ||
| Rassf5 | 1.9 | ||
| Dkk3 | 1.9 | ||
| Rab31 | 1.9 | ||
| Ptpn22 | 1.9 | ||
| Trex1 | 1.9 | ||
| Rin3 | 1.9 | ||
| Arhgap45 | 1.9 | ||
| Tpm3 | 1.8 | ||
| Spon1 | 1.8 | ||
| Rnase6 | 1.8 | ||
| Plekho1 | 1.8 | ||
| Lpcat1 | 1.8 | ||
| Cmtm7 | 1.8 | ||
| Pald1 | 1.8 | ||
| Tmsb10 | 1.8 | ||
| St8sia4 | 1.8 | ||
| Plod3 | 1.7 | ||
| Pfn1 | 1.7 | ||
| Zcchc24 | 1.7 | ||
| Parp12 | 1.7 | ||
| Rftn1 | 1.7 | ||
| Stat6 | 1.7 | ||
| Coro1c | 1.7 | ||
| Fkbp15 | 1.7 | ||
| Syngr2 | 1.7 | ||
| Reep4 | 1.6 | ||
| Itm2c | 1.6 | ||
| Tbc1d2b | 1.6 | ||
| Tapbp | 1.6 | ||
| Rpl12 | 1.6 | ||
| Fhod1 | 1.6 | ||
| Clic1 | 1.6 | ||
| Rpl10 | 1.6 | ||
| Atp8b2 | 1.6 | ||
| Rpl27 | 1.5 | ||
| Ikbkb | 1.5 | ||
| Mvp | 1.5 | ||
| Crip1 | 1.5 | ||
| Cd47 | 1.5 | ||
| Ccm2 | 1.5 | ||
| Psme2 | 1.5 | ||
| Serping1 | 1.5 | ||
| Rnf166 | 1.5 | ||
| Il17ra | 1.5 | ||
| Ppm1m | 1.5 | ||
DOCA, deoxycorticosterone acetate. *Defined as >1.5-fold higher with adjusted P value <0.05.
A similar trend was observed when transcripts for senescent-inducing genes, senescent markers, and the Reactome cellular senescence gene set were interrogated (Table 1). Compared with control mice, podocytes from DOCA-treated mice showed increased Cdkn1a/p21 (1.66-fold), Cdkn2a/p16 INK4A (5.36-fold), and SerpinB2 (8-fold). qRT-PCR validates this for p16 INK4A (Cdkn2a, variant 2), p19 ARF (Cdkn2a, variant 1), and Cdkn1a (p21) (Fig. 2A) and by in situ hybridization for p16 INK4A (Cdkn2a, variant 2) and p19 ARF (Cdkn2d, variant 1) (Fig. 2, B–E) in mice given DOCA. Moreover, immunostaining showed elevated protein levels for p16 INK4A (Cdkn2a, variant 2), p19 ARF (Cdkn2a, variant 1), and p21 (Cdkn1a) in DOCA-treated mice compared with age-matched controls (Fig. 2, F–N). Finally, glomerular staining for the senescent marker SA-β-gal was higher in DOCA-treated mice than in age-matched control mice (Fig. 2, O–Q).
Figure 2.

Age- and stress-associated senescent genes are increased in podocytes from young mice following deoxycorticosterone acetate (DOCA) treatment. A: qRT-PCR on isolated podocytes shows that the expression for Cdkn2a (p16 INK4A, variant 2), Cdkn2a (p19 ARF, variant 1), and Cdkn1a (p21) are higher in young DOCA-treated mice. B–E: in situ hybridization (ISH). ISH (brown color) was increased for Cdkn2a (p16 INK4A, variant 2) (B and C) and Cdkn2a (p19 ARF, variant 1) (D and E) in DOCA-treated mice. F–N: representative immunoperoxidase immunostaining (brown) and quantitation. Immunostaining validated the increases in p16 (F–H), p19 (I–K), and p21 (L–N). The senescent marker SA-β-galactosidase (SA-β-gal, blue) was higher in DOCA-treated mice (O–Q). Scale bar: 40 μm.
Like markers of senescence, analysis of genes/gene sets representing a senescent-associated secretory phenotype (SASP) were detected in DOCA-treated mice (Table 2). These included upregulated Timp1 (10.2-fold) and Tgfb1(3.62-fold), which were also validated at the protein level by immunostaining (Supplemental Fig. S3, A–C and P–R). Analysis of genes of the SASP Reactome and the Fridman senescence genes were also enriched in podocytes of mice treated with DOCA (Table 3) and included ubiquitin-conjugating enzyme E2C (Ube2c, increased 6-fold) that decreases mitotic cyclins thereby limiting cell cycle progression (41) and interleukin 1A (Il1a, increased 5.6-fold), which increases in podocytes in human glomerulonephritis (42). Finally, we also observed the enrichment of gene sets of the Tp53 and DNA repair pathways (Supplemental Fig. S4, A–D).
Table 2.
Senescent-associated secretory phenotype in DOCA-treated mice
| SASP Gene Set Identified in Ref. 27 |
Reactome_Senescence_Associated_Secretory_Phenotype_SASP |
||||
|---|---|---|---|---|---|
| Up Genes |
Down Genes |
||||
| Gene Name | *Fold Change | Gene Name | *Fold Change in DOCA vs. Control | Gene Name | *Fold Change in DOCA vs. Control |
| Timp1 | 10.2 | Ube2c | 6.1 | H3c15 | −1.8 |
| Igfbp2 | 6.6 | Il1a | 5.6 | Igfbp7 | −1.6 |
| Ccl2 | 4.4 | Cdkn2a | 5.4 | ||
| Cxcl1 | 3.0 | H4c11 | 4.1 | ||
| Irf5 | 2.7 | Il6 | 3.1 | ||
| Mmp12 | 2.6 | Ccna2 | 2.9 | ||
| Cebpb | 2.5 | Cebpb | 2.5 | ||
| Timp2 | 2.1 | Cdk6 | 2.5 | ||
| Ccl3 | 2.0 | Rps6ka1 | 2.1 | ||
| Jdp2 | 1.9 | Nfkb1 | 1.9 | ||
| Ccl5 | 1.5 | H4c9 | 1.9 | ||
| Mmp13 | 1.3 | Stat3 | 1.8 | ||
| Mmp2 | 1.3 | Cdkn1a | 1.7 | ||
| Igfbp3 | 1.1 | Rela | 1.7 | ||
| Hmgb1 | 1.1 | Cdkn2c | 1.6 | ||
| Igfbp4 | −1.1 | Cdk2 | 1.5 | ||
| Igfbp7 | −1.6 | ||||
| Igfbp5 | −2.6 | ||||
| Jchain | 3.5 | ||||
DOCA, deoxycorticosterone acetate; SASP, senescent-associated secretory phenotype. *Defined as >1.5-fold higher with adjusted P value <0.05.
