

Key Points
-
•
Generation of a lineage-specific STAT5BN642H transgenic mouse model that develops NK-cell leukemia.
-
•
Leukemic NK cells with a STAT5B gain-of-function mutation share a unique transcriptional profile in mice and human patients.
Visual Abstract
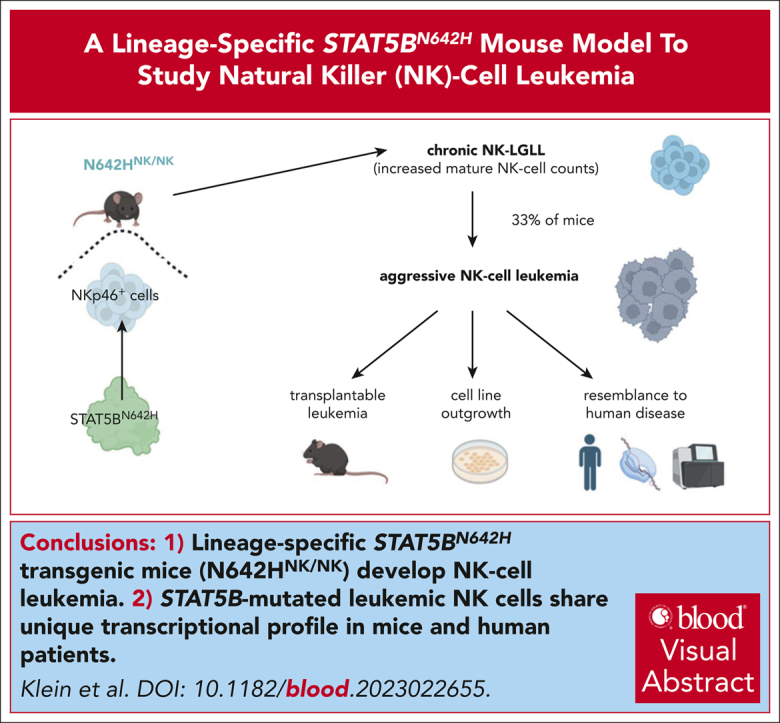
Abstract
Patients with T- and natural killer (NK)-cell neoplasms frequently have somatic STAT5B gain-of-function mutations. The most frequent STAT5B mutation is STAT5BN642H, which is known to drive murine T-cell leukemia, although its role in NK-cell malignancies is unclear. Introduction of the STAT5BN642H mutation into human NK-cell lines enhances their potential to induce leukemia in mice. We have generated a mouse model that enables tissue-specific expression of STAT5BN642H and have selectively expressed the mutated STAT5B in hematopoietic cells (N642Hvav/+) or exclusively in NK cells (N642HNK/NK). All N642Hvav/+ mice rapidly develop an aggressive T/NKT-cell leukemia, whereas N642HNK/NK mice display an indolent NK-large granular lymphocytic leukemia (NK-LGLL) that progresses to an aggressive leukemia with age. Samples from patients with NK-cell leukemia have a distinctive transcriptional signature driven by mutant STAT5B, which overlaps with that of murine leukemic N642HNK/NK NK cells. To our knowledge, we have generated the first reliable STAT5BN642H-driven preclinical mouse model that displays an indolent NK-LGLL progressing to aggressive NK-cell leukemia. This novel in vivo tool will enable us to explore the transition from an indolent to an aggressive disease and will thus permit the study of prevention and treatment options for NK-cell malignancies.
Introduction
Natural killer (NK)-cell malignancies are rare types of cancer that originate from the abnormal growth and proliferation of NK cells. They can be aggressive and challenging to treat. The World Health Organization distinguishes the following types of NK-cell neoplasms: extranodal NK/T-cell lymphoma (ENKL), aggressive NK-cell leukemia (ANKL), chronic active Epstein-Barr virus (EBV) infection of NK cells, and NK-large granular lymphocytic leukemia (NK-LGLL), formerly called chronic lymphoproliferative disorder of NK cells.1 ENKL and ANKL are EBV-positive and associated with a poor prognosis.1 NK-LGLL is usually EBV-negative, represents a subset of LGLL, and is largely an indolent disease that may develop into an aggressive NK-cell malignancy.1, 2, 3, 4, 5, 6 The factors that trigger the transformation of an indolent into an aggressive form of NK-cell leukemia are unknown.
Signal transducer and activator of transcription 5 (STAT5) is a crucial component of the Janus kinase (JAK)/STAT pathway essential for the survival, proliferation, and functionality of various hematopoietic cell types.7 In leukemia, STAT activity is often enhanced by aberrant upstream tyrosine kinase activation, copy number gains,8 or activating mutations within STAT3/5B proteins themselves.9, 10, 11, 12, 13, 14 STAT5 is the most frequently deregulated member of the JAK/STAT family in hematopoietic cancer.15, 16, 17 It comprises 2 individual genes, STAT5A and STAT5B, which express proteins with high homology.7,18, 19, 20 Although STAT5A and STAT5B have largely redundant functions, they both have some individual roles.18,21 STAT5B is the dominant gene product in T and NK cells and promotes their survival, proliferation, and cytotoxicity. Stat5b-deficient mice show reduced NK-cell numbers, and humans with STAT5B deficiency experience immunodeficiencies caused by impairment in T-, regulatory T-, and NK-cell differentiation and activation.18,22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32
Activating STAT5 mutations in hematological cancer predominantly occur in STAT5B.18 The most frequent STAT5B gain-of-function (GOF) mutation, STAT5BN642H, has been described in various forms of lymphoproliferative disorders, including T-cell lymphoma/leukemia, γδ–T-cell lymphoma, LGLL, and NK-cell malignancies.9,11,13,14,33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43 Activating STAT5B mutations have been detected in various NK-cell malignancies including cases of NK-LGLL and are linked to an aggressive clinical course.5,6,11,14,40,41,43, 44, 45, 46 STAT5BN642H gives a proliferative advantage to human NK cells,43 but whether it alone is sufficient to drive NK-cell leukemia remains unknown.
Compared with the wild-type allele, STAT5BN642H enhances dimer stability and causes elevated and prolonged STAT5B tyrosine phosphorylation. As a consequence, STAT5B target gene expression is increased.9, 10, 11,34,39,42 But even in the presence of activating STAT5B mutations, upstream cytokine signaling is necessary to activate JAK/STAT5 signaling.10,43,47 In immune cells, STAT5 signaling is induced by various cytokines, including interleukin-2 (IL-2), IL-7, and IL-15, which promote cell proliferation, survival, and maturation.48,49 IL-15 overexpression has been implicated in leukemogenesis because IL-15 transgenic mice develop NK- or NK T-cell (NKT-cell) leukemia.50, 51, 52 Another transgenic mouse model expressing human IL-15 and a transgenic mouse model expressing human STAT5BN642H in the hematopoietic system under the vav promoter (vav-N642H) develop a lethal CD8+ T-cell expansion.10,53 The rapid development of the highly aggressive CD8+ T-cell disease may mask and prevent the development of other malignancies because transplantation of NKT or γδ T cells from vav-N642H mice lead to leukemia development.9,54 To investigate the oncogenic potential of STAT5BN642H in other cell types, we generated a lox-stop-lox STAT5BN642H transgenic mouse model that enables lineage-restricted transgene expression driven by cell type-specific expression of the Cre recombinase. The use of Cre recombinase under an NKp46+ cell–specific promoter allows for us to study the role of STAT5BN642H in NK cells (N642HNK/NK mice). Here, the transcriptional changes associated with STAT5BN642H expression in leukemic NK cells closely resemble disease signatures of human NK-cell leukemia with STAT5B GOF mutations, enabling the assessment of further treatment options using the novel NK-cell leukemia model.
Materials and methods
Conditional N642H mouse generation
Rosa26 (R26)–targeted lox-stop-lox STAT5B and STAT5BN642H knockin mice were generated using a STOP-EGFP-ROSA-CAG targeting vector55 (obtained from Thomas Wunderlich, University of Cologne). This vector integrates into the R26 locus enabling transgene expression under the CAG promoter coupled to internal ribosome entry site (IRES)-controlled enhanced green fluorescent protein (eGFP) expression upon Cre recombinase–mediated excision of the floxed STOP cassette. C-terminally V5-tagged human STAT5B or STAT5BN642H transgenes were cloned downstream of the STOP cassette into the STOP-EGFP-ROSA-CAG targeting vector. A control construct lacking a transgene but containing IRES-controlled eGFP downstream of the STOP cassette was included. The 3 R26-LSL knockin lines B6-Gt(ROSA)26Sortm1(STAT5B-N642H)Biat, B6-Gt(ROSA)26Sortm2(STAT5B)Biat, and B6-Gt(ROSA)26Sortm3(EGFP)Biat were generated using the linearized vectors for electroporation into C57BL/6N embryonic stem (ES) cells (parental ES cell line C2; stock number: 011989-MU; Citation ID: RRID: MMRRC_011989-MU). Positively screened ES cell clones were injected into BALB/c blastocysts, transferred to pseudopregnant mice, and chimeric offspring were bred with C57BL/6N mice. Germ line transmission was confirmed by polymerase chain reaction (forward primers: 5'-GCACTTGCTCTCCCAAAGTCGCTC-3' [R26_wt_fw] and 5'-CGCCGACCACTACCAGCAGAACAC-3' [R26_EGFP_fw]; reverse primer: 5'-ACAACGCCCACACACCAGGTTAGC-3' [R26_wt_rev]) and selected for further crossing to Cre lines.
