

Abstract
Cognitive or motor impairment is common among individuals with neurofibromatosis type 1 (NF1), an autosomal dominant tumor‐predisposition disorder. As many as 70% of children with NF1 report difficulties with spatial/working memory, attention, executive function, and fine motor movements. In contrast to the utilization of various Nf1 mouse models, here we employ an NF1 +/ex42del miniswine model to evaluate the mechanisms and characteristics of these presentations, taking advantage of a large animal species more like human anatomy and physiology. The prefrontal lobe, anterior cingulate, and hippocampus from NF1 +/ex42del and wild‐type miniswine were examined longitudinally, revealing abnormalities in mature oligodendrocytes and astrocytes, and microglial activation over time. Imbalances in GABA: Glutamate ratios and GAD67 expression were observed in the hippocampus and motor cortex, supporting the role of disruption in inhibitory neurotransmission in NF1 cognitive impairment and motor dysfunction. Moreover, NF1 +/ex42del miniswine demonstrated slower and shorter steps, indicative of a balance‐preserving response commonly observed in NF1 patients, and progressive memory and learning impairments. Collectively, our findings affirm the effectiveness of NF1 +/ex42del miniswine as a valuable resource for assessing cognitive and motor impairments associated with NF1, investigating the involvement of specific neural circuits and glia in these processes, and evaluating potential therapeutic interventions.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
As many as 70% of individuals with neurofibromatosis type I (NF1) report experiencing cognitive and sociopsychological comorbidities and impaired motor function. The mechanisms underlying the dysfunctions are demonstrated in Nf1 +/− mice which show memory and spatial learning deficits and dysregulation of neural signal transduction in a simple model that fails to replicate the full extent of clinical features experienced by NF1 patients. The NF1 +/ex42del miniswine recapitulates human clinical phenotype and phenocopies of human disease.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study investigated the NF1 cognitive and motor dysfunctions at a cellular and behavioral level using the NF1 +/ex42del miniswine. This study also addressed how the NF1 disease phenotypes are dependent on sex and cutaneous neurofibroma burden.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Our results show that NF1 +/ex42del miniswine presents multiple cellular, cell signaling, and behavioral abnormalities which are highly dependent on sex and tumor burden. Aberrant glia responses and inhibitory signaling imbalances were shown in brain regions specific to memory and learning deficits in individuals with NF1. Additionally, we demonstrated that motor and memory/learning abnormalities are impacted by sex and neurofibroma burden and worsen over time.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
This study provides compelling evidence that the NF1 +/ex42del miniswine is a powerful, clinically relevant animal model to study NF1 disease. This valuable resource will be instrumental in evaluating disease detection methods and potential therapeutic interventions for NF1 patients.
INTRODUCTION
Neurofibromatosis type 1 (NF1) is an autosomal dominant disease that affects ~1 in 3500 individuals. The NF1 gene, which encodes the tumor‐suppressive RAS GTPase activating protein (GAP) called neurofibromin, contains 60 exons that are subject to over 1000 different disease‐associated mutations. 1 The diversity of mutations, including “second‐hit” losses of heterozygosity in individual cells as well as single‐nucleotide polymorphisms, 2 cause the disease to present in a heterogeneous manner, such that the development and severity of symptoms vary widely from individual to individual. The most notable diagnostic features of NF1 result from unregulated proliferation of neural‐crest‐derived cell populations: hyperpigmentation, café au lait macules, Lisch nodes in the iris, cutaneous neurofibromas (cNFs) throughout the nervous system, and at times, malignant peripheral nerve sheath tumors (MPNSTs).
Cognitive and sociopsychological comorbidities are also reported but are less understood, including speech and motor difficulties, learning disabilities, anxiety, and depression, among others. 3 Severity of neurocognitive abnormalities varies greatly among children, with more severe learning deficits associated with NF1 loss in developing neural stem cells. 4 Children with NF1 frequently encounter challenges related to learning and attention. Studies indicate that approximately 63% of children with NF1 struggle to sustain attention, 5 while others face difficulties in spatial learning tests. 6 Moreover, there is an increased prevalence of autism spectrum disorder (ASD) among NF1 individuals, with 13% exhibiting severe quantitative ASD traits. 7 Impaired motor function is another common feature, characterized by a gait pattern with prolonged step time, reduced velocity, cadence, and stride length. 8 Remarkably, a significant majority (81%) of individuals aged 4–15 years living with NF1 demonstrate below‐average or well below‐average motor proficiency. 9 These individuals can exhibit muscle weakness, reduced muscle size, and up to 50% diminished muscle strength compared with their healthy counterparts. 10
While the role of neurofibromin inactivation in tumor formation has been studied in detail, the effects of NF1 mutations on cognitive and motor abilities are less understood. Recent studies have made significant advancements in understanding the cognitive dysfunction associated with NF1, including demonstrations that Nf1 +/− mice exhibit deficits in memory and spatial learning. Notably, these deficits can be reversed by interventions that inhibit Ras function, the primary downstream target of neurofibromin. 11 Moreover, the increase in Ras and MAPK/ERK signaling in Nf1 deficient mouse models was found to promote phosphorylation of synapsin 1. This, in turn, leads to an excessive release of gamma‐aminobutyric acid (GABA) from inhibitory neurons in specific brain regions, such as the hippocampus, medial prefrontal cortex, and striatum. This dysregulation can then contribute to a reduction in long‐term potentiation, which is crucial for working memory. 11 , 12 Similarly, dysfunctional inhibitory signaling has been suggested to contribute to motor deficits of other NF1 models, with increased MAPK/ERK signaling implicated in neuronal migration abnormalities in the cerebellum and altered NF1‐cAMP pathways implicated in abnormal dopamine processing in the striatum. 13 , 14 , 15
While Nf1 mouse models have provided valuable insights into the mechanisms underlying cognitive and motor dysfunction in NF1, their simplicity as heterozygous models fall short in replicating the full spectrum of clinical features seen in NF1 patients. Notably, these models lack key presentations such as abnormal skin pigmentation and cNF/MPNST formation, limiting the translatability of murine models to some aspects of the human condition. To study the effects of NF1 mutation in an animal model that has greater similarity to human anatomy and physiology, including the necessary presence of a gyrencephalic brain, we characterized a novel miniswine model of NF1 bearing an orthologous mutation (deletion of exon 42) seen in individuals with NF1. NF1 +/ex42del miniswine effectively phenocopied the human disease and recapitulated several human clinical phenotypes that are lacking in Nf1 mutant mouse models, including café au lait spots and cNF formation. 16 Expanding on our initial characterization, we now delve into the potential of NF1 +/ex42del miniswine for studying cognitive and motor dysfunction at cellular and behavioral levels. Our findings reveal various cellular, memory/learning, and gait abnormalities in this miniswine model, with the phenotypes influenced by sex and cNF burden.
MATERIALS AND METHODS
Animals
Wild‐type and NF1 +/ex42del Yucatan miniswine were generated using methods described previously. 16 Longitudinal analysis was performed with age‐matched groups of 8, 12, 15, 18, and 24 months. For behavioral testing, male NF1 +/ex42del miniswine were grouped by either the presence or absence of cutaneous neurofibromas (cNFs) and as previously described, only male NF1 +/ex42del miniswine presented with cNFs during the study period. 16 A description of specific cNF presentation in each animal is described in Table S1. All miniswine were maintained at Exemplar Genetics under an approved IACUC protocol (# MRP2016‐009).
