

Abstract
Tuning the oxygen activity in perovskite oxides (ABO3) is promising to surmount the trade-off between activity and selectivity in redox reactions. However, this remains challenging due to the limited understanding in its activation mechanism. Herein, we propose the discovery that generating subsurface A-site cation (Lasub.) vacancy beneath surface Fe-O layer greatly improved the oxygen activity in LaFeO3, rendering enhanced methane conversion that is 2.9-fold higher than stoichiometric LaFeO3 while maintaining high syngas selectivity of 98% in anaerobic oxidation. Experimental and theoretical studies reveal that absence of Lasub.-O interaction lowered the electron density over oxygen and improved the oxygen mobility, which reduced the barrier for C-H bond cleavage and promoted the oxidation of C-atom, substantially boosting methane-to-syngas conversion. This discovery highlights the importance of A-site cations in modulating electronic state of oxygen, which is fundamentally different from the traditional scheme that mainly credits the redox activity to B-site cations and can pave a new avenue for designing prospective redox catalysts.
Subject terms: Heterogeneous catalysis, Porous materials, Density functional theory
Tuning the oxygen activity in perovskite oxides (ABO3) holds great potential for overcoming the trade-off between activity and selectivity in redox reactions. This paper highlights how A-site cations influence the electronic state of oxygen and how subsurface A-site vacancies enhance oxygen activity for improved methane conversion.
Introduction
Tuning the oxygen activity in metal oxides offers opportunities to manufacture prospective catalysts for reactions proceeding via the Mars−van Krevelen mechanism1–3. However, it was usually found that the oxides with high oxidizing capability display low selectivity, and vice versa, which poses huge obstacle for designing an efficient redox catalyst4,5. The perovskite oxides (structural formula: ABO3) has attracted particular attention in redox cycling reactions due to their capacity to accommodate different cations in single perovskite matrix6–8. The oxygen activity in ABO3 can be widely adjusted by selecting suitable A- and B-site cations, substantially endows these oxides outstanding redox properties and superior performance in various processes, e.g., selective methane oxidation9,10, dehydrogenation of light alkanes11,12, oxygen evolution reaction13–15, and CO oxidation16,17. Therefore, disclosing the underlying mechanism affecting the oxygen activity in perovskite oxides would be promising to surmount the trade-off between activity and selectivity during redox reactions.
The current understanding mainly credits the outstanding oxygen activity in perovskites to transition metals at the B-site, since the A-site is generally occupied by redox-inert alkaline, alkaline-earth, or lanthanide cations18–20. To this end, great effort, such as constructing asymmetric B1-O-B2 interaction, acid etching, and increase B/A ratio in perovskite oxides, has been taken to optimize the redox activity by regulating B-O interaction or terminating catalyst surface by more B-site cations21–23. Compared to the dazzling glory of B-site cations, the functionalization of A-site cations is normally explained by an indirect manner of generating oxygen vacancies, adjusting the valence state of B-site cations, or changing the crystal structure24–26. However, from the view of structure analysis (Fig. 1a), a lattice oxygen in the bulk (Obulk), taken cubic structure as an example, is coordinated to four A-site cations and two B-site cations, which provides possibilities for A-site cations to directly regulate the electronic state of oxygen. This hypothesis is validated by some recent observations. Gong et al.27 found that the substitution of La3+ by Ce3+ in LaxCe1-xFeO3 showed a pronounced effect on oxygen mobility without changing the concentration of oxygen vacancies, Fe valence state, and the crystalline structure. Tamai et al.28 showed that the local electronic structure around Sr in SrFeO3-δ obviously changed with the concentration of oxygen vacancies. These results highlight the essential role of A-site cation in modulating the oxygen activity, yet this long-standing issue have been overlooked to some content, which renders precious control of the oxygen activity remains a grand challenge due to the limited understanding in its activation mechanism.
Fig. 1. Perovskite structure and characterization of fresh samples.

a Unit cell of perovskite oxide (ABO3) with an ideal cubic structure (left) and corresponding coordination state of bulk (middle) and surface (right) lattice oxygen (perovskite surface terminated with Fe cations). EDS maps of b La1.03FeO3, c LaFeO3, and d La0.97FeO3, and corresponding HRTEM images of e La1.03FeO3, f LaFeO3, and g La0.97FeO3. h H2-TPR profiles of LaxFeO3 (x = 1.03, 1, and 0.97) oxides.
Herein, we show that the A-site La cation, even if located at the subsurface site beneath the surface Fe-O layer, can exert great influence on the redox performance of LaFeO3. Removing the subsurface La cation (Lasub.) greatly enhances oxygen mobility and lowers the electron density over O-atom due to the elimination of the Lasub.-O interaction, which highlights the essential role of redox-inert A-site cation in affecting oxygen activity. To exemplify the practicability of this hypothesis, chemical looping anaerobic oxidation of methane was applied as a probe reaction, wherein the lattice oxygen of metal oxides (known as oxygen carrier) was directly used for C-H bond activation and methane oxidation, substantially giving value-added syngas (H2/CO ratio of 2)29–31. It was found that both in-situ redox treatment of stoichiometric LaFeO3 and lowering La/Fe ratio could generate subsurface La vacancies (Lavac.), enabling pronounced methane conversion that is 2.9 times higher than stoichiometric LaFeO3 and exhibits excellent syngas selectivity (98%) and cyclic stability.
Results
Characterization of fresh redox catalysts
To investigate the role of A-site cation in adjusting the oxygen activity of perovskite oxides, a series of LaxFeO3 (x = 1.03, 1, and 0.97) catalysts with different La content was prepared. XRD results (Fig. S1) show that all reflection peaks of fresh samples are well indexed to the perovskite phase while HRTEM images (Fig. 1b–g) displayed well-resolved lattice fringes with homogeneous mapping of La, Fe, and O elements, which signifies the high purity and fine perovskite-type structure of these oxides. SEM images (Fig. S2) revealed that the particle size notably increased with reducing La content from 50 to 150 nm for La1.03FeO3 to 170–400 nm for La0.97FeO3, corresponding to a decreased specific surface area from 9.4 to 2.2 m2/g.