Table 3.
Senescent genes in podocytes of DOCA-treated mice
|
Reactome_Cellular_Senescence
|
Fridman Senescence Genes
|
||||
|---|---|---|---|---|---|
| Up Genes |
Down Genes |
||||
| Gene Name | *Fold Change in DOCA vs. Control | Gene Name | *Fold Change in DOCA vs. Control | Gene Name | *Fold Change in DOCA vs. Control |
| Ube2c | 6.1 | Mapk10 | −3.9 | Fn1 | 10.7 |
| Il1a | 5.6 | Id1 | −1.9 | Serpinb2 | 8.1 |
| Cdkn2a | 5.4 | H3c15 | −1.9 | Igfbp2 | 6.6 |
| H4c11 | 4.1 | Mapk11 | −1.8 | Cdkn2a | 5.4 |
| Ccne1 | 3.8 | Igfbp7 | −1.6 | Cxcl14 | 3.5 |
| Ccne2 | 3.4 | Cbx8 | −1.5 | Cd44 | 3.4 |
| Il6 | 3.1 | Tfdp2 | −1.5 | Tnfaip3 | 3.3 |
| Lmnb1 | 3.0 | Igfbp6 | 2.8 | ||
| Ccna2 | 2.9 | Irf7 | 2.8 | ||
| Kdm6b | 2.8 | Irf5 | 2.7 | ||
| Cebpb | 2.5 | Isg15 | 2.3 | ||
| Cdk6 | 2.5 | Stat1 | 2.3 | ||
| Ets2 | 2.4 | Thbs1 | 2.3 | ||
| Ago3 | 2.2 | Serpine1 | 2.2 | ||
| E2f1 | 2.2 | Vim | 2.1 | ||
| Rps6ka1 | 2.1 | Cyp1b1 | 1.9 | ||
| Nfkb1 | 1.9 | Filip1l | 1.9 | ||
| Mapkapk2 | 1.9 | Rab31 | 1.9 | ||
| H4c9 | 1.9 | Creg1 | 1.9 | ||
| Mapkapk3 | 1.8 | Cdkn1a | 1.7 | ||
| Mapk14 | 1.8 | Rgl2 | 1.6 | ||
| Stat3 | 1.8 | Hps5 | 1.5 | ||
| Tinf2 | 1.7 | Mdm2 | 1.5 | ||
| Ezh2 | 1.7 | ||||
| Hmga1 | 1.7 | ||||
| Ubn1 | 1.7 | ||||
| Cdkn1a | 1.7 | ||||
| Rela | 1.7 | ||||
| Rad50 | 1.7 | ||||
| Eed | 1.6 | ||||
| Cdkn2c | 1.6 | ||||
| Ep400 | 1.5 | ||||
| Mdm2 | 1.5 | ||||
| Ago4 | 1.5 | ||||
| Cdk2 | 1.5 | ||||
| Mdm4 | 1.5 | ||||
DOCA, deoxycorticosterone acetate. *Defined as >1.5-fold higher with adjusted P value <0.05.
Together, these results show that podocytes from young mice with hypertension resulting from DOCA administration showed hallmarks for premature aging and the acquisition of a senescent and a SASP phenotype. Importantly, this includes the expression of aging-inducing senescent genes (p16 and p19), stress-inducing senescent genes (p21 and p53), and an enriched DNA repair pathway.
Hypertension Decreases the Expression of Podocyte Canonical Genes and Podocyte Number
Besides the upregulation of inflammatory and senescent pathways, aging also results in a marked reduction of canonical podocyte genes (27, 33, 35, 43). Indeed, many canonical podocyte genes such as Podxl, Synpo, Nphs2, Vegfa, and Tpj1 were downregulated in podocytes of DOCA-treated mice compared with controls (Fig. 3A). Interestingly, the podocyte gene Ptpro/Glepp-1 was 1.6-fold higher in DOCA-treated mice, suggesting that injury may induce its expression (Fig. 3A). Importantly, these changes were confirmed by immunostaining for nephrin (Fig. 3, B–D), podocin (Fig. 3, E–G), synaptopodin (Fig. 3, H–J), Wilms’ tumor 1 (WT-1, Fig. 3, K–M), and vascular endothelial growth factor A (VEGFa, Fig. 3, N–P). Notably, these changes had physiological consequences as podocyte density, quantitated by immunostaining for the podocyte marker p57 (Fig. 3, Q–S) was significantly lower in DOCA-treated mice compared with control mice (329 vs. 223 p57+ cells/106 μm3), P = 0.04). Finally, immunostaining for the endothelial cell marker ERG (Supplemental Fig. S4, E and F) and the activated parietal epithelial cell marker CD44 (Supplemental Fig. S4, G and H) did not change in mice given DOCA. These results show that 6 wk of DOCA-induced hypertension in mice causes podocyte damage, including lower canonical gene expression, reduced podocyte density, and reduced expression of both canonical podocyte genes as well as genes coding for proteins key to glomerular function, such as Vegfa.
Figure 3.

Deoxycorticosterone acetate (DOCA) treatment lowers the expression of canonical podocyte proteins. A: RNA sequencing results. The transcript levels for individual podocyte genes decreases in DOCA-treated mice compared with controls. B–S: representative immunoperoxidase immunostaining (brown) for individual podocyte proteins and accompanying graph showing their quantitation. In DOCA-treated mice, there was a decrease in immunostaining nephrin (B–D), podocin (E–G), synaptopodin (H–J), Wilms’ tumor 1 (WT-1; K–M), vascular endothelial growth factor A (VEGFa; N–P), and p57 (Q–S). Scale bar: 40 μm.
High-Fat Diet Induces an Inflammatory Podocyte Phenotype
Next, we compared the impact of hypertension on podocyte health with the effects of obesity by providing 4-mo-old mice with an ad libitum HFD for 12 wk. Control mice fed a normal diet for 12 wk had an initial average body weight of 29.9 ± 1.7 g at 4 mo of age that increased by 17% to 35.1 ± 1.3 g (P < 0.0001) (Supplemental Fig. S5A). For reference, 6-mo-old male C57BL/6J mice had average body weight of 30.7 ± 2.0 g, according to The Jackson Laboratory Mouse Phenome Database (44). In the separate group of mice given a HFD, their initial weights at 4 mo of age were 30.4 ± 2.4 g. HFD-fed mice gained an average weekly increase of 3.9 ± 2.2% in body weight ending at a final average weight of 48.5 ± 6.5 g or a 58.4% increase from their initial weight (P < 0.0001) (Supplemental Fig. S5B). Furthermore, oil red O staining demonstrated increased lipid accumulation in the glomeruli of HFD-fed mice (Supplemental Fig. S5, C and D).