Histological preparation and immunohistochemistry
Animals were euthanized with pentobarbital at 14 months (n = 16; 8 WT (4F, 4M); 8 NF1 +/ex42del (4F, 4M)) and 18–24 months (n = 20; 8 WT (4F, 4M); 12 NF1 +/ex42del (4F, 4M with cNFs, 4M without cNFs)). Brain hemispheres were fixed in 10% formalin for 3 weeks. After fixation, the brains were blocked, cryoprotected in 30% sucrose, and sectioned at 45 μm thickness on a freezing microtome. Floating sections were immunolabeled following previously published procedures 17 using the following primary antibodies: ionized calcium‐binding adaptor molecule 1‐Iba1 (BioCare Medical 290; 1:2000), glial fibrillary acidic protein‐GFAP (Dako Z0334; 1:16000), myelin basic protein (MBP) (Millipore MAB386; 1:1000), Calbindin (Swant CB38; 1:2000), Parvalbumin (Swant PV27, 1:2500) and Calretinin (Swant CR7699/3H, 1:2000); and the following secondary antibodies: biotinylated anti‐rat (MBP) and biotinylated anti‐rabbit (Iba1, GFAP, Calbindin, Parvalbumin, and Calretinin).
Image acquisition and analysis
Triplicate sections of the prefrontal lobe, anterior cingulate, and hippocampus immunolabeled with anti‐MBP and anti‐GFAP antibodies were imaged with an Aperio slide scanner at 20× magnification. At least three images were extracted from each tissue slice (observer blinded to group assignment), 18 for at least nine images per animal/per marker/per region. These images were condensed by Adobe Photoshop and split into red/green/blue channels by ImageJ. Images were run through ImageJ batch processing using a rolling ball radius for background subtraction and analyzed by threshold analysis. Total pixel area data (%) was selected and analyzed using a one‐way ANOVA with Tukey's post hoc in GraphPad Prism 6.0.
Triplicate sections of the prefrontal lobe, anterior cingulate, and hippocampus immunolabeled with anti‐Iba1 antibodies were imaged with a Nikon 90i at 20× magnification. Multiple images were viewed from each section, which varied from 3 to 9 sections per animal in the anterior cingulate, 6–9 sections in the prefrontal lobe, and 6–19 sections in the hippocampus. Using Nikon NIS element software, a 500 × 500 μm box was drawn on each image and the number of ramified and amoeboid Iba+ microglia were counted for each image. The total number of ramified and amoeboid microglia was tallied for each miniswine and normalized to the number of sections for that animal. Cell counts were analyzed in GraphPad Prism 6.0 using unpaired t‐tests.
Triplicate sections of the hippocampus and motor cortex were labeled with anti‐parvalbumin, anti‐calbindin, and anti‐calretinin antibodies. Sections were mounted onto slides and imaged with the Nikon Ni‐E at 100× magnification. Images were split into multiple channels with ImageJ and the blue channel was used to adjust threshold and particle size settings to match the size and shape of individually labeled cell bodies. The particle size corresponded to the number of labeled cell bodies in each image. Cell count means were generated for each animal and statistical tests were run (two‐way ANOVA with uncorrected Fisher's LSD).
High‐performance liquid chromatography
Aliquots from the hippocampus and motor cortex were deproteinized by the addition of 450 μL of Isocratic Excitatory AA buffer (0.1 M sodium phosphate dibasic heptahydrate (Fisher Scientific), 0.13 mM EDTA (Research Products International) in 33% methanol (Sigma‐Aldrich); pH 5.88) and sonicated on ice using a tip sonicator. Glutamate and GABA neurotransmitter levels were quantified by high‐performance liquid chromatography (HPLC) in porcine brain homogenates following previously published procedures for the derivatization technique 19 and fluorescent detection methods that detailed specific excitation and emission wavelengths for GABA and glutamate. 20 Briefly, samples were derivatized using o‐phthalaldehyde (Fisher Scientific) and sodium sulfite (Sigma‐Aldrich) prior to separation on a C18 reverse phase column. Samples were eluted using an isocratic elution method and detected using a fluorescence detector (FLD) at 240 nm excitation wavelength and 450 nm emission wavelength. Peak area and external standards of individual and mixed amino acids were used to calculate relative amounts of each amino acid, and unpaired t‐tests were run in GraphPad Prism 6.0+.
Protein sample preparation and Western Blotting
Aliquots from the motor cortex and hippocampus were lysed in a lysis buffer and sonicated on ice using a tip sonicator. Cell lysates (80 μg) and phosphate‐buffered saline (Sigma‐Aldrich) were heated to 100°C for 10 min. And 50 μL of protein lysates were loaded into each well of a 4–15% precast gel (Mini‐PROTEAN® TGX™; BioI‐Rad) for electrophoresis. Separated proteins were transferred onto nitrocellulose membranes using Thermo Scientific Power blotter station (Invitrogen™). Membranes were incubated with anti‐glutamic acid decarboxylase 67‐GAD67 (BD BioSciences 611,604; 1:1000) and β‐actin antibodies – as a loading control (Cell Signaling 4967; 1:3000) overnight at 4°C. After washing, membranes were incubated for 2 h with either anti‐mouse or anti‐rabbit HRP secondary antibodies at 4°C. Adult bovine serum in tris‐buffered saline with 0.1% Tween‐20 was used for blocking and antibody incubation steps. Bands were then visualized with Clarity™ Western ECL HRP substrate (BioRad) and imaged using ChemDoc™ MP Imaging system (BioRad). Using Image J, a region of interest (ROI) was selected around each GAD67 and β‐actin band and below each band (for background) to measure mean gray values as a measure of pixel density. The background density was subtracted from its respective GAD67 or β‐actin band density to calculate a net β‐actin band value and a net GAD67 band value. The ratio of net GAD67 over net β‐actin for each NF1 +/ex42del and wild‐type miniswine (14, 18, and 24 M) was calculated and analyzed with GraphPad Prism 6.0 using unpaired t‐tests at each timepoint.
Neurobehavior testing
Simple T‐maze
We followed the methodology from our previous publication, 16 except for using dry animal pellets as a reward. Briefly, animals were placed in a simple T‐maze for two testing paradigms. During the acquisition phases, the animal learned which arm a food reward was located in. During the reversal phases, the reward was moved to the opposite arm, and the animal had to relearn the location of the reward. The test was video recorded, and the animals were tracked with the Anymaze software v4.99 (Stoelting Co., Wood Dale, IL). A blinded observer watched the T‐maze videos and recorded the arm chosen by each animal during the acquisition and reversal phases. The number of correct responses was analyzed with GraphPad Prism 6.0 using a one‐way ANOVA, Tukey post hoc.
Gait analysis
Motor performance was analyzed using a kinematic walkway as previously described. 21 Briefly, a 4.87 × 0.6 m Zeno Electronic Walkway (ZenoMetrics Peekskill, NY) was used for data acquisition. Animals were trained to walk by receiving food as a reward for each completed walk. Data were processed with PKMAS Software ver. 509C1 (Protokinetics LLC, Havertown, PA), and 186 parameters were collected per footprint. Five walks per subject were analyzed at each timepoint, with each walk consisting of eight consecutive footprints of the front legs. Walks in which the pig stopped or stepped out of the mat were eliminated. The 186 parameters were analyzed utilizing a multivariate principal component analysis (PCA) implemented in R utilizing the FactorMineR package to determine which principal components (PCs) accounted for the most variation in the data. The most significant variables contributing to the most variation in the PCs included: stride time, step time, stance [measured by % gait cycle (GC)], swing (%GC), stride length, step length, and integrated pressure.