The oxygen mobility of fresh catalysts was assessed by O2-TPD (Fig. S3) and H2-TPR (Fig. 1h) experiments. During the O2-TPD process, no signals assigned to O2 was detected. This should be induced by the low specific surface areas of these OCs, leading to the trace amount of O2 being adsorbed on surface oxygen vacancies. Besides, the lattice oxygen in LaFeO3 is stable due to the high oxygen vacancy formation energy (>3.62 eV)21. Therefore, the desorption of lattice oxygen to generate oxygen vacancy by simple thermal treatment in He is difficult. As for reduction in hydrogen (Fig. 1h), in general, two H2 consumption peaks, located at low-temperature zone from 300 to 500 oC and high temperature above 700 oC, respectively, could be observed over three samples. As for La1.03FeO3 and LaFeO3 samples, the H2 uptakes at low-temperature zone is low, which should be assigned to the reduction of surface-active oxygen species. In contrast, an obviously enhanced reduction peak is detected for the La0.97FeO3 sample, suggesting that partial lattice oxygen is activated upon the introduction of La vacancies into the perovskite matrix. The accumulated H2 consumption for La0.97FeO3 (La-deficiency of 126 μmol/g) at this zone reaches 190 μmol/g, corresponding to Oactivated/Lavac. ratio of 1.51, which highlights the important role of La vacancies in activating the lattice oxygen. Compared with the distinct difference at low-temperature zone, the reduction behavior of these samples at high temperature is almost similar, which should be induced by the analogous coordination structure of most oxygen atoms in the catalysts.
XPS analysis was conducted to evaluate the chemical state of surface O and Fe. As shown in Fig. 2a, the O 1s spectra can be deconvoluted into four secondary peaks, which were assigned to lattice oxygen O2- (528.8−529.1 eV), surface adsorbed oxygen O22− and O− (529.8−530.2 eV), carbonate CO32− and hydroxyl OH- (530.9−531.2 eV), and molecular water (532.4−532.7 eV), respectively32. It is noted that the proportion of CO32− and OH− notable reduced from 36.8% of La1.03FeO3 to 24.6% of La0.97FeO3, which coincides with the reduced concentration of alkaline La cations at the surface (Table S1) that leads to degraded adsorption of CO32− and OH−. Besides, the peak position of lattice oxygen gradually shifted to higher bonding energy with lowering the concentration of La3+ cations, suggesting that the formation of La vacancies renders a declined electron density over the lattice oxygen. Correspondingly, an obvious shift of Fe 2p3/2 peak to higher binding energy from 709.7 eV of La1.03FeO3 to 710.1 eV of La0.97FeO3 can be noted (Fig. 2b), suggesting that reduction of La content could induce improved valence state of Fe cations to maintain the charge balance in the oxides33,34. Overall, the above results illustrate that engineering the La concentration at A-site of perovskite ferrites is effective to alter chemical state of lattice oxygen, which is expected to exert great influence to the redox activity.
Fig. 2. XPS spectra of fresh samples.

a O 1s and b Fe 2p XPS spectra of LaxFeO3 (x = 1.03, 1, and 0.97) oxides.
Effect of La engineering on the oxygen activity
CH4-TPR experiments (Fig. 3a–c) was first conducted to evaluate the oxygen activity of these redox catalysts. As for La1.03FeO3, signals assigned to H2 and CO was detected at temperatures of 738 and 746 oC, respectively, which corresponds to the C-H bond cleavage of CH4 (CH4 → *C + 2H2) and selective oxidation of C-atom in CH4 by lattice oxygen (*C + OL → CO + [Ovac.]). Although bearing lowered specific surface area, it is noted that the onset temperature of syngas over LaFeO3 is notably reduced to 714 oC for the H2 profile and 718 oC for the CO profile, respectively, and further slightly declined to 712 oC for the H2 profile and 717 oC for CO profile over La0.97FeO3 sample. Besides, the intensity of syngas signals was also gradually enhanced. These results suggest that lowering La concentration at the A-site could greatly promote the intrinsic reactivity for methane anaerobic oxidation.
Fig. 3. Catalytic performance and stability test.

CH4-TPR profiles for a La1.03FeO3, b LaFeO3, and c La0.97FeO3. Reaction conditions: 100 mg catalyst treated with 5% CH4/He (30 mL/min) from 20 to 900 oC with a rate ramp of 10 oC/min. The kinetic curves for the methane partial oxidation step in the first cycle of d La1.03FeO3, e LaFeO3, and f La0.97FeO3. g The syngas yields of fresh LaxFeO3 (x = 1.03, 1 and 0.97) samples. h The syngas productivity during 20 redox cycles for LaxFeO3 samples (x = 1.03, 1, and 0.97). i The performance of CH4 partial oxidation step for LaFeO3 from 20th cycle to 250th cycle (8–108 h). Reaction conditions: 100 mg catalyst treated with 5% CH4/He (15 mL/min) for 8 min during CH4 partial oxidation step, 5% CO2/He (15 mL/min) for 10 min during CO2 regeneration step at 900 oC, and the reactor was purged with He for 4 min (20 ml/min) between partial oxidation and reoxidation step.
To assess the redox performance, these catalysts were subjected to isothermal redox reaction, wherein the catalysts was reduced by CH4 and regenerated by CO2 oxidation. The spent catalyst was marked as LaxFeO3-Y (Y represents the number of redox cycles). As deposited in Fig. 3d–f, upon feeding CH4 to these catalysts, obvious signals assigned to syngas with an H2/CO ratio of ca. 2.0 (Fig. S4) were observed immediately over all samples while signals of CO2 are negligible, which highlights the outstanding selectivity of perovskite ferrites towards syngas production. The syngas productivity (Fig. 3g) in the first redox cycle was gradually improved with the order of La1.03FeO3 (2.7 mmol/g) <LaFeO3 (4.7 mmol/g) <La0.97FeO3 (5.2 mmol/g), which is in line with CH4-TPR results. Although these catalysts remain a high syngas selectivity above 95% in subsequent cycling tests (Fig. S4), the reactivity differs greatly. As for La1.03FeO3-3 and LaFeO3-3, the activity notably reduced by 70% (0.8 mmol/g) and 63% (1.7 mmol/g), respectively, in the first three cycles. In contrast, the syngas productivity of La0.97FeO3-3 sample only slightly reduced by 9%, highlighting the promoting effect of La-deficiency in improving the redox performance (Fig. 3h). Besides, it is surprising to note that the reactivity of LaFeO3 is gradually recovered from 4th cycle to 20th cycle, substantially giving a steady syngas yield (4.6 mmol/g) in the following reaction until 250th cycle (Fig. 3h, i). A similar variation in CO2 conversion was also observed as shown in Fig. S5. To double-check this phenomenon, CH4-TPR experiments were conducted for fresh LaFeO3 and samples after three cycles and 20 cycles (Fig. S6). It was found that methane conversion was greatly suppressed over LaFeO3-3, as suggested by the reduced intensity for syngas production, while the reactivity of LaFeO3-20 was almost identical to that of fresh LaFeO3. These results suggest that surface reconstruction may occur for LaFeO3 samples during redox cycles, which induces the high reactivity matching that of La0.97FeO3.