As with DOCA-treated mice, we isolated podocytes and performed an unbiased analysis of transcriptomic changes comparing podocytes from obese versus non-obese mice (Fig. 4B). PCA analysis and volcano plot comparing the two conditions (control vs. HFD) again identified dramatic differences (Fig. 4D). Moreover, there was no overlap between mice on HFD and mice treated with DOCA (Fig. 4D). The volcano plot shows genes positively and negatively enriched by HFD (Fig. 4E). Hallmark pathway GSEA analysis showed that several inflammatory-associated pathways were enriched in podocytes from HFD mice, such as allograft rejection, IL-6-JAK-Stat signaling, inflammatory response, IFN-γ and IFN-α, reactive oxygen species, IL-2-Stat5, p53 pathway (Fig. 4, F and H), and epithelial-mesenchymal transition (Fig. 4F and Supplemental Fig. S6, A and B). Conversely, Hallmark pathways downregulated in podocytes from obese mice included apical proteins, KRAS signaling, cholesterol homeostasis, and oxidative phosphorylation (Fig. 4G and Supplemental Fig. S6, C and D). Noteworthy increases were genes of the NLRP3 inflammasome (e.g., Nlrp3 and its downstream mediator Casp1) and the PD-1 signaling pathway (e.g., Cd274, Pdcd1lg2, and Pdcd1) (Fig. 4I).
A Senescent and Aged Phenotype Accompanies High-Fat Diet-Induced Obesity in Mice
Next, we investigated whether obesity results in premature aging and senescence in mice on a 12-wk HFD. Analysis of the FRIDMAN_SENESCENCE_UP (45), cellular senescence Reactome (46), and RODWELL_AGING_KIDNEY_UP (40) gene sets were enriched in obese versus nonobese mice (Table 4). Examples include increased C-C motif chemokine ligand 5 (Ccl5), glycoprotein nonmetastatic melanoma protein b (Gpnmb), a marker of tubular injury (47), and stimulator of interferon genes (Sting1), a gene that has a key role in APOL1-associated podocytopathy (48).
Table 4.
Senescent and aging genes in podocytes of HFD mice
| Gene Name | *Fold Change in HFD vs. Controls | |
|---|---|---|
| FRIDMAN_SENESCENCE_UP Gene List | Hsp5 | 1.5 |
| Optn | −1.5 | |
| Senescence Reactome | Mdm4 | 1.7 |
| Mapkl4 | 1.6 | |
| Eed | 1.6 | |
| Mink1 | 1.5 | |
| RODWELL_AGING_KIDNEY_UP Gene List | C7 | 7.0 |
| Gpnmb | 5.7 | |
| Jchain | 4.9 | |
| Igkc | 4.4 | |
| Ms4a7 | 4.3 | |
| Lcp2 | 3.2 | |
| Slc15a3 | 2.9 | |
| Mpeg1 | 2.9 | |
| Ccl5 | 2.7 | |
| Col27a1 | 2.7 | |
| Vcam1 | 2.7 | |
| Cxcl16 | 2.5 | |
| Grn | 2.3 | |
| Birc3 | 2.3 | |
| Coro7 | 2.2 | |
| Arhgef6 | 2.2 | |
| Irak2 | 2.1 | |
| Sting1 | 2.0 | |
| Man2b1 | 1.9 | |
| Prag1 | 1.6 | |
| Syngr2 | 1.6 | |
| Coro1c | 1.5 | |
| Zcchc7 | 1.5 | |
| Dlgap1 | −3.8 | |
| Lix1 | −3.0 | |
| Tspan1 | −2.3 | |
| Tesc | −2.2 | |
| Wfdc2 | −2.1 | |
| Tspan18 | −1.9 | |
| Cdo1 | −1.8 | |
| Gpx8 | −1.7 | |
| Aebp1 | −1.7 |
HFD, high-fat diet.
*Defined as >1.5-fold and P value < 0.05.
In contrast to DOCA-treated hypertensive mice, the aging and senescence phenotype was not accompanied by statistically significant changes in the age-inducing senescent genes Cdkn2a, variant 2 (p16 INK4A), or Cdkn2a, variant 1 (p19 ARF), as measured by qRT-PCR (Fig. 5A), in situ hybridization (Fig. 5, B–E), or immunostaining (Fig. 5, F–K). However, immunostaining for the DNA repair pathway component Cdkn1a (p21) was increased (Fig. 5, L–N) as well as glomerular staining for the senescent marker SA-β-gal (Fig. 5, O–Q) in mice fed HFD compared with age-matched mice on a normal diet. Together, these results show that in obese mice, podocytes express markers consistent with a stress-induced senescence (p21, p53, and SA-β-gal-positive) phenotype.
Figure 5.

High-fat diet (HFD) induced obesity in young mice is accompanied by stress-induced senescence. A: qRT-PCR on isolated podocytes. There were no differences in the expression for Cdkn2a (p16 INK4A, variant 2), Cdkn2a (p19 ARF, variant 1), and Cdkn1a (p21) between control and HFD mice. B–E: in situ hybridization. Neither Cdkn2a (p16 INK4A, variant 2) (B and C) nor Cdkn2a (p19 ARF, variant 1) (D and E) increased in glomeruli of HFD mice. F–N: immunoperoxidase staining (brown) and quantitation. Immunostaining for p16 (F–H) and p19 (I–K) did not change in mice given a HFD. In contrast, staining for p21 was higher in HFD-treated mice, accompanied by an increase in the senescent maker SA-β-galactosidase (SA-β-gal) (blue) (O–Q). Scale bar: 40 μm.
High-Fat Diet Lowers Podocyte Canonical Gene Expression and Podocyte Density
Finally, we compared canonical podocyte gene expression between obese and nonobese mice. Indeed, the RNA-sequencing data demonstrated reduced transcript levels of many canonical genes such as Nphs2, Wt1, and Synpo (Fig. 6A). This was accompanied by reduced immunostaining for nephrin, podocin, synaptopodin, and VEGFa (Fig. 6, B–M). Interestingly, similar to DOCA-treated mice, Ptpro/Glepp-1 was increased in HFD-fed mice compared with controls and was also accompanied by increased levels of transient receptor potential cation channel 6 (Trpc6) (Fig. 6A). Finally, the density of podocytes was significantly lower in mice on a HFD diet compared with normal diet-fed mice (398 ± 32 vs. 302 ± 14 p57+ cells/106 μm3, P = 0.008) (Fig. 6, N–P).