RNA-Seq of aged mouse NK cells
Frozen liver samples from aged nondiseased and diseased mice (healthy Cre negative [neg; n = 3], GFPNK/NK [n = 2], STAT5BNK/NK [n = 3], and N642HNK/NK [n = 5]; diseased N642HNK/NK [n = 8]) were thawed, and (GFP+) CD3–NK1.1+ NK cells (and additionally CD3+NK1.1+ cells from number 8) were sorted into a SMARTSeq lysis buffer using a CytoFlex SRT Sorter (Beckman Coulter). Libraries were constructed using the SMART-SEQ3 method56 at the Vienna BioCenter Core facilities, member of the Vienna BioCenter, Vienna, Austria. Sequencing was performed on an Illumina NovaSeq 6000 system (50-bp paired-end; Illumina, San Diego, CA). Sequencing reads were quality controlled using the FastQC software (version 0.12.1).57 Detailed RNA sequencing (RNA-Seq) analysis was performed as described in the supplemental Methods, available on the Blood website. The RNA-Seq data reported in this article have been deposited in the ArrayExpress database (accession ID: E-MTAB-13797).
Human patient data
Primary samples were obtained from bone marrow (BM) or peripheral blood of patients diagnosed with NK-cell neoplasms (n = 64) or healthy donors (peripheral blood mononuclear cells) after informed consent. Three patients harbored activating STAT5B mutations previously described44 (1 with STAT5BN642Hmutation, a second with STAT5BQ706L mutation, and the third with both STAT5BY665F and STAT5BV712E mutations). DNA and RNA were isolated from total leukocytes, followed by whole-genome sequencing and RNA-Seq at the Munich Leukemia Laboratory as previously described.58 Reads were aligned to human reference genome (GRCh37, Ensembl annotation) using Isaac aligner (v3.16.02.19). Tumor-unmatched normal variant calling was performed with a pool of sex-matched DNA (Promega, Madison, WI) using Strelka (v.2.4.7). Variants were queried against the gnomAD database (v.2.1.1) to remove common germ line calls and annotated with Ensembl VEP. Analysis was restricted to protein-altering and canonical splice-site variants. For transcriptome analysis, the TruSeq Total Stranded RNA kit was used, starting with 250 ng of total RNA, to generate RNA libraries following the manufacturer’s recommendations (Illumina). A total of 2× 100-bp paired-end reads were sequenced on the NovaSeq 6000 (Illumina), with a median of 50 million reads per sample. Reads were mapped with STAR aligner (v2.5.0a) to the human reference genome hg19 (RefSeq annotation). Gene- and transcript-specific read abundance was calculated with Cufflinks (v2.2.1). For gene expression analysis, estimated read counts for each gene were normalized by trimmed mean of M-values normalization, and the resulting log2 counts per million were used.
Statistical analysis
The appropriate statistical method was used based on testing for normal distribution and homogeneity of variance. Tests were performed using GraphPad Prism. The statistical test is indicated in the corresponding figure legends.
Animal experiments were discussed and approved by the ethics and animal welfare committee of the University of Veterinary Medicine Vienna and the national authority (Austrian Federal Ministry of Education, Science and Research) in accordance with good scientific practice guidelines and national legislation, under licenses BMBWF-68.205/0103-WF/V/3b/2015, BMBWF-68.205/0010-V/3b/2019, BMBWF-68.205/0174-V/3b/20182022-0.762.012, and 2023-0.108.862, 2022-0.404.452.
Results
N642Hvav/+ mice develop a hematopoietic malignancy
We generated mice with a human V5-tagged STAT5BN642H transgene and IRES-eGFP under the CAG promoter downstream of a lox-stop-lox-cassette integrated into the Rosa26 locus. The animals were crossed to Vav-Cre mice59 (N642Hvav/+) to study the effects of STAT5BN642H on the hematopoietic system (Figure 1A). We validated the presence of the STAT5BN642H transgene by analyzing the expression of eGFP and the V5 tag in N642Hvav/+ mice (Figure 1B; supplemental Figure 1A-B). N642Hvav/+ BM cells displayed increased levels of tyrosine-phosphorylated STAT5 (pYSTAT5) compared with control BM cells, although reduced levels when compared with the pYSTAT5 levels of vav-N642H mice10 (Figure 1B). 8-week-old N642Hvav/+ mice had an elevated BM cellularity and an enlarged hematopoietic stem cell pool under homeostatic conditions (supplemental Figure 1C-F). Numbers of erythroid cells (Ter119+) and NK cells (CD3–NK1.1+) were reduced, whereas T cells (CD3+CD4+ or CD3+CD8+), B cells (CD19+), and myeloid cells (CD11b+Gr1+) were increased in the BM (supplemental Figure 1G). At age 8 weeks, N642Hvav/+ mice showed splenomegaly (Figure 1C) with significantly expanded myeloid and B-cell compartments (Figure 1D). The peripheral blood of N642Hvav/+ mice lacked any significant alterations in the composition of leukocytes, except for a decrease in the frequency of CD4+ T cells (supplemental Figure 1H).
Figure 1.

N642Hvav/+ mice develop a hematopoietic malignancy. (A) Schematic overview of the generation of N642Hvav/+ mice. (B) V5, pYSTAT5, and STAT5A/B immunoblot analysis of BM cells from vav-N642H (n = 1), N642Hvav/+ (n = 4), and control (N642HSTOP/+) mice (n = 4) (left). α-TUBULIN served as loading control. Quantification of fold change (fc) of pYSTAT5 relative to total STAT5A/B levels, based on immunoblot analysis, from 8-week-old control and N642Hvav/+ BM cells (n = 4 per genotype; mean ± standard deviation [SD]) (right). (C) Relative quantification of spleen weights from 8-week-old control and N642Hvav/+ mice (n = 4 per genotype; mean ± SD). (D) Relative quantification (percentages out of living cells) (left) and total numbers (right) of myeloid cells (CD11b+Gr1+), B cells (CD19+), CD4+ T cells (CD3+CD4+), CD8+ T cells (CD3+CD8+), and NK cells (CD3–NK1.1+) in the spleen of 8-week-old control and N642Hvav/+ mice (n = 4 per genotype; mean ± SD). (E) Survival analysis of aged N642Hvav/+ (142-363 days of survival) and control mice (n ≥ 5 per genotype). (F) Representative pictures of spleens and LNs of aged control (left) and diseased N642Hvav/+ mice (right). (G) Relative quantification (left) and total numbers (right) of myeloid cells (CD11b+Gr1+), B cells (CD19+), CD4+ T cells (CD3+CD4+), CD8+ T cells (CD3+CD8+), and NK cells (CD3-NK1.1+) in the spleen of aged control (∼360 days old) and diseased N642Hvav/+ mice (end point analysis; n ≥ 5 per genotype; mean ± SD). (H) Hemacolor Rapid staining of blood smears from control and diseased N642Hvav/+ mice (1 representative picture per genotype). (I) Hematoxylin and eosin staining of lung tissue from control and diseased N642Hvav/+ mice (1 representative picture per genotype). Levels of significance were calculated using the unpaired t test (B-D and G) and the Mantel-Cox test (E). ∗P < .05; ∗∗P < .01; ∗∗∗P < .001; ∗∗∗∗P < .0001. LNs, lymph nodes.
Upon aging, all N642Hvav/+ mice developed a hematopoietic malignancy with a median survival of 186 days (Figure 1E). The mice suffered from reduced body weight and enlarged spleen and lymph nodes (Figure 1F-G; supplemental Figure 1I-J). They had significantly elevated numbers of mature hematopoietic cell types in spleen, blood, and lymph nodes but not in the BM (Figure 1G; supplemental Figure 1K-M). Cell numbers were elevated in all lineages, and no cell type was dominantly expanded (Figure 1G; supplemental Figure 1K-M). Blood smears of the diseased N642Hvav/+ mice showed leukemic blast-like cells (Figure 1H). The immune cell infiltration in the lungs was associated with a disruption of the regular lung architecture (Figure 1I).
Leukemic N642Hvav/+ T/NKT cells expand upon transplantation
To test whether the hematopoietic malignancy is transplantable, we injected Ly5.2+ splenic cells of diseased N642Hvav/+ and healthy aged control mice into immunodeficient NOD scid gamma (NSG) recipients (NOD.Cg-Prkdcˢᶜⁱᵈ Il2rgᵗᵐ1ᵂʲˡ/SzJ). All recipients of N642Hvav/+ splenocytes developed a disease within 3 months (Figure 2A). Ly5.2+ N642Hvav/+ cells densely infiltrated the BM, spleen, and lungs of the recipients, indicating development of leukemia (Figure 2B; supplemental Figure 2A-C). The infiltrating cell types were either T or NKT cells (Figure 2C-D). N642Hvav/+ T and NKT cells expressed almost exclusively T-cell receptor β (TCRβ) but not TCRγδ (Figure 2E-F). CD4+ and CD8+ T/NKT cells expanded in the recipient mice (Figure 2E,G). This argues against the idea that a specific T/NKT-cell subtype is driving leukemia. The diseases that develop in N642Hvav/+ mice closely resemble the T- /NKT-cell diseases observed in patients harboring the STAT5BN642H mutation.9
Figure 2.