Structure evolution of catalysts during isothermal redox reactions
To better understand the parameters that determine the oxygen activity, the catalysts after isothermal redox reactions are subjected to detailed characterizations. The XRD results (Fig. S7) show that the perovskite structure is maintained for catalysts after 20 redox cycles, which suggests that no obvious phase separation that gives nano-sized La2O3 or FeOx as impurities occurs within this reaction period. The SEM results (Fig. S8) demonstrate that the particle size of La1.03FeO3-20, LaFeO3−3, and La1.03FeO3-20 in these samples all notably increased after undergoing oxidation-reduction treatment, which should account for the seriously degraded reactivity of La1.03FeO3 and LaFeO3−3 samples. However, for LaFeO3, as the number of cycles was extended to 20 or even 250 cycles, the growth of particle size slowed down (Fig. S8). Besides, it is noted that samples with higher La content maintain a smaller particle size after 20 redox cycles, indicating that surface La enrichment is beneficial for inhibiting the sintering of oxygen carrier particles. To further disclose the distinct redox performance of these samples, XPS analysis was conducted to monitor the surface structure evolution. As deposited in Fig. 4, no obvious changes were observed for O 1s and Fe 2p profiles of fresh La1.03FeO3 and La1.03FeO3-20, which indicates that surface reconstruction during redox cycles was negligible over the La-excess sample. In contrast, compared with the fresh samples, the ratio of surface carbonates notably improved by 15.8 and 59.6% for LaFeO3-20 and La0.97FeO3-20 samples (Table S1). Consequently, a slight shift of Fe 2p spectra to higher binding energy is observed. These results give a clue that partial La cations should be removed from the perovskite matrix and aggregates into oxide clusters during the redox reaction.
Fig. 4. XPS spectra of fresh and cycled samples.

a, b O 1s and Fe 2p for La1.03FeO3 and La1.03FeO3-20, c, d O 1s and Fe 2p for LaFeO3 and LaFeO3-20, e, f O 1s and Fe 2p of La0.97FeO3 and La0.97FeO3-20.
To double-check the above conclusion, the surface structure evolution of LaFeO3 during redox reaction was analyzed by LEIS, which is sensitive to the changes of the first layer atoms at the catalyst surface35,36. As shown in Fig. 5a and Fig. S9, the surface of fresh LaFeO3 is composed of 80% La cations and 20% Fe cations. This suggests that LaFeO3 is mainly terminated by La cations at A-site, which is in line with previous reports37. After three redox cycles, it is noted that the ratio of surface La notably reduced to 42%, rendering the first layer atoms mainly terminated by Fe cations (58%). This indicates that the redox reaction can result in aggregation of the outmost La cations, as elucidated in Fig. 5a, leading to Fe as the overwhelming cation at the surface. It is interesting to note that the ratio of surface La further increased to 50% after 20 cycles, suggesting that the continuous redox treatment can induce exsolution of more La cations (Lasub.) beneath surface Fe out of perovskite matrix. This conclusion can be also supported by the AC-HAADF-STEM result (Fig. 5b-left) of LaFeO3-20. The site with weakest La-atom intensity (marked by black arrow), where accommodating the fewest number of La-atoms, from first layer to fourth layer (Fig. 5b-right) differs greatly with each other. This result suggests that La cation vacancies were generated and randomly distributed at these layers. Besides, XRD results show that obvious signals assigned to La2O3 can be detected after 50 cycles, which should be induced by the sustained out-diffusion of La cations from the subsurface sites. Corresponding XRD and Raman analysis for LaFeO3-20 and LaFeO3-50 (Fig. S10) shows that phases of FeOx is not observed in these spent catalysts, indicating that the Fe cations are still stabilized in perovskite matrix rather than aggregates into FeOx nanoparticles after removal of subsurface La cations38–40. Overall, these results demonstrate that subsurface La ions in stoichiometric LaFeO3 can migrate out of the perovskite matrix during the redox reaction, which results in surface enrichment of La2O3 oxide and formation of subsurface La vacancies, substantially contributing to the sustained redox performance despite of gradually increased particle size of these catalysts.
Fig. 5. Characterization of subsurface La vacancies.

a The LEIS results for La and Fe atoms in the first layer of LaxFeO3-Y oxides and corresponding schematic representation of the surface structural evolution. b Aberration-corrected high-angle annular dark-field scanning transmission electron microscopy image of LaFeO3-20 along with corresponding showcasing diagram of La content in first four layers. (Lavac. stands for subsurface La vacancy).
The inherent reason for generating subsurface La vacancies was studied by analyzing the structural evolution of LaFeO3 during CH4-CO2 redox treatment. It was found that, as shown in Fig. S11, when LaFeO3 was reduced by CH4 (marked as CH4-Re), some Fe cations were reduced to metallic Fe0 with a production of La2O3 oxides. Subsequent oxidation by CO2 could recover the lattice oxygen. After 4 min of oxidation (marked as CO2-Ro-4min), it was found that no signals for Fe0 (or FeOx) could be observed while that of La2O3 can be clearly detected, suggesting that the access of Fe cations back to LaFeO3 perovskite matrix proceeds much faster than that of La cations. This is mainly due to the slow rate of A-site cation migration (cation size is much larger than that of B-site cation), a rate-controlling step for the formation of an ideal perovskite crystal structure with an A/B ratio of 141,42, which induced the formation of subsurface La vacancies in LaFeO3 during chemical looping processes.
Theoretical insights into the effect of subsurface La on oxygen activity
To gain insight into the effect of La cation engineering on oxygen activity, density functional calculations (DFT) were further conducted. There are two different possible surface structures for LaFeO3: the Fe-O terminated surface and the La-O terminated surface (Fig. S12). As shown in Table S2, although the La-O terminated surface is relatively more stable, it is almost inert for the methane partial oxidation reaction43. Considering the high performance of lanthanum ferrite, the Fe-O terminated surface was used as the model structure due to its much better activity than the La-O terminated surface for methane conversion. As for the location of La vacancies, there are four different sites could be identified (Fig. S13), i.e., La1vac, La2vac, La3vac, and La4vac, because of the two different O sites in the LaFeO3 structure, i.e., top O (coordinated with La1 and La2) and bottom O (coordinated with La3 and La4). According to the structural symmetry, La1vac and La3vac are equivalent to La2vac and La4vac, respectively. As tabulated in Table S3, the total energy of LaFeO3 with La1vac and total energy with La3vac are almost the same, indicating single La vacancy is possibly generated in both La1vac and La3vac sites. However, the top O shows lower O vacancy formation energy than the bottom O in a single La vacancy structure for LaFeO3, suggesting that the top O is conducive to activate methane (Table S4). Consequently, the LaFeO3 with La1vac and La2vac was used as the models for the following calculations. Additionally, we present the results calculated based on the bottom O (Figs. S14, 15 and Table S5) for better reference.