Figure 6.

Canonical podocyte genes are lower in obese mice fed a high-fat diet (HFD). A: RNA-sequencing. Table shows fold change decreases in gene expression for canonical podocyte genes in HFD-fed mice compared with controls. B–P: immunoperoxidase staining (brown) and quantitation. Compared with controls staining was lower in HFD-fed mice for nephrin (B–D), podocin (E–G), synaptopodin (H and J), VEGFa (K–M), and p57 (N–P).
DISCUSSION
DOCA and HFD treatments are well-established translational mouse models to study the pathophysiology and mechanisms of hypertension and obesity, respectively. Indeed, studies have shown that, like humans, they cause podocyte injury (12–14, 20, 23, 49). Our results confirmed the presence of podocyte injury in both models. This included a decrease in podocyte density, a reduction in the expression of key canonical podocyte genes and reduced expression of genes encoding proteins that are essential for glomerular maintenance and function, e.g., Vegfa. Nevertheless, the current study now extends on these findings. To our knowledge, we show for the first time that in young mice, a short duration of hypertension following DOCA administration for 6 wk or obesity upon feeding a HFD for 12 wk leads to the de novo expression of markers of inflammation, senescence, and aging and the establishment of a SASP in podocytes. To better understand the transcriptional changes associated with these phenotypes, we performed unbiased bulk RNA sequencing. In addition to the induction of genes representing senescence, a SASP, and aging, we observed a differential profile of senescence-inducing genes. Although podocytes from young mice treated with DOCA-salt exhibited expression of age-inducing senescent genes Cdkn2a (p16 INK4A, variant 2) and Cdkn2a (p19 ARF, variant 1), as well as stress-inducing senescent gene Cdkn1a (p21) and the Tp53 and DNA damage repair pathways, mice that developed obesity by feeding HFD showed increased expression/activation of Cdkn1a (p21), as well as the Tp53 and DNA damage repair pathways, but not age-inducing senescent genes Cdkn2a (p16 INK4A, variant 2) and Cdkn2a (p19 ARF, variant 1). In the context of an increase in the senescence marker SA-β-gal in young mice with hypertension or obesity, the increase in these senescent inducing genes in young mice suggests that they are likely causing the senescent/aged phenotype in podocytes.
Senescent proteins p16 and p19 typically cause cellular senescence in healthy aging (50). In fact, we and others have reported an increase in p16 and p19 in middle-aged and aged glomerular cells, including podocytes, and in aged tubular epithelial cells (27, 33, 35, 51, 52). More recently, we reported an increase in p16 following acute podocyte injury in an experimental model of FSGS (53). Conversely, p21 and p53 typically cause stress-induced senescence (but can contribute to age-associated senescence). Studies have shown expression of p21 and p53 in aged podocytes (27, 52) and p21 and p53 are also increased following stress-induced senescence in podocytes (54, 55), including podocytes injured by complement (56). In podocytes, p21 is required for TGF-β1-induced apoptosis (57) and limits proliferation in cultured podocytes exposed to mechanical stretch (58). In contrast, to a recent study (55), the stress-induced podocytes in the current study did not exhibit changes in the expression of HDAC1 and 2. Thus, our results are consistent with experimental hypertension and obesity being associated with the de novo development of a podocyte senescent and aged phenotype in mice. Yet, there are differences between hypertension and obesity-associated podocyte senescence. Both are stress-induced, but the one in hypertensive mice is superimposed by age-associated senescence form. The consequences of this potentially accelerated podocyte/glomerular aging during hypertension or obesity needs further study.
It is important to note that the connection between obesity and senescence has been reported in other organs. For example, obesity induces senescence in the glial cells of the lateral ventricle in mice (59) and increases p16 and p21 levels in the livers of rats (60). Moreover, cellular senescence has been implicated as a causal factor in obesity-related inflammation (61). In mice fed a HFD that developed obesity, renal tubular cells began to express the senescent markers p16 and p53 that were accompanied by a SASP phenotype (62).
It is interesting to note that endothelial senescence has been implicated in the pathogenesis of systemic hypertension (for a review, see Ref. 63) and pulmonary hypertension (64). Surprisingly, no published data show that systemic hypertension causes endothelial senescence. Similarly, we did not detect changes in the glomerular endothelial marker gene expression in the hypertensive mice in the current study.
As expected for a podocyte injury model, podocyte density was reduced in both mouse models compared with their age-matched controls. Although we have not directly investigated the underlying mechanism, several possible mechanisms for podocyte loss include apoptosis, pyroptosis, and p53 pathways, all of which were increased in our two mouse models. Among the highest enriched GSEA pathways in podocytes from hypertensive and obese mice were several related to inflammation. Increased NLRP3 activity has been reported in podocytes following HFD feeding (23). We have recently reported on the importance of PD-1 and NLRP3-inflammasome signaling (33, 35). Both pathways are increased in middle-aged and aged podocytes and under experimental FSGS conditions (53). Moreover, inhibiting either of these two forms of intracellular inflammation in middle-aged mice reduced the senescence/aged phenotype. Importantly, this led to a longer podocyte lifespan and improved health span.
In addition, the GSEA identified that the reactive oxygen species gene set is enriched, and the oxidative phosphorylation gene set is depleted in podocytes of obese mice. This agrees with the observation that oxidative stress has been reported in podocytes in mice with HFD (49). Moreover, administering the mitochondrial stabilizer SS-31 prevented the loss of podocytes in mice fed HFD, suggesting abnormal mitochondrial bioenergetics contribute to podocyte dysfunction/loss in obese mice (65). Finally, several canonical podocyte genes were decreased in DOCA-treated and HFD-fed mice. These included structural podocyte proteins such as nephrin, podocin, and synaptopodin, as well as VEGFa, which is often used as a proxy for podocyte function (66). It is interesting that podocyte damage preceded any damage to glomerular endothelial cells or activation of parietal epithelial cells within the glomerulus, as markers for these were not changed.
We acknowledge several limitations of the study. The study aimed to determine how these common clinical stressors (hypertension and obesity) impact podocytes qualitatively, but not to compare their injury responses quantitatively. Although we did not provide mechanisms for the inflammation/senescent/aging changes in injured podocytes, nor the other transcriptional changes, this body of work provides a wealth of information for future studies, including several candidate genes that may warrant in-depth analysis. For example, Mmp7, a downstream target of β-catenin, has been previously shown to be secreted from tubules, decrease nephrin levels, and increase proteinuria (67). Similarly, Prg4, a ligand for CD44 (68), is increased in parietal epithelial cells following podocyte injury and contributes to the fibrotic response during glomerular injury (69). Strain considerations are always important, as has been reported in mice on a HFD (70). Finally, our study was limited to male mice. Male mice were chosen for this study as hypertension and renal disease tend to be less severe in female mice due to the protective effects of estrogen (71). Comparable future studies in female mice should be considered. Although we have not been able to detect sex differences in podocytes during aging (27), the emerging data on sex-specific immune responses in a wide range of diseases warrants a similar comparative analysis for podocyte injury.