Leukemic N642Hvav/+ T/NKT cells expand upon transplantation. Splenic cells (Ly5.2+) from either aged control or diseased N642Hvav/+ mice were IV injected into NSG mice and analyzed. (A) Survival analysis (n ≥ 4 per genotype). (B) Quantification of GFP levels among transplanted Ly5.2+ cells in BM, spleen, and lung of NSG mice injected with control or N642Hvav/+ cells (n ≥ 3 per genotype; mean ± standard deviation). (C) Quantification of the leukemia type developed in the NSG recipients with N642Hvav/+ transplantation. (D) Relative quantification (percentages out of injected Ly5.2+ N642Hvav/+ cells) of N642Hvav/+ myeloid cells (CD11b+Gr1+), B cells (CD19+), T cells (CD3+NK1.1–), NKT cells (CD3+NK1.1+), and NK cells (CD3–NK1.1+) in BM, lung, and spleen of 4 to 5 of the diseased NSG mice with N642Hvav/+transplantation. (E) Representative FACS plots of TCRβ and TCRγδ gating starting from Ly5.2+ T cells (CD3+NK1.1–) or NKT cells (CD3+NK1.1+) in BM of NSG mice with N642Hvav/+ transplantation. (F) Relative quantification of TCRβ or TCRγδ expression on transplanted Ly5.2+ N642Hvav/+ T cells (CD3+NK1.1–) or NKT cells (CD3+NK1.1+) (n ≥ 4). (G) Relative quantification of CD4 or CD8 expression on transplanted Ly5.2+TCRβ+CD3+NK1.1– or Ly5.2+ TCRβ+CD3+NK1.1+ N642Hvav/+ cells (n ≥ 4). Levels of significance were calculated using the Mantel-Cox test (A) or the Mann-Whitney test (B). ∗P < .05; ∗∗P < .01. FACS, fluorescence-activated cell sorting.
STAT5BN642H promotes cytokine independence of human NK-cell lines
Despite the presence of an activating STAT5B mutation, N642Hvav/+ mice did not develop NK-cell leukemia. To investigate the oncogenic potential of STAT5BN642H in NK cells, we ectopically expressed human STAT5B or STATBN642H in 2 human NK-cell lines (IMC-1 and KHYG-1) that harbor TP53 mutations but lack mutations in the JAK/STAT3/5 pathway.40,60 Transduction with STAT5B or STAT5BN642H decreased cell growth in standard IL-2 culture (100 U/mL) but gave a growth advantage at limited IL-2 concentrations (25 U/mL; supplemental Figure 3A-F). In the absence of IL-2, STAT5BN642H was required for cytokine-independent growth (Figure 3A-B; supplemental Figure 3C,F). This prompted us to test whether STAT5BN642H enhances the disease-initiating potential of KHYG-1 and IMC-1 cells in vivo (Figure 3C). When injected into NSG mice, STAT5BN642H-expressing IMC-1 cells accelerated disease onset significantly compared with parental and nonmutant STAT5B-expressing cells (Figure 3D). In contrast, neither the parental nor the STAT5B-overexpressing KHYG-1 cells caused disease in NSG recipient mice. All STAT5BN642H-transduced KHYG1 cells induced leukemia within 21 to 25 days (Figure 3D). The disease primarily manifested in the BM and the liver (Figure 3E-G; supplemental Figure 3G), both typical sites of disease manifestation in patients with NK-cell leukemia.11,14,61,62
Figure 3.

STAT5BN642H promotes cytokine independence of leukemic human NK cells. (A) IMC-1 and (B) KHYG-1 cell lines were transduced with nonmutant STAT5B (+STAT5B) or STAT5BN642H (+STAT5BN642H). As a control, cells were transduced with the empty vector, carrying only IRES-controlled eGFP (+GFP). After initial culture in presence of IL-2, transduced cells were completely deprived of IL-2. The percentage of transduced (GFP+) cells depicted as log2 fc relative to day 0 was monitored over time after cytokine withdrawal. (C) Schematic overview for transplantation of cytokine-independent STAT5BN642H transduced, IL-2 dependent nonmutant STAT5B transduced, and nontransduced IMC-1 and KHYG-1 cells into immunodeficient NSG mice. (D) Survival analysis of NSG mice that received transplantation with IMC-1 (left) and KHYG-1 (right) (n ≥ 3 per cell line). (E) Relative quantification (percentages out of living cells) of nonmutant STAT5B or STAT5BN642H transduced (GFP+) cells in blood, spleen, liver, and BM of NSG mice that received transplantation (n ≥ 2 per genotype; mean ± standard deviation). (F) Representative histograms for GFP signal within living cells in the liver (left) and BM (right) of NSG mice that received transplantation. (G) Representative images of hematoxylin and eosin stained liver and BM tissue from untransplanted (no NK) NSG mice and NSG mice that received transplantation with nonmutant STAT5B (KHYG1 + STAT5B) or STAT5BN642H transduced KHYG-1 cells (KHYG1 + STAT5BN642H). Levels of significance were calculated using the Mantel-Cox test (D) and the Mann-Whitney test (E). ∗P < .05; ∗∗P < .01; ∗∗∗P < .001; ∗∗∗∗P < .0001.
An NKp46+ cell–specific mouse model to study the oncogenic role of STAT5BN642H in NK cells
To investigate the oncogenic role of STAT5BN642H in NK cells in detail, we crossed the B6-Gt(ROSA)26Sortm1(STAT5B-N642H) mice to Ncr1-iCreTg mice63 (N642HNK/NK). These mice express STAT5BN642H exclusively in NKp46+ cells, which mainly represent mature NK cells.64,65 A human STAT5B transgene-expressing mouse strain (STAT5BNK/NK) and a strain solely expressing eGFP (GFPNK/NK) were used as controls (Figure 4A). All Cre-positive litters expressed GFP in NK cells (supplemental Figure 4A). We confirmed the V5-tagged transgene expression and elevated STAT5 protein levels in STAT5BNK/NK and N642HNK/NK NK cells compared with GFPNK/NK NK cells (Figure 4B).
Figure 4.

An NKp46+ cell–specific mouse model to study the oncogenic role of STAT5BN642H in NK cells. (A) Schematic overview of the generation of N642HNK/NK, STAT5BNK/NK, and GFPNK/NK mice. (B) pYSTAT5, STAT5A/B, and V5 immunoblot analysis of IL-2–cultured NK cells from GFPNK/NK, STAT5BNK/NK, and N642HNK/NK mice. IL-2–cultured NK cells were either directly lysed (+IL-2), lysed after being starved off IL-2 for 3 hours (starv.), or lysed after IL-2 starvation and restimulation with IL-2 and IL-15 (restim.). β-actin served as a loading control. Absolute numbers of NK cells (CD3–NK1.1+NKp46+) in blood (C) and spleen (D) of 8- to 12-week-old GFPNK/NK, STAT5BNK/NK, N642HNK/NK, and Cre neg control mice (n ≥ 5 per genotype; mean ± standard deviation [SD]). (E) Absolute numbers of NK cells (lineage [Lin] negative [CD3–CD19–Gr1–Ter119–] CD122+ cells) in BM of 8- to 12-week-old GFPNK/NK, STAT5BNK/NK, N642HNK/NK, and Cre neg mice (n ≥ 6 per genotype; mean ± SD). (F) Representative gating of NK-cell developmental stages among Lin–CD122+ cells within the BM, including NK1.1–NKp46– NK-cell precursors (NKPs), NK1.1+NKp46– immature (iNKs), and NK1.1+NKp46+ mature NK cells (mNKs) (left). Percentages of NKPs, iNKs, and mNKs among Lin–CD122+ BM cells (n ≥ 6 per genotype; mean ± SD) (right). (G) Schematic overview on splenic NK-cell maturation stages based on CD27 and CD11b expression (left). Percentages of CD27+CD11b–, CD27+CD11b+ and CD27–CD11b+ NK cells in the spleens of GFPNK/NK, STAT5BNK/NK, N642HNK/NK, and Cre neg mice (n ≥ 6 per genotype; mean ± SD) (right). (H) Apoptosis staining of splenic NK cells from GFPNK/NK, STAT5BNK/NK, N642HNK/NK, and Cre neg mice (n ≥ 4 per genotype; mean ± SD). Levels of significance were calculated using the 1-way analysis of variance (C-H). ∗P < .05; ∗∗P < .01; ∗∗∗P < .001; ∗∗∗∗P < .0001.
STAT5BN642H molecules have an enhanced capacity for self-dimerization and a reduced susceptibility to inactivation by dephosphorylation.9 Compared with STAT5BNK/NK NK cells, N642HNK/NK splenic NK cells displayed enhanced pYSTAT5 levels ex vivo already in an unstimulated state and more pronounced after IL-15 stimulation (supplemental Figure 4C-D). Elevated pYSTAT5 levels were also observed in vitro in IL-2–cultured N642HNK/NK splenic NK cells (Figure 4B; supplemental Figure 4B). Upon cytokine withdrawal, pYSTAT5 dephosphorylation was delayed in N642HNK/NK compared with control NK cells (Figure 4B; supplemental Figure 4B-D).