Deformation of surface structure with a concentration of Lasub. cations were first studied and displayed in Fig. 6a–c. It is found that the Fesurface-O-Fesurface motif (coordinated with the top O) bears a bond angle of 171.6o with an average Fe-O bond length of 1.930 Å for stoichiometric LaFeO3 with two Lasub. cations located beneath the surface oxygen. Once one Lasub. was removed, out-of-plane movement occurs to the oxygen due to degraded Lasub.-O interaction, leading to a decreased Fesurface-O-Fesurface bond angle to 153.9o with reduced average Fe-O bond length of 1.880 Å. As for oxygen without adjacent Lasub. (two Lavac.), the bond angle further reduced to 126.1o while the average Fe-O bond length reduced to 1.806 Å. These results suggest that the Lasub. can exert great influence on the chemical state of surface Fe and O.
Fig. 6. Theoretical investigations on the effect of subsurface La vacancies.

a–c Surface structure changes with Lasub. vacancy concentration, d TDOS of Fe and O bands for different Lasub. vacancy concentration, e charge-transfer energy with corresponding band centers of unoccupied Fe 3d and occupied O 2p states for different Lasub. vacancy concentration, f–h computational model of oxygen vacancy formation in the bulk of different Lasub. vacancy concentration, and i comparison of energy profile for CH4 activation over oxygen coordinated with different number of Lasub. vacancies. Lavac. stands for subsurface La vacancy; Lasub. stands for subsurface La.
The evolution of La coordination on Fesurface-O interaction was verified by the analysis of the total density of state (TDOS), charge transfer energy from O 2p to Fe 3d band, and the Bader charge. As shown in Fig. 6d, the overlap of Fe 3d and O 2p orbitals in energy interval from −8 to 0 is detected, which is characteristic of the covalent feature of Fe-O bond. Corresponding charge-transfer energy was calculated based on the center of Fe and O bands44,45. It was found that the charge-transfer energy (Fig. 6e) was notably reduced from 4.87 eV (no Lavac.) to 4.31 eV (two Lavac. sites) with increasing the La vacancies, highlighting the enhanced Fe-O covalency due to weakened Lasub.-O interaction. Consequently, the Bader charge (Table S6) of Fe was gradually increased from +1.64 (no Lavac.) to +1.67 (two Lavac. sites), while that of O slightly decreased from −1.11 (no Lavac.) to −1.02 (two Lavac. sites), which is in line with the XPS analysis (Fig. 3). Oxygen mobility is one key parameter that determines the redox properties of metal oxides. To quantitatively evaluate the influence of subsurface La cations on oxygen mobility, the variation of oxygen vacancy formation energy (Ev) with the number of Lasub. was calculated. As for stoichiometric LaFeO3, Ev of 3.25 eV needs to be conquered to remove the surface lattice oxygen coordinated with two Fesurface and two Lasub. cations. When one Lavac. exists, the value of Ev is notably reduced to 1.37 eV, which is further lowered to 0.79 eV for lattice oxygen with two adjacent Lavac. sites (Fig. 6f–h). These results demonstrate that it is indeed the intimate Lasub.-O interaction, instead of Fesurface-O, plays a dominant role in determining the Ev value and renders a high stability of surface oxygen. Therefore, generating subsurface La vacancies would greatly improve the surface oxygen mobility.
The above results demonstrate that engineering the concentration of La cations at the subsurface is efficient in modulating the oxygen mobility and the Fesurface-O interaction, which would substantially influence the catalytic performance. Such evolution of the oxygen state on the methane activation is then evaluated. It was found that the H-atom of CH4 prefers to adsorb on the lattice oxygen, while the generated *CH3 is stabilized by the adjacent Fe cation. The adsorption energy H-atom, one main driving force of methane activation, notably increased with removing the Lasub. cations from −1.43 eV (no Lavac.) to −2.04 eV (two Lavac. sites), as seen in Table S6, suggesting that the reduction of electron density over O-atom can intensify the hydrogen adsorption process. Consequently, homolytic cleavage of the C-H bond, generally accepted as the rate-determining step for CH4 conversion, is preferred over La-deficient catalysts and the activation barrier notably reduced from 1.88 eV (no Lavac.) to 1.03 eV (two Lavac. sites), as deposited in Fig. 6i and Table S746,47. Besides, it was found that the energy barriers for cleavage of other C-H bonds in *CH3 or generation of syngas (H2 and CO) are obviously reduced after generating subsurface La vacancies, which should be ascribed to the reduced electron intensity over lattice oxygen and the declined oxygen vacancy formation energy (Fig. 6i and Table S7). Overall, the calculation results signify that the generation of subsurface La vacancies represents an effective method for altering the redox performance of perovskite catalysts and can potentially promote oxygen reactivity for methane conversion.
Discussion
According to previous studies, selective anaerobic oxidation of CH4 by redox catalysts proceeds through C-H bond cleavage (CH4 → *C + 2H2) and oxidization of C-atom by lattice oxygen (*C + OL → CO + [Ovac.]), substantially giving H2 and CO as the products21. Selection of suitable redox catalysts with balanced activity for breaking the C-H bond and oxidizing the deposited C-atom is the key to an efficient methane-to-syngas process5,48,49. The B-O redox pair in perovskite catalysts was generally regarded as the active centers for methane conversion, triggering a strong passion to promote the redox performance by terminating catalyst surface by more B-O pair (instead of A-O) or regulating the B-O interaction via asymmetric B1-O-B2 interaction.