In summary, this study provides tantalizing data on how a short period of hypertension or obesity can dramatically alter podocyte biology. In particular, the long-term implications and consequences of premature podocyte senescence and aging are a so far unrealized paradigm of glomerular biology. However, they will likely be a critical aspect to understand kidney health in the future.
DATA AVAILABILITY
The raw data supporting the conclusions of this article will be made available upon reasonable request.
SUPPLEMENTAL DATA
Supplemental Figs. S1–S6 and Supplemental Table S1: https://doi.org/10.6084/m9.figshare.25236832.v1.
GRANTS
S.J.S. and O.W. were supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants 5R01DK056799-10, 5R01DK056799-12, 1R01DK097598-01A1, UC2DK126006-2, and 1R01DK090358-12 and by Department of Defense Grant DOD PR180585/PR180585P1. T.C. was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants R01DK127634 and RC2DK125960 and by Cancer Prevention and Research Institute of Texas Grant RP220201.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
D.G.E., J.W.P., O.W., and S.J.S. conceived and designed research; S.R.M., N.K., R.A.S., C.C., D.G.E., and J.W.P. performed experiments; S.R.M., N.K., R.A.S., C.C., D.G.E., B.M.V.P., B.K., J.W.P., O.W., and S.J.S. analyzed data; S.R.M., N.K., R.A.S., C.C., D.G.E., J.W.P., O.W., and S.J.S. interpreted results of experiments; S.R.M., N.K., D.G.E., J.W.P., and S.J.S. prepared figures; S.R.M., N.K., J.W.P., O.W., and S.J.S. drafted manuscript; S.R.M., N.K., D.G.E., J.W.P., O.W., and S.J.S. edited and revised manuscript; S.R.M., N.K., J.W.P., O.W., and S.J.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the Histology and Imaging Core at the University of Washington for imaging assistance. Diagrams were created with BioRender.com.
REFERENCES
- 1. Muntner P, Miles MA, Jaeger BC, Hannon Iii L, Hardy ST, Ostchega Y, Wozniak G, Schwartz JE. Blood pressure control among US adults, 2009 to 2012 through 2017 to 2020. Hypertension 79: 1971–1980, 2022. doi: 10.1161/HYPERTENSIONAHA.122.19222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Engin A. The definition and prevalence of obesity and metabolic syndrome. Adv Exp Med Biol 960: 1–17, 2017. doi: 10.1007/978-3-319-48382-5_1. [DOI] [PubMed] [Google Scholar]
- 3. Kohagura K. The public health impact of hypertension and diabetes: a powerful tag team for the development of chronic kidney disease. Hypertens Res 46: 339–340, 2023. doi: 10.1038/s41440-022-01114-9. [DOI] [PubMed] [Google Scholar]
- 4. Bidani AK, Griffin KA. Pathophysiology of hypertensive renal damage: implications for therapy. Hypertension 44: 595–601, 2004. doi: 10.1161/01.HYP.0000145180.38707.84. [DOI] [PubMed] [Google Scholar]
- 5. Griffin KA, Bidani AK. Hypertensive renal damage: insights from animal models and clinical relevance. Curr Hypertens Rep 6: 145–153, 2004. doi: 10.1007/s11906-004-0091-8. [DOI] [PubMed] [Google Scholar]
- 6. Fan L, Gao W, Nguyen BV, Jefferson JR, Liu Y, Fan F, Roman RJ. Impaired renal hemodynamics and glomerular hyperfiltration contribute to hypertension-induced renal injury. Am J Physiol Renal Physiol 319: F624–F635, 2020. doi: 10.1152/ajprenal.00239.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang G, Lai FM, Kwan BC, Lai KB, Chow KM, Li PK, Szeto CC. Podocyte loss in human hypertensive nephrosclerosis. Am J Hypertens 22: 300–306, 2009. doi: 10.1038/ajh.2008.360. [DOI] [PubMed] [Google Scholar]
- 8. Naik AS, Le D, Aqeel J, Wang SQ, Chowdhury M, Walters LM, Cibrik DM, Samaniego M, Wiggins RC. Podocyte stress and detachment measured in urine are related to mean arterial pressure in healthy humans. Kidney Int 98: 699–707, 2020. doi: 10.1016/j.kint.2020.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Perez-Hernandez J, Olivares MD, Solaz E, Martinez F, Martinez-Hervas S, Pichler G, Chaves FJ, Redon J, Cortes R. Urinary podocyte-associated molecules and albuminuria in hypertension. J Hypertens 36: 1712–1718, 2018. doi: 10.1097/HJH.0000000000001747. [DOI] [PubMed] [Google Scholar]
- 10. Puelles VG, Cullen-McEwen LA, Taylor GE, Li J, Hughson MD, Kerr PG, Hoy WE, Bertram JF. Human podocyte depletion in association with older age and hypertension. Am J Physiol Renal Physiol 310: F656–F668, 2016. doi: 10.1152/ajprenal.00497.2015. [DOI] [PubMed] [Google Scholar]
- 11. Haruhara K, Sasaki T, de Zoysa N, Okabayashi Y, Kanzaki G, Yamamoto I, Harper IS, Puelles VG, Shimizu A, Cullen-McEwen LA, Tsuboi N, Yokoo T, Bertram JF. Podometrics in Japanese living donor kidneys: associations with nephron number, age, and hypertension. J Am Soc Nephrol 32: 1187–1199, 2021. doi: 10.1681/ASN.2020101486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yu M, Ledeboer MW, Daniels M, Malojcic G, Tibbitts TT, Coeffet-Le Gal M, Pan-Zhou XR, Westerling-Bui A, Beconi M, Reilly JF, Mundel P, Harmange JC. Discovery of a potent and selective TRPC5 inhibitor, efficacious in a focal segmental glomerulosclerosis model. ACS Med Chem Lett 10: 1579–1585, 2019. doi: 10.1021/acsmedchemlett.9b00430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tesch F, Siegerist F, Hay E, Artelt N, Daniel C, Amann K, Zimmermann U, Kavvadas P, Grisk O, Chadjichristos C, Endlich K, Chatziantoniou C, Endlich N. Super-resolved local recruitment of CLDN5 to filtration slits implicates a direct relationship with podocyte foot process effacement. J Cell Mol Med 25: 7631–7641, 2021. doi: 10.1111/jcmm.16519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang Y, Zhang N, Zou Y, Song C, Cao K, Wu B, You S, Lu S, Wang D, Xu J, Huang X, Zhang P, Fan Z, Liu J, Cheng Z, Zhang Z, Kong C, Cao L, Sun Y. Deacetylation of Septin4 by SIRT2 (silent mating type information regulation 2 homolog-2) mitigates damaging of hypertensive nephropathy. Circ Res 132: 601–624, 2023. doi: 10.1161/CIRCRESAHA.122.321591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. D'Agati VD, Chagnac A, de Vries AP, Levi M, Porrini E, Herman-Edelstein M, Praga M. Obesity-related glomerulopathy: clinical and pathologic characteristics and pathogenesis. Nat Rev Nephrol 12: 453–471, 2016. doi: 10.1038/nrneph.2016.75. [DOI] [PubMed] [Google Scholar]