Statistical analysis
All statistical tests were performed in GraphPad Prism version 6.0+ or equivalent unless otherwise noted. Raw data and complete data tables with exact p‐values are available from the authors upon reasonable request. Individual statistical test details are specified in figure legends. PCA was performed with the FactorMineR package in R version 4.0.1+. Outliers were removed using the ROUT method, Q = 0. Graphs as presented as Mean ± SEM, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
RESULTS
NF1 +/ex42del swine show aberrant myelination, altered astrocyte response, and increased microglia presence in several brain regions
Frontal lobe white matter tracts of NF1 patients have been shown to be volumetrically enlarged compared with healthy individuals when imaged through diffusion tensor imaging. 22 We were interested in whether our NF1 +/ex42del swine model exhibited similar myelination abnormalities. We chose to focus on prefrontal tracts and the anterior cingulate cortex due to their association with memory and learning defects in individuals with NF1 and mutant mice models. 23 , 24 Tissue samples were split by sex and examined at two age ranges: 14 months and 18–24 months. Using immunohistochemistry of MBP, a marker of mature myelinating oligodendrocytes, adult NF1 +/ex42del miniswine presented with significantly more MBP+ expression than wild‐type counterparts in both the prefrontal tracts (Figure 1a,b, Figure S1A) and anterior cingulate (Figure 1c,d). Female NF1 +/ex42del miniswine showed this increase in the prefrontal tracts at 14 and 18–24 months of age while male NF1 +/ex42del miniswine showed this increase only at the later time (Figure 1b). In contrast, an increase in MBP was only present in the anterior cingulate in 18–24 month male NF1 +/ex42del miniswine (Figure 1d).
FIGURE 1.
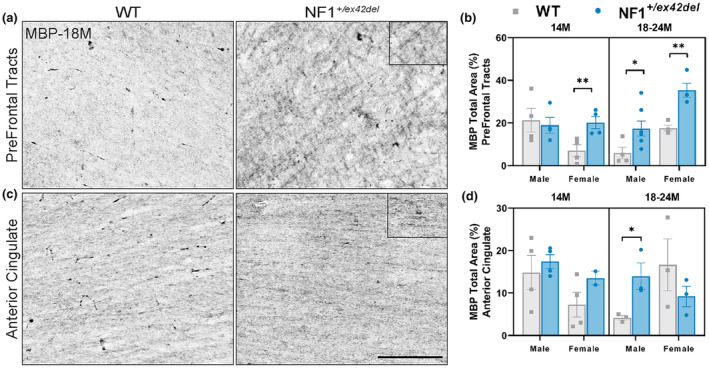
NF1 +/ex42del miniswine exhibit increased MBP+ mature oligodendrocytes in prefrontal tracts and anterior cingulate. (a) Representative images of MBP+ oligodendrocytes in the prefrontal tracts of wild‐type and NF1 +/ex42del miniswine at 18 months of age. (b) MBP+ analysis in prefrontal tracts indicating significant differences between NF1 +/ex42del miniswine and wild‐type counterparts at 14 and 18–24 months of age. (c) Representative images of MBP+ oligodendrocytes in the anterior cingulate of wild‐type and NF1 +/ex42del miniswine at 18 months of age. (d) MBP+ analysis in the anterior cingulate indicates significant differences between male NF1 +/ex42del and wild‐type counterparts at 18 months. Unpaired t‐tests. n = 3–4/group, *p < 0.05, **p < 0.01. Mean ± SEM. Scale bar = 200 μm.
Astrogliosis, characterized by the proliferation of GFAP+ astrocytes, has been observed in various NF1 models. However, the expression of GFAP in astrocytes shows variability depending on factors, such as age, animal model, and specific brain region. For example, in several Nf1 mutant mice, an increased number of GFAP‐labeled astrocytes has been detected in the mediolateral region of the periaqueductal gray and nucleus accumbens, irrespective of age. In contrast, GFAP expression in the hippocampus varied across all age groups, and no significant differences in GFAP expression were observed in the cortex or white matter tracts. 25 To determine if NF1 +/ex42del miniswine showed abnormal astrocyte phenotypes, regions associated with planning/decision‐making (prefrontal lobe and anterior cingulate) and memory/learning (hippocampus) were immunolabeled for GFAP+ astrocytosis. 11 Across several brain regions at 14 months of age, male and female NF1 +/ex42del miniswine showed no consistent differences in GFAP expression, including in the prefrontal lobe (Figure 2a,b, Figure S1B), anterior cingulate (Figure 2c,d, Figure S1C), and dentate gyrus of the hippocampus (Figure 2g,h, Figure S1D). A significant increase was found in male NF1 +/ex42del miniswine within the CA1 of the hippocampus (Figure 2e,f, Figure S1C). At 18–24 months, an increase in GFAP was seen in the prefrontal lobe (Figure 2a,b) in female NF1 +/ex42del miniswine, whereas a decrease was found in male NF1 +/ex42del miniswine in the dentate gyrus (Figure 2g,h, Figure S1D). Therefore, no consistent astrocyte response was observed in NF1 +/ex42del miniswine across time and within several brain regions.
FIGURE 2.
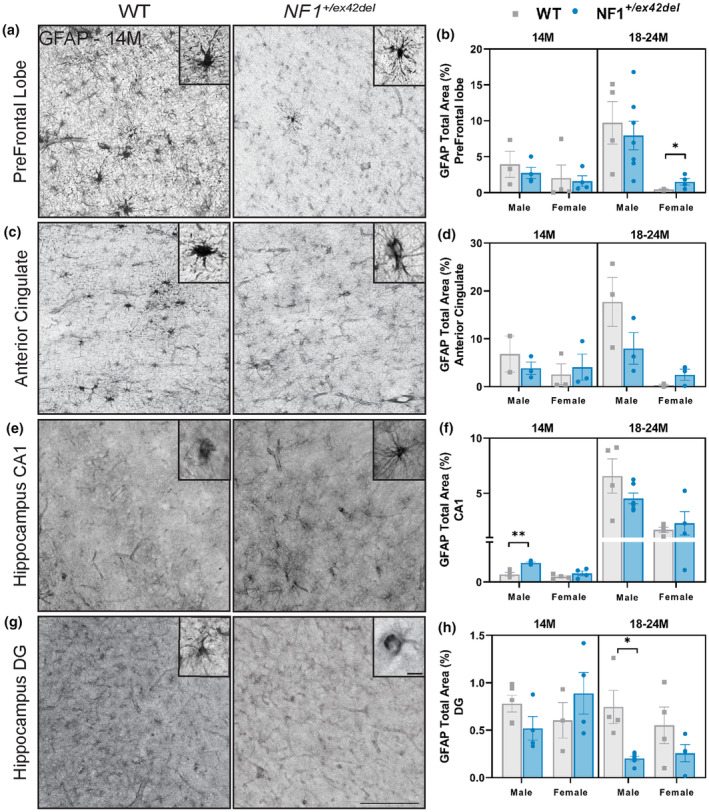
NF1 +/ex42del miniswine exhibit inconsistent GFAP+ astrocyte expression in the prefrontal lobe, anterior cingulate, and hippocampus. (a) Representative images of GFAP+ astrocytes in the prefrontal tracts of 14‐month‐old wild‐type and NF1 +/ex42del miniswine. (b) GFAP+ analysis of prefrontal tracts indicates significant increases in female NF1 +/ex42del miniswine at 18–24 months of age. (c) Representative images of GFAP+ astrocytes in the anterior cingulate of 14‐month‐old animals. (d) GFAP+ analysis of the anterior cingulate indicates no significant differences at either timepoint. (e) Representative images of GFAP+ astrocytes in the CA1 region of the hippocampus in 14‐month‐old animals. (f) GFAP+ analysis of hippocampus CA1 indicates an increase in astrocyte expression in 14‐month‐old male NF1 +/ex42del miniswine. (g) Representative images of GFAP+ astrocytes in the dentate gyrus of the hippocampus in 14‐month‐old animals. (h) GFAP+ analysis of the dentate gyrus indicates a decrease in male NF1 +/ex42del miniswine at 18 months of age. Unpaired t‐tests. n = 3‐4/group, *p < 0.05, **p < 0.01. Mean ± SEM. Scale bar = 200 μm.