As for lanthanum ferrite, the redox capability of the Fe-O pair is superior to the La-O pair, rendering the catalysts terminated mainly by Fe cations more active for methane conversion. This also explains the low reactivity of the La-excess La1.03FeO3 sample for methane anaerobic oxidation. However, it is noticed that the samples of LaFeO3−3, LaFeO3-20, and La0.97FeO3, although bearing similar percentages of Fe cations at the surface and specific surface area, display distinct performance. After three redox cycles, the percentage of surface Fe cations increased from 20% (fresh LaFeO3) to 58% (LaFeO3−3), while the syngas yield notably declined from 4.7 mmol/g (LaFeO3) to 1.8 mmol/g (LaFeO3-3), which was mainly induced by the notable decline of BET surface area (from 5.8 of LaFeO3 to 1.3 m2/g of LaFeO3-3) (Fig. S16 and Table S8). When prolonging the tests to 20 cycles, substantial change in specific surface area was insignificant (from 1.3 m2/g of LaFeO3-3 to 1.8 m2/g of LaFeO3−20), while a slight decline of surface Fe percentage from 58 to 50% occurs due to continuous removal of La cations from the perovskite matrix (Fig. 7). However, the syngas yield was notably improved by 2.5 times (from 1.8 mmol/g of LaFeO3-3 to 4.6 mmol/g of LaFeO3-3). Meanwhile, the La0.97FeO3 sample with 3% La-deficiency that bears 56% surface Fe cations (Fig. S17) and a similar surface area of 2.2 m2/g gives a syngas yield of 5.2 mmol/g, which is 2.9 times higher than the LaFeO3-3 sample. Combined with the experimental studies and DFT calculations, it is found that subsurface La vacancies can be generated by conducting redox treatment, such as the chemical looping process, or by lowering the La/Fe ratio. The absence of subsurface La (electron donor), on the one hand, reduces the electron density over surface oxygen (Table S6), which is favorable for hydrogen (from CH4) adsorption, lowering the energy barrier for homolytic cleavage of the C-H bond. On the other hand, the absence of strong Lasub.-O interaction greatly promotes oxygen mobility, which contributes to oxidizing the C-atom of CH4 to CO, substantially leading to significantly enhanced performance for methane anaerobic oxidation to syngas.
Fig. 7.
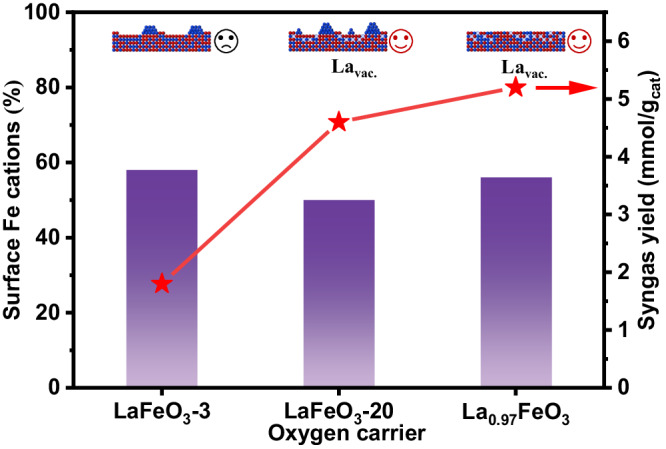
Correlation between surface Fe percentage and corresponding methane anaerobic oxidation performance for LaFeO3-3, LaFeO3−20, and La0.97FeO3. Lavac. stands for subsurface La vacancy.
In summary, we propose the finding that generating subsurface La vacancy for lanthanum ferrite can directly tune the electronic state of lattice oxygen, which ultimately activates the lattice oxygen to enhance the redox performance in methane anaerobic oxidation reaction. The syngas yield over the sample with subsurface La vacancy is 2.9 times larger than that of stoichiometric LaFeO3−3. Combined characterizations and DFT calculations show the formation of subsurface La vacancy can lower the electron density over surface lattice oxygen, which induces a facile homolytic cleavage of the C-H bond. Besides, the absence of Lasub.-O interaction could reduce the oxygen vacancy formation energy, contributing to improve the oxygen mobility and oxidation of deposited C-atom. Consequently, the methane-to-syngas conversion is notably promoted over the La-deficient lanthanum ferrites. This strategy of generating subsurface A-site vacancies would be useful for designing advanced redox catalysts towards reactions proceeded via the Mars−van Krevelen mechanism and chemical looping processes.
Methods
Catalyst preparation
The LaxFeO3 (x = 0.97, 1, 1.03) samples were prepared via the combustion method. Taking LaFeO3 as an example, firstly, 0.87 g of La(NO3)3·6H2O, 0.81 g of Fe(NO3)3·9H2O, and 0.60 g of glycine (the molar ratio of citric acid/total metal ions was 2) were dissolved in 1.2 g of water. The solution is then dried in an oven at a constant temperature of 60 °C for 1 h, which were then burned in a muffle furnace for 10 min at 500 °C. Finally, the obtained powders were crushed into powder and calcined at 850 °C for 4 h.
Characterization
The X-ray diffraction (XRD) patterns were acquired using a PANalytical X’Pert-Pro powder X-ray diffractometer (U = 40 kV, I = 40 mA) with Cu Kα radiation of λ = 0.15418 nm to analyze the crystal structure and phase composition of fresh samples. The Raman spectrum was collected by a Raman-atomic force microscope imaging analysis system with an incident wavelength of 532 nm and a scan range of 100–500 cm−1. The morphology of these catalysts was observed by field emission scanning electron microscopy (JEOL JSM-7800F SEM). The high-resolution transmission electron microscopy (HRTEM) images and elemental distribution of the samples were obtained by transmission electron microscopy (JEOL JEM-2100F TEM). Atomic column high-angle annular dark-field scanning transmission electron microscopy (AC-HAADF-STEM) analysis was conducted using a JEOL JEM-ARM200F microscope fitted with a CEOS probe corrector to achieve a resolution of 0.08 nm. The specific surface area was obtained by calculating the results of the QUADRSORB SI analyzer using the Brunauer–Emmett–Teller (BET) method. Low energy ion scattering (LEIS) was conducted on an ION-TOF Qtac100 for surface compositional analysis and depth profiling. Before the analysis, the sample was treated at a temperature of 300 °C for 1 h to remove the surface contaminant, and the charge neutralization technique was used for spectral acquisition and sputtering. With Ne+ as the detection source, different sputtering depths were calculated by adjusting the sputtering time. X-ray photoelectron spectroscopy (XPS) was tested on a Thermo Fisher Nexsa-type photoelectron spectrometer. To avoid the influence of air exposure on the analysis results, the samples after the chemical looping reaction (oxidized by CO2) were cooled down to room temperature in He atmosphere, which were then moved into a well-closed XPS transfer bin in the glove box (Ar protected). The elemental composition and ionic state of the surface samples were analyzed using a monochromatic Al target Kα, and the binding energy was calibrated according to the contaminated carbon-C 1s (284.8 eV). The oxygen activity of fresh samples was studied by H2 temperature-programed reduction (H2-TPR) over Micromeritics Autochem II 2920 apparatus. Specifically, 200 mg of sample was placed in a quartz tube and the temperature was gradually increased to 900 °C at a rising rate of 10 °C /min in a 10% H2/Ar mixture (50 mL/min). The CH4-TPR experiments were carried out in a fixed bed reactor with 100 mg of 60–80 mesh catalyst in a 6 mm quartz tube, ramped at 10 °C/min until 900 °C in 5% CH4/He (30 mL/min).