- 16. Chen HM, Liu ZH, Zeng CH, Li SJ, Wang QW, Li LS. Podocyte lesions in patients with obesity-related glomerulopathy. Am J Kidney Dis 48: 772–779, 2006. doi: 10.1053/j.ajkd.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 17. Navarro-Díaz M, Serra A, Romero R, Bonet J, Bayes B, Homs M, Pérez N, Bonal J. Effect of drastic weight loss after bariatric surgery on renal parameters in extremely obese patients: long-term follow-up. J Am Soc Nephrol 17: S213–S217, 2006. doi: 10.1681/ASN.2006080917. [DOI] [PubMed] [Google Scholar]
- 18. Sharma K, Ramachandrarao S, Qiu G, Usui HK, Zhu Y, Dunn SR, Ouedraogo R, Hough K, McCue P, Chan L, Falkner B, Goldstein BJ. Adiponectin regulates albuminuria and podocyte function in mice. J Clin Invest 118: 1645–1656, 2008. doi: 10.1172/JCI32691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Giannini G, Kopp JB, Rosenberg AZ. Podocytopathy in obesity: challenges of living large. Semin Nephrol 41: 307–317, 2021. doi: 10.1016/j.semnephrol.2021.06.003. [DOI] [PubMed] [Google Scholar]
- 20. Yu Y, Mo H, Zhuo H, Yu C, Liu Y. High fat diet induces kidney injury via stimulating Wnt/β-catenin signaling. Front Med (Lausanne) 9: 851618, 2022. doi: 10.3389/fmed.2022.851618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ye Y, Zhong X, Li N, Pan T. Protective effects of liraglutide on glomerular podocytes in obese mice by inhibiting the inflammatory factor TNF-α-mediated NF-κB and MAPK pathway. Obes Res Clin Pract 13: 385–390, 2019. doi: 10.1016/j.orcp.2019.03.003. [DOI] [PubMed] [Google Scholar]
- 22. Guo H, Wang B, Li H, Ling L, Niu J, Gu Y. Glucagon-like peptide-1 analog prevents obesity-related glomerulopathy by inhibiting excessive autophagy in podocytes. Am J Physiol Renal Physiol 314: F181–F189, 2018. doi: 10.1152/ajprenal.00302.2017. [DOI] [PubMed] [Google Scholar]
- 23. Boini KM, Xia M, Abais JM, Li G, Pitzer AL, Gehr TW, Zhang Y, Li PL. Activation of inflammasomes in podocyte injury of mice on the high fat diet: effects of ASC gene deletion and silencing. Biochim Biophys Acta 1843: 836–845, 2014. doi: 10.1016/j.bbamcr.2014.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guo S, Kowalewska J, Wietecha TA, Iyoda M, Wang L, Yi K, Spencer M, Banas M, Alexandrescu S, Hudkins KL, Alpers CE. Renin-angiotensin system blockade is renoprotective in immune complex-mediated glomerulonephritis. J Am Soc Nephrol 19: 1168–1176, 2008. doi: 10.1681/ASN.2007050607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pichaiwong W, Hudkins KL, Wietecha T, Nguyen TQ, Tachaudomdach C, Li W, Askari B, Kobayashi T, O'Brien KD, Pippin JW, Shankland SJ, Alpers CE. Reversibility of structural and functional damage in a model of advanced diabetic nephropathy. J Am Soc Nephrol 24: 1088–1102, 2013. doi: 10.1681/ASN.2012050445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lichtnekert J, Kaverina N, Eng D, Gross K, Kutz J, Pippin J, Shankland S. Renin-angiotensin-aldosterone system inhibition increases podocyte derivation from cells of renin lineage. J Am Soc Nephrol 27: 3611–3627, 2016. doi: 10.1681/ASN.2015080877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang Y, Eng DE, Kaverina NV, Loretz CJ, Koirala A, Akilesh S, Pippin JW, Shankland SJ. Global transcriptomic changes in aged mouse podocytes. Kidney Int 98: 1160–1173, 2020. doi: 10.1016/j.kint.2020.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14: 417–419, 2017. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Soneson C, Love MI, Robinson MD. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res 4: 1521, 2015. doi: 10.12688/f1000research.7563.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 550, 2014. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Marini F, Binder H. pcaExplorer: an R/Bioconductor package for interacting with RNA-seq principal components. BMC Bioinformatics 20: 331, 2019. doi: 10.1186/s12859-019-2879-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102: 15545–15550, 2005. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kaverina N, Schweickart RA, Chan GC, Maggiore JC, Eng DG, Zeng Y, McKinzie SR, Perry HS, Ali A, O'Connor C, Pereira BMV, Theberge AB, Vaughan JC, Loretz CJ, Chang A, Hukriede NA, Bitzer M, Pippin JW, Wessely O, Shankland SJ. Inhibiting NLRP3 signaling in aging podocytes improves their life- and health-span. Aging (Albany NY) 15: 6658–6689, 2023. doi: 10.18632/aging.204897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Venkatareddy M, Wang S, Yang Y, Patel S, Wickman L, Nishizono R, Chowdhury M, Hodgin J, Wiggins PA, Wiggins RC. Estimating podocyte number and density using a single histologic section. J Am Soc Nephrol 25: 1118–1129, 2014. doi: 10.1681/ASN.2013080859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pippin JW, Kaverina N, Wang Y, Eng DG, Zeng Y, Tran U, Loretz CJ, Chang A, Akilesh S, Poudel C, Perry HS, O'Connor C, Vaughan JC, Bitzer M, Wessely O, Sj S. Upregulated PD-1 signaling antagonizes glomerular health in aged kidneys and disease. J Clin Invest 132: e156250, 2022. doi: 10.1172/JCI156250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang J, Wise L, Fukuchi KI. TLR4 cross-talk with NLRP3 inflammasome and complement signaling pathways in Alzheimer’s disease. Front Immunol 11: 724, 2020. doi: 10.3389/fimmu.2020.00724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wnuk M, Hlushchuk R, Janot M, Tuffin G, Martiny-Baron G, Holzer P, Imbach-Weese P, Djonov V, Huynh-Do U. Podocyte EphB4 signaling helps recovery from glomerular injury. Kidney Int 81: 1212–1225, 2012. doi: 10.1038/ki.2012.17. [DOI] [PubMed] [Google Scholar]