Adult N642HNK/NK mice show increased NK-cell numbers in blood, spleen, and BM (Figure 4C-E; supplemental Figure 4E-G). Notably, an expansion of NK cells was already detectable in the blood of N642HNK/NK mice as early as age 4 weeks (supplemental Figure 4H). Furthermore, N642HNK/NK mice display more mature NK cells in the BM and spleen than control strains (Figure 4F-G; supplemental Figure 4I). Furthermore, STAT5BN642H expression in NK cells was associated with increased survival and reduced apoptosis ex vivo (Figure 4H). The data are consistent with the idea that STAT5B promotes NK-cell survival and maturation.32 We found enhanced levels of granzyme B and perforin in N642HNK/NK NK cells (supplemental Figure 4J), supporting the role of STAT5B in regulating the levels of cytolytic molecules.22,23,30,32
N642HNK/NK mice develop NK-cell leukemia
The oncogenic potential of STAT5BN642H in NK cells was assessed by aging of the animals. Although the majority of N642HNK/NK mice maintained an indolent expansion of NK cells, 8 of 24 (∼33%) developed disease symptoms within 17 months. One STAT5BNK/NK and 1 Cre neg control mouse (of 50 control mice) were euthanized due to unspecific age-related symptoms without any signs of leukemia after 486 and 518 days, respectively (Figure 5A). The diseased N642HNK/NK mice consistently displayed a leukemic phenotype and suffered from significant body weight loss, splenomegaly, and an expansion of GFP+ cells in various organs, including spleen, liver, BM, and blood (Figure 5B-C; supplemental Figure 5A-B; supplemental Table 1). NK cells were the predominantly expanded cell type in the spleen of 5 of 8 diseased N642HNK/NK mice (numbers 1-5). One of the diseased mice (number 8) displayed an expansion of CD3+NK1.1+ γδ T cells, whereas 2 other mice (numbers 6 and 7) had a predominant expansion of GFP+ cells lacking both NK- and T-cell markers (CD3– TCR–NK1.1– NKp46– cells) (Figure 5D-E; supplemental Figure 5C; supplemental Table 1). Flow cytometric analysis revealed a downregulation of CD11b, CD49b, and NKp46 and a partial increase in CD27, CD49a, and NKG2D expression in diseased N642HNK/NK NK cells. KLRG1 expression was significantly increased in diseased N642HNK/NK NK cells (Figure 5F-G; supplemental Figure 5D-F). Similar deregulations were partially observed in NK cells from nondiseased N642HNK/NK mice, which however more closely resembled control NK cells (Figure 5B,F-G; supplemental Figure 5D; supplemental Table 1).
Figure 5.

STAT5BN642H induces NK-cell leukemia in mice. Cre neg, GFPNK/NK, STAT5BNK/NK, and N642HNK/NK mice were aged and monitored for signs of disease development. (A) Survival analysis of Cre neg, GFPNK/NK, STAT5BNK/NK, and N642HNK/NK mice (n ≥ 12 per genotype). (B) Flow cytometric analysis of GFP+ cells in different tissues of Cre neg, GFPNK/NK, STAT5BNK/NK, nondiseased N642HNK/NK, and diseased N642HNK/NK mice (n ≥ 8 per group; mean ± standard deviation [SD]). (C) Body weight quantification of Cre neg, GFPNK/NK, STAT5BNK/NK, nondiseased N642HNK/NK, and diseased N642HNK/NK mice (n ≥ 8 per group; mean ± SD). (D) Quantification of the leukemia type developed by N642HNK/NK mice. (E) Relative quantification of CD3+NK1.1– T cells, CD3+NK1.1+ NKT cells, CD3–NK1.1+ NK cells and CD3–NK1.1– “undifferentiated” cells among GFP+ cells in the spleen of diseased N642HNK/NK mice (n = 8). (F) Flow cytometric analysis of GFP+ cells in the liver of Cre neg, GFPNK/NK, STAT5BNK/NK, nondiseased N642HNK/NK, and diseased N642HNK/NK mice (n ≥ 8 per group; mean ± SD). Heat map depicts percentage of GFP+ cells out of living cells, percentages of double-negative (DN; CD27–CD11b–), CD27+, double-positive (DP; CD27+CD11b+), CD11b+, CD49a+, CD49b+, and KLRG1+ cells out of GFP+ NK cells (CD3–NK1.1+) and median fluorescence intensity (MFI) of NKp46, KLRG1, and NKG2D on GFP+ NK cells. (G) Percentages of KLRG1+ (left) and MFI (right) of KLRG1 on GFP+ NK cells in the spleen and liver of Cre neg, GFPNK/NK, STAT5BNK/NK, nondiseased N642HNK/NK, and diseased N642HNK/NK mice (n ≥ 8 per group; mean ± SD). Levels of significance were calculated using the Mantel-Cox test (A) and the 1-way analysis of variance (B-C and G). ∗P < .05; ∗∗P < .01; ∗∗∗P < .001; ∗∗∗∗P < .0001.
To confirm the expansion of leukemic cells as the disease cause, we transplanted splenic cells from the diseased N642HNK/NK mice (numbers 1-4 and 6-8) into NSG mice (Figure 6A). Transplantation initiated a fast-progressing leukemia in all recipient mice. The diseased mice suffered from weight loss, hepatosplenomegaly, anemia, and multiple organ infiltration. A leukemia with NK-cell phenotype was observed in ∼70% of the mice who received transplantation (Figure 6B-E; supplemental Figure 6A-H; supplemental Table 2). The transplantation of splenocytes from the mouse that had a lethal expansion of CD3+NK1.1+ TCRγδ+ T cells (number 8) verified a disease driven by STAT5BN642H-expressing γδ T cells. The transplantation of splenic cells with an accumulation of GFP+ CD3– TCR–NK1.1– NKp46– cells (numbers 6 and 7) revealed that the mice suffered more likely from an NK-cell leukemia than an acute leukemia of T-cell origin because there was a pronounced NK1.1+ but not a CD3+ or TCR+ population upon transplantation (supplemental Figure 6A-H). We observed leukemic blast-like cells in the blood of all diseased recipient mice (Figure 6F). To gauge the potential for immune evasion of the transformed N642HNK/NK NK cells, we performed parallel transplantations into NSG and Ly5.1 mice (Figure 6G). The N642HNK/NK leukemic cells incited disease in both NSG and Ly5.1 mice within a similar time frame and showed comparable organ infiltration (Figure 6H; supplemental Figure 6I-M). Furthermore, we established stable NK-cell lines from diseased N642HNK/NK mice and tested their cytokine dependency. All tested cell lines exhibited a dependency on IL-2. One cell line (number 3) displayed a growth advantage under reduced IL-2 levels (supplemental Figure 6N-O). In summary, N642HNK/NK mice predominantly develop a transplantable NK-cell leukemia, which evades immune recognition.
Figure 6.

STAT5BN642H induces transplantable NK-cell leukemia in mice. (A) Schematic overview of the IV transplantation of splenocytes from diseased N642HNK/NK mice into NSG mice. (B) Survival analysis of NSG mice that received transplantation with splenocytes of diseased N642HNK/NK mice (numbers 1-4 and 6-8) (n = 7). (C) Quantification of body weight (left), spleen to body weight ratio (middle), and liver to body weight ratio (right) of diseased NSG mice with N642HNK/NK transplantation and untransplanted controls (n ≥ 4 per group; mean ± standard deviation). (D) Flow cytometric analysis of GFP+ cells in different tissues of NSG mice that received transplantation with splenocytes from the different diseased N642HNK/NK mice (first transplant). (E) Quantification of the leukemia type developed by NSG recipients of diseased N642HNK/NK splenocytes. (F) Representative images of hematoxylin and eosin stained blood smears from NSG mice that received transplantation with splenocytes from diseased N642HNK/NK mouse number 7 (left) and number 3 (right). (G) Schematic overview of the IV transplantation of splenocytes from diseased NSG mice with N642HNK/NK transplantation into another round of NSG recipients or immunocompetent WT (Ly5.1+) mice (second transplant). (H) Survival analysis of NSG and Ly5.1 recipients with N642HNK/NK transplantation (n = 4 per group). Levels of significance were calculated using the unpaired t test (C). ∗P < .05; ∗∗P < .01. WT, wild type.
Leukemic N642HNK/NK NK cells display molecular features of patients with NK-cell leukemia harboring STAT5B GOF mutations
To investigate the transcriptional changes in STAT5BN642H-driven NK-cell leukemia, we performed RNA-Seq of ex vivo sorted NK cells from the livers of diseased N642HNK/NK and aged nondiseased control (Cre neg and GFPNK/NK), STAT5BNK/NK and N642HNK/NK mice, and γδ T cells of the diseased N642HNK/NK mouse 8. Diseased N642HNK/NK NK cells displayed a distinct transcriptional profile (supplemental Figure 7A). The leukemic γδ T cells (number 8) clustered closely to the leukemic NK cells (supplemental Figure 7A). We identified significant differentially expressed genes (DEGs) in diseased N642HNK/NK NK cells compared with all genotypes (vs controls: 888 DEGs; vs STAT5BNK/NK: 997 DEGs; vs nondiseased N642HNK/NK: 1038 DEGs), mainly upregulated (supplemental Figure 7B-C; supplemental Table 3). We focused on the DEGs in NK cells from diseased N642HNK/NK mice vs controls for further analysis. To test the identified DEGs for similarities to patients with NK-cell leukemia harboring STAT5B GOF mutations, we analyzed RNA-Seq data from 64 patients with NK-cell leukemia, showing a different transcriptional profile to healthy controls (peripheral blood mononuclear cells) and other leukemias. We subdivided the patients with NK-cell leukemia according to their JAK/STAT mutations: 3 with STAT5B GOF mutations44 (NK-cell leukemia [STAT5B GOF]), 18 with STAT3 mutations (NK-cell leukemia [STAT3 mut]), 1 with a JAK1 mutation (NK-cell leukemia [JAK1 mut]), and 44 without JAK/STAT mutations (NK-cell leukemia) (Figure 7A). Comparison of the leukemic mutant STAT5B-driven DEGs between mouse (diseased N642HNK/NK vs control) and human (NK-cell leukemia [STAT5B GOF] vs NK-cell leukemia) identified a set of 135 common DEGs (Figure 7B; supplemental Table 4). Commonly upregulated genes included genes with oncogenic function (eg, Rras2 and Mybl1), whereas genes with tumor suppressive and proinflammatory activities were downregulated (eg, Tcf4, Dusp1, Fos, and Junb) (Figure 7C). Gene set enrichment analysis revealed 13 identical significant hallmark pathways in STAT5B GOF human and mouse leukemic NK cells (Figure 7D). All pathways were regulated in the same direction in the mouse and human STAT5B GOF comparisons. Significantly upregulated pathways were associated with cell cycle progression, whereas downregulated pathways were associated with apoptosis and inflammatory processes (Figure 7E; supplemental Figure 7D; supplemental Table 5).