An increase in microglia presence has been reported in human NF1‐associated astrocytomas and the optic nerve of Nf1 mutant mice, 26 and aberrant microglial function can contribute to cell death and cognitive dysfunction. To understand if aberrant microglia activity was evident in NF1 +/ex42del miniswine, brain regions were examined for the presence and morphology of ionized calcium‐binding adaptor molecule 1 positive (Iba1+) microglia, with active microglia taking a more amoeboid shape. 27 While we did not observe an increase in active, amoeboid microglia that would indicate chronic neuroinflammation, an increased number of ramified, non‐activated microglia were found in the prefrontal lobe of 18‐month‐old male NF1 +/ex42del miniswine (Figure S2A,B), and in the hippocampus of male NF1 +/ex42del miniswine at 14 and 18 months (Figure S3C,D). Female animals showed no microglial abnormalities in either region at any timepoint, and the anterior cingulate showed no differences in the number of microglia at any timepoint between either sex (data not shown).
NF1 +/ex42del swine show altered inhibitory signaling in the hippocampus and motor cortex
As imbalances in inhibitory signaling have been proposed to be one mechanism contributing to memory, learning, and motor delays in Nf1 mutant mice 11 , 12 , 24 as well as motor deficits in NF1 patients, 28 we probed into the balance of GABAergic and glutamatergic neurons in the hippocampus and motor cortex of NF1 +/ex42del miniswine. Using HPLC, on homogenates of the hippocampus and motor cortex, a significantly decreased GABA: Glutamate ratio was detected in 18–24‐month‐old female NF1 +/ex42del miniswine in both regions (Figure 3a,b), indicating an imbalance of excitatory to inhibitory signaling in a region important to memory/learning function and motor coordination. We also investigated the types of inhibitory neurons by immunolabeling the motor cortex and hippocampal sections with the GABAergic interneuron markers calbindin, calretinin, and parvalbumin. In the hippocampus at 14 months, greater numbers of calretinin+ interneurons were found in female NF1 +/ex42del animals, whereas less parvalbumin+ interneurons were found in male NF1 +/ex42del animals (Figure 4a,b, Figure S3A). No consistent differences were noted in other time ranges. In the motor cortex at 14 months, there were fewer calbindin+ interneurons in male NF1 +/ex42del animals (Figure 5a,b, Figure S3B). When measuring the relative protein level of GAD67, an enzyme that synthesizes GABA, in the hippocampus and motor cortex, no changes were detected at either time range (Figure S4A–D).
FIGURE 3.
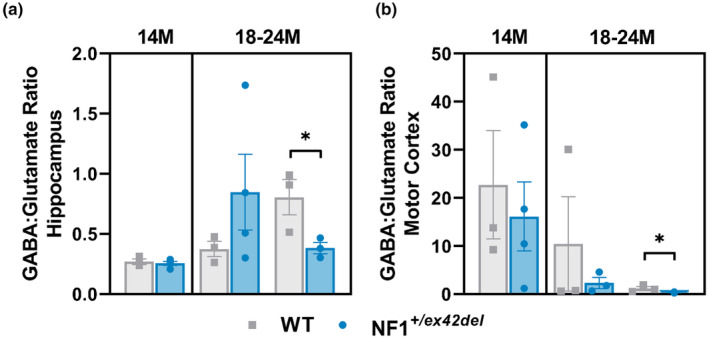
Decreased GABA signaling in the hippocampus and motor cortex of NF1 +/ex42del miniswine. (a) HPLC analysis of GABA: Glutamate indicates a decreased ratio in the hippocampus of female NF1 +/ex42del miniswine at 18–24 months. (b) HPLC analysis of GABA: Glutamate indicates a decreased ratio in the motor cortex of female NF1 +/ex42del miniswine at 18–24 months. Unpaired t‐tests. n = 3–4/group, *p < 0.05. Mean ± SEM.
FIGURE 4.
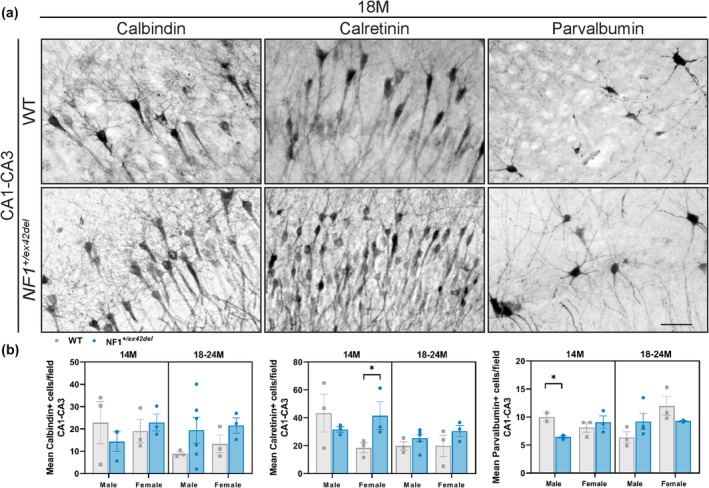
No consistent differences in GABAergic interneurons in hippocampus of NF1 +/ex42del miniswine. (a) Representative images of calbindin+, calretinin+, and parvalbumin+ interneurons in hippocampus of NF1 +/ex42del and wild‐type miniswine at 18 months of age. (b) Significantly greater number of calretinin+ interneurons in 14‐month‐old female NF1 +/ex42del animals, and decreased number of parvalbumin+ interneurons in 14‐month‐old male NF1 +/ex42del animals. Unpaired t‐tests. n = 3–4/group, *p < 0.05. Mean ± SEM. Scale bar = 100 μm.
FIGURE 5.
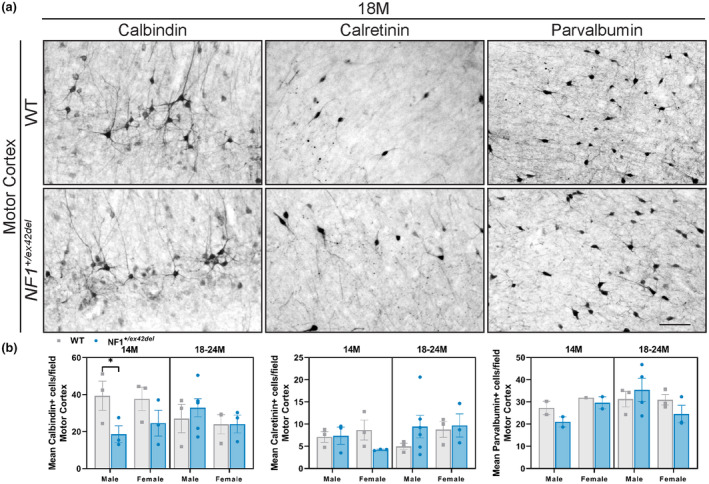
No consistent differences in GABAergic interneurons in the motor cortex of NF1 +/ex42del miniswine. (a) Representative images of calbindin+, calretinin+, and parvalbumin+ interneurons in the motor cortex of NF1 +/ex42del and wild‐type miniswine at 18 months of age. (b) Significantly lower number of calbindin+ interneurons in 14‐month‐old male NF1 +/ex42del animals. Unpaired t‐tests. n = 3‐4/group. *p < 0.05. Mean ± SEM. Scale bar = 100 μm.