- 38. Zhou L, Liu Y. Wnt/β-catenin signalling and podocyte dysfunction in proteinuric kidney disease. Nat Rev Nephrol 11: 535–545, 2015. doi: 10.1038/nrneph.2015.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schunk SJ, Floege J, Fliser D, Speer T. WNT-β-catenin signalling—a versatile player in kidney injury and repair. Nat Rev Nephrol 17: 172–184, 2021. doi: 10.1038/s41581-020-00343-w. [DOI] [PubMed] [Google Scholar]
- 40. Rodwell GE, Sonu R, Zahn JM, Lund J, Wilhelmy J, Wang L, Xiao W, Mindrinos M, Crane E, Segal E, Myers BD, Brooks JD, Davis RW, Higgins J, Owen AB, Kim SK. A transcriptional profile of aging in the human kidney. PLoS Biol 2: e427, 2004. doi: 10.1371/journal.pbio.0020427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hao Z, Zhang H, Cowell J. Ubiquitin-conjugating enzyme UBE2C: molecular biology, role in tumorigenesis, and potential as a biomarker. Tumour Biol 33: 723–730, 2012. doi: 10.1007/s13277-011-0291-1. [DOI] [PubMed] [Google Scholar]
- 42. Niemir ZI, Stein H, Dworacki G, Mundel P, Koehl N, Koch B, Autschbach F, Andrassy K, Ritz E, Waldherr R, Otto HF. Podocytes are the major source of IL-1 α and IL-1 β in human glomerulonephritides. Kidney Int 52: 393–403, 1997. doi: 10.1038/ki.1997.346. [DOI] [PubMed] [Google Scholar]
- 43. Shankland SJ, Wang Y, Shaw AS, Vaughan JC, Pippin JW, Wessely O. Podocyte aging: why and how getting old matters. J Am Soc Nephrol 32: 2697–2713, 2021. doi: 10.1681/ASN.2021050614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bogue MA, Ball RL, Philip VM, Walton DO, Dunn MH, Kolishovski G, Lamoureux A, Gerring M, Liang H, Emerson J, Stearns T, He H, Mukherjee G, Bluis J, Desai S, Sundberg B, Kadakkuzha B, Kunde-Ramamoorthy G, Chesler EJ. Mouse Phenome Database: towards a more FAIR-compliant and TRUST-worthy data repository and tool suite for phenotypes and genotypes. Nucleic Acids Res 51: D1067–D1074, 2023. doi: 10.1093/nar/gkac1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fridman AL, Tainsky MA. Critical pathways in cellular senescence and immortalization revealed by gene expression profiling. Oncogene 27: 5975–5987, 2008. doi: 10.1038/onc.2008.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Milacic M, Beavers D, Conley P, Gong C, Gillespie M, Griss J, Haw R, Jassal B, Matthews L, May B, Petryszak R, Ragueneau E, Rothfels K, Sevilla C, Shamovsky V, Stephan R, Tiwari K, Varusai T, Weiser J, Wright A, Wu G, Stein L, Hermjakob H, D’Eustachio P. The Reactome Pathway Knowledgebase 2024. Nucleic Acids Res 52: D672–D678, 2024. doi: 10.1093/nar/gkad1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Satirapoj B, Nast CC, Adler SG. Novel insights into the relationship between glomerular pathology and progressive kidney disease. Adv Chronic Kidney Dis 19: 93–100, 2012. doi: 10.1053/j.ackd.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 48. Wu J, Raman A, Coffey NJ, Sheng X, Wahba J, Seasock MJ, Ma Z, Beckerman P, Laczko D, Palmer MB, Kopp JB, Kuo JJ, Pullen SS, Boustany-Kari CM, Linkermann A, Susztak K. The key role of NLRP3 and STING in APOL1-associated podocytopathy. J Clin Invest 131: e136329, 2021. doi: 10.1172/JCI136329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sun Y, Ge X, Li X, He J, Wei X, Du J, Sun J, Li X, Xun Z, Liu W, Zhang H, Wang ZY, Li YC. High-fat diet promotes renal injury by inducing oxidative stress and mitochondrial dysfunction. Cell Death Dis 11: 914, 2020. doi: 10.1038/s41419-020-03122-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Huang W, Hickson LJ, Eirin A, Kirkland JL, Lerman LO. Cellular senescence: the good, the bad and the unknown. Nat Rev Nephrol 18: 611–627, 2022. doi: 10.1038/s41581-022-00601-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Li S, Livingston MJ, Ma Z, Hu X, Wen L, Ding HF, Zhou D, Dong Z. Tubular cell senescence promotes maladaptive kidney repair and chronic kidney disease after cisplatin nephrotoxicity. JCI Insight 8: e166643, 2023. doi: 10.1172/jci.insight.166643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Fang Y, Chen B, Liu Z, Gong AY, Gunning WT, Ge Y, Malhotra D, Gohara AF, Dworkin LD, Gong R. Age-related GSK3β overexpression drives podocyte senescence and glomerular aging. J Clin Invest 132: e141848, 2022. doi: 10.1172/JCI141848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Veloso Pereira BM, Zeng Y, Maggiore JC, Schweickart RA, Eng DG, Kaverina N, McKinzie SR, Chang A, Loretz CJ, Thieme K, Hukriede NA, Pippin JW, Wessely O, Shankland SJ. Podocyte injury at young age causes premature senescence and worsens glomerular aging. Am J Physiol Renal Physiol 326: F120–F134, 2023. doi: 10.1152/ajprenal.00261.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Tian X, Pedigo CE, Li K, Ma X, Bunda P, Pell J, Lek A, Gu J, Zhang Y, Medina Rangel PX, Li W, Schwartze E, Nagata S, Lerner G, Perincheri S, Priyadarshini A, Zhao H, Lek M, Menon MC, Fu R, Ishibe S. Profilin1 is required to prevent mitotic catastrophe in murine and human glomerular diseases. J Clin Invest 133: e171237, 2023. doi: 10.1172/JCI171237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Medina Rangel PX, Cross E, Liu C, Pedigo CE, Tian X, Gutierrez-Calabres E, Nagata S, Priyadarshini A, Lerner G, Bunda P, Perincheri S, Gu J, Zhao H, Wang Y, Inoue K, Ishibe S. Cell cycle and senescence regulation by podocyte histone deacetylase 1 and 2. J Am Soc Nephrol 34: 433–450, 2023. doi: 10.1681/ASN.2022050598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Shankland SJ, Floege J, Thomas SE, Nangaku M, Hugo C, Pippin J, Henne K, Hockenberry DM, Johnson RJ, Couser WG. Cyclin kinase inhibitors are increased during experimental membranous nephropathy: potential role in limiting glomerular epithelial cell proliferation in vivo. Kidney Int 52: 404–413, 1997. doi: 10.1038/ki.1997.347. [DOI] [PubMed] [Google Scholar]