Isothermal redox reactions
Experiments to evaluate the redox activity and stability of these catalysts were carried out on an atmospheric pressure fixed bed reactor. Typically, the fresh sample of 100 mg (60–80 mesh) was placed in a quartz tube with an inner diameter of 6 mm. A successive CH4 reduction-CO2 regeneration redox cycles were performed at 900 °C. The catalyst were reduced in a 5% CH4/He atmosphere for 8 min (15 mL/min) before being regenerated in a 5% CO2/He atmosphere for 10 min (15 mL/min). Between reduction and reoxidation reaction, the reactor was purged with He for 4 min (20 ml/min). In addition, the products were analyzed using an online quadrupole mass spectrometer (IPI GAM 200). Before the isothermal redox reaction, a standard gas with known composition is used to calibrate the mass spectrometer. The average CH4 conversion (XCH4), CO selectivity (SCO), syngas yield (Ysyngas), oxygen output (Ot), H2/CO ratio (R) for methane anaerobic oxidation step, and CO2 conversion (XCO2) for CO2 regeneration step was calculated by following equations:
| 1 |
| 2 |
| 3 |
| 4 |
| 5 |
| 6 |
The term nx represents the amount of the corresponding gas x (mol) flowing into or out of the reactor, and m is the mass of the catalyst used.
Density functional theory calculations
We used the density functional theory (DFT) calculations with the Vienna ab initio simulation package (VASP) software. For the core electrons, we utilized the projector augmented wave (PAW) method and the Perdew–Burke–Ernzerhof (PBE) exchange-correlation function to describe them50. The wavefunctions were expanded on a plane-wave basis, corresponding kinetic energy cutoff was set to 400 eV. We used a 3 × 3 × 1 gird to employ the gamma-centered k-point mesh. To deal with the strong Coulomb interactions of Fe 3d electrons, we carried out the DFT + U method to revise it51. The geometric optimization was known as convergent only when the energy change was smaller than 0.02 eV Å−1. Also, only the energy change was less than 10−5 eV, and the electron energy was considered self-consistent. When the energy and force were reached to the expected outcome simultaneously, the energy of optimized structures were considered obtained. To avoid the interaction between the periodic images in the z-direction, a vacuum gap of 15 Å was set. Apart from the top two layers, the others were fixed to ensure the geometry to be constant during the optimization process. The adsorption energy (Eads) for adsorbate can be calculated in the form below equation:
| 7 |
Ead/sub is the electronic energy of the optimized adsorbate/substrate system. The Ead and Esub denote the energies of the sole adsorbate and the clean substrate, respectively. Moreover, vacancy formation energy (Evac) was determined by equation:
| 8 |
where Edefect-surface, EO2, and Esurface represent the total energies of the defective surface system, the O2, and the complete surface system, respectively. The total orbital band center εtot was obtained through equation:
| 9 |
where ntot (ε) and ε indicate the value of the total orbital density of states (TDOS) and the energy of the total orbital. To find the ion’s relative valence, we calculated the Bader charge, and then through normalizing the number of valence electrons, we can obtain the relevant oxidation states. The relative distance between the occupied O 2p center and the unoccupied Fe 3d center were calculated to represent the dimension of Fe-O covalence, which is also can be proved by the relative size of the Bader charge. The climbing-image nudged elastic band (CI-NEB) method was employed to find the transition states (TS) roughly with a force convergence of 0.5 eV Å−1 and an energy convergence of 10−5 eV. Then, the precise outcomes were searched by the dimer method with a force convergence of 0.05 eV Å−1 and an energy convergence of 10−7 eV.
Supplementary information
Acknowledgements
Financial support from the National Natural Science Foundation of China (grant No. 21706254 (C.H.), 22378331 (Y.Z.), and 22178337 (X.W.)), NSFC Center for Single-Atom Catalysis (grant No. 22388102 (X.W.)), and Youth Innovation Promotion Association, CAS (grant No. 2023189 (C.H.)) are gratefully acknowledged. Furthermore, the authors would like to extend sincere gratitude to Engineer Xiaoli Pan for their invaluable assistance with the electron microscopy work.
Author contributions
J.H.: Investigation, methodology, data curation, validation, and writing—original draft. T.W.: DFT calculation, formal analysis, and software. X.B.: Data curation and formal analysis. Y.T.: Data curation and formal analysis. C.H.: Conceptualization, formal analysis, supervision, writing—review and editing, funding acquisition, and project administration. W.X.: Methodology and validation. Y.H.: Investigation and formal analysis. Z.W.: Software and investigation. B.J.: Methodology and formal analysis. Y.G.: Formal analysis. Y.Z.: Funding acquisition, methodology, supervision, review, and editing. X.W.: Funding acquisition, project administration, supervision, review, and editing.
Peer review
Peer review information
Nature Communications thanks Rajaram Bal and Zhuo Cheng for their contribution to the peer review of this work. A peer review file is available.
Data availability
The data that support the findings of this study are all available within the paper and its Supplementary Information.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Jiahui He, Tengjiao Wang.
Contributor Information
Chuande Huang, Email: huangchuande@dicp.ac.cn.
Bo Jiang, Email: bjiang@dlut.edu.cn.
Yanyan Zhu, Email: zhuyanyan@nwu.edu.cn.
Xiaodong Wang, Email: xdwang@dicp.ac.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-49776-y.