NF1 +/ex42del swine show sex and neurofibroma‐dependent memory/learning and motor deficits that worsen over time
We previously showed memory and learning deficits in an initial characterization of 9‐month‐old male NF1 +/ex42del animals. 16 To determine if these deficits were consistent over time, we measured memory and learning abilities at 8, 12, and 18 months of age using a simple T‐maze. Of note, a subset of male animals began forming cutaneous neurofibromas (cNFs) during the study period (male‐specific – see 16 , 29 , 30 and Table S1). Therefore, to determine if behavioral abnormalities were specific to the presence of cNFs, NF1 +/ex42del data were further split into animals that had cNFs (males only) or did not have cNFs (mix of males and females). Overall, there were no differences in learned responses (Acquisition Day 1 and 2) or new task responses (Reversal Day 1 and 2) at 8 months of age (Figure 6a–d). NF1 +/ex42del males with cNFs showed deficits in maintaining acquired learning (Acquisition Day 2) as well as in learning a new task (Reversal Day 1 and 2) beginning at 12 months of age, which progressively worsened by 18 months of age (Figure 6b–d). In fact, 18‐month‐old male animals with cNFs did not perform well enough in the 18‐month acquisition trials to be tested in the reversal trials at that age (see reduced n, Figure 6c,d), indicating the severity of the phenotype. There were no significant differences between wild‐type and NF1 +/ex42del animals without cNFs at any timepoint (mix of males and females), suggesting a cNF and male‐specific deficit. That result is partially consistent with NF1 male patients with symptoms of autism spectrum disorder 31 and in NF1 mouse models with male‐specific spatial learning deficits. 32
FIGURE 6.
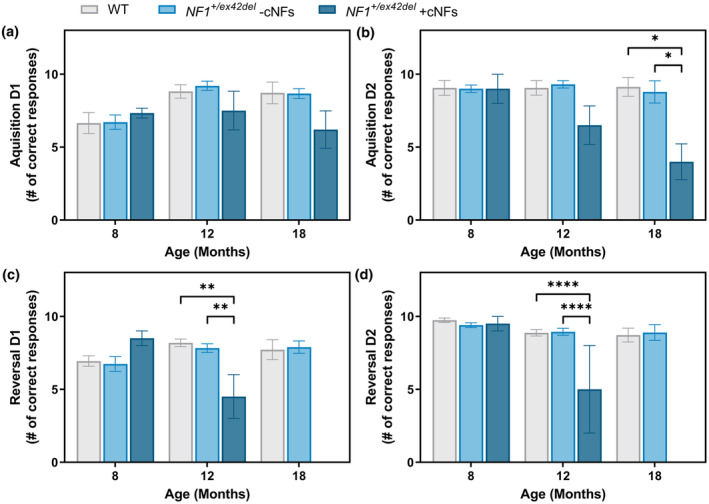
Simple T‐maze indicates delayed and sustained memory and learning deficits in male NF1 +/ex42del miniswine with cNFs. (a) T‐maze results for acquisition day 1, indicating no significant differences between wild‐type and NF1 +/ex42del miniswine at any age group. (b) T‐maze results for acquisition day 2, indicating learning delays in 18‐month‐old male NF1 +/ex42del miniswine with cNFs. (c) T‐maze results for reversal day 1, indicating learning delays between wild‐type and male NF1 +/ex42del miniswine with cNFs at 12 and 18 months of age. (d) T‐maze results for reversal day 2, indicating continued learning delays in 12‐ and 18‐month‐old male NF1 +/ex42del miniswine with cNFs. Mixed model ANOVA, Tukey post hoc. n = 3–9/group, *p < 0.05, **p < 0.01, ****p < 0.0001. Mean ± SEM.
To assess whether NF1 +/ex42del miniswine exhibits motor difficulties similar to those in patients with NF1, we utilized a kinematic walkway and measured 186 parameters from the forelegs of all miniswine as they walked naturally. A PCA was used to reduce the 186 parameters to the 20 most influential parameters, 33 , 34 which showed cNF‐burdened NF1 +/ex42del males to have significant gait abnormalities at 12 and 15 months of age (Figure 7a). These abnormalities were lost at 18 months of age, though this is likely due to fewer animals being available to test at this timepoint. Similarly, in the hindlegs, cNF‐burdened NF1 +/ex42del males had significant gait abnormalities at 12 and 15 months of age (Figure S5). In the forelegs, cNF‐burdened NF1 +/ex42del males took significantly longer to complete their stride (Figure 7b), took significantly longer to complete their individual steps (Figure 7c), completed their steps with more pressure (Figure 7d), and appeared to have impaired balance/coordination as demonstrated by an increased walk ratio (Figure 7e). Additionally, cNF‐burdened NF1 +/ex42del males took significantly fewer steps/minute (Figure 7f), had a decrease in walk velocity (Figure 7g), an increase in time spent with their feet on the ground (Figure 7h), and a decrease in time spent with their feet off the ground (Figure 7i). These phenotypes were not present in animals without cNFs (a mix of males and females).
FIGURE 7.
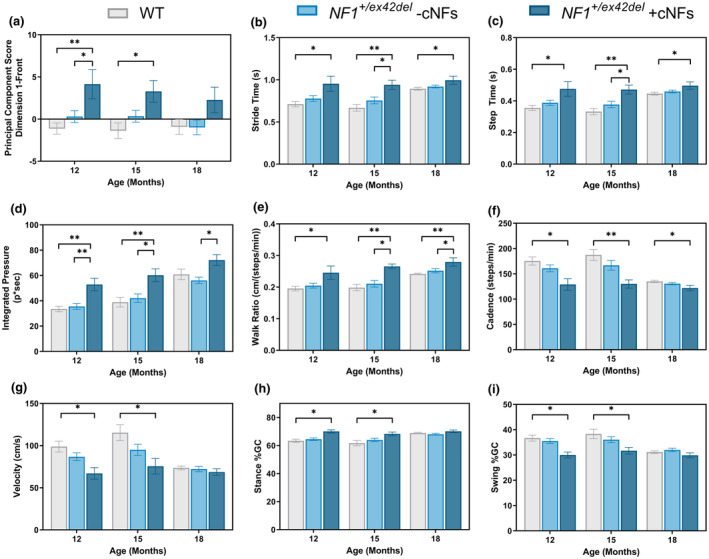
Male NF1 +/ex42del miniswine with cNFs show a cautious walking strategy in a kinematic gait analysis. (a) Principal component score of most significant variables indicates that male NF1 +/ex42del miniswine with cNFs have significant gait abnormalities at 12 and 15 months of age. There is lost at 18 months of age, likely due to a lower n. Among these top variables, cNF‐burdened NF1 +/ex42del males had (b) increased stride time, (c) increased step time, (d) increased integrated pressure, (e) increased walk ratio, (f) decreased cadence, (g) decreased walk velocity, (h) increase in stance (time with feet on ground), and (i) decrease swing (d time spent with feet off the ground). PCA implemented in R utilizing the FactorMineR package. n = 3–4 NF1 +/ex42del + cNFs; n = 6–22 NF1 +/ex42del ‐cNFs; n = 7–17 WT. One‐way ANOVA, Tukey post hoc. *p < 0.05, **p < 0.01. Mean ± SEM.