Figure 7.

Leukemic N642HNK/NK NK cells display molecular features of patients with NK-cell leukemia harboring STAT5B GOF mutations. RNA-Seq data of NK cells from Cre neg, GFPNK/NK, STAT5BNK/NK, nondiseased and diseased N642HNK/NK mice and patients with NK-cell leukemia. (A) Principal component analysis of RNA-Seq data of control (peripheral blood mononuclear cells), B-cell precursor acute lymphoblastic leukemia (BCP-ALL), T-cell acute lymphoblastic leukemia (T-ALL), patients with NK-cell leukemia: without JAK/STAT mutations (NK-cell leukemia, n = 44), with mutated STAT3 (NK-cell leukemia [STAT3 mut], n = 16), with mutated JAK1 (NK-cell leukemia [JAK1 mut], n = 1), or with STAT5B GOF mutations (NK-cell leukemia [STAT5B GOF], n = 3). One patient with STAT5B GOF harbors a STAT5BN642H, 1 harbors a STAT5BQ706L, and 1 patient harbors a STAT5BY665F and a STAT5BV712E comutation. (B) Venn diagram illustrating common DEGs from the comparisons diseased N642HNK/NK (n = 8) vs control (GFPNK/NK or Cre neg mice; n = 5) (adjusted P < .1) and NK-cell leukemia (STAT5B GOF) (n = 3) vs NK-cell leukemia (n = 44) (FDR < 0.1). This analysis considered exclusively DEGs with available human-mouse orthologues. (C) Heat map illustrating expression of the commonly regulated DEGs from the comparisons: diseased N642HNK/NK (n = 8) vs control (n = 5) and NK-cell leukemia (STAT5B GOF) (n = 3) vs NK-cell leukemia (n = 44). (D) Venn diagram illustrating significant (FDR < 0.1) hallmark pathways from the comparisons diseased N642HNK/NK (n = 8) vs control (n = 5) and NK-cell leukemia (STAT5B GOF) (n = 3) vs NK-cell leukemia (n = 44). (E) Quantification of the normalized enrichment score of the 13 common hallmark pathways identified (D). FDR, false discovery rate; GSEA, gene set enrichment analysis.
Our findings show that leukemic N642HNK/NK NK cells exhibit transcriptional patterns resembling those found in STAT5B-mutated human NK-cell leukemia, underlining the translational validity of the mouse model.
Discussion
STAT5B is a prominent driver of hematopoietic diseases.18 The STAT5BN642H mutation is primarily found in diseases arising from T/NKT cells.27 Previously, a vav-STAT5BN642H mouse model was reported to develop an aggressive CD8+ T-cell lymphoma.10 We now describe a mouse model (N642Hvav/+ mice; Figure 1., Figure 2.) that develops slowly progressing CD4+, CD8+ T- or NKT-cell leukemia. N642Hvav/+ mice display lower pYSTAT5 levels than vav-N642H mice. The different pYSTAT5 levels could stem from a difference in transgene expression levels or might reflect the more progressive CD8+ T-cell disease in young vav-N642H mice. Variations in disease type and onset may result from different promoters driving transgene expression (CAG vs Vav1). N642Hvav/+ mice not only develop a CD8+ T-cell leukemia but also display diverse disease phenotypes, making them a closer representation of patients with STAT5B GOF mutations.9 Our mouse model allows for lineage- or tissue-specific transgene expression to study the impact of STAT5BN642H in different cellular and disease contexts.
We focused on using the model to decipher STAT5BN642H’s function in NK cells. STAT5BN642H expression in NK cells results in hyperactive STAT5B signaling, elevated cell numbers, decreased apoptosis, increased maturation, and higher levels of lytic granzymes. The increased count of mature NK cells in 8- to 12-week-old N642HNK/NK mice, which do not display disease symptoms, is indicative of an indolent NK-LGLL phenotype.66,67 This finding aligns with the indolent phenotype of patients with CD4+ T- and NK-LGLL carrying STAT5B mutations.12,44,46 One-third of the N642HNK/NK mice develop an aggressive disease, suggesting that indolent cases of NK-cell malignancies can transform into aggressive phenotypes, as reported in 1 patient with NK-LGLL with a STAT5BN642H mutation.11,68
Restricting STAT5BN642H expression to NKp46+ cells63 was crucial for the establishment of a STAT5BN642H-driven NK-cell leukemia model, because vav-N642H10,54 and N642Hvav/+ mice develop T-/NKT-cell but not NK-cell leukemia. NKp46 expression marks mature NK cells64,65 and highlights them as the origin of NK-cell leukemia in N642HNK/NK mice. In line, human indolent and aggressive NK-cell neoplasms display a mature cytotoxic phenotype.1,69 Limited data prevent the assessment of whether STAT5B GOF mutations in patients with NK-cell leukemia are acquired in mature NK cells or at earlier developmental stages. In 1 patient with NK-LGLL, STAT5BN642H was detected in both NK cells and a subset of NKT cells,11 indicating its occurrence at a common progenitor state.
In addition to NK-cell leukemia, 1 N642HNK/NK mouse developed γδ T-cell leukemia, consistent with NKp46 expression in subsets of γδ T cells70 and the oncogenic potential of STAT5BN642H in γδ T cells.9 Two diseased N642HNK/NK mice showed an expansion of GFP+ cells lacking CD3, TCR, and NK1.1 expression, indicating an “undifferentiated” leukemia subtype. Aberrant expression of NK-cell markers has been observed in human cases of mature NK-cell malignancies,71,72 and dedifferentiation is a common feature in several tumor types.73
Our mouse model resembles human disease as demonstrated by comparative transcriptional analysis. The analyzed human NK-cell leukemia cohort included patients with STAT3 mutations, which occur more frequent than STAT5B mutations.33,36,39,41,43,44,46,68,74, 75, 76, 77, 78, 79, 80, 81, 82, 83 STAT5B GOF cases exhibited a unique transcriptional profile distinct from STAT3 mutant cases. Although both STAT3 and STAT5 act as oncogenes in hematopoietic cancers,84, 85, 86, 87, 88 STAT3 mutations in NK-LGLL associate with more symptomatic cases and an expansion of cytotoxic NK cells.46,67,76,80,82 However, STAT3 GOF mutations alone cannot induce LGLL in mouse models,89 unlike STAT5BN642H, which represents a potent oncogenic driver.9,10,54 The scarcity of STAT5B mutated cases might relate to a stronger negative feedback regulation. Excessive STAT5 activation by mutations or cytokines can induce cell death or senescence, and chronic exposure to STAT5-activating cytokines can have a negative impact on NK cells.90, 91, 92, 93, 94 The initial growth/survival disadvantage observed upon STAT5BN642H overexpression in human NK-cell lines supports this idea (supplemental Figure 3A-F) and might explain the low prevalence of aggressive leukemia cases in N642HNK/NK mice. Further exploration is necessary to fully grasp the distinct roles of STAT3 and STAT5B mutations in human NK-cell leukemia and murine disease models.
STAT5B mutations have also been described in EBV+ NK-cell malignancies, such as ENKL and ANKL.33,36,43,61,95 Although N642HNK/NK mice do not mimic the contribution of EBV infection, understanding the involvement of STAT5B mutations in NK-cell transformation is relevant for EBV+ NK-cell malignancies. Crossing our N642HNK/NK mice with mouse models displaying conditional expression of EBV proteins96,97 may further elucidate the interaction between EBV infection and STAT5B GOF mutations in NK-cell transformation.
Conventional therapies targeting aberrant STAT5B signaling involve JAK inhibitors. Inhibition of JAKs has drawbacks because it affects additional signaling cascades leading to unintended side effects.98 Targeting STAT5B directly using specialized STAT inhibitors or proteolysis targeting chimeras remain challenging due to the lack of an enzymatic activity in STAT5B and its structural similarity to other STAT proteins. This underscores the importance of identifying feasible therapeutic targets downstream of mutant STAT5B. Notably, we observed a significant increase in KLRG1 on the surface of leukemic N642HNK/NK NK cells, suggesting a potential therapeutic target. Targeting KLRG1 with monoclonal antibodies could selectively deplete malignant clones.99 Additionally, our findings indicate immune evasion of transplanted leukemic N642HNK/NK cells, highlighting potential therapeutic implications, particularly in the context of immunotherapies being explored in NK-cell malignancies.100
Overall, our STAT5BN642H-driven NK-cell leukemia mouse model closely mirrors human NK-cell leukemia representing a resource for a better understanding of NK-cell transformation, the transition from indolent to aggressive disease, and for exploring novel therapeutic interventions.