References
- 1.Kitano M, et al. Low-temperature synthesis of perovskite oxynitride-hydrides as ammonia synthesis catalysts. J. Am. Chem. Soc. 2019;141:20344–20353. doi: 10.1021/jacs.9b10726. [DOI] [PubMed] [Google Scholar]
- 2.Ruiz Puigdollers A, Schlexer P, Tosoni S, Pacchioni G. Increasing oxide reducibility: the role of metal/oxide interfaces in the formation of oxygen vacancies. ACS Catal. 2017;7:6493–6513. doi: 10.1021/acscatal.7b01913. [DOI] [Google Scholar]
- 3.Zhu X, et al. A tailored multi-functional catalyst for ultra-efficient styrene production under a cyclic redox scheme. Nat. Commun. 2021;12:1329–11339. doi: 10.1038/s41467-021-21374-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fan Q, et al. Breaking the stoichiometric limit in oxygen-carrying capacity of Fe-based oxygen carriers for chemical looping combustion using the Mg-Fe-O solid solution system. ACS Sustain. Chem. Eng. 2022;10:7242–7252. doi: 10.1021/acssuschemeng.2c00271. [DOI] [Google Scholar]
- 5.Liu Y, et al. Near 100% CO selectivity in nanoscaled iron-based oxygen carriers for chemical looping methane partial oxidation. Nat. Commun. 2019;10:5503–5508. doi: 10.1038/s41467-019-13560-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jie Yang, et al. On the ensemble requirement of fully selective chemical looping methane partial oxidation over La-Fe-based perovskites. Appl. Catal. B. 2022;301:120788–120792. doi: 10.1016/j.apcatb.2021.120788. [DOI] [Google Scholar]
- 7.Hwang J, et al. Perovskites in catalysis and electrocatalysis. Science. 2017;358:751–756. doi: 10.1126/science.aam7092. [DOI] [PubMed] [Google Scholar]
- 8.Wang K, et al. Perovskite oxide catalysts for advanced oxidation reactions. Adv. Funct. Mater. 2021;31:2102089. doi: 10.1002/adfm.202102089. [DOI] [Google Scholar]
- 9.Neal LM, Shafiefarhood A, Li FX. Dynamic methane partial oxidation using a Fe2O3@La1.8Sr0.2FeO3-δ core-shell redox catalyst in the absence of gaseous oxygen. ACS Catal. 2014;4:3560–3569. doi: 10.1021/cs5008415. [DOI] [Google Scholar]
- 10.Sun Z, et al. Chemical looping-based energy transformation via lattice oxygen modulated selective oxidation. Prog. Energy Combust. Sci. 2023;96:101045. doi: 10.1016/j.pecs.2022.101045. [DOI] [Google Scholar]
- 11.Chen S, et al. Modulating lattice oxygen in dual-functional Mo−V−O mixed oxides for chemical looping oxidative dehydrogenation. J. Am. Chem. Soc. 2019;141:18653–18657. doi: 10.1021/jacs.9b09235. [DOI] [PubMed] [Google Scholar]
- 12.Wang C, et al. Near 100% ethene selectivity achieved by tailoring dual active sites to isolate dehydrogenation and oxidation. Nat. Commun. 2021;12:5447. doi: 10.1038/s41467-021-25782-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jia C, et al. Shifting oxygen evolution reaction pathway via activating lattice oxygen in layered perovskite oxide. Adv. Funct. Mater. 2023;33:2301981. doi: 10.1002/adfm.202301981. [DOI] [Google Scholar]
- 14.Min Lu, et al. Artificially steering electrocatalytic oxygen evolution reaction mechanism by regulating oxygen defect contents in perovskites. Sci. Adv. 2022;8:eabq3563. doi: 10.1126/sciadv.abq3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Han M, et al. Tuning of oxygen electrocatalysis in perovskite oxide nanoparticles by the cationic composition. ACS Catal. 2023;13:5733–5743. doi: 10.1021/acscatal.3c00461. [DOI] [Google Scholar]
- 16.Wu J, et al. Calcium and copper substitution in stoichiometric and La-deficient LaFeO3 compositions: a starting point in next generation of Three-Way-Catalysts for gasoline engines. Appl. Catal. B. 2021;282:119621. doi: 10.1016/j.apcatb.2020.119621. [DOI] [Google Scholar]
- 17.Meier M, et al. CO oxidation by Pt2/Fe3O4: metastable dimer and support configurations facilitate lattice oxygen extraction. Sci. Adv. 2022;8:eabn4580. doi: 10.1126/sciadv.abn4580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Grabowska E. Selected perovskite oxides: characterization, preparation and photocatalytic properties—a review. Appl. Catal. B. 2016;186:97–126. doi: 10.1016/j.apcatb.2015.12.035. [DOI] [Google Scholar]
- 19.Jiang B, et al. Iron–oxygen covalency in perovskites to dominate syngas yield in chemical looping partial oxidation. J. Mater. Chem. A. 2021;9:13008–13018. doi: 10.1039/D1TA02103F. [DOI] [Google Scholar]
- 20.Gao Y, et al. A-site vacancy induced electronic engineering of perovskite for synergistic modulation of redox activity and magnetocaloric effect. Nano Energy. 2023;117:108912. doi: 10.1016/j.nanoen.2023.108912. [DOI] [Google Scholar]
- 21.Chang W, et al. Asymmetric coordination activated lattice oxygen in perovskite ferrites for selective anaerobic oxidation of methane. J. Mater. Chem. A. 2023;11:4651–4660. doi: 10.1039/D2TA09187A. [DOI] [Google Scholar]
- 22.Si W, Wang Y, Peng Y, Li J. Selective dissolution of A‐site cations in ABO3 perovskites: a new path to high‐performance catalysts. Angew. Chem. Int. Ed. 2015;54:7954–7957. doi: 10.1002/anie.201502632. [DOI] [PubMed] [Google Scholar]
- 23.Liu F, et al. Promoting nitrate electroreduction to ammonia over A-site deficient cobalt-based perovskite oxides. J. Mater. Chem. A. 2023;11:10596–10604. doi: 10.1039/D3TA01063E. [DOI] [Google Scholar]
- 24.Chang H, et al. Effects of oxygen mobility in La–Fe-based perovskites on the catalytic activity and selectivity of methane oxidation. ACS Catal. 2020;10:3707–3719. doi: 10.1021/acscatal.9b05154. [DOI] [Google Scholar]
- 25.Donat F, Müller CR. CO2-free conversion of CH4 to syngas using chemical looping. Appl. Catal. B. 2020;278:119328. doi: 10.1016/j.apcatb.2020.119328. [DOI] [Google Scholar]
- 26.He F, et al. The use of La1−xSrxFeO3 perovskite-type oxides as oxygen carriers in chemical-looping reforming of methane. Fuel. 2013;108:465–473. doi: 10.1016/j.fuel.2012.11.035. [DOI] [Google Scholar]