DISCUSSION
Here we demonstrate multiple cellular, cell signaling, and behavioral abnormalities in NF1 +/ex42del miniswine, with many phenotypes dependent on sex and cNF burden. We previously reported that male NF1 +/ex42del miniswine develop cNFs and present with poor quality of life measurements that correlated with tumor burden, while their female counterparts do not. 29 , 30 In our current study, we describe similar male‐specific changes in astrocyte reactivity, increases in microglial populations, decreased numbers of parvalbumin+ and calbindin+ interneurons, deficits in memory/learning tasks, and abnormal, cautious walking strategies. This is consistent with some aspects of the human disease, as male individuals with NF1 have been reported to develop cNFs earlier than females and have shown higher rates of ASD. 35 Nf1 mutant mice have similarly shown male‐specific memory/learning abnormalities. 36 In contrast, female individuals with NF1 and Nf1 mutant mice have more severe optic gliomas and visual deficits, with female‐specific retinal cell loss and thinning. 2 , 32 , 37 Additionally, internal tumors, that may transform into MPNSTs, have been reported to develop earlier in female patients and increase during puberty, with several studies supporting a link between puberty or pregnancy and cNF presentation in female NF1 individuals (singular cNFs and pregnancy 38 and mouse models). 35
Interestingly, female NF1 +/ex42del miniswine presented a less prominent disease phenotype although they still display defects consistent with NF1. Key female‐specific features observed in our mutant pig model include increased expression of mature oligodendrocytes, irregular inhibitory signaling (decreased ratio of GABA: Glutamate), and abnormalities in reaction to thermal stimulation. 30 It is possible that cNFs and other NF1‐associated presentations did not develop due to a lack of hormonal components that are present in female individuals with NF1. Therefore, further research is necessary to explore the relationship of sex hormones on NF1 presentations in female NF1 +/ex42del miniswine to determine the full utility of this swine model.
Microglia are known for their reactive states in neurodegenerative and cognitive diseases, such as Alzheimer's, 39 Parkinson's, 40 and Huntington's disease, in which severity of the disease correlates with an increase in activated microglia. 41 Although we were surprised to find no significant difference in the number of reactive amoeboid Iba+ microglia in NF1 +/ex42del miniswine compared with wild‐type control, we found an increase in the total number of Iba1+ microglia in the prefrontal cortex and hippocampus of male NF1 +/ex42del miniswine. In injury models, increased numbers of microglia have been described as one of the first responses following injury, including optic nerve injury 27 and sciatic nerve injury, 42 and a similar response may be occurring in our swine model. Notably, these reported increases in microglial activation in the brain occur after a peripheral sciatic nerve injury, suggesting a possible explanation for the increased microglial population in male animals. Indeed, as female NF1 +/ex42del miniswine show no cNF presentation, and additionally no aberrant microglial presence, perhaps similar pathways contribute to microglial proliferation and cNF formation in NF1 miniswine.
Cognitive and motor abnormalities in Nf1 mouse models have also been related to an imbalance of inhibitory signaling, 11 , 12 , 13 which is similar to the inhibitory imbalances that we demonstrated in our miniswine via decreased GABA: Glutamate ratios. While other measurements of inhibitory imbalances (such as interneuron counts) did not show strong changes in our study, our results concur with previously described changes in both individuals with NF1 and mouse models and should be explored with a larger miniswine sample size. For example, the trend toward a reduction in GABA: Glutamate ratio in the motor cortex of NF1 +/ex42del miniswine mimics the decreased cortical GABA concentration and GABA receptors in people with NF1 43 and is suggested to contribute to cortical dysregulation. Whereas in the hippocampus, an increased GABA: Glutamate ratio is similar to the excitatory‐inhibitory imbalance seen in ASD, 44 within a Nf1 mouse model of autism, 24 and in the hippocampus of juvenile female Nf1 mice. 36 Altered inhibitory signaling is not unique to NF1 cognitive dysfunction. Imbalanced excitatory to inhibitory neurotransmission in the motor cortex of NF1 children has been linked to motor abnormalities, 45 and high releases of GABA have been reported in Alzheimer's disease 46 and Parkinson's disease. Specifically, the GABAergic output from the basal ganglia to neurons in the thalamus and cortex has been proposed to be overactive in Parkinson's disease, causing GABAergic inhibition in the cortico‐basal ganglia loop and subsequent decreases in velocity and overall movement. 47 Interestingly, we report that male NF1 +/ex42del miniswine exhibit gait abnormalities resembling an early Parkinsonian gait characterized by longer step time and reduced cadence, 8 suggesting the possibility that enhanced inhibition in the thalamocortical loop of the cortico‐basal ganglia circuit is involved in NF1 motor abnormalities.
We found an increase in “integrated pressure” when cNF‐burdened male NF1 +/ex42del miniswine steps, which could reflect foot stomping as the animal walks. This may be indicative of peripheral neuropathy or steppage gait issues, where abnormalities stem from muscle weakness. The specific stomping characteristics seen in our study are correlated to deficits of proprioception (limb and joint positioning) that are commonly seen in a sensory ataxic gait, 48 where individuals cannot determine where their feet are located in relationship to the ground, the stance is widened, the gait is unsteady, and the foot stomps down. However, as gait abnormalities were preferentially found in male NF1 +/ex42del miniswine with cNFs, it is unclear whether the differences in gait are true motor differences or the result of increased pain response to a tumor. Indeed, we previously reported an enhanced sensitization to mechanical stimulation, poor sleep, and enhanced resting activity in cNF‐burdened NF1 +/ex42del miniswine. 30 Further research is needed to parse out motor deficits from responses to pain.
Taken together, we report cellular, cell signaling, and behavioral abnormalities in NF1 +/ex42del miniswine that are consistent with NF1 disease phenotypes seen in NF1 patients and mouse models. We suggest that the NF1 +/ex42del miniswine represent a powerful, clinically relevant animal model to study the NF1 disease, including the precise mechanisms contributing to cognitive and gait dysfunction as well as the interplay between sex, cognitive dysfunction, tumor growth, pain, and nervous/muscular function. Ultimately, this unique platform may prove instrumental in developing improved methods for disease detection and monitoring while enabling therapeutic testing, in an animal that more closely phenocopies humans, to identify better treatments for NF1 patients.
Future considerations and limitations
One of the logistical challenges in the characterization of a large animal model is to find applicable brain regions that can be sectioned, labeled, and imaged with minimal to no fragmentation or generation of artifacts. Unfortunately, the anterior cingulate was more fragile than any of the other brain regions we studied and resulted in the loss of tissue or fragmentation to an extent that analysis was not possible. This resulted in an n = 2 in the MBP‐labeled tissues from 14 M female NF1 +/ex42del miniswine and n = 2 in the GFAP‐labeled tissues from 14 M male wild‐type miniswine. Future studies will need to consider using thicker tissue sections or increasing the number of sections per experiment to prevent loss and fragmentation, therefore increasing the overall animal numbers per experiment.
Large animal models of disease are complicated by the lack of species‐specific tools, such as antibodies, protocols, and behavior equipment. We have tried to optimize markers for oligodendrocyte precursor cells (Olig2), in the brains of our swine models. Though we were able to validate that these markers did label the oligodendrocytes, we also had nonspecific labeling of neurons and in many of the brain tissues, there was background. 49 A huge challenge in developing more translatable large animal models of diseases will persist if appropriate reagents are not readily available.