Conflict-of-interest disclosure: G.H. and W.W. report employment by MLL Munich Leukemia Laboratory. C.G.M. received research funding from Pfizer and AbbVie; serves on the Illumina advisory board; and holds royalties in Cyrus and stocks in Amgen. The remaining authors declare no competing financial interests.
Acknowledgments
The authors thank Sabine Fajmann, Petra Kudweis, and Philipp Jodl for their experimental support; Michaela Prchal-Murphy for help and administration regarding animal experiments and ethical permits and appreciation is extended to all animal caretakers for their work; Bettina Wagner, Lill Anderson, and Tina Bernthaler for their help in the generation of the new NK cell–specific STAT5BN642H transgenic mouse model; and Stephan Hutter (MLL Munich Leukemia Laboratory) for bioinformatics support in the analysis of primary patient samples. The authors also acknowledge the Next Generation Sequencing Facility at Vienna BioCenter Core Facilities, a member of the Vienna BioCenter, Austria, for their support. BioRender.com was used for the graphical illustration of some figures.
This work was funded in part by the Austrian Science Fund (FWF) Special Research Program SFB-F6107 (grant DOI: 10.55776/F61), the PhD program “Inflammation and Immunity” FWF W1212, Austrian Academy of Sciences funds DOC 32-B28 (grant DOI: 10.55776/DOC32), and the FWF ZK-81B (grant DOI: 10.55776/ZK81). The work was also supported by the Fellinger Cancer Research association, the City of Vienna (Stadt Wien Kultur) MA7 grant, and the University of Veterinary Medicine Vienna. K.K. is a recipient of a DOC Fellowship of the Austrian Academy of Sciences at the University of Veterinary Medicine.
Authorship
Contribution: K.K., V.S., and D.G. conceived the study; T.R. and K.K. generated the mouse model; K.K., S.K., A.H., M.R., J.L., J.T., J.K., and D.G. performed the experiments; K.K., S.K., and D.G. analyzed the data; A.W.-S., C.A.B., and B.M. established methods and helped with the experiments and analysis of the data; R.M. and C.G.M. contributed to experimental design and scientific discussions; R.G., T.K., J.K., and S.K. analyzed sequencing data; G.H., W.W., and C.G.M. provided bioinformatic patient data analysis; D.G., K.K., S.K., and V.S. wrote the manuscript; D.G., and V.S. provided reagents and supervised the study; and all authors revised the manuscript.
Footnotes
K.K. and S.K. contributed equally to this study.
V.S. and D.G. contributed equally to this study.
The RNA sequencing data reported in this article have been deposited in the ArrayExpress database (accession ID: E-MTAB-13797).
All other relevant data that support the conclusions of the study are available on request from the corresponding author, Dagmar Gotthardt (dagmar.gotthardt@vetmeduni.ac.at).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Supplementary Material
References
- 1.Matutes E. The 2017 WHO update on mature T- and natural killer (NK) cell neoplasms. Int J Lab Hematol. 2018;40(suppl 1):97–103. doi: 10.1111/ijlh.12817. [DOI] [PubMed] [Google Scholar]
- 2.El Hussein S, Medeiros LJ, Khoury JD. Aggressive NK cell leukemia: current state of the art. Cancers. 2020;12(10):2900-16. doi: 10.3390/cancers12102900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sokol L, Loughran TP. Large granular lymphocyte leukemia. Oncol. 2006;11(3):263–273. doi: 10.1634/theoncologist.11-3-263. [DOI] [PubMed] [Google Scholar]
- 4.Nicolae A, Ganapathi KA, Pham THT, et al. EBV-negative aggressive NK-cell leukemia/lymphoma: clinical, pathological and genetic features. Am J Surg Pathol. 2017;41(1):67–74. doi: 10.1097/PAS.0000000000000735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koo M, Olevsky O, Ruchalski K, Song S. Primary intestinal NK-cell lymphoma, EBV-negative: a case report and literature review. Hum Pathol Case Reports. 2019;17 [Google Scholar]
- 6.Tang YT, Wang D, Luo H, et al. Aggressive NK-cell leukemia: clinical subtypes, molecular features, and treatment outcomes. Blood Cancer J. 2017;7(12):660. doi: 10.1038/s41408-017-0021-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stark GR, Darnell JE. The JAK-STAT pathway at twenty. Immunity. 2012;36(4):503–514. doi: 10.1016/j.immuni.2012.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sorger H, Dey S, Vieyra-Garcia PA, et al. Blocking STAT3/5 through direct or upstream kinase targeting in leukemic cutaneous T-cell lymphoma. EMBO Mol Med. 2022;14(12) doi: 10.15252/emmm.202115200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Araujo ED, Erdogan F, Neubauer HA, et al. Structural and functional consequences of the STAT5B N642H driver mutation. Nat Commun. 2019;10(1) doi: 10.1038/s41467-019-10422-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pham HTT, Maurer B, Prchal-Murphy M, et al. STAT5BN642H is a driver mutation for T cell neoplasia. J Clin Invest. 2018;128(1):387–401. doi: 10.1172/JCI94509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rajala HLM, Eldfors S, Kuusanmäki H, et al. Discovery of somatic STAT5b mutations in large granular lymphocytic leukemia. Blood. 2013;121(22):4541–4550. doi: 10.1182/blood-2012-12-474577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andersson EI, Tanahashi T, Sekiguchi N, et al. High incidence of activating STAT5B mutations in CD4-positive T-cell large granular lymphocyte leukemia. Blood. 2016;128(20):2465–2468. doi: 10.1182/blood-2016-06-724856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bandapalli OR, Schuessele S, Kunz JB, et al. The activating STAT5B N642H mutation is a common abnormality in pediatric T-cell acute lymphoblastic leukemia and confers a higher risk of relapse. Haematologica. 2014;99(10):e188–e192. doi: 10.3324/haematol.2014.104992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rajala HLM, Porkka K, MacIejewski JP, Loughran TP, Mustjoki S. Uncovering the pathogenesis of large granular lymphocytic leukemia—novel STAT3 and STAT5b mutations. Ann Med. 2014;46(3):114–122. doi: 10.3109/07853890.2014.882105. [DOI] [PubMed] [Google Scholar]
- 15.Thomas SJ, Snowden JA, Zeidler MP, Danson SJ. The role of JAK/STAT signaling in the pathogenesis, prognosis and treatment of solid tumors. Br J Cancer. 2015;113(3):365–371. doi: 10.1038/bjc.2015.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pencik J, Pham HTT, Schmoellerl J, et al. JAK-STAT signaling in cancer: from cytokines to non-coding genome. Cytokine. 2016;87:26–36. doi: 10.1016/j.cyto.2016.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Berger A, Sexl V, Valent P, Moriggl R. Inhibition of STAT5: a therapeutic option in BCR-ABL1-driven leukemia. Oncotarget. 2014;5(20):9564–9576. doi: 10.18632/oncotarget.2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Maurer B, Kollmann S, Pickem J, Hoelbl-Kovacic A, Sexl V. STAT5A and STAT5B-twins with different personalities in hematopoiesis and leukemia. Cancers. 2019;11(11) doi: 10.3390/cancers11111726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu H, Jove R. The STATs of cancer--new molecular targets come of age. Nat Rev Cancer. 2004;4(2):97–105. doi: 10.1038/nrc1275. [DOI] [PubMed] [Google Scholar]
- 20.Steelman LS, Pohnert SC, Shelton JG, Franklin RA, Bertrand FE, McCubrey JA. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia. 2004;18(2):189–218. doi: 10.1038/sj.leu.2403241. [DOI] [PubMed] [Google Scholar]
- 21.Kollmann S, Grausenburger R, Klampfl T, et al. A STAT5B-CD9 axis determines self-renewal in hematopoietic and leukemic stem cells. Blood. 2021;138(23):2347–2359. doi: 10.1182/blood.2021010980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vargas-Hernández A, Witalisz-Siepracka A, Prchal-Murphy M, et al. Human STAT5b mutation causes dysregulated human natural killer cell maturation and impaired lytic function. J Allergy Clin Immunol. 2020;145(1):345–357.e9. doi: 10.1016/j.jaci.2019.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Villarino A V, Sciumè G, Davis FP, et al. Subset- and tissue-defined STAT5 thresholds control homeostasis and function of innate lymphoid cells. J Exp Med. 2017;214(10):2999–3014. doi: 10.1084/jem.20150907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bagger FO, Sasivarevic D, Sohi SH, et al. BloodSpot: a database of gene expression profiles and transcriptional programs for healthy and malignant hematopoiesis. Nucleic Acids Res. 2016;44(D1):D917–D924. doi: 10.1093/nar/gkv1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kofoed EM, Hwa V, Little B, et al. Growth hormone insensitivity associated with a STAT5b mutation. N Engl J Med. 2003;349(12):1139–1147. doi: 10.1056/NEJMoa022926. [DOI] [PubMed] [Google Scholar]
- 26.Bezrodnik L, Di Giovanni D, Caldirola MS, Azcoiti ME, Torgerson T, Gaillard MI. Long-term follow-up of STAT5B deficiency in three Argentinian patients: clinical and immunological features. J Clin Immunol. 2015;35(3):264–272. doi: 10.1007/s10875-015-0145-5. [DOI] [PubMed] [Google Scholar]