- 57. Wada T, Pippin JW, Terada Y, Shankland SJ. The cyclin-dependent kinase inhibitor p21 is required for TGF-β1-induced podocyte apoptosis. Kidney Int 68: 1618–1629, 2005. doi: 10.1111/j.1523-1755.2005.00574.x. [DOI] [PubMed] [Google Scholar]
- 58. Petermann AT, Hiromura K, Blonski M, Pippin J, Monkawa T, Durvasula R, Couser WG, Shankland SJ. Mechanical stress reduces podocyte proliferation in vitro. Kidney Int 61: 40–50, 2002. doi: 10.1046/j.1523-1755.2002.00102.x. [DOI] [PubMed] [Google Scholar]
- 59. Ogrodnik M, Zhu Y, Langhi LGP, Tchkonia T, Kruger P, Fielder E, Victorelli S, Ruswhandi RA, Giorgadze N, Pirtskhalava T, Podgorni O, Enikolopov G, Johnson KO, Xu M, Inman C, Palmer AK, Schafer M, Weigl M, Ikeno Y, Burns TC, Passos JF, von Zglinicki T, Kirkland JL, Jurk D. Obesity-induced cellular senescence drives anxiety and impairs neurogenesis. Cell Metab 29: 1061–1077 e1068, 2019. [Erratum in Cell Metab 29: 1233, 2019]. doi: 10.1016/j.cmet.2018.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang X, Zhou D, Strakovsky R, Zhang Y, Pan YX. Hepatic cellular senescence pathway genes are induced through histone modifications in a diet-induced obese rat model. Am J Physiol Gastrointest Liver Physiol 302: G558–G564, 2012. doi: 10.1152/ajpgi.00032.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Palmer AK, Xu M, Zhu Y, Pirtskhalava T, Weivoda MM, Hachfeld CM, Prata LG, van Dijk TH, Verkade E, Casaclang-Verzosa G, Johnson KO, Cubro H, Doornebal EJ, Ogrodnik M, Jurk D, Jensen MD, Chini EN, Miller JD, Matveyenko A, Stout MB, Schafer MJ, White TA, Hickson LJ, Demaria M, Garovic V, Grande J, Arriaga EA, Kuipers F, von Zglinicki T, LeBrasseur NK, Campisi J, Tchkonia T, Kirkland JL. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 18: e12950, 2019. doi: 10.1111/acel.12950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kim SR, Jiang K, Ogrodnik M, Chen X, Zhu XY, Lohmeier H, Ahmed L, Tang H, Tchkonia T, Hickson LJ, Kirkland JL, Lerman LO. Increased renal cellular senescence in murine high-fat diet: effect of the senolytic drug quercetin. Transl Res 213: 112–123, 2019. doi: 10.1016/j.trsl.2019.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Afsar B, Afsar RE. Hypertension and cellular senescence. Biogerontology 24: 457–478, 2023. doi: 10.1007/s10522-023-10031-4. [DOI] [PubMed] [Google Scholar]
- 64. Lawrie A, Francis SE. Frataxin and endothelial cell senescence in pulmonary hypertension. J Clin Invest 131: e149721, 2021. doi: 10.1172/JCI149721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Szeto HH, Liu S, Soong Y, Alam N, Prusky GT, Seshan SV. Protection of mitochondria prevents high-fat diet-induced glomerulopathy and proximal tubular injury. Kidney Int 90: 997–1011, 2016. doi: 10.1016/j.kint.2016.06.013. [DOI] [PubMed] [Google Scholar]
- 66. Eremina V, Quaggin SE. The role of VEGF-A in glomerular development and function. Curr Opin Nephrol Hypertens 13: 9–15, 2004. doi: 10.1097/00041552-200401000-00002. [DOI] [PubMed] [Google Scholar]
- 67. Tan RJ, Li Y, Rush BM, Cerqueira DM, Zhou D, Fu H, Ho J, Beer Stolz D, Liu Y. Tubular injury triggers podocyte dysfunction by β-catenin-driven release of MMP-7. JCI Insight 4: e122399, 2019. doi: 10.1172/jci.insight.122399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Qadri MM. Targeting CD44 receptor pathways in degenerative joint diseases: involvement of proteoglycan-4 (PRG4). Pharmaceuticals (Basel) 16: 1425, 2023. doi: 10.3390/ph16101425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Smeets B, Stucker F, Wetzels J, Brocheriou I, Ronco P, Grone HJ, D'Agati V, Fogo AB, van Kuppevelt TH, Fischer HP, Boor P, Floege J, Ostendorf T, Moeller MJ. Detection of activated parietal epithelial cells on the glomerular tuft distinguishes early focal segmental glomerulosclerosis from minimal change disease. Am J Pathol 184: 3239–3248, 2014. doi: 10.1016/j.ajpath.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wicks SE, Nguyen TT, Breaux C, Kruger C, Stadler K. Diet-induced obesity and kidney disease—in search of a susceptible mouse model. Biochimie 124: 65–73, 2016. doi: 10.1016/j.biochi.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bairey Merz CN, Dember LM, Ingelfinger JR, Vinson A, Neugarten J, Sandberg KL, Sullivan JC, Maric-Bilkan C, Rankin TL, Kimmel PL, Star RA;. participants of the National Institute of Diabetes and Digestive and Kidney Diseases Workshop on “Sex and the Kidneys”. Sex and the kidneys: current understanding and research opportunities. Nat Rev Nephrol 15: 776–783, 2019. doi: 10.1038/s41581-019-0208-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figs. S1–S6 and Supplemental Table S1: https://doi.org/10.6084/m9.figshare.25236832.v1.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available upon reasonable request.