Though all behavior assays have their limitations, rescue experiments would need to be designed with the specific social behavior of swine in mind, which is considerably different than that of rodent models. For example, we have tried to assay social anxiety in our previous NF1 +/ex42del miniswine studies and found no significant differences. 16 Our team has utilized the simple T‐maze and gait walkway in multiple miniswine studies and has optimized these methods to produce repeatable and consistent results. However, we have had mixed results with the FitBark activity monitor, which may be due to the short time that the device was worn by each animal, differences between genotypes, or differences in other factors such as environment. 16 , 30 , 33 , 50 Utilizing the Fitbark for longer periods of time (2 weeks or greater) in future longitudinal miniswine studies might improve data quality, reduce variability, and add an important component to these behavior studies.
AUTHOR CONTRIBUTIONS
V.J.S., K.A.W., P.L.N., T.B.J., and H.G.L. wrote the manuscript; V.J.S., K.A.W., D.K.M., D.E.Q., R.K., C.S.R, and J.M.W. designed the research; V.J.S., K.A.W., D.K.M., P.L.N., T.B.J., H.G.L., R.D.D. M.J.R. performed the research; V.J.S., K.A.W., P.L.N., T.B.J., H.G.L., and D.K.M. analyzed the data; and D.E.Q., R.K., C.S.R, and J.M.W. contributed new reagents/analytical tools.
FUNDING INFORMATION
Synodos for NF1 program at the Children's Tumor Foundation to JMW and NIH P20GM103620.
CONFLICT OF INTEREST STATEMENT
JMW is an employee of Amicus Therapeutics and holds an interest in the form of stock‐based compensation. The study presented here is unrelated to the interests of Amicus. CSR is an employee of Exemplar, a company that is commercializing the NF1 minipig model. All other authors declare no conflicts of interest for this work.
Supporting information
Data S1:
Swier VJ, White KA, Negrão de Assis PL, et al. NF1+/ex42del miniswine model the cellular disruptions and behavioral presentations of NF1‐associated cognitive and motor impairment. Clin Transl Sci. 2024;17:e13858. doi: 10.1111/cts.13858
Vicki J. Swier and Katherine A. White are co‐first authors and contributed equally to this study.
REFERENCES
- 1. Messiaen LM, Callens T, Mortier G, et al. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum Mutat. 2000;15:541‐555. doi: [DOI] [PubMed] [Google Scholar]
- 2. Warrington NM, Sun T, Luo J, et al. The cyclic AMP pathway is a sex‐specific modifier of glioma risk in type I neurofibromatosis patients. Cancer Res. 2015;75:16‐21. doi: 10.1158/0008-5472.CAN-14-1891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Torres Nupan MM, Velez Van Meerbeke A, López Cabra CA, Herrera Gomez PM. Cognitive and behavioral disorders in children with neurofibromatosis type 1. Front Pediatr. 2017;5:227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang Y, Kim E, Wang X, et al. ERK inhibition rescues defects in fate specification of Nf1‐deficient neural progenitors and brain abnormalities. Cell. 2012;150:816‐830. doi: 10.1016/j.cell.2012.06.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hyman SL, Shores A, North KN. The nature and frequency of cognitive deficits in children with neurofibromatosis type 1. Neurology. 2005;65:1037‐1044. doi: 10.1212/01.wnl.0000179303.72345.ce [DOI] [PubMed] [Google Scholar]
- 6. Ullrich NJ, Ayr L, Leaffer E, Irons MB, Rey‐Casserly C. Pilot study of a novel computerized task to assess spatial learning in children and adolescents with neurofibromatosis type 1. J Child Neurol. 2010;25:1195‐1202. doi: 10.1177/0883073809358454 [DOI] [PubMed] [Google Scholar]
- 7. Morris SM, Acosta MT, Garg S, et al. Disease burden and symptom structure of autism in Neurofibromatosis type 1: a study of the international NF1‐ASD consortium team (INFACT). JAMA Psychiatry. 2016;73:1276‐1284. doi: 10.1001/jamapsychiatry.2016.2600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Champion JA, Rose KJ, Payne JM, Burns J, North KN. Relationship between cognitive dysfunction, gait, and motor impairment in children and adolescents with neurofibromatosis type 1. Dev Med Child Neurol. 2014;56:468‐474. doi: 10.1111/dmcn.12361 [DOI] [PubMed] [Google Scholar]
- 9. Johnson BA, MacWilliams BA, Carey JC, Viskochil DH, D'Astous JL, Stevenson DA. Motor proficiency in children with neurofibromatosis type 1. Pediatric Physical Therapy. 2010;22:344‐348. doi: 10.1097/PEP.0b013e3181f9dbc8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cornett KM, North KN, Rose KJ, Burns J. Muscle weakness in children with neurofibromatosis type 1. Dev Med Child Neurol. 2015;57:733‐736. doi: 10.1111/dmcn.12777 [DOI] [PubMed] [Google Scholar]
- 11. Costa RM, Federov NB, Kogan JH, et al. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature. 2002;415:526‐530. doi: 10.1038/nature711 [DOI] [PubMed] [Google Scholar]
- 12. Cui Y, Costa RM, Murphy GG, et al. Neurofibromin regulation of ERK signaling modulates GABA release and learning. Cell. 2008;135:549‐560. doi: 10.1016/j.cell.2008.09.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim E, Wang Y, Kim SJ, et al. Transient inhibition of the ERK pathway prevents cerebellar developmental defects and improves long‐term motor functions in murine models of neurofibromatosis type 1. eLife. 2014;3:e05151. doi: 10.7554/eLife.05151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sanchez‐Ortiz E, Cho W, Nazarenko I, Mo W, Chen J, Parada LF. NF1 regulation of RAS/ERK signaling is required for appropriate granule neuron progenitor expansion and migration in cerebellar development. Genes Dev. 2014;28:2407‐2420. doi: 10.1101/gad.246603.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sutton LP, Muntean BS, Ostrovskaya O, et al. NF1‐cAMP signaling dissociates cell type‐specific contributions of striatal medium spiny neurons to reward valuation and motor control. PLoS Biol. 2019;17:e3000477. doi: 10.1371/journal.pbio.3000477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. White KA, Swier VJ, Cain JT, et al. A porcine model of neurofibromatosis type 1 that mimics the human disease. JCI Insight. 2018;3:e120402. doi: 10.1172/jci.insight.120402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bible E, Gupta P, Hofmann SL, Cooper JD. Regional and cellular neuropathology in the palmitoyl protein thioesterase‐1 null mutant mouse model of infantile neuronal ceroid lipofuscinosis. Neurobiol Dis. 2004;16:346‐359. doi: 10.1016/j.nbd.2004.02.010S0969996104000440 [DOI] [PubMed] [Google Scholar]
- 18. Meyerholz DK, Beck AP. Principles and approaches for reproducible scoring of tissue stains in research. Lab Investig. 2018;98:844‐855. doi: 10.1038/s41374-018-0057-0 [DOI] [PubMed] [Google Scholar]
- 19. Monge‐Acuna AA, Fornaguera‐Trias J. A high performance liquid chromatography method with electrochemical detection of gamma‐aminobutyric acid, glutamate and glutamine in rat brain homogenates. J Neurosci Methods. 2009;183:176‐181. doi: 10.1016/j.jneumeth.2009.06.042 [DOI] [PubMed] [Google Scholar]