- 27.Hwa V. STAT5B deficiency: Impacts on human growth and immunity. Growth Horm IGF Res. 2016;28:16–20. doi: 10.1016/j.ghir.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanai T, Jenks J, nadeau KC. The STAT5b pathway defect and autoimmunity. Front Immunol. 2012;3:234. doi: 10.3389/fimmu.2012.00234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bernasconi A, Marino R, Ribas A, et al. Characterization of immunodeficiency in a patient with growth hormone insensitivity secondary to a novel STAT5b gene mutation. Pediatrics. 2006;118(5):e1584–e1592. doi: 10.1542/peds.2005-2882. [DOI] [PubMed] [Google Scholar]
- 30.Imada K, Bloom ET, Nakajima H, et al. Stat5b is essential for natural killer cell-mediated proliferation and cytolytic activity. J Exp Med. 1998;188(11):2067–2074. doi: 10.1084/jem.188.11.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Villarino A, Laurence A, Robinson GW, et al. Signal transducer and activator of transcription 5 (STAT5) paralog dose governs T cell effector and regulatory functions. Elife. 2016;5 doi: 10.7554/eLife.08384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gotthardt D, Putz EM, Grundschober E, et al. STAT5 is a key regulator in NK cells and acts as a molecular switch from tumor surveillance to tumor promotion. Cancer Discov. 2016;6(4):414–429. doi: 10.1158/2159-8290.CD-15-0732. [DOI] [PubMed] [Google Scholar]
- 33.Song TL, Nairismägi ML, Laurensia Y, et al. Oncogenic activation of the STAT3 pathway drives PD-L1 expression in natural killer/T-cell lymphoma. Blood. 2018;132(11):1146–1158. doi: 10.1182/blood-2018-01-829424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nicolae A, Xi L, Pittaluga S, et al. Frequent STAT5B mutations in γδ hepatosplenic T-cell lymphomas. Leuk. 2014;28(11):2244–2248. doi: 10.1038/leu.2014.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKinney M, Moffitt AB, Gaulard P, et al. The genetic basis of hepatosplenic T-cell lymphoma. Cancer Discov. 2017;7(4):369–379. doi: 10.1158/2159-8290.CD-16-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang L, Liu D, Wang N, et al. Integrated genomic analysis identifies deregulated JAK/STAT-MYC-biosynthesis axis in aggressive NK-cell leukemia. Cell Res. 2017;28(2):172–186. doi: 10.1038/cr.2017.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kiel MJ, Velusamy T, Rolland D, et al. Integrated genomic sequencing reveals mutational landscape of T-cell prolymphocytic leukemia. Blood. 2014;124(9):1460–1472. doi: 10.1182/blood-2014-03-559542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kiel MJ, Sahasrabuddhe AA, Rolland DCM, et al. Genomic analyses reveal recurrent mutations in epigenetic modifiers and the JAK–STAT pathway in Sézary syndrome. Nat Commun. 2015;6(1):8470. doi: 10.1038/ncomms9470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dufva O, Kankainen M, Kelkka T, et al. Aggressive natural killer-cell leukemia mutational landscape and drug profiling highlight JAK-STAT signaling as therapeutic target. Nat Commun. 2018;9(1):1567. doi: 10.1038/s41467-018-03987-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao LM, Zhao S, Liu WP, et al. Clinicopathologic characterization of aggressive natural killer cell leukemia involving different tissue sites. Am J Surg Pathol. 2016;40(6):836–846. doi: 10.1097/PAS.0000000000000634. [DOI] [PubMed] [Google Scholar]
- 41.Jiang L, Gu Z, Yan Z, et al. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nat Genet. 2015;47(9):1061–1066. doi: 10.1038/ng.3358. [DOI] [PubMed] [Google Scholar]
- 42.Kontro M, Kuusanmäki H, Eldfors S, et al. Novel activating STAT5B mutations as putative drivers of T-cell acute lymphoblastic leukemia. Leukemia. 2014;28(8):1738–1742. doi: 10.1038/leu.2014.89. [DOI] [PubMed] [Google Scholar]
- 43.Küçük C, Jiang B, Hu X, et al. Activating mutations of STAT5B and STAT3 in lymphomas derived from γδ-T or NK cells. Nat Commun. 2015;6:6025. doi: 10.1038/ncomms7025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baer C, Kimura S, Rana MS, et al. CCL22 mutations drive natural killer cell lymphoproliferative disease by deregulating microenvironmental crosstalk. Nat Genet. 2022;54(5):637–648. doi: 10.1038/s41588-022-01059-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xiong J, Cui BW, Wang N, et al. Genomic and transcriptomic characterization of natural killer T cell lymphoma. Cancer Cell. 2020;37(3):403–419.e6. doi: 10.1016/j.ccell.2020.02.005. [DOI] [PubMed] [Google Scholar]
- 46.Pastoret C, Desmots F, Drillet G, et al. Linking the KIR phenotype with STAT3 and TET2 mutations to identify chronic lymphoproliferative disorders of NK cells. Blood. 2021;137(23):3237–3250. doi: 10.1182/blood.2020006721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Waldmann TA, Chen J. Disorders of the JAK/STAT pathway in T cell lymphoma pathogenesis: implications for immunotherapy. Annu Rev Immunol. 2017;35:533–550. doi: 10.1146/annurev-immunol-110416-120628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu Y, Tian Z, Wei H. Developmental and functional control of natural killer cells by cytokines. Front Immunol. 2017;8 doi: 10.3389/fimmu.2017.00930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rani A, Murphy JJ. STAT5 in cancer and immunity. J Interferon Cytokine Res. 2016;36(4):226–237. doi: 10.1089/jir.2015.0054. [DOI] [PubMed] [Google Scholar]
- 50.Yokohama A, Mishra A, Mitsui T, et al. A novel mouse model for the aggressive variant of NK cell and T cell large granular lymphocyte leukemia. Leuk Res. 2010;34(2):203–209. doi: 10.1016/j.leukres.2009.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fehniger TA, Suzuki K, VanDeusen JB, Cooper MA, Freud AG, Caligiuri MA. Fatal leukemia in interleukin-15 transgenic mice. Blood Cells Mol Dis. 2001;27(1):223–230. doi: 10.1006/bcmd.2001.0379. [DOI] [PubMed] [Google Scholar]
- 52.Fehniger TA, Suzuki K, Ponnappan A, et al. Fatal leukemia in interleukin 15 transgenic mice follows early expansions in natural killer and memory phenotype CD8+ T cells. J Exp Med. 2001;193(2):219–231. doi: 10.1084/jem.193.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sato N, Sabzevari H, Fu S, et al. Development of an IL-15–autocrine CD8 T-cell leukemia in IL-15–transgenic mice require the cis expression of IL-15Rα. Blood. 2011;117(15):4032–4040. doi: 10.1182/blood-2010-09-307504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Klein K, Witalisz-Siepracka A, Maurer B, et al. STAT5BN642H drives transformation of NKT cells: a novel mouse model for CD56+ T-LGL leukemia. Leukemia. 2019;33(9):2336–2340. doi: 10.1038/s41375-019-0471-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Belgardt BF, Husch A, Rother E, et al. PDK1 deficiency in POMC-expressing cells reveals FOXO1-dependent and -independent pathways in control of energy homeostasis and stress response. Cell Metab. 2008;7(4):291–301. doi: 10.1016/j.cmet.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 56.Hagemann-Jensen M, Ziegenhain C, Chen P, et al. Single-cell RNA counting at allele and isoform resolution using smart-seq3. Nat Biotechnol. 2020;38(6):708–714. doi: 10.1038/s41587-020-0497-0. [DOI] [PubMed] [Google Scholar]
- 57.Babraham Bioinformatics. https://www.bioinformatics.babraham.ac.uk/projects/fastqc/
- 58.Stengel A, Shahswar R, Haferlach T, et al. Whole transcriptome sequencing detects a large number of novel fusion transcripts in patients with AML and MDS. Blood Adv. 2020;4(21):5393–5401. doi: 10.1182/bloodadvances.2020003007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ogilvy S, Metcalf D, Gibson L, Bath ML, Harris AW, Adams JM. Promoter elements of vav drive transgene expression in vivo throughout the hematopoietic compartment. Blood. 1999;94(6):1855–1863. [PubMed] [Google Scholar]
- 60.Parri E, Kuusanmäki H, Bulanova D, Mustjoki S, Wennerberg K. Selective drug combination vulnerabilities in STAT3- and TP53-mutant malignant NK cells. Blood Adv. 2021;5(7):1862–1875. doi: 10.1182/bloodadvances.2020003300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gao LM, Zhao S, Liu WP, et al. Clinicopathologic characterization of aggressive natural killer cell leukemia involving different tissue sites. Am J Surg Pathol. 2016;40(6):836–846. doi: 10.1097/PAS.0000000000000634. [DOI] [PubMed] [Google Scholar]
- 62.Kameda K, Yanagiya R, Miyatake Y, et al. The hepatic niche leads to aggressive natural killer cell leukemia proliferation through the transferrin–transferrin receptor 1 axis. Blood. 2023;142(4):352–364. doi: 10.1182/blood.2022018597. [DOI] [PubMed] [Google Scholar]
- 63.Eckelhart E, Warsch W, Zebedin E, et al. A novel Ncr1-Cre mouse reveals the essential role of STAT5 for NK cell survival and development. Blood. 2011;117(5):1565–1573. doi: 10.1182/blood-2010-06-291633. [DOI] [PubMed] [Google Scholar]
- 64.Gotthardt D, Prchal-Murphy M, Seillet C, et al. NK cell development in bone marrow and liver: site matters. Genes Immun. 2014;15(8):584–587. doi: 10.1038/gene.2014.55. [DOI] [PubMed] [Google Scholar]
- 65.Ma S, Caligiuri MA, Yu J. A four-stage model for murine natural killer cell development in vivo. J Hematol Oncol. 2022;15(1):31. doi: 10.1186/s13045-022-01243-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Poullot E, Zambello R, Leblanc F, et al. Chronic natural killer lymphoproliferative disorders: characteristics of an international cohort of 70 patients. Ann Oncol. 2014;25(10):2030–2035. doi: 10.1093/annonc/mdu369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kurt H, Jorgensen JL, Amin HM, et al. Chronic lymphoproliferative disorder of NK-cells: a single-institution review with emphasis on relative utility of multimodality diagnostic tools. Eur J Haematol. 2018;100(5):444–454. doi: 10.1111/ejh.13038. [DOI] [PubMed] [Google Scholar]
- 68.Gasparini VR, Binatti A, Coppe A, et al. A high definition picture of somatic mutations in chronic lymphoproliferative disorder of natural killer cells. Blood Cancer J. 2020;10(4):42. doi: 10.1038/s41408-020-0309-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Alaggio R, Amador C, Anagnostopoulos I, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumors: lymphoid neoplasms. Leukemia. 2022;36(7):1720–1748. doi: 10.1038/s41375-022-01620-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Walzer T, Bléry M, Chaix J, et al. Identification, activation, and selective in vivo ablation of mouse NK cells via NKp46. Proc Natl Acad Sci U S A. 2007;104(9):3384–3389. doi: 10.1073/pnas.0609692104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang J, Li P, Piao Y, et al. CD56-negative extranodal natural killer/T-cell lymphoma: a retrospective study in 443 patients treated by chemotherapy with or without asparaginase. Front Immunol. 2022;13 doi: 10.3389/fimmu.2022.829366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Guerreiro M, Príncipe F, Teles MJ, et al. CD56-negative aggressive NK cell leukemia relapsing as multiple cranial nerve palsies: case report and literature review. Case Rep Hematol. 2017;2017:3724017. doi: 10.1155/2017/3724017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li J, Stanger BZ. How tumor cell dedifferentiation drives immune evasion and resistance to immunotherapy. Cancer Res. 2020;80(19):4037–4041. doi: 10.1158/0008-5472.CAN-20-1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ishida F. Aggressive NK-cell leukemia. Front Pediatr. 2018;6:292. doi: 10.3389/fped.2018.00292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jerez A, Clemente MJ, Makishima H, et al. STAT3 mutations unify the pathogenesis of chronic lymphoproliferative disorders of NK cells and T cell large granular lymphocyte leukemia. Blood. 2012;120(15):3048–3057. doi: 10.1182/blood-2012-06-435297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kawakami T, Sekiguchi N, Kobayashi J, et al. STAT3 mutations in natural killer cells are associated with cytopenia in patients with chronic lymphoproliferative disorder of natural killer cells. Int J Hematol. 2019;109(5):563–571. doi: 10.1007/s12185-019-02625-x. [DOI] [PubMed] [Google Scholar]
- 77.Fasan A, Kern W, Grossmann V, et al. STAT3 mutations are highly specific for large granular lymphocytic leukemia. Leukemia. 2013;27(7):1598–1600. doi: 10.1038/leu.2012.350. [DOI] [PubMed] [Google Scholar]
- 78.Lee S, Park HY, Kang SY, et al. Genetic alterations of JAK/STAT cascade and histone modification in extranodal NK/T-cell lymphoma nasal type. Oncotarget. 2015;6(19):17764–17776. doi: 10.18632/oncotarget.3776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tse E, Kwong YL. The diagnosis and management of NK/T-cell lymphomas. J Hematol Oncol. 2017;10(1) doi: 10.1186/s13045-017-0452-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Barilà G, Calabretto G, Teramo A, et al. T cell large granular lymphocyte leukemia and chronic NK lymphocytosis. Best Pract Res Clin Haematol. 2019;32(3):207–216. doi: 10.1016/j.beha.2019.06.006. [DOI] [PubMed] [Google Scholar]
- 81.Barilà G, Teramo A, Calabretto G, et al. Stat3 mutations impact on overall survival in large granular lymphocyte leukemia: a single-center experience of 205 patients. Leukemia. 2020;34(4):1116–1124. doi: 10.1038/s41375-019-0644-0. [DOI] [PubMed] [Google Scholar]
- 82.Olson TL, Cheon HJ, Xing JC, et al. Frequent somatic TET2 mutations in chronic NK-LGL leukemia with distinct patterns of cytopenias. Blood. 2021;138(8):662–673. doi: 10.1182/blood.2020005831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Teramo A, Barilà G, Calabretto G, et al. Insights into genetic landscape of large granular lymphocyte leukemia. Front Oncol. 2020;10 doi: 10.3389/fonc.2020.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wingelhofer B, Maurer B, Heyes EC, et al. Pharmacologic inhibition of STAT5 in acute myeloid leukemia. Leukemia. 2018;32(5):1135–1146. doi: 10.1038/s41375-017-0005-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Orlova A, Wagner C, De Araujo ED, et al. Direct targeting options for STAT3 and STAT5 in cancer. Cancers. 2019;11(12) doi: 10.3390/cancers11121930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Brachet-Botineau M, Deynoux M, Vallet N, et al. A novel inhibitor of STAT5 signaling overcomes chemotherapy resistance in myeloid leukemia cells. Cancers. 2019;11(12) doi: 10.3390/cancers11122043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Igelmann S, Neubauer HA, Ferbeyre G. STAT3 and STAT5 activation in solid cancers. Cancers. 2019;11(10) doi: 10.3390/cancers11101428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Verhoeven Y, Tilborghs S, Jacobs J, et al. The potential and controversy of targeting STAT family members in cancer. Semin Cancer Biol. 2020;60:41–56. doi: 10.1016/j.semcancer.2019.10.002. [DOI] [PubMed] [Google Scholar]
- 89.Dutta A, Yan D, Hutchison RE, Mohi G. STAT3 mutations are not sufficient to induce large granular lymphocytic leukemia in mice. Br J Haematol. 2018;180(6):911–915. doi: 10.1111/bjh.14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Felices M, Lenvik AJ, McElmurry R, et al. Continuous treatment with IL-15 exhausts human NK cells via a metabolic defect. JCI Insight. 2018;3(3) doi: 10.1172/jci.insight.96219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Alvarez M, Simonetta F, Baker J, et al. Regulation of murine NK cell exhaustion through the activation of the DNA damage repair pathway. JCI Insight. 2019;5(14) doi: 10.1172/jci.insight.127729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Elpek KG, Rubinstein MP, Bellemare-Pelletier A, Goldrath AW, Turley SJ. Mature natural killer cells with phenotypic and functional alterations accumulate upon sustained stimulation with IL-15/IL-15Rα complexes. Proc Natl Acad Sci U S A. 2010;107(50):21647–21652. doi: 10.1073/pnas.1012128107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mallette FA, Gaumont-Leclerc MF, Ferbeyre G. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev. 2007;21(1):43–48. doi: 10.1101/gad.1487307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Epling-Burnette PK, Liu JH, Catlett-Falcone R, et al. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J Clin Invest. 2001;107(3):351–362. doi: 10.1172/JCI9940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jiang L, Gu Z-H, Yan Z-X, et al. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nat Genet. 2015;47(9):1061–1066. doi: 10.1038/ng.3358. [DOI] [PubMed] [Google Scholar]
- 96.Wirtz S, Neufert C, Weigmann B, Neurath MF. Chemically induced mouse models of intestinal inflammation. Nat Protoc. 2007;2(3):541–546. doi: 10.1038/nprot.2007.41. [DOI] [PubMed] [Google Scholar]
- 97.Zhang B, Kracker S, Yasuda T, et al. Immune surveillance and therapy of lymphomas driven by Epstein-Barr virus protein LMP1 in a mouse model. Cell. 2012;148(4):739–751. doi: 10.1016/j.cell.2011.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shawky AM, Almalki FA, Abdalla AN, Abdelazeem AH, Gouda AM. A comprehensive overview of globally approved JAK inhibitors. Pharmaceutics. 2022;14(5) doi: 10.3390/pharmaceutics14051001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Assatova B, Willim R, Trevisani C, et al. KLRG1 cell depletion as a novel therapeutic strategy in patients with mature T-cell lymphoma subtypes. Clin Cancer Res. 2024:OF1–OF17. doi: 10.1158/1078-0432.CCR-23-3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.He L, Chen N, Dai L, Peng X. Advances and challenges of immunotherapies in NK/T cell lymphomas. iScience. 2023;26(11) doi: 10.1016/j.isci.2023.108192. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.