- 27.Zhang XH, et al. FeO6 octahedral distortion activates lattice oxygen in perovskite ferrite for methane partial oxidation coupled with CO2 splitting. J. Am. Chem. Soc. 2020;142:11540–11549. doi: 10.1021/jacs.0c04643. [DOI] [PubMed] [Google Scholar]
- 28.Tamai K, et al. Dynamics of the lattice oxygen in a Ruddlesden–Popper-type Sr3Fe2O7−δ catalyst during NO oxidation. ACS Catal. 2020;10:2528–2537. doi: 10.1021/acscatal.9b03857. [DOI] [Google Scholar]
- 29.Yang Q, et al. Boosted carbon resistance of ceria-hexaaluminate by in-situ formed CeFexAl1−xO3 as oxygen pool for chemical looping dry reforming of methane. Appl. Catal. B. 2023;330:122636. doi: 10.1016/j.apcatb.2023.122636. [DOI] [Google Scholar]
- 30.Zeng D, et al. Spatially controlled oxygen storage materials improved the syngas selectivity on chemical looping methane conversion. Appl. Catal. B. 2021;281:119472. doi: 10.1016/j.apcatb.2020.119472. [DOI] [Google Scholar]
- 31.Zeng D, et al. Tuning the support properties toward higher CO2 conversion during a chemical looping scheme. Environ. Sci. Technol. 2020;54:12467–12475. doi: 10.1021/acs.est.0c01702. [DOI] [PubMed] [Google Scholar]
- 32.Huang CD, et al. In situ encapsulation of iron(0) for solar thermochemical syngas production over iron-based perovskite material. Commun. Chem. 2018;1:55. doi: 10.1038/s42004-018-0050-y. [DOI] [Google Scholar]
- 33.Nandi S, et al. Mn- or Cu- substituted LaFeO3-based three-way catalysts: highlighting different catalytically operating modes of La0.67Fe0.8M0.2O3 (M=Cu, Mn) Appl. Catal. B. 2021;296:120330. doi: 10.1016/j.apcatb.2021.120330. [DOI] [Google Scholar]
- 34.Faye J, et al. Influence of lanthanum stoichiometry in La1−xFeO3−δ perovskites on their structure and catalytic performance in CH4 total oxidation. Appl. Catal. B. 2012;126:134–143. doi: 10.1016/j.apcatb.2012.07.001. [DOI] [Google Scholar]
- 35.Gao Y, Haeri F, He F, Li F. Alkali metal-promoted LaxSr2–xFeO4−δ redox catalysts for chemical looping oxidative dehydrogenation of ethane. ACS Catal. 2018;8:1757–1766. doi: 10.1021/acscatal.7b03928. [DOI] [Google Scholar]
- 36.Kang BS, Matsuda J, Ju YW, Kim HH, Ishihara T. Nano strain induced double columnar oxide as highly active oxygen-dissociation electrode for Ni-Fe metal supported solid oxide fuel cells. Nano Energy. 2019;56:382–390. doi: 10.1016/j.nanoen.2018.11.074. [DOI] [Google Scholar]
- 37.Druce J, et al. Surface termination and subsurface restructuring of perovskite-based solid oxide electrode materials. Energy Environ. Sci. 2014;7:3593–3599. doi: 10.1039/C4EE01497A. [DOI] [Google Scholar]
- 38.Gong S, et al. Highly active and humidity resistive perovskite LaFeO3 based catalysts for efficient ozone decomposition. Appl. Catal. B. 2019;241:578–587. doi: 10.1016/j.apcatb.2018.09.041. [DOI] [Google Scholar]
- 39.Weber MC, et al. Raman spectroscopy of rare-earth orthoferrites RFeO3 (R=La, Sm, Eu, Gd, Tb, Dy) Phys. Rev. B. 2016;94:214103. doi: 10.1103/PhysRevB.94.214103. [DOI] [Google Scholar]
- 40.Xia X, et al. Oxygen activity tuning via FeO6 octahedral tilting in perovskite ferrites for chemical looping dry reforming of methane. ACS Catal. 2022;12:7326–7335. doi: 10.1021/acscatal.2c00920. [DOI] [Google Scholar]
- 41.Buscaglia MT, Bassoli M, Buscaglia V, Vormberg R. Solid‐state synthesis of nanocrystalline BaTiO3: reaction kinetics and powder properties. J. Am. Ceram. Soc. 2008;91:2862–2869. doi: 10.1111/j.1551-2916.2008.02576.x. [DOI] [Google Scholar]
- 42.Huang C, Wang X, Liu X, Tian M, Zhang T. Extensive analysis of the formation mechanism of BaSnO3 by solid-state reaction between BaCO3 and SnO2. J. Eur. Ceram. Soc. 2016;36:583–592. doi: 10.1016/j.jeurceramsoc.2015.11.001. [DOI] [Google Scholar]
- 43.Wu M, Ma S, Chen S, Xiang W. Fe–O terminated LaFeO3 perovskite oxide surface for low temperature toluene oxidation. J. Clean. Prod. 2020;277:123224. doi: 10.1016/j.jclepro.2020.123224. [DOI] [Google Scholar]
- 44.Sun Y, et al. Unfolding B-O-B bonds for an enhanced ORR performance in ABO3‐type perovskites. Small. 2018;15:1803513. doi: 10.1002/smll.201803513. [DOI] [PubMed] [Google Scholar]
- 45.Hong WT, et al. Charge-transfer-energy-dependent oxygen evolution reaction mechanisms for perovskite oxides. Energy Environ. Sci. 2017;10:2190–2200. doi: 10.1039/C7EE02052J. [DOI] [Google Scholar]
- 46.Wang Y, Hu P, Yang J, Zhu Y-A, Chen D. C–H bond activation in light alkanes: a theoretical perspective. Chem. Soc. Rev. 2021;50:4299–4358. doi: 10.1039/D0CS01262A. [DOI] [PubMed] [Google Scholar]
- 47.Fung V, Polo-Garzon F, Wu Z, Jiang D-E. Exploring perovskites for methane activation from first principles. Catal. Sci. Technol. 2018;8:702–709. doi: 10.1039/C7CY01791J. [DOI] [Google Scholar]
- 48.Mihai O, Chen D, Holmen A. Chemical looping methane partial oxidation: the effect of the crystal size and O content of LaFeO3. J. Catal. 2012;293:175–185. doi: 10.1016/j.jcat.2012.06.022. [DOI] [Google Scholar]
- 49.Mihai O, Chen D, Holmen A. Catalytic consequence of oxygen of lanthanum ferrite perovskite in chemical looping reforming of methane. Ind. Eng. Chem. Res. 2010;50:2613–2621. doi: 10.1021/ie100651d. [DOI] [Google Scholar]
- 50.Singh S, et al. Study of higher discharge capacity, phase transition, and relative structural stability in Li2FeSiO4 cathode upon lithium extraction using an experimental and theoretical approach and full cell prototype study. ACS Appl. Energy Mater. 2019;2:6584–6598. doi: 10.1021/acsaem.9b01145. [DOI] [Google Scholar]
- 51.Janesko BG. Replacing hybrid density functional theory: motivation and recent advances. Chem. Soc. Rev. 2021;50:8470–8495. doi: 10.1039/D0CS01074J. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are all available within the paper and its Supplementary Information.