- 20. Perucho J, Gonzalo‐Gobernado R, Bazan E, et al. Optimal excitation and emission wavelengths to analyze amino acids and optimize neurotransmitters quantification using precolumn OPA‐derivatization by HPLC. Amino Acids. 2015;47:963‐973. doi: 10.1007/s00726-015-1925-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Beraldi R, Chan CH, Rogers CS, et al. A novel porcine model of ataxia telangiectasia reproduces neurological features and motor deficits of human disease. Hum Mol Genet. 2015;24:6473‐6484. doi: 10.1093/hmg/ddv356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Karlsgodt KH, Rosser T, Lutkenhoff ES, Cannon TD, Silva A, Bearden CE. Alterations in white matter microstructure in neurofibromatosis‐1. PLoS One. 2012;7:e47854. doi: 10.1371/journal.pone.0047854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shilyansky C, Karlsgodt KH, Cummings DM, et al. Neurofibromin regulates corticostriatal inhibitory networks during working memory performance. Proc Natl Acad Sci USA. 2010;107:13141‐13146. doi: 10.1073/pnas.1004829107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Goncalves J, Violante IR, Sereno J, et al. Testing the excitation/inhibition imbalance hypothesis in a mouse model of the autism spectrum disorder: in vivo neurospectroscopy and molecular evidence for regional phenotypes. Mol Autism. 2017;8:47. doi: 10.1186/s13229-017-0166-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rizvi TA, Akunuru S, de Courten‐Myers G, Switzer RC III, Nordlund ML, Ratner N. Region‐specific astrogliosis in brains of mice heterozygous for mutations in the neurofibromatosis type 1 (Nf1) tumor suppressor. Brain Res. 1999;816:111‐123. doi: 10.1016/s0006-8993(98)01133-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Simmons GW, Pong WW, Emnett RJ, et al. Neurofibromatosis‐1 heterozygosity increases microglia in a spatially and temporally restricted pattern relevant to mouse optic glioma formation and growth. J Neuropathol Exp Neurol. 2011;70:51‐62. doi: 10.1097/NEN.0b013e3182032d37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Davis BM, Salinas‐Navarro M, Cordeiro MF, Moons L, De Groef L. Characterizing microglia activation: a spatial statistics approach to maximize information extraction. Sci Rep. 2017;7:1576. doi: 10.1038/s41598-017-01747-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Silva G, Duarte IC, Bernardino I, Marques T, Violante IR, Castelo‐Branco M. Oscillatory motor patterning is impaired in neurofibromatosis type 1: a behavioural, EEG and fMRI study. J Neurodev Disord. 2018;10:11. doi: 10.1186/s11689-018-9230-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Uthoff J, Larson J, Sato TS, et al. Longitudinal phenotype development in a minipig model of neurofibromatosis type 1. Sci Rep. 2020;10:5046. doi: 10.1038/s41598-020-61251-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Khanna R, Moutal A, White KA, et al. Assessment of nociception and related quality‐of‐life measures in a porcine model of neurofibromatosis type 1. Pain. 2019;160:2473‐2486. doi: 10.1097/j.pain.0000000000001648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Adviento B, Corbin IL, Widjaja F, et al. Autism traits in the RASopathies. J Med Genet. 2014;51:10‐20. doi: 10.1136/jmedgenet-2013-101951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Diggs‐Andrews KA, Brown JA, Gianino SM, Rubin JB, Wozniak DF, Gutmann DH. Sex is a major determinant of neuronal dysfunction in neurofibromatosis type 1. Ann Neurol. 2014;75:309‐316. doi: 10.1002/ana.24093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Swier VJ, White KA, Johnson TB, et al. A novel porcine model of CLN2 batten disease that recapitulates patient phenotypes. Neurotherapeutics. 2022;3:1905‐1919. doi: 10.1007/s13311-022-01296-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Johnson TB, Brudvig JJ, Lehtimäki KK, et al. A multimodal approach to identify clinically relevant biomarkers to comprehensively monitor disease progression in a mouse model of pediatric neurodegenerative disease. Prog Neurobiol. 2020;189:101789. doi: 10.1016/j.pneurobio.2020.101789 [DOI] [PubMed] [Google Scholar]
- 35. Sbidian E, Duong TA, Valeyrie‐Allanore L, Wolkenstein P. Neurofibromatosis type 1: neurofibromas and sex. Br J Dermatol. 2016;174:402‐404. doi: 10.1111/bjd.13966 [DOI] [PubMed] [Google Scholar]
- 36. Santos S, Martins B, Sereno J, Martins J, Castelo‐Branco M, Gonçalves J. Neurobehavioral sex‐related differences in Nf1+/− mice: female show a “camouflaging”‐type behavior. Biol Sex Differ. 2023;14:24. doi: 10.1186/s13293-023-00509-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Toonen JA, Solga AC, Ma Y, Gutmann DH. Estrogen activation of microglia underlies the sexually dimorphic differences in Nf1 optic glioma‐induced retinal pathology. J Exp Med. 2017;214:17‐25. doi: 10.1084/jem.20160447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Well L, Jaeger A, Kehrer‐Sawatzki H, et al. The effect of pregnancy on growth‐dynamics of neurofibromas in Neurofibromatosis type 1. PLoS One. 2020;15:e0232031. doi: 10.1371/journal.pone.0232031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McGeer PL, Itagaki S, Tago H, McGeer EG. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA‐DR. Neurosci Lett. 1987;79:195‐200. doi: 10.1016/0304-3940(87)90696-3 [DOI] [PubMed] [Google Scholar]
- 40. Gerhard A, Pavese N, Hotton G, et al. In vivo imaging of microglial activation with [11C](R)‐PK11195 PET in idiopathic Parkinson's disease. Neurobiol Dis. 2006;21:404‐412. doi: 10.1016/j.nbd.2005.08.002 [DOI] [PubMed] [Google Scholar]
- 41. Pavese N, Gerhard A, Tai YF, et al. Microglial activation correlates with severity in Huntington disease: a clinical and PET study. Neurology. 2006;66:1638‐1643. doi: 10.1212/01.wnl.0000222734.56412.17 [DOI] [PubMed] [Google Scholar]
- 42. Barcelon EE, Cho WH, Jun SB, Lee SJ. Brain microglial activation in chronic pain‐associated affective disorder. Front Neurosci. 2019;13:213. doi: 10.3389/fnins.2019.00213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Violante IR, Patricio M, Bernardino I, et al. GABA deficiency in NF1: a multimodal [11C]‐flumazenil and spectroscopy study. Neurology. 2016;87:897‐904. doi: 10.1212/WNL.0000000000003044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Inui T, Kumagaya S, Myowa‐Yamakoshi M. Neurodevelopmental hypothesis about the etiology of autism Spectrum disorders. Front Hum Neurosci. 2017;11:354. doi: 10.3389/fnhum.2017.00354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Doherty AC, Huddleston DA, Horn PS, et al. Motor function and physiology in youth with Neurofibromatosis type 1. Pediatr Neurol. 2023;143:34‐43. doi: 10.1016/j.pediatrneurol.2023.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wu Z, Guo Z, Gearing M, Chen G. Tonic inhibition in dentate gyrus impairs long‐term potentiation and memory in an Alzheimer's [corrected] disease model. Nat Commun. 2014;5:4159. doi: 10.1038/ncomms5159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 1990;13:281‐285. doi: 10.1016/0166-2236(90)90110-v [DOI] [PubMed] [Google Scholar]
- 48. Baker JM. Gait Disorders. Am J Med. 2018;131:602‐607. doi: 10.1016/j.amjmed.2017.11.051 [DOI] [PubMed] [Google Scholar]
- 49. Swier VJ, White KA, Meyerholz DK, et al. Validating indicators of CNS disorders in a swine model of neurological disease. PLoS One. 2020;15:e0228222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Swier VJ, White KA, Johnson TB, et al. A novel porcine model of CLN3 batten disease recapitulates clinical phenotypes. Dis Model Mech. 2023;16:dmm050038. doi: 10.1242/dmm.050038 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1: